Synthesis of Gold Catalysts Supported on Mesoporous Silica Materials: Recent Developments

Abstract
: Mesoporous silica materials (MSM) with ordered and controllable porous structure, high surface area, pore volume and thermal stability are very suitable catalyst supports, because they provide high dispersion of metal nanoparticles and facilitate the access of the substrates to the active sites. Since the conventional wet-impregnation and deposition-precipitation methods are not appropriate for the incorporation of gold nanoparticles (AuNPs) into MSM, considerable efforts have been made to develop suitable methods to synthesize Au/MSM catalysts, because the incorporation of AuNPs into the channel system can prevent their agglomeration and leaching. In this review, we summarize the main methods to synthesize active gold catalysts supported on MSM. Examples and details of the preparative methods, as well as selected applications are provided. We expect this article to be interesting to researchers due to the wide variety of chemical reactions that can be catalyzed by gold supported catalysts.1. Introduction
After the pioneering works of Hutchings [1] and Haruta et al. [2], the high activity of gold catalysts has demonstrated that gold can be the catalyst of choice for an important number of chemical reactions such as selective oxidations and hydrogenations of organic substrates [2–6], water-gas shift reaction [7–9], acetylene hydrochlorination [10,11], direct synthesis of hydrogen peroxide [12], reduction of NO to N2 [13,14] and the addition of nucleophiles to acetylenes [15], among others [16,17]. Although gold has been sometimes alloyed with other metals such as Pd, Cu and Ag, in most cases, gold alone exhibits high and exceptional catalytic activity.
Notwithstanding the origin of the active sites associated to the supported gold catalysts is still under debate because of their very complex nature, and still remains challenging for the homogeneous control of particle size distribution of the supported gold nanoparticles, heterogeneous gold catalysis can be currently considered as a mature topic. As illustrated in Figure 1, the published papers on the topic of gold catalysis have augmented exponentially in the last 20 years, and the rate of publication shows no declining signs. Moreover, the numerous reviews covering a wide range of applications of gold catalysts and the most relevant aspects of gold chemistry [17–30] have provided a huge and fascinating understanding of catalysis by gold. From all this available literature, nowadays it is well known that the catalytic activity of gold catalysts is highly influenced by [31–33]: (i) the size and shape of gold particles; (ii) the catalyst synthesis methods; (iii) the nature of support; (iv) the gold-support interface interactions; and (v) the oxidation state(s) of gold in the synthesized catalysts. Furthermore, it has been reported that the synthesis of gold catalysis by the conventional incipient wetness impregnation method yields AuNPs larger than 30 nm, due to the weak interaction of the most commonly used gold precursor (HAuCl4) with the metal oxide support. The chloride remaining on the support promotes the aggregation of AuNPs and may poison the active sites of the catalyst [18,22,34]. Consequently, numerous papers describing different methods to incorporate AuNPs on a variety of metal oxides, including TiO2, Al2O3, Fe2O3, CeO2, Co3O4, ZrO2, MgO and SiO2 have been published by different research groups to overcome this problem [18,35,36]. Depending on the metal oxide support, gold catalysts can be synthesized mainly by deposition-precipitation, co-precipitation, colloidal dispersion and gas- and liquid-phase grafting of organo-gold complexes. Deposition-precipitation has been the most used method for the preparation of gold catalysts supported on metal oxides having high IEP, such as TiO2, Fe2O3, Al2O3, ZrO2 and CeO2 [36]. However, in the case of supports having low IEP, such as SiO2 (IEP ∼2), the deposition-precipitation method is not appropriate for the incorporation of AuNPs, because under the high pH conditions required to hydrolyze the HAuCl4, commonly used as gold precursor, the weak interaction between the negatively charged silica surface and the [Au(OH)nCl4−n]− species hinders the gold adsorption and facilitates the mobility of AuNPs, which can sinter easily during the synthesis process, especially during the calcination step, yielding low gold loadings and inactive catalysts [37].
Mesoporous silica materials with ordered and controllable porous structure, excellent mechanical properties, high surface area, pore volume and thermal stability are very suitable catalyst supports, because they provide high dispersion of metal nanoparticles and facilitate the access of the substrates to the active sites [39]. Consequently, considerable efforts have been made for developing suitable methods to synthesize gold catalysts supported on these materials, because the incorporation of AuNPs into the channel system can prevent their agglomeration and leaching.
In principle, gold can be loaded into MSM during or after the synthesis of the mesoporous silica support [40]. The most used methods for preparing Au/MSM catalysts include the modification of the MSM support with organic functional groups by post-grafting or co-condensation before gold loading [41,42], one-pot synthesis by the incorporation of both gold and the coupling agent containing functional groups into the MSM synthesis [43], the use of cationic gold complexes such as [Au(en)2]Cl3 (en = ethylenediamine) [44], chemical vapor deposition using expensive organometallic gold precursors [45], synthesis of the MSM in the presence of gold colloids [46,47], dispersion of gold colloids protected by ligands or polymers onto SiO2 [46,47], and modification of the mesoporous SiO2 supports with other metal oxides, such as TiO2, Al2O3 and CeO2 to prepare SiO2-based gold catalysts [48,49].
Although there are several published reviews on different aspects of gold catalysis [17–30], as mentioned previously, to the best of our knowledge the synthesis of gold catalysts supported on MSM have not been extensively reviewed. Only two interesting contributions focusing on the preparation of Au/SiO2 catalysts, mainly for CO oxidation, have been published by Ma and Dai [35] and Ma et al. [36]. Herein we reviewed the main methods to synthesize gold catalysts supported on MSM, as well as some selected examples of their catalytic applications, in order to contribute to the fascinating topic of catalysis by gold. Because of the large number of available literature and applications, neither the characterization methods of the synthesized gold catalysts nor the physical chemistry of the catalytic reactions is covered in this review. Readers interested on these topics can find useful information in this special issue of Catalysts, in the recent thematic issue of the Chemical Society Reviews (2008 Gold: Chemistry, Materials and Catalysis issue), and in the current specific literature [5,35,40,50].
2. Methods to Synthesize Active Gold Catalyst Supported on MSM
2.1. Postsynthetic Functionalization of MSM (Grafting) Before Gold Loading
The subsequent modification of the silica surface with organic functional groups (commonly named grafting [51]) prior to the gold loading, has been a surface-engineering strategy to promote the interaction between the frequently used HAuCl4 gold precursor and the mesoporous silica support, which lead to avoid the mobility and aggregation of the AuNPs on the silica surfaces. After functionalization, the MSM adsorb easily the AuCl4− ions, by the formation of a monolayer of positively charged groups on the pore surface. Upon calcination or chemical reduction with NaBH4, the metallic gold precursor evolves in highly dispersed metal AuNPs. The general procedure to synthesize gold catalysts following this approach, summarized in Figure 2, consists normally of two general steps: (i) the grafting of the stabilizing ligands into the inner surfaces of the mesoporous silica material; and (ii) the gold loading. The first step consists in the reaction of a suitable organic functional group (usually organosilanes containing amine or thiol groups) with the mesoporous silica support using an appropriate solvent (normally toluene or ethanol) in a refluxing system under nitrogen [20,52,53]. The resulting solids are subsequently recovered by filtration, washed with the solvent and dried. In the second step, the functionalized MSM are dispersed in a yellow solution of the gold precursor (commonly HAuCl4) under vigorous stirring. Then, the solution turns colorless, while the solids become yellow, indicating that the ion-exchange between the gold precursor solution and the functionalized MSM support has been attained. After filtration, the gold catalysts are washed with abundant deionized water to remove the residual chloride ions, dried and subsequently calcined to remove the functional ligands, and to reduce the oxidized Au3+ species to metallic Au0 gold particles strongly attached to the support [18]. The catalysts prepared under this approach, can also be reduced in H2 or chemically using NaBH4 solution [54].
This method has been used for the synthesis of numerous gold catalysts supported on different mesoporous silicas such as MCM-41, MCM-48, SBA-11, SBA-12, SBA-15, SBA-16, HMM-2 and HMS, as summarized in Table 1 and illustrated in Figure 3. Some relevant catalysts synthesized by this method and their catalytic applications are highlighted below.
Chi et al. [54] synthesized AuNPs confined in the channels of MCM-41, MCM-48 and SBA-15 by previous modification of their surface with APS. To do this, the as-synthesized MCM-41 and MCM-48 materials were first acidified with nitric acid solution (pH < 1.0), and then, the as-synthesized SBA-15 and the acidified MCM-41 and MCM-48 silicas were modified with APS in a reflux system for 3–6 h at about 70 °C, using ethanol as solvent. After filtration, washing and drying, the resulting surface-functionalized solids were mixed with HAuCl4 aqueous solution, and the slurry was chemically reduced with NaBH4 solution. The catalysts (Au-loading of 4%wt), activated by calcination at 560 °C with subsequent heating to 600 °C under H2/N2 flow for 1 h, were active for CO oxidation, and showed an average Au particle sizes of 5.1, 6.9 and 5.5 nm on MCM-41, MCM-48 and SBA-15 supports, respectively. The smaller the Au particle size, the higher the catalyst activity. Moreover, the XPS analysis revealed that AuNPs were present on the catalysts in the metallic Au0 state.
Richards group [57] used APS and MPTS as stabilizing ligands, to immobilize AuNPs into mesoporous SBA-15. The resulting gold catalysts (Au-loading ∼4%wt) were highly active for the aerobic oxidation of cyclohexane, and conversions ranging between 21 and 32% after 6 h reaction at 150 °C and 1.5 MPa of oxygen pressure were attained. Although the synthesized Au/SBA-15 catalysts showed smaller surface area and pore size than pure SBA-15, they retained the characteristic isotherm shape of the mesoporous materials, demonstrating that SBA-15 support was stable to the surface modification and gold loading. AuNPs along the channels of the silica walls with mean particle sizes of 2.8 and 6.5 nm were obtained when using APS and MPTS, respectively. According to these authors, the smallest AuNPs were obtained using APS, because the aqueous solution precursor AuCl3OH−can interact strongly with the amine groups, preventing the agglomeration of particles during the synthesis process.
The preparation of dispersed AuNPs in mesoporous SBA-15 using TPTAC as functionalizing agent has been reported by Yang et al. [41,58]. Gold was loaded on the functionalized SBA-15 using HAuCl4 solution as precursor, and the slurry was chemically reduced with NaBH4 aqueous solution. After filtration, washing with water and acetone, and drying in ambient conditions, the catalysts (Au loading of 4.8%wt) showed dispersed metallic (Au0) AuNPs located in the SBA-15 channels, with a mean size of approximately 4.5 nm, which were active for CO oxidation in the temperature interval between 30 and 90 °C. The catalysts reduced under H2 at 200 °C did not show catalytic activity in CO oxidation at room temperature.
Recently, Wei et al. [84] developed a modified external passivation route to control the location and the distribution of AuNPs in periodic mesoporous organosilica. In this approach, the catalysts synthesis method consisted in three main steps: (i) one external passivation of the PMOs with a long chain of hydrophobic n-octadecyltrimethoxysilane, to obtain C18TMS-PMOs material; (ii) the internal amino-functionalization of the C18TMS-PMOs material with aminopropyl groups (APS); and (iii) the Au impregnation of the obtained NH2-C18TMS-PMOs support using HAuCl4, followed by reduction under H2 at 100 °C for 2 h. The synthesized catalysts displayed the typical mesoporous structure and narrow pore size distribution of the PMOs, and were active for the reduction of methylene blue. In this interesting method of synthesis, to avoid the adsorption of AuNPs onto the PMOs external surface, the functionalization with C18TMS was made, because it has no affinity to gold complex. However, to introduce AuNPs into the channels of the PMOs, the internal surface was modified with APS, which significantly enhanced the adsorption of AuNPs with particle sizes between 3 and 5 nm inside the pores of the silica material. This approach is a good example of how the surface-engineering can be used in order to control the location of the AuNPs into MSM.
Since the MSM have not the same structural properties, the Dai's group [70] evaluated the influences of the synthesis conditions and mesoporous structures on the AuNPs supported on different mesoporous silicas (HMS, MCM-41, MCM-48, SBA-15 and SBA-16). The MSM were modified with two different amine ligands (APTS and TPED) using toluene as solvent. The functionalized silica materials were washed several times with a large quantity of ethanol and toluene, and then vacuum-dried at 80 °C for 6 h. Gold was incorporated into the mesopores by mixing HAuCl4 dissolved in water or ethanol with the silica hosts, followed by sonication for 30 min. The resulting materials were finally filtered, vacuum-dried at room temperature and calcined at 200 or 550 °C for 1 h. On the basis of the obtained materials characterization, the following conclusions were drawn: (i) Bigger AuNPs were generated when using diamine instead of monoamine ligand, and when ethanol was used as solvent. Although the explanation of this behavior was related to the fact that the use of diamine ligand could have facilitated a higher uptake of gold and that ethanol could have facilitated the reduction of gold cations, unfortunately the amount of loaded gold on the synthesized materials was not reported; (ii) The gold particle size increases with the calcination temperature, but the extent of sintering is different with different mesoporous hosts; (iii) The size of nanopores may play a role in determining the thermal stability of AuNPs against sintering; (iv) Bicontinuous pore structures such as HMS, MCM-48 and SBA-16 materials could lead to the sintering of gold nanoparticles, whereas one-dimensional pore channels such as MCM-41 and SBA-15 materials could mitigate the sintering of AuNPs. These results were in agreement with the previous works carried out by Bore et al. [56] and Gabaldon et al. [55], who studied the incorporation of gold on a variety of mesoporous silica materials (MCM-48, SBA-11, SBA-12, SBA-15 and HMM-2) with different mesoporous architectures, and concluded that 1-D mesoporous architectures were most effective for controlling Au sizes, than cubic and 3-D mesoporous architectures, because the easier access and connectivity of the 3-D structures facilitates the particle growth, while when using 1-D mesoporous silicas, the amount of free gold that can participate in Ostwald ripening is limited. Moreover, these authors pointed out that among the 1-D pores, the curved pores were better for confining Au particles than the 1-D straight pores, and that small pore sizes are not sufficient to control the Au particle size.
The post grafting functionalization has been also used for synthesize supported bimetallic gold catalysts. For example, a four step grafting method was used by Ma et al. [59] to modify mesoporous SBA-15 before gold loading. Firstly, the as-synthesized SBA-15 was grafted with MPS, and after removing the surfactant template by ethanol extraction and subsequent calcination at 300 °C for 4 h, the MPS functionalized SBA-15 was grafted again with APTS. This double modified material was loaded with both Au and Au-Pd particles, using HAuCl4 and Pd(NO3)2 aqueous solutions, respectively, and calcined at 200 °C for 2 h. Gold and palladium particles penetrate the pores of SBA-15 owing to the high affinity of the –NH groups to the binding metals [59]. The resulting Au/SBA-15 and Au-Pd/SBA-15 catalysts conserved the mesoporous structure of the support, and were active for the oxidation of benzyl alcohol under air flow at 80 °C in alkaline media. According to these authors, the addition of Pd to Au/SBA-15 catalyst decreased the size of AuNPs from 10 to 5 nm, contributed to their uniform dispersion, increased the activity and retained high selectivity towards benzaldehyde. Yang et al. [60] immobilized Au and Au-In nanoparticles on SBA-15 using APS as functionalizing agent. The spherical Au and Au-In particles with average size of around 2 nm were homogeneously dispersed on the mesopore walls of SBA-15, and were highly active and selective for the hydrogenation of crotonaldehyde to crotyl alcohol in liquid phase. Yen et al. [89] used APS to functionalize the surface of mesoporous MCM-41 for the synthesis of bimetallic Au-Ag/MCM-41 catalysts. The catalysts, activated by calcination with subsequent hydrogen reduction at 600 °C, showed Au-Ag bimetallic nanoparticles with sizes ranging between 4 to 6 nm, and exhibited high activity in catalysis for low-temperature CO oxidation with high stability. Moreover, these catalysts were resistant to moisture over a long storage time. Liu et al. [90] prepared Au-Cu alloy nanoparticles with sizes of around 3 nm in the confined space of mesoporous SBA-15 using APTS to functionalize the support surface. The calcined (at 500 °C for 6 h) and subsequently reduced under H2 (at 550 °C for 2 h) catalysts, showed much better performance than monometallic gold particles in catalyzing CO oxidation. Recently, Chen et al. [80] modified the SBA-16 surface with APTS to prepare Au-Pd/SBA-16 catalysts. The highly dispersed Au-Pd bimetallic nanoparticles with a mean diameter ∼6 nm were active for the selective solvent-free oxidation of a variety of aromatic alcohols and maintained good catalytic stability after recycling.
The above mentioned examples demonstrate that the postsynthetic functionalization of MSM with organic moieties containing polar groups such as –NH2, –SH, etc. before gold loading, may be a good way to synthesize Au/MSM catalysts. However, this method not allows the homogeneous distribution of the organic functional groups in the pore surface, because it depends on numerous parameters, such as the number of the surface silanol residual groups, the diffusion of reagents through the pores channels, and steric factors [91]. Consequently the AuNPs are not homogeneously distributed after gold loading [92]. Moreover, the grafting procedure lead to a reduction of the specific surface area and the pore volume of the MSM [91], which are normally further reduced after gold incorporation.
2.2. Self-Assembly Functionalization of MSM Before Gold Loading
The self-assembly functionalization of MSM before gold loading consists in the co-hydrolysis and polycondensation of the silica source with the organic functional groups (mainly organosilanes, RSi(OR')3) in the presence of a structure-directing agent. This method of synthesis enables the functional groups to be anchored covalently with regular distribution inside the pore walls of the MSM [51,91]. Moreover, pore blocking is not a problem, because the functional organic groups are direct components of the silica matrix [51,91]. The gold loading is carried out in a subsequent step, mainly by deposition-precipitation using HAuCl4 as gold precursor. After filtration, the synthesized gold catalysts are washed with abundant deionized water to remove the residual chloride ions, dried and subsequently calcined to remove the functional ligands and to reduce the oxidized Au3+ species to metallic Au0 gold particles. The catalysts prepared following this approach can also be reduced in H2 or chemically using NaBH4 solution [54]. Figure 4 presents a general scheme of this synthesis procedure. In Table 2 we present the characteristics of the Au/MSM catalysis prepared following this method of synthesis, whereas some interesting papers are briefly discussed below.
Ghosh et al. [93] synthesized Au/MCM-41 hybrid materials by direct functionalization of the silica material with APS and MPS in a one-pot process. The surfactant of the functionalized MCM-41-NH2 and MCM-41-SH supports was removed by solvent extraction under reflux for 24 h, with a mixture of solvents containing 85 g of methanol and 2 g of HCl (35.5%). Subsequently, gold (3.0–3.5%) was loaded by mixing the aforementioned supports with HAuCl4 aqueous solution for 96 h. After filtration, washing and vacuum drying at room temperature, the resulting catalysts retained the ordered mesoporous structure characteristic of the pristine MCM-41, but a decrease of approximately 20% and 22% in the surface areas of Au/MCM-41-NH2 and Au/MCM-41-SH, respectively, was observed. Although the pore volumes of the catalysts also decreased as a consequence of the guest species incorporation, the average pore diameters of samples did not change considerably after gold loading. The mean diameters of the AuNPs were approximately 3.4 ± 0.5 and 3.2 ± 0.5 nm for Au/MCM-41-NH2 and Au/MCM-41-SH, respectively. The synthesized catalysts were active and completely selective for the hydrogenation of different olefinic substrates (cyclohexene, 1-hexene and 1-octene), and fairly comparable catalytic activity was found for both Au/MCM-41-NH2 and Au/MCM-41-SH samples.
Guari et al. [42,94] described a methodology to synthesize Au/SBA-15, preparing first the support by co-condensation of MPS with TEOS in the presence of Pluronic 123. After surfactant removal by Soxhlet extraction, the functionalized support was vacuum dried at 120 °C. The gold loading was performed by using two gold precursors: HAuCl4 and AuCl(THT). When HAuCl4 was employed, the functionalized support was suspended in water and heated under reflux in the presence of the gold precursor. Once the yellowish solution color was transferred onto the solid, sodium citrate was added. When [AuCl(THT)] was utilized, the functionalized support was suspended in tetrahydrofurane, and sodium acetylacetonate hydrate was added after the gold precursor addition. According to these authors, upon chemical reduction with NaBH4 ethanolic solution at 60 °C, metallic AuNPs (2.5 and 4.5 nm, respectively) were formed within the channels of the silica matrix. However, the typical ordered SBA-15 pore channels cannot be identified from the published TEM images, and unfortunately the catalytic activity of the synthesized materials was not evaluated.
Rombi et al. [61] functionalized SBA-15 with MPS by co-condensation using a TEOS:MPS molar ratio of 10, and after surfactant removal by Soxhlet extraction with a mixture of ethanol and HCl, gold (3%wt) was loaded using HAuCl4 as precursor. Once the solution became colorless, indicating the deposition of gold on the SBA-15 surface, sodium citrate was poured into the mixture to reduce the Au3+ to Au+ species. Then, the solid was recovered by filtration and dried at 120 °C for 12 h. Subsequently, NaBH4 1%wt ethanol solution was mixed with the solid during 18 h at 60 °C in order to obtain metallic gold. Finally, the suspension was filtered and the solid was dried at 120 °C overnight. Calcination of the catalysts in air at 300 and 560 °C led to the formation of small Au spherical nanocrystals homogeneously dispersed inside the SBA-15 channels with average sizes of about 2–3 and ca. 5 nm, respectively. Subsequent reduction under H2/He 600 °C led to an increase in the size of AuNPs up to ca. 6.5 nm. However, despite their large particle size, remarkable catalytic activity for CO oxidation was observed for the catalysts that were first calcined at 560 °C and then reduced under H2/He 600 °C, whereas the as-made catalysts were no active for CO oxidation at low temperatures. Moreover, gold was found mainly in metallic Au0 state, although because of the strong interaction between the mercapto functional groups and gold, positive Auδ+ species were also present.
Besson et al. [97] described a soft method allowing the incorporation of monodisperse AuNPs with average sizes of about 2–3 nm within the framework of 2D-hexagonal mesoporous silicas without any thermal treatment. In this approach, the framework of the silica material was functionalized by co-condensation of a hydrophilic bis-silylated precursor containing disulfide (prepared by reaction between 3-isocyanatopropyltriethoxysilane and 2,2'-diaminediethyldisulfide in THF) and TEOS in the presence of pluronic acid (P123) as template. After reduction of disulfide groups into thiol groups with a solution of triphenylphosphine, AuNPs were anchored in the support by impregnation and subsequent reduction of the organometallic gold precursor AuCl(THT) with ethanolic NaBH4. The XRD analysis revealed that the ordered structure of the host materials was preserved after the gold loading. Moreover, the observed broad diffraction peaks at 38.24, 43.81, 64.41 and 78.05° in the XRD high-angle region (2θ = 20 – 90°) confirmed the presence of metallic Au0 nanoparticles. In addition, the size of AuNPs was found to depend slightly on the percentage of organic groups in the framework. The AuNPs were very small (<2 nm) in materials containing low quantities of SH groups, while for the more concentrated material in SH groups the AuNPs size was no more monodisperse. Although this method seems to be interesting for the incorporation of AuNPs into MSM, the catalytic activity of the synthesized materials was not evaluated. Moreover, the authors pointed out that AuNPs were stable as long as the organic groups remained intact (up to about 200 °C). Beyond this temperature, there was a significant loss of organic moieties which involved the sintering of AuNPs. However, the presence of both the organic moieties and large AuNPs could have a negative effect on the catalytic activity of these composite materials.
Kumagi and Yano [98] prepared functionalized core/shell monodispersed mesoporous silica spheres, by the co-condensation of the silica source (TMOS) with MPS and propyl trimethoxysilane in the presence of CTACl in alkaline media. After the surfactant removal by acid extraction performed in an ethanol mixture of concentrated hydrochloric acid, the samples were filtered off, washed with ethanol, and dried in air. This resulting material was impregnated with a solution of HAuCl4 in hydrochloric acid to obtain gold-loaded core/shell mesoporous silica spheres containing propyl group in the shell. AuNPs with particle size distributions ranging from 1 to ca. 15 nm were formed, but no catalytic activity was reported. A similar method of synthesis, without the addition of propyl trimethoxysilane had been previously reported by Nakamura et al. [99] for the preparation of gold-containing nanoporous silica spheres, with AuNPs sizes in the range of 4.8 nm. However, the catalytic performance of this material was not reported either.
Hérault et al. [95] recently obtained SBA-15 mesoporous organosilicas containing mannitol derivative in the framework, by co-hydrolysis and poly-condensation of a bis-silylated mannitol precursor (3,4-Di-O-[3-(triethoxysilylpropyl)carbamate]-1,2:5,6-di-O-isopropylidene-D-mannitol, prepared by coupling of 1,2:5,6-di-O-isopropylidene-D-mannitol with 3-(triethoxysilylpropyl)isocyanate) with TEOS in the presence of triblock co-polymer P123 as structure-directing agent in acidic media. After stirring for 24 h at 30 °C, the mixture was heated at 110 °C for 24 h. Then, the temperature was diminished to room temperature and the reaction mixture was left without stirring for 3 days. The resulting solids were filtered and washed with ethanol, followed by Soxhlet extractions with ethanol (15 h) and then with acetone (15 h), prior to drying under vacuum. From the three different TEOS: bis-silylated mannitol precursor ratios studied (R9, R19 and R39), it was found that: (i) the highest polycondensation degree (80%) was obtained in the case of R39; (ii) only the R39 sample exhibit the three low-angle XRD diffraction peaks, characteristic of well ordered SBA-15 type materials; (iii) the BET surface areas, the total pore volumes and the pore size distributions ranged from 530 to 630 m2/g, 0.60 to 1.40 cm3/g and 4.4 to 9.0 nm, respectively. On the other hand, during the hydrolysis and poly-condensation processes, some OH functional groups were released, which were used for the stabilization of AuNPs using HAuCl4 or AuCl(THT) as gold precursors, involving or not the use of reducing (NaBH4) and destabilizing (trisodium citrate and sodium acetylacetonate monohydrate) agents. From the characterization analysis it was pointed out that: (i) AuNPs were not formed when using the organogold precursor AuCl(THT); (ii) the absence of destabilizing agent and reducing agent led to the formation of large AuNPs (15–20 nm) overlapping several channels of mesostructured material; (iii) the presence of reducing agent (NaBH4) allowed the formation of small AuNPs (average size of 6 nm); (iv) the best results, in terms of dispersion, size (3 nm) and location of AuNPs within the walls of the 2D-hexagonal structure of the MSM, were obtained when using trisodium citrate and NaBH4 as destabilizing and reducing agents, respectively. Unfortunately the catalytic activity of the catalysts synthesized under this approach was not evaluated.
Yu et al. [39] functionalized different periodic mesoporous organosilicas (MCM-41, MSU and SBA-15) with an organic bridged silsesquioxane precursor containing disulfide-ionic liquid moieties in one-pot synthesis procedure. Subsequently AuNPs were loaded using aqueous HAuCl4 solution at pH values of 7.0–8.0. The resulting Au/MCM-41, Au/MSU and Au/SBA-15 catalysts showed well dispersed AuNPs, as presented in Figure 5(a–c), with mean particle sizes of 2.1, 4.7 and 2.0 nm, respectively, indicating that porous supports with higher pore connectivity, such as MSU with 3-D worm-like pore structure, should be more vulnerable to the aggregation of AuNPs. The catalytic activity, evaluated for the epoxidation of various olefins (α-pinene, cyclohexene, 4-chlorostyrene, α-methylstyrene, 1-hexene and 1-octene) using H2O2 as oxidant, was inversely related to the Au particle size, following the order Au/SBA-15 > Au/MCM-41 > Au/MSU. Moreover, after eight consecutive runs, all catalysts showed quite good reusability with no significant losses in both α-pinene conversion and α-pinene oxide selectivity.
Despite the fact that the self-assemble functionalization of MSM before gold loading has permitted the synthesis of Au/MSM materials having well dispersed AuNPs within the channels, as presented in Figure 5, the main disadvantages of this method lie in the care that must be taken to avoid the destruction of the functional groups during the surfactant removal and in the difficulty of maintaining the mesostructure of the functionalized silica materials, because the degree of their mesoscopic order decreases with increasing the concentration of functional groups [36,51]. Moreover, given that the functional group is quite hydrophilic, the functionalization could occur partially within the framework. Then, the organic functional groups have to be sufficiently lipophilic to enter in the core micelle and not too bulky to avoid bursting [101,102].
2.3. Direct Synthesis
This method of synthesis, also called co-condensation or one-pot synthesis, is a simple procedure to introduce AuNPs in MSM, but not necessarily placed within the pores. It consists in the copolymerization of the silica source with the gold precursor in the presence of a structure-directing agent, so that the formation of mesostructures and the gold anchoring occur simultaneously. Organosilane coupling agents (RSi(OR')3) are frequently added into the gel synthesis in order to functionalize the silica surface for enhancing the gold adsorption. This approach is often preferred, because as mentioned in Section 2.2, it allows regular distribution of the functional groups inside the channel pores, and therefore uniform distribution of gold, as well as a loading control within the limits of the content supported by the micelle [101]. Although this procedure presents similar shortcomings than the self-assembly functionalization of MSM before gold loading, the direct synthesis method has been largely employed for the preparation of gold catalysts supported on different MSM, as summarized in Table 3. Some relevant applications are discussed below.
Lu et al. [103,104] synthesized Au/MCM-41 and Au/SBA-15 catalysts by using a direct synthesis process in which the silica and gold precursors (TEOS and aqueous HAuCl4, respectively) were added to the dissolved surfactants (cetylpyridinium bromide for MCM-41, and P123 for SBA-15) in acidic media. After the hydrothermal treatments, the solids were filtered, washed with deionized water, dried and calcined in air at 550 °C for 4.5 h. The gold loading (0.13–1.21%wt) had only a modest effect on the surface area of the supports, and the synthesized catalysts maintained the typical structure of mesoporous materials. Gold was anchored both inside the channels and deposited on the external surface of the mesoporous structures, and it was mainly present in the metallic Au0 state. These catalysts were active for the oxidation of cyclohexane, but unfortunately the size of AuNPs was not reported.
Wu et al. [116] recently synthesized SBA-15-supported AuNPs (2–4 nm) by means of a one-pot synthesis method in the presence of different amounts of MPS as functionalizing agent. Subsequent to dissolution of the surfactant (P123) in acidic media, different mixtures of TEOS:MPS were slowly added prior the introduction of HAuCl4 solution (gold loading of 1%wt). After stirring (40 °C for 24 h) and hydrothermally treatment (100 °C for 24 h), the resulting solids were filtered, washed, dried under vacuum and calcined at 500 °C during 8 h, to remove the surfactant and the organic functional group, and to decompose the gold species into metallic Au0. From the analysis performed in this study, it can be highlighted that: (i) the functionalizing agent contributed to coordinate the gold precursor via thiol ligands, and to form covalent bonds with the silica matrix. During the synthesis, the silane moiety co-condensed with the silicon precursor to form the silica framework, and the alkanethiol end interacted with AuCl4− to form Au (I)—thiolate complexes, which firmly attached the gold species on the silica framework; (ii) The amount of MPS played a critical role in the loading and dispersion of AuNPs. The larger the amounts of MPS introduced, the higher the Au loading efficiency and dispersion. However, the AuNPs began to aggregate and unevenly distribute with the decrease of added MPS. Withal, when the concentration of MPS in the gel increased up to 20% (molar ratio TEOS:MPS is 32:8), the 2D hexagonal ordered structure decreased significantly, indicating that high concentrations of MPS hindered the TEOS condensation to form an ordered framework; (iii) Because of the formation of void defects on the pore walls after removal of functional groups by calcination, AuNPs were partially intercalated in the pore walls of the mesoporous SBA-15, and the catalysts showed values of surface area higher than the pure SBA-15; (iv) The catalysts were active for the selective oxidation of cyclohexane with molecular oxygen, and the increase of MPS led to an increase on the conversion of cyclohexane up to 21.5% with selectivities towards cyclohexanone and cyclohexanol of up to ∼95%. Moreover, the catalyst containing 10%MPS exhibited no obvious activity loss after six runs, demonstrating its good stability; (v) Au0 was designed as the active site for the cyclohexane oxidation.
In a previous study, Wu et al. [62] employed VTES as functionalizing agent to prepare Au/SBA-15 catalysts by a one-pot method, using a TEOS:VTES molar ratio of 20, and HAuCl4 solution as gold precursor. After stirring for 24 h at 40 °C and hydrothermal treatment at 100 °C for 24 h, the solid was filtered, washed and dried, prior to template removal by extraction with ethanol at 70 °C for 6 h and reduction under H2 at 250 °C for 2 h. The resulting catalyst (Au loading of 1%wt) retained the ordered mesostructure of SBA-15, and the AuNPs (∼5 nm), present as metallic Au0, were anchored and evenly dispersed in the functionalized SBA-15. The high activity and selectivity for the solvent-free selective oxidation of cyclohexane using this catalyst indicated that VTES may be a good functional group to prepare Au/MSM catalysts by direct synthesis. Moreover, the catalytic activity and selectivity of this catalyst was much better than the Au/SBA-15 catalyst prepared by post-grafting with the same functional group, whose average Au particle size was about 9 nm.
Glomm et al. [40] prepared Au/MCM-48, Au/SBA-15 and Au/SBA-16 materials by one-pot process using a gold-modified precursor (HAuCl4 functionalized with TMPDA), which was added to the synthesis solutions after precipitation of each MSM. The resulting gold-containing MSM were recovered after stirring for 24 h, and were subsequently filtered, washed and calcined in air at 550 °C for 5 h. Although the catalytic activity was not evaluated, the following conclusions were highlighted from the characterization analysis: (i) This in situ method resulted in high retention of gold species in all MSM, indicating the important role of the amine functionalizing agent; (ii) The surface areas of the mesoporous SBA-15 and SBA-16 materials were significantly reduced after incorporation of gold, whereas the Au loading did not significantly affect the surface area of the MCM-48; (iii) The three MSM retained their long-range order upon incorporation of gold; (iv) AuNPs ranging between 5 to 20, 5 to 50 and 10 to 100 nm, were present on the synthesized materials supported on MCM-48, SBA-15 and SBA-16, respectively; (v) Even though a significant fraction of the particles was incorporated within the pores, the majority of AuNPs were found to reside on the external surface of the silica materials.
The feasibility of other MSM for retaining AuNPs using amine functionalizing agents has been also evaluated. For example, Lee et al. [126] prepared gold-containing HMS, MSU and PMOs materials by a co-synthesis sol-gel method, using HAuCl4 as gold precursor, and TMPTA and TMPDA as bifunctional silane ligands for HMS and PMOs, and MSU materials, respectively. The structure-directing templates (DDA for HMS and PMOs, and Triton X-100 for MSU) were removed by ethanol extraction at room temperature for 6 h, and the Au(III) precursor was reduced to metallic AuNPs by heating in Ar/H2 atmosphere (4% H2), or by calcination. The resulting materials displayed AuNPs uniformly distributed inside the mesopores, with sizes smaller than 5 nm. Although the authors mentioned that this synthesis procedure may be used to prepare AuNPs supported on other mesoporous materials, except for those formed in acidic media due to the protonation of the amine functional groups, the catalytic activity of the synthesized materials was not reported.
Recently, we have used BTESPTS to synthesize active gold crystallites (3–14 nm) dispersed on mesoporous silica, which were mainly present in the metallic Au0 state, and displayed high catalytic activity (100% lactose conversion after 100 min reaction) and 100% selectivity towards lactobionic acid, when a catalyst containing 0.7% Au was used at a catalyst/lactose ratio of 0.2 under alkaline (pH 9.0) and mild reaction temperature (65 °C). In general, as indicated by Wu et al. [118], during the synthesis procedure the thioether groups incorporated into the silica walls by co-condensation in the presence of the surfactant (P123), form a complex with the tetrachloroauric anions (AuCl4−) leading to a good dispersion of gold on the MSM. Under the hydrothermal treatment, part of AuCl4−—thioether complexes decomposed to form Au clusters stabilized by thioether groups. The subsequent calcination of the resulting solids at high temperature (500 °C for 5 h) allows the removal of the template and functional groups, at the same time that gold species are reduced to AuNPs. This simple procedure has been also employed to immobilize gold within mesoporous silica thin films [124] and mesoporous silica [64,118–122]. The resulting composite materials have shown high activity and selectivity in the oxidation of benzyl alcohol [119], n-hexadecane [64,120] and cyclohexane [118], as well as in the plasma-assisted total oxidation of trichloroethylene [121,122]. However, the main drawback of this method lies to the fact that the structural ordering of the mesoporous material suffers from disturbance at high BTESPTS loading levels. Therefore catalysts show irregular shapes and pores. When using a BTESPTS molar concentration of about 7%, we found that gold catalysts with Au loading of 0.4, 0.5, 0.7, 0.8 and 1.0% presented a wormhole-like framework structure containing interconnected 3D-mesopores (Figure 6(g)), suitable for the minimization of diffusion limitation phenomena often encountered in adsorption and catalytic reactions [4]. Conversely, when the molar concentration of BTESPTS was decreased to 2%, ordered mesoporous silica was obtained, but the gold loading was significantly reduced, the size of AuNPs increased as illustrated in Figure 6(h), and the catalyst became inactive. These results were in agreement with the recent findings of Wu et al. [118] who reported that molar concentrations of BTESPTS higher than 2.5% led to a decrease in the regularity of the 2D hexagonal ordered structure of SBA-15, but lowering this concentration the AuNPs exhibited average sizes up to 24.4 nm.
The direct synthesis has been also carried out to synthesize bimetallic gold catalysts. Sobczak et al. prepared Au/MCM-41 [105] and Au/Nb-MCM-41 [106,107] catalysts (Au loading 1%wt) by co-precipitation, adding the gold precursor (HAuCl4) into the gel containing the Si source (sodium silicate) and the template (CTACl) following the conventional method of synthesis of the MCM-41. The template was removed by calcination at 550 °C for 2 h under He, and in air under static conditions for 14 h. The resulting gold catalysts exhibited hexagonally ordered mesopores and Au crystallites in the range of 3–18 nm. Moreover, this approach allowed the formation of gold in two forms: metallic and surrounded (bonded) by chloride. These chloride ions served as a catalytically active basic species in the acetonylacetone (AcAc) cyclization/dehydration in the gas phase, and took part as promoters in the electron transfer to oxygen in the NO reduction with propene in presence of oxygen.
As discussed above, the direct synthesis provides a simple pathway to prepare stable Au/MSM catalysts using different functional moieties, which, as briefly mentioned by Ma et al. [36] can be employed to synthesize gold onto other mesoporous supports such as mesoporous TiO2, for example. Although the gold loading and Au particle size are correlated with the amount of organic functional ligand, special care must be taken in order to find the optimal conditions of the ratio silica precursor/OFL to obtain ordered structures and well distributed small AuNPs.
2.4. Synthesis of Au/MSM Using Cationic Gold Complex [Au(en)2]Cl3 (en = Ethylenediamine)
The synthesis procedures mentioned above normally imply the addition of organic functional ligands to incorporate the AuNPs within MSM, when tetrachloroauric acid is used as gold precursor. Although these approaches allowed the preparation of active Au/MSM catalysts, the grafting of OFL may generate some defects on the mesoporous structure, and the ligands removal at high temperatures may conduce to a decrease in the catalytic activity because of the increase of the gold particle size.
In the early 50's, Block and Bailar [127] found that tetrachloroauric acid reacts with ethylenediamine (en) to form the gold complex [Au(en)2]Cl3 after precipitation with ethanol. This complex may act as acid by losing a proton from the coordinated amine group, under basic conditions. Based on this principle, the synthesis of Au/MSM catalysts becomes easier by deposition-precipitation of this gold complex, given that in alkaline media the negative-charged surface of the MSM can readily adsorb the [Au(en)2]3+ cations, by deprotonation reaction of ethylenediamine ligands [128].
Dai and coworkers [44,129] synthesized gold catalysts supported on mesoporous SBA-15 by mixing the support with an aqueous solution of [Au(en)2]Cl3 in alkaline media (pH range between 6.0 and 10.0). After agitation of the slurry for 2 h, the yellowish solid was filtrated, vacuum dried and reduced under H2/Ar (4%) at 150 °C for 1 h. This procedure led to obtain small (∼4.9 nm) and uniform AuNPs confined within the SBA-15 mesopores. Moreover, when the pH increased up to ∼10, the gold loading increased (2.70 to 9.08%wt) and the gold particle size decreased (from 5.4 to 4.9 nm). However, only the catalysts synthesized at pH value higher than 8 exhibited high catalytic activity for low-temperature CO oxidation (below 0 °C), prior to their activation by calcination in air at 400 °C for 1 h. From these results, it can be concluded that the pH of synthesis plays an important role on the catalyst activity, and ethylenediamine moieties may form a strong bond with the surface of AuNPs, interfering with the catalytic activity. This work was a valuable reference for other groups for synthesizing gold catalysts supported on mesoporous silicas, as presented in Table 4. For example, Guan and Hensen [130] prepared gold supported on different MSM (SBA-15, SBA-16 MCM-41) using this approach. After adding the supports to an aqueous solution of [Au(en)2]Cl3, the pH was adjusted to a constant value of 12. The suspension was stirred for 2 h, followed by filtration, washing with deionized water, drying at 110 °C overnight and calcination at 400 °C for 4 h. The resulting catalysts, showing average Au particle sizes of 4.9 ± 1.3, 7.3 ± 1.6 and 6.7 ± 2.1 nm for SBA-15, SBA-16 and MCM-41 materials, respectively, were active for ethanol oxidation. Parreira et al. [131] synthesized gold catalysts supported on pure and metal-modified hexagonal mesoporous silica (HMS, HMS-M, where M = Ce, Ti, Fe), using [Au(en)2]Cl3 as gold precursor. The HMS and the metal-modified HMS, synthesized by direct synthesis via co-precipitation using appropriate metal salts, were mixed with an aqueous solution of [Au(en)2]Cl3 maintaining the pH value at ca. 10. The catalysts were calcined at 300 °C for 1.5 h, and subsequently reduced in hydrogen for 1.5 h. The resulting AuNPs with average Au particle sizes ranging between 4.1 and 5.9 nm were active for the aerobic oxidative esterification of benzyl alcohol, and displayed gold in metallic Au0 and Auδ+ and Au+ oxidation states. Recently, we have used [Au(en)2]Cl3 to synthesize gold clusters supported on mesoporous silica decorated by ceria (SBA-15-CeO2) [132]. The support and the gold precursor were mixed in alkaline media (pH 10), and after vacuum filtration, the solid was thoroughly washed with deionized water, vacuum dried overnight at 100 °C, and calcined at 500 °C for 5 h. The resulting catalysts showed a regular array of mesopores, as well as well dispersed and not aggregated spherical AuNPs of about 5 nm. However, in agreement with the low-angle XRD analysis, the hexagonal order of the mesoporous SBA-15-CeO2 support was somewhat affected after gold loading, as it can be depicted in Figure 7(d,e). The N2 physisorption and XRD analysis revealed that support possess ordered hexagonal mesoporous structure, high surface area and large pore volume, similar to SBA-15, whereas BET surface area and pore volume of catalysts were significantly decreased upon impregnation. Moreover, XPS analysis revealed the coexistence of metallic and oxidized species on the catalyst, with relative surface concentrations of Au0 (78.17%) > Au+ (13.08%) > Au3+ (8.76%), and the presence of both Ce3+ and Ce4+ oxidation states. The catalytic activity of the synthesized catalysts was evaluated on the partial oxidation of lactose for selective synthesis of lactobionic acid (LBA) for therapeutic, pharmaceutical and food grade applications. After 100 min of reaction, the 0.7% Au/SBA-15-CeO2 catalyst showed high catalytic activity (100% lactose conversion) and a 100% selectivity towards LBA, when it was used at a catalyst/lactose ratio of 0.2 under alkaline (pH 9.0) and mild reaction temperature (65 °C).
The use of the cationic gold complex [Au(en)2]Cl3 as precursor for the synthesis of monometallic [129,133–139] and bimetallic [140–143] gold catalysts supported amorphous SiO2, as well as for the preparation of Au/SiO2-based catalysts [136], has demonstrated that amorphous silica can be also employed as a support to prepare active Au/SiO2 catalysts. Although this synthesis method has not been intensively investigated, it can be regarded as highly efficient for the preparation of gold catalysts supported on MSM. However, it should be noted that the [Au(en)2]Cl3 precursor must be kept protected from light to prevent its decomposition [129,133–139].
2.5. Chemical Vapor Deposition
Chemical vapor deposition of dimethyl gold acetylacetonate was first used by Okumura et al. [37,45,144,145] to deposit AuNPs on SiO2 and MCM-41, demonstrating that silica may be a suitable support for the preparation of active gold catalysts. To do this, the supports were first evacuated in vacuum at 200 °C for 4 h to remove the adsorbed water, and then treated with O2 at 200 °C for 30 min to remove organic residue from the surface by oxidation. Subsequently, the gold precursor was gradually evaporated at 33 °C on the supports. The resulting materials were calcined in air for 4 h at high temperature (200–500 °C) to decompose the gold precursor into metallic AuNPs on the support surface. The synthesized catalysts exhibited remarkably catalytic activities for the oxidation of CO and H2, and showed a large number of gold particles smaller than 10 nm (mean diameters of 6.6 and 4.2 nm for SiO2 and MCM-41, respectively), which were three times smaller than those of Au/SiO2 prepared by impregnation method. From the characterization analysis, it was concluded that the interaction between the gold precursor and the support surface occurred mainly between the oxygen atoms of Me2Au(acac) and the OH groups of the SiO2 surface; and that temperatures above 300 °C are needed to prepare highly active Au catalysts following this preparation method.
Schimpf et al. [146] synthesized Au/SiO2 (Au loading of 2.4%wt) by CVD of Me2Au(acac), following the method proposed by Okumura et al. [147]. Highly dispersed AuNPs with mean value of 1.4 nm were obtained. Moreover, the synthesized catalysts were active for the low-temperature oxidation of CO. Araki et al. [148] prepared Au/FSM-16 by CVD of AuMe2(HFA). The gold precursor was adsorbed on the support under reduced pressure at room temperature for 4 h. The resulting white powder turned to pale purple after UV irradiation under reduced pressure for 4 h, indicating the formation of metallic gold. The obtained Au nano-wires (2.5 × 17.1 nm) in the mesopores of FSM-16 were active in the oxidation of CO.
Notwithstanding the CVD allowed the synthesis of highly active Au/SiO2 and Au/MSM catalysts, the main drawbacks of this preparation method relate to the high cost of the organometallic gold precursor and to the requirements of special equipments for the catalysts synthesis, Moreover, the amount of metal that can be incorporated following this approach is limited by the pore volume of the support [47,54].
2.6. Synthesis by Dispersion of Gold Colloids or Presynthesized AuNPs
Gold colloids and nanoparticles have been prepared by different procedures and used as precursors to prepare gold supported on TiO2, ZnO, ZrO2, Al2O3, carbon and SiO2 materials [32,149–157]. The synthesis of gold supported on MSM using gold colloids or presynthesized AuNPs can be achieved by using two different strategies: (i) dispersing the presynthesized gold precursors on the MSM support and (ii) synthesizing the MSM in the presence of presynthesized gold colloids or AuNPs. Even though suitable, this Au/MSM catalyst synthesis procedure has not been extensively applied in catalytic applications. The main advantage of this method lies to the easy control of AuNPs size throughout the gold sol synthesis, and therefore it offers the possibility to tailor the size of gold particles before their deposition on the support. This could be also attractive to control the AuNP aggregation, because the particle size is normally preserved during the immobilization step [150,158]. Some relevant applications are presented below.
Ma et al. [159] recently reported the synthesis of gold catalysts supported on extra-large mesoporous silica material (EP-FDU-12), which were active and highly selective in the gas-phase oxidation of benzyl alcohol to benzaldehyde. The EP-FDU-12 support was prepared using Pluronic F127 as template agent and TEOS as silica precursor, in the presence of 1,3,5-trimethylbenzene and KCl. After stirring at 15 °C during 24 h, the mixture was hydrothermally treated for 24 h at temperatures ranging between 100 and 220 °C. The solids were filtered and dried at room temperature, prior to the template removal by microwave digestion using 30% H2O2 and 15 M HNO3. The Au catalysts (Au loading of 0.5%wt) were obtained by adding the EP-FDU-12 support to a chloroform solution of AuNPs, prepared from AuPPh3Cl as described by Zheng et al. [160]. Subsequent to stirring, the solid product was centrifuged, dried in air and calcined at 350 °C for 5 h. From this method of synthesis, it is interesting to pull out these two observations: (i) though the Au particle size was almost the same (11.3 ± 2.7 nm vs. 10.3 ± 2.7 nm) after reaction, carbon deposition was at least 10-fold less on catalysts with pore size of 36 nm than on those with pore size of 23 nm, suggesting that large pores facilitate the diffusion of organic products and therefore diminishing the coke formation; (ii) The pore size can be tuned by modifying the KCl:F127 concentration and the hydrothermal temperature, allowing different applications to these materials.
Tai et al. [161] prepared dodecanethiol-capped AuNPs following the method proposed by Brust et al. [162], which were adsorbed on a mesoporous silica wet-gel (size of mesopores of maximum 15 nm), synthesized by hydrolysis and subsequent condensation of TMOS in methanol using ammonia as catalyst. The gel was immersed in the AuNPs solution using different solvents (toluene, THF and toluene:THF 1:1). After sufficient contact (more than one day), supercritical CO2 was used for drying the wet-gel gold containing silica. In the resulting material, the Au particle size distribution was almost identical to that of the original dodecanethiol-capped AuNPs (average 2.4 nm vs. average 2.6 nm, respectively). Moreover, AuNPs were homogeneously distributed in the gel formed in THF, whereas when using toluene the AuNPs were present only in the peripheral part of the gel, indicating that the spatial distribution of the AuNPs inside the gels can be controlled by changing the polarity of the solvents. Nevertheless, the catalytic activity of this composite aerogel was not evaluated. A similar method for synthesizing mesoporous colloidal gold aerogels, using AuNPs (sized at either 5 to 28 nm) prepared by citrate reduction of HAuCl4 has been reported by Anderson et al. [163], but the catalytic behavior was not evaluated.
Glomm et al. [40] used citrate-coated AuNPs of 5 and 10 nm for the synthesis of Au/MCM-48, Au/SBA-15 and Au/SBA-16. To do this, the as-synthesized silica supports were first suspended in water, followed by pH adjustment to 9.0 using 2 M NaOH. Then, the AuNPs were added, and after 24 h stirring at room temperature, the gold-containing materials were filtrated, washed, dried and calcined at 550 °C in air during 5 h. Although the three mesoporous structures retained their long-range order upon incorporation of AuNPs, their surface area was significantly reduced after Au loading. Moreover, most of the AuNPs were found on the external surface of the silica materials, regardless a significant fraction of the particles was incorporated within the pores. Unfortunately, no data of catalytic activity of these materials were reported.
The second strategy to prepare Au/MSM using gold colloids or presynthesized AuNPs involves mainly two steps: the synthesis of AuNPs in the presence of a block copolymer, and the synthesis of the MSM using the gold-nanoparticle copolymer unit as template [46]. Notwithstanding the catalytic activity was not evaluated, from the interesting works of the Somorjai group [46,47,164], who have used this approach to encapsulate AuNPs of different sizes (2, 5, 10 and 20 nm) into the channels of SBA-15, MCM-41 and MCM 48 using P123, hexadecylamine and cetylbenzyldimethylammonium chloride as templates, respectively, it can be highlighted that: (i) the presence of small Au nanocrystals (2–10 nm) influences only slightly the well-ordered structure but changes the lattice spacing of the MSM; (ii) although the mesopore channels of the MSM expand when small AuNPs are included, the narrow pore size distribution is preserved; (iii) when high amounts of AuNPs or large AuNPs (20 nm) are used, the structure of the materials remains porous, but their crystallinity decreases and a worm like structure appears, instead the hexagonal arrangement; (iv) after calcination, the small AuNPs are incorporated within the channels of the mesoporous host matrixes, but nanoparticles whose diameter is larger than the MSM pore size (20 nm fro SBA-15 and 10 nm for MCM-41 and MCM-48), cannot be inserted and they remains exclusively outside of the channels; (v) when using bimodal AuNPs (2 and 5 nm, or 2 and 10 nm), the resultant pore size of the SBA-15 material is controlled by the larger size of nanoparticles; (vi) the AuNPs incorporated inside the pores are accessible to reactant molecules, as confirmed by XANES used in combination with the adsorption of thiols.
Lin et al. [165] prepared AuNPs by mixing HAuCl4 aqueous solution with CTABr, followed by chemical reduction with aqueous NaBH4. The silica precursors (sodium silicate and sodium aluminates) were added into the Au-surfactant solution, and after neutralization (pH value of the gel solution ∼8–10), the gel solution was hydrothermally treated at 100 °C for 2 days. The solids were then filtrated, washed, dried and calcined at 560 °C. The resulting catalysts with Au loadings of 4 to 12%wt, showing less ordered pore structure than pure MCM-41, and the AuNPs sized between 5 and 15 nm displayed relatively low reactivity (less than 16% conversion) in CO oxidation, probably due to the large size of Au nanoparticles.
Employing the conventional conditions for the synthesis of MCM-41, Aprile et al. [166] prepared Au/meso-SiO2 catalysts using colloidal AuNPs (2–5 nm) stabilized with a quaternary ammonium ion ligand having at one end a long alkyl chain (N-[3-(triethoxysilyl)propyl] O-2(dicetylmethylammonium)ethyl urethane) and TEOS as gold and silica precursors, respectively, in the presence of CTABr as structure directing agent. The resulting catalysts, calcined at 500 °C for 5 h, were active for the oxidation of primary and secondary alcohols (3,4-dimethoxybenzyl alcohol and 1-phenylethanol) at atmospheric pressure. Moreover, these catalysts showed higher activity than those prepared by IWI of the tetraoctylammonium stabilized colloidal AuNPs in a pre-formed SBA-15. However, in aqueous media, the Au/meso-SiO2 catalysts where complete deactivated by collapse of the mesoporous structure.
The encapsulation of metal nanoparticles (Au and Ag) during the growth of an organic-inorganic hybrid gel was recently reported by Wichner et al. [167], using phenylethylthiol-coated metal nanoparticles dissolved in THF, which were added to a sol-gel mixture of phenyltriethoxysilane, water and THF in acidic media (pH 1.2). The suspension was stirred for 1 h, and then TMOS was added. After 3 h, the pH was increased with NH3(aq) to 6.0–7.0 to accelerate the condensation. Once the gelation was complete, the organic components were removed by calcination at 600 °C in air for 6 h. When the synthesis pH value was raised to 7.0 mesopores were formed with mesopore diameter of about 3.5 nm, whereas at lower pH values the gel matrices were microporous. The resulting catalysts with average diameter of metal particles of 6 nm were catalytically active in CO oxidation, although relatively high activation temperatures of at least 330 °C were needed.
Ferrara et al. [168] synthesized mesoporous silica films doped with gold, by the dispersion of stable AuNPs (8–9 nm) functionalized by dodecanethiol chains in an acid-catalyzed sol-gel silica solution. Even though the size, shape and crystalline domains of the AuNPs remain unchanged during the synthesis process, the catalytic activity was not evaluated. Chen et al. [169] prepared mesoporous gold-silica nanocomposites (Au loading from 6.9 to 11.4%wt) using a simple one-step method involving the sol-gel reaction of the silica precursor (TEOS) with a gold sol (Liquid Bright Gold 5154, containing 5%wt metallo-organic gold compound in cyclohexanone) in the presence of DBTA as a nonsurfactant template. After removal of DBTA by calcination at 550 °C during 16 h, the mesoporous worm-hole-like structure of the gold-silica nanocomposites, with AuNPs ranging between 2 and 8 nm dispersed throughout the silica porous matrix, showed high surface area (up to 630 m2/g), large pore volume (∼0.5 cm3/g) and pore diameters of 3–5 nm with relatively narrow pore size distributions. However, the catalytic activity of this material was not reported.
Bönnemann et al. [170] prepared monometallic Au and bimetallic Au-Pd catalyst by embedding pre-synthesized tetraalkylammonium bromide-stabilized Au and Pd-Au alloy particles in a solid silica matrix, following a modified sol-gel process. The colloidal Au and Pd-Au particles were prepared by the co-reduction of Pd and Au salts (palladium acetate and gold chloride) with tetraoctylammonium triethylhydroborate in dry THF under argon at ambient temperature during 16 h. After solvent removal by evaporation under vacuum, the surfactant was removed by dipping the colloidal powder in diethyl ether followed by precipitation with an ethanol/methanol mixture. The embedding of the resulting Au and Pd-Au colloids was carried out in THF using TEOS as silica precursor. The sol was stirred at 70 °C under reflux until the gelification was complete (2 days). The resulting gel was dried at 110 °C, calcined in air at 450 °C, and subsequent reduced in H2 at 450 °C. The resulting materials showed mesoporous structure with a sharp pore diameter distribution and channels randomly distributed and no tubes. Particle sizes ranging between 3.0 and 4.9 nm for both Au and Pd-Au catalysts were observed. The monometallic Au catalyst displayed very low activity in the selective semi-hydrogenation of 3-hexyn-1-ol, whereas the bimetallic Au-Pd catalyst exhibited high activity and was remarkably resistant to deactivation. Some years later, Parvulescu et al. [171] evaluated the catalytic activity of the before mentioned catalysts in the selective hydrogenation of 3-hexyn-1-ol, cinnamaldehyde, and styrene. Their results showed that alloying Pd with Au in bimetallic colloids led to enhanced activity and improved selectivity. Moreover, the bimetallic Au-Pd catalysts were very stable against poisoning, as was evidenced for the hydrogenation of styrene in the presence of thiophene.
Liu et al. [172] prepared Au-Ag alloy particles (<10 nm) supported on mesoporous MCM-41 which were synthesized by one-pot approach using CTABr both as a stabilizing agent for AuNPs and as a template for assembling the MCM-41 structure. After template removal by calcination at 560 °C and subsequent reduction at 600 °C with 10% H2/N2 the alloy nanoparticles were uniformly dispersed on the silica support, but their size was enlarged (>30 nm). Moreover, the increase of Ag concentration led to a bigger size of the alloy particles, suggesting that the presence of Ag provokes the aggregation more severely, because of the Ag lower melting point. Besides, mesoporous silica particles were very disordered. However, the bimetallic Au-Ag/MCM-41 catalysts (Au/Ag molar ratios of 3/1, 5/1 and 10/1) were active for CO oxidation at 25 °C, while pure Ag/MCM-41 did not exhibit any catalytic activity, and Au/MCM-41 catalyst only had a low CO conversion. These results indicate that the size of bimetallic Au-Ag nanoparticles is not a critical factor in the CO conversion.
2.7. Deposition-Precipitation
As mentioned in Section 1, the deposition-precipitation method requires the pH adjustment of the gold precursor solution to high values in order to generate [Au(OH)nCl4−n]− species with little chloride as possible. However, under alkaline conditions the surface of the MSM becomes negatively charged and the interaction between the gold and the silica surface is very weak, hindering the gold adsorption and facilitating the mobility and sintering of AuNPs [22,37]. For this reason, it has been reported that DP is not a suitable method for the incorporation of gold on silica materials, and other strategies have been attempted, as it was described in the preceding sections. However, some gold catalysts supported on MSM have been synthesized using this approach, as summarized in Table 5. For example, Hereijgers and Weckhuysen [173] prepared Au/SBA-15 by DP from a dilute solution of HAuCl4 in HCl at pH of 9.5 using ammonia solution. After calcination, AuNPs with average value of 4.0 nm were obtained, but the structure of the SBA-15 support was slightly damaged because of the high pH effect. This catalyst did not show good catalytic performance in the selective oxidation of cyclohexane to cyclohexanone. Ma et al. [59] synthesized monometallic Au/SBA-15 and bimetallic Au-Pd/SBA-15 by DP using chloroauric acid solution and 1 M NaOH solution for adjusting the solution pH to 7.0. After adding the SBA-15 support, the slurry was aged at room temperature for 12 h, followed by washing, drying at 70 °C for 5 h and calcination at 200 °C for 2 h. The surface area and the pore size of the resulting catalysts were reduced two folds, in comparison with the SBA-15 support, but the pore volume and the adsorption isotherms of both catalysts were almost identical to those of SBA-15. Moreover, the Au/SBA-15 catalyst showed well-developed parallel pore channels with well dispersed AuNPs (in the range of 10–50 nm) on the intra-surface of SBA-15, as illustrated in Figure 8(c), while the Au-Pd nanoparticles dispersed better compared to Au/SBA-15 catalyst but did not enter into the channels of SBA-15 as depicted in Figure 8(d). Albeit both catalysts were active for the oxidation of benzyl alcohol, the Au-Pd/SBA-15 was more active than the Au/SBA-15, but its selectivity towards benzaldehyde was slightly smaller (99 vs. 96%). However, in comparison with bimetallic Au-Pd/SBA-15 catalysts prepared by post-grafting modification of SBA-15 with MPS and APTS, the activity of the bimetallic catalysts synthesized by DP was smaller and decreased with time, indicating that the interaction between Au-Pd nanoparticles could be weaker when using the DP method. Zhou et al. [71] synthesized Au/SBA-15 by DP using HAuCl4 aqueous solution whose pH was adjusted to 7.0. After filtration, washing, freeze-drying and calcination in air at 200 °C for 4 h, the pink colored catalyst conserved the 2D hexagonal ordered structure of the SBA-15, but most of the AuNPs (average size ranging between 10 to 15 nm) formed spherical aggregates outside the channels of SBA-15, as it can be depicted in Figure 8(a). Li et al. [63] prepared Au/MCM-41 (Au loading 1%wt) by pouring the as-synthesized support into a HAuCl4 solution. After mixing the slurry for 24 h at 80 °C, the solids were filtered, washed repeatedly in cycles of water and ethanol, and dried at room temperature. Then, the template was removed by refluxing the solid in HCl/EtOH solution at 78 °C for 24 h. The resulting material was reduced in a flow of 5% H2/Ar at 200 °C for 4 h. This catalyst displayed poor dispersion of AuNPs (particle size approximately in the range of 60–100 nm), which were located on the external surface of the MCM-41 support. Moreover, the catalyst showed low activity for the cyclohexane oxidation. Under comparable reaction conditions, cyclohexane conversion was less than that obtained with the 1%Au/SBA-15 catalysts prepared by post-grafting of the SBA-15 support with APS (<1% vs. 15.5%). Liu et al. [174] synthesized Au/SBA-15 by DP, by the addition of the SBA-15 support to an aqueous solution of HAuCl4 whose pH was adjusted to 10 by adding ammonia solution. After stirring for 2 h at room temperature, the water was evaporated, and the sample was freeze dried for more than 24 h, and finally the solids were calcined at 200 °C for 2 h in vacuum. The resulting catalyst, with AuNPs of about 10 nm, was active for the oxidation of benzyl alcohol.
Mokhonoana et al. [111] incorporated AuNPs into MCM-41, using ethylenediamine which served as both a base and complexing agent for the Au(III) species, through a modified DP method. An aqueous solution of HAuCl4 was mixed separately with both the as-synthesized MCM-41 (still containing CTABr) and the calcined MCM-41 for 1 h, and then the pH was adjusted to 10 using 1 M ethylenediamine solution. The slurry was stirred at room temperature for 13 h, and the recovered solids were washed with warm water to remove the chloride ions, dried and calcined at 500 °C during 12 h (for the as-synthesized MCM-41) or 400 °C during 4 h (for the calcined MCM-41). The resulting catalysts (4 and 5% nominal Au loading, respectively), containing average AuNPs of 12 and 10 nm, respectively, retained the ordered structure and high surface area of the MCM-41 material. However, upon calcination at 500 °C, the AuNPs aggregated and migrated to the surface. Nevertheless, both catalysts were active in the CO oxidation at T > 250 °C.
Au/HMM-2 (Au 2%wt) was prepared by DP of HAuCl4 aqueous solution in alkaline conditions (pH 11.7) and the resulting solid sample was reduced under H2 at 200 °C for 2 h [176]. The obtained purple powder with homogeneously dispersed AuNPs (mean diameter of 3.2 ± 0.5 nm) retained the 3D-hexagonal structure of the HMM-2 after gold loading. However, the catalytic activity of this material was not evaluated.
2.8. Incipient Wetness Impregnation
Incipient wetness impregnation with HAuCl4 solution has been considered as an inappropriate method for the synthesis of gold catalysts because it yields larger AuNPs (>30 nm), and for the reason that the residual chloride present on the surface may poison the active sites of the catalysts [18,22,34,36]. To circumvent this problem, Delannoy et al. [177] proposed a modified IWI method for the synthesis of gold catalysts supported on different oxides, including silica, involving a washing step with ammonia 1 M or with ammonium chloride 0.25 M, followed by calcination at high temperature (300 °C). The ammonia washing removes the chloride ligands responsible for the aggregation of AuNPs, and leads to the formation of an amino-hydroxo-aquo cationic gold complex [Au(NH3)2(H2O)2−x(OH)x](3−x)+, which interacts with the support surface either electrostatically or through grafting, preventing the gold leaching. Moreover, during calcinations, small AuNPs are formed. Nevertheless, this method has been hardly exploited for the Au/MSM catalyst formulation. On the other hand, it is important to underline that after the washing step with ammonia, some fulminating gold can be formed, and since it violently decomposes around 210 °C, a drying pretreatment must be introduced before calcination [178].
Gold catalysts supported on MCM-41 (Au loading of 1%wt) were prepared by Grams and Sobczak [105,106] by IWI of the support with HAuCl4. After mixing, the solids were dried at 100 °C for 5 h followed by calcination in air at 550 °C for 3 h. The resulting catalysts exhibited hexagonally ordered mesopores, as well as surface area, pore diameter and pore volume almost identical to the pristine MCM-41 support. However, the metallic AuNPs showed lower dispersion in comparison to the same catalysts prepared by co-precipitation in the presence of CTACl, and displayed diverse dimensions and shapes, making the estimation of their size difficult to carry out. Moreover, it was found that the presence of chloride ions in the surrounding of gold centers was approximately one order of magnitude lower for the catalysts synthesized by IWI than for those prepared by co-precipitation. This was attributed to the interaction of the gold precursor with the CTACl, leading to the formation of Au-Cl species incorporated into the walls of the MCM-41 material. Although both catalysts showed similar activity in the acetonylacetone (AcAc) cyclization (35 vs. 38% conversion), the selectivity towards methylcyclopentenone was higher for the catalysts prepared by co-precipitation, which was attributed to the presence of the chloride ions.
Liu et al. [174] prepared Au/SBA-15 by impregnation of the support with an aqueous solution of HAuCl4. The slurry was stirred for 2 h at room temperature, and after evaporating water; the solids were dried for more than 24 h using a freeze drier, and then calcined under vacuum at 200 °C for 2 h. The resulting catalyst containing AuNPs of about 10 nm was active for the oxidation of benzyl alcohol, but its activity was slightly lower than that obtained with the Au/SBA-15 synthesized by deposition-precipitation, which displayed similar AuNPs size.
Araki et al. [148] impregnated FSM-16 with HAuCl4 in aqueous solutions at various pH values (5, 7 and 10). The FSM-16 was added to the HAuCl4 solution, and the mixture was stirred at room temperature for 24 h. After evaporation and washing with water, the Au3+/FSM-16 (Au loading of 2.5%wt) samples were reduced with H2 at 400 °C for 2 h or by photoreduction by irradiation with UV light for 24 h after exposition to water vapor (20 Torr) for 2 h and then to methanol vapor (100 Torr) for 2 h. After reduction in H2, the samples impregnated at pH 5 gave a mixture of Au nano-wires (mean diameter of 2.5 nm and a mean length of 18.1 nm) and nanoparticles (10 nm). However, only AuNPs (mean diameter of 19.2 nm) were obtained on the external surface of the FSM-16 support, when UV was used. Moreover, as the pH value increased, only small AuNPs were formed, for both reduction methods. For example, after reduction in H2, the samples impregnated at pH 10 gave homogeneously dispersed AuNPs in the mesopores with mean diameter of 1.7 nm, whereas the photoreduction of the samples prepared at pH 10 gave AuNPs with a mean diameter of 2.5 nm in the mesopores. Althoug these AuNPs showed high catalytic activity in the CO oxidation reaction, the rate constant of the Au/FSM-16 synthesized at pH 10 was 40–80 times higher than those of other Au/FSM-16 catalysts, because of the high dispersion of small AuNPs.
The IWI method was used recently by Wu et al. [62] to prepare gold catalysts supported on SBA-15 and organically functionalized SBA-15 with VTES. The supports were dispersed in HAuCl4 solution after stirring for 2 h at room temperature; the slurries were sonicated for 20 min. The resulting products were then evaporated, dried at 60 °C for 48 h, and reduced under H2 at 250 °C for 2 h. From the characterization analysis it was found that: (i) the structure of the catalyst prepared with the functionalized SBA-15 collapsed during the impregnation process, while the catalyst synthesized using the unmodified SBA-15 as support exhibited the small-angle XRD profile typical of the well ordered mesostructures; (ii) the XPS analysis revealed that both catalysts presented AuNPs as metallic gold (Au0); (iii) the average diameters of the AuNPs were 8 and 9 nm for the catalysts synthesized using the unmodified and the functionalized SBA-15, respectively. These AuNPs were larger than those present on the catalysts prepared by one-pot synthesis process (∼5 nm), indicating that during the direct synthesis, the VTES led to the evenly dispersion of the AuNPs in the mesoporous silica support; (iv) both catalysts displayed almost the same activity and selectivity in the oxidation of cyclohexane, with a conversion of about 8% after 2 h of reaction. However, this activity was practically doubled when the catalyst prepared by one-pot synthesis process was used, due to the presence of smaller AuNPs.
Metallic AuNPs uniformly dispersed inside the pores of monolithic porous silica with average diameter of about 4 nm were obtained by Shi et al. [179] and Cai et al. [180] after more than two weeks of soaking the mesoporous support into HAuCl4 solution (0.03–0.05 M) at room temperature, followed by reduction in H2 at 700 °C for 1 h. However, the catalytic activity of this material was not evaluated.
Tsung et al. [181] incorporated AuNPs with diameters in the range of 5–25 nm within the mesoporous silica nanofibers, by impregnation overnight with HAuCl4 and subsequent reduction with H2 at temperatures ranging between 55 and 100 °C. In this procedure, dichloromethane was added before the reduction step, to induce the gold precursor adsorbed on the outer surfaces of the nanofibers to move into the pore channels. However, no catalytic activity was reported.
Lotz and Fröba [182] incorporated AuNPs into the mesopores of SBA-15 using the cluster compound Au55(PPh3)12Cl6 (diameter of 2.1 nm) as gold precursor. The cluster was dissolved in dichloromethane. After addition of the SBA-15 support, the slurry was stirred for 30 min at room temperature, the solvent was evaporated and the solids were dried under vacuum, followed by annealing in air at 150–250 °C. The resulting material retained the structure of the SBA-15, and the Au55 clusters decomposed to metallic gold within the pores by annealing. However, the catalytic activity was not given.
AuNPs (2.5 ± 0.3 nm) closely packed in the mesopores of MCM-41-type mesoporous silica films were prepared by impregnation with HAuCl4 by Fukuoka et al. [176,183]. The silica materials were immersed in aqueous solution of HAuCl4. The mixtures were irradiated with ultrasonic wave for 30 s under reduced pressure, and the samples were left in the solution for additional 24 h. After washing with deionized water and drying under vacuum for 24 h, the resulting solids were reduced by H2 at 400 °C for 2 h or by UV-visible irradiation. With both reduction methods, the samples retained the structure of the original supports. However, the catalytic activity of these materials was not investigated.
The IWI method has been also employed for the synthesis of bimetallic Au catalysts. For example, Venezia et al. [184] prepared bimetallic AuPd/HMS catalyst by co-impregnating the HMS support with aqueous solutions of the two metal chlorides, PdCl2 and AuCl3, followed by a drying step at 120 °C and calcination at 400 °C for 2 h. The resulting catalysts, containing PdO nanoparticles (7 nm) and large AuNPs (18 nm), were active for methane oxidation. Li et al. [185] dispersed bimetallic Au-Pd nanoparticles within monolithic mesoporous silica by immersion of the silica host into HAuCl4 solution at room temperature. After soaking for two weeks, the samples were annealed in N2-H2 mixed gas at 500 °C for 2 h to reduce gold ions and form AuNPs within the pores of the silica host. The Au/silica samples were then immersed into a PdCl2 solution at room temperature for two weeks, and subsequently they were taken out and washed with deionized water, before annealing in the N2-H2 mixed gas starting at 500 °C for 1 h. With this method, an assembly of AucorePdshell nanoparticles into monolithic mesoporous silica was obtained, with an Aucore of about 4 nm. However, the catalytic activity of this material was not reported.
Tsoncheva et al. [175] used HAuCl4 and AuCl(PPh3), and Fe2O3 as gold and iron precursors, respectively, to synthesize Au/SBA-15 and AuFe/SBA-15 materials. After impregnation of the support with the gold and iron precursors, the solids were calcined in air at 200–400 °C for 2 h, and the resulting solids contained 2% Au and 12% Fe. From the analysis performed in this work, it was pointed out that: (i) The monometallic Au materials showed similar surface area than the pristine SBA-15, but the surface area of the bimetallic materials was reduced about 50% after the metals loading. A similar tendency was observed for the total pore volume of the synthesized materials; (ii) The low angle XRD patterns of the SBA-15-based materials were typical for SBA-15 structure and no substantial changes were observed after the modification with Au and Fe; (iii) The temperature of calcination had a significant effect on the AuNPs size. A broad size distribution of the AuNPs (small, about 6–9 nm and larger ones, 20–50 nm) was observed after calcination 200 °C, whereas predominantly larger particles (50–70 nm) were found after calcination at higher temperatures (300–400 °C). On the other hand, no reflections of Fe2O3 crystalline phase were observed by XRD analysis, indicating the formation of highly dispersed iron oxide phase; (iv) The monometallic and bimetallic catalysts were active for the ethylacetate combustion. However, the catalytic activity of monometallic catalysts was higher when AuCl(PPh3) was used as gold precursor, whereas the catalytic activity of bimetallic catalysts was higher when HAuCl4 was used as gold precursor. Unfortunately, neither the role of the different gold precursors nor the comparison of the catalytic activity of monometallic and bimetallic catalysts was discussed.
2.9. SiO2-Based Gold Catalysts
Another strategy to incorporate AuNPs into mesoporous silica is to modify the SiO2 materials with inorganic additives and synthesize SiO2-based gold catalysts. This approach allows the increase of the isoelectric point of the support, which facilitates the deposition of the Au(OH)xCl4−x− species under alkaline conditions required for DP method. At the same time, the sintering is minimized by the separation of AuNPs from one another. Moreover, when adding inorganic additives, such as metal “active” oxides for example, they can also work as co-catalysts taking part in the reaction [186]. Furthermore, the “active” oxides may be responsible of the electronic interactions and structural defects, factors that could represent the key issues in the formation and stabilization of small AuNPs, which may enhance the catalysts activity [187]. Therefore, silica-based materials have been largely employed as supports for AuNPs, as summarized in Table 6. In Figure 9 the distribution of AuNPs on different mesoporous SiO2-based supports is depicted, while some synthesis procedures and catalytic applications are briefly discussed below.
In the late of 90s, the Haruta's group initiated the investigation of the catalytic activity of gold deposited on titanium-containing mesoporous MCM-41 and MCM-48 materials, for the partial oxidation of propylene to propylene oxide, in the presence of oxygen and hydrogen [188–194,218–223]. They prepared Ti-doped MCM-41 and MCM-48 by means of a sol-gel process using tetrabutylorthotitanate [Ti(OC4H9)4] as titanium source, and then gold (4–12%wt) was loaded on these supports by DP (T = 70 °C, pH = 6.5–7.0) or by liquid grafting methods using HAuCl4 or (CH3)2Au(O2C5H7) as gold precursors, respectively. After calcination at 300 °C for 4 h, AuNPs with diameters ranging between 2.0 and 5.0 nm, were homogeneously dispersed on the Ti-modified MCM supports. Although these catalysts showed low propylene conversion (∼5%), their selectivity towards propylene oxide was very high (>90%). Moreover, Au/Ti-MCM-48 catalysts showed better performance in comparison with the Au/Ti-MCM-41 catalyst, due to the three-dimensional pore system of the MCM-48 material. On the other hand, is was pointed out that under the same reaction conditions, catalysts with the same gold loading on pure MCM silica, and samples of Ti-MCM without gold loading were catalytically inactive, indicating the existence of synergetic effects between gold and titanium in these catalysts.
Ruszel et al. [198] synthesized Au/Ti-SBA-15 catalysts (1%wt Au) impregnating first the mesoporous SBA-15 with titanium isopropoxide in isopropanol, followed by filtration, drying, and calcination at 400 °C. The modified Ti-SBA-15 support was loaded with gold by DP at room temperature and pH values of 9.5–10, using an aqueous solution of AuCl3 as gold precursor. After filtration, washing and drying, the solids were calcined at 350 °C for 4 h. The catalytic activity of the resulting catalysts was evaluated in the CO oxidation of at low temperature, including “preferential” oxidation of CO in the presence of H2 (PROX). From the analysis carried out in this study, it was concluded that the increase of the Ti content, leads to the decrease of the amount of the smallest AuNPs (1–2 nm), and simultaneously contributes to the formation of large AuNPs (5–8 nm). Consequently, the Ti content had a major influence on the catalytic activity. The high activity was observed for the catalysts containing 0.13–1.32%wt of Ti, for which AuNPs of sizes between 1 and 4 nm were dominating. However, catalysts without Ti were inactive. Moreover, these authors reported that the performance of the best Au/Ti-SBA-15 catalyst in the PROX CO oxidation was better than that of the Au/TiO2 reference catalyst, distributed by World Gold Council.
A layer-by-layer approach was reported by Yan et al. [199,200], who modified the surface of mesoporous SBA-15 with TiO2 via hydrolytic surface sol-gel process, by mixing the SBA-15 with proper amounts of titanium (IV) butoxide in a mixture of anhydrous toluene and anhydrous methanol. After vacuum filtration, washing with anhydrous methanol and vacuum drying, the monolayer of titanium oxide was hydrolyzed with an excess of deionized water. A second TiO2 layer was coated following the same procedure, after washing thoroughly with anhydrous methanol and drying. Subsequently gold was loaded on the surface of the modified support using the DP method at pH 10 and 60 °C with HAuCl4 solution. After stirring for 2 h, the precipitates were separated by centrifugation, washed with deionized water and dried at 40 °C overnight. The resulting catalysts showed uniform ultra small AuNPs (0.8–1.0 nm) assembled inside the ordered mesopores. These AuNPs, present as oxidized (Au3+) and metallic (Au0) gold species, displayed high catalytic activity for low-temperature CO oxidation.
Titania has been also used to modify the surface of HMS before the loading of AuNPs through an in situ process, using DDA as template, TEOS, tetrabutylorthotitanate as silica and titanium sources, and HAuCl4 as gold source [209]. After template removal by ethanol extraction at room temperature, the dried powder was calcined at 400 °C. The resulting Au/Ti-HMS catalyst (Au loading of 3.9%wt), containing AuNPs of sizes <20 nm, showed high activity in the direct synthesis of H2O2 from H2 and O2 in methanol solvent. Additionally, high removal rates of bulky sulfur compounds were obtained using the in situ generated H2O2 over the synthesized Au/Ti-HMS catalyst.
Chiang et al. [201,202] incorporated AuNPs over an acidic Al-SBA-15 thin film supports, prepared by mixing pluronic P123 with sodium aluminate in water, followed by the addition of a surfactant mixture solution including CTABr and SDS. The pH and temperature of this mixture were adjusted in the range of 5–6, and 40–50 °C, respectively. The as-synthesized Al-SBA-15 materials were functionalized with APS by ethanol refluxing for 12–24 h at 80–90 °C. Subsequently, gold was loaded using HAuCl4 aqueous solution as gold precursor. After stirring, the solids were filtered and dried at room temperature. Finally, the oxidized Au3+ species were chemically reduced by means of NaBH4 aqueous solution. Before the catalytic tests in CO oxidation, the catalyst were activated by heating to 600 °C under 10% H2/N2 flow for 1 h, followed by cooling step to room temperature under N2. The resulting catalysts (Au loading of 16.22–19.83%wt), containing AuNPs of about 2.7 nm uniformly distributed within the channels of the support, exhibited high activity for CO oxidation at 80 °C, and this activity attained the highest value of ca. 97% CO conversion when using a Si/Al molar ratio of 39. Moreover, it was reported that most of the Al was present in the tetrahedral framework well-dispersed site, which contributed not only to generate acidity, but also to limit the Au particle size. In an attempt to elucidate the effects of different aluminosilicate supports, the same authors prepared Au supported on Al-SBA-15 thin films and on Al-SBA-15 normal form (Au loading of ∼17%wt), using APS and MPS as functionalizing agents [202]. After H2 reduction at 600 °C, the size of AuNPs was almost similar for all the supports (∼3.0 nm). However, they found that hydrogen reduction is necessary to activate the catalysts supported on Al-SBA-15 thin films, whereas for the normal Al-SBA-15 support the activation must include a previous calcination step at 560 °C for 6 h before reduction under H2, to obtain good catalytic performance of CO oxidation at 80 °C.
Mixed mesoporous silica samples (MCM-41 and aerosol-derived silica) with Co, Al and Fe as heteroelements (Si/heteroelement molar ratio of 30) were synthesized by Bore et al. [195]. These modified materials were subsequently functionalized with amine groups (APS) before gold loading (5%wt) with HAuCl4 at neutral pH. After reduction under H2 at 200 °C for 2 h, the resulting SiO2-based gold catalysts were active for CO oxidation. The size of the AuNPs of these catalysts was less than 2 nm for all samples, except for Au/Co-MCM-41 and Au/Fe-Aerosol silica. These authors concluded that the addition of heteroelements did not lead to a significant improvement in the thermal stability of catalysts, but contributed to increase the catalyst activity compared to the pure silica support. Among the evaluated catalysts, the Au/Co-doped aerosol silica and the Au/Co-MCM-41 showed the highest and lowest reactivity for CO oxidation, respectively.
Zepeda et al. [210] modified hexagonal mesoporous silica (HMS) with Fe3+, Ce4+ and Ti4+ ions prior to the incorporation of Au species by deposition-precipitation (nominal Au loading of 3.5%wt). The added heteroatoms were incorporated principally into the HMS framework, and contributed to the stabilization of AuNPs, which were located mainly in the internal pore network of the support. Gold particle sizes of 4.6, 5.2 and 7.4 nm were obtained for Au/HMS-Fe, Au/HMS-Ti and Au/HMS-Ce, respectively. The catalytic activity of the H2 reduced samples (300 °C, 2 h) for total and preferential CO oxidation followed the trend: Au/HMS-Fe > Au/HMS-Ce > Au/HMS-Ti.
Recently, Zepeda's group [217] proposed a “green-synthesis” process for the synthesis of monometallic Au (Au loading of 2.05%wt) and bimetallic Au-Ag (Au and Ag loadings of 0.88 and 1.32%wt, respectively) catalysts supported on commercial mesoporous SiO2-Al2O3. In their approach, Camellia sinensis plant extract was used as biological reducing agent. The catalysts were synthesized by IWI of the support with HAuCl4 and AgNO3. After addition of each one of metal precursor solutions, Camellia sinensis extract (2 mL) was added to the slurry, followed by vigorous stirring for 48 h at room temperature. Then, the samples were dried at 100 °C in air flow. For both catalysts, it was observed that after few minutes of the Camellia sinensis extract addition, the color of the solutions changed drastically, indicating the reduction of both Au3+ and Ag1+ ions. The resulting catalysts preserved the structural parameters of the SiO2-Al2O3 support. Moreover, from TEM images, it was observed that in both catalysts the metallic particle sizes varied between 1.5 and 6.5 nm, with a mean value of about 4.5 nm. However, the bimetallic catalyst presented higher amounts of AuNPs of size lower that 4.5 nm, in comparison with the monometallic catalyst, indicating that the presence of Ag contributes to the regular morphology and dispersion of metallic particles. On the other hand, metallic Au0 (81%) and oxidized Au+1 (19%) species were found in the monometallic catalysts, whereas only metallic Au0 species were present in the bimetallic catalyst, which also contained metallic Ag0 (93%) and oxidized Ag+1 (7%) species, demonstrating that the use of Camellia sinensis led to the formation of metallic nanoparticles. After catalytic activity evaluation in CO oxidation, it was found that bimetallic catalyst displayed higher activity than the monometallic sample. At a reaction temperature of 270 °C, 100% CO conversion was obtained with the bimetallic Au-Ag catalyst, whereas the CO conversion for the monometallic sample was 85%. Moreover, both catalysts were more active than the monometallic catalyst synthesized by DP and calcined at 473 °C for 3 h. Conversely, the activity of the monometallic Au catalyst in the CO hydrogenation was 1.6 times more active than the bimetallic catalyst, and 2 times more active than the monometallic catalyst synthesized by DP. In this interesting “green” approach study, the high activity of the bio-reduced catalysts was mainly attributed to the small metallic particles obtained, because of the absence of calcination or reduction steps at high temperature. Moreover, it was hypothesized that hydroxyl and carboxylic acid functional groups present the Camellia sinensis extract, could be responsible of the bio-reduction process and the stabilization and protection as capping agent of the AuNPs, respectively.
2.10. Other Methods of Synthesis
In the previous sections we presented the main methods for the synthesis of gold catalysts supported on MSM. However, because of the advances in nanotechnology, other less-conventional methods, using different technologies and gold precursors, have been published. Although these methods have not been largely employed, and the catalytic activity of the synthesized materials has not always been investigated, it is worth to mention some of them.
Chatterjee et al. [224] deposited gold into the channels of MCM-48 (Au loading of ca. 5%wt) through a process involving H2-assisted reduction of HAuCl4 (aqueous solution) in supercritical CO2. The resulting catalysts, containing AuNPs varying between 2 and 25 nm, were active for the selective hydrogenation of crotonaldehyde. Moreover, it was reported that the size of AuNPs can be controlled by tuning the CO2 pressure, without disturbing the mesoporous structure. Similarly, Gupta et al. [225] loaded pre-synthesized Au nanocrystals (mean diameter of 2.2 ± 0.3 nm) dispersed in toluene into mesoporous SBA-15 and MCM-41 (2%wt), using supercritical CO2 as antisolvent. It was estimated that approximately 10–100 Au nanocrystals per pore were infused inside the mesoporous silica. However, the catalytic activity of these materials was not evaluated.
Campelo et al. [226] synthesized Au/SBA-12 catalyst (Au loading of 1.2%wt) in a conventional microwave oven, using gold bromide dissolved in ethanol-acetone mixture (1:1v/v), as Au precursor. They obtained well and homogeneously dispersed AuNPs of ca. 1.9 nm, highly active for styrene oxidation.
Corma et al. [227] prepared Au/MCM-41 (loading of 0.20–30 mmol-Au/g support) using a gold-carbene complex [(carbene)AuCl] as Au precursor. The resulting catalysts were active for the hydrogenation of alkenes and the Suzuki cross-coupling reaction between halobenzene and arylboronic acids.
Small Au clusters (∼1 nm) have been deposited on mesoporous SBA-15, MCF and HMS using triphenylphosphine-protected Au11 (Au11:TPP) clusters as gold precursor [174,228]. To do this, the Au11:TPP clusters and the support were dispersed in CH2Cl2/C2H5OH (80/20) and stirred for 2 h. After filtration, the resulting solids were calcined at 200 °C for 2 h under vacuum. The resulting catalysts were active, highly selective and reusable for the oxidation of various alcohols with H2O2, such as benzyl alcohol, under microwave heating.
Silver-catalyzed electroless deposition was used to incorporate AuNPs with sizes of 5.0 to 9.0 [229] and ca. 6.4 nm [230] within the channels of mesoporous SBA-15, but the catalytic activity of the obtained materials was not reported.
Chen et al. [231–235] and Sun et al. [236] have reported a sonochemical method for the incorporation of AuNPs into mesoporous silica at room temperature. The AuNPs ranging from 2 to 9 nm were dispersed uniformly within the pores of the silica support by the immersion of the host material into a chloroauric acid solution (0.8 mmol/L) containing 0.2 mol/L of isopropanol during 2 to 3 weeks. The mixture was subsequently irradiated for various periods from 600 to 7200 s with 40 kHz ultrasonic waves at 100 W output power. The filtered solid was washed with distilled water and dried at 120 °C for 30 min. After ultrasonic irradiation, the AuNPs were formed within pores of the mesoporous silica. However, no catalytic activity was reported.
The bio-assisted synthesis of AuNPs supported on mesoporous silica tubes was recently reported by Jan et al. [237], who deposited synthetic polypeptides (PLL and PLT) on the walls of polycarbonate membrane pores to form multilayer films for in-situ mediating the formation of AuNPs embedded in porous silica nanostructures. After calcination at 400–500 °C for 3–10 h, the resulting materials having AuNPs of ∼6.5 nm, displayed promising catalytic activity for the reduction of p-nitrophenol.
Recently, Deng et al. [238] prepared multifunctional microspheres with a core of nonporous silica-protected magnetite particles, a transition layer of active gold nanoparticles, and an outer shell of ordered mesoporous silica with perpendicularly aligned pore channels. These catalytic materials showed excellent catalytic activity for the reduction of 4-nitrophenol as well as highly selective epoxidation of styrene using t-butyl hydroperoxide as oxidant. Moreover, the resulting catalysts containing nanocrystals of 12.2 and 8.3 nm for Au and magnetite nanoparticles, respectively, presented the surface evenly covered by AuNPs and displayed the three low-angle XRD diffraction peaks typical of 2-D hexagonal mesostructures.
Lee et al. [239] fabricated a nanoreactor framework of a Au/SiO2 yolk/shell structure through selective etching of metal cores from Au/SiO2 by treatment with KCN. Although gold core diameters showed high values (43–104 nm) depending on the added amount of KCN, these Au/SiO2 yolk/shell particles were active for the catalytic reduction of p-nitrophenol by NaBH4.
3. Conclusions and Outlook
The high catalytic activity of gold at the nanoscale discovered in the late 1980s has been one of the most exciting discoveries in the field of catalysis and, since then, gold catalysts have been synthesized on a variety of metal oxides supports. Particularly, silica was considered an inappropriate support for AuNPs, until the revolutionary work of Haruta's group [37,45,144,145] confirmed that silica materials could be suitable supports for AuNPs using appropriate synthesis methods. Since the chemical vapor deposition approach employed in those studies requires special equipment and expensive gold precursors, extensive research has been developed to synthesize gold catalysts supported on mesoporous silicas, such as MCM-41, MCM-48, SBA-15 and SBA-16, because of the excellent properties of these materials as catalytic supports. Mesoporous SBA-15 has proven to be a better support for gold catalyst in comparison with other mesoporous silica materials, including HMS, MCM-41, MCM-48, SBA-16, because it has a two-dimensional (2D) hexagonal pore structure, large pore size and strong pore wall, allowing the immobilization and stability of AuNPs inside the pores [56,62,70].
Although the catalytic activity of Au/MSM catalysts has been evaluated in numerous catalytic reactions, as illustrated in Tables 1–6, from the published results one can say that gold catalysts supported on mesoporous silicas would have a significant impact on the redox type reactions in liquid phase, in which the mesoporous architecture could enhance the diffusivity of bulky reactants and products, while the AuNPs are kept inside the pores. However, the main problem of the synthesis of Au/MSM catalysts lies in obtaining very small (about 2 nm) and well dispersed AuNPs. To overcome these problems, the main strategies and their related drawbacks can be summarized as follows:
Because of the low isoelectric point of silica (∼2.0), the conventional IWI and DP methods are not appropriate for the incorporation of gold nanoparticles (AuNPs) into MSM, although some strategies such as washing with a base or reduction under hydrogen at high temperatures have been employed to synthesize active gold catalysts following these procedures. However, these approaches have not been largely employed, because the AuNPs are usually formed both inside and outside the channels, and the control of shape and particle size is not possible. Despite the fact that the use of pre-synthesized capped AuNPs contributes to circumvent this problem, and seems to be a promising approach in which the size and shape of gold particles can be controlled before the gold loading, this strategy has not been extensively applied in catalytic applications.
A general trend consists then in the modification of the silica materials either by direct or post-grafting functionalization with amine or thiol groups. These groups can act not only as ligands enhancing the interaction between the metal precursors and the silica surface, but also as mild reductants under basic conditions for in situ reduction of Au(III). Even if several active Au/MSM have been prepared using functionalized mesoporous silicas by direct or subsequent incorporation of the gold precursor, as summarized in Tables 1 to 3, the main problems related to these procedures lie in the followings facts: (i) The gold loading and particle size are correlated with the amount of organic functional ligands, but high concentrations of organic functional groups may hinder the condensation of silica precursors to form an ordered framework, when using direct functionalization, or significantly reduced surface area, pore diameter and pore volume, when using the post-grafting functionalization; (ii) The post-synthetic functionalization of MSM does not allow the homogeneous distribution of the organic functional groups on the pore surface, and consequently, the AuNPs are not homogeneously distributed after gold loading; (iii) The calcination step required for the removal of functional groups, may contribute to the formation of large AuNPs, with the inherent negative effect on the catalytic activity; (iv) When using the simple way of direct incorporation of the gold precursor during the mesoporous silica synthesis, the AuNPs are not necessarily placed within the pores, which can lead to metal leaching and poor life of catalysts.
The modification of silica supports with other metal oxides to produce SiO2-based gold catalysts, is also an interesting approach, because it allows modifying the isoelectric point of the silica, facilitating the incorporation and dispersion of the AuNPs by simple deposition-precipitation of gold precursor, as summarized in Table 6, while simultaneously the functionalizing oxides may take part in the reaction as co-catalysts for the enhancement of the catalytic activity.
Despite the fact that rapid advances in nanotechnology have allowed the emergence of new sophisticated methods for preparing Au/MSM catalysts, it is worth considering the feasibility of the synthesis procedures for applications other than at a laboratory scale. Some disadvantages of various preparation methods lie in the fact that the experimental procedures are not suitable for general applications because of the requirements of special equipment and/or expensive or complicated synthesis of the gold precursors. Moreover, in many cases, the catalytic activity of such catalysts has unfortunately not always been evaluated.
Thus, considering that the success of the introduction of AuNPs into MSM requires the fulfillment of three criteria [43]: (i) retaining the order structure of the MSM; (ii) achieving very small uniform and well dispersed AuNPs; and (iii) attaining high and controlled gold loading; the use of gold complex Au(en)2Cl3 (en = ethylenediamine) can be regarded as a promising precursor for the synthesis of not only Au/MSM, but also of gold supported on amorphous silicas. Although this precursor has not been widely used, the catalysts prepared using Au(en)2Cl3 have displayed not only small and well dispersed AuNPs, but also high catalytic activities in both gas and liquid phase oxidation reactions [44,132].
Finally, it is true to say that gold catalyst has become a hot topic for chemical reactions; however, there are still challenges ahead for the development of Au/MSM satisfying the main requirements of any catalyst: a synthesis method as easy and inexpensive as possible with high activity, selectivity and stability. Moreover, the identification of the active gold species is still a challenging task for most of the chemical reactions catalyzed by gold.










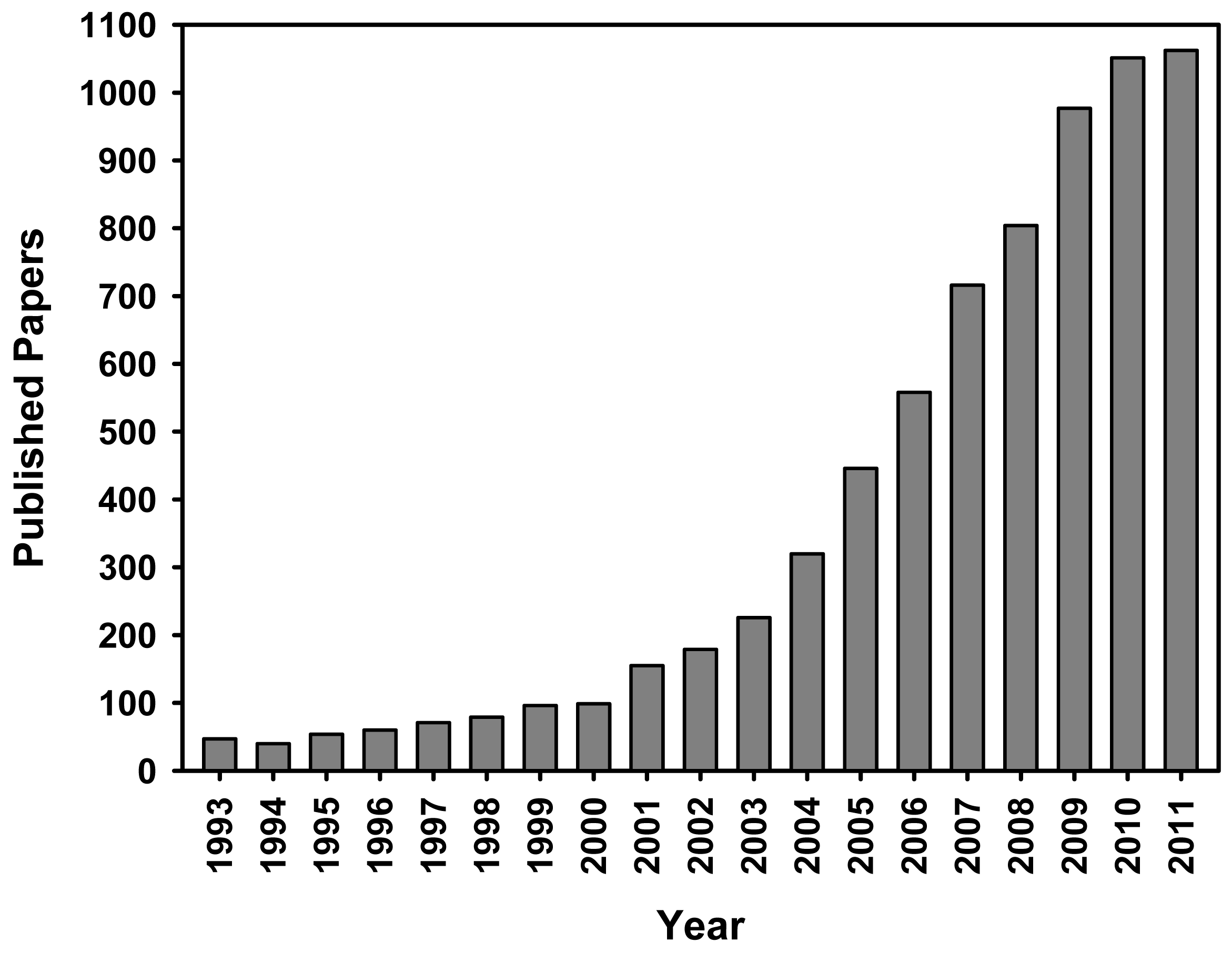{kind=link}
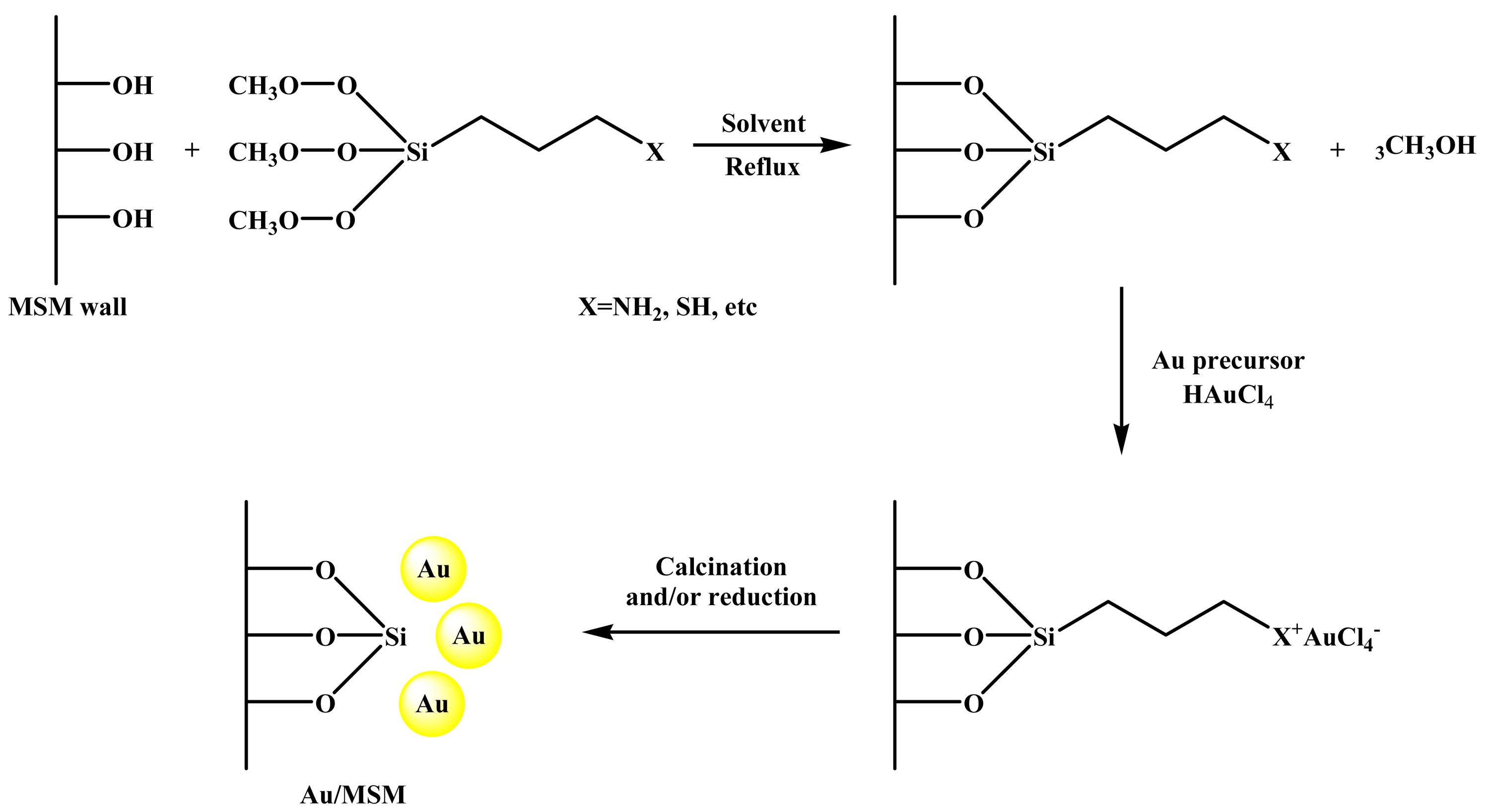{kind=link}
{kind=link}
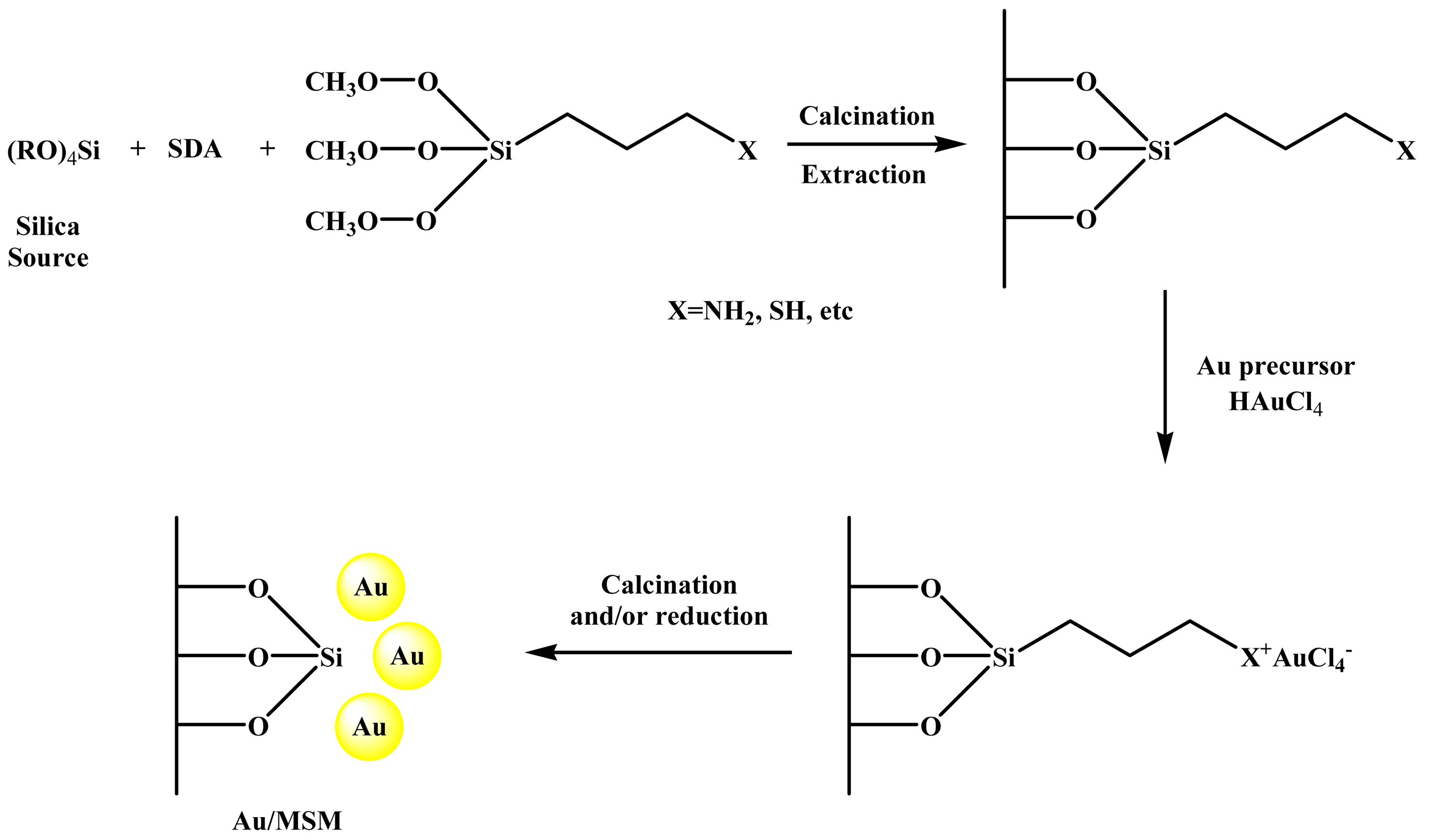{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
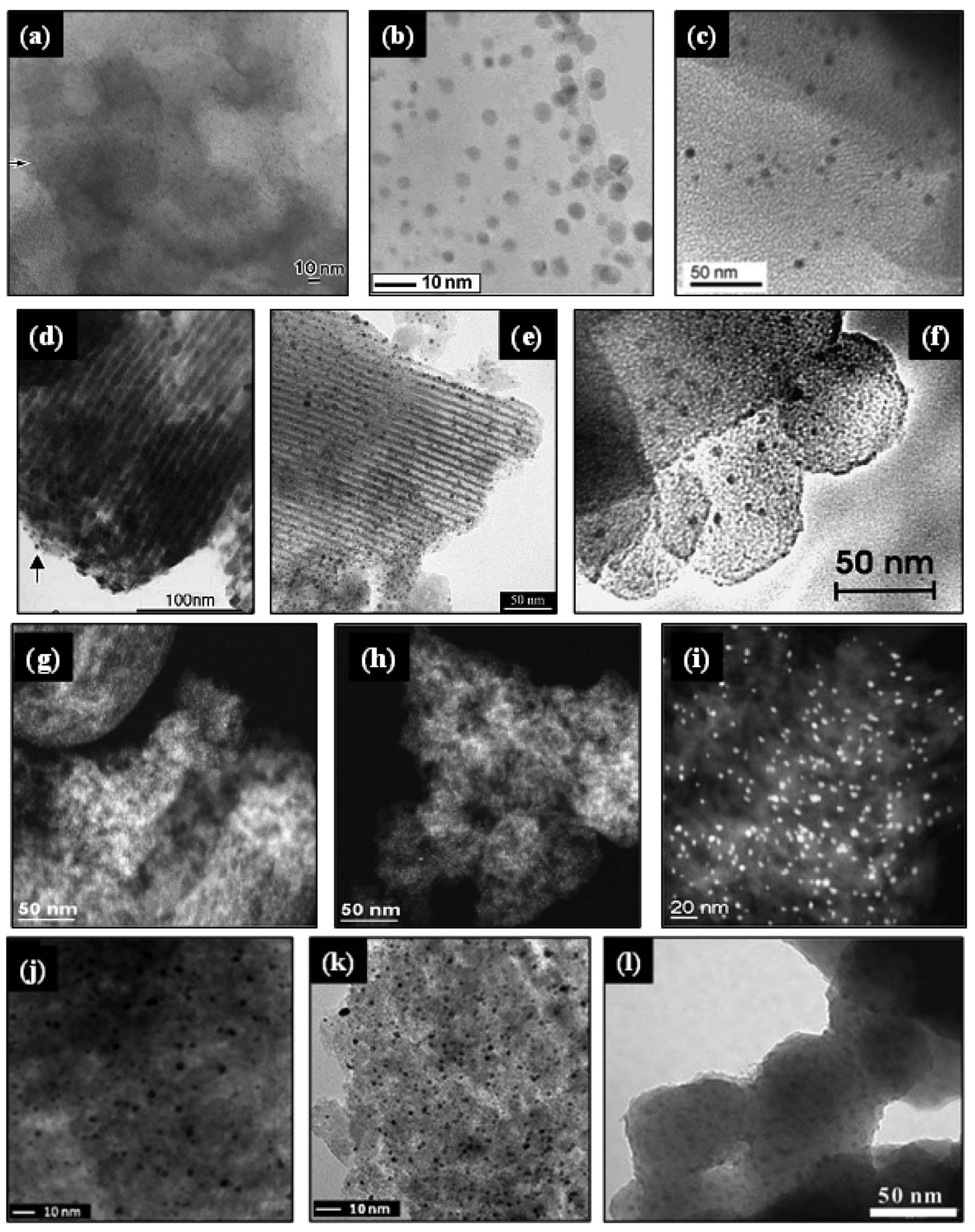{kind=link}
| Support | Functional ligand | Au Precursor | Au loading (%wt) | Au size (nm) | Catalytic application | Reference |
|---|---|---|---|---|---|---|
| SBA-11 | APS | HAuCl4 | 5.0 | 6.0 ± 1.9 | CO oxidation | [55,56] |
| SBA-12 | APS | HAuCl4 | 5.0 | 4.2 ± 0.8 | CO oxidation | [55,56] |
| SBA-15 | OBSQ | HAuCl4 | 1.4 | 1.6 | Epoxidation of olefins | [39] |
| SBA-15 | APS | HAuCl4 | 5.0 | 4.1 ± 0.8 | CO oxidation | [55,56] |
| SBA-15 | APS | HAuCl4 | 4.0 | 5.8 | CO oxidation | [54] |
| SBA-15 | APS | HAuCl4 | 3.86 | 2.8 | Cyclohexane oxidation | [57] |
| SBA-15 | MPTS | HAuCl4 | 4.53 | 6.5 | Cyclohexane oxidation | [57] |
| SBA-15 | TPTAC | HAuCl4 | 4.8 | 4.5 | CO oxidation | [58] |
| SBA-15 | MPS and APTS | HAuCl4 | 4.0 | 10.0 | Benzyl alcohol oxidation | [59] |
| SBA-15 | MPS and APTS | HAuCl4 | 4.0 | 5.0 | [59] | |
| Pd(NO3)2 | 0.5 | |||||
| SBA-15 | APS | HAuCl4 | 5.0 | 1.0–4.0 | Hydrogenation of crotonaldehyde | [60] |
| SBA-15 | APS | HAuCl4 | 4.6–4.9 | 1.0–5.0 | [60] | |
| In(NO3)3 | 0.5–1.5 | |||||
| SBA-15 | MPS | HAuCl4 | 2.0 | 3.4–6.6 | CO oxidation | [61] |
| SBA-15 | VTES | HAuCl4 | 1.0 | 9.0 | Cyclohexane oxidation | [62] |
| SBA-15 | APS | HAuCl4 | 0.5–1.5 | 3.0–8.0 | Cyclohexane oxidation | [63] |
| SBA-15 | MPTS | HAuCl4 | 4.53 | 5.0–8.0 | n-Hexadecane oxidation | [64] |
| SBA-15 | APTS | HAuCl4 | 1.0 | 4.0–13.0 | Benzyl alcohol oxidation | [65] |
| SBA-15 | PAMAM | HAuCl4 | 10.83–12.51 | 1.0–6.0 | Oxidation of alcohols | [66] |
| SBA-15 | APS | HAuCl4 | 18.0 | 3.0–5.0 | Glucose oxidation | [67] |
| SBA-15 | TPED | HAuCl4 | 2.6 | 2.0–3.0 | Cyclohexene oxidation | [68] |
| SBA-15 | Py | HAuCl4 | 2.7 | 2.0–3.0 | Cyclohexene oxidation | [68] |
| SBA-15 | MPS | HAuCl4 | 3.0 | 2.7–6.4 | CO oxidation | [69] |
| SBA-15 | APTS | HAuCl4 | - | 2.1–10.3 | - | [70] |
| SBA-15 | APTS | HAuCl4 a | - | 4.5–19.4 | - | [70] |
| SBA-15 | TPED | HAuCl4 | - | 5.8–11.7 | - | [70] |
| SBA-15 | TPED | HAuCl4 a | - | 4.5–23.3 | - | [70] |
| SBA-15 | TPTAC | HAuCl4 | 1.7 | 3.0 | - | [71] |
| SBA-15 | MPS | Au colloids | 2.1 | 5.0–7.0 | - | [72] |
| SBA-15 | APS and Glycidol | HAuCl4 | 9.0 | 5.0–10.0 | - | [73] |
| SBA-15 | NH3 | HAuCl4 | - | - | - | [74] |
| SBA-15 | TPTAC | HAuCl4 | 4.0–9.0 | 1.0–5.0 | - | [75] |
| SBA-15 | APTS | HAuCl4 | 6.0 | 3.0–5.0 | - | [76] |
| SBA-15 | TPTAC | HAuCl4 | 2.7–3.0 | <7.0 | - | [77] |
| SBA-15 | G4-PAMAM | HAuCl4 | 0.5% mol | 1.5 | Microwave-assisted Suzuki-Miyaura coupling reaction | [78] |
| K2PdCl4 | ||||||
| SBA-15 | APTS | HAuCl4 | 6.0 | 2.9 | CO oxidation | [79] |
| AgNO3 | ||||||
| SBA-16 | APTS | HAuCl4 | 1.0 | 2.0–9.0 | Benzyl alcohol oxidation | [65] |
| SBA-16 | APTS | HAuCl4 | 0.89 | 3.0–11.0 | Benzyl alcohol oxidation | [80] |
| SBA-16 | APTS | HAuCl4 | 0.24–1.15 | 2.0–12.0 | Benzyl alcohol oxidation | [80] |
| PdCl2 | 0.24–0.55 | |||||
| SBA-16 | APTS | HAuCl4 | - | 2.4–11.4 | - | [70] |
| MCM-41 | APS | HAuCl4 | 4.0 | 5.1 | CO oxidation | [54] |
| MCM-41 | APS | HAuCl4 | - | 3.5 ± 0.5 | Styrene hydrogenation | [81] |
| MCM-41 | APS | HAuCl4 | 5.0 | 3.9 ± 0.7 | CO oxidation | [55,56] |
| MCM-41 | APTS | HAuCl4 | 1.0 | 2.0–11.0 | Benzyl alcohol oxidation | [65] |
| MCM-41 | APS | HAuCl4 | 5.0 | 3.9 ± 0.9 | CO oxidation | [82] |
| MCM-41 | MPTS | Au colloids | - | 52.4 | - | [83] |
| MCM-41 | APTS | HAuCl4 | - | 1.9–21.0 | - | [70] |
| MCM-48 | APS | HAuCl4 | 4.0 | 6.9 | CO oxidation | [54] |
| MCM-48 | APTS | HAuCl4 | - | 1.4–27.2 | - | [70] |
| HMM-2 | APS | HAuCl4 | 5.0 | 4.7 ± 0.7 | CO oxidation | [55,56] |
| PMOs | APS | HAuCl4 | - | 50.0 | Reduction of methylene blue | [84] |
| PMOs | C18TMS-APS | HAuCl4 | - | 3.0–5.0 | CO oxidation | [84] |
| Aerosol derived SiO2 | APS | HAuCl4 | 5.0 | 1.4 ± 0.2 | [55] | |
| Aerosol silica | APS | HAuCl4 | 5.0 | 1.4 ± 0.2 | CO oxidation | [82] |
| Nanosilica | MPS | Au colloids | 1.0–5.0 | 6.3 | Cyclohexane oxidation | [85] |
| HMS | APS | HAuCl4 | - | 80.0 | Glucose oxidation | [86] |
| HMS | MPS | HAuCl4 | - | 80.0 | Glucose oxidation | [86] |
| FDU-12 | APTS | Au nanocrystals | 15.0 | 4.0 | - | [87] |
| MCF | C10H23ClSi | 15.0 | 4.0 | - | [87] | |
| MTFs | APS or TPED | HAuCl4 | 16.2 | 7.0 | - | [88] |
| HMS | APTS | HAuCl4 | - | 2.1–22.4 | - | [70] |
| HMS | APTS | HAuCl4 a | - | 2.3–18.3 | - | [70] |
| HMS | TPED | HAuCl4 | - | 8.7–20.8 | - | [70] |
| HMS | TPED | HAuCl4 a | - | 4.7–22.3 | - | [70] |
aDissolved in ethanol.
| Support | Functional ligand | Au precursor | Au loading (%wt) | Au size (nm) | Catalytic application | Reference |
|---|---|---|---|---|---|---|
| MCM-41 | APS | HAuCl4 | 3.1–3.2 | 3.4 ± 0.5 | Hydrogenation of olefinic substrates | [93] |
| MCM-41 | MPS | HAuCl4 | 3.2–3.5 | 3.2 ± 0.5 | [93] | |
| MCM-41 | OBSQ | HAuCl4 | 2.1 | 2.1 | Epoxidation of olefins | [39] |
| SBA-15 | MPS | HAuCl4 | 1.05–16.1 | 2.5 | - | [42,94] |
| SBA-15 | MPS | AuCl(THT) | 1.05–16.1 | 4.5 | - | [42,94] |
| SBA-15 | Mannitol derivative | HAuCl4 | - | 3.0–6.0 | - | [95] |
| SBA-15 | MPS | HAuCl4 | 3.0 | 2.0–6.5 | CO oxidation | [61] |
| SBA-15 | OBSQ | HAuCl4 | 1.0–5.5 | 1.8 | Styrene epoxidation | [92,96] |
| SBA-15 | OBSQ | HAuCl4 | 1.0–5.5 | 2.2 ± 0.5 | Benzyl alcohol oxidation | [92,96] |
| SBA-15 | OBSQ | HAuCl4 | 1.4–2.1 | 2.0 | Epoxidation of olefins | [39] |
| PMOs | Bis-silylated with disulfide unit | AuCl(THT) | 5.0–20.0 a | 2.0 | - | [97] |
| MSS | MPS and PrTMS | HAuCl4 | 1.0 | 1.0–15.0 | - | [98] |
| MSS | MPS | HAuCl4 | 14.0 | 4.8 | - | [99] |
| MSU | OBSQ | HAuCl4 | 2.0 | 4.7 | Epoxidation of olefins | [39] |
| KIT-5 | APS | HAuCl4 | ∼10.0 | 2.0–5.0 | - | [100] |
aAu/S molar ratio × 100.
| Support | Functional ligand | Au Precursor | Au loading (%wt) | Au size (nm) | Catalytic application | Reference |
|---|---|---|---|---|---|---|
| MCM-41 | - | HAuCl4 | 0.13–1.21 | - | Cyclohexane oxidation | [103,104] |
| MCM-41 | - | HAuCl4 | 1.0 | 3.0–18.0 | Acetonylacetone cyclization | [105] |
| MCM-41 | - | HAuCl4 | 1.0 | - | Methanol oxidation | [106] |
| MCM-41 | - | HAuCl4 | 1.0 | - | Methanol oxidation | [106] |
| C2NbO4 | ||||||
| MCM-41 | - | HAuCl4 | 1.0 | - | Methanol oxidation | [107] |
| C2NbO4 | ||||||
| MCM-41 | - | HAuCl4 | 0.386–2.02 | 3.0–6.0 | - | [108,109] |
| MCM-41 | - | HAuCl4 | 1.0 | - | Acetonylacetone cyclization and methanol oxidation | [110] |
| MCM-41 | - | HAuCl4 | 1.3 | 20.0 | CO oxidation | [111] |
| MCM-41 | - | HAuCl4 | 1.0 | - | Methanol oxidation | [112] |
| VOSO4 | ||||||
| MCM-41 | - | HAuCl4 | 1.0 | - | Methanol oxidation | [112] |
| VOSO4 | ||||||
| C4H4NNbO9 | ||||||
| MCM-41 | AAPTS | HAuCl4 | 0.7–7.0 | 2.0–5.0 | - | [43] |
| MCM-41 | TMPTA | HAuCl4 | - | - | - | [113] |
| MCM-41 | TMPTA | HAuCl4 | 2.2 | 2.0–3.0 | CO oxidation | [114] |
| MCM-48 | TMPDA | HAuCl4 | - | 5.0–20.0 | - | [40] |
| MCM | - | HAuCl4 | 8.0 | 6.7 | CO oxidation | [31] |
| MCM | - | HAuCl4 | 8.0 | 20.0–30.0 | CO oxidation | [31,115] |
| AgNO3 | 0/1–1/0 b | |||||
| SBA-15 | - | HAuCl4 | 0.77 | - | Cyclohexane oxidation | [103,104] |
| SBA-15 | - | HAuCl4 | 1.0 | 3.0–8.0 | Cyclohexane oxidation | [63] |
| SBA-15 | TMPDA | HAuCl4 | - | 5.0–50.0 | - | [40] |
| SBA-15 | MPS | HAuCl4 | 1.0 | 2.0–4.0 | Cyclohexane oxidation | [116] |
| SBA-15 | VTES | HAuCl4 | 1.0 | 5.0 | Cyclohexane oxidation | [62] |
| SBA-16 | TMPDA | HAuCl4 | - | 10–100 | - | [40] |
| ZSM-5 | - | HAuCl4 | 0.51–1.30 | - | Cyclohexane oxidation | [117] |
| ZSM-5 | - | HAuCl4 | 0.17–1.16 | 2.0–5.0 | - | [109] |
| MS | BTESPTS | HAuCl4 | 0.4–1.0 | 3.0–14.0 | Lactose oxidation | [4] |
| MS | BTESPTS (0.625) | HAuCl4 | 0.75 | 4.0–15.0 | Cyclohexane oxidation | [118] |
| MS | BTESPTS (1.25) | HAuCl4 | 0.94 | 3.0–8.0 | Cyclohexane oxidation | [118] |
| MS | BTESPTS (2.5) | HAuCl4 | 1.07 | 3.0–8.0 | Cyclohexane oxidation | [118] |
| MS | BTESPTS (5.0) | HAuCl4 | 1.10 | 4.0–12.0 | Cyclohexane oxidation | [118] |
| MS | BTESPTS | HAuCl4 | 0.25–10.0 | ∼3.0 | Oxidation of benzyl alcohol | [119] |
| MS | BTESPTS | HAuCl4 | 2.0–10.0 | ∼2.0 | n-hexadecane oxidation | [64,120] |
| MS | BTESPTS | HAuCl4 | 0.5–10.0 | - | Trichloroethylene oxidation | [121,122] |
| MS | MPS | HAuCl4 | 1.5–5.0 | CO oxidation | [123] | |
| MSTF | BTESPTS | Au(en)2Cl3 | 2.1–5.1 | 3.0–7.0 | - | [124] |
| LSX | - | Au salt | 0.646 | ∼3.0 | - | [109] |
| HSN | APS | HAuCl4 | 1.7 | 2.8–4.5 | 4-nitrophenol reduction | [125] |
| WSS | TMPTA | HAuCl4 | 1.6 | 2.6 ± 0.8 | CO oxidation | [114] |
| HMS | TMPTA | HAuCl4 | 0.5–1.0 a | 1.7–5.0 | - | [126] |
| PMOs | TMPTA | HAuCl4 | 0.5–1.1 a | 1.5–2.5 | - | [126] |
| MSU | TMPDA | HAuCl4 | 0.4 * | 1.7–5.0 | - | [126] |
aAu/Si molar ratio × 100;bAu/Ag molar ratio.
| Support | pH of synthesis | Au loading (%wt) | Au size (nm) | Catalytic application | Reference |
|---|---|---|---|---|---|
| SBA-15 | 6.0 | 2.70 | 5.4 | CO oxidation | [44,129] |
| SBA-15 | 7.4 | 5.98 | 5.2 | CO oxidation | [44,129] |
| SBA-15 | 8.5 | 6.90 | 4.9 | CO oxidation | [44,129] |
| SBA-15 | 9.6 | 9.08 | 4.9 | CO oxidation | [44,129] |
| SBA-15 | ∼10.0 | 2.20 | 2.0–8.0 | CO oxidation | [44,129] |
| SBA-15 | 12.0 | 2.00 | 4.9 ± 1.3 | Ethanol oxidation | [130] |
| SBA-16 | 12.0 | 2.00 | 7.3 ± 1.6 | Ethanol oxidation | [130] |
| MCM-41 | 12.0 | 2.00 | 6.7 ± 2.1 | Ethanol oxidation | [130] |
| HMS | ∼10.0 | 2.19 | 5.4 ± 0.2 | Aerobic oxidative esterification of benzyl alcohol | [131] |
| HMS-Ce | ∼10.0 | 2.94 | 5.9 ± 0.5 | [131] | |
| HMS-Ti | ∼10.0 | 2.96 | 4.8 ± 0.3 | [131] | |
| HMS-Fe | ∼10.0 | 2.87 | 4.1 ± 0.4 | [131] | |
| SBA-15-CeO2 | 10.0 | 0.7 | ∼5.0 | Lactose oxidation | [132] |
| Support | pH of synthesis | Au Precursor | Au loading (%wt) | AuNP size (nm) | Catalytic application | Reference |
|---|---|---|---|---|---|---|
| SBA-15 | 9.5 | HAuCl4 | 1.0 | 2.0–12.0 | Cyclohexane oxidation | [173] |
| SBA-15 | 7.0 | HAuCl4 | 4.0 | 10.0–50.0 | Benzyl alcohol Oxidation | [59] |
| SBA-15 | 7.0 | HAuCl4 | 4.0 | - | [59] | |
| Pd(NO3)2 | 0.5 | |||||
| SBA-15 | 7.0 | HAuCl4 | 0.39 | 10.0–15.0 | - | [71] |
| SBA-15 | - | HAuCl4 | 2.0 | 4.0–9.0 | Ethylacetate combustion | [175] |
| SBA-15 | - | HAuCl4 | 2.0 | - | [175] | |
| Fe(NO3)3 | 12.0 | |||||
| SBA-15 | 10.0 | HAuCl4 | 0.16–0.20 | - | Benzyl alcohol Oxidation | [174] |
| MCM-41 | - | HAuCl4 | 1.0 | 60.0–100.0 | Cyclohexane oxidation | [63] |
| MCM-41 | 10.0 | HAuCl4 | 5.0 | 9.0–13.0 | CO oxidation | [111] |
| MCM-41 | 10.0 * | HAuCl4 | 3.7 | 11.0–15.0 | CO oxidation | [111] |
| MCM-41 | 6.4 | HAuCl4 | 1.312 | 2.0–3.0 | - | [108] |
| HMM-2 | 11.7 | HAuCl4 | 2.0 | 3.2 ± 0.5 | - | [176] |
*pH adjusted using ethylenediamine.
| Support | pH of synthesis | Au Precursor | Au loading (%wt) | AuNP size (nm) | Catalytic application | Reference |
|---|---|---|---|---|---|---|
| Ti-MCM-41 | 7.0 | HAuCl4 a | - | 2.8 ± 1.0 | CO oxidation | [55] |
| Co-MCM-41 i | 7.0 | HAuCl4 a | - | 3.9 ± 0.8 | CO oxidation | [55] |
| Al-MCM-41 i | 7.0 | HAuCl4 a | - | 1.7 ± 0.2 | CO oxidation | [55] |
| Nb-MCM-41 | - | HAuCl4 b | 1.0 | - | Methanol oxidation and acetonylacetone cyclization | [107] |
| Ti-MCM-41 | 7.0 | HAuCl4 a | 4.0–12.0 | 2.0 | Propylene oxidation | [188] |
| Ti-MCM-41 | 7.0 | HAuCl4 a | 1.0 | 2.2 ± 0.5 | Propylene oxidation | [189] |
| Ti-MCM-41 | 7.0 | HAuCl4 a | 0.015–0.021 | - | Propylene oxidation | [190] |
| Ti-MCM-41 | 7.0 | HAuCl4 a | 0.19–0.37 | 2.8–3.8 | Propylene oxidation | [191] |
| Ti-MCM-41 | 7.0 | HAuCl4 a | 0.42 | 2.1 | Propylene oxidation | [192] |
| Ti-MCM-41 | 6.5 | HAuCl4 a | 0.36 | 3.0 ± 1.0 | Propylene oxidation | [193] |
| Ti-MCM-41 | 7.0 | HAuCl4 a | 0.3 ± 1.0 | 2.0–5.0 | Propylene oxidation | [194] |
| Fe-MCM-41 i | 7.0 | HAuCl4 a | 5.0 | <2.0 | CO oxidation | [195] |
| Co-MCM-41 i | 7.0 | HAuCl4 a | 5.0 | >2.0 | CO oxidation | [195] |
| Al-MCM-41 i | 7.0 | HAuCl a | 5.0 | <2.0 | CO oxidation | [195] |
| Ti-MCM-48 | 7.0 | (CH3)2Au(O2C5H7) c | 0.37–0.57 | 3.3–5.2 | Propylene oxidation | [190] |
| Ti-MCM-48 | 7.0 | HAuCl4 a | 0.01–0.32 | 2.0–2.6 | Propylene oxidation | [192] |
| Ti-MCM-48 | 6.5 | HAuCl4 a | 0.32 | 3.0 ± 1.0 | Propylene oxidation | [193] |
| Ti-MCM-48 | 7.0 | HAuCl4 a | 0.3 ± 1.0 | 2.0–5.0 | Propylene oxidation | [194] |
| Ti-MCM-48 | 7.0 | HAuCl4 a | ∼3.0 | ∼3.0 | CO oxidation | [49] |
| Ti-MCM-48 | 7.0 | HAuCl4 a | 3.0–7.5 | <1.0 | CO oxidation | [196,197] |
| Ti-SBA-15 | - | HAuCl4 a | 0.62 | 5.1 ± 2.9 | CO oxidation | [128] |
| Ti-SBA-15 | 1.5 | Au colloids d | 2.47 | 2.7 | CO oxidation | [128] |
| Ti-SBA-15 | - | AuNPs e | 1.47 | 5.7 ± 2.1 | CO oxidation | [128] |
| Ti-SBA-15 | 9.5–10.0 | AuCl3 a | 1.0 | 1.0–8.0 | CO oxidation | [198] |
| Ti-SBA-15 | ∼10.0 | HAuCl4 a | - | 0.8–1.0 | CO oxidation | [199] |
| Ti-SBA-15 | ∼10.0 | HAuCl4 a | - | 0.8–1.0 | CO oxidation | [200] |
| Al-SBA-15 g | - | HAuCl4 | 16.22–19.83 | 2.6–3.0 | CO oxidation | [201] |
| Al-SBA-15 g | - | HAuCl4 | 17.97 | 3.0 ± 0.8 | CO oxidation | [202] |
| Al-SBA-15 h | - | HAuCl4 | 17.61 | 3.0 ± 1.0 | CO oxidation | [202] |
| Al-SBA-15 i | - | HAuCl4 | 17.73 | 3.2 ± 0.7 | CO oxidation | [202] |
| Al-SBA-15 j | - | HAuCl4 | 17.07 | 2.6 ± 0.4 | CO oxidation | [202] |
| Ti-SBA-15 | ∼10.0 | HAuCl4 a | - | 0.8–1.0 | CO oxidation | [203] |
| Co-SBA-15 | 7.0 | HAuCl a | 1.0–4.0 | ∼5.0 | CO oxidation | [204] |
| Ti-SBA-15 k | 9.5 | HAuCl4 a | 1.0 | 2.0–9.0 | Propene epoxidation | [205] |
| Ti-SBA-15 l | 9.5 | HAuCl4 a | 1.0 | 3.0–12.0 | Propene epoxidation | [205] |
| Ti-SBA-15 | 10.0 | HAuCl4 a | 5.0–10.0 | 1.9–2.9 | - | [206] |
| Ti(10)-SBA-15 | 11.0 | HAuCl4 a | 3.6 | 4.0 ± 1.5 | CO oxidation | [207] |
| Ti(20)-SBA-15 | 11.0 | HAuCl4 a | 1.7 | 4.3 ± 1.5 | CO oxidation | [207] |
| Ti(40)-SBA-15 | 11.0 | HAuCl4 a | 3.0 | 7.2 ± 3.2 | CO oxidation | [207] |
| Ti-SBA-15 | 9.0 | HAuCl4 a | 1.0 | 2.0–15.0 | Propene oxidation | [208] |
| SBA-15-CeO2 | - | Au colloids d | 1.76 | 2.7 | CO oxidation | [128] |
| SBA-15-CeO2 | 10.0 | [Au(en)2]Cl3 | 0.7 | ∼5.0 | Lactose oxidation | [132] |
| Ce-HMS | ∼10.0 | [Au(en)2]Cl3 | 2.94 | 5.9 ± 0.5 | Aerobic oxidative | [131] |
| Ti-HMS | ∼10.0 | [Au(en)2]Cl3 | 2.96 | 4.8 ± 0.3 | esterification of benzyl | [131] |
| Fe-HMS | ∼10.0 | [Au(en)2]Cl3 | 2.87 | 4.1 ± 0.4 | alcohol | [131] |
| Ti-HMS | - | HAuCl4 f | 3.9 | <20 | Direct synthesis of H2O2 and in situ-H2O2-ODS | [209] |
| Ti-HMS | - | HAuCl4 a | 4.6 | - | [209] | |
| Fe-HMS | 9.0 | HAuCl4 a | 2.84 | 4.6 | CO oxidation | [210] |
| Ce-HMS | 9.0 | HAuCl4 a | 3.44 | 7.4 | CO oxidation | [210] |
| Ti-HMS | 9.0 | HAuCl4 a | 3.02 | 5.2 | CO oxidation | [210] |
| Ti-HMS | - | HAuCl4 f | 2.3–2.4 | 4.5–12.6 | Direct synthesis of H2O2 and in situ-H2O2-ODS | [211] |
| Co-UVM-7 | 9.0 | HAuCl4 a | 1.07–1.21 | 3.0–4.0 | CO and propane | [212] |
| Fe-AerosolSi i | 7.0 | HAuCl4 a | 5.0 | >2.0 | CO oxidation | [195] |
| Co-AerosolSi i | 7.0 | HAuCl4 a | 5.0 | <2.0 | CO oxidation | [195] |
| Al-AerosolSi i | 7.0 | HAuCl4 a | 5.0 | <2.0 | CO oxidation | [195] |
| Co-AerosolSi i | 7.0 | HAuCl4 a | - | <2.0 | CO oxidation | [55] |
| Al-AerosolSi i | 7.0 | HAuCl4 a | - | <2.0 | CO oxidation | [55] |
| Ti-Silica Aerogel | - | Thiol-capped AuNPs | 0.14–20.0 | 1.0–4.0 | CO oxidation | [213] |
| Ti-Silica Aerogel | - | Thiol-capped AuNPs | 3.4–6.6 | 1.4–6.4 | CO oxidation | [214] |
| TiO2-SiO2 | 5.0–5.5 | HAuCl4 a | 0.2–3.2 | 2.1–3.2 | CO oxidation | [215] |
| TiO2-SiO2 | 7.0 | HAuCl4 a | 5.0–10.0 | 1.8–1.9 | - | [206] |
| TiO2-SiO2 | 7.0 | HAuCl4 a | - | 1.8–6.4 | CO oxidation | [216] |
| SiO2-Al2O3 | - | HAuCl4 b | 2.05 | ∼4.5 | Oxidation and hydrogenation of CO | [217] |
| SiO2-Al2O3 | - | HAuCl4 b-AgNO3 | 0.88–1.32 | ∼4.5 | [217] | |
| SiO2-Al2O3 | 9.0 | HAuCl4 a | 2.5 | - | [217] |
aDeposition-Precipitation;bIWI;cLiquid grafting method;dIn presence of polyvinylalcohol (PVA) as stabilizer;eIn presence of poly(diallyldimethylammonium) chloride (PDDA);fOne-pot synthesis;gThin film functionalized with APS;hThin film functionalized with MPS;iFunctionalized with APS;jFunctionalized with MPS;kTi incorporated by direct synthesis (Si/Ti molar ratio: 20);lTi incorporated by post-grafting (Si/Ti molar ratio: 20).
Acknowledgments
Luis-Felipe Gutiérrez acknowledges the Universidad Nacional de Colombia for granting study leave and financial support for his Ph.D. studies. Authors acknowledge also “le Fonds Québécois de la Recherche sur la Nature et les Technologies, FQRNT-Équipe” for financial support for this project.
References
- Hutchings, G.J. Vapor-phase hydrochlorination of acetylene—Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. [Google Scholar]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon-monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar]
- Della Pina, C.; Falletta, E.; Prati, L.; Rossi, M. Selective oxidation using gold. Chem. Soc. Rev. 2008, 37, 2077–2095. [Google Scholar]
- Gutiérrez, L.-F.; Hamoudi, S.; Belkacemi, K. Selective production of lactobionic acid by aerobic oxidation of lactose over gold crystallites supported on mesoporous silica. Appl. Catal. A Gen. 2011, 402, 94–103. [Google Scholar]
- Claus, P. Heterogeneously catalysed hydrogenation using gold catalysts. Appl. Catal. A Gen. 2005, 291, 222–229. [Google Scholar]
- Corma, A.; Serna, P. Chemoselective hydrogenation of nitro compounds with supported gold catalysts. Science 2006, 313, 332–334. [Google Scholar]
- Fu, Q.; Weber, A.; Flytzani-Stephanopoulos, M. Nanostructured Au-CeO2 catalysts for low-temperature water-gas shift. Catal. Lett. 2001, 77, 87–95. [Google Scholar]
- Idakiev, V.; Tabakova, T.; Tenchev, K.; Yuan, Z.Y.; Ren, T.Z.; Vantomme, A.; Su, B.L. Gold nanoparticles supported on ceria-modified mesoporous-macroporous binary metal oxides as highly active catalysts for low-temperature water-gas shift reaction. J. Mater. Sci. 2009, 44, 6637–6643. [Google Scholar]
- Flytzani-Stephanopoulos, M.; Fu, Q.; Saltsburg, H. Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 2003, 301, 935–938. [Google Scholar]
- Nkosi, B.; Coville, N.J.; Hutchings, G.J.; Adams, M.D.; Friedl, J.; Wagner, F.E. Hydrochlorination of acetylene using gold catalysts—A study of catalyst deactivation. J. Catal. 1991, 128, 366–377. [Google Scholar]
- Conte, M.; Carley, A.F.; Heirene, C.; Willock, D.J.; Johnston, P.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Hydrochlorination of acetylene using a supported gold catalyst: A study of the reaction mechanism. J. Catal. 2007, 250, 231–239. [Google Scholar]
- Edwards, J.K.; Solsona, B.E.; Landon, P.; Carley, A.F.; Herzing, A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using TiO2-supported Au-Pd catalysts. J. Catal. 2005, 236, 69–79. [Google Scholar]
- Ueda, A.; Oshima, T.; Haruta, M. Reduction of nitrogen monoxide with propene in the presence of oxygen and moisture over gold supported on metal oxides. Appl. Catal. B Environ. 1997, 12, 81–93. [Google Scholar]
- Nguyen, L.Q.; Salim, C.; Hinode, H. Roles of nano-sized au in the reduction of NOx by propene over Au/TiO2: An in situ drifts study. Appl. Catal. B Environ. 2010, 96, 299–306. [Google Scholar]
- Teles, J.H.; Brode, S.; Chabanas, M. Cationic gold(I) complexes: Highly efficient catalysts for the addition of alcohols to alkynes. Angew. Chem. Int. Ed. 1998, 37, 1415–1418. [Google Scholar]
- Corma, A.; Carrettin, S.; Guzman, J. Supported gold catalyzes the homocoupling of phenylboronic acid with high conversion and selectivity. Angew. Chem. Int. Ed. 2005, 44, 2242–2245. [Google Scholar]
- Hashmi, A.S.K.; Rudolph, M. Gold catalysis in total synthesis. Chem. Soc. Rev. 2008, 37, 1766–1775. [Google Scholar]
- Haruta, M. Catalysis of gold nanoparticles deposited on metal oxides. Cattech 2002, 6, 102–115. [Google Scholar]
- Hashmi, A.S.K.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar]
- Daniel, M.C.; Astruc, D. Gold nanoparticles: Assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar]
- Hutchings, G.J.; Brust, M.; Schmidbaur, H. Gold—An introductory perspective. Chem. Soc. Rev. 2008, 37, 1759–1765. [Google Scholar]
- Hutchings, G.J.; Haruta, M. A golden age of catalysis: A perspective. Appl. Catal. A Gen. 2005, 291, 2–5. [Google Scholar]
- Thompson, D.T.; Bond, G.C. Catalysis by gold. Catal. Rev. 1999, 41, 319–388. [Google Scholar]
- Hutchings, G.J. Catalysis by gold. Catal. Today 2005, 100, 55–61. [Google Scholar]
- Gates, B.C.; Fierro-Gonzalez, J.C. Catalysis by gold dispersed on supports: The importance of cationic gold. Chem. Soc. Rev. 2008, 37, 2127–2134. [Google Scholar]
- Pyykko, P. Theoretical chemistry of gold. III. Chem. Soc. Rev. 2008, 37, 1967–1997. [Google Scholar]
- Jansen, M. The chemistry of gold as an anion. Chem. Soc. Rev. 2008, 37, 1824–1835. [Google Scholar]
- Corma, A.; Garcia, H. Supported gold nanoparticles as catalysts for organic reactions. Chem. Soc. Rev. 2008, 37, 2096–2126. [Google Scholar]
- Rossi, M.; Della Pina, C.; Falletta, E.; Prati, L. Selective oxidation using gold. Chem. Soc. Rev. 2008, 37, 2077–2095. [Google Scholar]
- Haruta, M.; Date, M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A Gen. 2001, 222, 427–437. [Google Scholar]
- Wang, A.Q.; Liu, J.H.; Lin, S.D.; Lin, T.S.; Mou, C.Y. A novel efficient Au-Ag alloy catalyst system: Preparation, activity, and characterization. J. Catal. 2005, 233, 186–197. [Google Scholar]
- Zheng, N.F.; Stucky, G.D. A general synthetic strategy for oxide-supported metal nanoparticle catalysts. J. Am. Chem. Soc. 2006, 128, 14278–14280. [Google Scholar]
- Guzman, J.; Gates, B.C. Catalysis by supported gold: Correlation between catalytic activity for CO oxidation and oxidation states of gold. J. Am. Chem. Soc. 2004, 126, 2672–2673. [Google Scholar]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon-monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar]
- Ma, Z.; Dai, S. Development of novel supported gold catalysts: A materials perspective. Nano Res. 2011, 4, 3–32. [Google Scholar]
- Ma, Z.; Overbury, S.H.; Dai, S. Gold nanoparticles as chemical catalysts. In Nanomaterials: Inorganic and Bioinorganic Perspectives; Lukehart, C.M., Scott, R.A., Eds.; John Wiley & Sons: Chichester, UK, 2008; pp. 247–266. [Google Scholar]
- Okumura, M.; Nakamura, S.; Tsubota, S.; Nakamura, T.; Azuma, M.; Haruta, M. Chemical vapor deposition of gold on Al2O3, SiO2, and TiO2 for the oxidation of CO and of H2. Catal. Lett. 1998, 51, 53–58. [Google Scholar]
- Gold Catalysts; Citation Report; Web of ScienceSM, Thomson Reuters: New York, NY, USA; 23; November; 2011.
- Liu, S.B.; Yu, N.Y.; Ding, Y.; Lo, A.Y.; Huang, S.J.; Wu, P.H.; Liu, C.; Yin, D.H.; Fu, Z.H.; Yin, D.L.; et al. Gold nanoparticles supported on periodic mesoporous organosilicas for epoxidation of olefins: Effects of pore architecture and surface modification method of the supports. Microporous Mesoporous Mater. 2011, 143, 426–434. [Google Scholar]
- Glomm, W.R.; Oye, G.; Walmsley, J.; Sjoblom, J. Synthesis and characterization of gold nanoparticle-functionalized ordered mesoporous materials. J. Disper. Sci. Technol. 2005, 26, 729–744. [Google Scholar]
- Yang, C.M.; Liu, P.H.; Ho, Y.F.; Chiu, C.Y.; Chao, K.J. Highly dispersed metal nanoparticles in functionalized SBA-15. Chem. Mater. 2003, 15, 275–280. [Google Scholar]
- Guari, Y.; Thieuleux, C.; Mehdi, A.; Reye, C.; Corriu, R.J.P.; Gomez-Gallardo, S.; Philippot, K.; Chaudret, B. In situ formation of gold nanoparticles within thiol functionalized HMS-C16 and SBA-15 type materials via an organometallic two-step approach. Chem. Mater. 2003, 15, 2017–2024. [Google Scholar]
- Zhu, H.G.; Lee, B.; Dai, S.; Overbury, S.H. Coassembly synthesis of ordered mesoporous silica materials containing au nanoparticles. Langmuir 2003, 19, 3974–3980. [Google Scholar]
- Zhu, H.G.; Liang, C.D.; Yan, W.F.; Overbury, S.H.; Dai, S. Preparation of highly active silica-supported Au catalysts for CO oxidation by a solution-based technique. J. Phys. Chem. B 2006, 110, 10842–10848. [Google Scholar]
- Okumura, M.; Tsubota, S.; Iwamoto, M.; Haruta, M. Chemical vapor deposition of gold nanoparticles on MCM-41 and their catalytic activities for the low-temperature oxidation of CO and of H2. Chem. Lett. 1998, 315–316. [Google Scholar]
- Zhu, J.; Konya, Z.; Puntes, V.F.; Kiricsi, I.; Miao, C.X.; Ager, J.W.; Alivisatos, A.P.; Somorjai, G.A. Encapsulation of metal (Au, Ag, Pt) nanoparticles into the mesoporous SBA-15 structure. Langmuir 2003, 19, 4396–4401. [Google Scholar]
- Konya, Z.; Puntes, V.F.; Kiricsi, I.; Zhu, J.; Ager, J.W.; Ko, M.K.; Frei, H.; Alivisatos, P.; Somorjai, G.A. Synthetic insertion of gold nanoparticles into mesoporous silica. Chem. Mater. 2003, 15, 1242–1248. [Google Scholar]
- Yan, W.F.; Mahurin, S.M.; Chen, B.; Overbury, S.H.; Dai, S. Effect of supporting surface layers on catalytic activities of gold nanoparticles in CO oxidation. J. Phys. Chem. B 2005, 109, 15489–15496. [Google Scholar]
- Bandyopadhyay, M.; Korsak, O.; van den Berg, M.W.E.; Grunert, W.; Birkner, A.; Li, W.; Schuth, F.; Gies, H. Gold nano-particles stabilized in mesoporous MCM-48 as active CO-oxidation catalyst. Microporous Mesoporous Mater. 2006, 89, 158–163. [Google Scholar]
- Alcaide, B.; Almendros, P.; Alonso, J.M. Gold-catalyzed cyclizations of alkynol-based compounds: Synthesis of natural products and derivatives. Molecules 2011, 16, 7815–7843. [Google Scholar]
- Froba, M.; Hoffmann, F.; Cornelius, M.; Morell, J. Silica-based mesoporous organic-inorganic hybrid materials. Angew. Chem. Int. Ed. 2006, 45, 3216–3251. [Google Scholar]
- Sayari, A.; Hamoudi, S. Periodic mesoporous silica-based organic—Inorganic nanocomposite materials. Chem. Mater. 2001, 13, 3151–3168. [Google Scholar]
- Kumar, R.; Ghosh, A.; Patra, C.R.; Mukherjee, P.; Sastry, M. Gold nanoparticles formed within ordered mesoporous silica and on amorphous silica. In Nanotechnology in Catalysis; Springer: New York, NY, USA, 2004; pp. 111–136. [Google Scholar]
- Chi, Y.S.; Lin, H.P.; Mou, C.Y. CO oxidation over gold nanocatalyst confined in mesoporous silica. Appl. Catal. A Gen. 2005, 284, 199–206. [Google Scholar]
- Gabaldon, J.P.; Bore, M.; Datye, A.K. Mesoporous silica supports for improved thermal stability in supported Au catalysts. Top. Catal. 2007, 44, 253–262. [Google Scholar]
- Bore, M.T.; Pham, H.N.; Switzer, E.E.; Ward, T.L.; Fukuoka, A.; Datye, A.K. The role of pore size and structure on the thermal stability of gold nanoparticles within mesoporous silica. J. Phys. Chem. B 2005, 109, 2873–2880. [Google Scholar]
- Zhu, K.K.; Hu, J.C.; Richards, R. Aerobic oxidation of cyclohexane by gold nanoparticles immobilized upon mesoporous silica. Catal. Lett. 2005, 100, 195–199. [Google Scholar]
- Yang, C.M.; Kalwei, M.; Schuth, F.; Chao, K.J. Gold nanoparticles in SBA-15 showing catalytic activity in CO oxidation. Appl. Catal. A Gen. 2003, 254, 289–296. [Google Scholar]
- Ma, C.Y.; Dou, B.J.; Li, J.J.; Cheng, J.; Hu, Q.; Hao, Z.P.; Qiao, S.Z. Catalytic oxidation of benzyl alcohol on Au or Au-Pd nanoparticles confined in mesoporous silica. Appl. Catal. B Environ. 2009, 92, 202–208. [Google Scholar]
- Pei, Y.; Yang, Q.Y.; Zhu, Y.; Tian, L.; Xie, S.H.; Li, H.; Li, H.X.; Qiao, M.H.; Fan, K.N. Preparation and characterization of Au-In/APTMS-SBA-15 catalysts for chemoselective hydrogenation of crotonaldehyde to crotyl alcohol. Appl. Catal. A Gen. 2009, 369, 67–76. [Google Scholar]
- Rombi, E.; Cutrufello, M.G.; Cannas, C.; Casu, M.; Gazzoli, D.; Occhiuzzi, M.; Monacia, R.; Ferinoa, I. Modifications induced by pretreatments on Au/SBA-15 and their influence on the catalytic activity for low temperature CO oxidation. Phys. Chem. Chem. Phys. 2009, 11, 593–602. [Google Scholar]
- Wu, P.P.; Bai, P.; Loh, K.P.; Zhao, X.S. Au nanoparticles dispersed on functionalized mesoporous silica for selective oxidation of cyclohexane. Catal. Today 2010, 158, 220–227. [Google Scholar]
- Li, L.; Jin, C.; Wang, X.C.; Ji, W.J.; Pan, Y.; van der Knaap, T.; van der Stoel, R.; Au, C.T. Cyclohexane oxidation over size-uniform Au nanoparticles (SBA-15 hosted) in a continuously stirred tank reactor under mild conditions. Catal. Lett. 2009, 129, 303–311. [Google Scholar]
- Chen, L.F.; Hu, J.C.; Richards, R.M. Intercalation of aggregation-free and well-dispersed gold nanoparticles into the walls of mesoporous silica as a highly active catalyst for n-alkane and alcohols oxidation. J. Am. Chem. Soc. 2009, 131, 914–915. [Google Scholar]
- Yang, Y.H.; Sun, H.; Tang, Q.H.; Du, Y.; Liu, X.B.; Chen, Y. Mesostructured SBA-16 with excellent hydrothermal, thermal and mechanical stabilities: Modified synthesis and its catalytic application. J. Colloid Interface Sci. 2009, 333, 317–323. [Google Scholar]
- Cao, R.; Li, H.F.; Zheng, Z.L.; Cao, M.N. Stable gold nanoparticle encapsulated in silica-dendrimers organic-inorganic hybrid composite as recyclable catalyst for oxidation of alcohol. Microporous Mesoporous Mater. 2010, 136, 42–49. [Google Scholar]
- Bai, Y.; Yang, H.; Yang, W.W.; Li, Y.C.; Sun, C.Q. Gold nanoparticles-mesoporous silica composite used as an enzyme immobilization matrix for amperometric glucose biosensor construction. Sens. Actuator B Chem. 2007, 124, 179–186. [Google Scholar]
- Xiao, F.S.; Wang, L.; Wang, H.; Hapala, P.; Zhu, L.F.; Ren, L.M.; Meng, X.J.; Lewis, J.P. Superior catalytic properties in aerobic oxidation of olefins over Au nanoparticles on pyrrolidone-modified SBA-15. J. Catal. 2011, 281, 30–39. [Google Scholar]
- Cutrufello, M.G.; Rombi, E.; Cannas, C.; Casu, M.; Virga, A.; Fiorilli, S.; Onida, B.; Ferino, I. Synthesis, characterization and catalytic activity of Au supported on functionalized SBA-15 for low temperature CO oxidation. J. Mater. Sci. 2009, 44, 6644–6653. [Google Scholar]
- Lee, B.; Ma, Z.; Zhang, Z.T.; Park, C.; Dai, S. Influences of synthesis conditions and mesoporous structures on the gold nanoparticles supported on mesoporous silica hosts. Microporous Mesoporous Mater. 2009, 122, 160–167. [Google Scholar]
- Liu, H.L.; Zhou, L.H.; Hu, J.; Xie, S.G. Dispersion of active Au nanoparticles on mesoporous SBA-15 materials. Chin. J. Chem. Eng. 2007, 15, 507–511. [Google Scholar]
- Petkov, N.; Stock, N.; Bein, T. Gold electroless reduction in nanosized channels of thiol-modified SBA-15 material. J. Phys. Chem. B 2005, 109, 10737–10743. [Google Scholar]
- Okubo, T.; Gu, J.; Fan, W.; Shimojima, A. Microwave-induced synthesis of highly dispersed gold nanoparticles within the pore channels of mesoporous silica. J. Solid State Chem. 2008, 181, 957–963. [Google Scholar]
- Liu, Z.M.; Zhao, Y.F.; Qi, Y.; Zhang, Y.Y.; Zhang, S.G. Nano-Au/silica composite synthesized using nitrided SBA-15 as a host. Mater. Lett. 2008, 62, 1197–1199. [Google Scholar]
- Chao, K.J.; Cheng, M.H.; Ho, Y.F.; Liu, P.H. Preparation and characterization of highly dispersed gold nanoparticles within channels of mesoporous silica. Catal. Today 2004, 97, 49–53. [Google Scholar]
- Richards, R.; Li, Z.; Kubel, C.; Parvulescu, V.I. Size tunable gold nanorods evenly distributed in the channels of mesoporous silica. ACS Nano 2008, 2, 1205–1212. [Google Scholar]
- Liu, P.H.; Chang, Y.P.; Phan, T.H.; Chao, K.J. The morphology and size of nanostructured Au in Au/SBA-15 affected by preparation conditions. Mater. Sci. Eng. C 2006, 26, 1017–1022. [Google Scholar]
- Cao, R.; Zheng, Z.L.; Li, H.F.; Liu, T.F. Monodisperse noble metal nanoparticles stabilized in SBA-15: Synthesis, characterization and application in microwave-assisted suzuki-miyaura coupling reaction. J. Catal. 2010, 270, 268–274. [Google Scholar]
- Zhang, T.; Liu, X.Y.; Wang, A.Q.; Yang, X.F.; Mou, C.Y.; Su, D.S.; Li, J. Synthesis of thermally stable and highly active bimetallic Au-Ag nanoparticles on inert supports. Chem. Mater. 2009, 21, 410–418. [Google Scholar]
- Yang, Y.H.; Chen, Y.T.; Lim, H.M.; Tang, Q.H.; Gao, Y.T.; Sun, T.; Yan, Q.Y. Solvent-free aerobic oxidation of benzyl alcohol over Pd monometallic and Au-Pd bimetallic catalysts supported on SBA-16 mesoporous molecular sieves. Appl. Catal. A Gen. 2010, 380, 55–65. [Google Scholar]
- Mukherjee, P.; Patra, C.R.; Kumar, R.; Sastry, M. Entrapment and catalytic activity of gold nanoparticles in amine-functionalized MCM-41 matrices synthesized by spontaneous reduction of aqueous chloroaurate ions. PhysChemComm 2001, 4, 24–25. [Google Scholar]
- Bore, M.T.; Pham, H.N.; Ward, T.L.; Datye, A.K. Role of pore curvature on the thermal stability of gold nanoparticles in mesoporous silica. Chem. Commun. 2004, 1, 2620–2621. [Google Scholar]
- Sen, T.; Jana, S.; Koner, S.; Patra, A. Energy transfer between confined dye and surface attached au nanoparticles of mesoporous silica. J. Phys. Chem. C 2010, 114, 707–714. [Google Scholar]
- Wei, Q.; Zhong, Z.X.; Nie, Z.R.; Li, J.L.; Wang, F.; Li, Q.Y. Catalytically active gold nanoparticles confined in periodic mesoporous organosilica (PMOs) by a modified external passivation route. Microporous Mesoporous Mater. 2009, 117, 98–103. [Google Scholar]
- Xie, J.; Wang, Y.; Li, Y.; Wei, Y. Self-assembly preparation of Au/SiO2 catalyst and its catalysis for cyclohexane oxidation with air. React. Kinet. Mech. Catal. 2011, 102, 143–154. [Google Scholar]
- Yu, J.J.; Lu, S.; Li, J.W.; Zhao, F.Q.; Zeng, B.Z. Characterization of gold nanoparticles electrochemically deposited on amine-functioned mesoporous silica films and electrocatalytic oxidation of glucose. J. Solid State Electronchem. 2007, 11, 1211–1219. [Google Scholar]
- Zhao, L.; Zhu, G.S.; Zhang, D.L.; Chen, Y.; Qiu, S.L. A surface modification scheme for incorporation of nanocrystals in mesoporous silica matrix. J. Solid State Chem. 2005, 178, 2980–2986. [Google Scholar]
- Gu, J.L.; Shi, J.L.; You, G.J.; Xiong, L.M.; Qian, S.X.; Hua, Z.L.; Chen, H.R. Incorporation of highly dispersed gold nanoparticles into the pore channels of mesoporous silica thin films and their ultrafast nonlinear optical response. Adv. Mater. 2005, 17, 557–560. [Google Scholar]
- Mou, C.Y.; Yen, C.W.; Lin, M.L.; Wang, A.Q.; Chen, S.A.; Chen, J.M. CO oxidation catalyzed by Au-Ag bimetallic nanoparticles supported in mesoporous silica. J. Phys. Chem. C 2009, 113, 17831–17839. [Google Scholar]
- Zhang, T.; Liu, X.Y.; Wang, A.Q.; Wang, X.D.; Mou, C.Y. Au-Cu alloy nanoparticles confined in SBA-15 as a highly efficient catalyst for CO oxidation. Chem. Commun. 2008, 27, 3187–3189. [Google Scholar]
- Corriu, R.J.P.; Mehdi, A.; Reye, C. Molecular chemistry and nanosciences: On the way to interactive materials. J. Mater. Chem. 2005, 15, 4285–4294. [Google Scholar]
- Jin, Y.; Wang, P.J.; Yin, D.H.; Liu, J.F.; Qiu, H.Y.; Yu, N.Y. Gold nanoparticles stabilized in a novel periodic mesoporous organosilica of SBA-15 for styrene epoxidation. Microporous Mesoporous Mater. 2008, 111, 569–576. [Google Scholar]
- Ghosh, A.; Patra, C.R.; Mukherjee, P.; Sastry, M.; Kumar, R. Preparation and stabilization of gold nanoparticles formed by in situ reduction of aqueous chloroaurate ions within surface-modified mesoporous silica. Microporous Mesoporous Mater. 2003, 58, 201–211. [Google Scholar]
- Guari, Y.; Theiuleux, C.; Mehdi, A.; Reye, C.; Corriu, R.J.P.; Gomez-Gallardo, S.; Philippot, K.; Chaudret, B.; Dutartre, R. In situ formation of gold nanoparticles within functionalised ordered mesoporous silica via an organometallic 'chimie douce' approach. Chem. Commun. 2001, 15, 1374–1375. [Google Scholar]
- Mehdi, A.; Herault, D.; Cerveau, G.; Corriu, R.J.P. New mesostructured organosilica with chiral sugar derived structures: Nice host for gold nanoparticles stabilisation. Dalton Trans. 2011, 40, 446–451. [Google Scholar]
- Zhuang, D.Y.; Jin, Y.; Yu, N.Y.; Qin, L.S.; Liu, J.F.; Yin, D.H.; Yang, C.Q. Preparation and catalytic performance of gold nanoparticles supported on periodic mesoporous organosilica of SBA-15. Chin. J. Catal. 2009, 30, 896–900. [Google Scholar]
- Mehdi, A.; Besson, E.; Reye, C.; Corriu, R.J.P. Soft route for monodisperse gold nanoparticles confined within SH-functionalized walls of mesoporous silica. J. Mater. Chem. 2009, 19, 4746–4752. [Google Scholar]
- Kumagi, H.; Yano, K. Synthesis and characterization of Au-loaded core/shell mesoporous silica spheres containing propyl group in the shell. Chem. Mater. 2010, 22, 5112–5118. [Google Scholar]
- Nakamura, T.; Yamada, Y.; Yano, K. Direct synthesis of monodispersed thiol-functionalized nanoporous silica spheres and their application to a colloidal crystal embedded with gold nanoparticles. J. Mater. Chem. 2007, 17, 3726–3732. [Google Scholar]
- Hsu, Y.T.; Chen, W.L.; Yang, C.M. Co-condensation synthesis of aminopropyl-functionalized KIT-5 mesophases using carboxy-terminated triblock copolymer. J. Phys. Chem. C 2009, 113, 2777–2783. [Google Scholar]
- Boullanger, A.; Alauzun, J.; Mehdi, A.; Reye, C.; Corriu, R.J.P. Generic way for functionalised well-ordered cubic mesoporous silica via direct synthesis approach. New J. Chem. 2010, 34, 738–743. [Google Scholar]
- Rosenholm, J.M.; Linden, M. Wet-chemical analysis of surface concentration of accessible groups on different amino-functionalized mesoporous SBA-15 silicas. Chem. Mater. 2007, 19, 5023–5034. [Google Scholar]
- Lu, G.M.; Zhao, R.; Qian, G.; Qi, Y.X.; Wang, X.L.; Suo, J.S. A highly efficient catalyst Au/MCM-41 for selective oxidation cyclohexane using oxygen. Catal. Lett. 2004, 97, 115–118. [Google Scholar]
- Lu, G.M.; Ji, D.; Qian, G.; Qi, Y.X.; Wang, X.L.; Suo, J.S. Gold nanoparticles in mesoporous materials showing catalytic selective oxidation cyclohexane using oxygen. Appl. Catal. A Gen. 2005, 280, 175–180. [Google Scholar]
- Sobczak, I.; Kusior, A.; Grams, J.; Ziolek, M. The role of chlorine in the generation of catalytic active species located in Au-containing MCM-41 materials. J. Catal. 2007, 245, 259–266. [Google Scholar]
- Grams, J.; Sobczak, I. Application of tof-sims to the study of surfactant removal from AuNbMCM-41 and AuMCM-41 materials. Int. J. Mass Spectrom. 2010, 289, 138–143. [Google Scholar]
- Sobczak, I.; Kusior, A.; Grams, J.; Ziolek, M. Novel AuNbMCM-41 catalyst for methanol oxidation. In Studies in Surface Science and Catalysis; Ruren, X., Zi, G., Jiesheng, C., Wenfu, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 170, pp. 1300–1306. [Google Scholar]
- Akolekar, D.B.; Foran, G.; Bhargava, S.K. X-ray absorption spectroscopic studies on gold nanoparticles in mesoporous and microporous materials. J. Synchrotron Radiat. 2004, 11, 284–290. [Google Scholar]
- Akolekar, D.B.; Bhargava, S.K. Investigations on gold nanoparticles in mesoporous and microporous materials. J. Mol. Catal. A Chem. 2005, 236, 77–86. [Google Scholar]
- Wojtaszek, A.; Sobczak, I.; Ziolek, M.; Tielens, F. Gold grafted to mesoporous silica surfaces, a molecular picture. J. Phys. Chem. C 2009, 113, 13855–13859. [Google Scholar]
- Mokhonoana, M.P.; Coville, N.J.; Datye, A.K. Small Au nanoparticles supported on MCM-41 containing a surfactant. Catal. Lett. 2010, 135, 1–9. [Google Scholar]
- Sobczak, I.; Kieronczyk, N.; Trejda, M.; Ziolek, M. Gold, vanadium and niobium containing MCM-41 materials-catalytic properties in methanol oxidation. Catal. Today 2008, 139, 188–195. [Google Scholar]
- Hagaman, E.W.; Zhu, H.G.; Overbury, S.H.; Dai, S. C-13 NMR characterization of the organic constituents in ligand-modified hexagonal mesoporous silicas: Media for the synthesis of small, uniform-size gold nanoparticles. Langmuir 2004, 20, 9577–9584. [Google Scholar]
- Overbury, S.H.; Ortiz-Soto, L.; Zhu, H.G.; Lee, B.; Amiridis, M.D.; Dai, S. Comparison of Au catalysts supported on mesoporous titania and silica: Investigation of au particle size effects and metal-support interactions. Catal. Lett. 2004, 95, 99–106. [Google Scholar]
- Mou, C.Y.; Wang, A.Q.; Chang, C.M. Evolution of catalytic activity of Au-Ag bimetallic nanoparticles on mesoporous support for CO oxidation. J. Phys. Chem. B 2005, 109, 18860–18867. [Google Scholar]
- Wu, P.P.; Bai, P.; Lei, Z.B.; Loh, K.P.; Zhao, X.S. Gold nanoparticles supported on functionalized mesoporous silica for selective oxidation of cyclohexane. Microporous Mesoporous Mater. 2011, 141, 222–230. [Google Scholar]
- Zhao, R.; Ji, D.; Lv, G.M.; Qian, G.; Yan, L.; Wang, X.L.; Suo, J.S. A highly efficient oxidation of cyclohexane over Au/ZSM-5 molecular sieve catalyst with oxygen as oxidant. Chem. Commun. 2004, 7, 904–905. [Google Scholar]
- Wu, P.; Xiong, Z.; Loh, K.P.; Zhao, X.S. Selective oxidation of cyclohexane over gold nanoparticles supported on mesoporous silica prepared in the presence of thioether functionality. Catal. Sci. Technol. 2011, 1, 285–294. [Google Scholar]
- Hu, J.C.; Chen, L.F.; Zhu, K.K.; Suchopar, A.; Richards, R. Aerobic oxidation of alcohols catalyzed by gold nano-particles confined in the walls of mesoporous silica. Catal. Today 2007, 122, 277–283. [Google Scholar]
- Hu, J.C.; Chen, L.F.; Richards, R. Intercalation of aggregation-free and well-dispersed gold nanoparticles into the walls of mesoporous silica as a robust “Green” Catalyst for n-alkane oxidation. J. Am. Chem. Soc. 2009, 131, 914–915. [Google Scholar]
- Magureanu, M.; Mandache, N.B.; Hu, J.C.; Richards, R.; Florea, M.; Parvulescu, V.I. Plasma-assisted catalysis total oxidation of trichloroethylene over gold nano-particles embedded in SBA-15 catalysts. Appl. Catal. B Environ. 2007, 76, 275–281. [Google Scholar]
- Piroi, D.; Magureanu, M.; Mandache, N.B.; Richards, R.; Parvulescu, V.I. Plasma-assisted catalysis for total oxidation of trichloroethylene over mesoporous Au-Sba catalysts. Proceedings of the XXVIII International Conference on Phenomena in Ionized Gases (ICPIG), Prague, Czech Republic, 15–20 July 2007; pp. 1342–1345.
- Budroni, G.; Corma, A. Gold-organic-inorganic high-surface-area materials as precursors of highly active catalysts. Angew. Chem. Int. Ed. 2006, 45, 3328–3331. [Google Scholar]
- Gu, J.L.; Xiong, L.M.; Shi, J.L.; Hua, Z.L.; Zhang, L.X.; Li, L. Thioether moiety functionalization of mesoporous silica films for the encapsulation of highly dispersed gold nanoparticles. J. Solid State Chem. 2006, 179, 1060–1066. [Google Scholar]
- Mou, C.Y.; Wu, S.H.; Tseng, C.T.; Lin, Y.S.; Lin, C.H.; Hung, Y. Catalytic nano-rattle of Au@hollow silica: Towards a poison-resistant nanocatalyst. J. Mater. Chem. 2011, 21, 789–794. [Google Scholar]
- Lee, B.; Zhu, H.G.; Zhang, Z.T.; Overbury, S.H.; Dai, S. Preparation of bicontinuous mesoporous silica and organosilica materials containing gold nanoparticles by co-synthesis method. Microporous Mesoporous Mater. 2004, 70, 71–80. [Google Scholar]
- Block, B.P.; Bailar, J.C. The reaction of gold(III) with some bidentate coordinating groups. J. Am. Chem. Soc. 1951, 73, 4722–4725. [Google Scholar]
- Beck, A.; Horvath, A.; Stefler, G.; Katona, R.; Geszti, O.; Tolnai, G.; Liotta, L.F.; Guczi, L. Formation and structure of Au/TiO2 and Au/CeO2 nanostructures in mesoporous SBA-15. Catal. Today 2008, 139, 180–187. [Google Scholar]
- Yin, H.F.; Ma, Z.; Zhu, H.G.; Chi, M.F.; Dai, S. Evidence for and mitigation of the encapsulation of gold nanoparticles within silica supports upon high-temperature treatment of Au/SiO2 catalysts: Implication to catalyst deactivation. Appl. Catal. A Gen. 2010, 386, 147–156. [Google Scholar]
- Guan, Y.J.; Hensen, E.J.M. Ethanol dehydrogenation by gold catalysts: The effect of the gold particle size and the presence of oxygen. Appl. Catal. A Gen. 2009, 361, 49–56. [Google Scholar]
- Parreira, L.A.; Bogdanchikova, N.; Pestryakov, A.; Zepeda, T.A.; Tuzovskaya, I.; Farias, M.H.; Gusevskaya, E.V. Nanocrystalline gold supported on Fe-, Ti- and Ce-modified hexagonal mesoporous silica as a catalyst for the aerobic oxidative esterification of benzyl alcohol. Appl. Catal. A Gen. 2011, 397, 145–152. [Google Scholar]
- Gutiérrez, L.-F.; Hamoudi, S.; Belkacemi, K. Selective oxidation of lactose to high value lactobionic acid over gold clusters supported on mesoporous silica decorated by Ceria. Proceedings of the 22nd Meeting of the North American Catalysis Society, Detroit, MI, USA, 5–10 June 2011.
- Dmowski, W.; Yin, H.F.; Dai, S.; Overbury, S.H.; Egami, T. Atomic structure of Au nanoparticles on a silica support by an X-ray PDF study. J. Phys. Chem. C 2010, 114, 6983–6988. [Google Scholar]
- Wu, Z.L.; Zhou, S.H.; Zhu, H.G.; Dai, S.; Overbury, S.H. Drifts-QMS study of room temperature CO oxidation on Au/SiO2 catalyst: Nature and role of different Au species. J. Phys. Chem. C 2009, 113, 3726–3734. [Google Scholar]
- Zanella, R.; Sandoval, A.; Gomez-Cortes, A.; Diaz, G.; Saniger, J.M. Gold nanoparticles: Support effects for the WGS reaction. J. Mol. Catal. A Chem. 2007, 278, 200–208. [Google Scholar]
- Zhu, H.G.; Ma, Z.; Clark, J.C.; Pan, Z.W.; Overbury, S.H.; Dai, S. Low-temperature CO oxidation on Au/fumed SiO2-based catalysts prepared from Au(en)2Cl3 precursor. Appl. Catal. A Gen. 2007, 326, 89–99. [Google Scholar]
- Zanella, R.; Sandoval, A.; Santiago, P.; Basiuk, V.A.; Saniger, J.M. New preparation method of gold nanoparticles on SiO2. J. Phys. Chem. B 2006, 110, 8559–8565. [Google Scholar]
- Yin, H.F.; Ma, Z.; Overbury, S.H.; Dai, S. Promotion of Au(en)2Cl3-derived Au/fumed SiO2 by treatment with KMnO4. J. Phys. Chem. C 2008, 112, 8349–8358. [Google Scholar]
- Morales-Saavedra, O.G.; Zanella, R. Structural and photophysical evaluation of Au-NPs/SiO2-based inorganic-inorganic sonogel hybrid composites. Mater. Chem. Phys. 2010, 124, 816–830. [Google Scholar]
- Reifsnyder, S.N.; Lamb, H.H. Characterization of silica-supported Pd-Au clusters by X-ray absorption spectroscopy. J. Phys. Chem. B 1999, 103, 321–329. [Google Scholar]
- Yuan, G.; Lopez, J.L.; Louis, C.; Delannoy, L.; Keane, M.A. Remarkable hydrodechlorination activity over silica supported nickel/gold catalysts. Catal. Commun. 2005, 6, 555–562. [Google Scholar]
- Bauer, J.C.; Mullins, D.; Li, M.J.; Wu, Z.L.; Payzant, E.A.; Overbury, S.H.; Dai, S. Synthesis of silica supported Au-Cu nanoparticle catalysts and the effects of pretreatment conditions for the CO oxidation reaction. Phys. Chem. Chem. Phys. 2011, 13, 2571–2581. [Google Scholar]
- Xu, Q.; Jiang, H.L.; Umegaki, T.; Akita, T.; Zhang, X.B.; Haruta, M. Bimetallic Au-Ni nanoparticles embedded in SiO2 nanospheres: Synergetic catalysis in hydrolytic dehydrogenation of ammonia borane. Chem. Eur. J. 2010, 16, 3132–3137. [Google Scholar]
- Okumura, M.; Nakamura, S.-i.; Tsubota, S.; Nakamura, T.; Haruta, M. Deposition of gold nanoparticles on Silica by CVD of gold acethylacetonate. In Studies in Surface Science and Catalysis; Delmon, B., Jacobs, P.A., Maggi, R., Martens, R., Grange, J.A., Poncelet, G., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; Volume 118, pp. 277–284. [Google Scholar]
- Okumura, M.; Tsubota, S.; Haruta, M. Preparation of supported gold catalysts by gas-phase grafting of gold acethylacetonate for low-temperature oxidation of CO and of H2. J. Mol. Catal. A Chem. 2003, 199, 73–84. [Google Scholar]
- Schimpf, S.; Lucas, M.; Mohr, C.; Rodemerck, U.; Bruckner, A.; Radnik, J.; Hofmeister, H.; Claus, P. Supported gold nanoparticles: In-depth catalyst characterization and application in hydrogenation and oxidation reactions. Catal. Today 2002, 72, 63–78. [Google Scholar]
- Okumura, M.; Tanaka, K.; Ueda, A.; Haruta, M. The reactivities of dimethylgold(III) beta-diketone on the surface of TiO2—A novel preparation method for Au catalysts. Solid State Ionics 1997, 95, 143–149. [Google Scholar]
- Araki, H.; Fukuoka, A.; Sakamoto, Y.; Inagaki, S.; Sugimoto, N.; Fukushima, Y.; Ichikawa, M. Template synthesis and characterization of gold nano-wires and -particles in mesoporous channels of FSM-16. J. Mol. Catal. A Chem. 2003, 199, 95–102. [Google Scholar]
- Chou, J.; Franklin, N.R.; Baeck, S.H.; Jaramillo, T.F.; McFarland, E.W. Gas-phase catalysis by micelle derived Au nanoparticles on oxide supports. Catal. Lett. 2004, 95, 107–111. [Google Scholar]
- Porta, F.; Prati, L.; Rossi, M.; Coluccia, S.; Martra, G. Metal sols as a useful tool for heterogeneous gold catalyst preparation: Reinvestigation of a liquid phase oxidation. Catal. Today 2000, 61, 165–172. [Google Scholar]
- Comotti, M.; Li, W.C.; Spliethoff, B.; Schuth, F. Support effect in high activity gold catalysts for CO oxidation. J. Am. Chem. Soc. 2006, 128, 917–924. [Google Scholar]
- Tsubota, S.; Nakamura, T.; Tanaka, K.; Haruta, M. Effect of calcination temperature on the catalytic activity of Au colloids mechanically mixed with TiO2 powder for CO oxidation. Catal. Lett. 1998, 56, 131–135. [Google Scholar]
- Pietron, J.J.; Stroud, R.M.; Rolison, D.R. Using three dimensions in catalytic mesoporous nanoarchitectures. Nano Lett. 2002, 2, 545–549. [Google Scholar]
- Xu, B.Q.; Shi, H.; Xu, N.; Zhao, D. Immobilized PVA-stabilized gold nanoparticles on silica show an unusual selectivity in the hydrogenation of cinnamaldehyde. Catal. Commun. 2008, 9, 1949–1954. [Google Scholar]
- Pawelec, B.; Venezia, A.M.; La Parola, V.; Thomas, S.; Fierro, J.L.G. Factors influencing selectivity in naphthalene hydrogenation over Au- and Pt-Au-supported catalysts. Appl. Catal. A Gen. 2005, 283, 165–175. [Google Scholar]
- Zheng, N.; Stucky, G.D. Methods of Generating Supported Nanocatalysts and Compositions Thereof. WIPO Patent Application WO 2009/089513 A2 2009. [Google Scholar]
- Botella, P.; Corma, A.; Navarro, M.T. Single gold nanoparticles encapsulated in monodispersed regular spheres of mesostructured silica produced by pseudomorphic transformation. Chem. Mater. 2007, 19, 1979–1983. [Google Scholar]
- Grunwaldt, J.D.; Kiener, C.; Wogerbauer, C.; Baiker, A. Preparation of supported gold catalysts for low-temperature CO oxidation via “Size-controlled” Gold colloids. J. Catal. 1999, 181, 223–232. [Google Scholar]
- Ma, G.C.; Yan, X.Q.; Li, Y.L.; Xiao, L.P.; Huang, Z.J.; Lu, Y.P.; Fan, J. Ordered nanoporous silica with periodic 30–60 nm pores as an effective support for gold nanoparticle catalysts with enhanced lifetime. J. Am. Chem. Soc. 2010, 132, 9596–9597. [Google Scholar]
- Zheng, N.; Fan, J.; Stucky, G.D. One-step one-phase synthesis of monodisperse noble-metallic nanoparticles and their colloidal crystals. J. Am. Chem. Soc. 2006, 128, 6550–6551. [Google Scholar]
- Tai, Y.; Watanabe, M.; Murakami, J.; Tajiri, K. Composite formation of thiol-capped Au nanoparticles and mesoporous silica prepared by a sol-gel method. J. Mater. Sci. 2007, 42, 1285–1292. [Google Scholar]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiol-derivatized gold nanoparticles in a 2-phase liquid-liquid system. J. Chem. Soc. Chem. Commun. 1994, 7, 801–802. [Google Scholar]
- Anderson, M.L.; Morris, C.A.; Stroud, R.M.; Merzbacher, C.I.; Rolison, D.R. Colloidal gold aerogels: Preparation, properties, and characterization. Langmuir 1999, 15, 674–681. [Google Scholar]
- Konya, Z.; Puntes, V.F.; Kiricsi, I.; Zhu, J.; Alivisatos, A.P.; Somorjai, G.A. Nanocrystal templating of silica mesopores with tunable pore sizes. Nano Lett. 2002, 2, 907–910. [Google Scholar]
- Lin, H.P.; Chi, Y.S.; Lin, J.N.; Mou, C.Y.; Wan, B.Z. Direct synthesis of MCM-41 mesoporous aluminosilicates containing Au nanoparticles in aqueous solution. Chem. Lett. 2001, 11, 1116–1117. [Google Scholar]
- Aprile, C.; Abad, A.; Hermenegildo, G.A.; Corma, A. Synthesis and catalytic activity of periodic mesoporous materials incorporating gold nanoparticles. J. Mater. Chem. 2005, 15, 4408–4413. [Google Scholar]
- Rothenberg, G.; Wichner, N.M.; Beckers, J.; Koller, H. Preventing sintering of Au and Ag nanoparticles in silica-based hybrid gels using phenyl spacer groups. J. Mater. Chem. 2010, 20, 3840–3847. [Google Scholar]
- Ferrara, M.C.; Mirenghi, L.; Mevoli, A.; Tapfer, L. Synthesis and characterization of sol-gel silica films doped with size-selected gold nanoparticles. Nanotechnology 2008, 19. [Google Scholar] [CrossRef]
- Cheng, S.; Wei, Y.; Feng, Q.W.; Qiu, K.Y.; Pang, J.B.; Jansen, S.A.; Yin, R.; Ong, K. Facile synthesis of mesoporous gold-silica nanocomposite materials via sol-gel process with nonsurfactant templates. Chem. Mater. 2003, 15, 1560–1566. [Google Scholar]
- Bonnemann, H.; Endruschat, U.; Tesche, B.; Rufinska, A.; Lehmann, C.W.; Wagner, F.E.; Filoti, G.; Parvulescu, V.; Parvulescu, V.I. An SiO2-embedded nanoscopic Pd/Au alloy colloid. Eur. J. Inorg. Chem. 2000, 2000, 819–822. [Google Scholar]
- Parvulescu, V.I.; Parvulescu, V.; Eudruschat, U.; Filoti, G.; Wagner, F.E.; Kubel, C.; Richards, R. Characterization and catalytic-hydrogenation behavior of SiO2-embedded nanoscopic Pd, Au, and Pd-Au alloy colloids. Chem. Eur. J. 2006, 12, 2343–2357. [Google Scholar]
- Mou, C.Y.; Liu, J.H.; Wang, A.Q.; Chi, Y.S.; Lin, H.P. Synergistic effect in an Au-Ag alloy nanocatalyst: CO oxidation. J. Phys. Chem. B 2005, 109, 40–43. [Google Scholar]
- Hereijgers, B.P.C.; Weckhuysen, B.M. Aerobic oxidation of cyclohexane by gold-based catalysts: New mechanistic insight by thorough product analysis. J. Catal. 2010, 270, 16–25. [Google Scholar]
- Tsukuda, T.; Liu, Y.M.; Tsunoyama, H.; Akita, T. Preparation of similar to 1 nm gold clusters confined within mesoporous silica and microwave-assisted catalytic application for alcohol oxidation. J. Phys. Chem. C 2009, 113, 13457–13461. [Google Scholar]
- Tsoncheva, T.; Tvanova, L.; Lotz, A.R.; Smatt, J.H.; Dimitrov, M.; Paneva, D.; Mitov, I.; Linden, M.; Minchev, C.; Froba, M. Gold and iron nanoparticles in mesoporous silicas: Preparation and characterization. Catal. Commun. 2007, 8, 1573–1577. [Google Scholar]
- Fukuoka, A.; Araki, H.; Kimura, J.; Sakamoto, Y.; Higuchi, T.; Sugimoto, N.; Inagaki, S.; Ichikawa, M. Template synthesis of nanoparticle arrays of gold, platinum and palladium in mesoporous silica films and powders. J. Mater. Chem. 2004, 14, 752–756. [Google Scholar]
- Delannoy, L.; El Hassan, N.; Musi, A.; Le To, N.N.; Krafft, J.M.; Louis, C. Preparation of supported gold nanoparticles by a modified incipient wetness impregnation method. J. Phys. Chem. B 2006, 110, 22471–22478. [Google Scholar]
- Damin, A.; Budnyk, A.P.; Agostini, G.; Zecchina, A. Gold nanoparticle aggregates immobilized on high surface area silica substrate for efficient and clean sers applications. J. Phys. Chem. C 2010, 114, 3857–3862. [Google Scholar]
- Shi, H.Z.; Zhang, L.D.; Cai, W.P. Preparation and optical absorption of gold nanoparticles within pores of mesoporous silica. Mater. Res. Bull. 2000, 35, 1689–1695. [Google Scholar]
- Cai, W.P.; Hofmeister, H.; Rainer, T.; Chen, W. Optical properties of Ag and Au nanoparticles dispersed within the pores of monolithic mesoporous silica. J. Nanopart. Res. 2001, 3, 443–453. [Google Scholar]
- Tsung, C.K.; Hong, W.B.; Shi, Q.H.; Kou, X.S.; Yeung, M.H.; Wang, J.F.; Stucky, G.D. Shape- and orientation-controlled gold nanoparticles formed within mesoporous silica nanofibers. Adv. Funct. Mater. 2006, 16, 2225–2230. [Google Scholar]
- Lotz, A.R.; Froba, M. Synthesis and characterization of Au-55 clusters within mesoporous silica. Z. Anorg. Allg. Chem. 2005, 631, 2800–2805. [Google Scholar]
- Fukuoka, A.; Araki, H.; Sakamoto, Y.; Sugimoto, N.; Tsukada, H.; Kumai, Y.; Akimoto, Y.; Ichikawa, M. Template synthesis of nanoparticle arrays of gold and platinum in mesoporous silica films. Nano Lett. 2002, 2, 793–795. [Google Scholar]
- Venezia, A.M.; Murania, R.; Pantaleo, G.; Deganello, G. Pd and PdAu on mesoporous silica for methane oxidation: Effect of SO2. J. Catal. 2007, 251, 94–102. [Google Scholar]
- Cai, W.P.; Li, C.C.; Kan, C.X.; Fu, G.H. Synthesis and optical characterization of Pd-Au bimetallic nanoparticles dispersed within monolithic mesoporous silica. Scr. Mater. 2004, 50, 1481–1486. [Google Scholar]
- Ma, Z.; Zaera, F. Heterogeneos catalysis by metals. In Encyclopedia of Inorganic Chemistry, 2nd ed.; King, R.B., Ed.; Wiley: Chichester, UK, 2005; p. 1768. [Google Scholar]
- Guczi, L.; Beck, A.; Frey, K. Role of promoting oxide morphology dictating the activity of Au/SiO2 catalyst in co oxidation. Gold Bull. 2009, 42, 5–12. [Google Scholar]
- Kalvachev, Y.A.; Hayashi, T.; Tsubota, S.; Haruta, M. Selective partial oxidation of propylene to propylene oxide on Au/Ti-MCM catalysts in the presence of hydrogen and oxygen. Stud. Surf. Sci. Catal. 1997, 110, 965–972. [Google Scholar]
- Uphade, B.S.; Okumura, M.; Tsubota, S.; Haruta, M. Effect of physical mixing of CsCl with Au/Ti-MCM-41 on the gas-phase epoxidation of propene using H2 and O2: Drastic depression of H2 consumption. Appl. Catal. A Gen. 2000, 190, 43–50. [Google Scholar]
- Qi, C.X.; Akita, T.; Okumura, M.; Kuraoka, K.; Haruta, M. Effect of surface chemical properties and texture of mesoporous titanosilicates on direct vapor-phase epoxidation of propylene over Au catalysts at high reaction temperature. Appl. Catal. A Gen. 2003, 253, 75–89. [Google Scholar]
- Sinha, A.K.; Seelan, S.; Akita, T.; Tsubota, S.; Haruta, M. Vapor-phase epoxidation of propene over Au/Ti-MCM-41 catalysts: Influence of Ti content and Au content. Catal. Lett. 2003, 85, 223–228. [Google Scholar]
- Uphade, B.S.; Akita, T.; Nakamura, T.; Haruta, M. Vapor-phase epoxidation of propene using H2 and O2 over Au/Ti-MCM-48. J. Catal. 2002, 209, 331–340. [Google Scholar]
- Sinha, A.K.; Seelan, S.; Okumura, M.; Akita, T.; Tsubota, S.; Haruta, M. Three-dimensional mesoporous titanosilicates prepared by modified sol-gel method: Ideal gold catalyst supports for enhanced propene epoxidation. J. Phys. Chem. B 2005, 109, 3956–3965. [Google Scholar]
- Sinha, A.K.; Seelan, S.; Tsubota, S.; Haruta, M. Catalysis by gold nanoparticles: Epoxidation of propene. Top. Catal. 2004, 29, 95–102. [Google Scholar]
- Datye, A.K.; Bore, M.T.; Mokhonoana, M.P.; Ward, T.L.; Coville, N.J. Synthesis and reactivity of gold nanoparticles supported on transition metal doped mesoporous silica. Microporous Mesoporous Mater. 2006, 95, 118–125. [Google Scholar]
- Grunert, W.; van den Berg, M.W.E.; de Toni, A.; Bandyopadhyay, M.; Gies, H. CO oxidation with Au/TiO2 aggregates encapsulated in the mesopores of MCM-48: Model studies on activation, deactivation and metal-support interaction. Appl. Catal. A Gen. 2011, 391, 268–280. [Google Scholar]
- Grunert, W.; de Toni, A.; Gies, H. The impact of water on co oxidation with Au/TiO2 catalysts: Poison or promotor? A study with an Au/TiO2/MCM-48 model catalyst. Catal. Lett. 2011, 141, 1282–1287. [Google Scholar]
- Ruszel, M.; Grzybowska, B.; Laniecki, M.; Wojtowski, M. Au/Ti-SBA-15 catalysts in CO and preferential (PROX) CO oxidation. Catal. Commun. 2007, 8, 1284–1286. [Google Scholar]
- Yan, W.F.; Chen, B.; Mahurin, S.M.; Hagaman, E.W.; Dai, S.; Overbury, S.H. Surface sol-gel modification of mesoporous silica materials with TiO2 for the assembly of ultrasmall gold nanoparticles. J. Phys. Chem. B 2004, 108, 2793–2796. [Google Scholar]
- Yan, W.F.; Petkov, V.; Mahurin, S.M.; Overbury, S.H.; Dai, S. Powder XRD analysis and catalysis characterization of ultra-small gold nanoparticles deposited on titania-modified SBA-15. Catal. Commun. 2005, 6, 404–408. [Google Scholar]
- Chiang, C.W.; Wang, A.Q.; Wan, B.Z.; Mou, C.Y. High catalytic activity for CO oxidation of gold nanoparticles confined in acidic support Al-SBA-15 at low temperatures. J. Phys. Chem. B 2005, 109, 18042–18047. [Google Scholar]
- Chiang, C.W.; Wang, A.Q.; Mou, C.Y. CO oxidation catalyzed by gold nanoparticles confined in mesoporous aluminosilicate Al-SBA-15: Pretreatment methods. Catal. Today 2006, 117, 220–227. [Google Scholar]
- Yan, W.; Overbury, S.H.; Dai, S. Gold catalysts supported on nanostructured materials: Support effects. In Nanotechnology in Catalysis; Zhou, B., Han, S., Raja, R., Somorjai, G.A., Eds.; Springer-Verlag: Berlin, Germany, 2007; Volume 3, pp. 55–71. [Google Scholar]
- Xu, X.; Li, J.; Hao, Z.; Zhao, W.; Hu, C. Characterization and catalytic performance of Co/SBA-15 supported gold catalysts for CO oxidation. Mater. Res. Bull. 2006, 41, 406–413. [Google Scholar]
- Weckhuysen, B.M.; Sacaliuc, E.; Beale, A.M.; Nijhuis, T.A. Propene epoxidation over Au/Ti-SBA-15 catalysts. J. Catal. 2007, 248, 235–248. [Google Scholar]
- Schwartz, V.; Mullins, D.R.; Yan, W.F.; Zhu, H.G.; Dai, S.; Overbury, S.H. Structural investigation of Au catalysts on TiO2-SiO2 supports: Nature of the local structure of Ti and Au atoms by EXAFS and XANES. J. Phys. Chem. C 2007, 111, 17322–17332. [Google Scholar]
- Nava, R.; Peza-Ledesma, C.L.; Escamilla-Perea, L.; Pawelec, B.; Fierro, J.L.G. Supported gold catalysts in SBA-15 modified with TiO2 for oxidation of carbon monoxide. Appl. Catal. A Gen. 2010, 375, 37–48. [Google Scholar]
- Weckhuysen, B.M.; Sacaliuc-Parvulescu, E.; Friedrich, H.; Palkovits, R.; Nijhuis, T.A. Understanding the effect of postsynthesis ammonium treatment on the catalytic activity of Au/Ti-SBA-15 catalysts for the oxidation of propene. J. Catal. 2008, 259, 43–53. [Google Scholar]
- Li, G.; Song, H.Y.; Wang, X.S. In situ synthesis of Au/T-HMS and its catalytic performance in oxidation of bulky sulfur compounds using in situ generated H2O2 in the presence of H2/O2. Microporous Mesoporous Mater. 2009, 120, 346–350. [Google Scholar]
- Zepeda, T.A.; Martinez-Hernandez, A.; Guil-Lopez, R.; Pawelec, B. Preferential CO oxidation in excess of hydrogen over Au/HMS catalysts modified by Ce, Fe and Ti oxides. Appl. Catal. B Environ. 2010, 100, 450–462. [Google Scholar]
- Li, G.; Song, H.Y.; Wang, X.S.; Chen, Y.Y. Characterization and catalytic performance of Au/Ti-HMS for direct generation of H2O2 and in situ-H2O2-ODS from H2 and O2: An in situ-reduction synthesis and a recycle study of catalyst. Microporous Mesoporous Mater. 2011, 139, 104–109. [Google Scholar]
- Perez-Cabero, M.; El Haskouri, J.; Solsona, B.; Vazquez, I.; Dejoz, A.; Garcia, T.; Alvarez-Rodriguez, J.; Beltran, A.; Beltran, D.; Amoros, P. Stable anchoring of dispersed gold nanoparticles on hierarchic porous silica-based materials. J. Mater. Chem. 2010, 20, 6780–6788. [Google Scholar]
- Tai, Y.; Tajiri, K. Preparation, thermal stability, and CO oxidation activity of highly loaded Au/titania-coated silica aerogel catalysts. Appl. Catal. A Gen. 2008, 342, 113–118. [Google Scholar]
- Tai, Y.; Yamaguchi, W.; Tajiri, K.; Kageyama, H. Structures and CO oxidation activities of size-selected Au nanoparticles in mesoporous titania-coated silica aerogels. Appl. Catal. A Gen. 2009, 364, 143–149. [Google Scholar]
- Husing, N.; Geserick, J.; Froschl, T.; Kucerova, G.; Makosch, M.; Diemant, T.; Eckle, S.; Behm, R.J. Molecular approaches towards mixed metal oxides and their behaviour in mixed oxide support Au catalysts for CO oxidation. Dalton Trans. 2011, 40, 3269–3286. [Google Scholar]
- Dai, S.; Zhu, H.G.; Pan, Z.W.; Chen, B.; Lee, B.; Mahurin, S.M.; Overbury, S.H. Synthesis of ordered mixed titania and silica mesostructured monoliths for gold catalysts. J. Phys. Chem. B 2004, 108, 20038–20044. [Google Scholar]
- Zepeda, T.A.; Vilchis-Nestor, A.R.; Avalos-Borja, M.; Gomez, S.A.; Hernandez, J.A.; Olivas, A. Alternative bio-reduction synthesis method for the preparation of Au(AgAu)/SiO2-Al2O3 catalysts: Oxidation and hydrogenation of CO. Appl. Catal. B Environ. 2009, 90, 64–73. [Google Scholar]
- Qi, C.; Okumura, M.; Akita, T.; Haruta, M. Vapor-phase epoxidation of propylene using H2/O2 mixture over gold catalysts supported on non-porous and mesoporous titania-silica: Effect of preparation conditions and pretreatments prior to reaction. Appl. Catal. A Gen. 2004, 263, 19–26. [Google Scholar]
- Sinha, A.K.; Seelan, S.; Tsubota, S.; Haruta, M. A three-dimensional mesoporous titanosilicate support for gold nanoparticles: Vapor-phase epoxidation of propene with high conversion. Angew. Chem. Int. Ed. 2004, 43, 1546–1548. [Google Scholar]
- Haruta, M.; Uphade, B.S.; Tsubota, S.; Miyamoto, A. Selective oxidation of propylene over gold deposited on titanium-based oxides. Res. Chem. Intermed. 1998, 24, 329–336. [Google Scholar]
- Haruta, M.; Chowdhitry, B.; Bravo-Suarez, J.J.; Date, M.; Tsubota, S. Trimethylamine as a gas-phase promoter: Highly efficient epoxidation of propylene over supported gold catalysts. Angew. Chem. Int. Ed. 2006, 45, 412–415. [Google Scholar]
- Hayashi, T.; Tanaka, K.; Haruta, M. Selective vapor-phase epoxidation of propylene over Au/TiO2 catalysts in the presence of oxygen and hydrogen. J. Catal. 1998, 178, 566–575. [Google Scholar]
- Uphade, B.S.; Okumura, M.; Yamada, N.; Tsubota, S.; Haruta, M. Vapor-phase epoxidation of propene using H2 and O2 over Au/Ti-MCM-41 and Au/Ti-MCM-48. In Studies in Surface Science and Catalysis; Corma, A., Melo, F.V., Mendioroz, S., Fierro, J.L.G., Eds.; Elsevier: Granada, Spain, 2000. [Google Scholar]
- Chatterjee, M.; Ikushima, Y.; Hakuta, Y.; Kawanami, H. In situ synthesis of gold nanoparticles inside the pores of MCM-48 in supercritical carbon dioxide and its catalytic application. Adv. Synth. Catal. 2006, 348, 1580–1590. [Google Scholar]
- Gupta, G.; Shah, P.S.; Zhang, X.G.; Saunders, A.E.; Korgel, B.A.; Johnston, K.P. Enhanced infusion of gold nanocrystals into mesoporous silica with supercritical carbon dioxide. Chem. Mater. 2005, 17, 6728–6738. [Google Scholar]
- Campelo, J.M.; Conesa, T.D.; Gracia, M.J.; Jurado, M.J.; Luque, R.; Marinas, J.M.; Romero, A.A. Microwave facile preparation of highly active and dispersed SBA-12 supported metal nanoparticles. Green Chem. 2008, 10, 853–858. [Google Scholar]
- Corma, A.; Gutierrez-Puebla, E.; Iglesias, M.; Monge, A.; Perez-Ferreras, S.; Sanchez, F. New heterogenized gold(I)-heterocyclic carbene complexes as reusable catalysts in hydrogenation and cross-coupling reactions. Adv. Synth. Catal. 2006, 348, 1899–1907. [Google Scholar]
- Tsukuda, T.; Liu, Y.M.; Tsunoyama, H.; Akita, T. Size effect of silica-supported gold clusters in the microwave-assisted oxidation of benzyl alcohol with H2O2. Chem. Lett. 2010, 39, 159–161. [Google Scholar]
- Asefa, T.; Lennox, R.B. Synthesis of gold nanoparticles via electroless deposition in SBA-15. Chem. Mater. 2005, 17, 2481–2483. [Google Scholar]
- Gu, J.L.; Shi, J.L.; Xiong, L.M.; Chen, H.R.; Li, L.; Ruan, M.L. A new strategy to incorporate high density gold nanowires into the channels of mesoporous silica thin films by electroless deposition. Solid State Sci. 2004, 6, 747–752. [Google Scholar]
- Chen, W.; Cai, W.P.; Zhang, Z.P.; Zhang, L. A convenient synthetic route to gold nanoparticles dispersed within mesoporous silica. Chem. Lett. 2001, 152–153. [Google Scholar]
- Chen, W.; Zhang, J.Y.; Cai, W.P. Sonochemical preparation of Au, Ag, Pd/SiO2 mesoporous nanocomposites. Scr. Mater. 2003, 48, 1061–1066. [Google Scholar]
- Chen, W.; Cai, W.P.; Zhang, L.; Wang, G.Z.; Zhang, L.D. Sonochemical processes and formation of gold nanoparticles within pores of mesoporous silica. J. Colloid Interface Sci. 2001, 238, 291–295. [Google Scholar]
- Chen, W.; Cai, W.P.; Chen, Z.X.; Zhang, L.D. Structural change of mesoporous silica with sonochemically prepared gold nanoparticles in its pores. Ultrason. Sonochem. 2001, 8, 335–339. [Google Scholar]
- Chen, W.; Cai, W.P.; Liang, C.H.; Zhang, L.D. Synthesis of gold nanoparticles dispersed within pores of mesoporous silica induced by ultrasonic irradiation and its characterization. Mater. Res. Bull. 2001, 36, 335–342. [Google Scholar]
- Sun, W.; Fu, G.; Cai, W. Ambient effect on optical absorption of Au nanoparticles dispersed within pores of mesoporous SiO2. Philos. Mag. 2007, 87, 337–353. [Google Scholar]
- Jan, J.S.; Chen, P.J.; Ho, Y.H. Bioassisted synthesis of catalytic gold/silica nanotubes using layer-by-layer assembled polypeptide templates. J. Colloid Interface Sci. 2011, 358, 409–415. [Google Scholar]
- Zhao, D.Y.; Deng, Y.H.; Cai, Y.; Sun, Z.K.; Liu, J.; Liu, C.; Wei, J.; Li, W.; Liu, C.; Wang, Y. Multifunctional mesoporous composite microspheres with well-designed nanostructure: A highly integrated catalyst system. J. Am. Chem. Soc. 2010, 132, 8466–8473. [Google Scholar]
- Lee, J.; Park, J.C.; Song, H. A nanoreactor framework of a Au@SiO2 yolk/shell structure for catalytic reduction of p-nitrophenol. Adv. Mater. 2008, 20, 1523–1528. [Google Scholar]
List of Acronyms
| AAPTS: | 3-(2-Aminoethylamino) Propyltrimethoxysilane |
| APS: | 3-Aminopropyltrimethoxysilane |
| APTS: | 3-Aminopropyltriethoxysilane |
| AuCl(THT): | Chloro-(thiophene)gold(I) |
| AuMe2(HFA): | Dimethyl(hexafluoroacetylacetoato) gold(III) |
| AuNPs: | Gold Nanoparticles |
| BET: | Brunauer-Emmett-Teller |
| BTESPTS: | bis-[3-(triethoxysilyl) propyl] tetrasulfide |
| C10H23ClSi: | Chlorodimethyloctylsilane |
| C18TMS: | n-Octadecyltrimethoxysilane |
| CTABr: | Cetyltrimethylammonium Bromide |
| CTACl: | Cetyltrimethylammonium Chloride |
| CVD: | Chemical Vapor Deposition |
| DBTA: | Dibenzoyl Tartaric Acid |
| DDA: | Dodecylamine |
| DP: | Deposition-Precipitation |
| en: | Ethylenediamine |
| EtOH: | Ethanol |
| F127: | Pluronic Block Copolymer Surfactant |
| FDU: | Fudan University in Shanghai Marerials |
| G4-PANAM: | Polyamidoamine Dendrimers Fourth Generation |
| HAADF: | Higher Angle Annular Dark Field Scanning Transmission |
| STEM: | Electron Microscopy |
| HMM: | Hiroshima Mesoporous Material |
| HMS: | Hexagonal Mesoporous Silica |
| HSN: | Hollow Silica Nanospheres |
| IEP: | Isoelectric Point |
| IWI: | Incipient Wet Impregantion |
| KIT-5: | Large Mesopore Fm3m Silica |
| MCF: | Mesostructured Cellular Foam |
| MCM: | Mobile Crystalline Material |
| Me2Au(acac): | Dimethyl Gold Acetylacetonate |
| MPS: | 3-Mercaptopropyltrimethoxysilane |
| MPTS: | 3-Mercaptopropyltriethoxysilane |
| MS: | Mesoporous Silica |
| MSM: | Mesoporous Silica Materials |
| MSS: | Mesoporous Silica Spheres |
| MSTF: | Mesoporous Silica Thin Films |
| MSU: | Michigan State University Materials |
| MTFs: | Mesoporous Thin Films |
| OBSQ: | Organic Bridged Silsesquioxane |
| OFL: | Organic Functional Ligand |
| P123: | Pluronic Acid Block Copolymer |
| PAMAM: | Polyamidoamine Dendrimers |
| PLL: | Poly(l-lysine) |
| PLT: | Poly(l-tyrosine) |
| PMOs: | Periodic Mesoporous Organosilica |
| PROX: | Preferential Oxidation of CO in the Presence of H2 |
| PrTMS: | Propyl Trimethoxysilane |
| Py: | (s)-(–)-2-Pyrrolidinone-5-carboxylic acid (Py) |
| SBA: | Santa Barbara Amorphous |
| SDA: | Structure-Directing Agent |
| SDS: | Sodium Dodecyl Sulfate |
| TEM: | Transmission Electron Microscopy |
| TEOS: | Tetraethoxysilane |
| THF: | Tetrahydrofuran |
| TMOS: | Tetramethoxysilane |
| TMPDA: | 3-(Trimethoxysilyl)propyl]diethylenediamine |
| TMPTA: | 3-(Trimethoxysilyl)propyl]diethylenetriamine |
| TPED: | N-[3-(Trimethoxysilyl)propyl]ethylenediamine |
| TPTAC: | N-Trimethoxysilylpropyl-N,N,N-trimethylammonium Chloride |
| VTES: | Vinyl Triethoxysilane |
| WSS: | Wormhole Silica Support |
| XANES: | X-ray Absorption Near-Edge Spectroscopy |
| XPS: | X-Ray Photoelectron Spectroscopy |
| XRD: | Powder X-Ray Diffraction |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gutiérrez, L.-F.; Hamoudi, S.; Belkacemi, K. Synthesis of Gold Catalysts Supported on Mesoporous Silica Materials: Recent Developments. Catalysts 2011, 1, 97-154. https://doi.org/10.3390/catal1010097
Gutiérrez L-F, Hamoudi S, Belkacemi K. Synthesis of Gold Catalysts Supported on Mesoporous Silica Materials: Recent Developments. Catalysts. 2011; 1(1):97-154. https://doi.org/10.3390/catal1010097
Chicago/Turabian StyleGutiérrez, Luis-Felipe, Safia Hamoudi, and Khaled Belkacemi. 2011. "Synthesis of Gold Catalysts Supported on Mesoporous Silica Materials: Recent Developments" Catalysts 1, no. 1: 97-154. https://doi.org/10.3390/catal1010097