Sulfur Tolerant Magnesium Nickel Silicate Catalyst for Reforming of Biomass Gasification Products to Syngas

Abstract
:1. Introduction
2. Results and Discussion
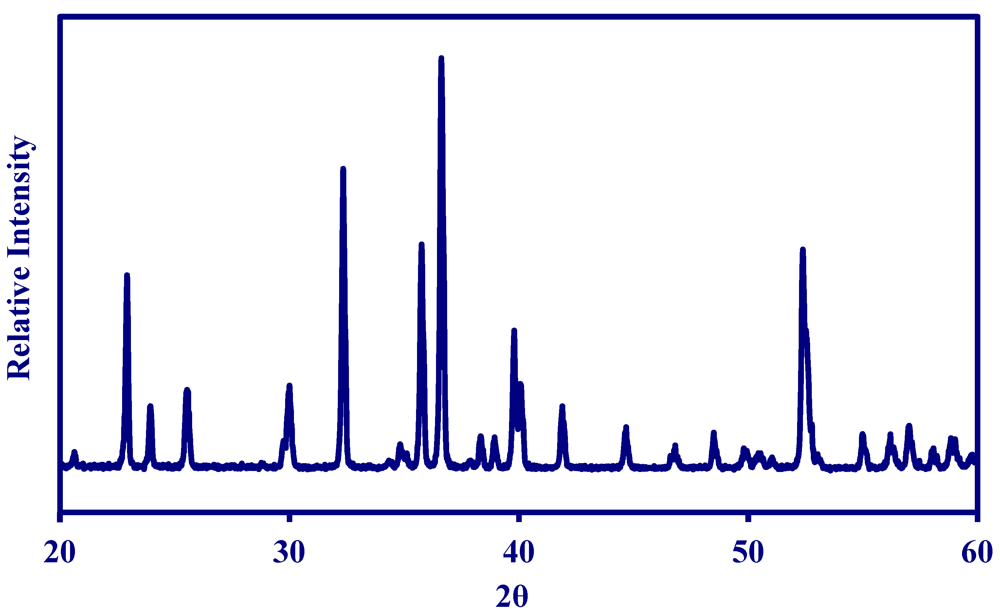
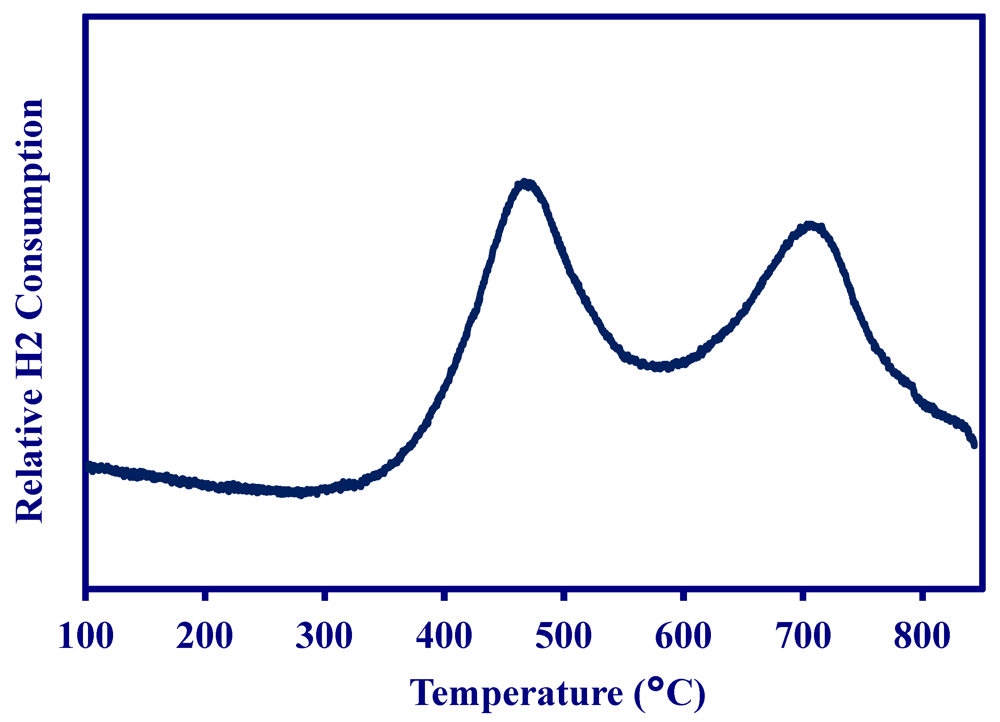
2.1. Properties of MNS Catalyst


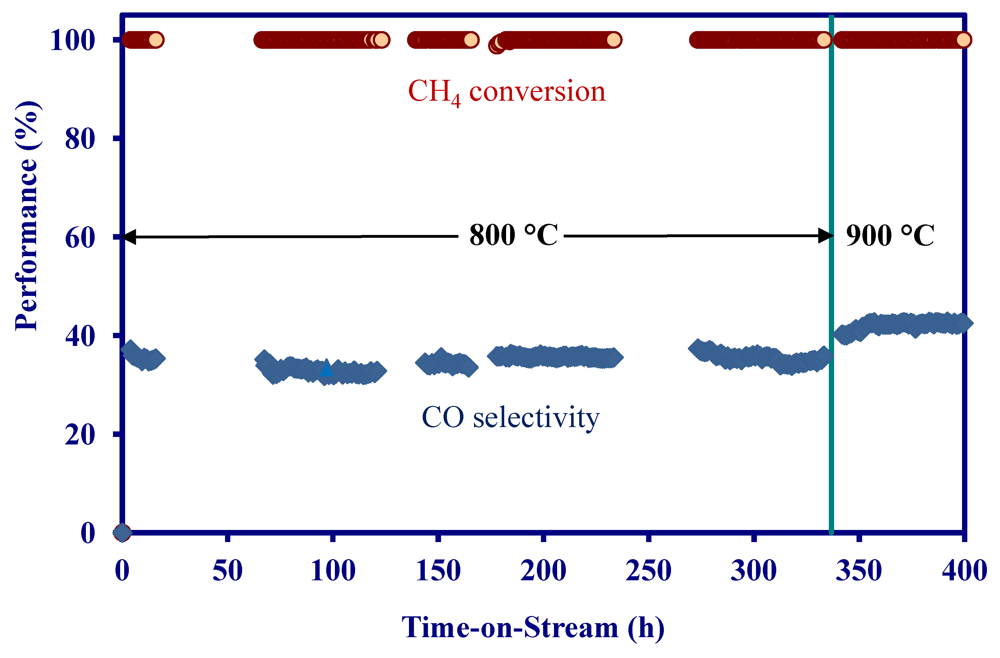
2.2. Methane Reforming on Granulated Catalyst


2.3. Catalytic Performance of MNS Extruded Monolith


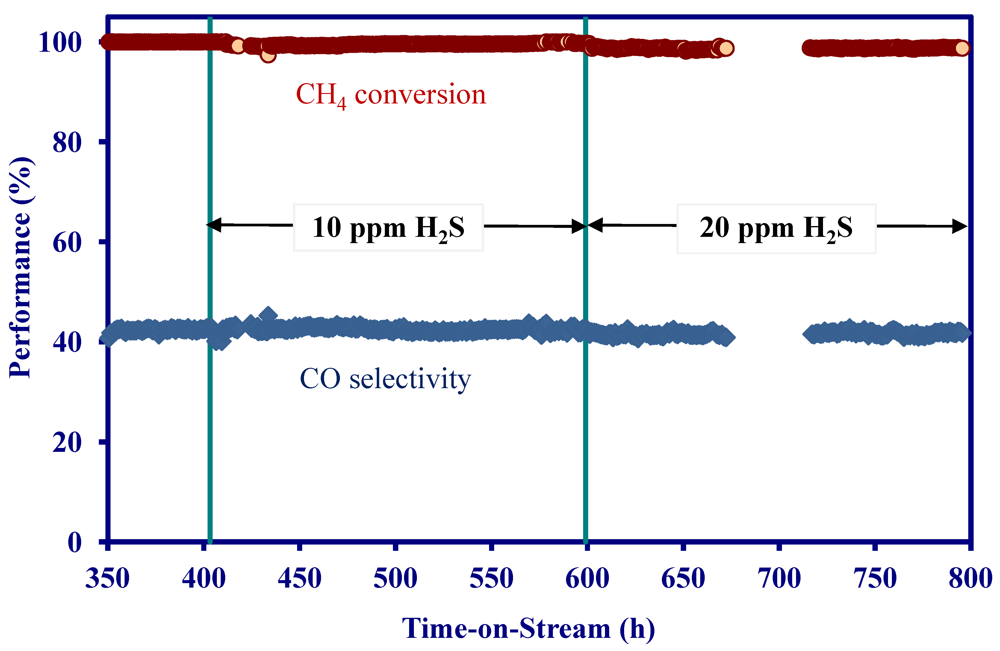
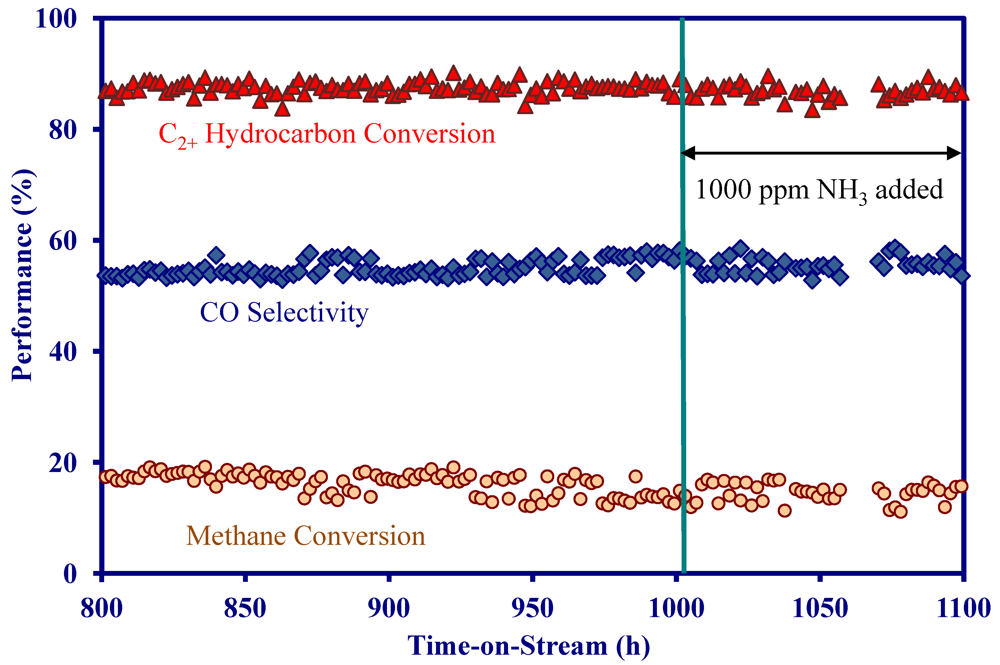
2.4. Catalytic Performance of MNS Washcoated Cordierite Monolith


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monolith Temperature (°C) | Space Velocity (h−1) | C2+ Conversion (%) | Methane Conversion (%) |
|---|---|---|---|
| 900 | 5000 | 100 | 37 |
| 875 | 3000 | 100 | 27 |
| 850 | 1800 | 99 | 20 |

3. Experimental Section
3.1. Catalyst Preparation and Characterization
3.2. Catalyst Performance Testing

4. Conclusions
- Granulated MNS catalysts achieved complete methane conversion at 900 °C and a space velocity of 24,000 mL/g/h in a simulated biomass gasification stream. Addition of 10–20 ppm H2S to the stream did not significantly change the catalyst performance.
- The MNS-extruded monolith exhibited poor activity in biomass reforming, which was attributed to a significant decrease in surface area during the monolith manufacturing process.
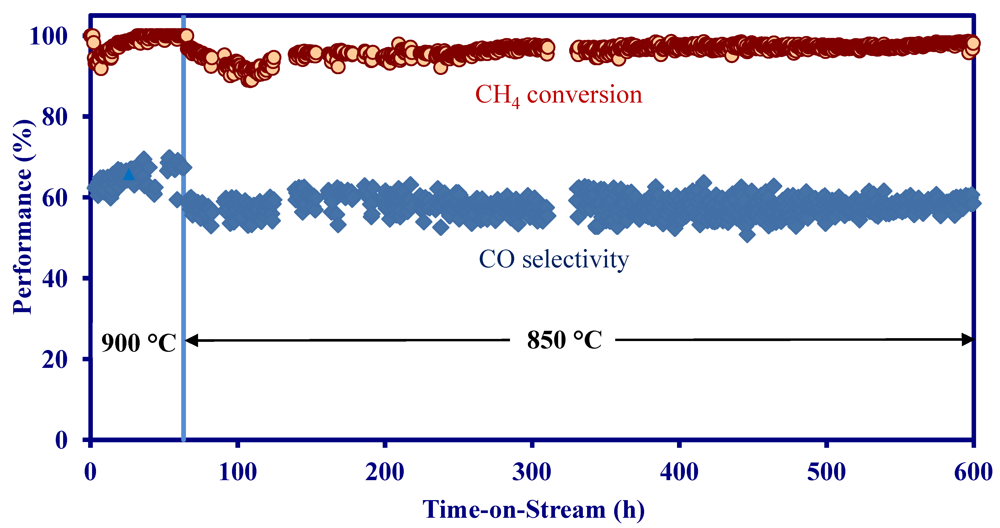
- The MNS catalyst washcoated on MNS-extruded monolith achieved 90–100% CH4 conversion at 850–900 °C and a space velocity of 10,000 h−1 in a simulated biomass gasification stream without H2S.
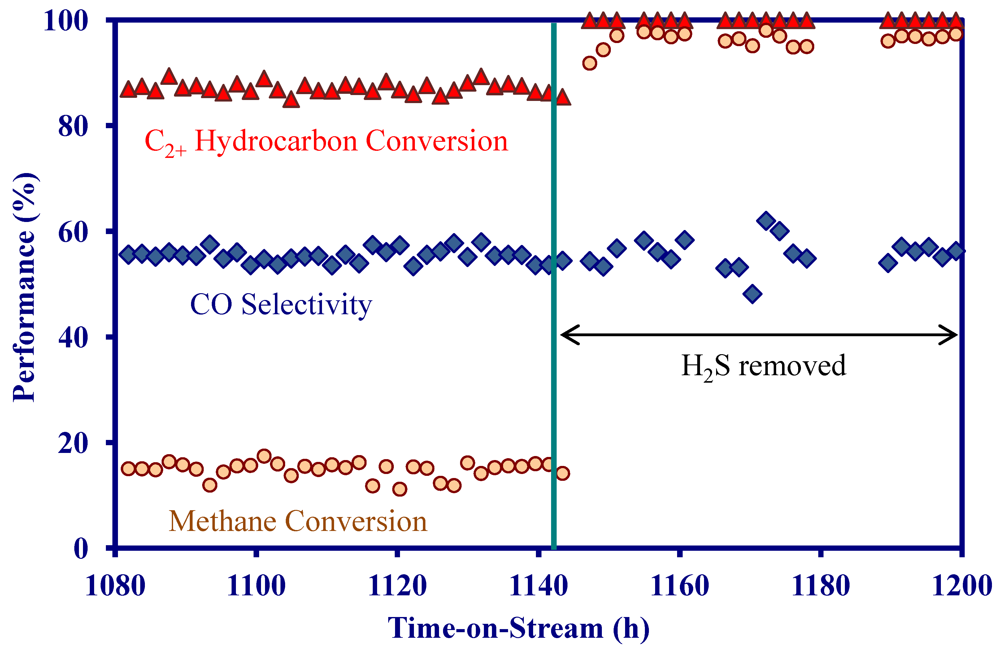
- On a MNS-washcoated cordierite monolith, the addition of 22 ppm H2S to a simulated biomass gasification stream decreased the initial activity at 900 °C, but the activity was constant for 1200 h in the presence of H2S.
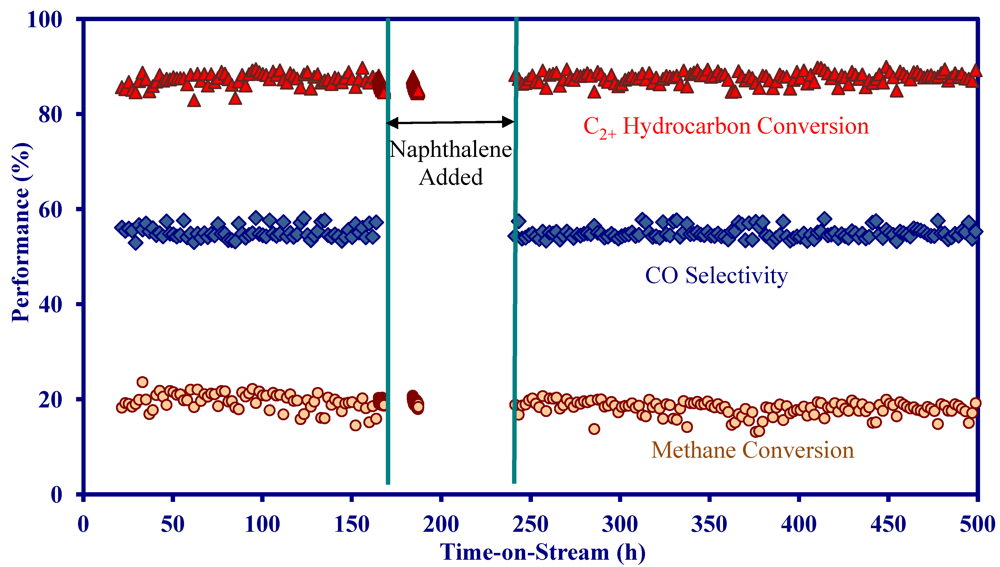
- On the MNS washcoated monolith, the reforming activity decreased in a sequence of C3 hydrocarbons > C2 hydrocarbons > methane.
- On the MNS-washcoated monolith, complete C2+ hydrocarbon conversion could be achieved at 900 °C and a GHSV of 5000 h−1 in the presence of H2S.
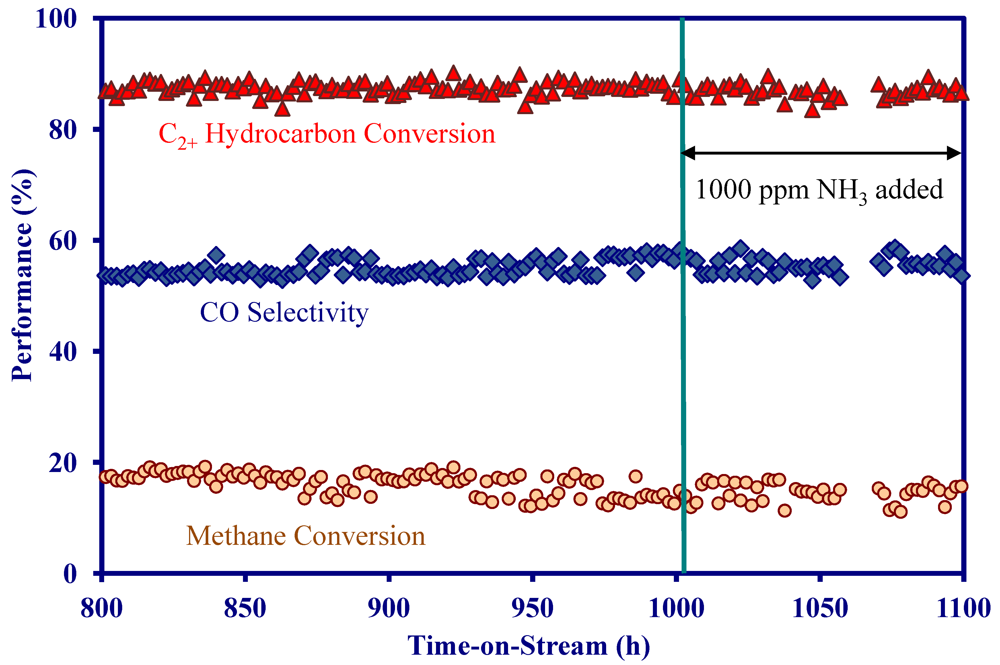
- The addition of 1000 ppm NH3 did not impact the catalytic performance at 900 °C in the presence and absence of H2S, indicating the MNS catalyst was immune to NH3.
Acknowledgements
References
- Bulushev, D.A.; Ross, J.R.H. Catalysis for conversion of biomass to fuels via pyrolysis and gasification: A review. Catal. Today 2011, 171, 1–13. [Google Scholar] [CrossRef]
- Anis, S.; Zainal, Z.A. Tar reduction in biomass producer gas via mechanical, catalytic and thermal methods: A review. Renew. Sustain. Energy Rev. 2011, 15, 2355–2377. [Google Scholar] [CrossRef]
- Corella, J.; Aznar, M.P.; Gil, J.; Caballero, M.A. Biomass gasification in fluidized bed: where to locate the dolomite to improve gasification? Energy Fuels 1999, 13, 1122–1127. [Google Scholar] [CrossRef]
- Perez, P.; Aznar, P.M.; Caballero, M.A.; Gil, J.; Martin, J.A.; Corella, J. Hot gas cleaning and upgrading with a calcined dolomite located downstream a biomass fluidized bed gasifier operating with steam–oxygen mixtures. Energy Fuels 1997, 11, 1194–1203. [Google Scholar] [CrossRef]
- Srinakruang, J.; Sato, K.; Vitidsant, T.; Fujimoto, K. A highly efficient catalyst for tar gasification with steam. Catal. Commun. 2005, 6, 437–440. [Google Scholar] [CrossRef]
- Wang, T.; Chang, J.; Lv, P. Novel catalyst for cracking of biomass tar. Energy Fuels 2005, 19, 22–27. [Google Scholar] [CrossRef]
- Wang, T.J.; Chang, J.C.; Wu, C.Z.; Fu, Y.; Chen, Y. The steam reforming of naphthalene over a nickel-dolomite cracking catalyst. Biomass Bioenergy 2005, 28, 508–514. [Google Scholar] [CrossRef]
- Devi, L.K.; Ptasinski, J.; Janssen, F.J.J.G. Decomposition of naphthalene as a biomass tar over pretreated olivine: effect of gas composition, kinetic approach, and reaction scheme. Ind. Eng. Chem. Res. 2005, 44, 9096–9104. [Google Scholar]
- Devi, L.; Craje, M.; Thune, P.; Ptasinski, K.J.; Janssen, F.J.J.G. Olivine as tar removal catalyst for biomass gasifiers: Catalyst characterization. Appl. Catal. A 2005, 294, 68–79. [Google Scholar] [CrossRef]
- Rapagnà, S.; Jand, N.; Kiennemann, A.; Foscolo, P.U. Steam-gasification of biomass in a fluidised-bed of olivine particles. Biomass Bioenergy 2000, 19, 187–197. [Google Scholar] [CrossRef]
- Courson, C.; Makaga, E.; Petit, C.; Kiennemann, A. Development of Ni catalysts for gas production from biomass gasification. Reactivity in steam- and dry-reforming. Catal. Today 2000, 63, 427–437. [Google Scholar]
- Courson, C.; Udron, L.; Świerczyński, D.; Petit, C.; Kiennemann, A. Hydrogen production from biomass gasification on nickel catalysts. Catal. Today 2002, 76, 75–86. [Google Scholar]
- Swierczynski, D.; Libs, S.; Courson, C.; Kiennermann, A. Steam reforming of tar from a biomass gasification process over Ni/olivine catalyst using toluene as a model compound. Appl. Catal. B 2007, 74, 211–222. [Google Scholar] [CrossRef]
- Kuhn, J.N.; Zhao, Z.; Felix, L.G.; Slimane, R.B.; Choi, C.W.; Ozkan, U.S. Olivine catalysts for methane- and tar-steam reforming. Appl. Catal. B 2008, 81, 14–26. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.; Brown, R.C. Steam reforming of tar compounds over Ni/olivine catalysts doped with CeO2. Energy Convers. Manag. 2007, 48, 68–77. [Google Scholar] [CrossRef]
- Li, D.; Wang, L.; Koike, M.; Nakagawa, Y.; Tomishige, K. Steam reforming of tar from pyrolysis of biomass over Ni/Mg/Al catalysts prepared from hydrotalcite-like precursors. Appl. Catal. B 2011, 102, 528–538. [Google Scholar] [CrossRef]
- Wang, L.; Li, D.; Koike, M.; Nakagawa, Y.; Xu, Y.; Tomishige, K. Catalytic performance and characterization of Ni-Fe catalysts for the steam reforming of tar from biomass pyrolysis to synthesis gas. Appl. Catal. A 2011, 392, 248–255. [Google Scholar] [CrossRef]
- Min, Z.; Asadullah, M.; Yimsiri, P.; Zhang, S.; Wu, H.; Li, C. Catalytic reforming of tar during gasification. Part I. Steam reforming of biomass tar using ilmenite as a catalyst. Fuel 2011, 90, 1847–1854. [Google Scholar] [CrossRef]
- Lamacz, A.; Krzton, A.; Djega-Mariadassou, G. Steam reforming of model gasification tars compounds on nickel based ceria-zirconia catalysts. Catal. Today 2011, 176, 347–351. [Google Scholar]
- Tomishige, K.; Miyazawa, T.; Asadullah, M.; Ito, S.; Kunimori, K. Catalyst performance in reforming of tar derived from biomass over noble metal catalysts. Green Chem. 2003, 5, 399–403. [Google Scholar] [CrossRef]
- Schmidt, S.; Giesa, S.; Drochner, A.; Vogel, H. Catalytic tar removal from bio syngas-Catalyst development and kinetic studies. Catal. Today 2011, 175, 442–449. [Google Scholar]
- Mendiara, T.; Johansen, J.M.; Utrilla, R.; Jensen, A.D.; Glarborg, P. Evaluation of different oxygen carriers for biomass tar reforming (II): Carbon deposition in experiments with methane and other gases. Fuel 2011, 90, 1370–1382. [Google Scholar]
- Kong, M.; Fei, J.; Wang, S.; Lu, W.; Zheng, X. Influence of supports on catalytic behavior of nickel catalysts in carbon dioxide reforming of toluene as a model compound of tar from biomass gasification. Bioresour. Technol. 2011, 102, 2004–2008. [Google Scholar] [CrossRef]
- Park, H.J.; Park, S.H.; Sohn, J.M.; Park., J.H.; Jeone, J.K.; Kim, S.S.; Park, Y.K. Steam reforming of biomass gasification tar using benzene as a model compound over various Ni supported metal oxide catalysts. Bioresour. Technol. 2010, 101, S101–S103. [Google Scholar]
- Sato, K.; Fujimoto, K. Development of new nickel based catalyst for tar reforming with superior resistance to sulfur poisoning and coking in biomass gasification. Catal. Commun. 2007, 8, 1697–1701. [Google Scholar] [CrossRef]
- Yang, X.Q.; Xu, X.P.; Xu, X.L.; Liu, X.D.; Liu, C.H. Nickel supported on modified olivine catalysts for steam reforming of biomass gasification tar. Catal. Commun. 2010, 11, 383–386. [Google Scholar] [CrossRef]
- Caballero, M.A.; Corella, J.; Aznar, M.P.; Jil, J. Biomass gasification with air in fluidized bed—hot gas cleanup with selected commercial and full-size nickel-based catalysts. Ind. Eng. Chem. Res. 2000, 39, 1143–1154. [Google Scholar] [CrossRef]
- Caballero, M.A.; Aznar, M.P.; Jil, J.; Martin, J.A.; Frances, E.; Corella, J. Commercial steam reforming catalysts to improve biomass gasification with steam−oxygen mixtures. 1. Hot gas upgrading by the catalytic reactor. Ind. Eng. Chem. Res. 1997, 36, 5227–5239. [Google Scholar] [CrossRef]
- Pfeifer, C.; Hofbauer, H. Development of catalytic tar decomposition downstream from a dual fluidized bed biomass steam gasifier. Powder Technol. 2008, 180, 9–16. [Google Scholar] [CrossRef]
- Srinakruang, J.; Sato, K.; Vitidsant, T.; Fujimoto, K. Highly efficient sulfur and coking resistance catalysts for tar gasification with steam. Fuel 2006, 85, 2419–2426. [Google Scholar] [CrossRef]
- Miyazawa, T.; Kimura, T.; Nishikawa, J.; Kado, S.; Kunimori, K.; Tomishige, K. Catalytic performance of supported Ni catalysts in partial oxidation and steam reforming of tar derived from the pyrolysis of wood biomass. Catal. Today 2006, 115, 254–262. [Google Scholar] [CrossRef]
- Li, C.; Hirabayashi, D.; Suzuki, K. A crucial role of O2− and O22− on mayenite structure for biomass tar steam reforming over Ni/Ca12Al14O33. Appl. Catal. B 2009, 88, 351–360. [Google Scholar] [CrossRef]
- Xu, C.; Donald, J.; Byambajav, E.; Ohtsuka, Y. Recent advances in catalysts for hot-gas removal of tar and NH3 from biomass gasification. Fuel 2010, 89, 1784–1795. [Google Scholar] [CrossRef]
- Kimura, T.; Miyazawa, T.; Nishikawa, J.; Kado, S.; Okumura, K.; Miyao, T. Development of Ni catalysts for tar removal by steam gasification of biomass. Appl. Catal. B 2006, 68, 160–170. [Google Scholar] [CrossRef]
- Sutton, D.; Kelleher, B.; Doyle, A.; Ross, J.R.H. Investigation of nickel supported catalysts for the upgrading of brown peat derived gasification products. Bioresour. Technol. 2001, 80, 111–116. [Google Scholar]
- Furusawa, T.; Tsutsumi, A. Comparison of Co/MgO and Ni/MgO catalysts for the steam reforming of naphthalene as a model compound of tar derived from biomass gasification. Appl. Catal. A 2005, 278, 207–212. [Google Scholar] [CrossRef]
- Engelen, K.; Zhang, Y.; Draelants, D.J.; Baron, G.V. A novel catalytic filter for tar removal from biomass gasification gas: Improvement of the catalytic activity in presence of H2S. Chem. Eng. Sci. 2003, 58, 665–670. [Google Scholar]
- Burattin, P.; Che, M.; Louis, C. Ni/SiO2 materials prepared by deposition-precipitation: influence of the reduction conditions and mechanism of formation of metal particles. J. Phys. Chem. B 2000, 104, 10482–10489. [Google Scholar] [CrossRef]
- Tobiasen, L.; Sarbæk, L. Advanced Gasification in a Low Temperature Circulating Fluidised Bed Gasifier, February 2002. The European Foundation for Power Engineering (EFPE) Website. Available online: http://www.efpe.org/theses/Tobiasen_Sarbaek.pdf (accessed on 28 February 2001).
- Yung, M.M.; Magrini-Bair, K.A.; Parent, Y.O.; Carpenter, D.L.; Feik, C.J.; Gaston, K.R.; Pomeroy, M.D.; Phillips, S.D. Demonstration and characterization of Ni/Mg/K/AD90 used for pilot-scale conditioning of biomass-derived syngas. Catal. Lett. 2010, 134, 242–249. [Google Scholar] [CrossRef]
- Hepole, J.; Simell, P. Sulphur poisoning of nickel-based hot gas cleaning catalysts in synthetic gasification gas. II. Chemisorption of hydrogen sulphide. Appl. Catal. B 1997, 14, 305–321. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Long, R.Q.; Monfort, S.M.; Arkenberg, G.B.; Matter, P.H.; Swartz, S.L. Sulfur Tolerant Magnesium Nickel Silicate Catalyst for Reforming of Biomass Gasification Products to Syngas. Catalysts 2012, 2, 264-280. https://doi.org/10.3390/catal2020264
Long RQ, Monfort SM, Arkenberg GB, Matter PH, Swartz SL. Sulfur Tolerant Magnesium Nickel Silicate Catalyst for Reforming of Biomass Gasification Products to Syngas. Catalysts. 2012; 2(2):264-280. https://doi.org/10.3390/catal2020264
Chicago/Turabian StyleLong, Richard Q., Scott M. Monfort, Gene B. Arkenberg, Paul H. Matter, and Scott L. Swartz. 2012. "Sulfur Tolerant Magnesium Nickel Silicate Catalyst for Reforming of Biomass Gasification Products to Syngas" Catalysts 2, no. 2: 264-280. https://doi.org/10.3390/catal2020264