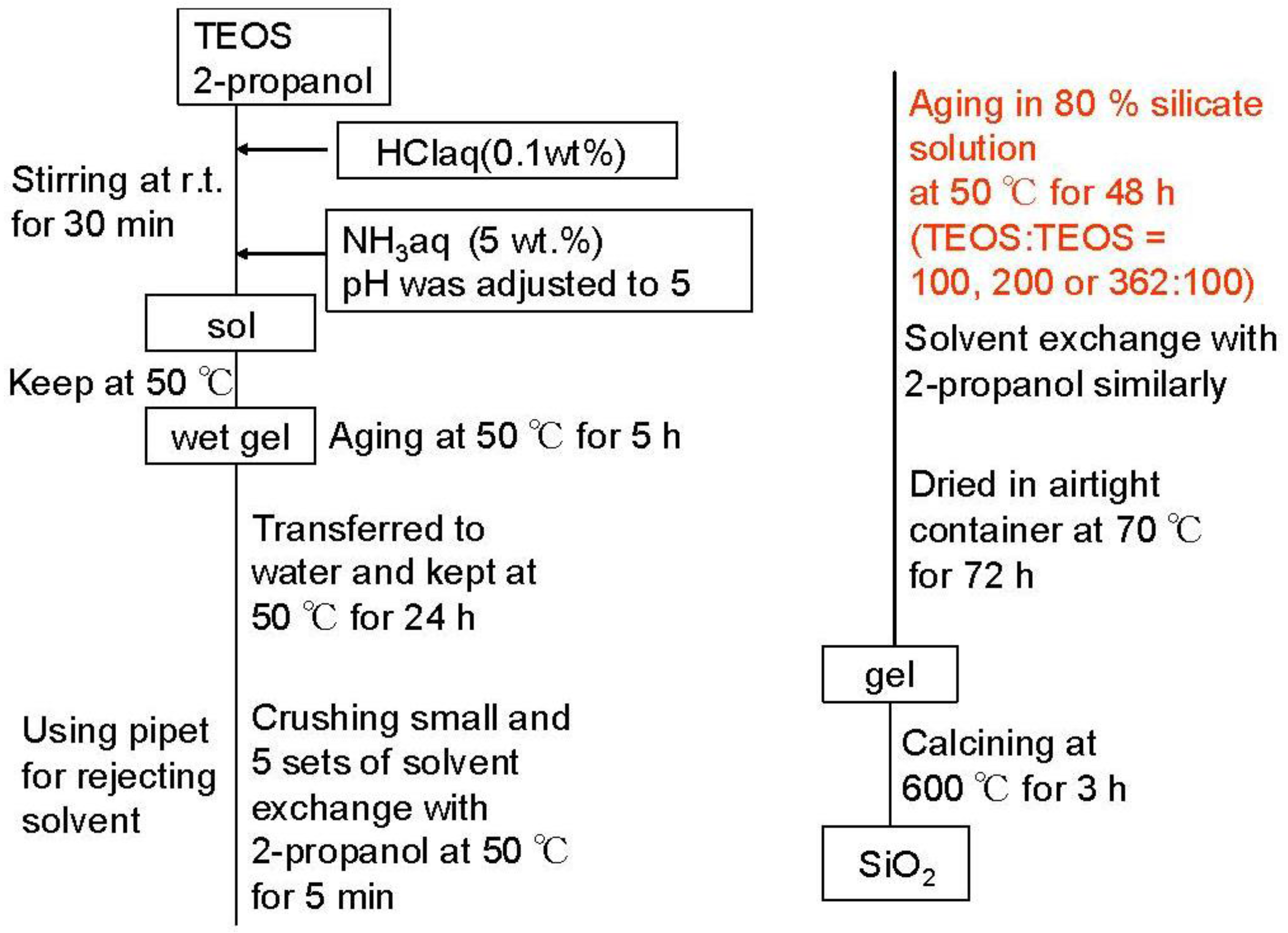
3.1. Characterization of Silicas and Silica-Aluminas with Large Mesopore
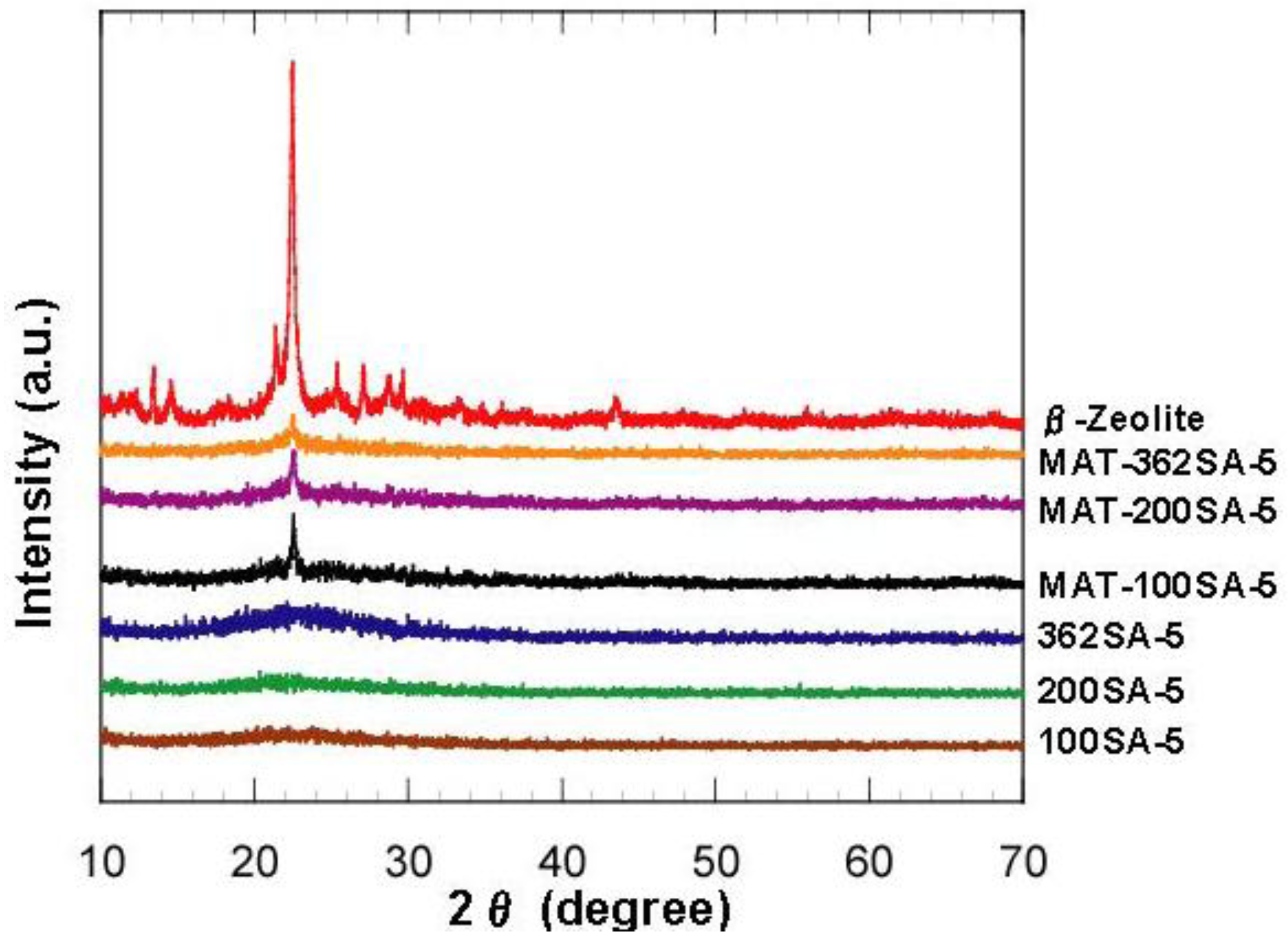
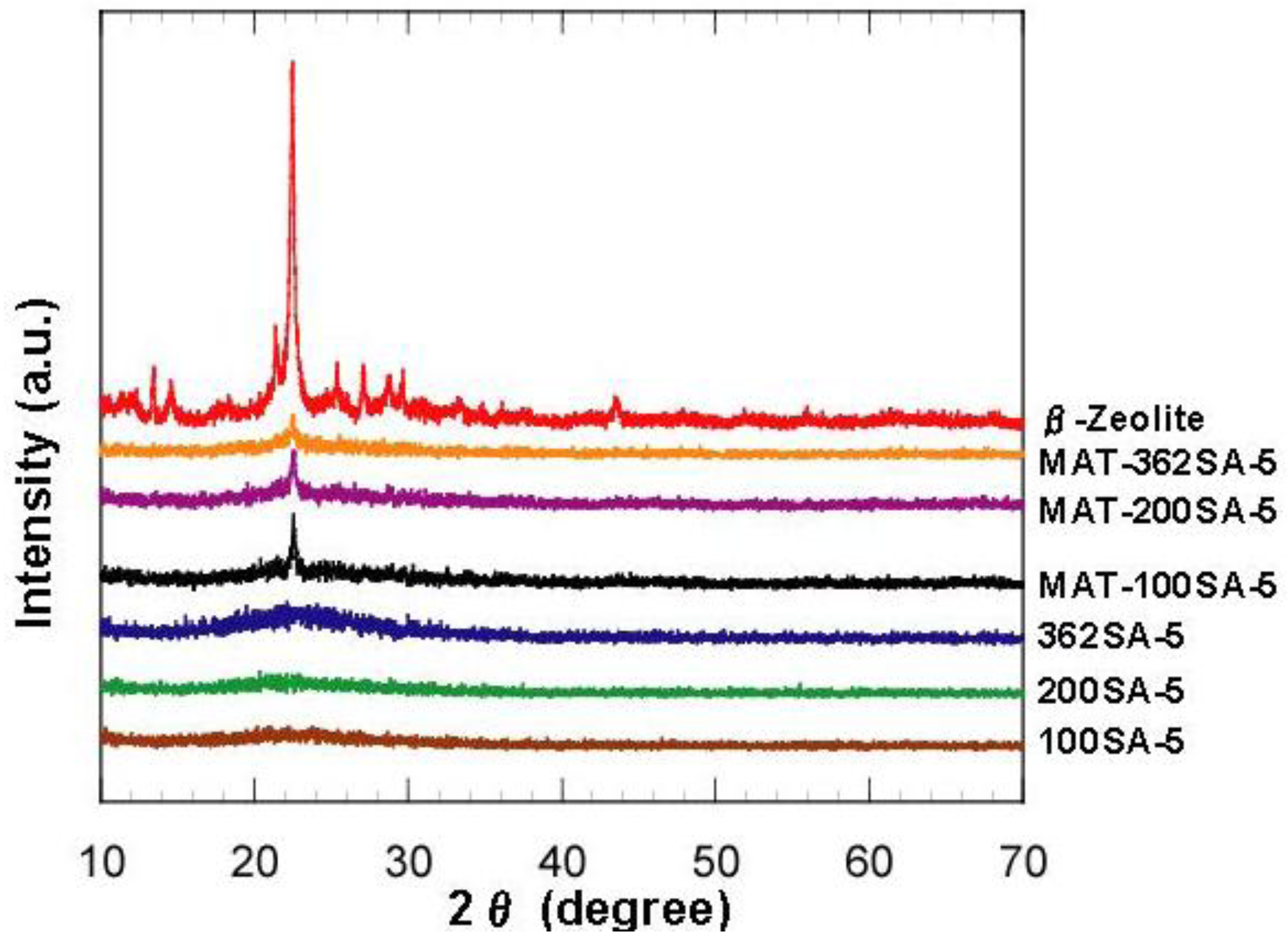
Figure 2 shows the XRD patterns of zeolite, mixed catalysts and silica-aluminas. There is no peak in XRD patterns of synthesized silica and silica-alumina, and it was confirmed that they were amorphous. XRD patterns of the mixed catalysts showed the peaks of zeolite, indicating that the crystal structure of zeolite was maintained. Results from N
2 adsorption are summarized in
Table 1.
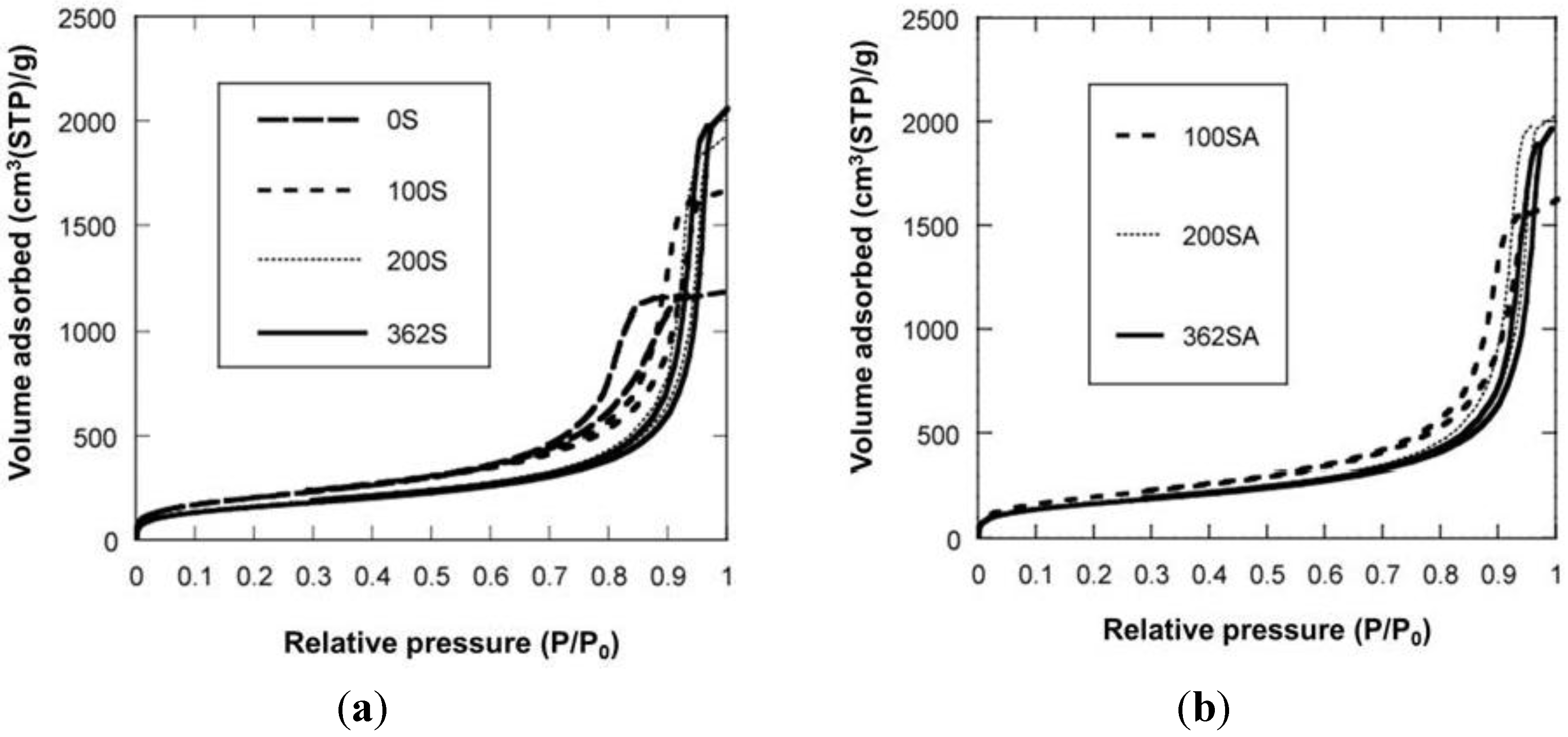
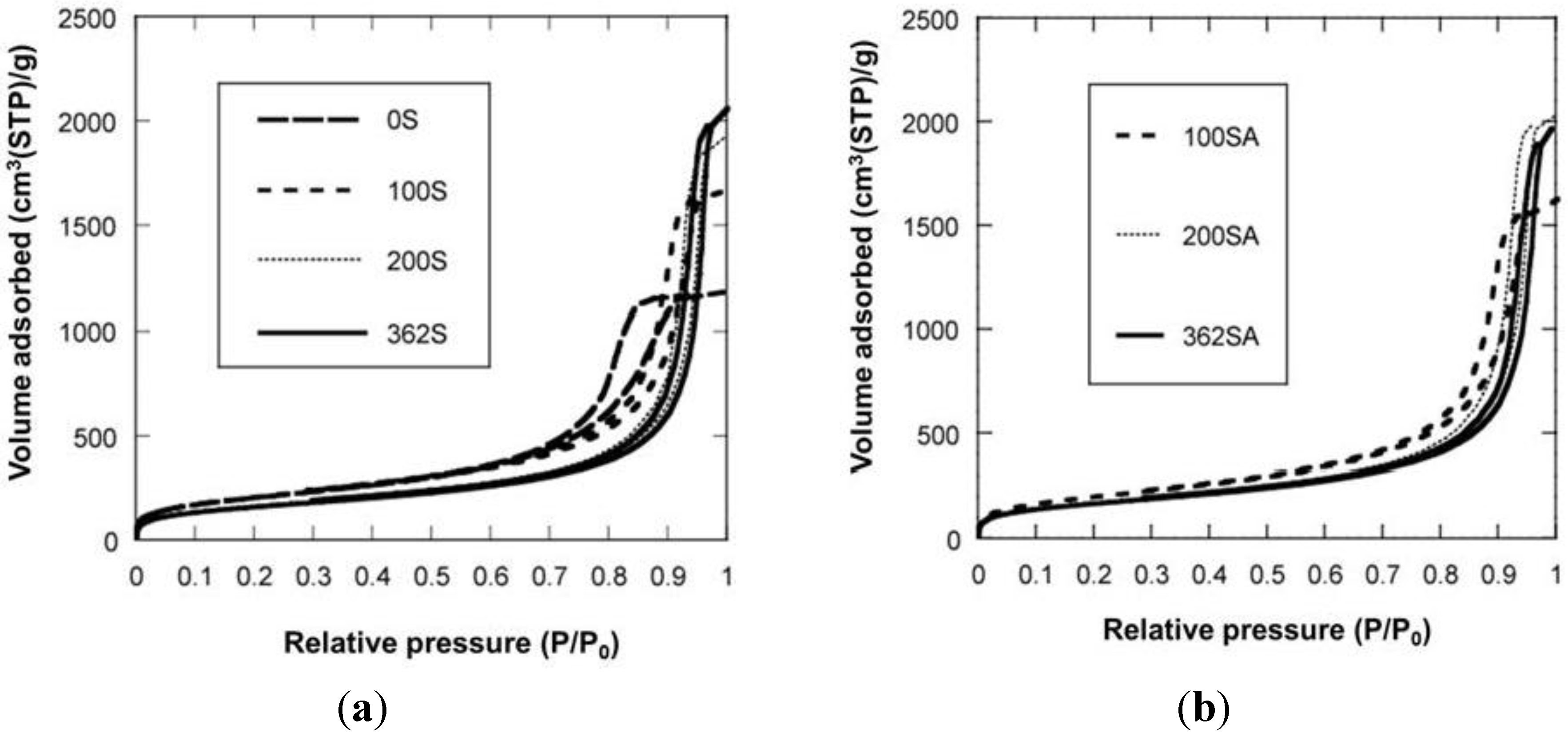
Figure 3 shows the N
2 adsorption-desorption isotherms. When the gel was not reinforced (0SiO
2), resultant SiO
2 showed the typical IV type isotherm, indicating that mesopore has been formed. When the gel was reinforced (100, 200 or 362SiO
2), a resultant SiO
2 showed an isotherm between IV type and III type. There was hysteresis in the isotherm, indicating that mesopore has also been formed, although the adsorption ability for an adsorbent may be lower than that of 0SiO
2. H When silica-gel was reinforced by TEOS solution, silica with large mesopores was prepared. For example, BJH surface area, pore volume and pore diameter (PD) for 100SiO
2 were 721 m
2/g, 2.51 cm
3/g and 18.5 nm, respectively, and were maintained after impregnation of Al species. Without the gel skeletal reinforcement, these three values were 703 m
2/g, 1.30 cm
3/g and 7.1 nm (PD), respectively. The porosity of the material can be increased considerably by removing water completely before aging in 2-propanol solvent. However, surface areas, pore volumes and pore diameters of silica and silica-alumina prepared with the gel skeletal reinforcement were considerably large compared with those for conventional silica and silica-alumina. For example, three values of ref.SA (JRC-SAH-1 supplied from Catalysis Society of Japan), which is prepared by a conventional method using water glass and aluminum sulfate, were 537 m
2/g, 0.86 cm
3/g, and 6.2 nm(PD), respectively.
Figure 2.
XRD patterns of zeolite, mixed catalysts and silica-aluminas.
Figure 2.
XRD patterns of zeolite, mixed catalysts and silica-aluminas.
Table 1.
Surface areas, pore volumes and pore diameters of silica, silica-alumina, zeolite and mixed catalysts. In naming samples, the weight ratio of 100, 200 or 362 of reinforcing TEOS against 100 of TEOS silica precursor was represented in front of SiO2. For silica-alumina samples, SA was used instead of SiO2. For the mixed catalyst with zeolite and the binder, MAT- was represented in front of sample name of SA.
Table 1.
Surface areas, pore volumes and pore diameters of silica, silica-alumina, zeolite and mixed catalysts. In naming samples, the weight ratio of 100, 200 or 362 of reinforcing TEOS against 100 of TEOS silica precursor was represented in front of SiO2. For silica-alumina samples, SA was used instead of SiO2. For the mixed catalyst with zeolite and the binder, MAT- was represented in front of sample name of SA.
| Catalyst | BET | | BJH |
|---|
| Surface area (m2/g) | Pore volume (cm3/g) | Pore diameter (nm) | Surface area (m2/g) | Pore volume (cm3/g) | Pore diameter (nm) |
|---|
| 0SiO2 | 837 | 1.43 | 6.8 | | 703 | 1.3 | 7.1 |
| 100SiO2 | 754 | 2.57 | 13.7 | | 721 | 2.51 | 18.5 |
| 200SiO2 | 598 | 2.94 | 19.7 | | 578 | 2.9 | 28.3 |
| 362SiO2 | 588 | 3.14 | 21.4 | | 555 | 3.09 | 32.6 |
| 100SA-5 | 737 | 2.49 | 13.5 | | 719 | 2.43 | 18.5 |
| 200SA-5 | 569 | 3.11 | 21.8 | | 634 | 3.08 | 28.3 |
| 362SA-5 | 621 | 3.04 | 19.6 | | 560 | 2.96 | 28.3 |
| MAT-100SA-5 | 567 | 1.5 | 10.6 | | 467 | 1.42 | 12.1 |
| MAT-200SA-5 | 530 | 1.6 | 12.1 | | 394 | 1.52 | 24.5 |
| MAT-362SA-5 | 512 | 1.8 | 14.1 | | 376 | 1.7 | 32.6 |
| ref.SA | 457 | 0.87 | 7.6 | | 537 | 0.86 | 6.2 |
| beta-Zeolite | 613 | 0.32 | 2.1 | | 24 | 0.07 | 3.3 |
| MAT-(ref.SA) | 765 | 0.64 | 5.7 | | 340 | 0.57 | 6.2 |
Figure 3.
N2 adsorption-desorption isotherm. (a) Silica; (b) Silica-alumina.
Figure 3.
N2 adsorption-desorption isotherm. (a) Silica; (b) Silica-alumina.
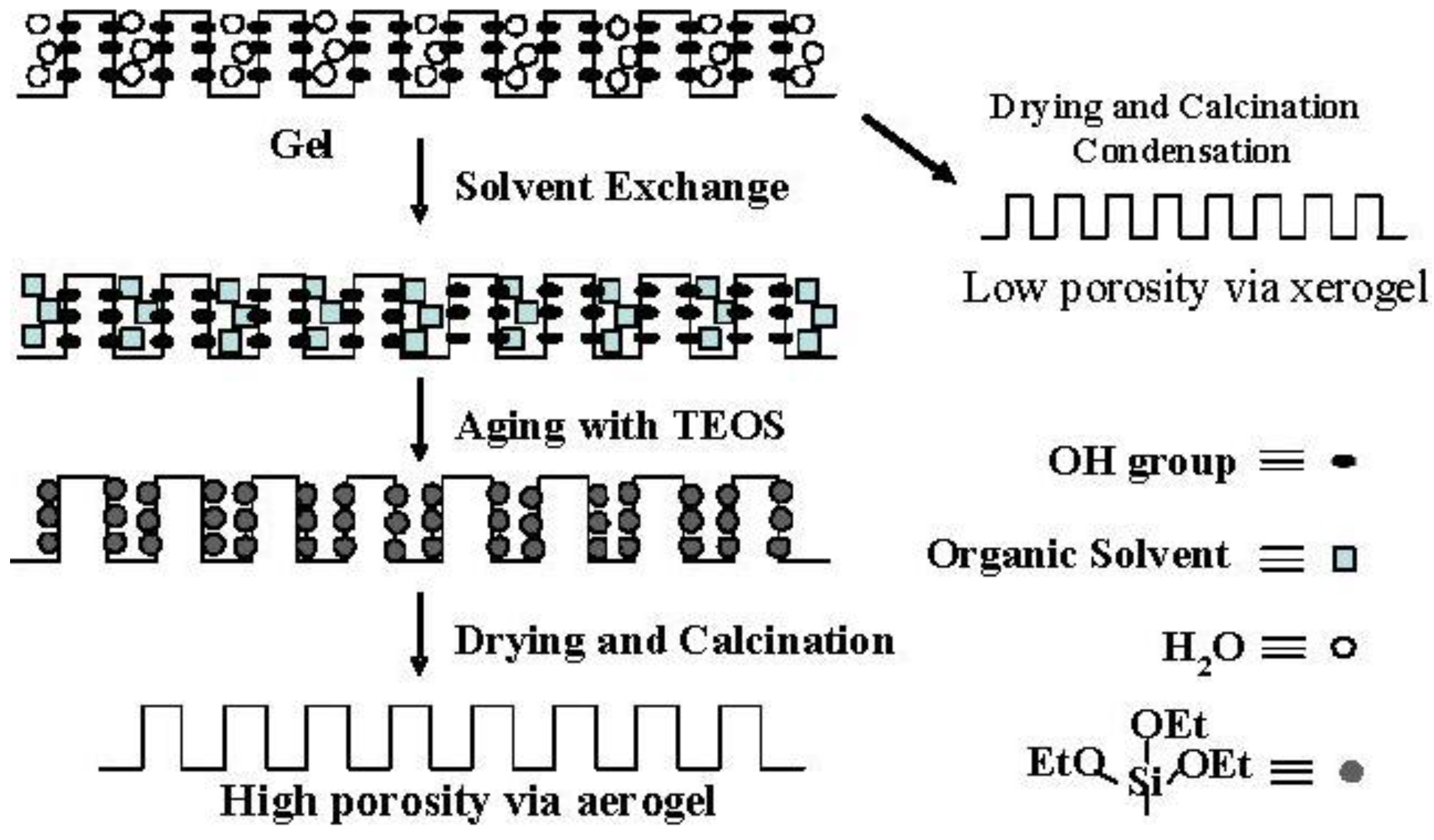
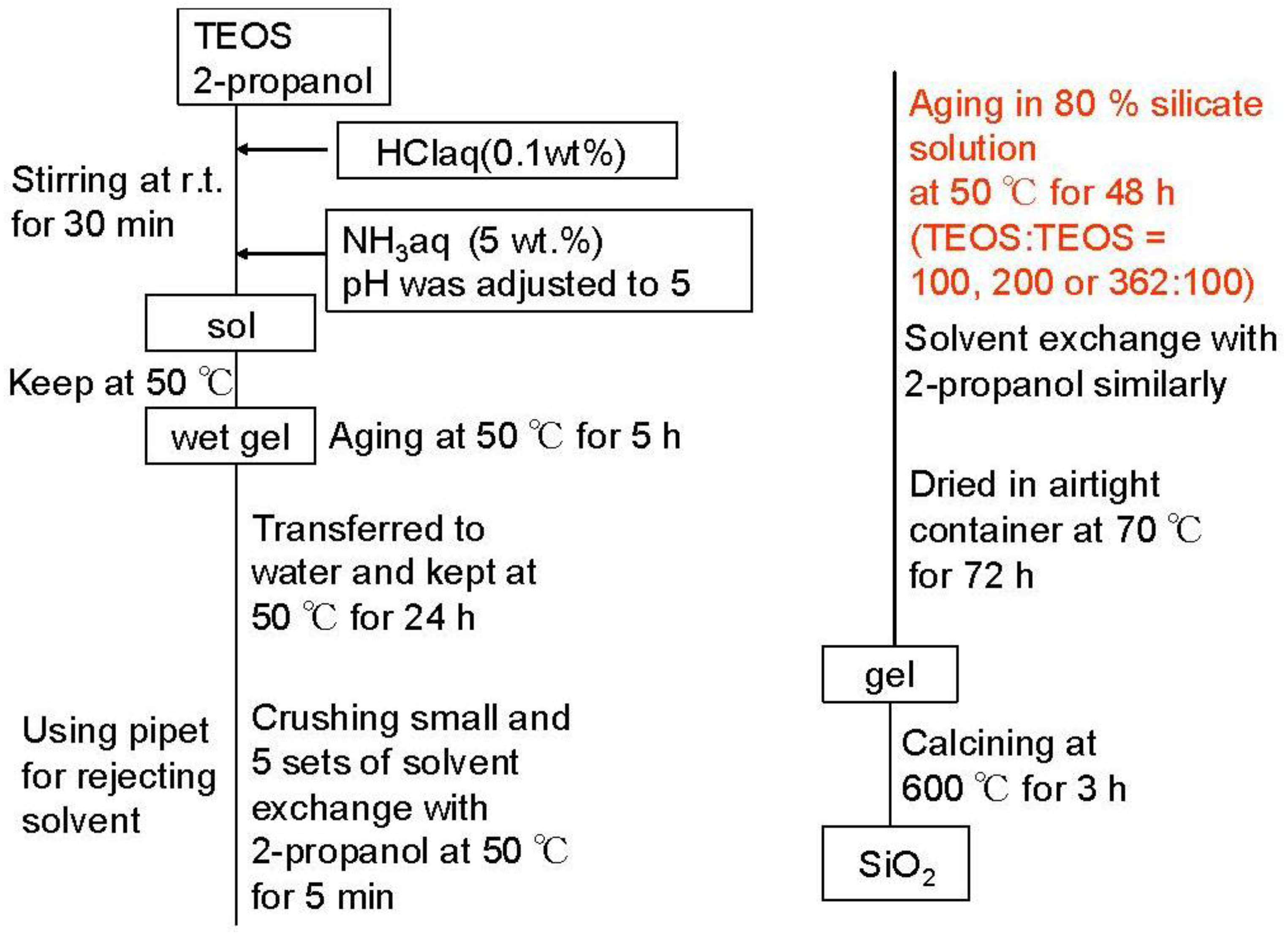
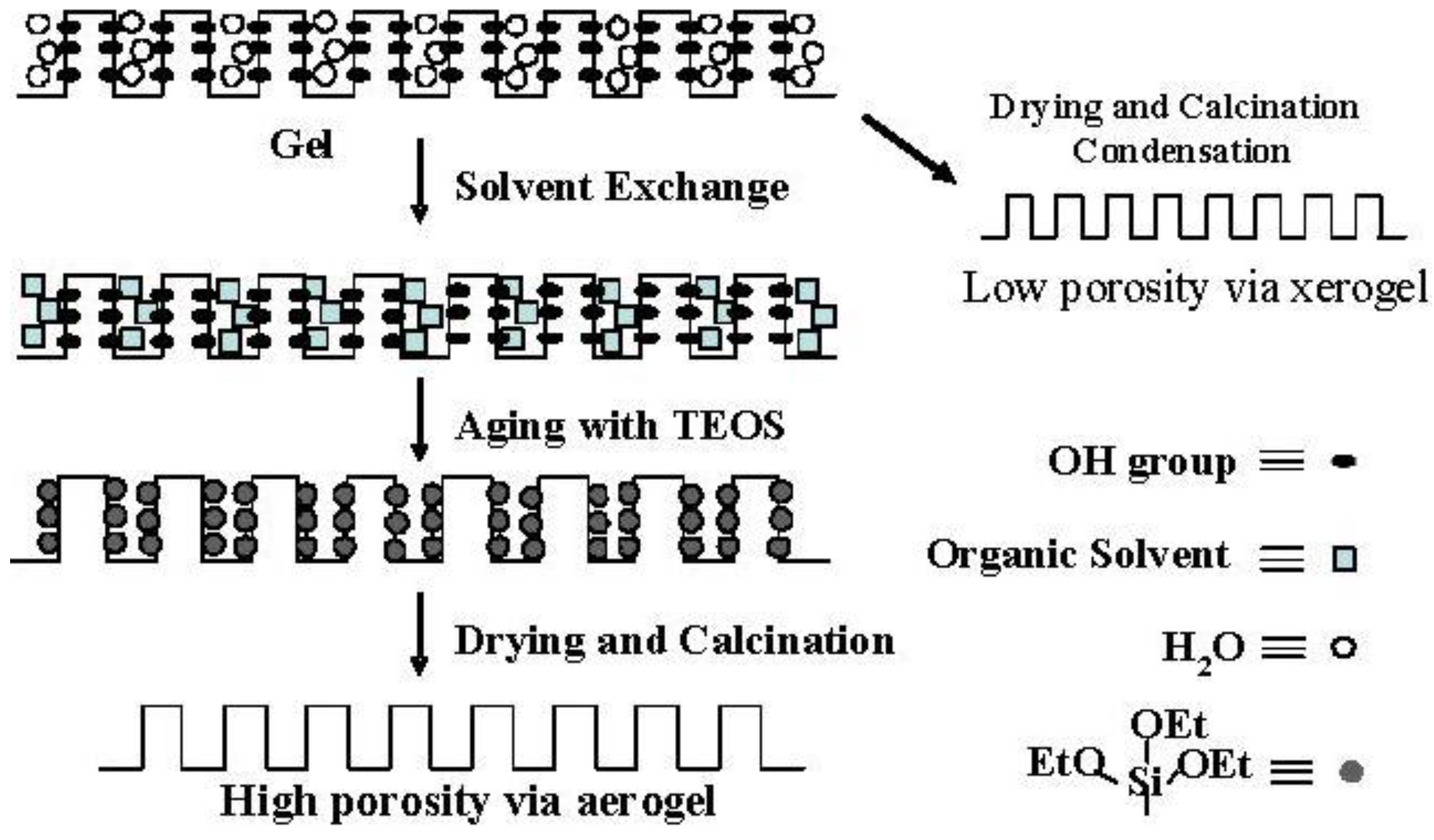
Figure 4.
Conceptual scheme of the gel skeletal reinforcement.
Figure 4.
Conceptual scheme of the gel skeletal reinforcement.
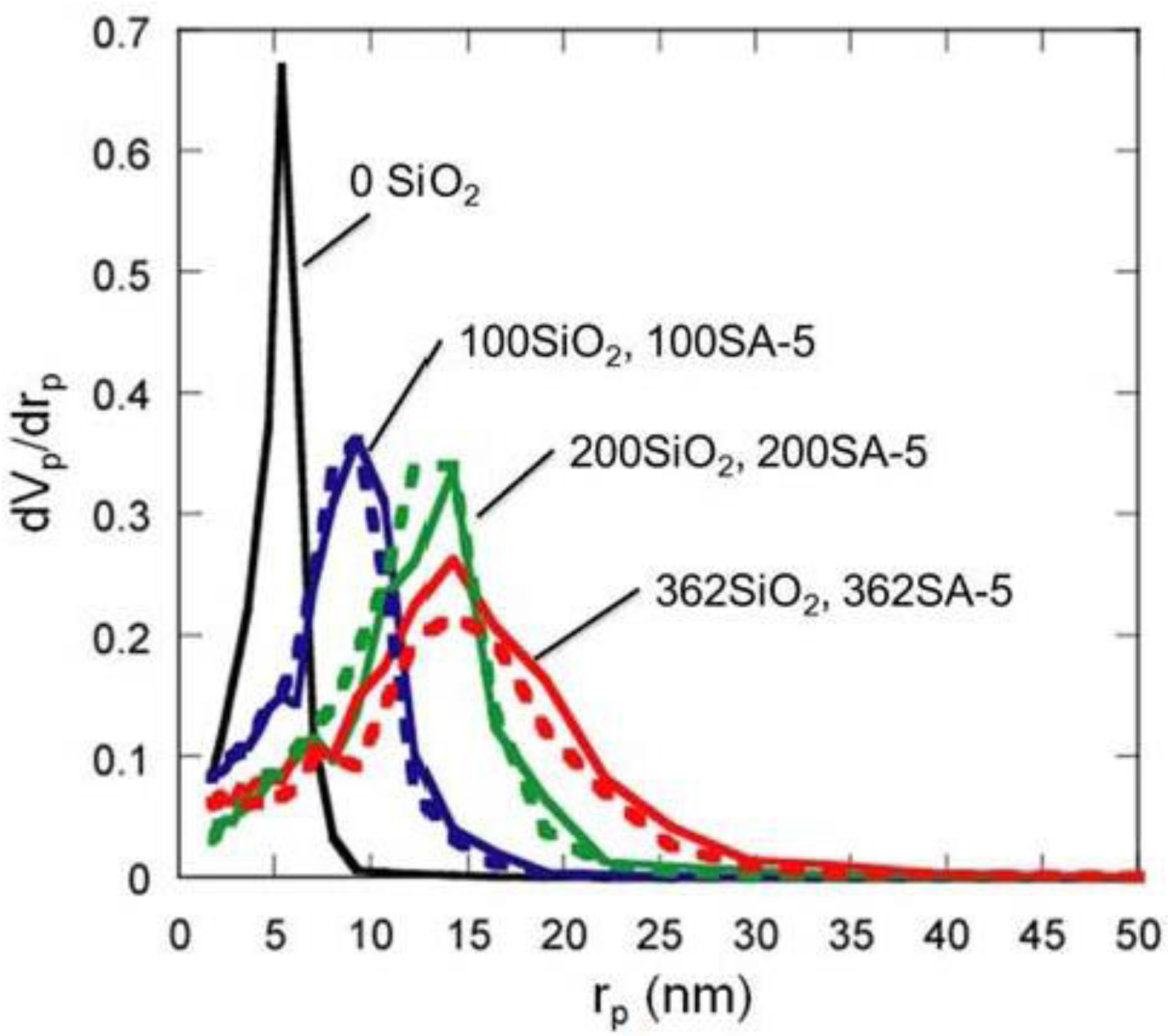
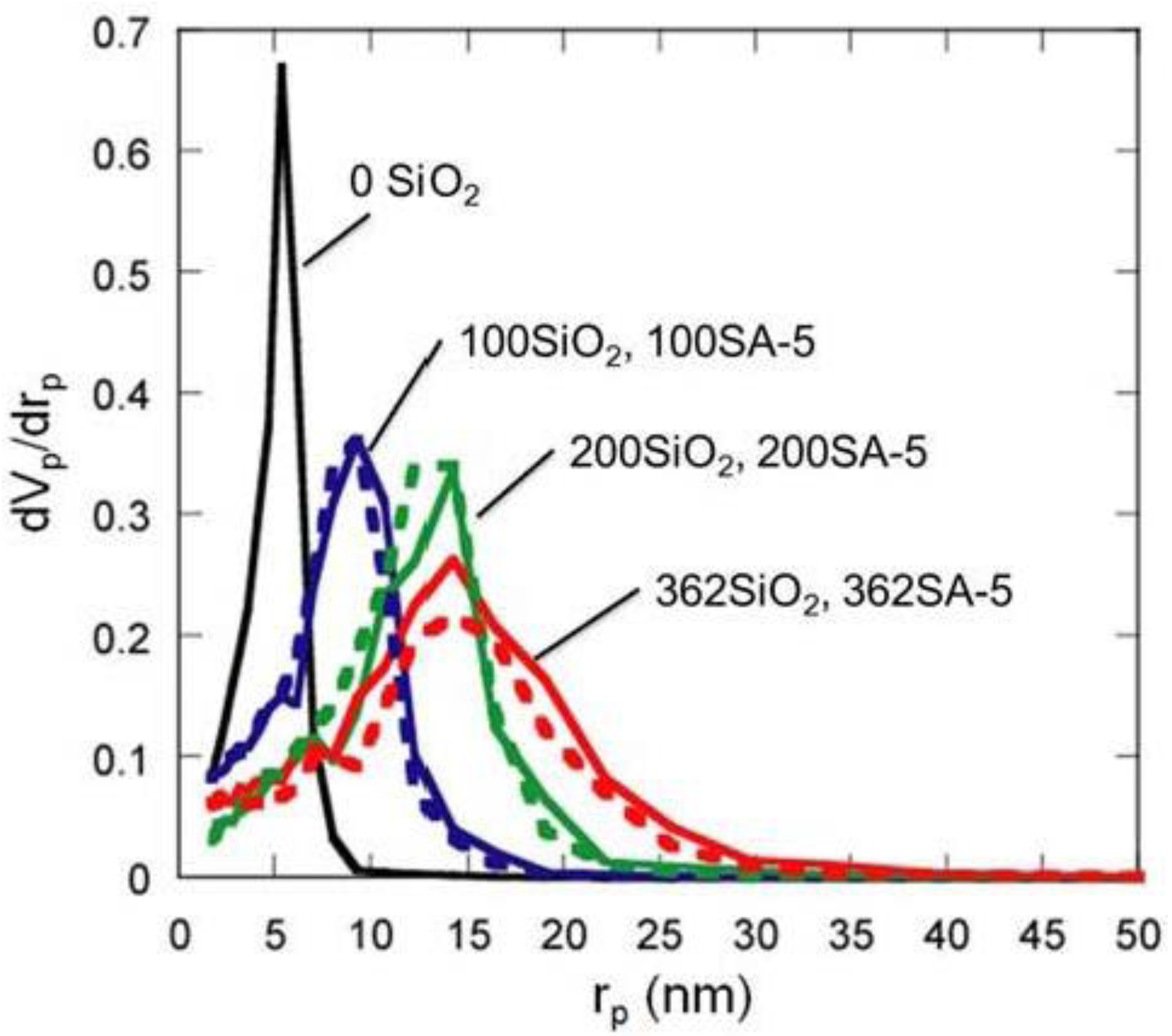
Figure 5.
BJH pore-size distribution of silica and silica-alumina. Solid line: silica; Dotted line: silica-alumina; rp: pore radius.
Figure 5.
BJH pore-size distribution of silica and silica-alumina. Solid line: silica; Dotted line: silica-alumina; rp: pore radius.
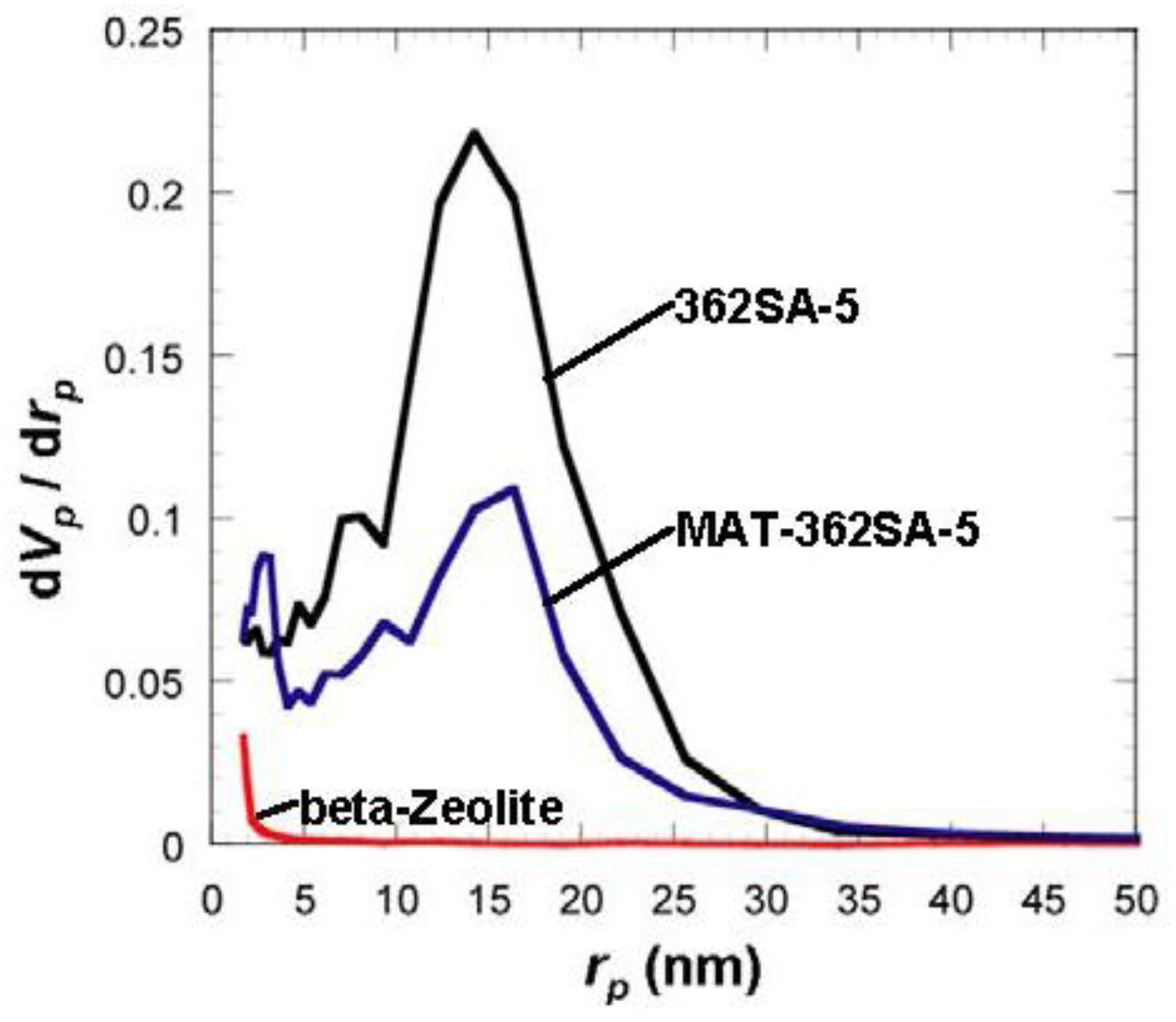
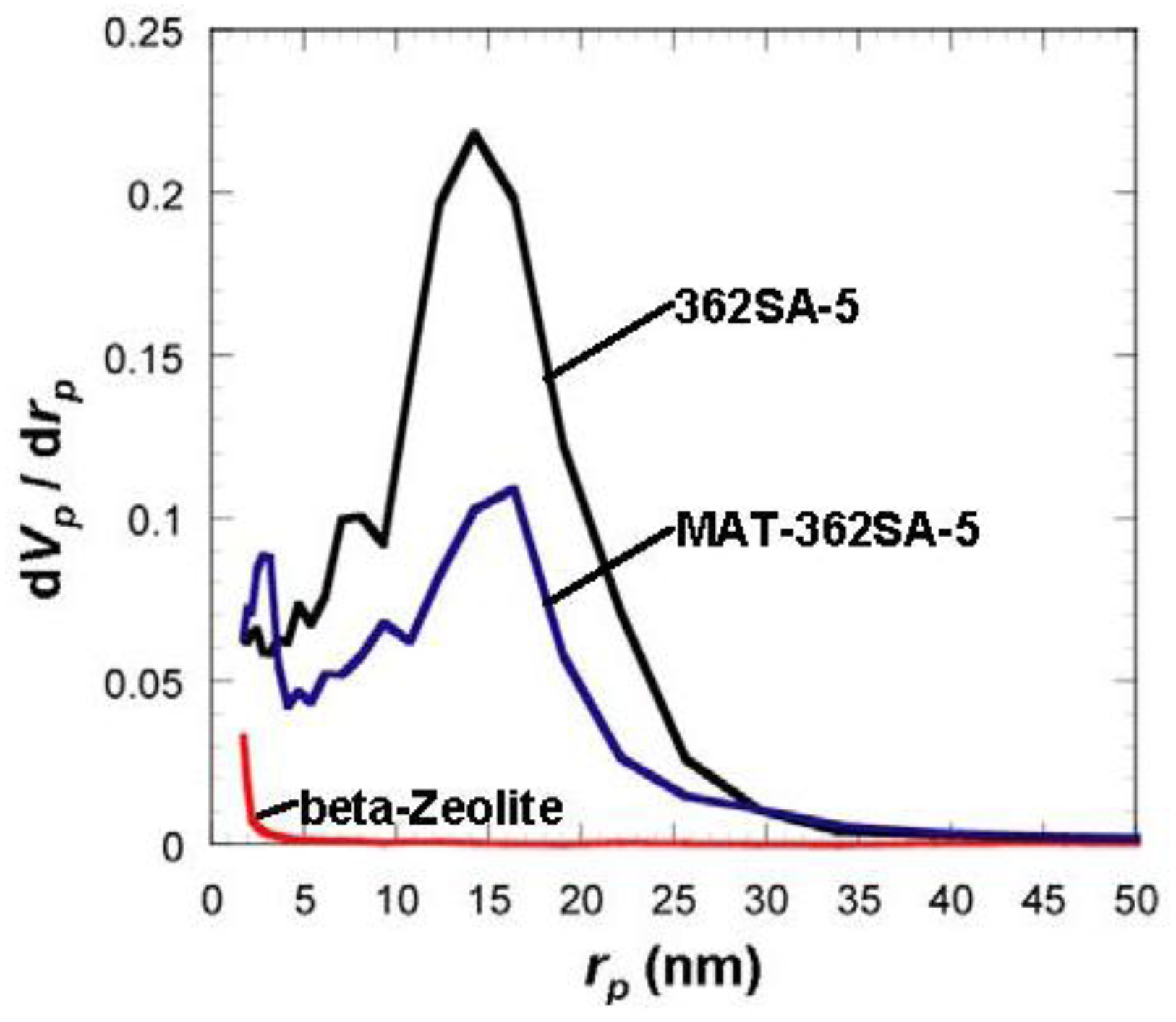
Figure 6.
Comparison of BJH pore-size distribution for silica-alumina, mixed catalyst and zeolite. rp: pore radius.
Figure 6.
Comparison of BJH pore-size distribution for silica-alumina, mixed catalyst and zeolite. rp: pore radius.
Although the Ostwald ripening effect to strengthen a gel surface is often described [
12,
13,
14,
15,
16,
17,
18,
19], our results suggest that the thorough removal of water in the inside of the gel and the gel skeletal reinforcement are essential for making large mesopores.
Figure 4 shows the conceptual scheme of the gel skeletal reinforcement. The thorough removal of water in the inside of the gel in solvent exchange inhibits the hydrolysis of reinforcing TEOS/2-propanol solution. The surface silanol is modified by TEOS to form the new surface covered by triethoxysilyl groups which inhibit the further condensation of the gel skeletal structure in the absence of water. Surface areas and pore volumes by the BET method were almost the same as those by the BJH method, indicating that most pores of silicas and silica-aluminas prepared by the gel skeletal reinforcement consist of mesopores.
Figure 5 shows the BJH pore size distribution of silica and silica-alumina. Silica and silica-alumina had larger and broader pore size distribution with increasing amounts of TEOS solution for reinforcement. When 362 wt% TEOS was used for reinforcement, the pore volume and pore diameter measured by the BJH method reached about 3 cm
3/g and 30 nm, respectively. To our knowledge, such high values for materials calcined at 600 °C for 3 h have never been obtained with any other methods. Although some papers reported higher porosity of materials [
6,
7,
8], thermal histories for the materials were about 200 °C even at maximum, which is much lower than our condition: 600 °C for 3 h. When silica-aluminas were mixed with zeolite and alumina sol (binder), surface areas and pore volumes of mesopores measured by the BJH method decreased; however almost the same pore diameters and pore distributions were maintained, as shown in
Figure 6.
3.2. Catalytic Cracking Performance of the Mixed Catalysts with Zeolite
The catalytic cracking was performed and the results are shown in
Table 2.
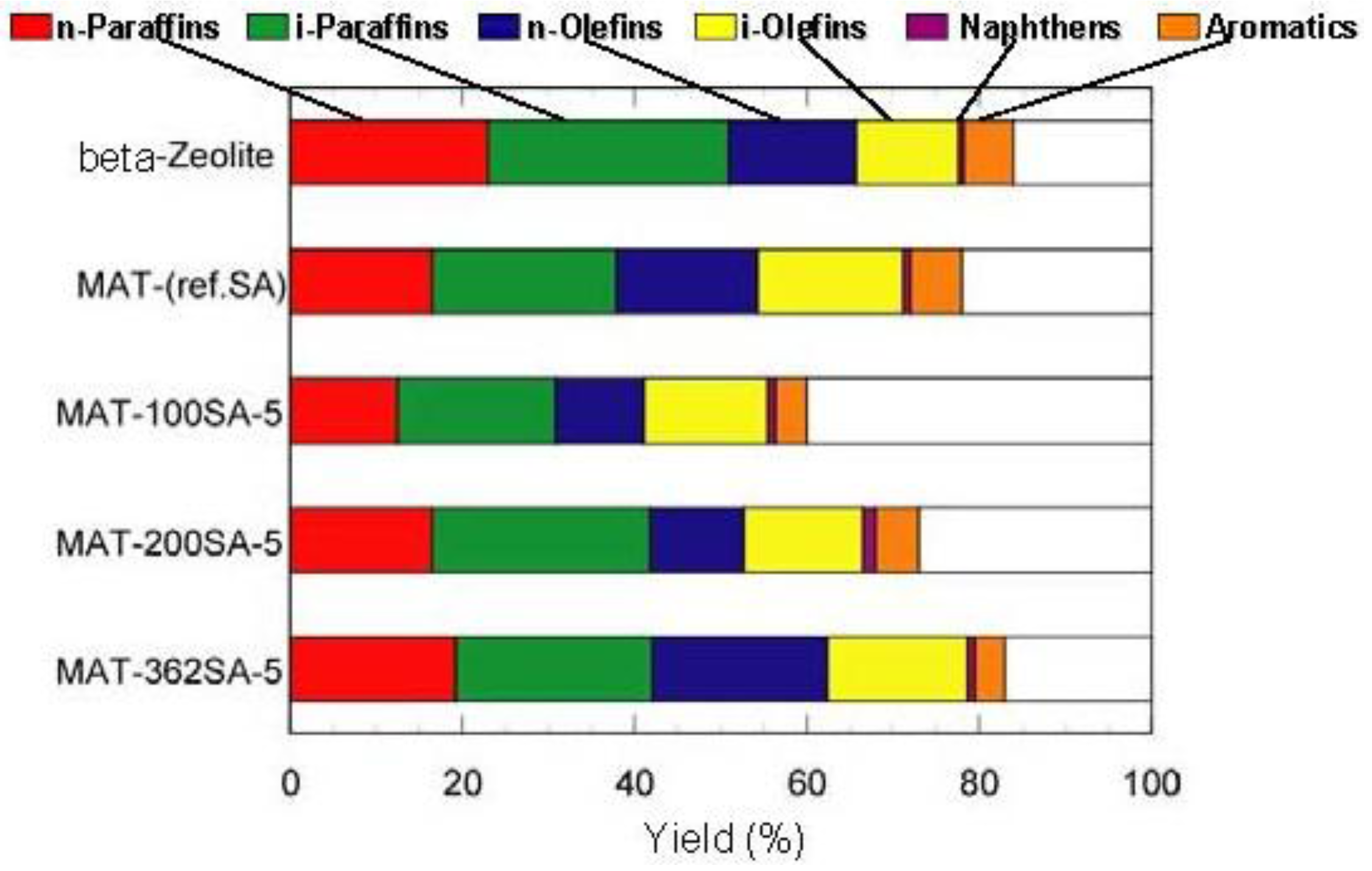
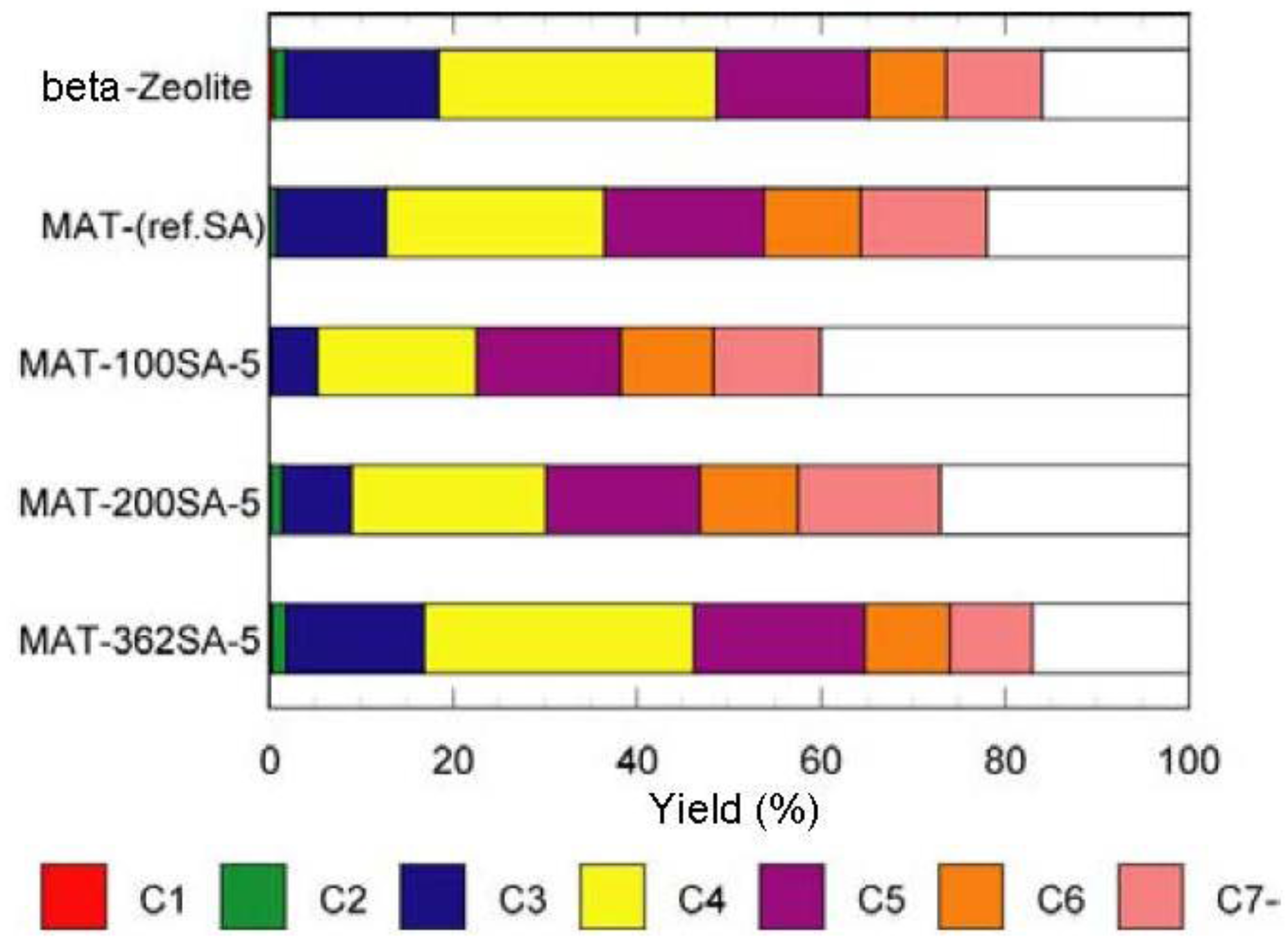
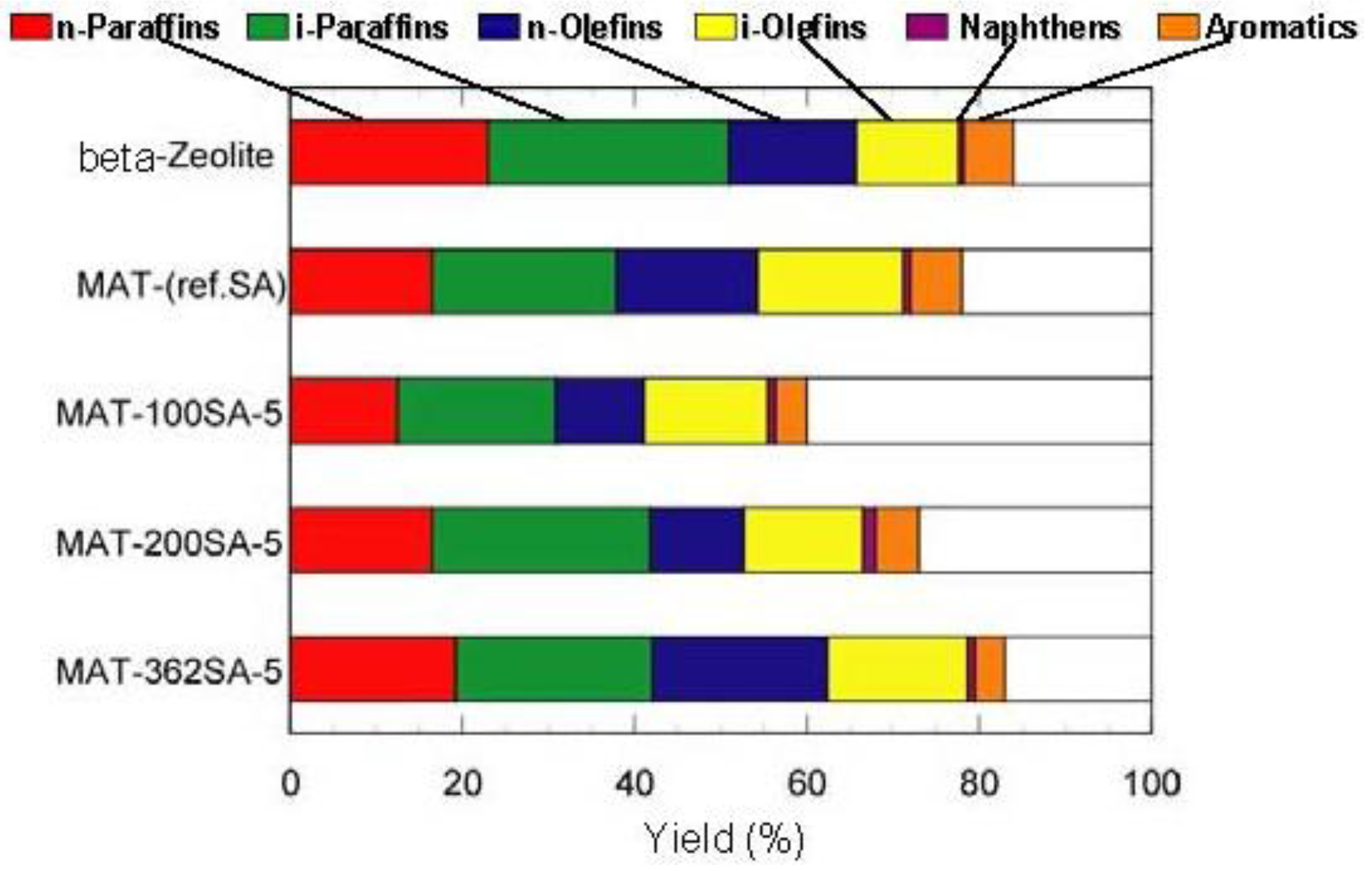
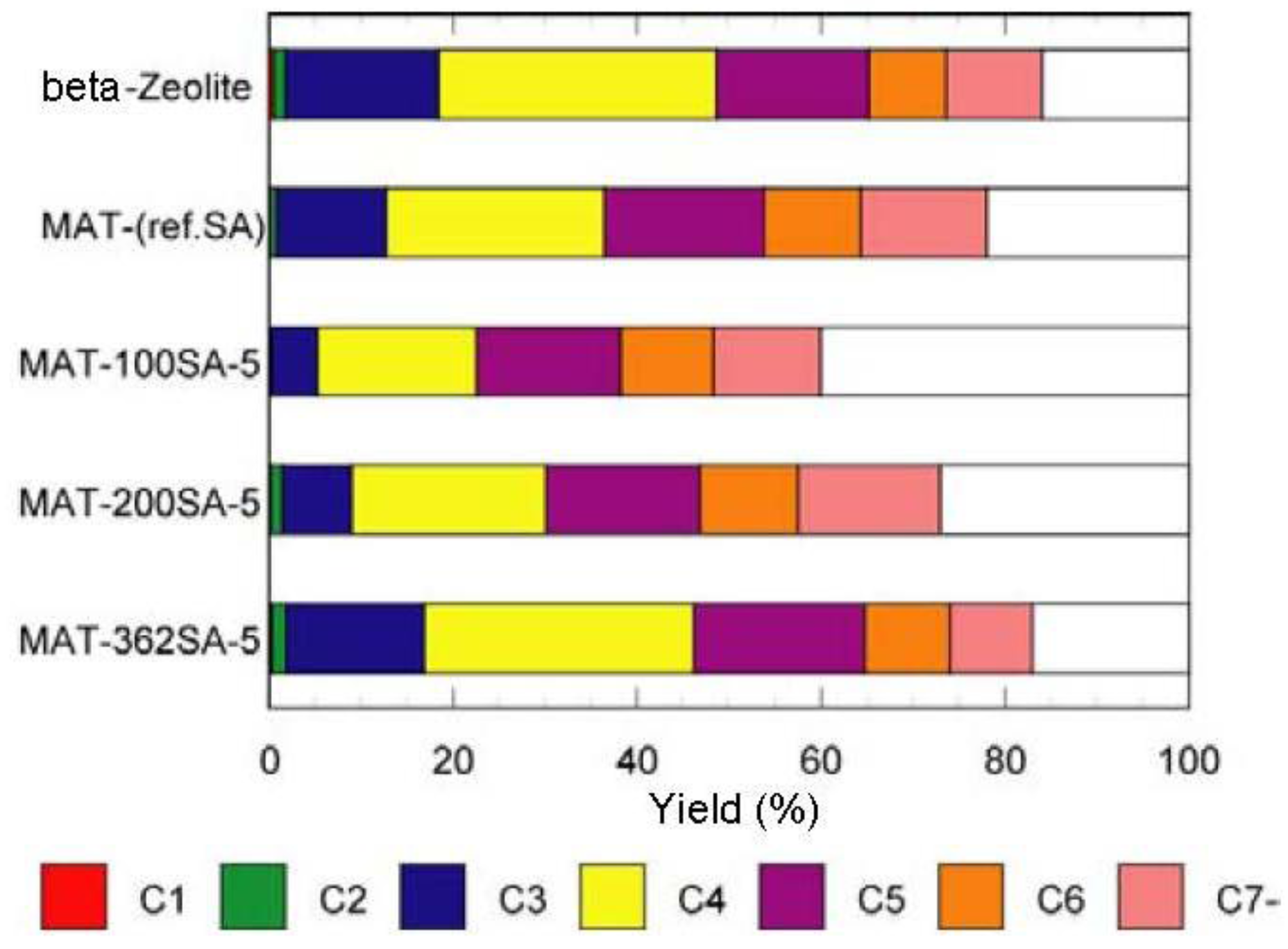
Figure 7 and
Figure 8 show the yields for paraffins, olefins, naphthenes and aromatics on catalytic cracking, and the distribution of carbon numbers on catalytic cracking, respectively. The conversion of
n-dodecane with single zeolite was 84%, and the yield of paraffins was much larger than those of olefins and aromatics as shown in
Figure 7. This shows that significant hydrogen transfer to olefins in the formation of aromatics proceeded. When silica-alumina was used in cracking of
n-dodecane, the conversion was only a few percent and the selectivity for olefin was rather high for each catalyst (see supplemental materials). The results show that the ability of cracking and hydrogen transfer in the catalytically active sites is limited to an extremely low level for single silica-alumina. When mixed catalysts were prepared with zeolite and silica-alumina with large mesopore, the comparable activities were obtained in cracking of
n-dodecane, although the mixed catalyst included only 26 wt% of zeolite. The conversion of dodecane for the mixed catalyst, MAT-362SA-5, was 83%, which was almost the same as that for single zeolite, indicating that the presence of larger mesopores in the mixed catalyst promoted the diffusion of reactant molecules effectively. As shown in
Figure 7, the difference between the product distributions of the mixed catalysts and the single zeolite was not so large although the yield of paraffins for the mixed catalysts was slightly less than that of the single zeolite. To clarify the difference between the mixed catalysts and the single zeolite or the mixed catalyst using a conventional silica-alumina (refSA), the product distribution of gas (C1-C4) and gasoline (C5-C11) fractions and some parameters in the gasoline fraction were compared and the results are summarized in
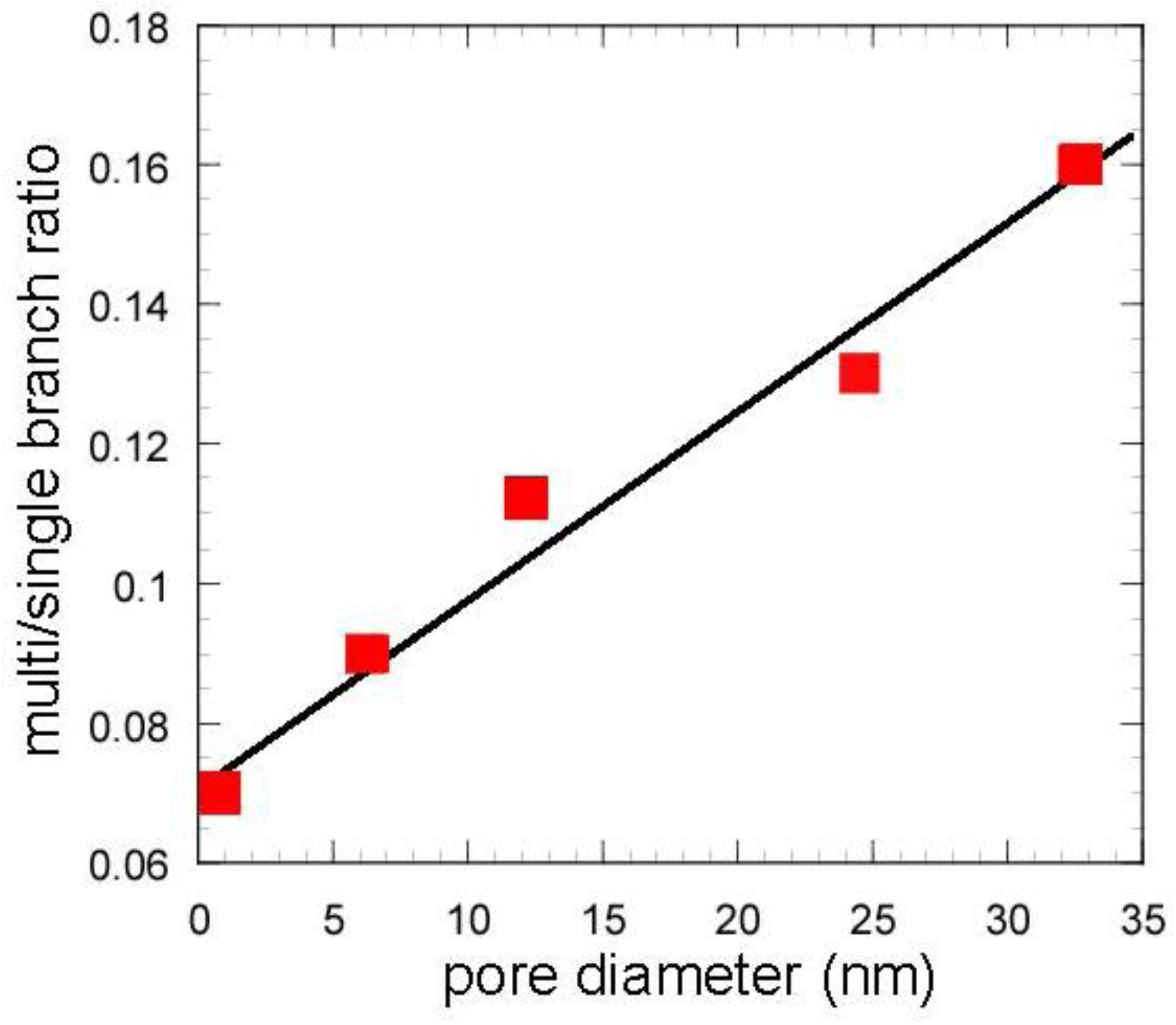
Table 2. In the mixed catalysts using the gel skeletal reinforcement, MAT-100SA-5 and MAT-200SA-5, the distribution of gasoline increased although the conversions slightly decreased. The product distribution of the mixed catalyst, MAT-362SA-5, was very similar to that of single zeolite, probably because the conversion was similar to that of a single zeolite. However, the major differences were observed in the yields of multi-branched products and single-branched products (
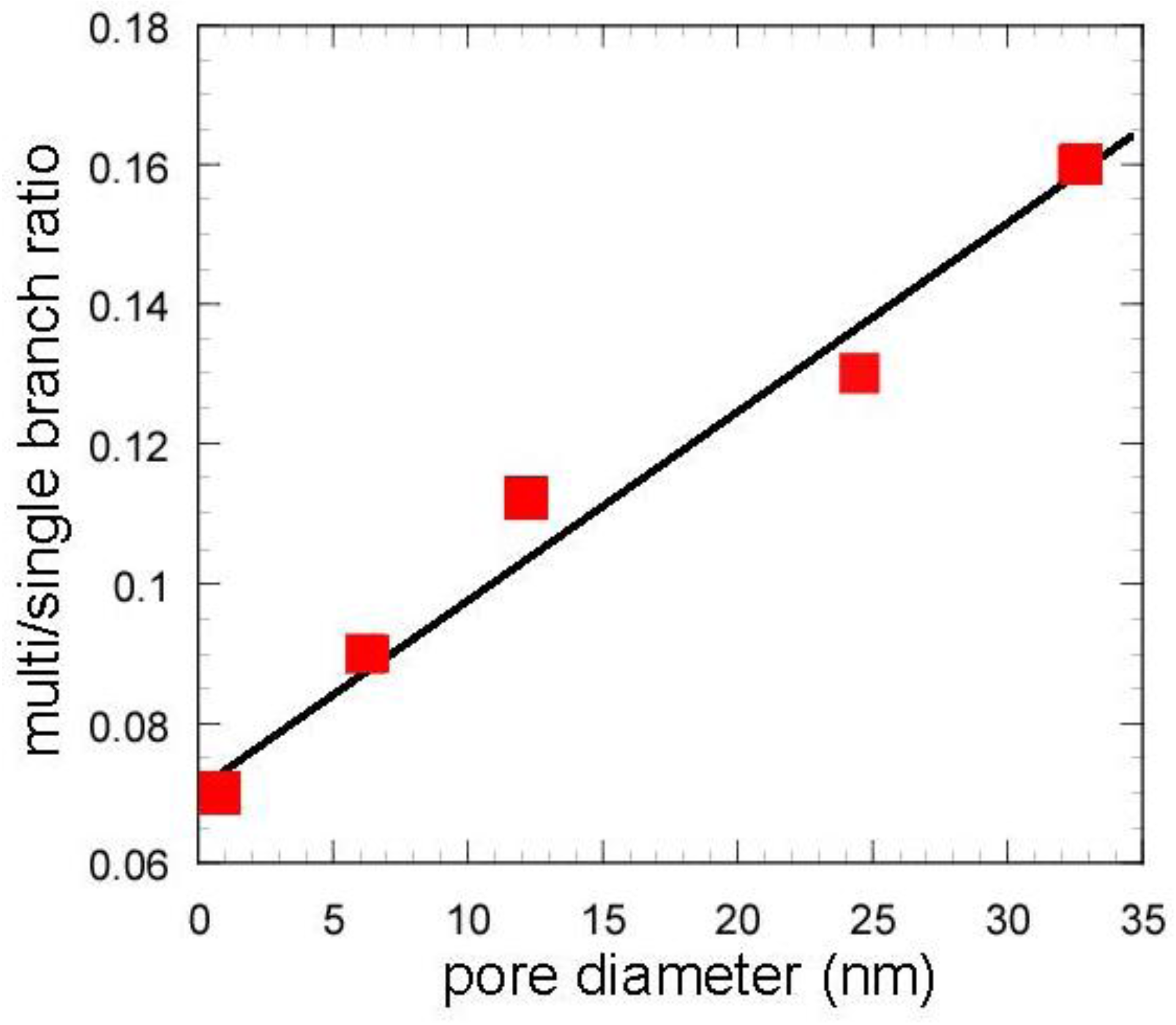
Table 2) and the ratio of those yields (m/s) (
Figure 9) for the mixed catalysts, which are important factors to increase the research octane number (RON) [
1]. In particular, the m/s ratios of the mixed catalysts using the gel skeletal reinforcement were much larger than those for single zeolite and the mixed catalyst with conventional silica-alumina (
Figure 9). The approximate linear relationship can be found between the m/s ratios and the values of pore diameters of these catalysts in
Figure 9. These results suggest that the presence of larger mesopores in the matrix of the cracking catalysts would promote not only the diffusion of reactant molecules to approach acid sites of zeolite and to produce bulky branched products, but also the effective elimination of the branched products to prevent the excess cracking of those products.
Table 2.
Product distribution and catalytic properties of catalyst. Temperature 500 °C, Catalyst 1 g, n-dodecane, 1.3 mL/min, 80 s, WHSV 58.5 h−1.
Table 2.
Product distribution and catalytic properties of catalyst. Temperature 500 °C, Catalyst 1 g, n-dodecane, 1.3 mL/min, 80 s, WHSV 58.5 h−1.
| Catalyst | Product distribution (%) | | Parameters in gasoline fraction |
|---|
| C1-C4 | Gasoline (C5-C11) | C12- | Conv. (%) | NH3b (10−4 mol/g) | Olefin/Paraffin | iso-/n- | Yield of single branch (%) | Yield of multi branch (%) |
|---|
| beta-Zeolite | 58 | 42 | 0.5 | 84(90) | 6.2(8.9) | 0.46 | 2.2 | 19 | 1.2 |
| MAT (ref.SA) | 47 | 53 | 0.6 | 78(81) | 5.8(8.4) | 0.76 | 1.9 | 21 | 1.8 |
| MAT-100SA-5 | 38 | 62 | 0.2 | 60(62) | 5.1(4.9) | 0.67 | 2 | 21 | 2.4 |
| MAT-200SA-5 | 41 | 58 | 0.5 | 73(75) | 5.4(4.9) | 0.55 | 2.4 | 23 | 3 |
| MAT-362SA-5 | 56 | 44 | 0.1 | 83(85) | 4.1(4.7) | 0.58 | 2 | 19 | 3 |
Figure 7.
Yields for Paraffins, Olefins, Naphthenes and Aromatics on catalytic cracking. Temperature 500 °C, Catalyst 1 g, n-dodecane 1.3 mL/min, 80 s, WHSV 58.5 h−1.
Figure 7.
Yields for Paraffins, Olefins, Naphthenes and Aromatics on catalytic cracking. Temperature 500 °C, Catalyst 1 g, n-dodecane 1.3 mL/min, 80 s, WHSV 58.5 h−1.
Figure 8.
Distribution of carbon numbers on catalytic cracking. Temperature 500 °C, Catalyst 1 g, n-dodecane 1.3 mL/min, 80 s, WHSV 58.5 h−1.
Figure 8.
Distribution of carbon numbers on catalytic cracking. Temperature 500 °C, Catalyst 1 g, n-dodecane 1.3 mL/min, 80 s, WHSV 58.5 h−1.
Figure 9.
Effect of pore diameter on multi/single branch ratio. Except zeolite, values in the horizontal line represent peaks of BJH pore diameter.
Figure 9.
Effect of pore diameter on multi/single branch ratio. Except zeolite, values in the horizontal line represent peaks of BJH pore diameter.
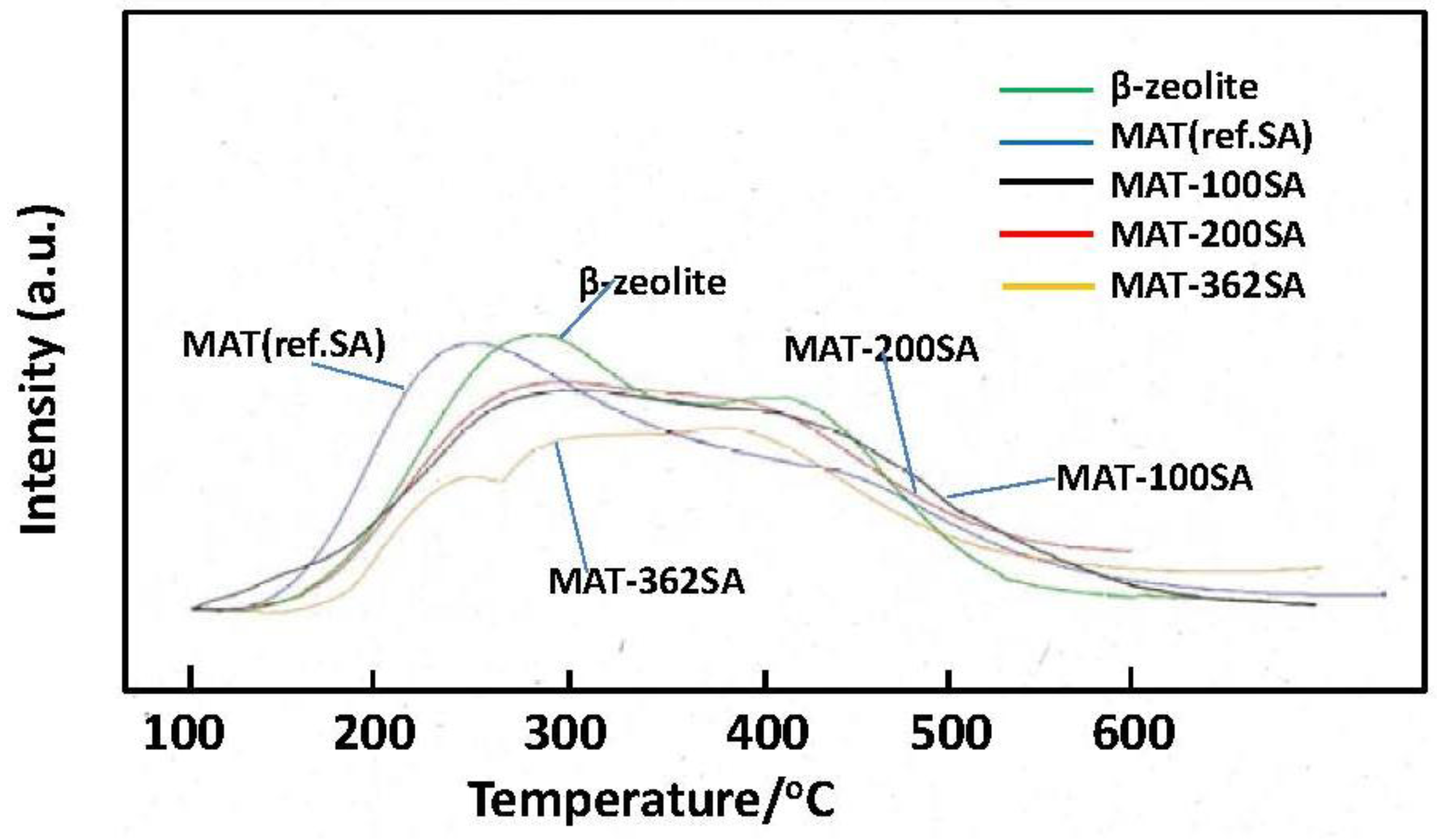
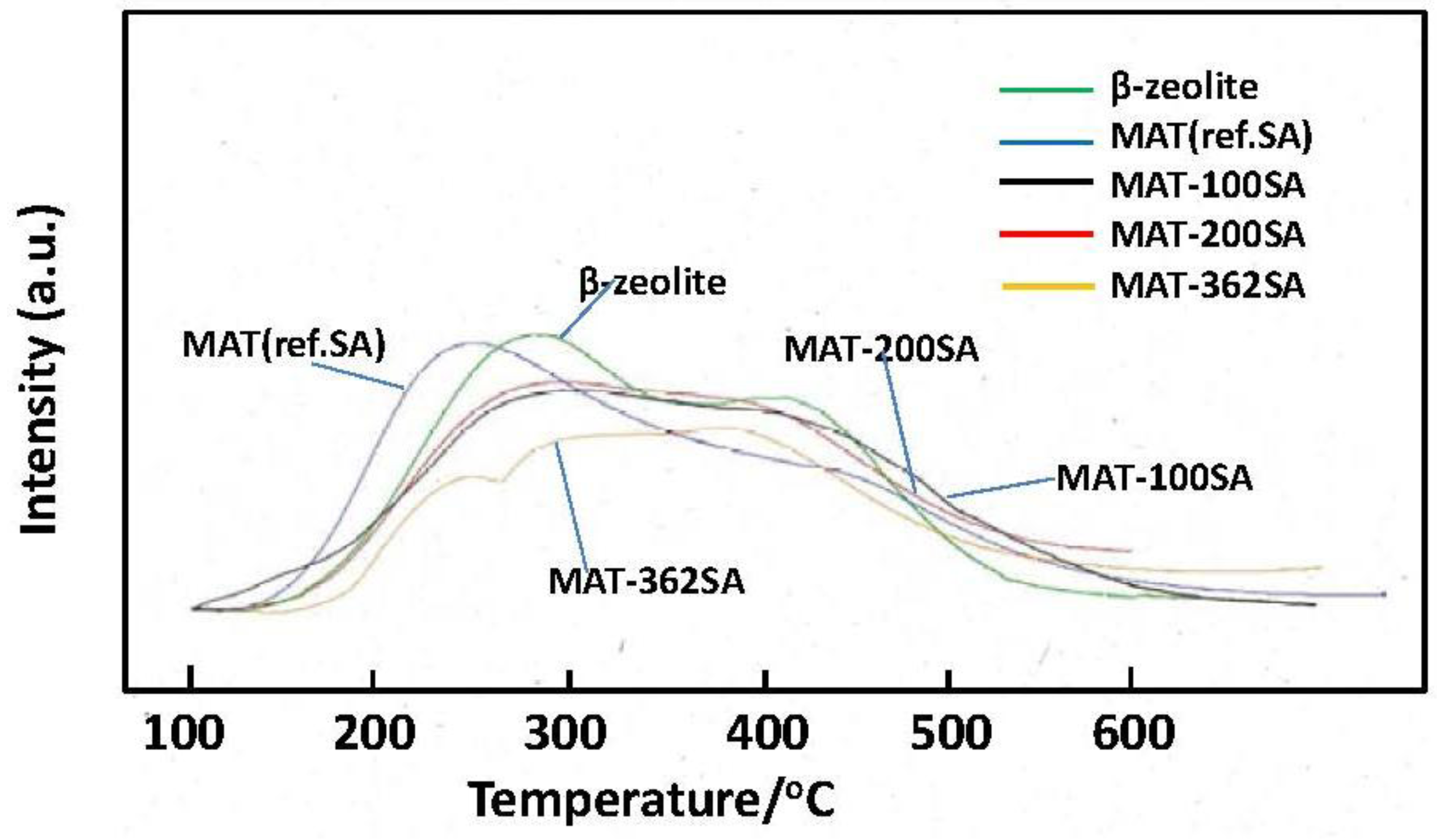
Figure 10 shows the NH
3-TPD profiles of zeolite and the mixed catalysts. Two peaks are usually observed and a peak observed at the higher temperature (about 400 °C) is assigned to NH
3 chemisorbed at acid sites [
22]. It seems that the height of this peak of MAT-362SA at the higher temperature was higher than those of other peaks of this catalyst. In contrast, the peaks of other catalysts observed at the higher temperature were lower than those at the lower temperature. The amounts of NH
3 desorbed from the mixed catalysts were slightly lower than that for zeolite as shown in
Figure 10 and
Table 2. Further, the tailing in a TPD profile was observed above 500 °C for the mixed catalyst. Although the relationship with the activity and selectivity is not clear at the present time, these profiles may be related to catalytic performance.
Figure 10.
NH3-TPD profiles of beta zeolite and mixed catalysts.
Figure 10.
NH3-TPD profiles of beta zeolite and mixed catalysts.
Figure 11 shows TEM images of silica, mixed catalyst and β-zeolite. Silica with large pore (362SiO
2) did not show a diffractive pattern, while the mixed catalyst showed a slightly obscure diffractive pattern and zeolite clear one, indicating that the crystal structure of zeolite is maintained in the mixed catalyst. Further, the TEM images of silica and mixed catalyst show the round shape of each particle and it can be considered that the space between these particles represents mesopores, while the TEM image of zeolite did not show a round shape, indicating that zeolite is well mixed and dispersed with silica-alumina.
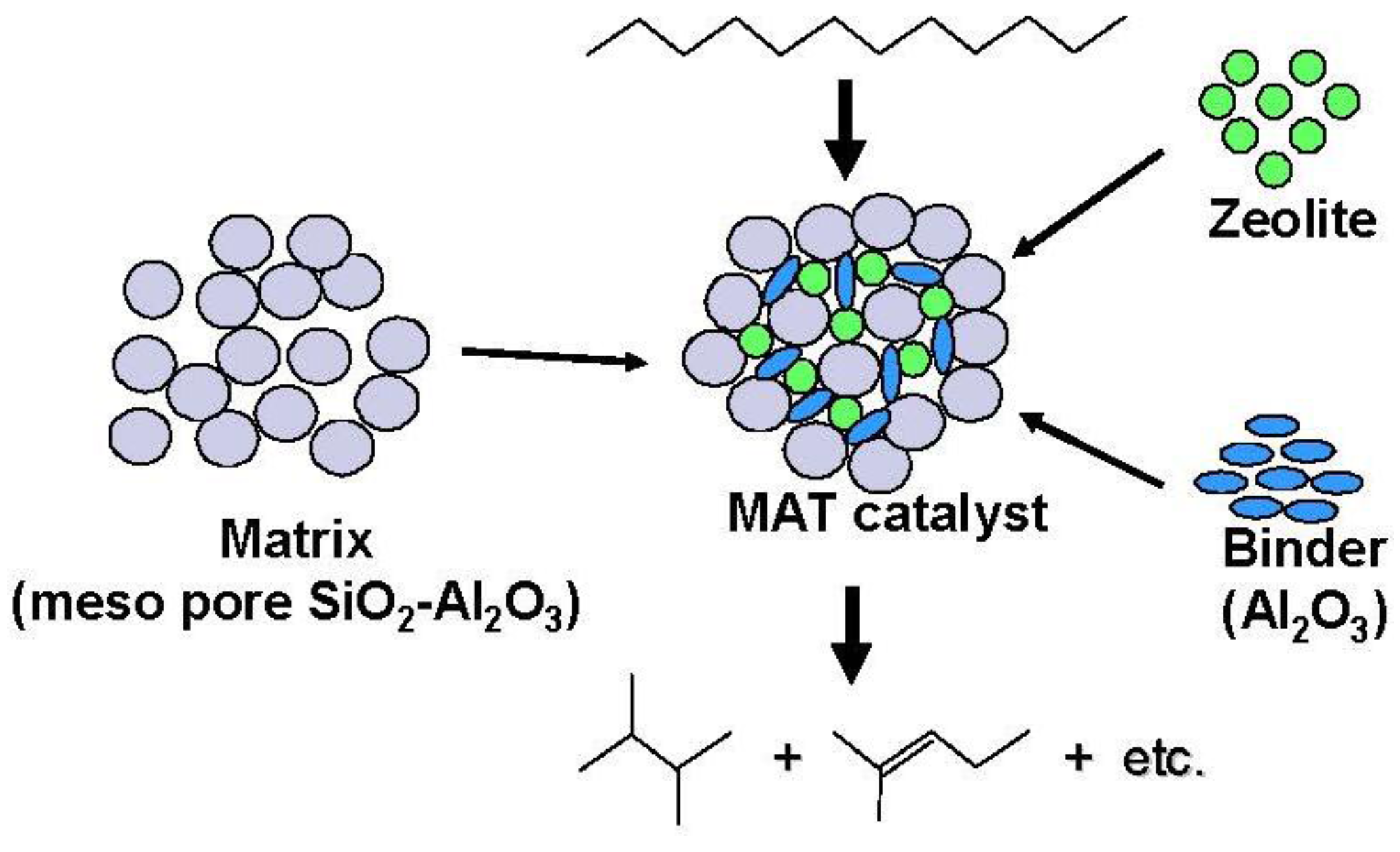
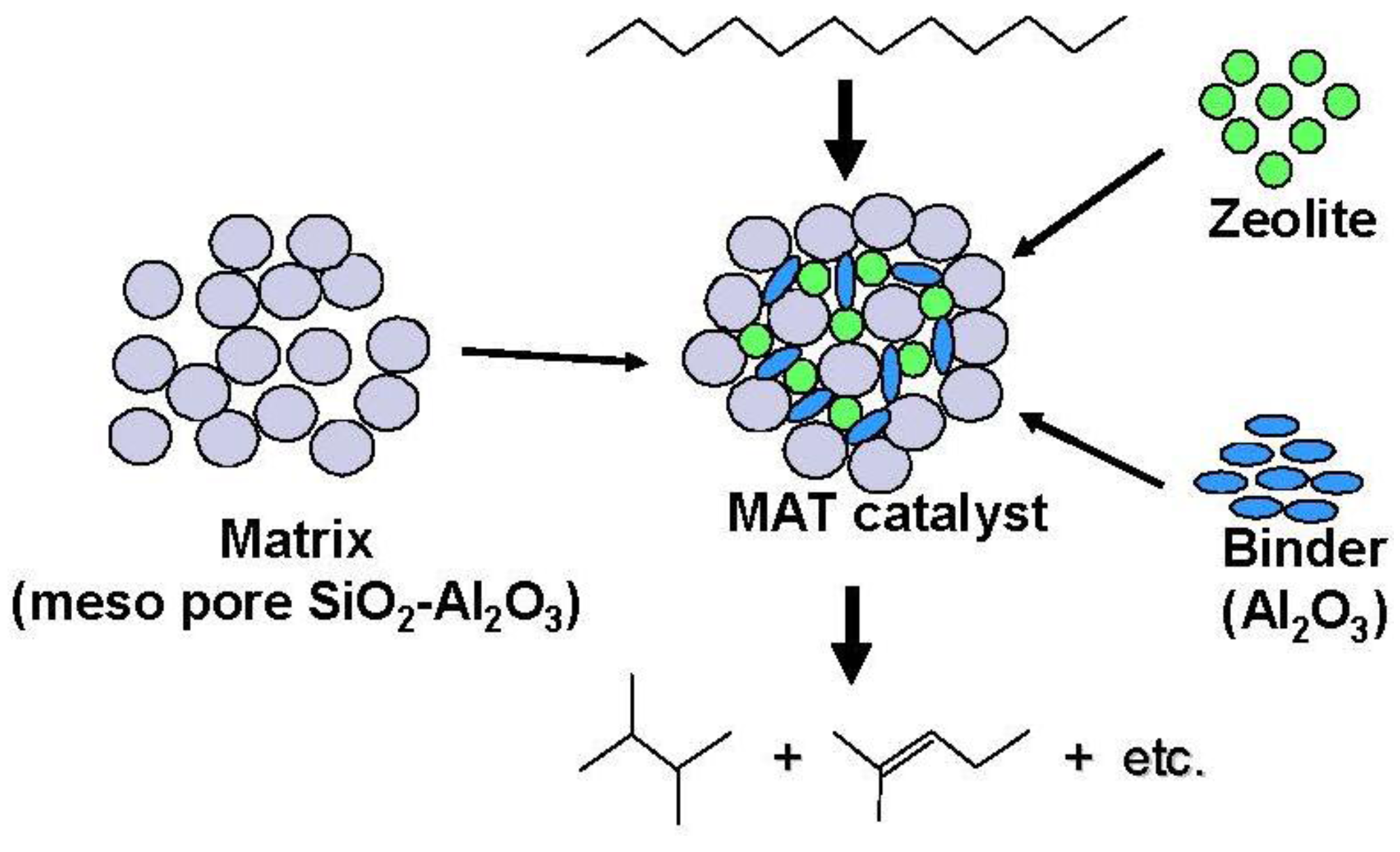
A proposed model structure of a catalytic cracking catalyst is illustrated in
Figure 12 [
1,
2]. A smaller amount of zeolite is embedded into the larger amount of matrix and acid sites on the matrix are effectively considered to crack saturated hydrocarbons into reactive olefins. The olefins formed were small, but may be adsorbed on acid sites of zeolite to effectively inhibit the rapid reaction of other reactive molecules, such as aromatics and the formation of coke by polymerization. If the amount of zeolite is large or zeolite is located in the upper bed, excess amount of olefin will be formed, which may lead to the rapid deactivation of the catalyst system. It has been reported that if the appropriate amount of reactive molecules such as olefins is present in a feed, the cracking of saturated hydrocarbons is promoted [
3]. Therefore, primary cracking of
n-dodecane on silica-alumina is likely to promote secondary cracking on zeolite and inhibit coke formation, even if the activity of silica-alumina is quite low. The distance for transfer of hydrocarbons inside the catalyst may be longer if the pore size of the matrix is larger. Further, the presence of larger spaces in the larger pores facilitates isomerization. Acid sites with the appropriate amount and strength on the matrix could also promote isomerization, as well as inhibit overcracking of olefins and branched paraffins. In our previous report, it was suggested that the primary reaction occurred in acid sites of the matrix because the reactivity of the mixed catalyst was very similar to that in the experiment where the matrix bed and the zeolite bed were separated and the former was put on the next upper site of the latter (that is, silica-alumina→zeolite (SA→Z)) [
1]. Therefore, dodecane is initially decomposed to produce olefin and paraffin on the acid sites of the matrix. Olefins interact with acid sites of zeolite to produce carbenium ions which undergo further cracking and related reactions. This reaction may effectively inhibit direct reaction of paraffin with acid sites of zeolite and decrease the coke formation. A similar example using amorphous silica and Y zeolite was also reported [
9]. When amorphous silica-alumina was put on the upper side of the zeolite in the cracking of 1,3,5-triisopropylbenzene, the catalyst system showed higher activity, lower coke formation and lower CO generation than a physical mixture of zeolite and silica-alumina or 100% zeolite. When only zeolite with strong acid sites was used, deactivation occurred because of coke formation. It is considered that when silica-alumina was used at the upper side to zeolite, primary cracking occurred to produce propylene which reacted with acid sites on zeolite and inhibited the direct reaction of aromatic compounds with effectively strong acid sites. These results suggest the important role of primary reaction on the matrix component.
Figure 11.
TEM images of silica, mixed catalyst and β-zeolite.
Figure 11.
TEM images of silica, mixed catalyst and β-zeolite.
It was reported that new acid sites were formed by embedding wet zeolite into alumina and silica-alumina and that the recrystallization of the zeolite component resulted in high hydrothermal stability and catalytic activity [
8]. This catalyst system exhibited higher activity than the physical mixture of zeolite and matrix components. In the preceding paragraph, it was mentioned that the SA→Z and mixed catalyst systems exhibited very similar selectivities and properties for products [
1]. The activity of the latter was higher than that of the former, indicating that embedding of zeolite into silica-alumina increased the activity.
Figure 12.
Structure and reactivity of a mixed catalyst (MAT catalyst) using a matrix amorphous silica-alumina, zeolite and binder alumina.
Figure 12.
Structure and reactivity of a mixed catalyst (MAT catalyst) using a matrix amorphous silica-alumina, zeolite and binder alumina.
On the other hand, the promotion of diffusion in the inside of the pore of catalyst is very important. In the present study, the formation of the branched products was promoted with the use of the matrix with larger pore size, indicating that this effect may be derived from the relative ease of the elimination of branched product. The rapid transfer of bulky branched products, which are easier to be cracked, effectively inhibits further cracking.
3.3. Concerning Reaction Mechanisms
The proposed reaction pathway of catalytic cracking is illustrated in
Figure 13 [
1,
2]. The
n-paraffin molecule undergoes cracking to form paraffin and olefin molecules at the ratio 1:1. The olefin may undergo dehydrocyclization and eliminate H
+ and H
− to give an aromatic compound. The eliminated H
+ and H
− are supplied to another olefin in the system to form another paraffin. This route is called hydrogen transfer. If the initially formed paraffin is cracked again, another paraffin and another olefin are formed at 1:1. Cracking of the initially formed olefin forms two olefin molecules. If there is no hydrogen transfer, the formation of aromatic and olefin molecules will increase. However, a catalyst with high ability for hydrogen transfer increases the formation of paraffin molecules. Since the hydrogen transfer occurred appropriately the o/p ratio in the gasoline fraction was lower than 1 in our system. Isomerization may occur with each process of cracking, hydrogen transfer, dehydrogenation, and cyclization. Not only the amount and strength of acid sites, but also the steric effect related to pore structure (size and shape) is very important for this isomerization. From this point of view, the mixed catalysts in our system exhibited a higher ability for hydrogen transfer to form larger amounts of paraffin as the o/p ratios for these catalysts were relatively lower in
Table 2. Further, the presence of the larger pores and appropriate acid sites in these catalysts also promoted isomerization and increased the iso/n ratio and multi-branched products as shown in
Table 2.
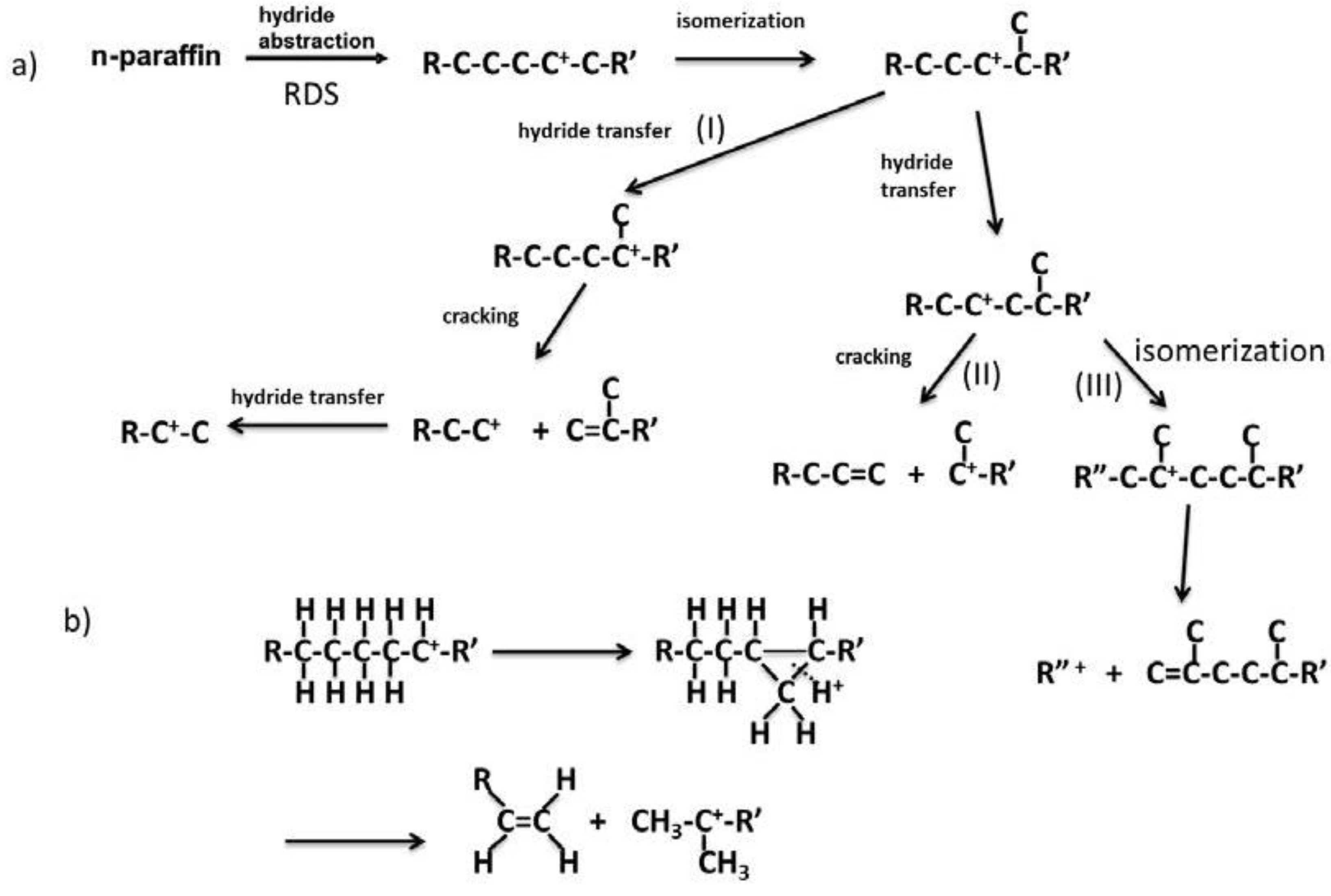
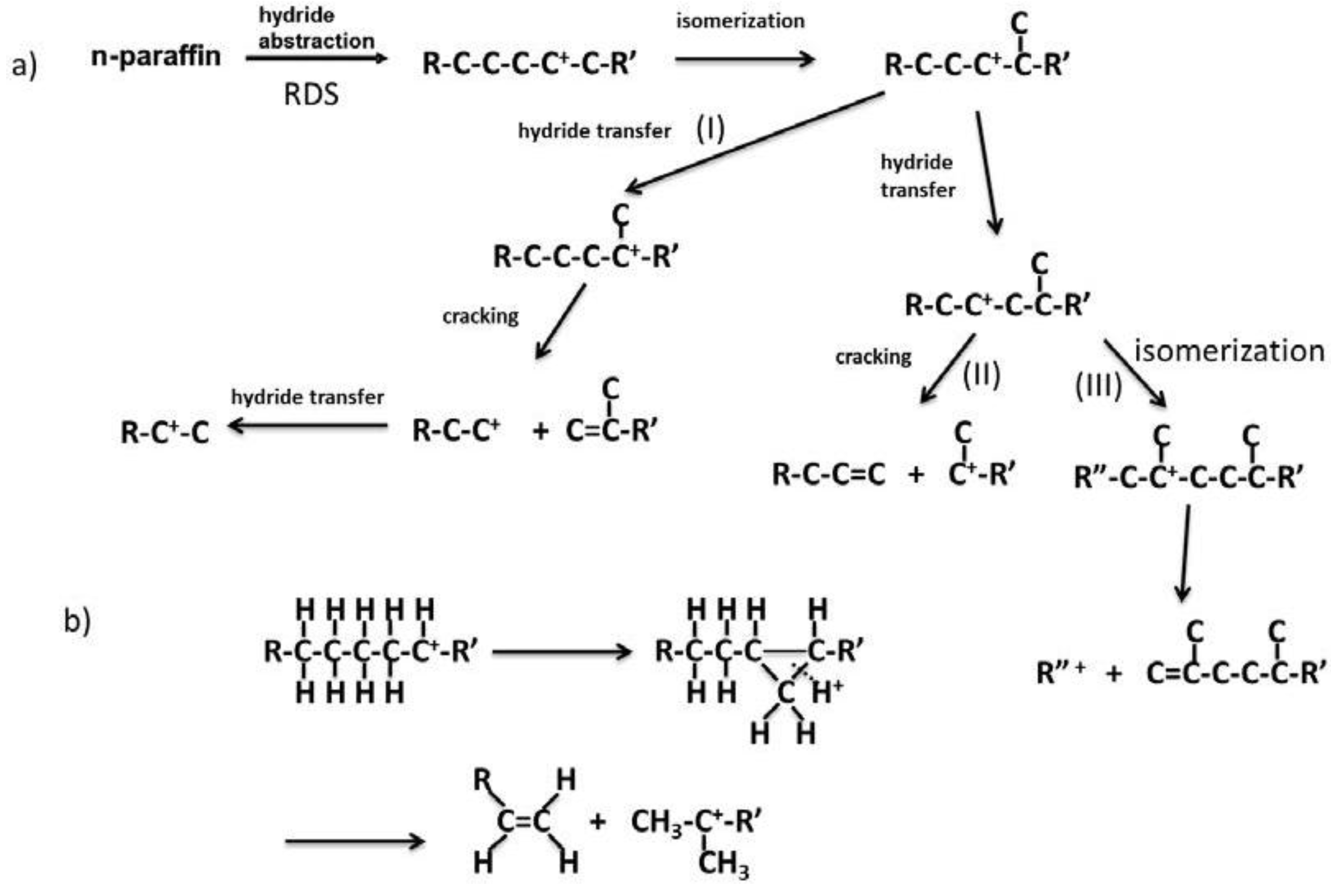
Figure 14 shows the reaction pathways of carbenium ions in the catalytic cracking of
n-paraffin which has generally been accepted [
3]. In
Figure 14a, hydride abstraction initially forms a carbenium ion which undergoes cracking to form an olefin and a primary carbenium ion (route I). Although the primary carbenium ion hardly undergoes cracking, isomerization or hydride transfer occurs to produce a secondary or tertiary carbenium ion, which undergoes further cracking to give olefin and primary carbenium ion [
3]. In contrast to this mechanism, it was stated that the primary carbenium was not formed because its stability is very low [
23,
24]. In that case, since cracking occurs so as to form a secondary or tertiary carbenium ion, a tertiary carbon or a quarternary carbon at β position of the carbenium ion is cracked (route II in
Figure 14a). This means that branched products are lost with increasing cracking rate. However, it has generally been accepted that hydrocarbons with higher molecular weight are cracked faster and produce larger amounts of branched products [
3,
25,
26]. In the mechanism with the formation of the primary carbenium ion in catalytic cracking (
Figure 14a route I and III), branched products do not decrease with cracking. On the other hand, in order to explain the formation of branched products without the formation of the primary carbenium ion, it is also suggested that cracking and isomerization occur simultaneously through the reaction pathway, including 1,3-hydride shift and 1,2-hydride shift, as well as β scission (
Figure 14b) [
23,
24]. We believe both routes I and II exist because branched products increased first and then decreased with increasing pore size, as shown in
Table 2. The maximum yield of branched products was obtained in MAT-200SA-5 which exhibited medium conversion.
Figure 13.
Reaction pathways in catalytic cracking of n-paraffin.
Figure 13.
Reaction pathways in catalytic cracking of n-paraffin.
Corma reported that amorphous silica-alumina with mesopores had lower hydrothermal stability and the acid property was also weak [
27]. However, simultaneous usage of zeolite and amorphous silica-alumina maintained the crystallinity of zeolite and recrystallized zeolite, indicating that amorphous silica-alumina still has great potential for use in catalytic cracking [
8]. On the other hand, growing zeolite crystals with mesopores was tried, using zeolite with micropores as a seed crystal or by assembling zeolite precursor [
10,
28,
29,
30,
31,
32]. Surface-active agents, such as tetraalkylammonium salts were used with the seed crystals to grow the zeolite structure, and higher hydrothermal stability was obtained, compared with MCM-41, which had lower hydrothermal stability. However, insufficient information is available for catalytic cracking. For example, although direct catalytic cracking of heavy oil was sometimes performed, the differences between catalysts are difficult to identify. In many cases, only cracking of alkylbenzene was performed and the cracking of saturated hydrocarbons, of which large amounts are included in heavy oil, has hardly been discussed. In our results, the cracking reactivity of saturated hydrocarbon on mesoporous catalysts, including amorphous silica-alumina and zeolite, can be investigated and described. It was shown that the presence of both large mesopores and an appropriate amount of aluminum component in matrix promoted the formation of branched products.
Figure 14.
Reaction pathway of carbenium ion in catalytic cracking of n-paraffin.
Figure 14.
Reaction pathway of carbenium ion in catalytic cracking of n-paraffin.
In our studies, the formation of branched paraffin was especially noted. When the cracking of VGO and AR is considered, the gasoline produced will undergo further hydrodesulfurization. The olefins in the gasoline are expected to be hydrogenated at hydrodesulfurization and the RON value of the gasoline will decrease. To overcome this situation, the catalyst must be designed to increase the RON value by increasing branched paraffin formation. Although the desired formulation of gasoline is unknown, this must be an important factor in the development to increase the RON value by branched paraffin formation. The development of catalysts, with selectivity for branched paraffins, will also be very important to obtain the stability of diesel oil at lower temperature. The technology can be applied to hydrocracking of WAX produced in the GTL process.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}