Hydrogen Production from Semiconductor-based Photocatalysis via Water Splitting


Abstract
:1. Alternative Energies and Hydrogen
1.1. Some Concerns about Hydrogen
1.2. Hydrogen Production
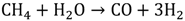

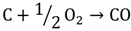
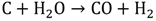
1.3. Hydrogen Production by Solar Energy
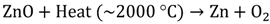
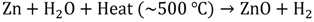
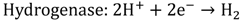
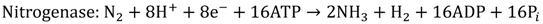
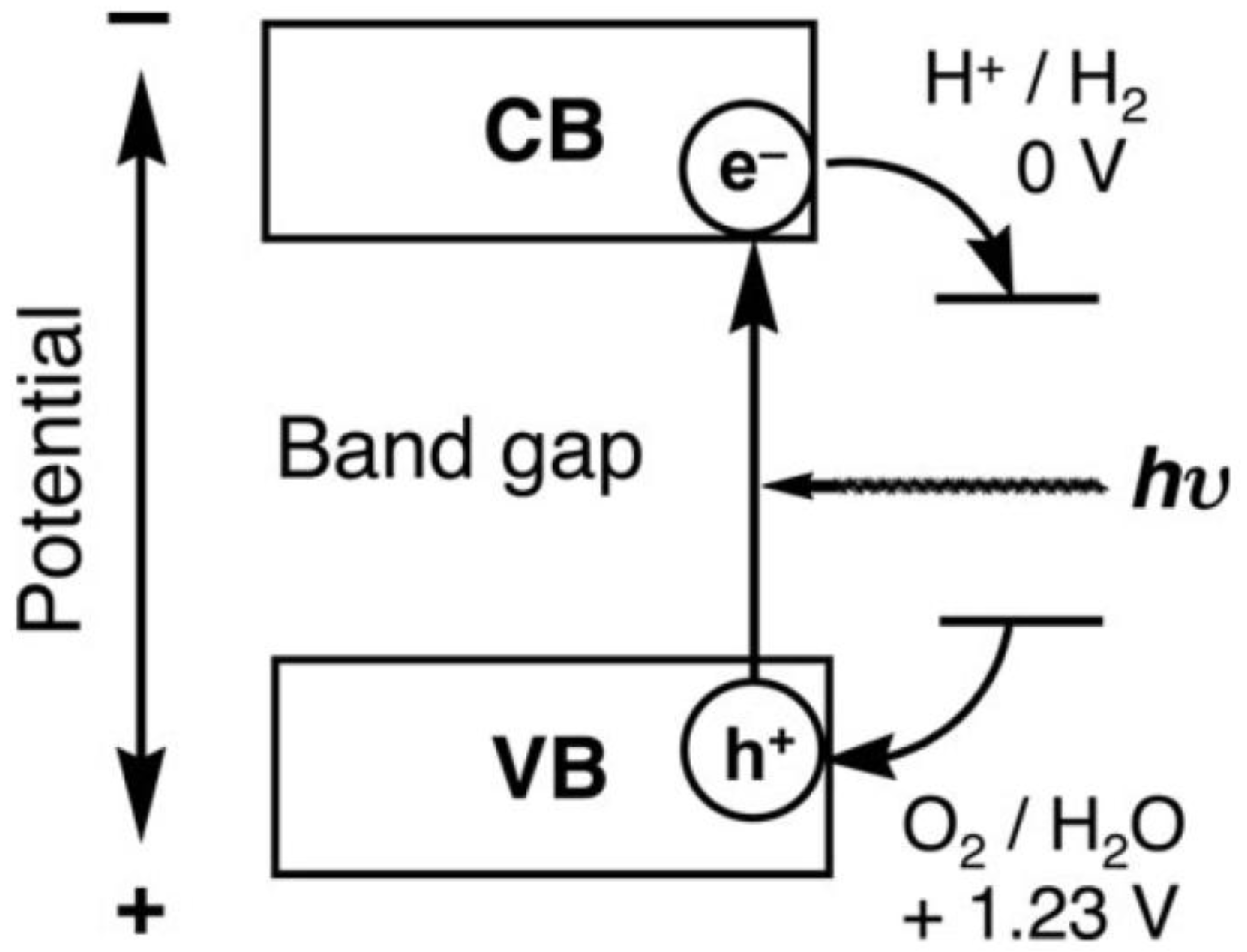
2. Photocatalytic Water Splitting
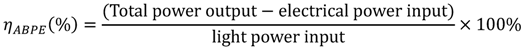
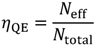
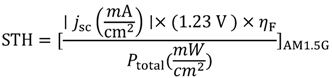
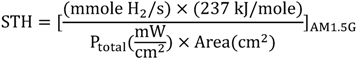
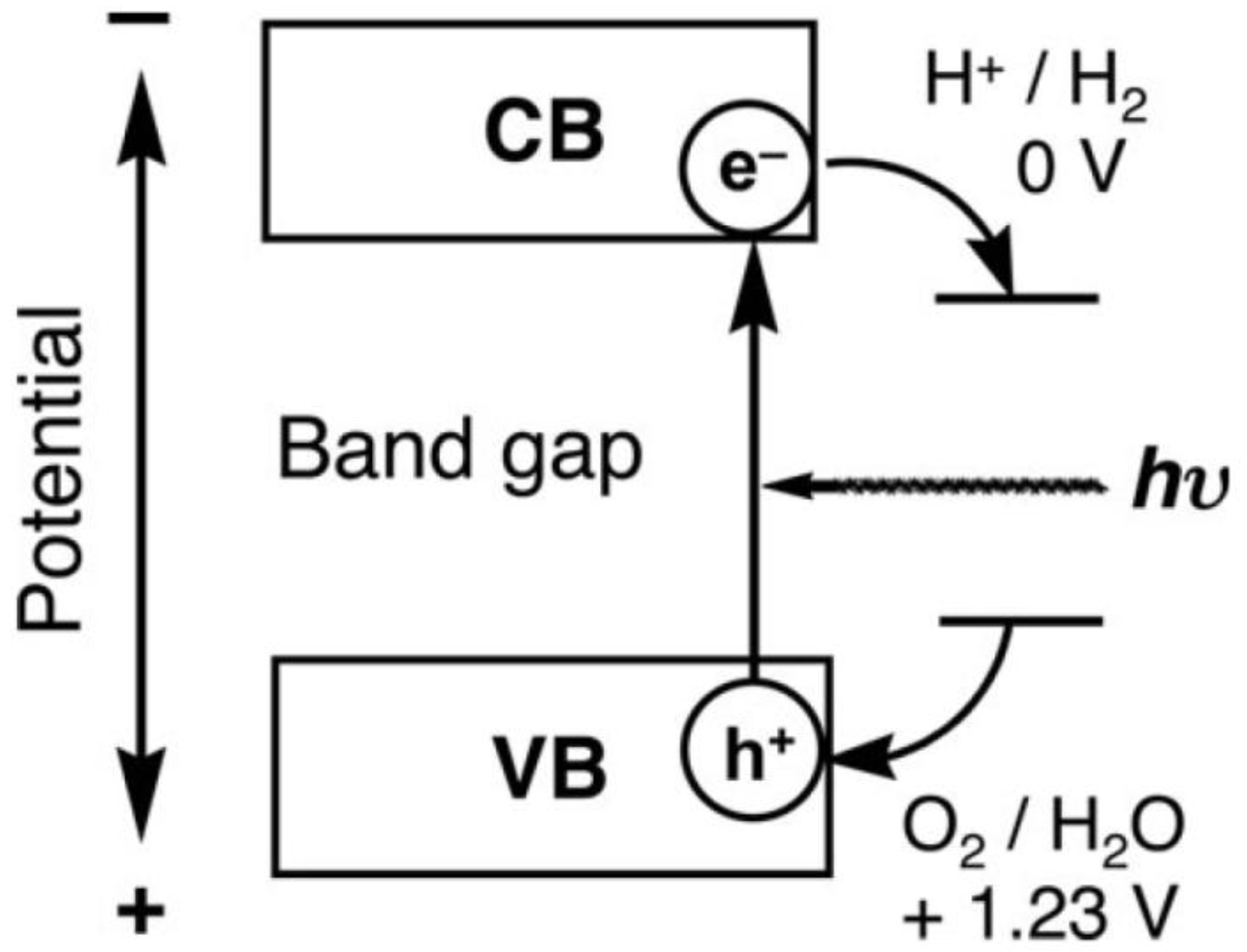
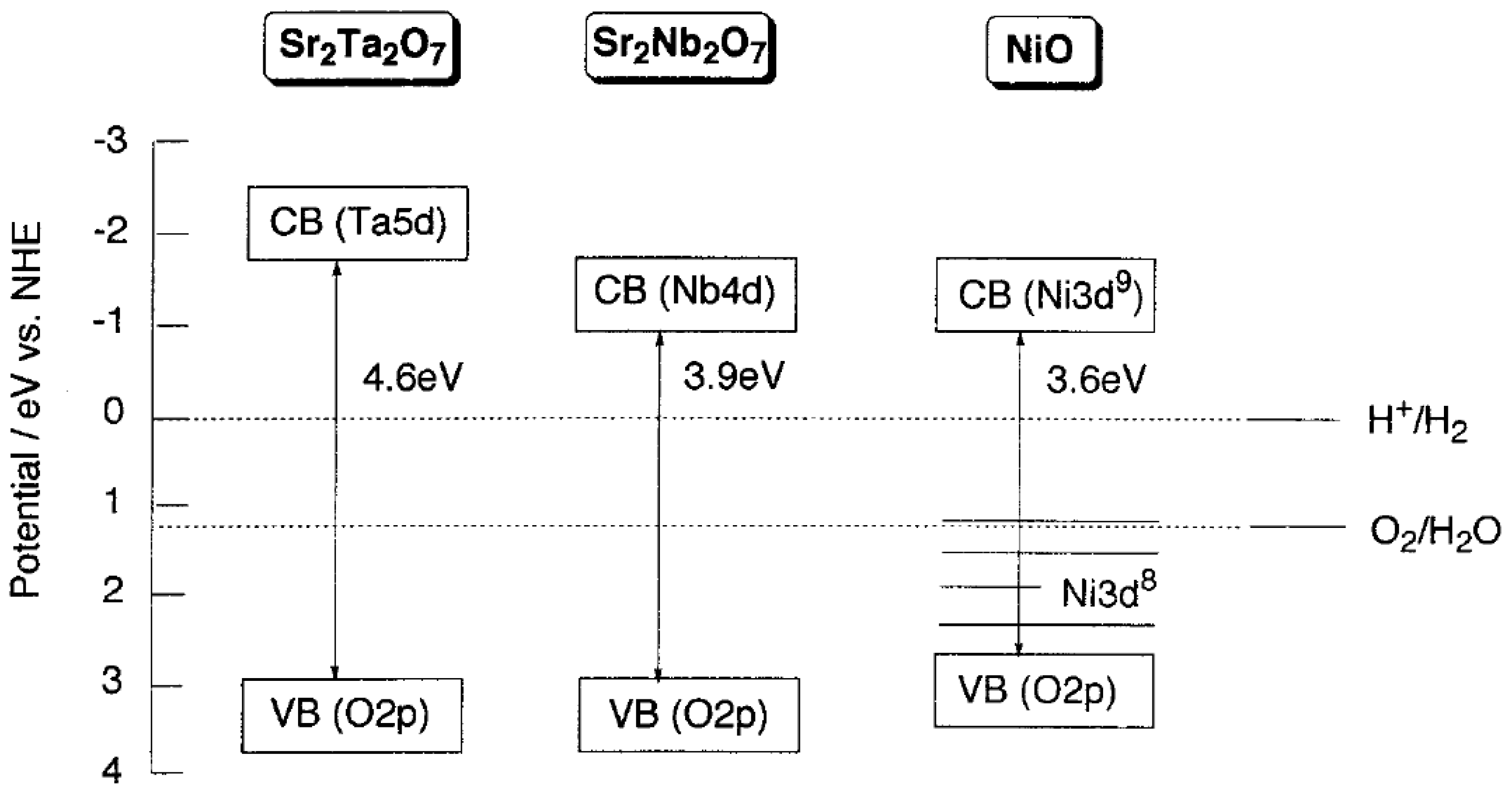
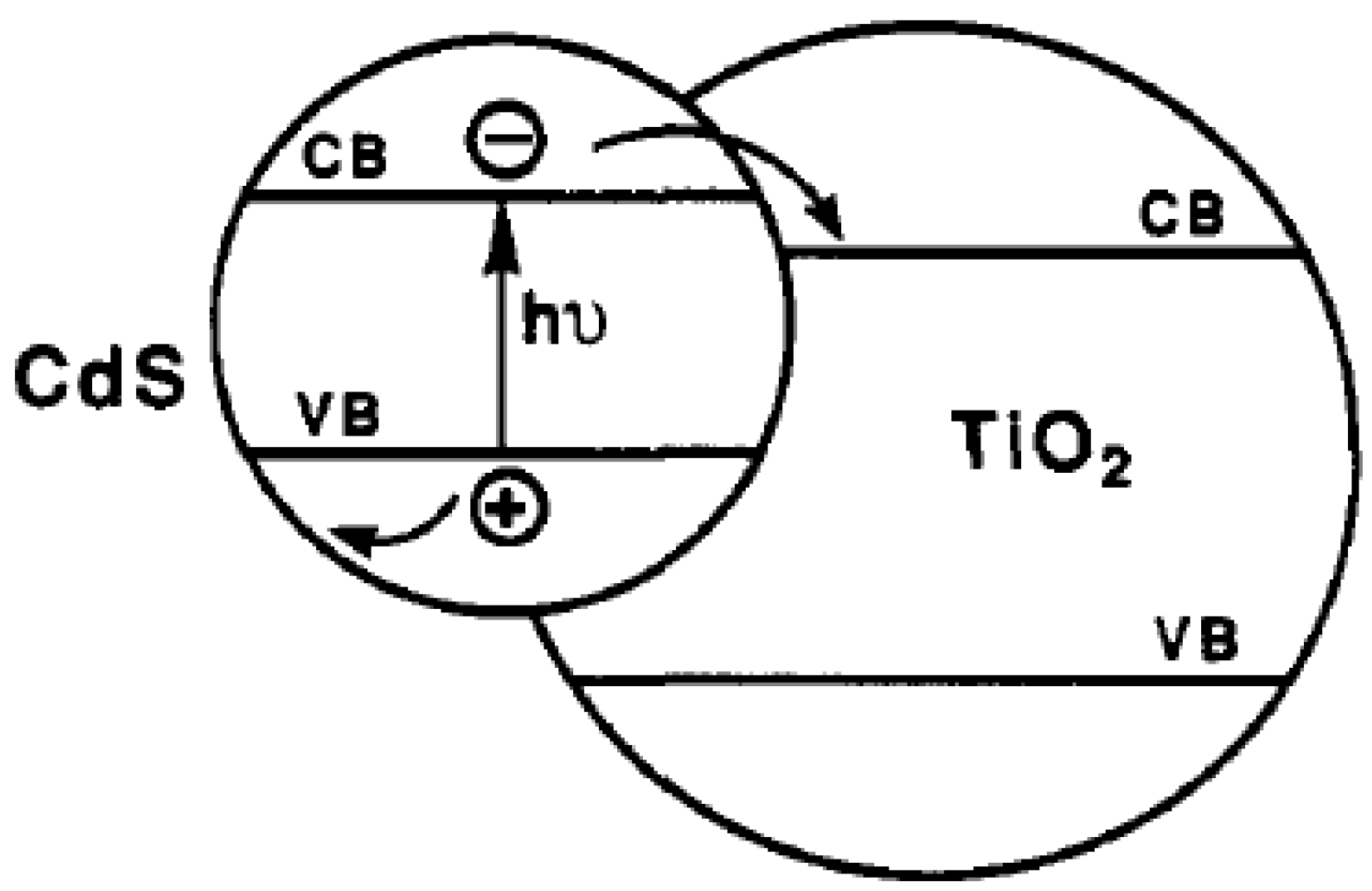
2.1. How to Improve the Photoactivity of TiO2
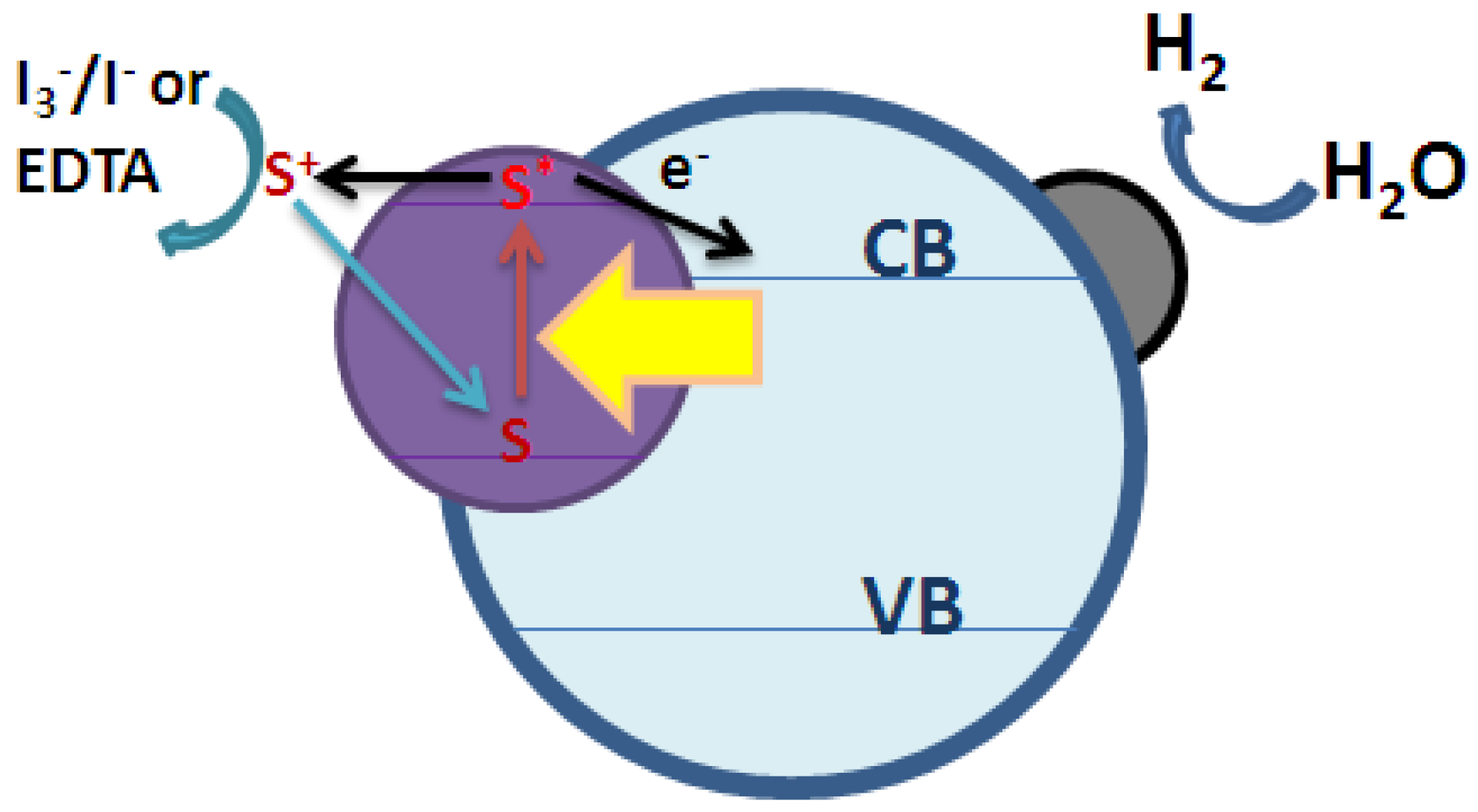
2.2. High-Efficient Photocatalytic System for Water Splitting
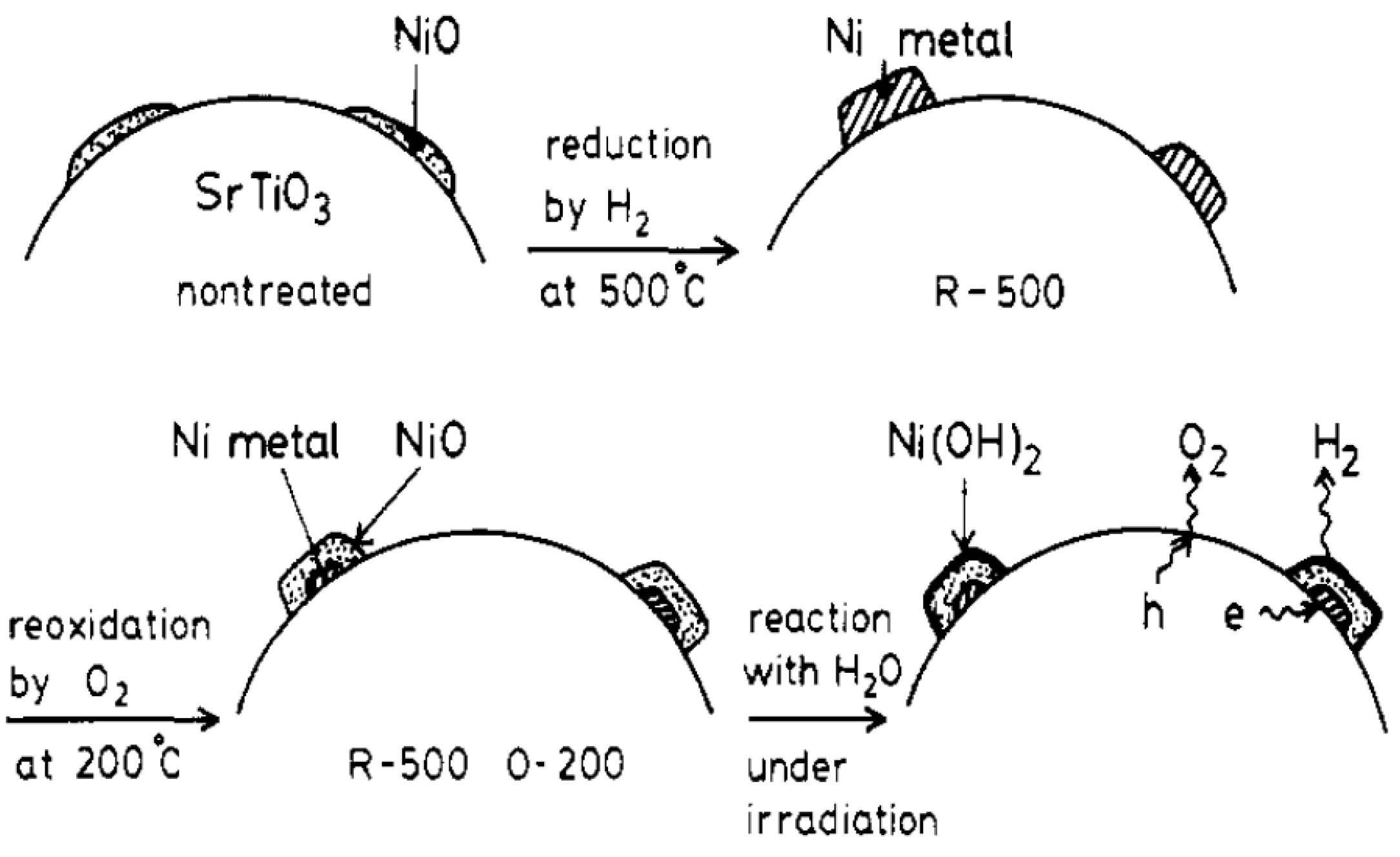
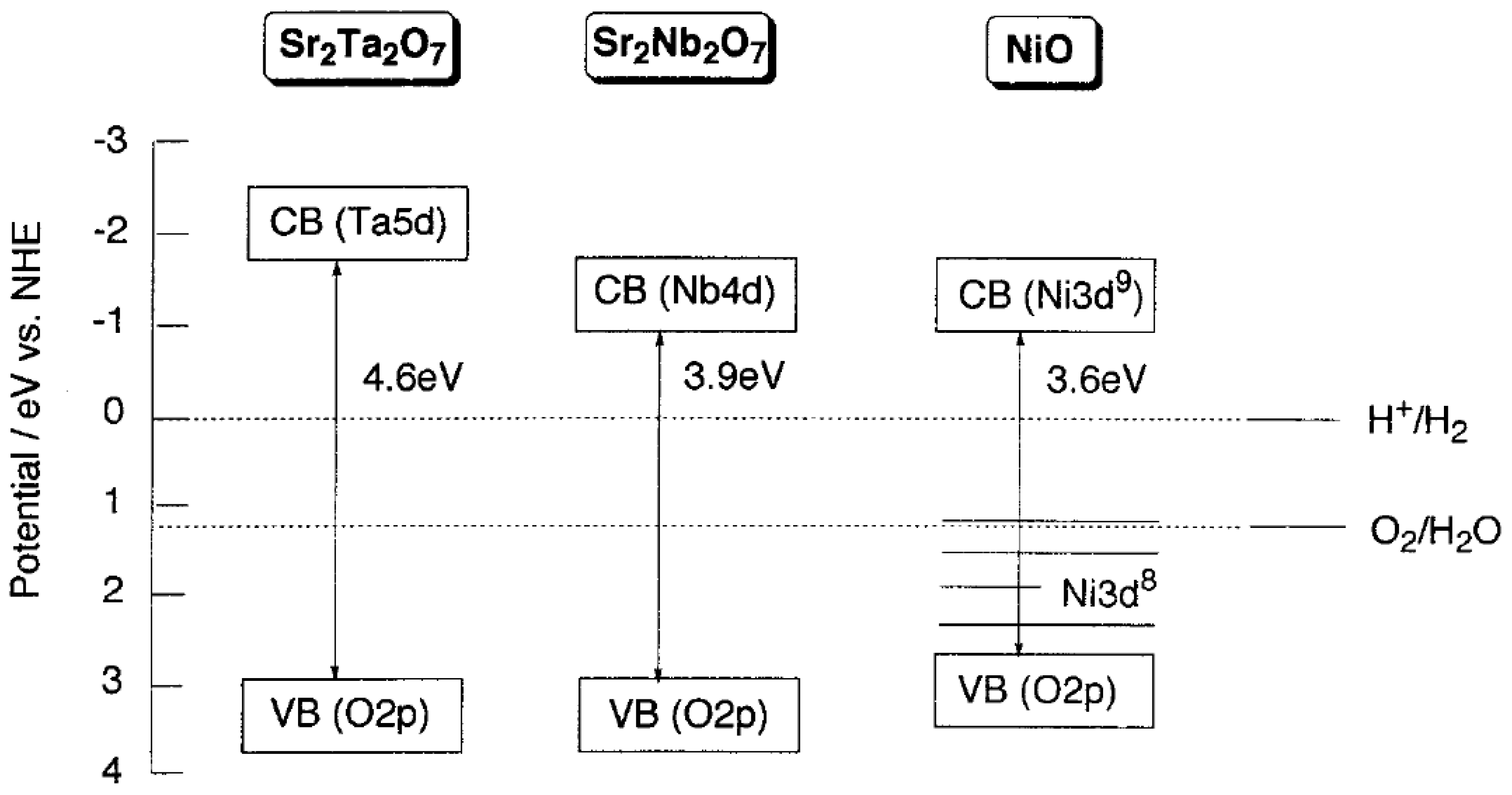
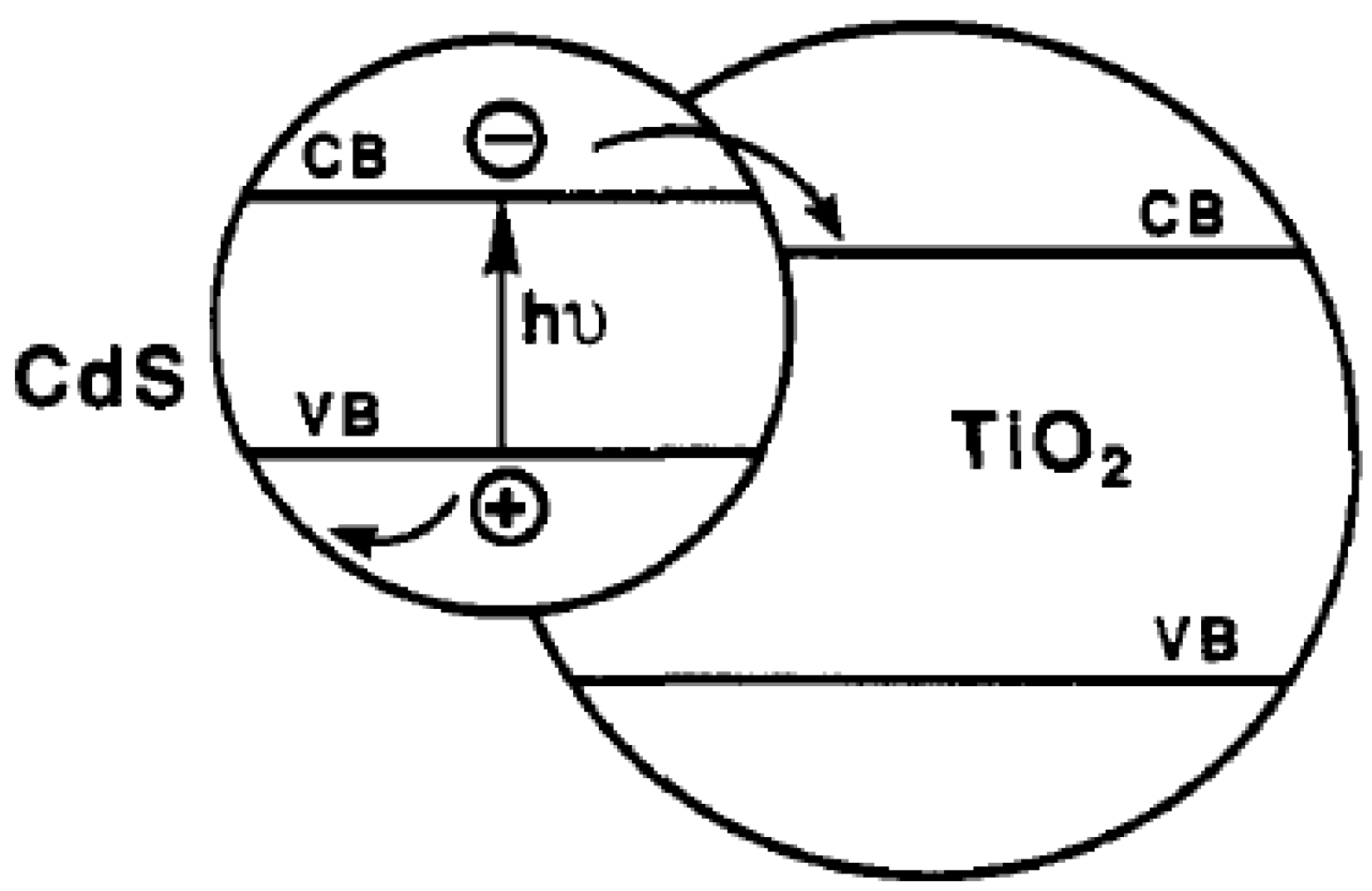
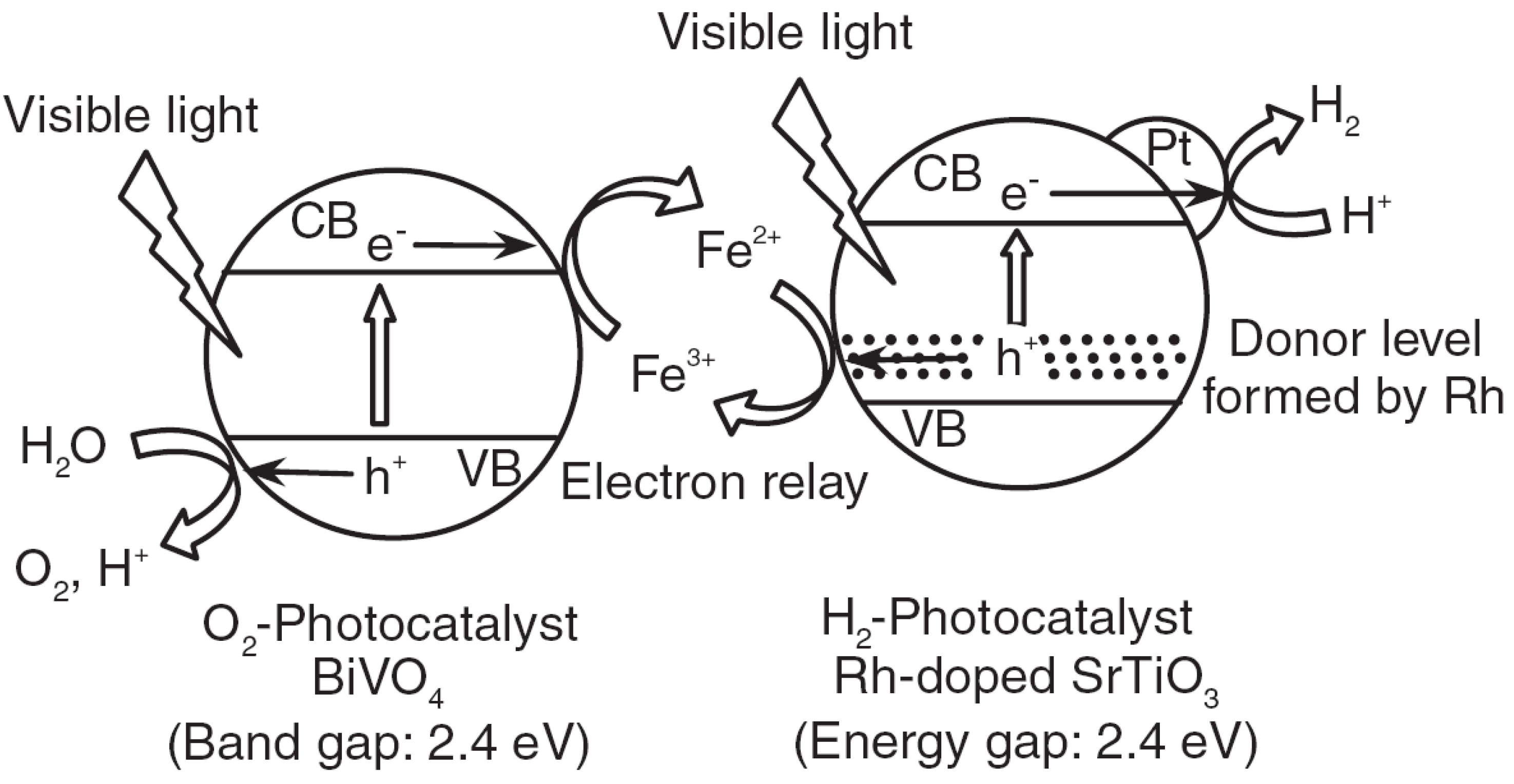
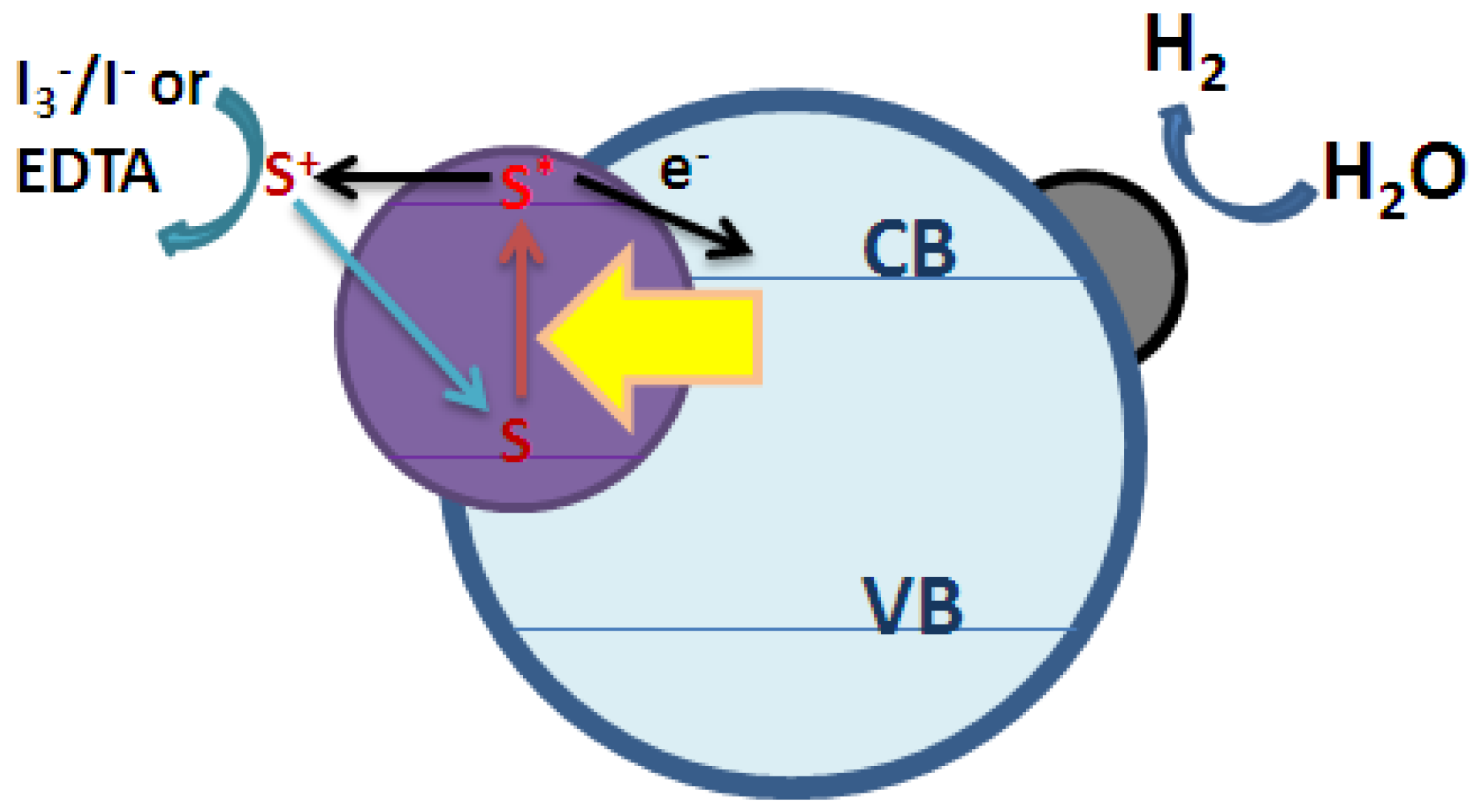
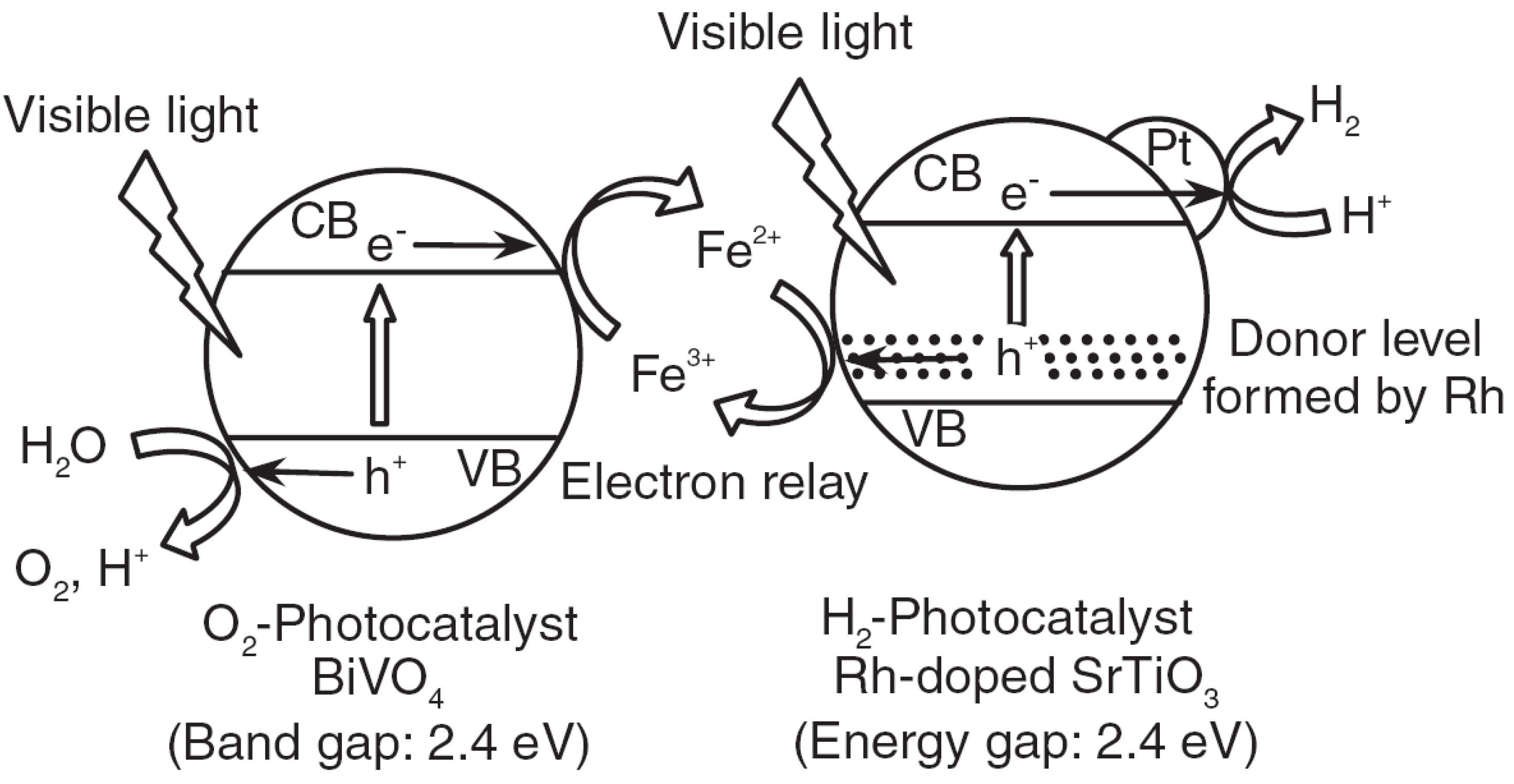
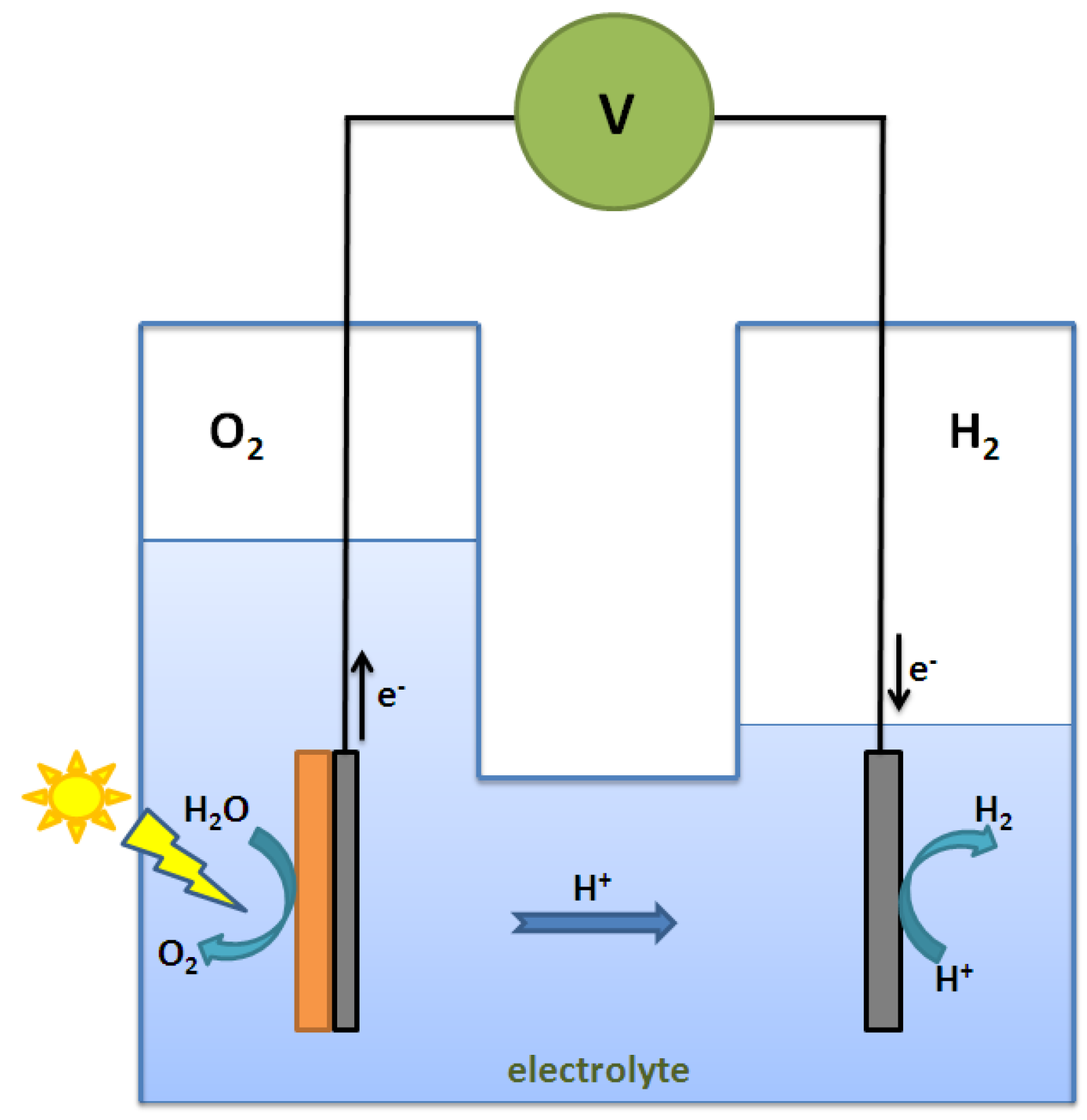
2.3. Types of Photocatalytic Water-Splitting Reaction
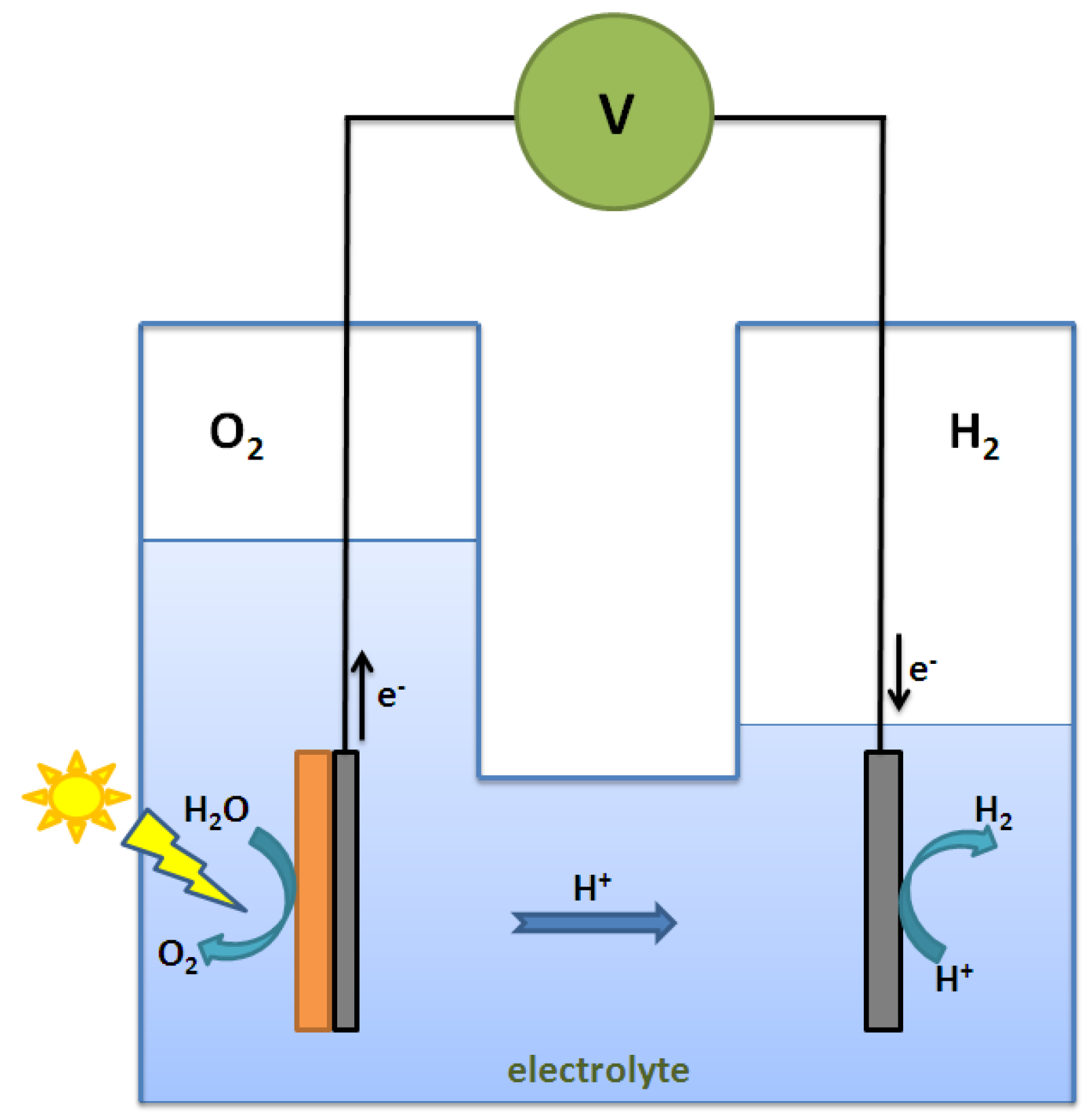
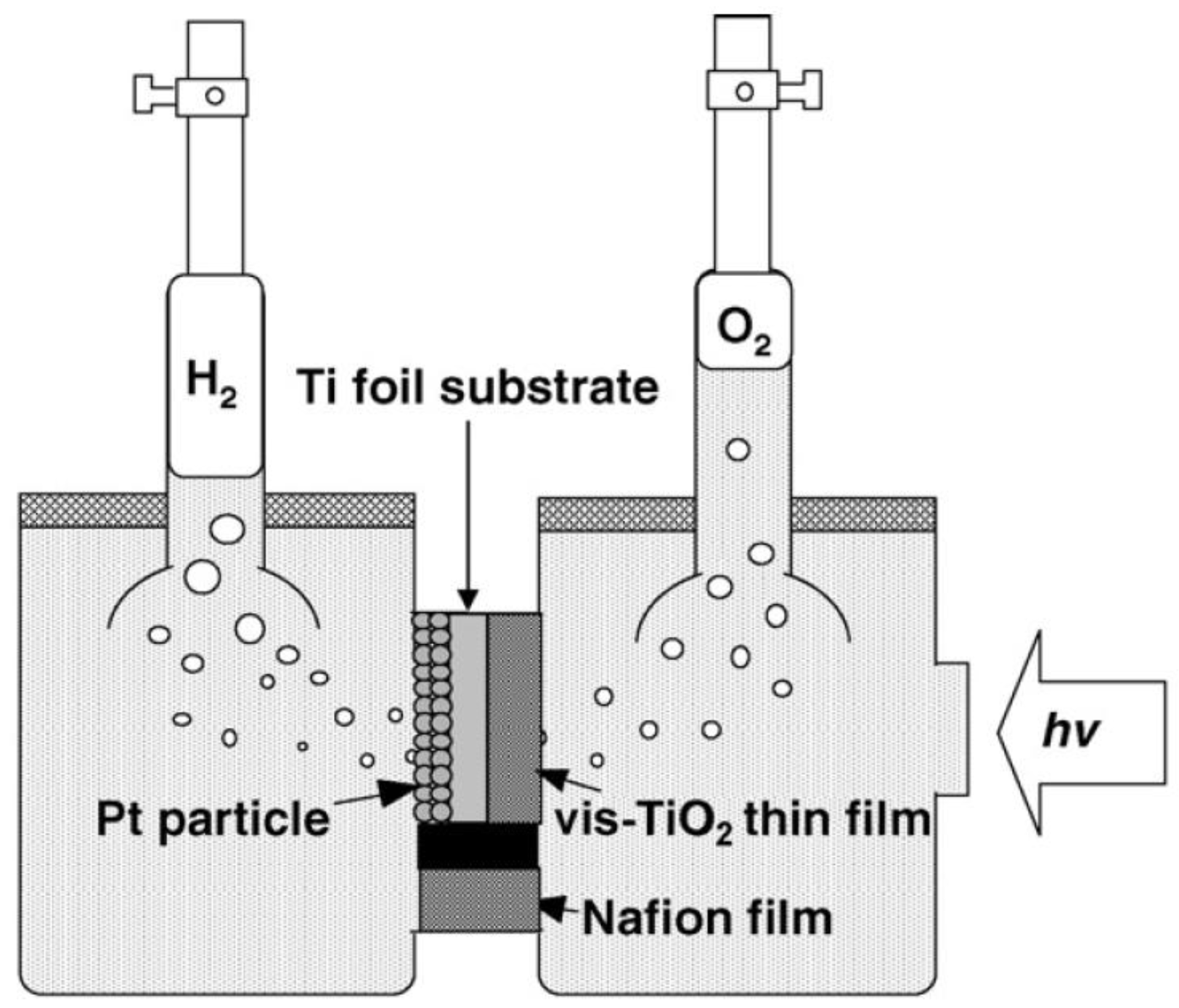

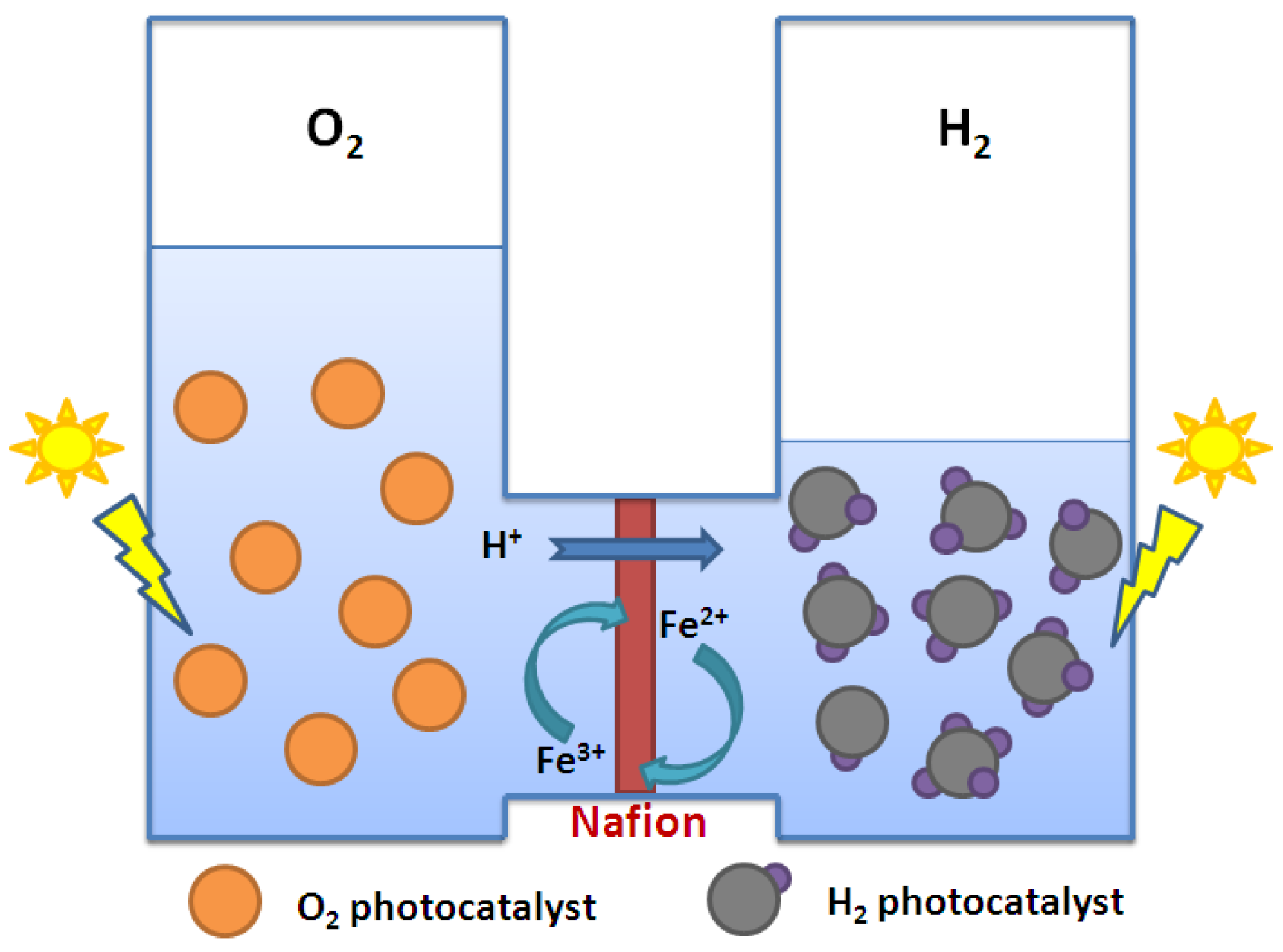

2.4. Summary of Photocatalytic Water Splitting

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalyst | Weight | Reaction solution | Light source | Rate of evolution (μmol h−1) | Reference | |
|---|---|---|---|---|---|---|
| H2 | O2 | |||||
| Pt/TiO2 | 0.3 g | 2.17M Na2CO3 | 400 W Hg lamp | 568 | 287 | [23] |
| ZrO2 | 1 g | distilled water | 400 W Hg lamp | 72 | 36 | [24] |
| ZrO2 | 1 g | 1.09M Na2CO3 | 400 W Hg lamp | 142 | 75 | [24] |
| Pt/ZrO2 | 1 g | 0.94M NaHCO3 | 400 W Hg lamp | 120 | 61 | [24] |
| Ru2O/ZrO2 | 1 g | distilled water | 400 W Hg lamp | 11 | 5 | [24] |
| Cu/ZrO2 | 1 g | distilled water | 400 W Hg lamp | 14 | 6 | [24] |
| NiO/Sr2Nb2O7 | 1 g | distilled water | 400 W Hg lamp | 110 | 36 | [50] |
| NiO/Sr2Ta2O7 | 1 g | distilled water | 400 W Hg lamp | 1000 | 480 | [50] |
| (Tetra)BaTa2O6 | 1 g | distilled water | 400 W Hg lamp | 21 | 10 | [51] |
| (Ortho)BaTa2O6 | 1 g | distilled water | 400 W Hg lamp | 33 | 15 | [51] |
| (Ortho)BaTa2O6 | 1 g | 0.0005 M Ba(OH)2 | 400 W Hg lamp | 126 | 59 | [51] |
| (Ortho)BaTa2O6 | 1 g | 0.001M KOH | 400 W Hg lamp | 24 | 11 | [51] |
| (Ortho)BaTa2O6 | 1 g | 0.0005 M BaCl2 | 400 W Hg lamp | 15 | 6 | [51] |
| NiO/BaTa2O6 | 1g | distilled water | 400 W Hg lamp | 629 | 303 | [51] |
| Ni/Rb4Nb6O17 | 1 g | distilled water | 400 W Hg lamp | 936 | 451 | [53] |
| Ni/K4Nb6O17 | 1 g | distilled water | 400 W Hg lamp | 403 | 197 | [53] |
| Pt/TiO2TiO2 | 12 mg | 2 M KBr6.5 mM FeCl2 | 500 W Hg | 2.8 | 1.3 | [57] |
| Pt-TaONPt-WO3 | 0.2 g | 5 mM NaI | 300 W Xe lamp with filters: λ > 420 nm | 24 | 12 | [45] |
| Pt/BaTaO2NPt/WO3 | 0.1 g | 5 mM NaI | 300 W Xe lamp with filters: λ > 420 nm | 6.6 | 3.1 | [59] |
| Pt/SrTiO3:Rh, BiVO4 | 0.1 g | 2 mM FeCl3 | 300W Xe with filter: λ > 420 nm | 15 | 7.2 | [60] |
| Pt/SrTiO3:Rh, Bi2MoO6 | 0.1 g | 2 mM FeCl3 | 300 W Xe with filter: λ > 420 nm | 19 | 8.9 | [60] |
| Pt/SrTiO3:Rh, WO3 | 0.1 g | 2 mM FeCl3 | 300 W Xe with filter: λ > 420 nm | 7.8 | 4.0 | [60] |
| K4Nb6O17 | 1 g | H2O | 450 W Hg lamp | 8 | 1 | [64] |
| NiO/ K4Nb6O17 | 1 g | H2O | 450 W Hg lamp | 77 | 37 | [64] |
| Pt/SrTiO3:RhWO3 | 0.3 | 2 mM FeCl2/FeCl3 | 500 W halogen lamp | 1.6 | 0.8 | [69] |
| Pt/SrTiO3:RhBiVO4 | 0.4 | 5mM FeCl2/FeCl3 | 300 W Xe lamp | 0.8 | 0.4 | [70] |
| Photoelectrode | Surface area (cm2) | Electrolyte | Light source | Efficiency/H2 yield | Applied bias (V) | Reference |
|---|---|---|---|---|---|---|
| TiO2 | 1 | Fe3+ solution | 500 W Xenon lamp | QE = 10% | N/A | [61] |
| SrTiO3 | 0.25 | 9.5 M NaOH | Argon ion laser (351 nm) | QE = 11% | N/A | [62] |
| SrTiO3 | 1.539 | 1 M NaOH | 150 W halogen lamp (340 nm) | QE = 3.5% | 0.5 | [63] |
| TiO2 | 2 | 0.5 M H2SO4/1 M NaOH | UV light with intensity of 25 mW/cm2 | 60 μmol in 8 h | N/A | [65] |
| Vis-WO3/vis-TiO2 | 2 | 0.025 M H2SO4/0.05 M NaOH | UV light with intensity of 2.5 mW/cm2 | 39 μmol in 8 h | N/A | [66] |
| Vis-WO3/vis-TiO2 | 2 | 0.025 M H2SO4/0.05 M NaOH | AM 1. 5 | 6 μmol in 8 h | N/A | [66] |
| Pillar TiO2 | 2 | 0.5 M H2SO4/1M NaOH | UV light with intensity of 25 mW/cm2 | 37 μmol in 8 h | N/A | [67] |
| WSe2 | 0.0125 | 1 M KI+ 0.05 M I2 | 60 mW/cm2 tungsten lamp | ABPE = 17.1% | N/A | [71] |
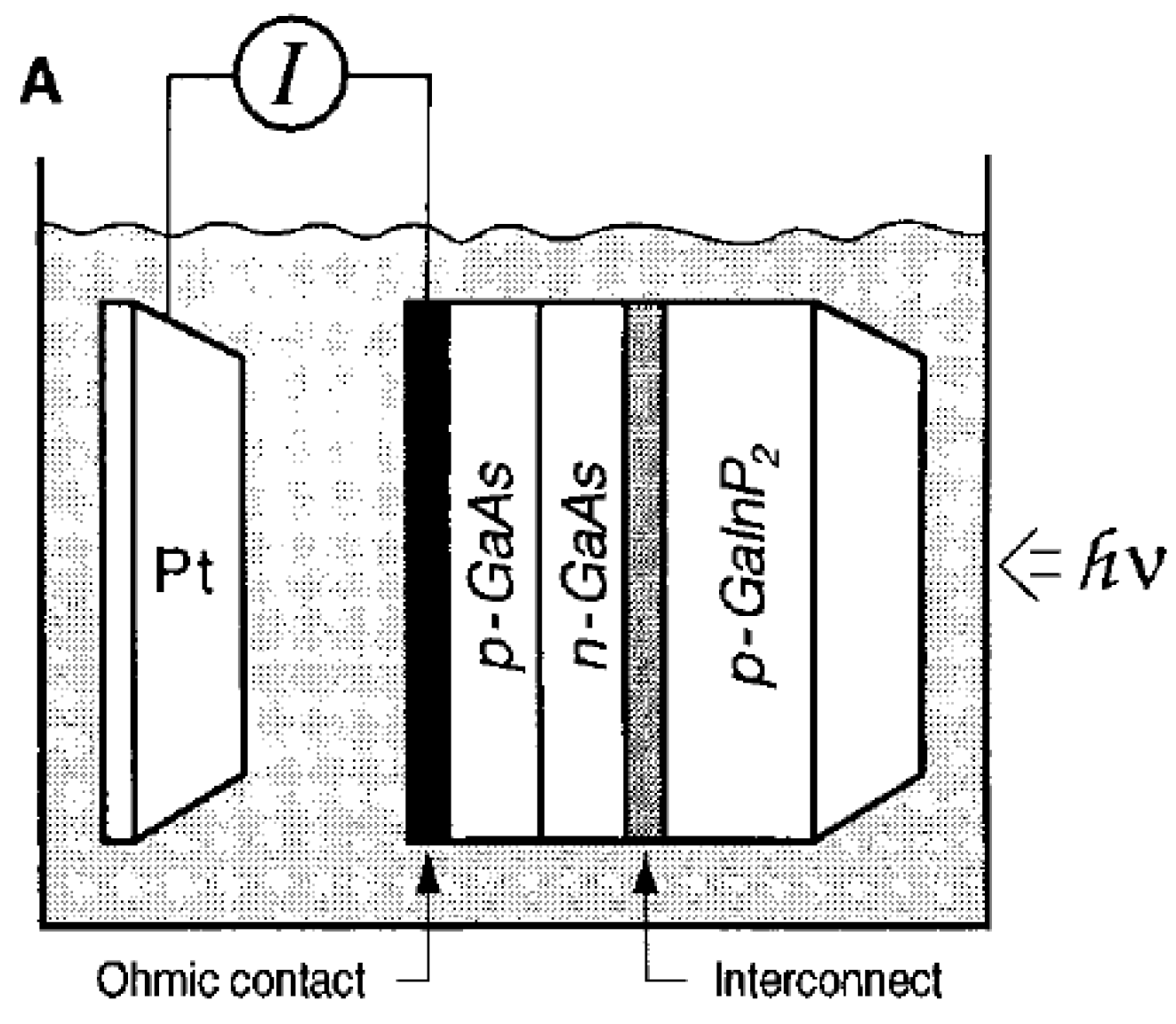
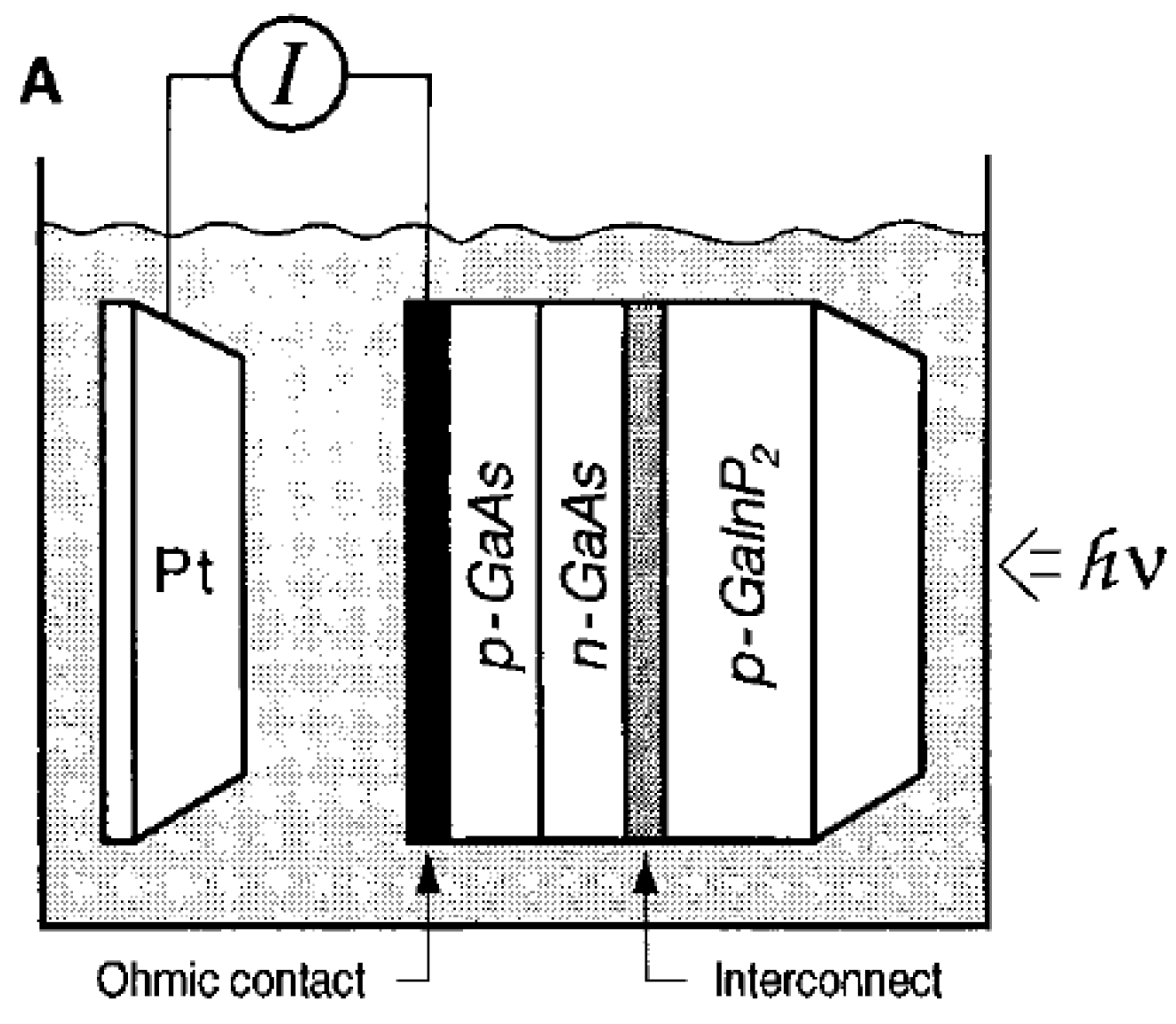
| p-GaAs/ n-GaAs/p-GaInP2 | 0.2 | 3 M H2SO4 | 150 W tungsten-halogen lamp | ABPE = 12.4% | 0.3 | [73] |
| GaInP2/GaAs | 0.5 | 2 M KOH | 75 W Xe lamp | ABPE = 16.5% | N/A | [75] |
| Triple a-Si | 0.3 | 2 M KOH | 75 W Xe lamp | ABPE = 7.8% | N/A | [75] |
| AlGaAs/Si | 0.22 | 1 M HClO4 | 50 W tungsten-halogen lamp | ABPE = 18.3% | N/A | [77] |
| InGaP/GaAs/Ge | 2 | 0.5 M H2SO4/1 M NaOH | AM 1.5 | 440 μmol in 8 h | N/A | [78] |
| CM n-TiO2 | 0.2 | 5 M KOH | 150 W Xe lamp | ABPE = 8.35% | 0.3 | [18] |
| n-TiO2 | 0.2 | 5 M KOH | 150 W Xe lamp | ABPE = 1.08% | 0.6 | [18] |
3. Current Challenges and Future Prospects of Photocatalytic Water Splitting
References
- Solomon, S.; Plattner, G.K.; Knutti, R.; Friedlingstein, P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sc. USA 2009, 106, 1704–1709. [Google Scholar]
- Primio, R.D.; Horsfield, B.; Guzman-Vega, M.A. Determining the temperature of petroleum formation from the kinetic properties of petroleum asphaltenes. Nature 2000, 406, 173–176. [Google Scholar] [CrossRef]
- Chiari, L.; Zecca, A. Constraints of fossil fuels depletion on global warming projections. Energy Policy 2011, 39, 5026–5034. [Google Scholar] [CrossRef]
- Dincer, F. The analysis on wind energy electricity generation status, potential and policies in the world. Renew. Sustain. Energy Rev. 2011, 15, 5135–5142. [Google Scholar] [CrossRef]
- Yuksel, I. Hydropower for sustainable water and energy development. Renew. Sustain. Energy Rev. 2010, 14, 462–469. [Google Scholar] [CrossRef]
- Parida, B.; Iniyan, S.; Goic, R. A review of solar photovoltaic technologies. Renew. Sustain. Energy Rev. 2011, 15, 1625–1636. [Google Scholar] [CrossRef]
- Xie, W.T.; Dai, Y.J.; Wang, R.Z.; Sumathy, K. Concentrated solar energy applications using Fresnel lenses: A review. Renew. Sustain. Energy Rev. 2011, 15, 2588–2606. [Google Scholar] [CrossRef]
- Barbier, E. Geothermal energy technology and current status: an overview. Renew. Sustain. Energy Rev. 2002, 6, 3–65. [Google Scholar] [CrossRef]
- Midilli, A.; Ay, M.; Dincer, I.; Rosen, M.A. On hydrogen and hydrogen energy strategies I: Current status and needs. Renew. Sustain. Energy Rev. 2005, 9, 255–271. [Google Scholar] [CrossRef]
- Hou, K.H.; Hughes, R. The kinetics of methane steam reforming over a Ni/alpha-Al2O catalyst. Chem. Eng. J. 2001, 82, 311–328. [Google Scholar] [CrossRef]
- Nowotny, J.; Sorrell, C.C.; Sheppard, L.R.; Bak, T. Solar-hydrogen: Environmentally safe fuel for the future. Int. J. Hydrog. Energy 2005, 30, 521–544. [Google Scholar] [CrossRef]
- Czernik, S.; Evans, R.; French, R. Hydrogen from biomass-production by steam reforming of biomass pyrolysis oil. Catal. Today 2007 129, 265–268.
- Ni, M.; Leung, D.Y.C.; Leung, M.K.H.; Sumathy, K. An overview of hydrogen production from biomass. Fuel Process. Tech. 2006, 87, 461–472. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar hydrogen production via a two-step water-splitting thermochemical cycle based on Zn/ZnO redox reactions. Int. J. Hydrog. Energy 2002, 27, 611–619. [Google Scholar] [CrossRef]
- Akkerman, I.; Janssen, M.; Rocha, J.; Wijffels, R.H. Photobiological hydrogen production: photochemical efficiency and bioreactor design. Int. J. Hydrog. Energy 2002, 27, 1195–1208. [Google Scholar] [CrossRef]
- Das, D.; Veziroglu, T.N. Advances in biological hydrogen production processes. Int. J. Hydrog. Energy 2008, 33, 6046–6057. [Google Scholar] [CrossRef]
- Guan, Y.F.; Deng, M.C.; Yu, X.J.; Zhang, W. Two-stage photo-biological production of hydrogen by marine green alga Platymonas subcordiformis. Biochem. Eng. J. 2004, 19, 69–73. [Google Scholar] [CrossRef]
- Khan, S.U.M.; Al-Shahry, M.; Ingler, W.B. Efficient photochemical water splitting by a chemically modified n-TiO2. Science 2002, 297, 2243–2245. [Google Scholar] [CrossRef]
- Bak, T.; Nowotny, J.; Rekas, M.; Sorrell, C.C. Photo-electrochemical hydrogen generation from water using solar energy. Materials-related aspects. Int. J. Hydrog. Energy 2002, 27, 991–1022. [Google Scholar] [CrossRef]
- Chen, Z.B.; Jaramillo, T.F.; Deutsch, T.G.; Kleiman-Shwarsctein, A.; Forman, A.J.; Gaillard, N.; Garland, R.; Takanabe, K.; Heske, C.; Sunkara, M.; et al. Accelerating materials development for photoelectrochemical hydrogen production: Standards for methods, definitions, and reporting protocols. J. Mater. Res. 2010, 25, 3–16. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Li, Y.X.; Lu, G.X.; Li, S.B. Photocatalytic production of hydrogen in single component and mixture systems of electron donors and monitoring adsorption of donors by in situ infrared spectroscopy. Chemosphere 2003, 52, 843–850. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Effect of carbonate salt addition on the photocatalytic decomposition of liquid water over Pt-TiO2 catalyst. J. Chem. Soc.-Faraday Trans. 1997, 93, 1647–1654. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Photocatalytic decomposition of water and photocatalytic reduction of carbon-dioxide over ZrO2 catalyst. J.Phys. Chem. 1993, 97, 531–533. [Google Scholar] [CrossRef]
- Subramanian, V.; Wolf, E.E.; Kamat, P.V. Catalysis with TiO2/gold nanocomposites. Effect of metal particle size on the Fermi level equilibration. J. Am. Chem. Soc. 2004, 126, 4943–4950. [Google Scholar]
- Bamwenda, G.R.; Tsubota, S.; Nakamura, T.; Haruta, M. Photoassisted hydrogen production from a water-ethanol solution: a comparison of activities of Au-TiO2 and Pt-TiO2. J. Photochem. Photobiol. A 1995, 89, 177–189. [Google Scholar] [CrossRef]
- Murdoch, M.; Waterhouse, G.I.N.; Nadeem, M.A.; Metson, J.B.; Keane, M.A.; Howe, R.F.; Llorca, J.; Idriss, H. The effect of gold loading and particle size on photocatalytic hydrogen production from ethanol over Au/TiO2 nanoparticles. Nature Chem. 2011, 3, 489–492. [Google Scholar]
- Anpo, M.; Takeuchi, M. The design and development of highly reactive titanium oxide photocatalysts operating under visible light irradiation. J. Catal. 2003, 216, 505–516. [Google Scholar] [CrossRef]
- Merlen, A.; Gadenne, V.; Romann, J.; Chevallier, V.; Patrone, L.; Valmalette, J.C. Surface enhanced Raman spectroscopy of organic molecules deposited on gold sputtered substrates. Nanotechnology 2009, 20. [Google Scholar] [CrossRef]
- Primo, A.; Corma, A.; Garcia, H. Titania supported gold nanoparticles as photocatalyst. Phys. Chem. Chem. Phys. 2011, 13, 886–910. [Google Scholar]
- Primo, A.; Marino, T.; Corma, A.; Molinari, R.; Garcia, H. Efficient Visible-Light Photocatalytic Water Splitting by Minute Amounts of Gold Supported on Nanoparticulate CeO2 Obtained by a Biopolymer Templating Method. J. Am. Chem. Soc. 2012, 133, 6930–6933. [Google Scholar]
- Awazu, K.; Fujimaki, M.; Rockstuhl, C.; Tominaga, J.; Murakami, H.; Ohki, Y.; Yoshida, N.; Watanabe, T. A plasmonic photocatalyst consisting of sliver nanoparticles embedded in titanium dioxide. J. Am. Chem. Soc. 2008, 130, 1676–1680. [Google Scholar]
- Kowalska, E.; Abe, R.; Ohtani, B. Visible light-induced photocatalytic reaction of gold-modified titanium(IV) oxide particles: Action spectrum analysis. Chem. Commun. 2009. [Google Scholar] [CrossRef]
- Silva, C.G.; Juarez, R.; Marino, T.; Molinari, R.; Garcia, H. Influence of Excitation Wavelength (UV or Visible Light) on the Photocatalytic Activity of Titania Containing Gold Nanoparticles for the Generation of Hydrogen or Oxygen from Water. J. Am. Chem. Soc. 2011, 133, 595–602. [Google Scholar] [CrossRef]
- Gurunathan, K.; Maruthamuthu, P.; Sastri, M.V.C. Photocatalytic hydrogen production by dye-sensitized Pt/SnO2 AND Pt/SnO2/RuO2 in aqueous methyl viologen solution. Int. J. Hyd. Energy 1997, 22, 57–62. [Google Scholar] [CrossRef]
- Ni, M.; Leung, M.K.H.; Leung, D.Y.C.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sustain. Energy Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Jing, D.; Guo, L. WS2 sensitized mesoporous TiO2 for efficient photocatalytic hydrogen production from water under visible light irradiation. Catal. Commun. 2007, 8, 795–799. [Google Scholar] [CrossRef]
- Sauve, G.; Cass, M.E.; Coia, G.; Doig, S.J.; Lauermann, I.; Pomykal, K.E.; Lewis, N.S. Dye sensitization of nanocrystalline titanium dioxide with osmium and ruthenium polypyridyl complexes. J. Phys. Chem. B 2000, 104, 6821–6836. [Google Scholar] [CrossRef]
- Chen, Y.S.; Li, C.; Zeng, Z.H.; Wang, W.B.; Wang, X.S.; Zhang, B.W. Efficient electron injection due to a special adsorbing group’s combination of carboxyl and hydroxyl: dye-sensitized solar cells based on new hemicyanine dyes. J. Mater. Chem. 2005, 15, 1654–1661. [Google Scholar] [CrossRef]
- Chen, C.P.; Qi, X.Y.; Zhou, B.M. Photosensitization of colloidal TiO2 with a cyanine dye. J. Photochem. Photobiol. 1997, 109, 155–158. [Google Scholar] [CrossRef]
- Chu, W.; Chan, K.H.; Jafvert, C.T.; Chan, Y.S. Removal of phenylurea herbicide monuron via riboflavin-mediated photo sensitization. Chemosphere 2007, 69, 177–183. [Google Scholar] [CrossRef]
- Choi, W.; Termin, A.; Hoffmann, M.R. The Role of Metal Ion Dopants in Quantum-Sized TiO2: Correlation between Photoreactivity and Charge Carrier Recombination Dynamics. J. Phys. Chem. 1994, 98, 13669–13679. [Google Scholar] [CrossRef]
- Litter, M.I. Heterogeneous photocatalysis: Transition metal ions in photocatalytic systems. Appl. Cataly. B 1999, 23, 89–114. [Google Scholar] [CrossRef]
- Asahi, R.; Morikawa, T.; Ohwaki, T.; Aoki, K.; Taga, Y. Visible-light photocatalysis in nitrogen-doped titanium oxides. Science 2001, 293, 269–271. [Google Scholar] [CrossRef]
- Abe, R.; Takata, T.; Sugihara, H.; Domen, K. Photocatalytic overall water splitting under visible light by TaON and WO3 with an IO3−/I− shuttle redox mediator. Chem. Commun. 2005, 38, 29–3831. [Google Scholar]
- Kobayakawa, K.; Murakami, Y.; Sato, Y. Visible-light active N-doped TiO2 prepared by heating of titanium hydroxide and urea. J. Photochem. Photobiol. A 2005, 170, 177–179. [Google Scholar] [CrossRef]
- Mrowetz, M.; Balcerski, W.; Colussi, A.J.; Hoffmann, M.R. Oxidative power of nitrogen-doped TiO2 photocatalysts under visible illumination. J. Phys. Chem. B 2004, 108, 17269–17273. [Google Scholar] [CrossRef]
- Torres, G.R.; Lindgren, T.; Lu, J.; Granqvist, C.-G.; Lindquist, S.-E. Photoelectrochemical Study of Nitrogen-Doped Titanium Dioxide for Water Oxidation. J. Phys. Chem. B 2004, 108, 5995–6003. [Google Scholar]
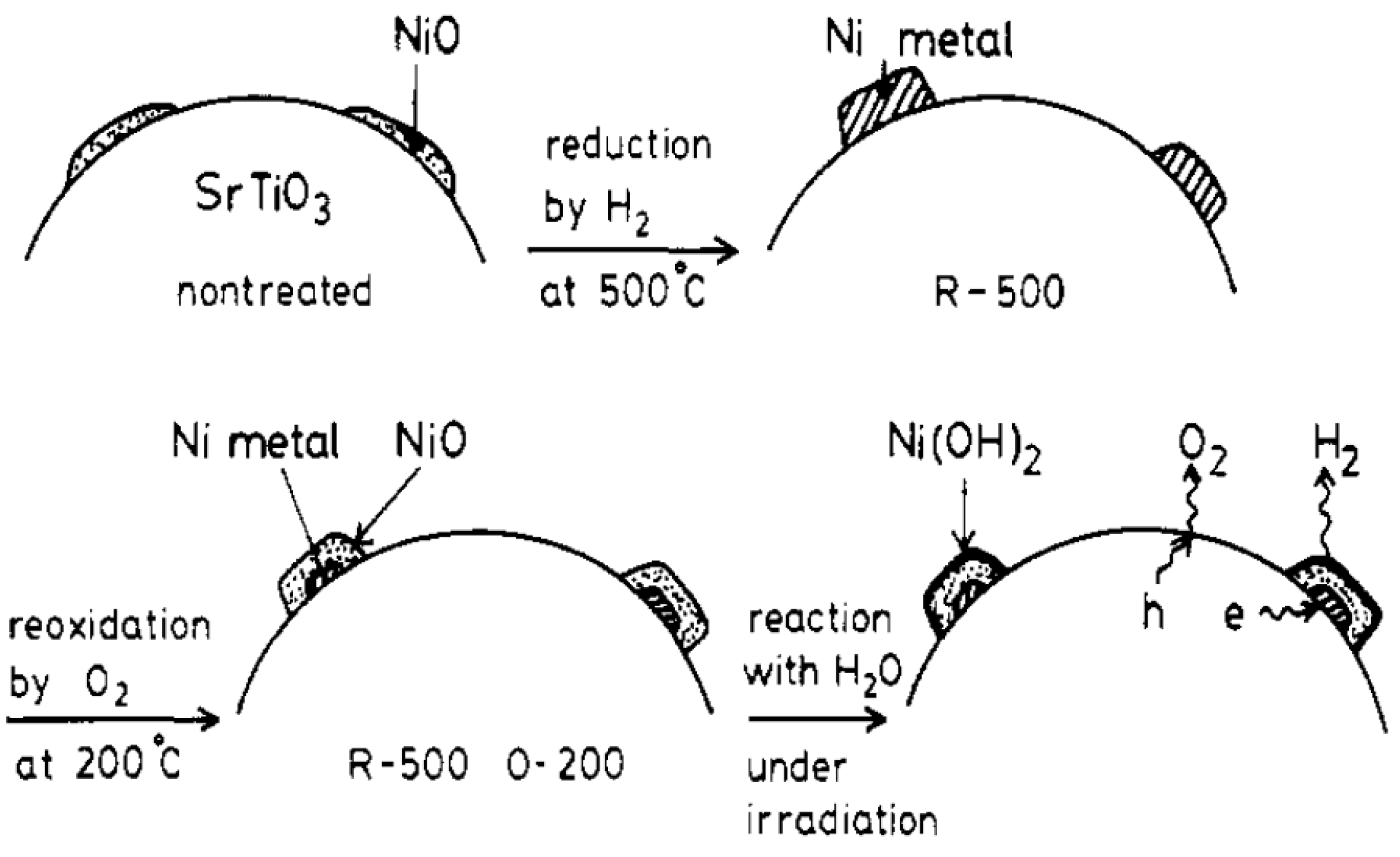
- Domen, K.; Kudo, A.; Onishi, T.; Kosugi, N.; Kuroda, H. Photocatalytic decomposition of water into hydrogen and oxygen over nickel(II) oxide-strontium titanate (SrTiO3) powder. 1. Structure of the catalysts. J. Phys. Chem. 1986, 90, 292–295. [Google Scholar]
- Kudo, A.; Kato, H.; Nakagawa, S. Water Splitting into H2 and O2 on New Sr2M2O7 (M = Nb and Ta) Photocatalysts with Layered Perovskite Structures: Factors Affecting the Photocatalytic Activity. J. Phys. Chem. B 1999, 104, 571–575. [Google Scholar]
- Kato, H.; Kudo, A. New tantalate photocatalysts for water decomposition into H2 and O2. Chem. Phys. Lett. 1998, 295, 487–492. [Google Scholar] [CrossRef]
- Sato, J.; Saito, N.; Nishiyama, H.; Inoue, Y. Photocatalytic Activity for Water Decomposition of Indates with Octahedrally Coordinated d10 Configuration. I. Influences of Preparation Conditions on Activity. J. Phys. Chem. B 2003, 107, 7965–7969. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H.; Domen, K. Photocatalytic water splitting on nickel intercalated A(4)Ta(x)Nb(6−x)O(17) (A = K, Rb). Cataly. Today 1996, 28, 175–182. [Google Scholar]
- Yamada, S.; Nosaka, A.Y.; Nosaka, Y. Fabrication of US photoelectrodes coated with titania nanosheets for water splitting with visible light. J. Electroanal. Chem. 2005, 585, 105–112. [Google Scholar] [CrossRef]
- Gopidas, K.R.; Bohorquez, M.; Kamat, P.V. Photophysical and photochemical aspects of coupled semiconductors: Charge-transfer processes in colloidal cadmium sulfide-titania and cadmium sulfide-silver(I) iodide systems. J. Phy. Chem. 1990, 94, 6435–6440. [Google Scholar] [CrossRef]
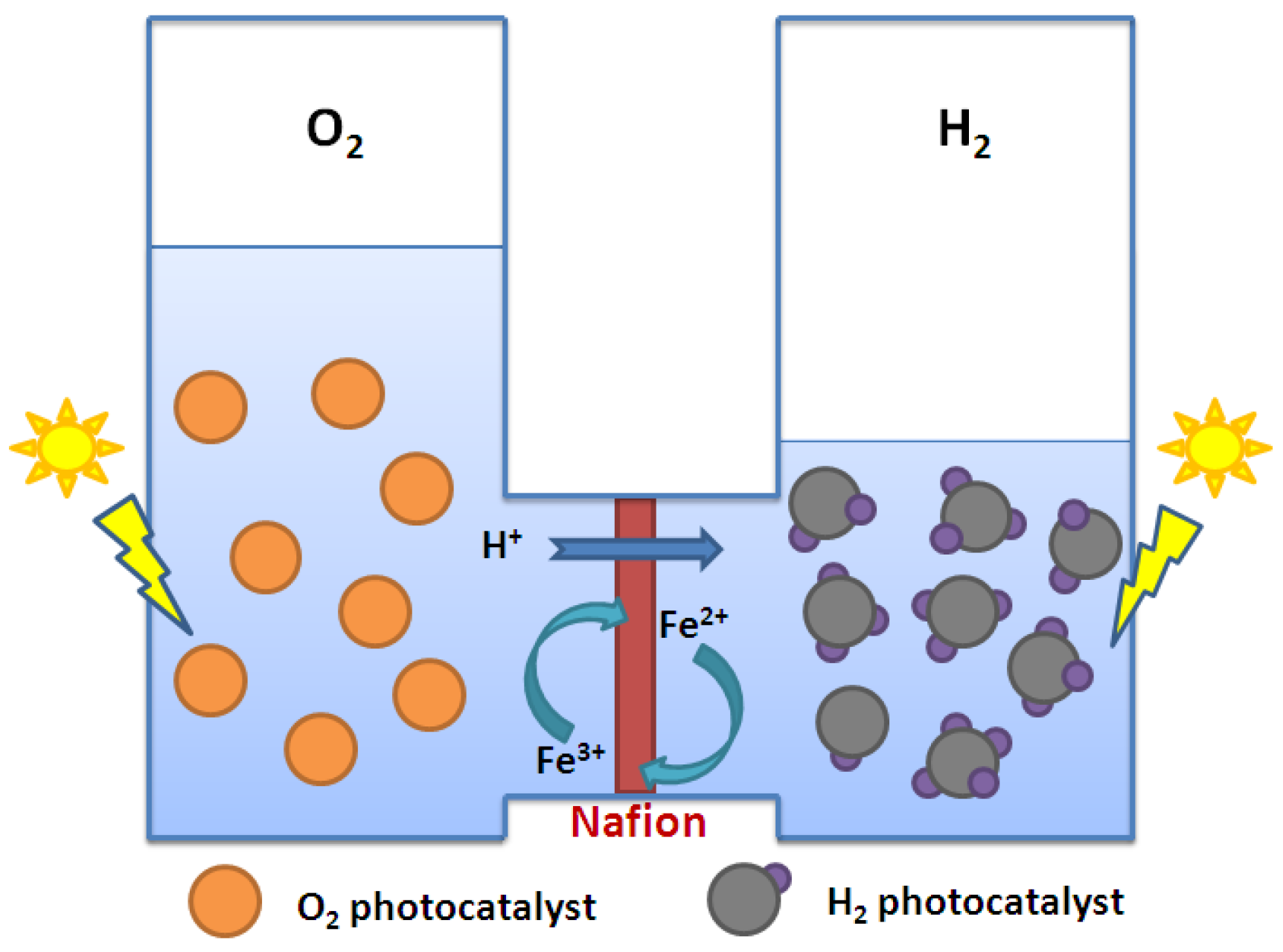
- Sasaki, Y.; Iwase, A.; Kato, H.; Kudo, A. The effect of co-catalyst for Z-scheme photocatalysis systems with an Fe3+/Fe2+ electron mediator on overall water splitting under visible light irradiation. J. Catal. 2008, 259, 133–137. [Google Scholar]
- Fujihara, K.; Ohno, T.; Matsumura, M. Splitting of water by electrochemical combination of two photocatalytic reactions on TiO2 particles. J. Chem. Soci. Faraday Trans. 1998, 94, 3705–3709. [Google Scholar] [CrossRef]
- Sayama, K.; Mukasa, K.; Abe, R.; Abe, Y.; Arakawa, H. A new photocatalytic water splitting system under visible light irradiation mimicking a Z-scheme mechanism in photosynthesis. J. Photochem. Photobiol. 2002, 148, 71–77. [Google Scholar] [CrossRef]
- Higashi, M.; Abe, R.; Takata, T.; Domen, K. Photocatalytic Overall Water Splitting under Visible Light Using ATaO2N (A = Ca, Sr, Ba) and WO3 in a IO3−/I− Shuttle Redox Mediated System. Chem.Mater. 2009, 21, 1543–1549. [Google Scholar] [CrossRef]
- Kato, H.; Hori, M.; Konta, R.; Shimodaira, Y.; Kudo, A. Construction of Z-scheme Type Heterogeneous Photocatalysis Systems for Water Splitting into H2 and O2 under Visible Light Irradiation. Chem. Lett. 2004, 33, 1348–1349. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Wrighton, M.S.; Ellis, A.B.; Wolczanski, P.T.; Morse, D.L.; Abrahamson, H.B.; Ginley, D.S. Strontium-titanate photoelectrodes-efficient photoassisted electrolysis of water at zero applied potential. J. Am. Chem. Soc. 1976, 98, 2774–2779. [Google Scholar] [CrossRef]
- Ki, H.Y.; Tae, H.K. Photoeffects in undoped and doped SrTiO3 ceramic electrodes. J. Solid State Chem. 1987, 67, 359–363. [Google Scholar] [CrossRef]
- Matsuoka, M.; Kitano, M.; Takeuchi, M.; Tsujimaru, K.; Anpo, M.; Thomas, J.M. Photocatalysis for new energy production: Recent advances in photocatalytic water splitting reactions for hydrogen production. Catal. Today 2007, 122, 51–61. [Google Scholar]
- Huang, C.W.; Liao, C.H.; Wu, J.C.S.; Liu, Y.C.; Chang, C.L.; Wu, C.H.; Anpo, M.; Matsuoka, M.; Takeuchi, M. Hydrogen generation from photocatalytic water splitting over TiO2 thin film prepared by electron beam-induced deposition. Int. J. Hydrog. Energy 2010, 35, 12005–12010. [Google Scholar]
- Liao, C.-H.; Huang, C.-W.; Wu, J.C.S. Novel dual-layer photoelectrode prepared by RF magnetron sputtering for photocatalytic water splitting. Int. J. Hydrog. Energy 2012, 37, 11632–11639. [Google Scholar] [CrossRef]
- Liao, Y.-T.; Huang, C.-W.; Liao, C.-H.; Wu, J.C.S.; Wu, K.C.W. Synthesis of mesoporous titania thin films (MTTFs) with two different structures as photocatalysts for generating hydrogen from water splitting. Appl. Energy 2012. [Google Scholar] [CrossRef]
- Matsuoka, M.; Kitano, M.; Fukumoto, S.; Iyatani, K.; Takeuchi, M.; Anpo, M. The effect of the hydrothermal treatment with aqueous NaOH solution on the photocatalytic and photoelectrochemical properties of visible light-responsive TiO2 thin films. Catal. Today 2008, 132, 159–164. [Google Scholar]
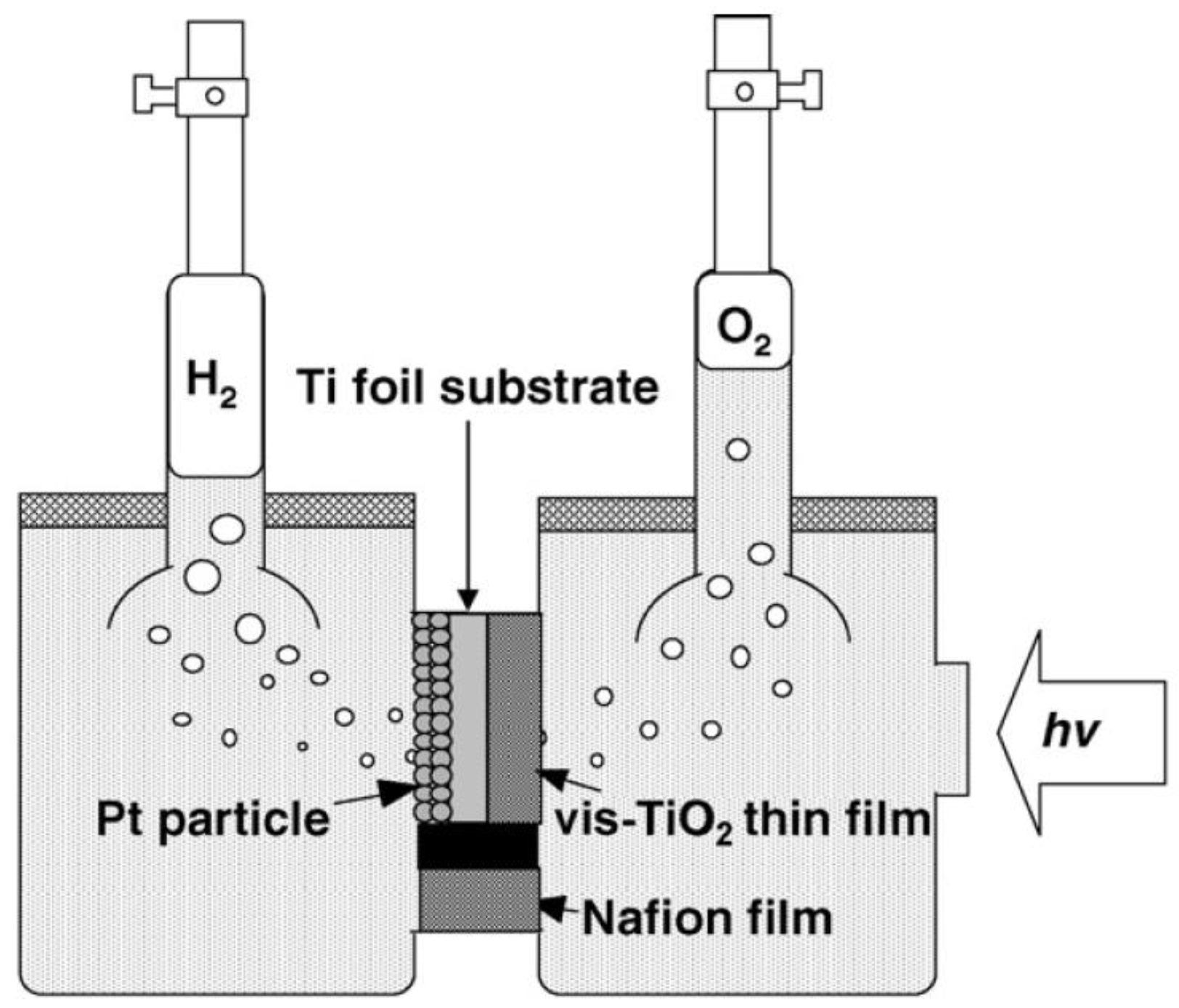
- Lo, C.-C.; Huang, C.-W.; Liao, C.-H.; Wu, J.C.S. Novel twin reactor for separate evolution of hydrogen and oxygen in photocatalytic water splitting. Int. J. Hydrog. Energy 2010, 35, 1523–1529. [Google Scholar]
- Yu, S.C.; Huang, C.W.; Liao, C.H.; Wu, J.C.S.; Chang, S.T.; Chen, K.H. A novel membrane reactor for separating hydrogen and oxygen in photocatalytic water splitting. J. Membr. Sci. 2011, 382, 291–299. [Google Scholar] [CrossRef]
- Prasad, G.; Chandra Babu, K.S.; Srivastava, O.N. Structural and photoelectrochemical studies of In2O3-TiO2 and WSe2 photoelectrodes for photoelectrochemical production of hydrogen. Int. J. Hydrog. Energy 1989, 14, 537–544. [Google Scholar] [CrossRef]
- Licht, S.; Wang, B.; Mukerji, S.; Soga, T.; Umeno, M.; Tributsch, H. Over 18% solar energy conversion to generation of hydrogen fuel; theory and experiment for efficient solar water splitting. Int. J. Hydrog. Energy 2001, 26, 653–659. [Google Scholar] [CrossRef]
- Khaselev, O.; Turner, J.A. A monolithic photovoltaic-photoelectrochemical device for hydrogen production via water splitting. Science 1998, 280, 425–427. [Google Scholar] [CrossRef]
- Peharz, G.; Dimroth, F.; Wittstadt, U. Solar hydrogen production by water splitting with a conversion efficiency of 18%. Int. J. Hydrog. Energy 2007, 32, 3248–3252. [Google Scholar] [CrossRef]
- Khaselev, O.; Bansal, A.; Turner, J.A. High-efficiency integrated multijunction photovoltaic/electrolysis systems for hydrogen production. Int. J. Hydrog. Energy 2001, 26, 127–132. [Google Scholar] [CrossRef]
- Miller, E.L.; Rocheleau, R.E.; Khan, S. A hybrid multijunction photoelectrode for hydrogen production fabricated with amorphous silicon/germanium and iron oxide thin films. Int. J. Hydrog. Energy 2004, 29, 907–914. [Google Scholar] [CrossRef]
- Licht, S.; Wang, B.; Mukerji, S.; Soga, T.; Umeno, M.; Tributsch, H. Efficient solar water splitting, exemplified by RuO2-catalyzed AlGaAs/Si photoelectrolysis. J. Phys. Chem. B 2000, 104, 8920–8924. [Google Scholar] [CrossRef]
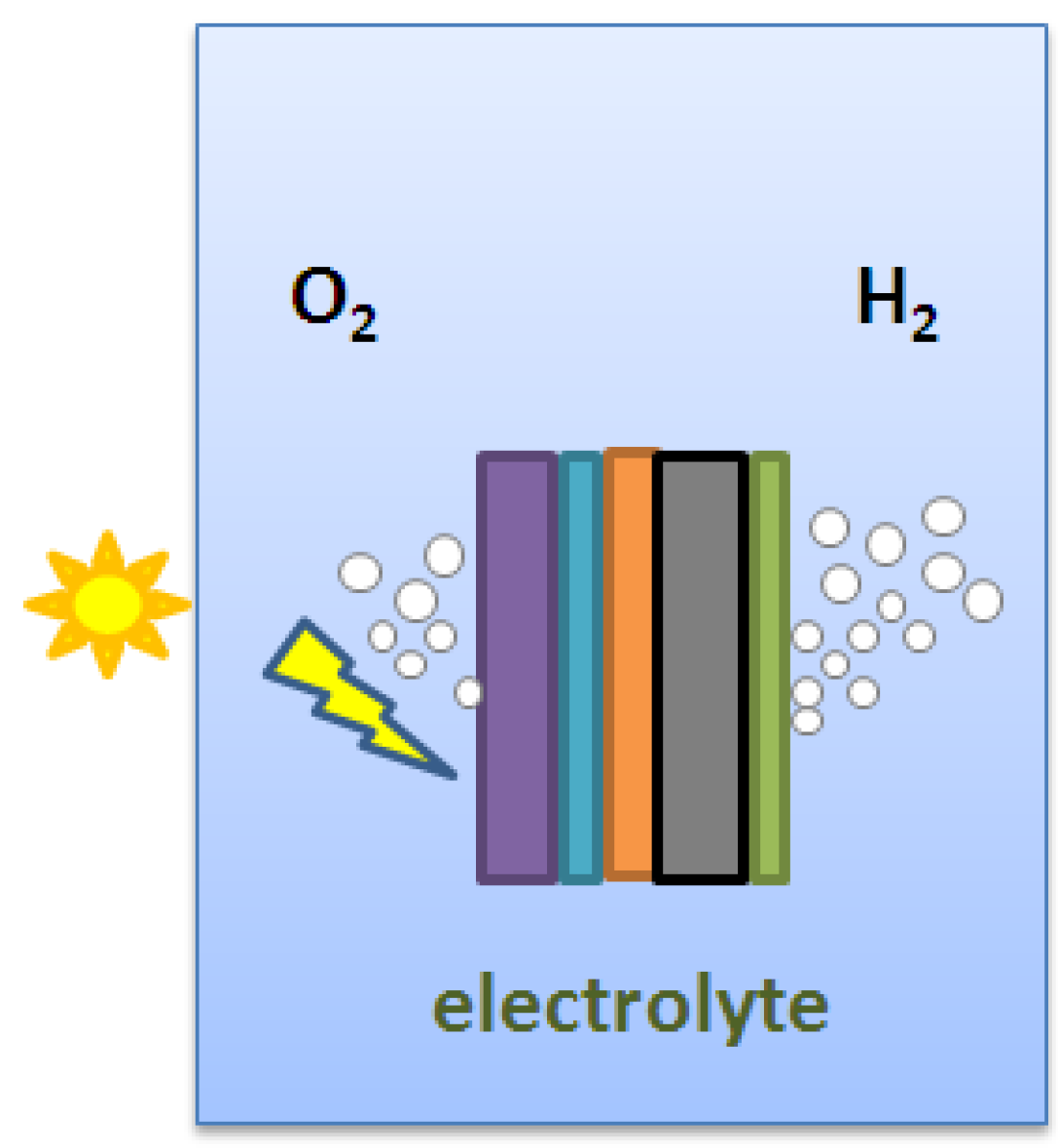
- Huang, C.-W.; Liao, C.-H.; Wu, C.-H.; Wu, J.C.S. Photocatalytic water splitting to produce hydrogen using multi-junction solar cell with different deposited thin films. Sol. Energy Mater. Sol. Cells 2012. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liao, C.-H.; Huang, C.-W.; Wu, J.C.S. Hydrogen Production from Semiconductor-based Photocatalysis via Water Splitting. Catalysts 2012, 2, 490-516. https://doi.org/10.3390/catal2040490
Liao C-H, Huang C-W, Wu JCS. Hydrogen Production from Semiconductor-based Photocatalysis via Water Splitting. Catalysts. 2012; 2(4):490-516. https://doi.org/10.3390/catal2040490
Chicago/Turabian StyleLiao, Chi-Hung, Chao-Wei Huang, and Jeffrey C. S. Wu. 2012. "Hydrogen Production from Semiconductor-based Photocatalysis via Water Splitting" Catalysts 2, no. 4: 490-516. https://doi.org/10.3390/catal2040490