Catalyst-Controlled Site-Selectivity Switching in Pd-Catalyzed Cross-Coupling of Dihaloarenes

Abstract
: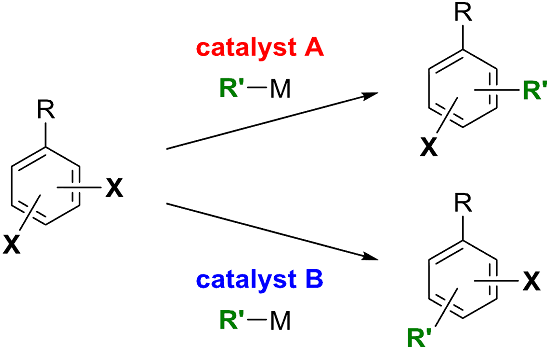
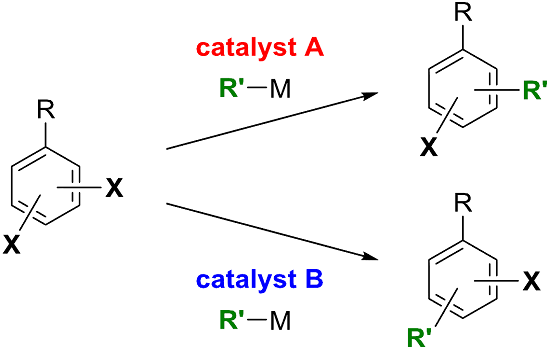
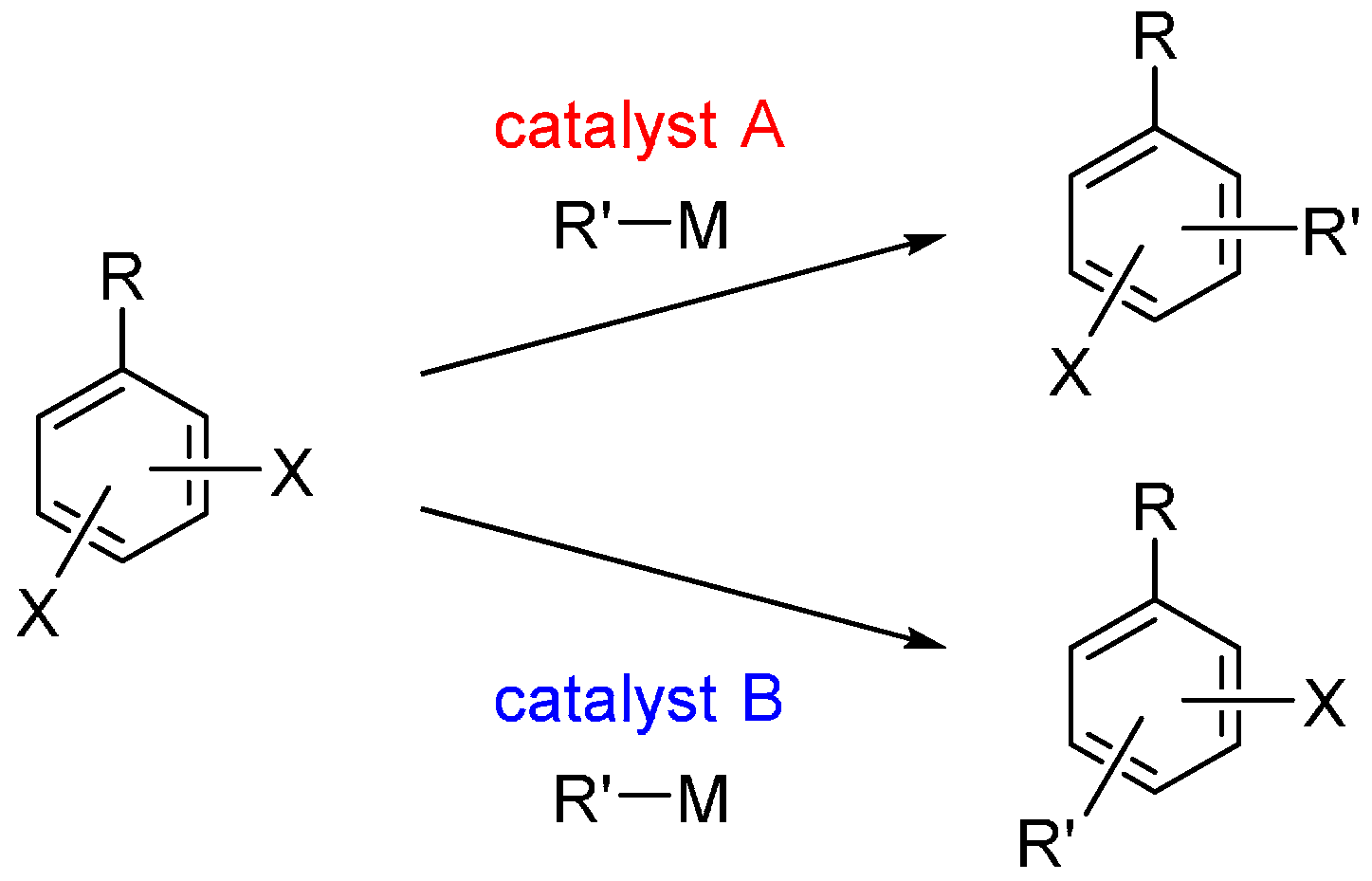
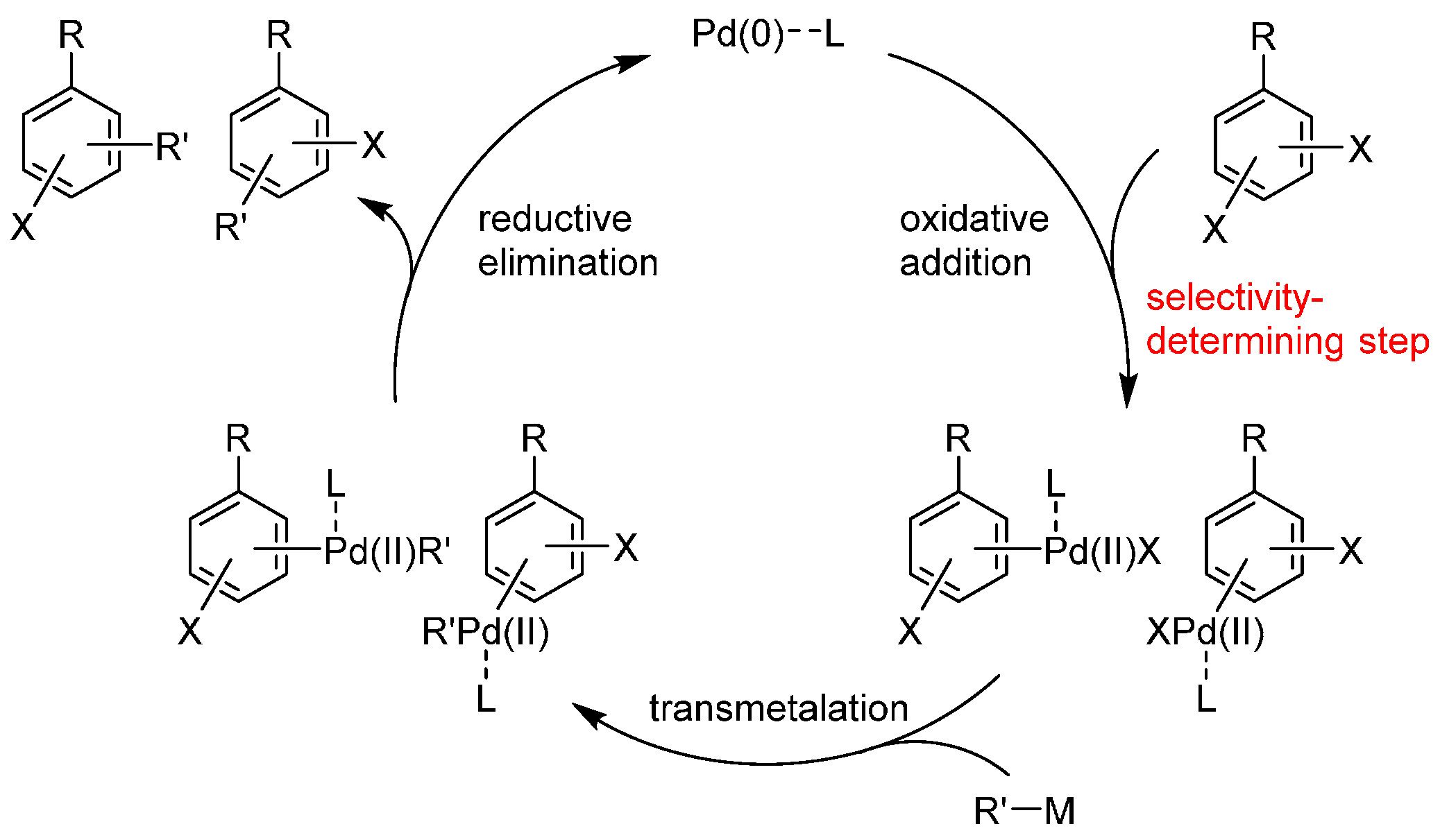
1. Introduction


2. Dihalobenzenes
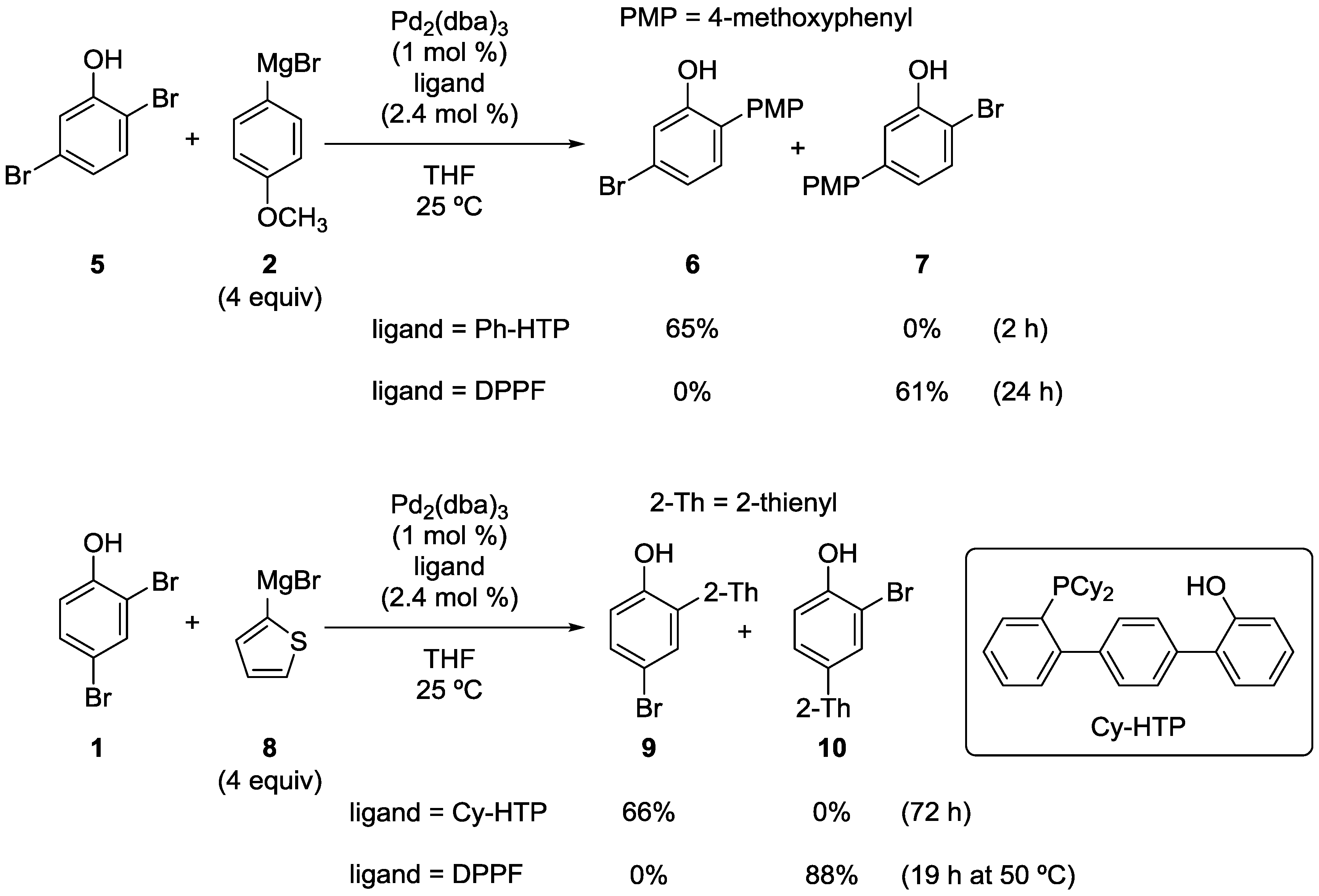
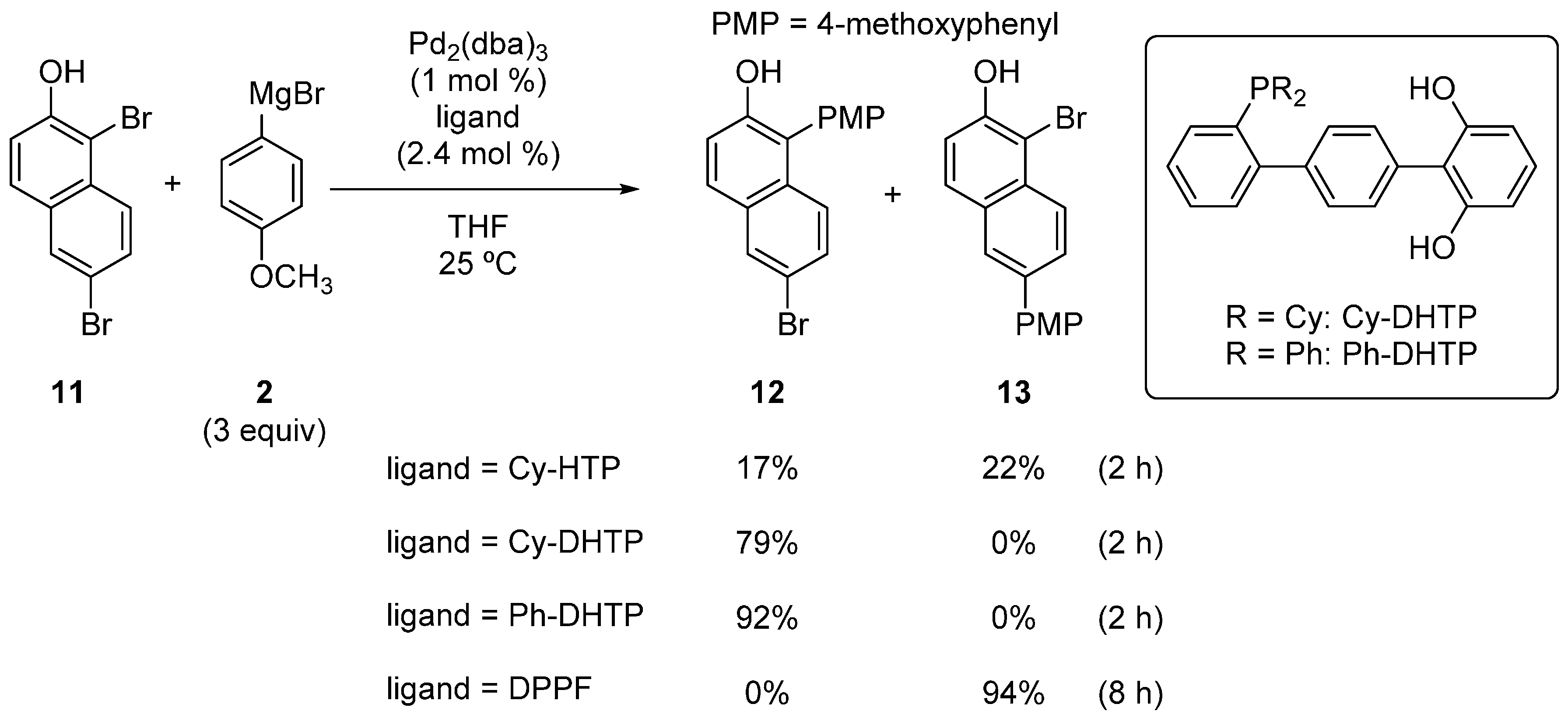
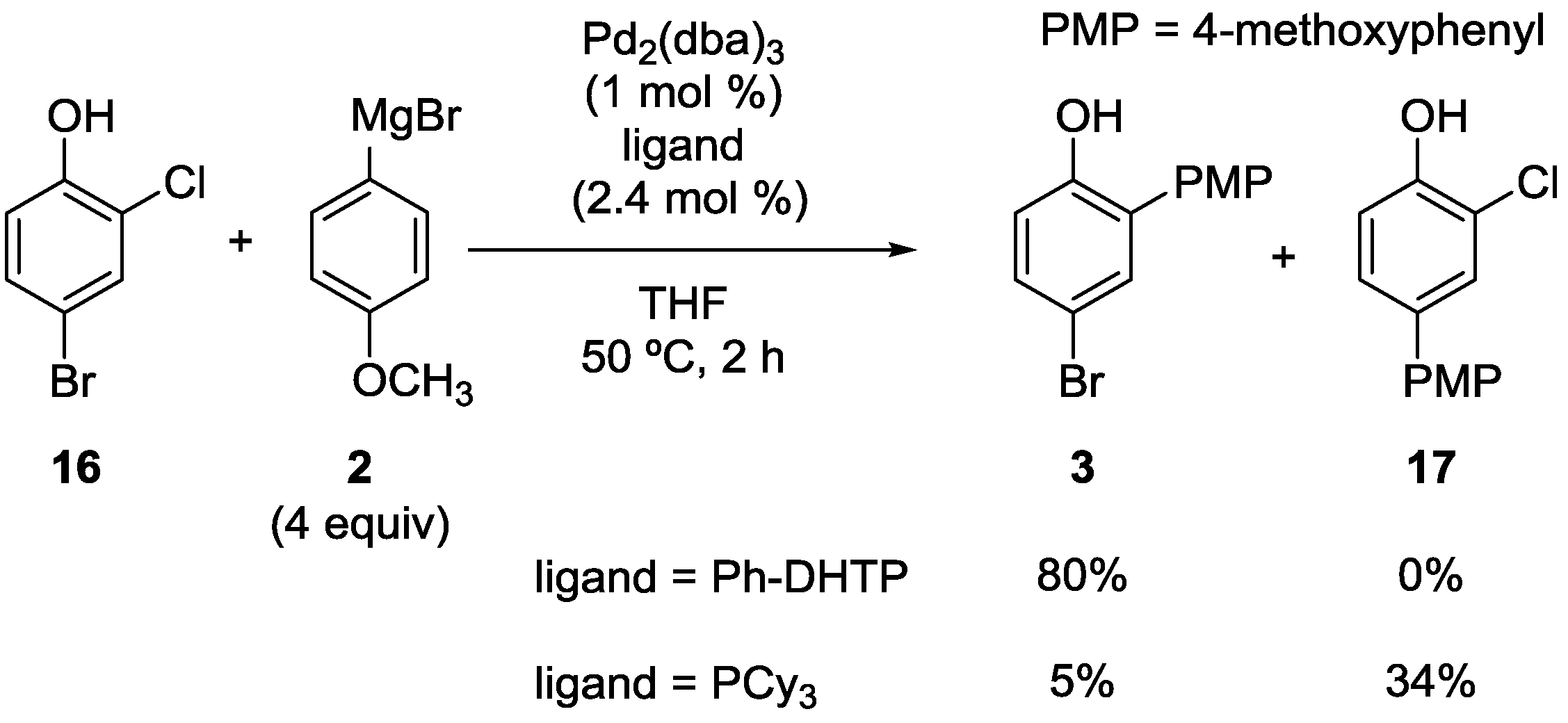
2.1. Phenol Derivatives



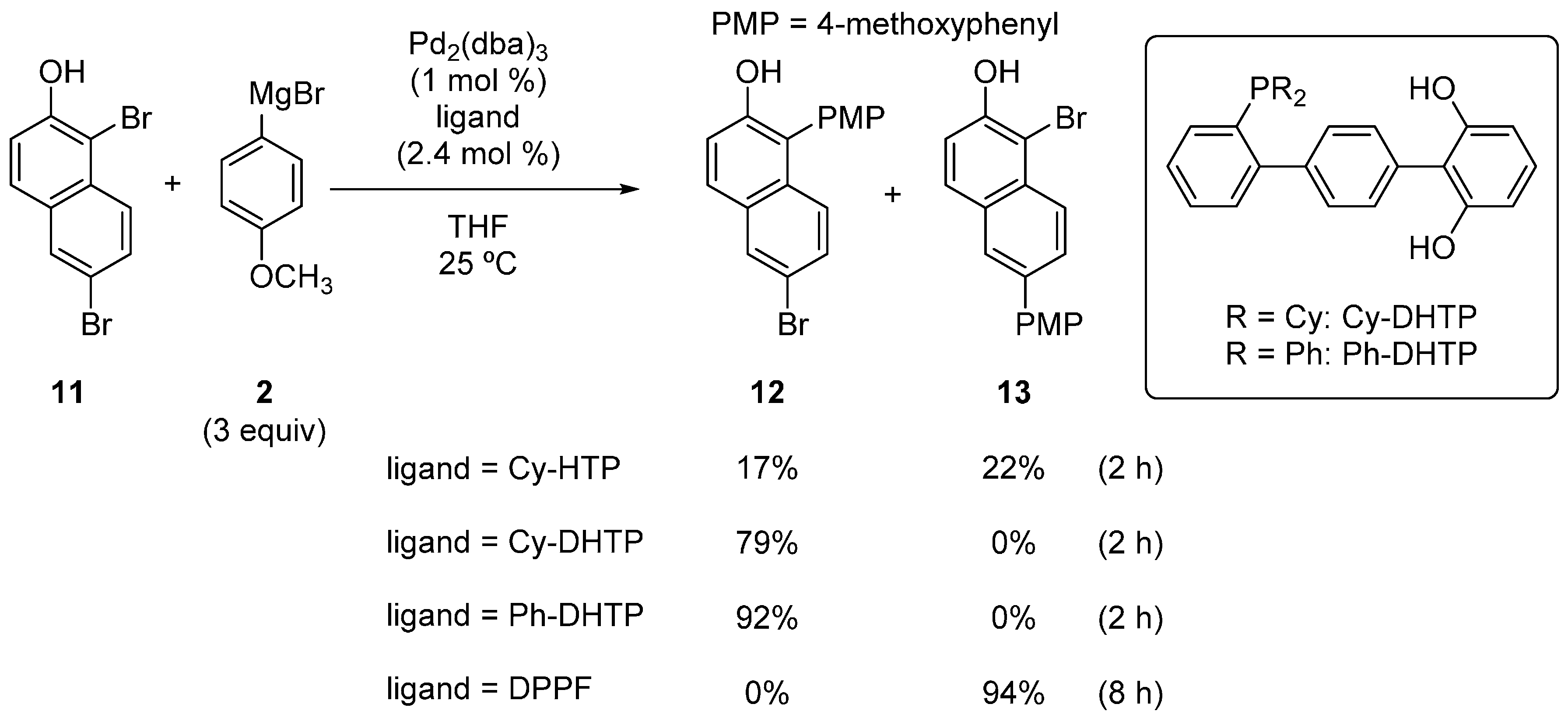


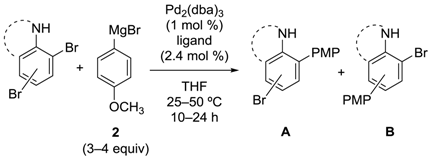
2.2. Aniline Derivatives
2.3. Benzoic Acid Derivatives

| Entry | Substrate | Ligand | Yield (%) | |
|---|---|---|---|---|
| A | B | |||
| 1 |  18 18 | Ph-DHTP | 90 | 0 |
| 2 | DPPF | 9 | 15 | |
| 3 |  19 19 | Ph-HTP | 63 | 0 |
| 4 | DPPF | 15 | 32 | |
| 5 |  20 20 | Ph-DHTP | 70 | 0 |
| 6 | DPPF | 29 | 21 | |
| 7 | 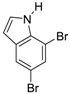 21 21 | Ph-DHTP | 81 | 0 |
| 8 | DPPF | 0 | 37 | |

3. Dihaloheteroarenes
3.1. Pyrone Derivatives

| Entry | CuI (equiv) | Conditions | Yield (%) | |
|---|---|---|---|---|
| 28 | 29 | |||
| 1 | 0 | toluene, 100 °C, 0.5 h | 81 | trace |
| 2 | 0.1 | toluene, 100 °C, 0.5 h | 94 | trace |
| 3 | 1.0 | toluene, 100 °C, 2 h | 71 | 6 |
| 4 | 0 | DMF, 50 °C, 4 days | 41 | 2 |
| 5 | 0.1 | DMF, 50 °C, 5 h | 34 | 20 |
| 6 | 0.5 | DMF, 50 °C, 2.5 h | trace | 64 |
| 7 | 1.0 | DMF, 50 °C, 2 h | trace | 75 |
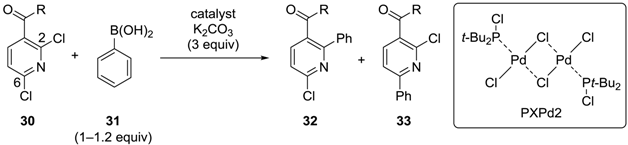
3.2. Pyridine Derivatives
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
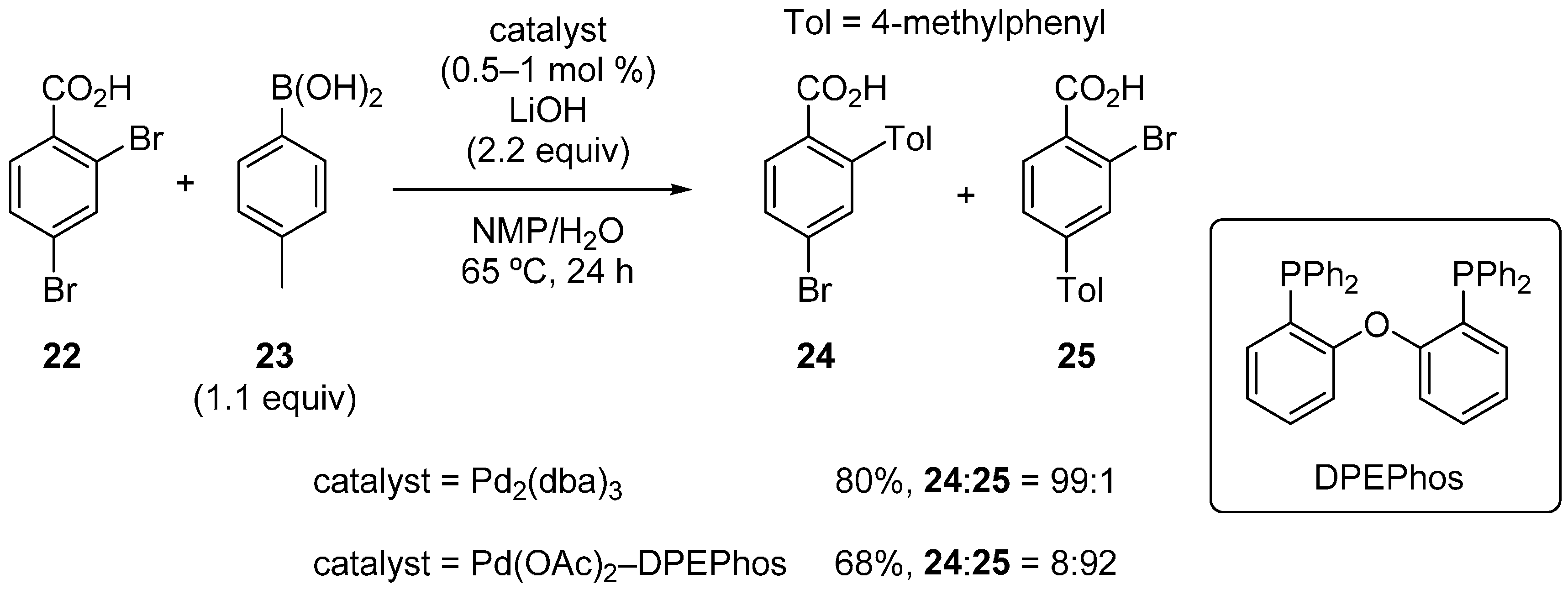{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R | Catalyst (mol %) | Conditions | 32:33 |
|---|---|---|---|---|
| 1 | OMe | Pd(PPh3)4 (5) | THF, reflux, 16 h | 1:5 * |
| 2 | OMe | PXPd2 (1) | MeOH, reflux, 30 min | 2.5:1 * |
| 3 | NHCH2CH2OPh | PXPd2 (1) | MeOH, 55 °C, 1 h | 9:1 ** |

| Entry | Ligand | Base | Solvent | Yield (%) | |
|---|---|---|---|---|---|
| 35 | 36 | ||||
| 1 | DPPF | Cs2CO3 | dioxane | 0 | 90 |
| 2 | Q-Phos | KF | toluene | 36 | 15 |
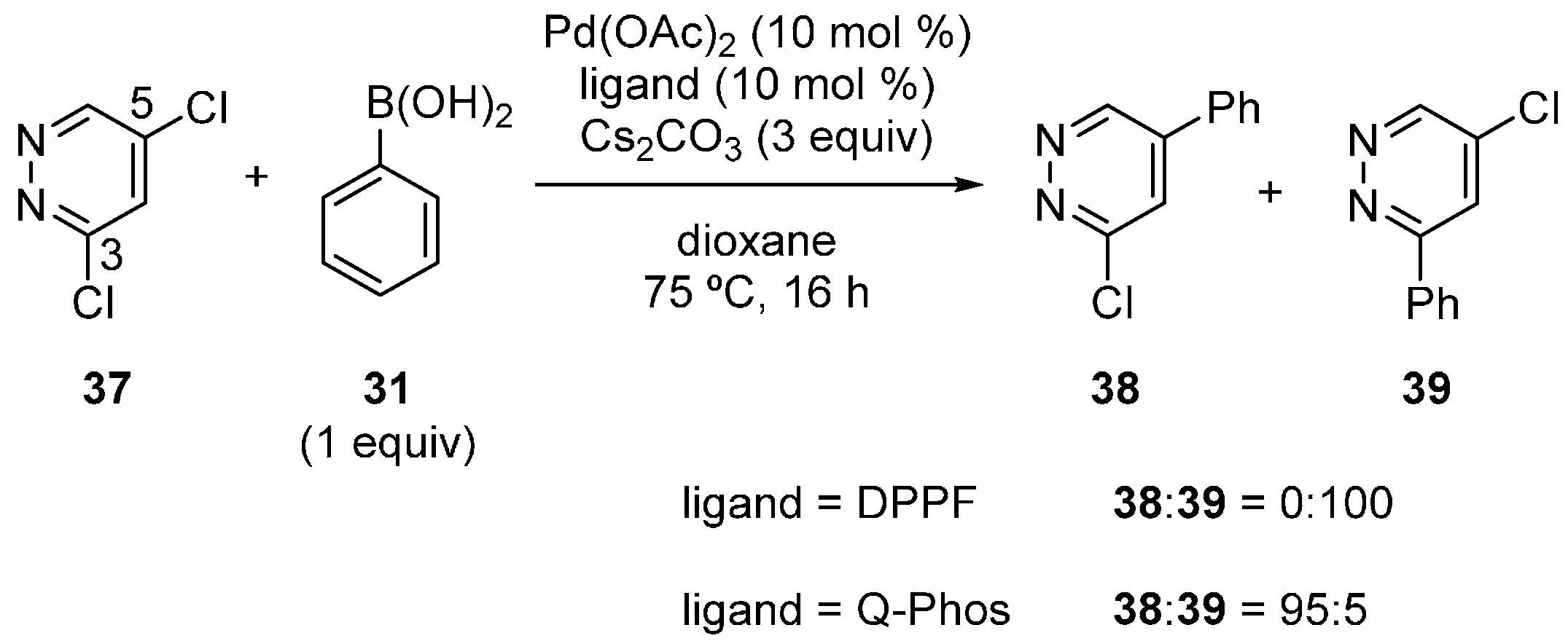
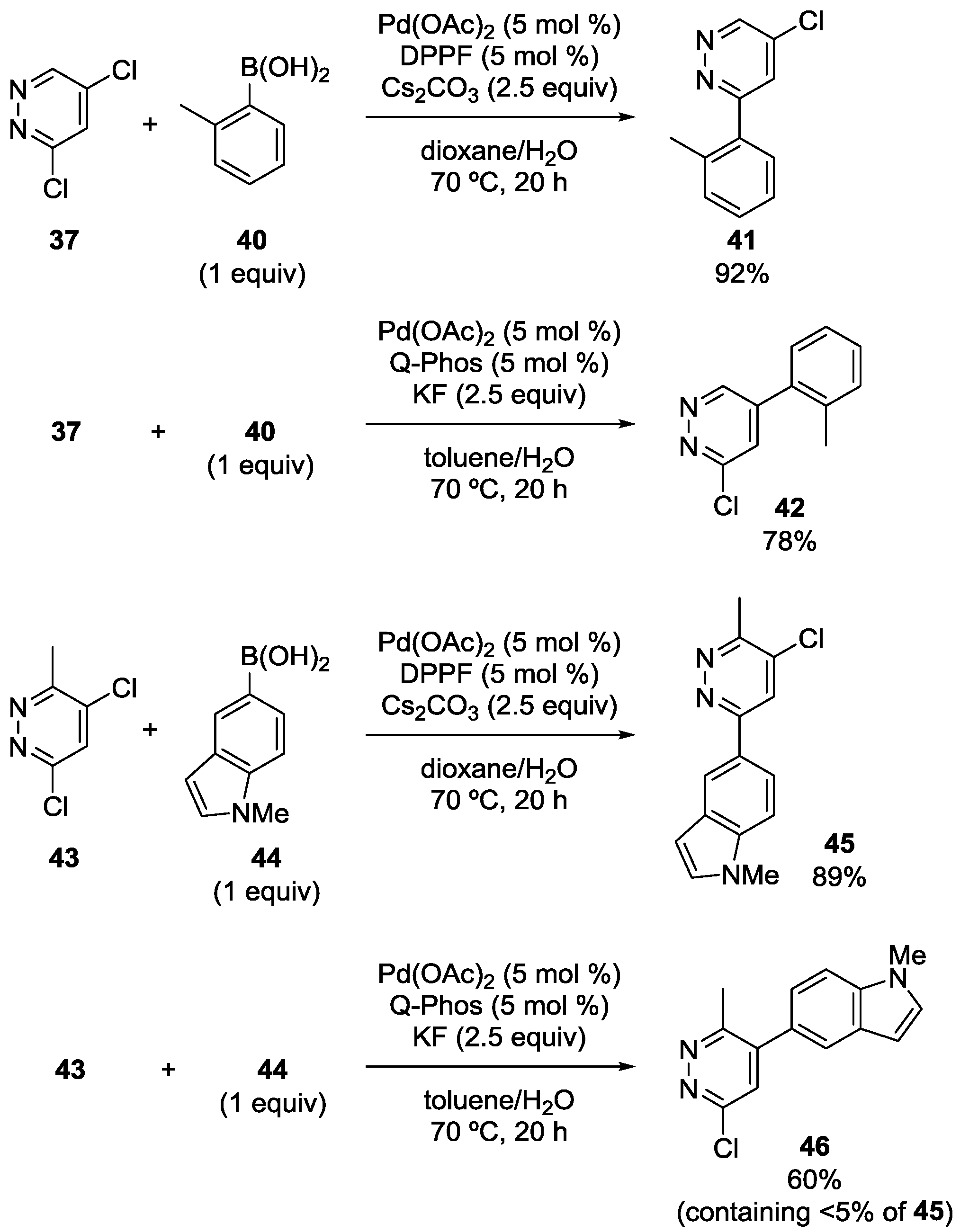
3.3. Pyridazine Derivatives


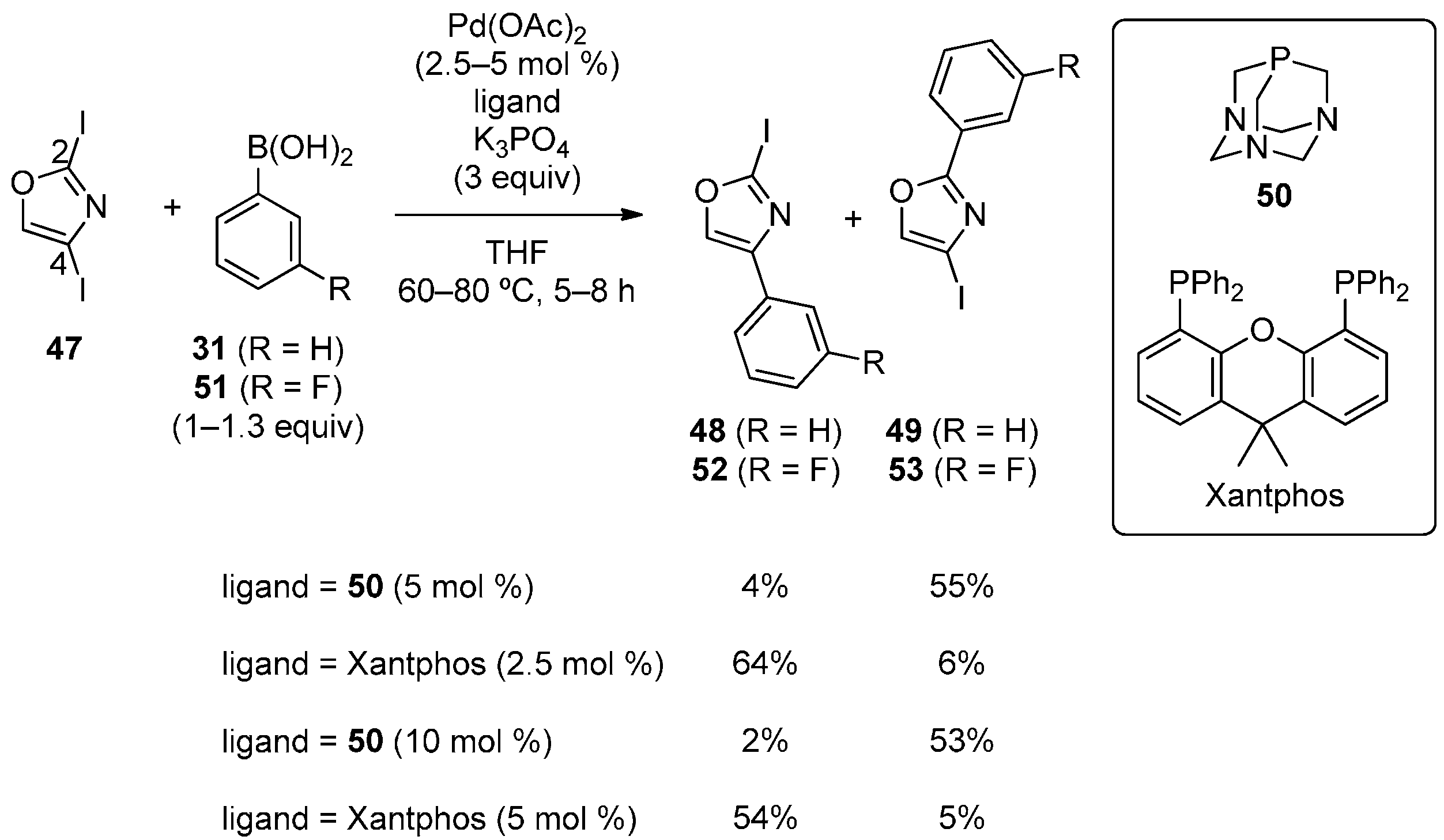
3.4. Oxazole Derivatives

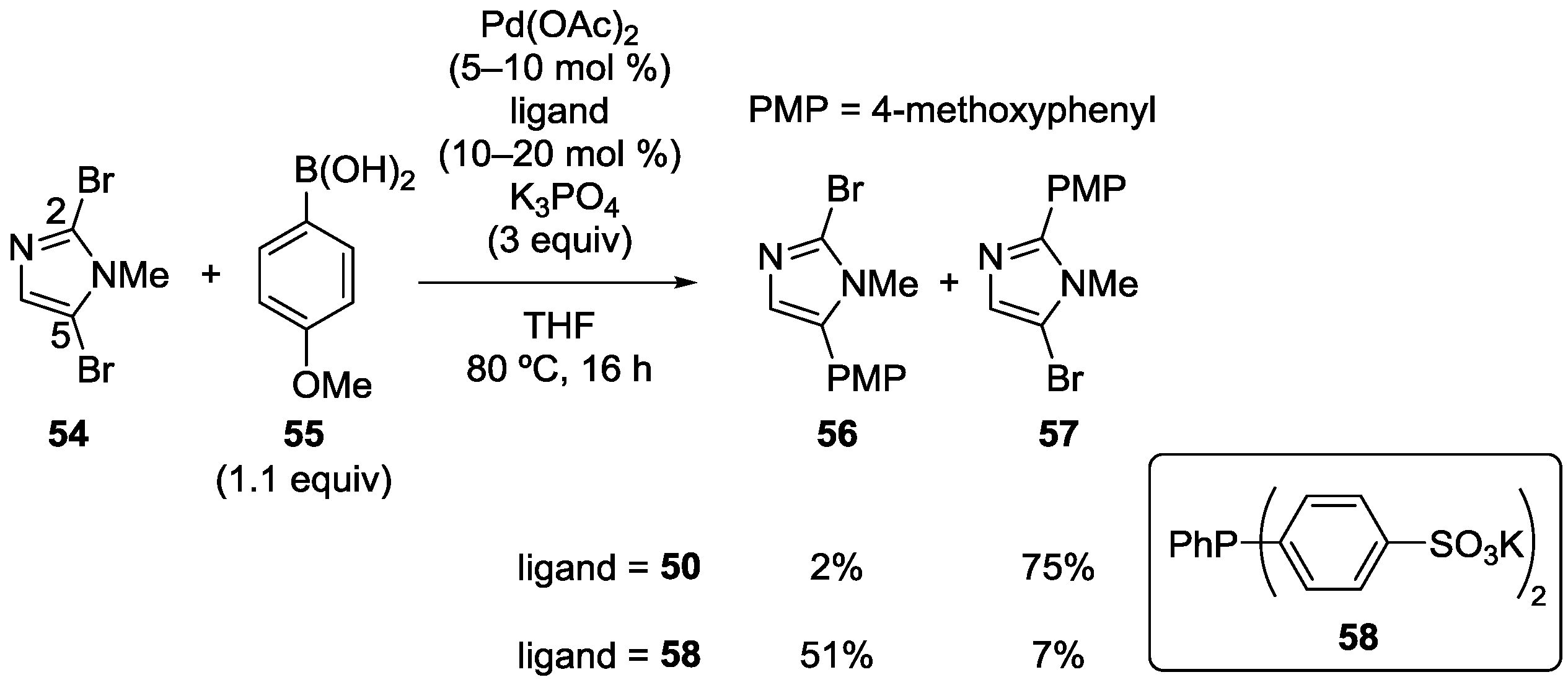
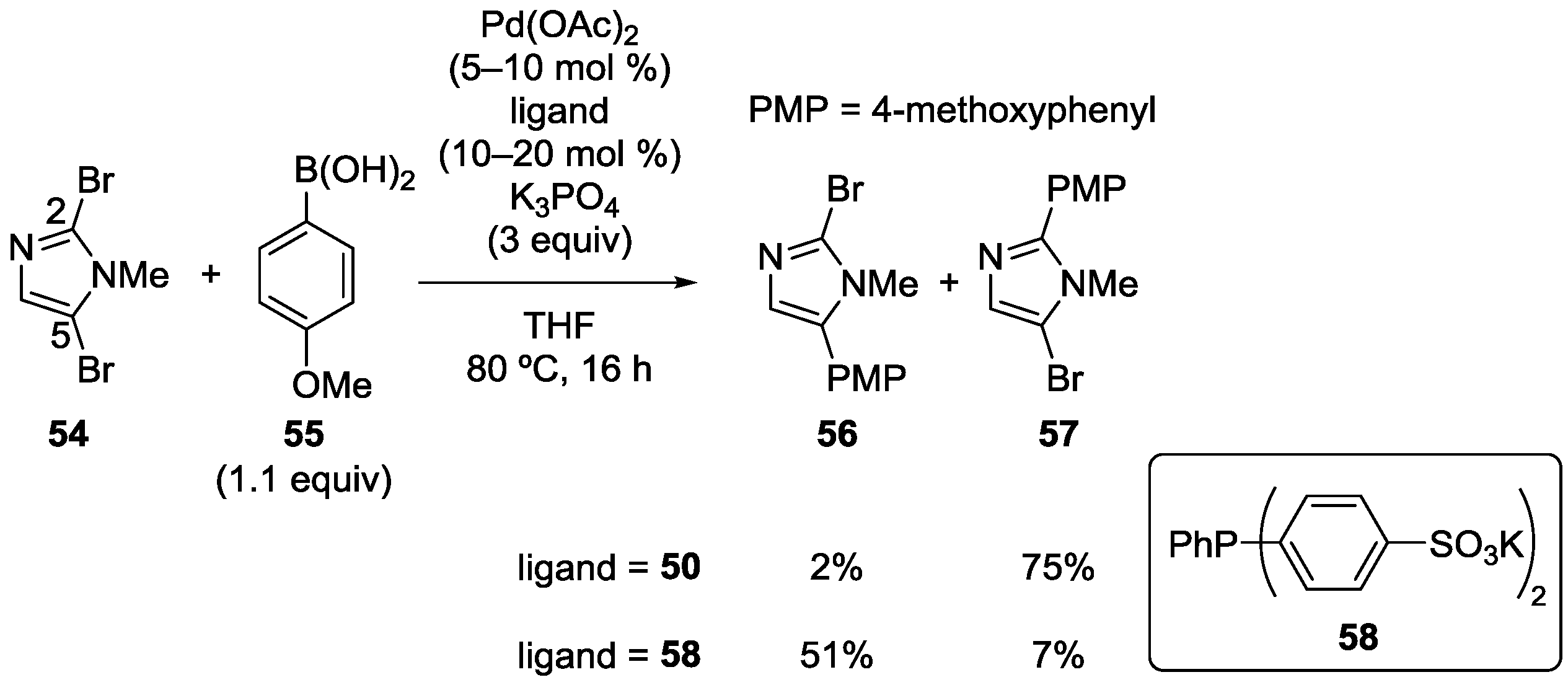
3.5. Imidazole Derivatives
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Negishi, E.-i. Handbook of Organopalladium. Chemistry for Organic Synthesis; John Wiley & Sons Ltd.: New York, NY, USA, 2002. [Google Scholar]
- De Meijere, A.; Diederich, F. Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Tsuji, J. Palladium Reagents and Catalysts; John Wiley & Sons: West Sussex, UK, 2004. [Google Scholar]
- Unrau, C.M.; Campbell, M.G.; Snieckus, V. Directed ortho metalation-suzuki cross coupling connections. convenient regiospecific routes to functionalized m- and p-Teraryls and m-Quinquearyls. Tetrahedron Lett. 1992, 33, 2773–2776. [Google Scholar]
- Goodby, J.W.; Hird, M.; Lewis, R.A.; Toyne, K.J. 5-Bromo-2-iodopyrimidine: A novel, useful intermediate in selective palladium-catalysed cross-coupling reactions for efficient convergent syntheses. Chem. Commun. 1996, 2719–2720. [Google Scholar] [CrossRef]
- Heinrich, A.C.J.; Thiedemann, B.; Gates, P.J.; Staubitz, A. Dual selectivity: Electrophile and nucleophile selective cross-coupling reactions on a single aromatic substrate. Org. Lett. 2013, 15, 4666–4669. [Google Scholar] [CrossRef] [PubMed]
- Montoir, D.; Tonnerre, A.; Duflos, M.; Bazin, M.-A. Differential functionalization of 1,6-Naphthyridin-2(1H)-ones through sequential one-pot suzuki–miyaura cross-couplings. Eur. J. Org. Chem. 2014, 1487–1495. [Google Scholar] [CrossRef]
- Kamikawa, T.; Hayashi, T. Control of reactive site in palladium-catalyzed grignard cross-coupling of arenes containing both bromide and triflate. Tetrahedron Lett. 1997, 38, 7087–7090. [Google Scholar] [CrossRef]
- Littke, A.F.; Dai, C.; Fu, G.C. Versatile catalysts for the suzuki cross-coupling of arylboronic acids with aryl and vinyl halides and triflates under mild conditions. J. Am. Chem. Soc. 2000, 122, 4020–4028. [Google Scholar] [CrossRef]
- Ashcroft, C.P.; Fussell, S.J.; Wilford, K. Catalyst controlled regioselective suzuki cross-coupling of 2-(4-bromophenyl)-5-chloropyrazine. Tetrahedron Lett. 2013, 54, 4529–4532. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Seko, T.; Nemoto, H. New mmethod for the synthesis of boron-10 containing nucleoside derivatives for neutron-capture therapy via palladium-catalyzed reaction. J. Org. Chem. 1989, 54, 4734–4736. [Google Scholar] [CrossRef]
- Schröter, S.; Stock, C.; Bach, T. Regioselective cross-coupling reactions of multiple halogenated nitrogen-, oxygen-, and sulfur-containing heterocycles. Tetrahedron 2005, 61, 2245–2267. [Google Scholar]
- Fairlamb, I.J.S. Regioselective (site-selective) functionalization of unsaturated halogenated nitrogen, oxygen and sulfur heterocycles by pd-catalysed cross-couplings and direct arylation processes. Chem. Soc. Rev. 2007, 36, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-R.; Manabe, K. Transition-Metal-Catalyzed site-selective cross-coupling of di- and polyhalogenated compounds. Synthesis 2009, 1405–1427. [Google Scholar]
- Hassan, Z.; Patonay, T.; Langer, P. Regioselective suzuki–miyaura reactions of aromatic bis-triflates: Electronic versus steric effects. Synlett 2013, 24, 412–423. [Google Scholar] [CrossRef]
- Singh, R.; Just, G. Rates and regioselectivities of the palladium-catalyzed ethynylation of substituted bromo- and dibromobenzenes. J. Org. Chem. 1989, 54, 4453–4457. [Google Scholar] [CrossRef]
- Handy, S.T.; Zhang, Y. A simple guide for predicting regioselectivity in the coupling of polyhaloheteroaromatics. Chem. Commun. 2006, 299–301. [Google Scholar]
- Legault, C.Y.; Garcia, Y.; Merlic, C.A.; Houk, K.N. Origin of regioselectivity in palladium-catalyzed cross-coupling reactions of polyhalogenated heterocycles. J. Am. Chem. Soc. 2007, 129, 12664–12665. [Google Scholar] [CrossRef] [PubMed]
- Garcia, Y.; Schoenebeck, F.; Legault, C.Y.; Merlic, C.A.; Houk, K.N. Theoretical bond dissociation energies of halo-heterocycles: Trends and relationships to regioselectivity in palladium-catalyzed cross-coupling reactions. J. Am. Chem. Soc. 2009, 131, 6632–6639. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.G.; Lautens, M. The role of reversible oxidative addition in selective palladium(0)-catalyzed intramolecular cross-couplings of polyhalogenated substrates: Synthesis of brominated indoles. J. Am. Chem. Soc. 2010, 132, 11416–11417. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Manabe, K. Ortho-Selective cross coupling of dibromophenols and dibromoanilines with grignard reagents in the presence of palladium catalysts bearing hydroxylated oligoarene-type phosphine. Chem. Lett. 2007, 36, 1304–1305. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon–carbon bond formation by cross-coupling of grignard reagents with organic halides. Catalysis by nickel–phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar]
- Corriu, R.J.P.; Masse, J.P. Activation of grignard reagents by transition-metal complexes. A new and simple synthesis of trans-stilbenes and polyphenyls. J. Chem. Soc. Chem. Commun. 1972, 144a. [Google Scholar]
- Ishikawa, S.; Manabe, K. Oligoarene strategy for catalyst development. hydroxylated oligoarene-type phosphines for palladium-catalyzed cross coupling. Chem. Lett. 2007, 36, 1302–1303. [Google Scholar]
- Ishikawa, S.; Manabe, K. Synthesis of hydroxylated oligoarene-type phosphines by a repetitive two-step method. Tetrahedron 2010, 66, 297–303. [Google Scholar] [CrossRef]
- Ishikawa, S.; Manabe, K. DHTP ligands for the highly ortho-selective, palladium-catalyzed cross-coupling of dihaloarenes with grignard reagents: A conformational approach for catalyst improvement. Angew. Chem. Int. Ed. 2010, 49, 772–775. [Google Scholar] [CrossRef]
- Ishikawa, S.; Manabe, K. Hydroxylated terphenylphosphine ligands for palladium-catalyzed ortho-selective cross-coupling of dibromophenols, dibromoanilines, and their congeners with grignard reagents. Tetrahedron 2011, 67, 10156–10163. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Katsumata, H.; Manabe, K. One-Pot synthesis of substituted benzo[b]furans from mono- and dichlorophenols using palladium catalysts bearing dihydroxyterphenylphosphine. J. Org. Chem. 2013, 78, 9270–9281. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Manabe, K. One-Pot synthesis of 2,4-disubstituted indoles from n-tosyl-2,3-dichloroaniline using palladium-dihydroxyterphenylphosphine catalyst. Org. Lett. 2014, 16, 2386–2389. [Google Scholar] [CrossRef] [PubMed]
- Boymond, L.; Rottländer, M.; Cahiez, G.; Knochel, P. Preparation of highly functionalized grignard reagents by an iodine–magnesium exchange reaction and its application in solid-phase synthesis. Angew. Chem. Int. Ed. 1998, 37, 1701–1703. [Google Scholar] [CrossRef]
- Suzuki, A. Cross-Coupling reactions of organoboranes: An easy way to construct C–C bonds (Nobel Lecture). Angew. Chem. Int. Ed. 2011, 50, 6722–6737. [Google Scholar] [CrossRef]
- Houpis, I.N.; Huang, C.; Nettekoven, U.; Chen, J.G.; Liu, R.; Canters, M. Carboxylate directed cross-coupling reactions in the synthesis of trisubstituted benzoic acids. Org. Lett. 2008, 10, 5601–5604. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, M.; Sasazawa, K.; Shimizu, Y.; Migita, T. Reactions of allyltin compounds III. Allylation of aromatic halides with allyltributyltin in the presence of Tetrakis(triphenylphosphine)palladium(0). Chem. Lett. 1977, 6, 301–302. [Google Scholar]
- Milstein, D.; Stille, J.K. A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. [Google Scholar]
- Kim, W.-S.; Kim, H.-J.; Cho, C.-G. Regioselectivity in the stille coupling reactions of 3,5-dibromo-2-pyrone. J. Am. Chem. Soc. 2003, 125, 14288–14289. [Google Scholar] [PubMed]
- Yang, W.; Wang, Y.; Corte, J.R. Efficient synthesis of 2-Aryl-6-chloronicotinamides via PXPd2-Catalyzed regioselective suzuki coupling. Org. Lett. 2003, 5, 3131–3134. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y. Catalysis using phosphine oxide and phosphine sulfide complexes with Pd and Ni for the synthesis of biaryls and arylamines. WO2002000574A2, 3 January 2002. [Google Scholar]
- Dai, X.; Chen, Y.; Garrell, S.; Liu, H.; Zhang, L.-K.; Palani, A.; Hughes, G.; Nargund, R. Ligand-Dependent site-selective suzuki cross-coupling of 3,5-dichloropyridazines. J. Org. Chem. 2013, 78, 7758–7763. [Google Scholar] [PubMed]
- Shelby, Q.; Kataoka, N.; Mann, G.; Hartwig, J. Unusual in situ ligand modification to generate a catalyst for room temperature aromatic C−O bond formation. J. Am. Chem. Soc. 2000, 122, 10718–10719. [Google Scholar] [CrossRef]
- Strotman, N.A.; Chobanian, H.R.; He, J.; Guo, Y.; Dormer, P.G.; Jones, C.M.; Steves, J.E. Catalyst-controlled regioselective suzuki couplings at both positions of dihaloimidazoles, dihalooxazoles, and dihalothiazoles. J. Org. Chem. 2010, 75, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Manabe, K.; Yamaguchi, M. Catalyst-Controlled Site-Selectivity Switching in Pd-Catalyzed Cross-Coupling of Dihaloarenes. Catalysts 2014, 4, 307-320. https://doi.org/10.3390/catal4030307
Manabe K, Yamaguchi M. Catalyst-Controlled Site-Selectivity Switching in Pd-Catalyzed Cross-Coupling of Dihaloarenes. Catalysts. 2014; 4(3):307-320. https://doi.org/10.3390/catal4030307
Chicago/Turabian StyleManabe, Kei, and Miyuki Yamaguchi. 2014. "Catalyst-Controlled Site-Selectivity Switching in Pd-Catalyzed Cross-Coupling of Dihaloarenes" Catalysts 4, no. 3: 307-320. https://doi.org/10.3390/catal4030307