
Solid-Liquid Phase C-Alkylation of Active Methylene Containing Compounds under Microwave Conditions




Abstract
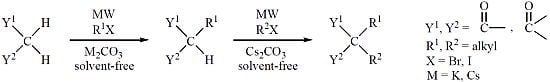
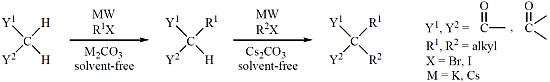
:
1. Introduction
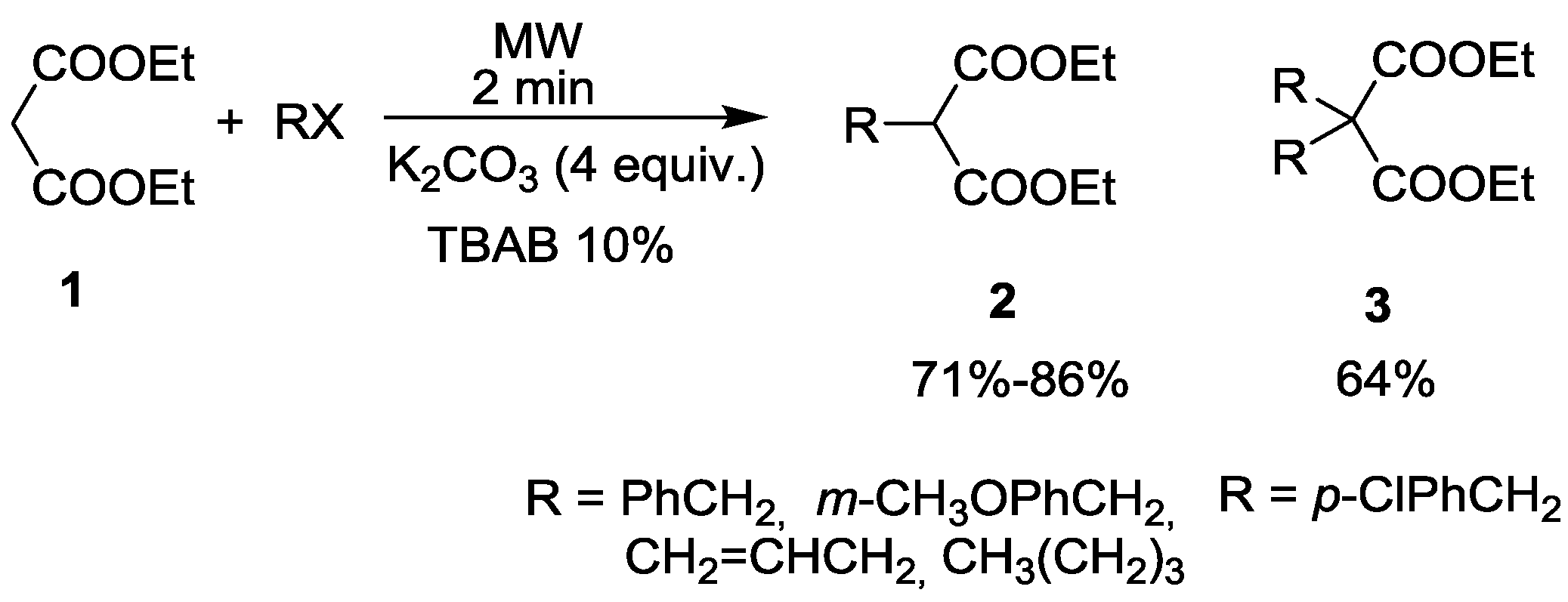
2. Alkylation of Active Methylene Containing Substrates under MW Conditions







{kind=link}
{kind=link}
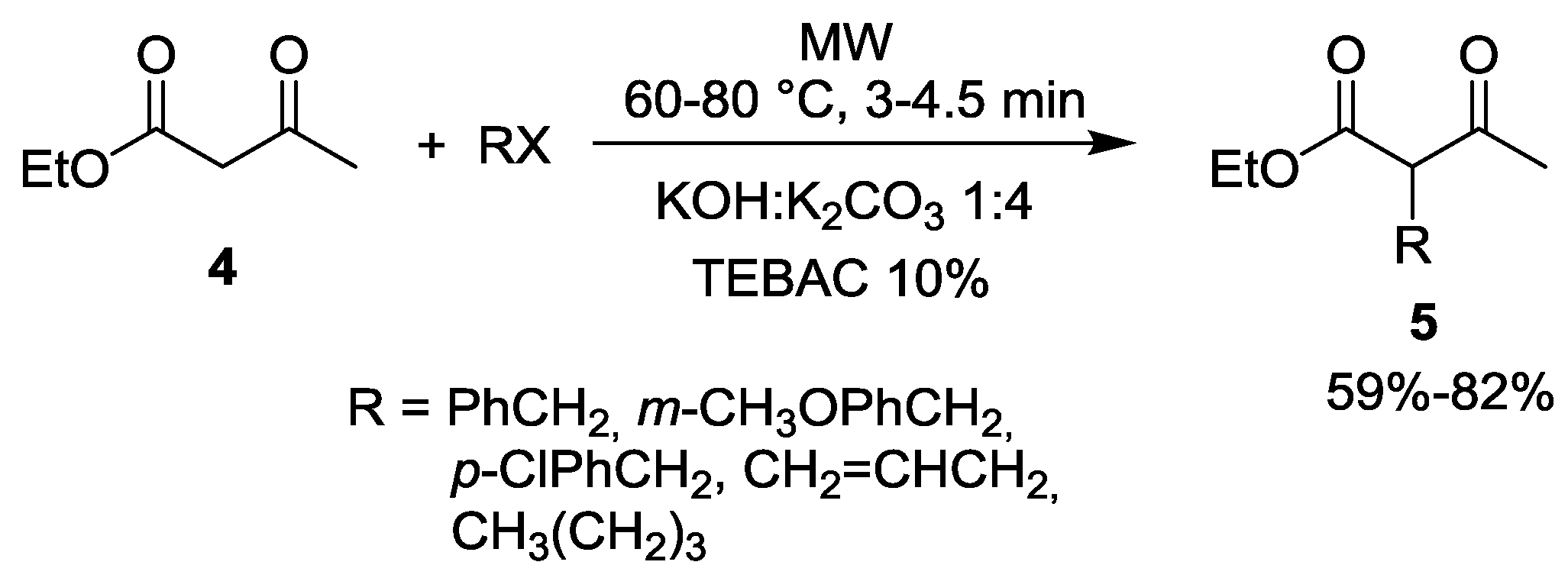{kind=link}
{kind=link}
{kind=link}
{kind=link}
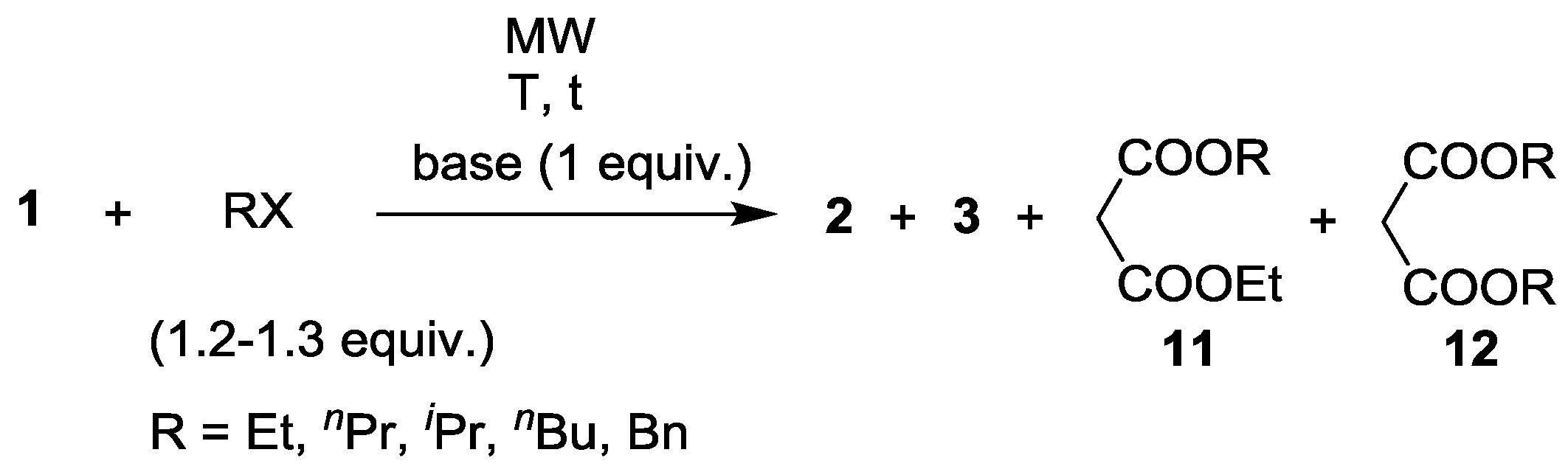{kind=link}
{kind=link}
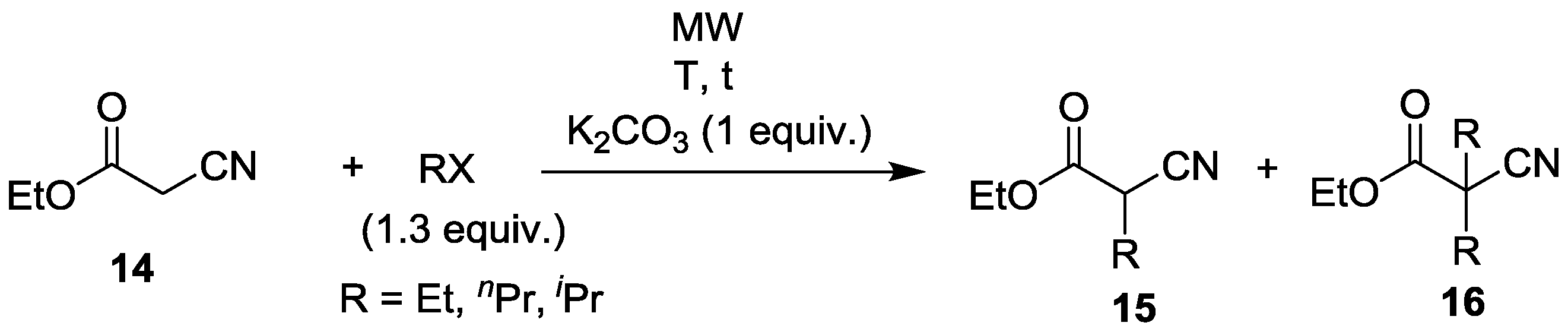{kind=link}
{kind=link}
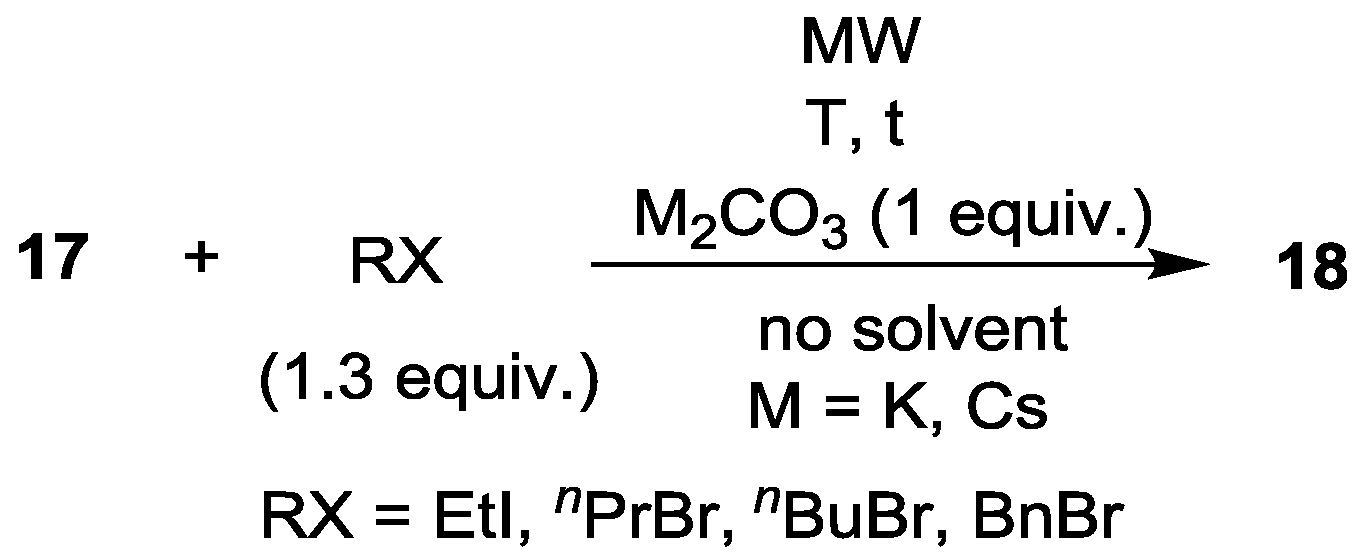{kind=link}
{kind=link}
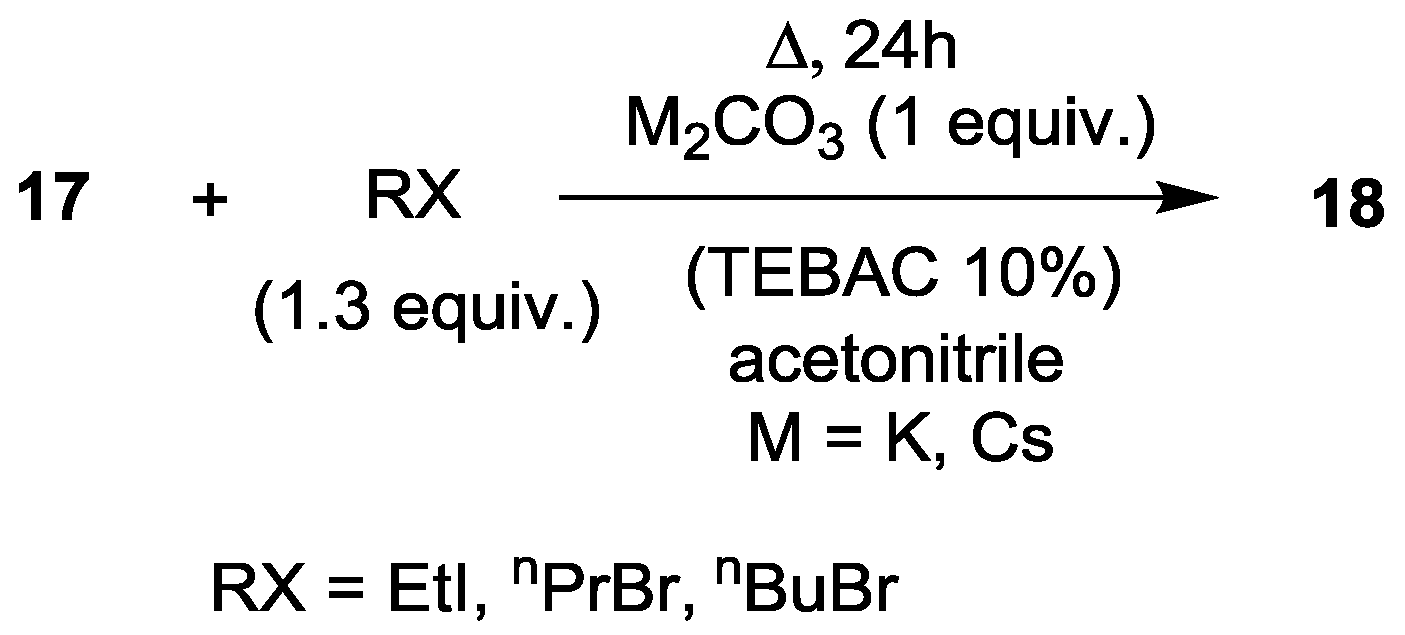{kind=link}
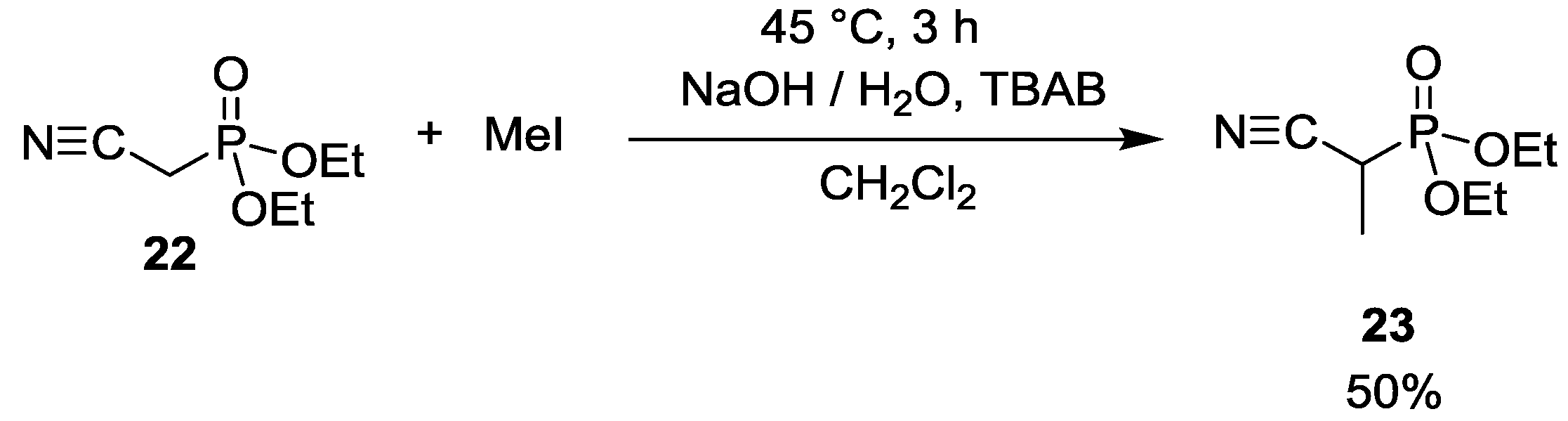{kind=link}
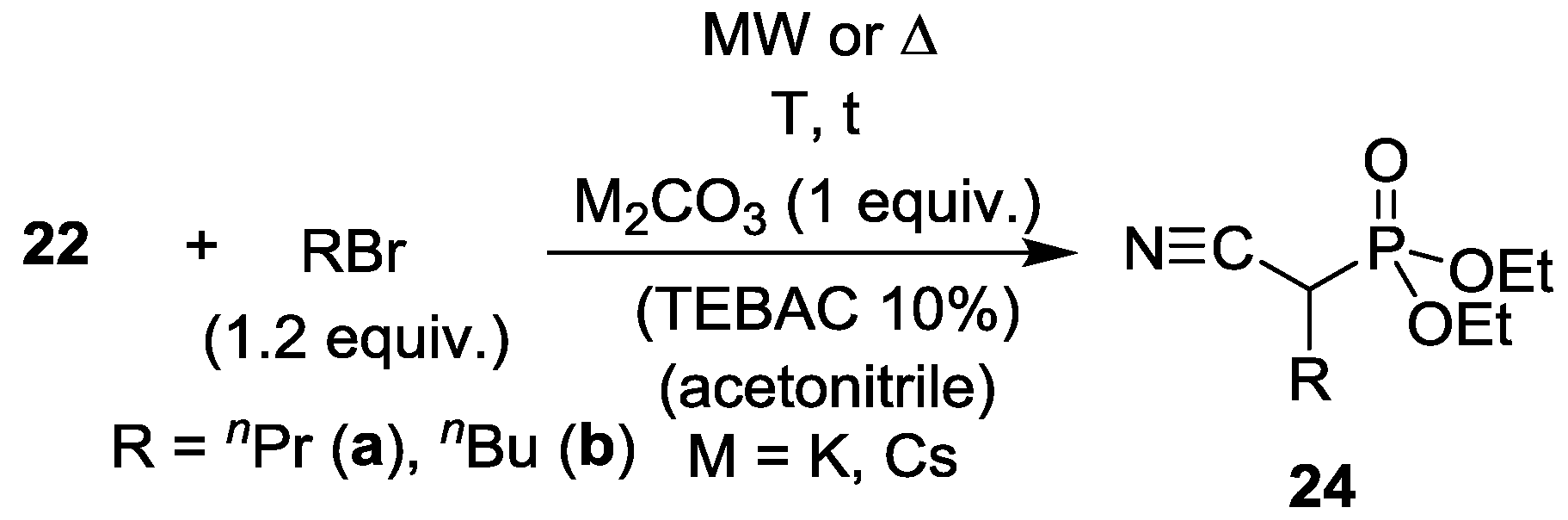{kind=link}
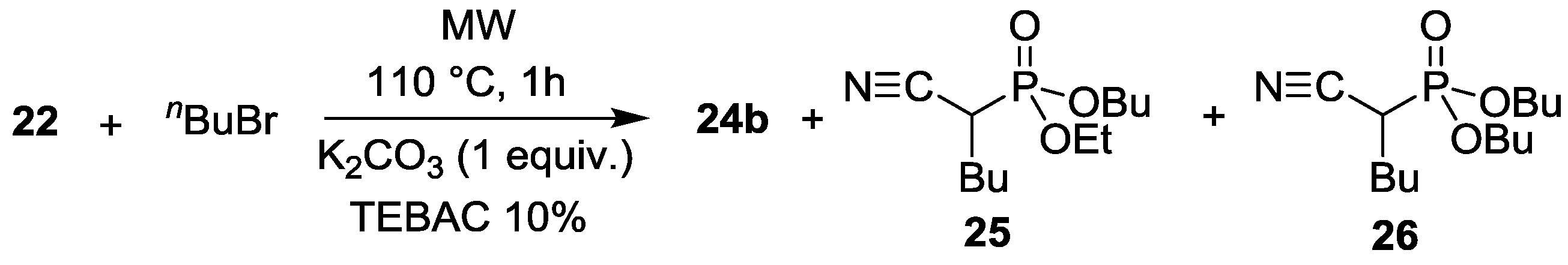{kind=link}
{kind=link}
{kind=link}
{kind=link}
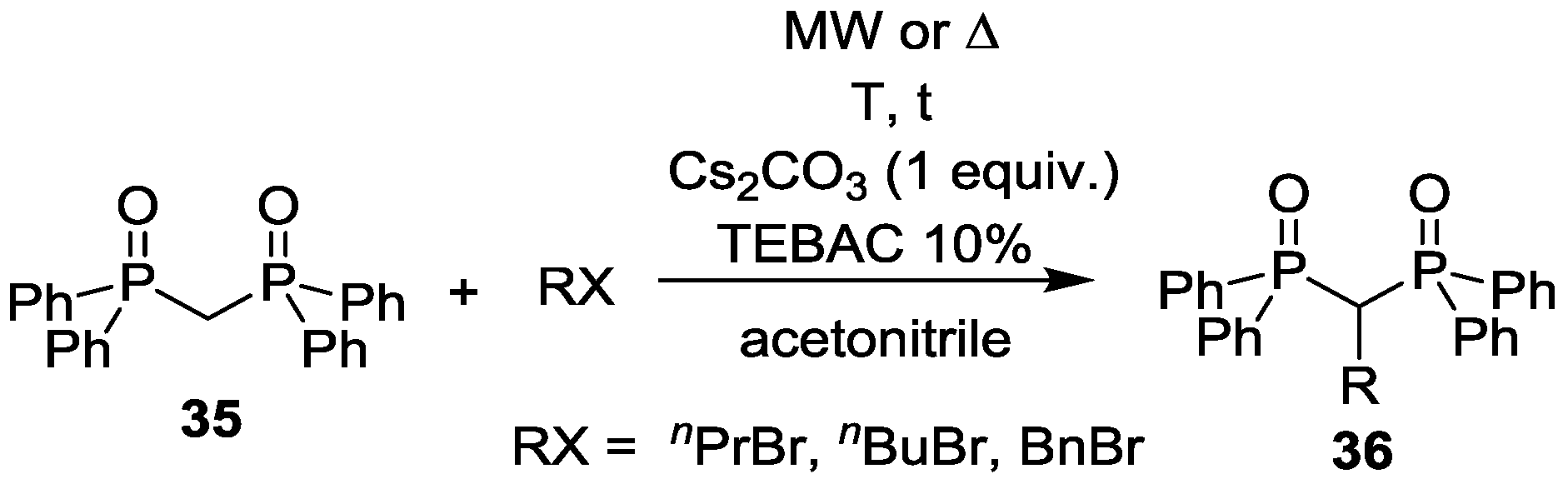{kind=link}
{kind=link}
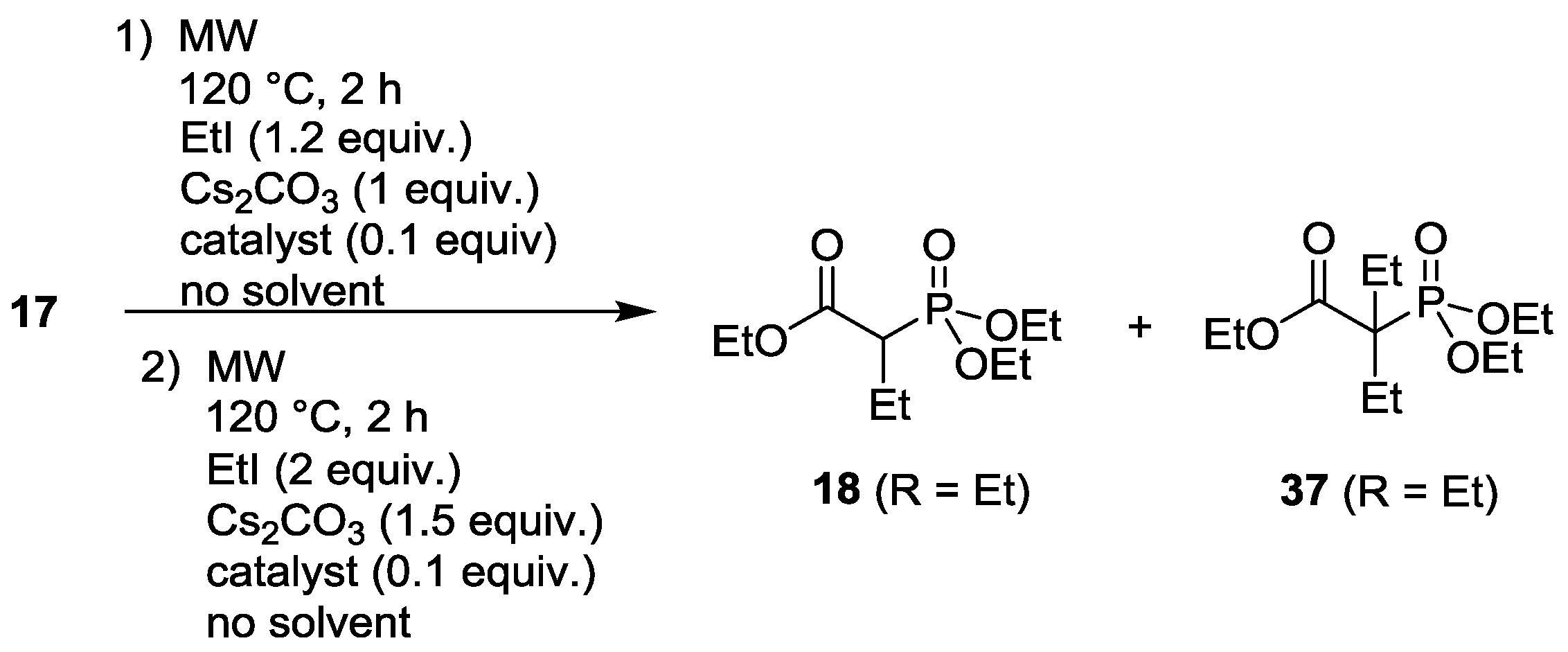{kind=link}
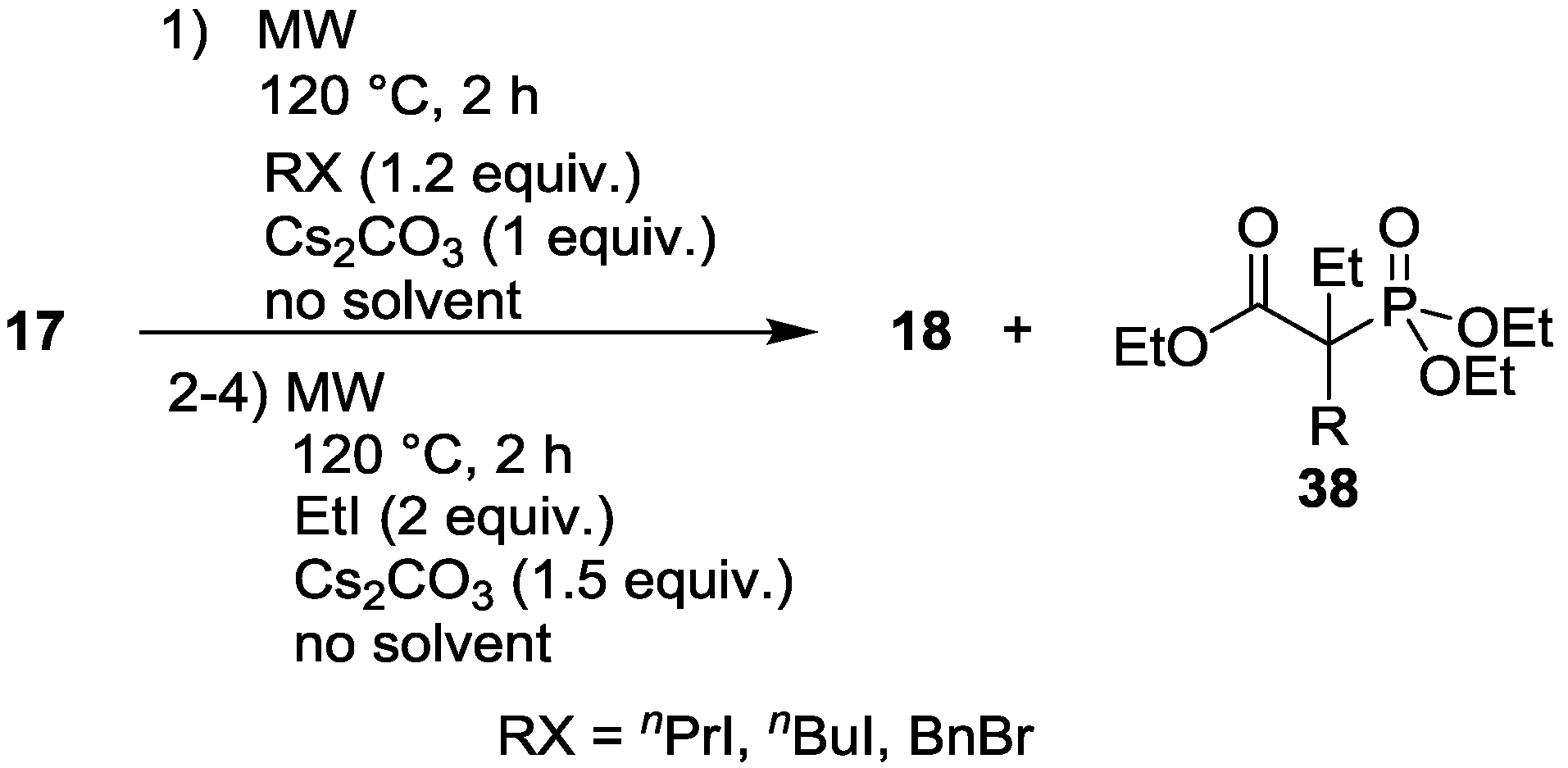{kind=link}
| Entry | RX | Base | T (°C) | t (min) | Yield of 2 (%) | By-products | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | EtI | K2CO3 | 160 | 45 | 93 | 3% 3 (R = Et) | 18 |
| 2 | EtI | Cs2CO3 | 140 | 90 | 97 | 3% 3 (R = Et) | 19 |
| 3 | nPrBr | K2CO3 | 185 | 45 | 97 | 2% 3 (R = nPr) | 18 |
| 4 | nPrBr | Cs2CO3 | 120 | 240 | 57 | 33% 11 (R = nPr) 10% 12 (R = nPr) | 19 |
| 5 | iPrBr | K2CO3 | 185 | 60 | 92 | - | 18 |
| 6 | nBuBr | K2CO3 | 185 | 45 | 88 | 5% 3 (R = nBu) | 18 |
| 7 | BnBr | K2CO3 | 180 | 45 | 68 | 1% 3 (R = Bn) | 18 |
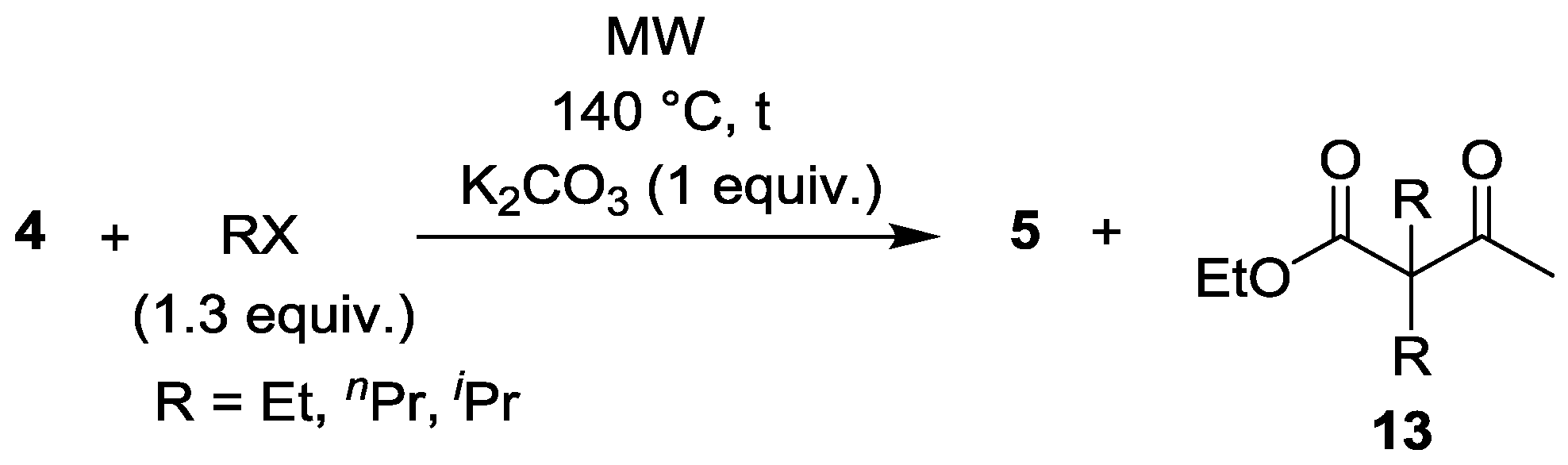
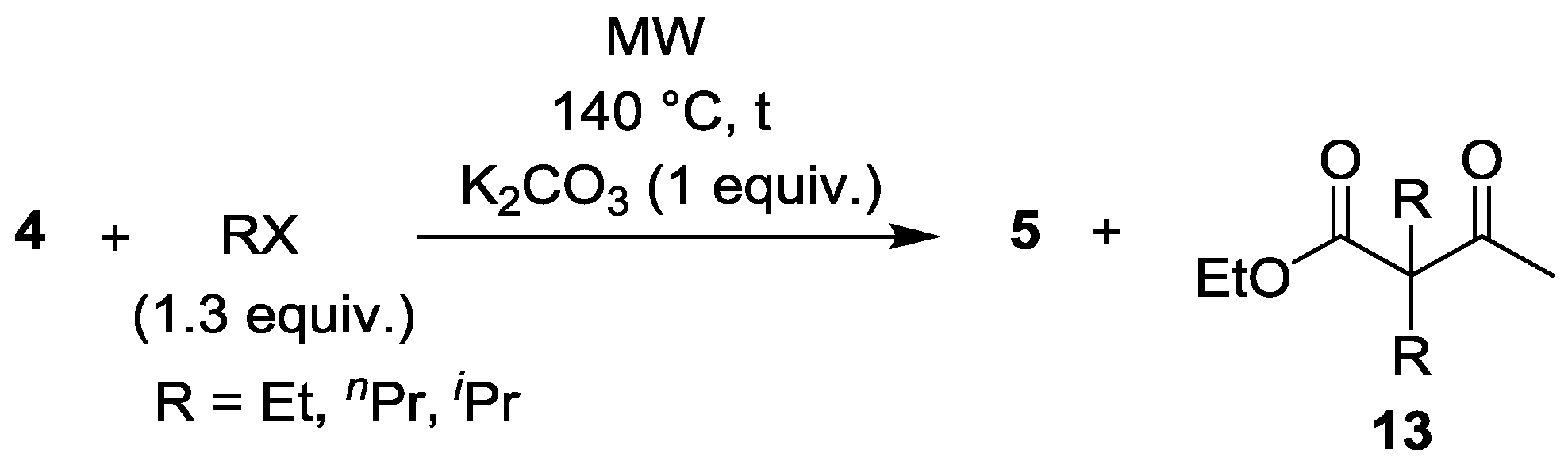
| Entry | RX | t (min) | Yield of 5 (%) | By-products |
|---|---|---|---|---|
| 1 | EtI | 30 | 85 | 5% 13 (R = Et) |
| 2 | nPrBr | 30 | 87 | 2% 13 (R = nPr) |
| 3 | iPrBr | 45 | 83 | - |

| Entry | RX | T (°C) | t (min) | Yield of 15 (%) | By-products |
|---|---|---|---|---|---|
| 1 | EtI | 100 | 60 | 78 | 12% 16 (R = Et) |
| 2 | EtI | 120 | 45 | 76 | 23% 16 (R = Et) |
| 3 | nPrBr | 120 | 45 | 82 | 10% 16 (R = nPr) 5% NCCHnPrCO2nPr |
| 4 | iPrBr | 140 | 45 | 86 | <1% 16 (R = iPr) 13% NCCHiPrCO2iPr 1% NCC(iPr)2CO2iPr |
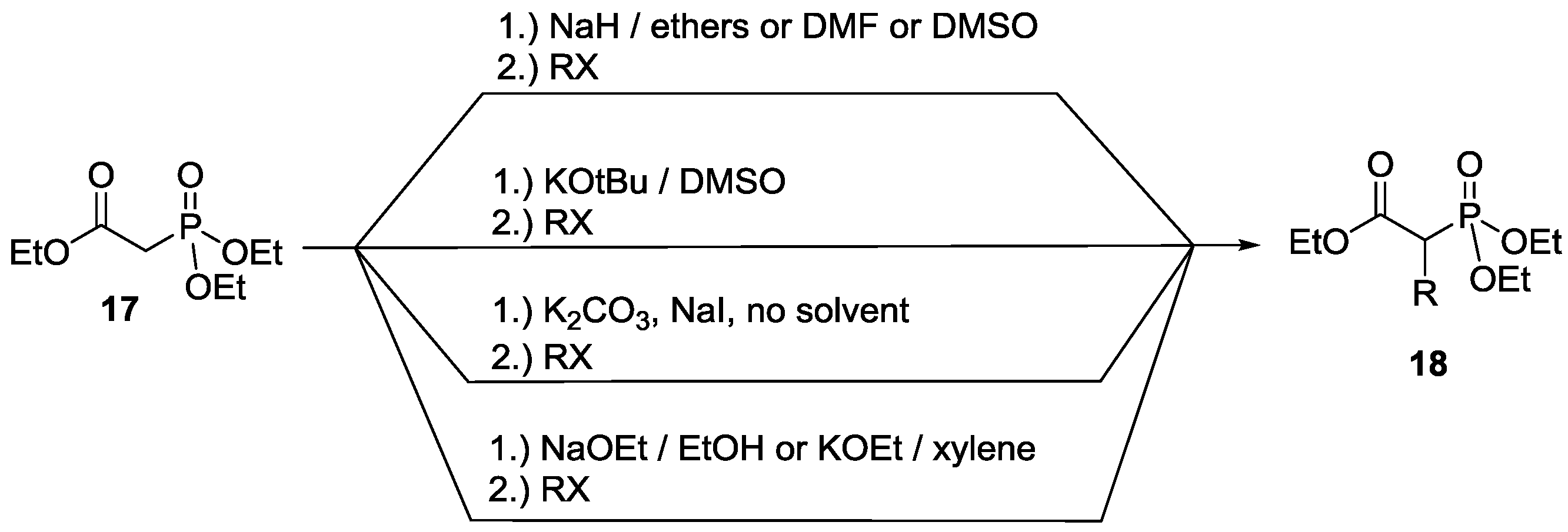
3. Alkylation of Active Methylene Containing Compounds with P=O Function under MW Conditions
3.1. Alkylation of Diethyl Ethoxycarbonylmethylphosphonate under MW and Solvent-Free Conditions


| Entry | RX | M2CO3 | T (°C) | t (h) | Composition (%) | ||
|---|---|---|---|---|---|---|---|
| 17 | 18 | Other | |||||
| 1 | EtI | K2CO3 | 120 | 2 | 13 | 82 (R = Et) | 5 |
| 2 | EtI | Cs2CO3 | 120 | 2 | 5 | 86 (R = Et) | 9 |
| 3 | nPrBr | K2CO3 | 130 | 2 | 28 | 57 (R = nPr) | 15 |
| 4 | nPrBr | Cs2CO3 | 120 | 2 | 10 | 79 (R = nPr) | 11 |
| 5 | nBuBr | Cs2CO3 | 120 | 2 | 7 | 83 (R = nBu) | 10 |
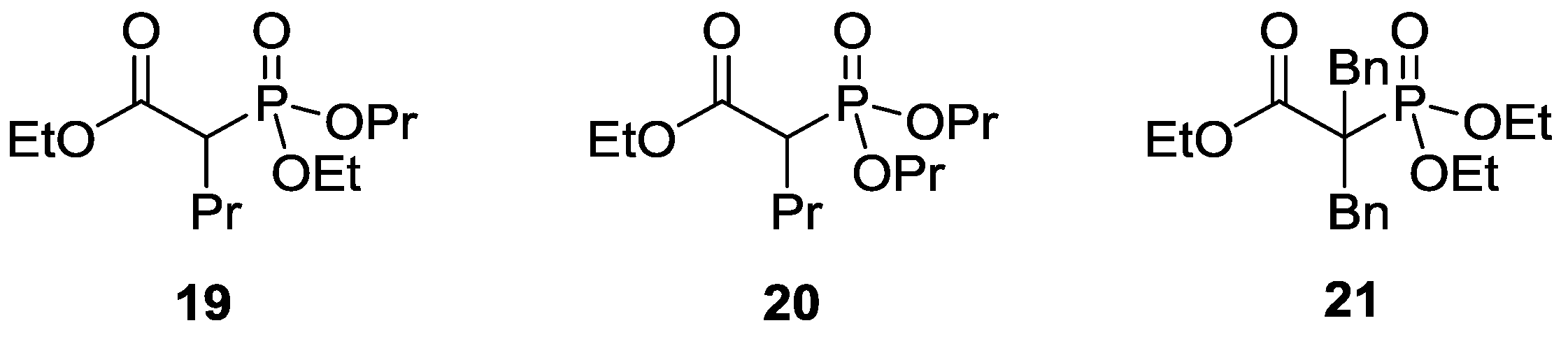
| 6 | BnBr | K2CO3 | 120 | 2 | 18 | 63 (R = Bn) | 19 a |
| 7 | BnBr | K2CO3 | 130 | 3 | 16 | 63 (R = Bn) | 21 b |
| 8 | BnBr | Cs2CO3 | 120 | 3 | 27 | 59 (R = Bn) | 14 c |


| Entry | RX | M2CO3 | TEBAC (10%) | Composition (%) | ||
|---|---|---|---|---|---|---|
| 17 | 18 | Other | ||||
| 1 | EtI | K2CO3 | + | 79 | 11 (R = Et) | 10 |
| 2 | nPrBr | K2CO3 | + | 80 | 14 (R = nPr) | 6 |
| 3 | nBuBr | K2CO3 | + | 72 | 28 (R = nBu) | - |
| 4 | EtI | Cs2CO3 | − | 24 | 74 (R = Et) | 2 a |
| 5 | nPrBr | Cs2CO3 | − | 55 | 45 (R = nPr) | - b |
| 6 | nBuBr | Cs2CO3 | − | 31 | 69 (R = nBu) | - c |
3.2. Alkylation of Diethyl Cyanomethylphosphonate under MW and Solvent-Free Conditions


| Entry | RX | M | Mode of heating | T (°C) | t (h) | Solvent | TEBAC (10%) | Composition (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 22 | 24 | Other | ||||||||
| 1 | nPrBr | K | Δ | 82 | 24 | MeCN | − | 53 | 45 (R = nPr) | 2 |
| 2 | nPrBr | K | Δ | 82 | 24 | MeCN | + | 26 | 37 (R = nPr) | 37 |
| 3 | nPrBr | Cs | Δ | 82 | 24 | MeCN | − | 7 | 84 (R = nPr) | 9 |
| 4 | nPrBr | Cs | Δ | 82 | 24 | MeCN | + | 2 | 67 (R = nPr) | 31 |
| 5 | nPrBr | K | MW | 100 | 2 | - | − | 13 | 70 (R = nPr) | 17 |
| 6 | nPrBr | K | MW | 120 | 1 | - | − | 20 | 58 (R = nPr) | 22 |
| 7 | nPrBr | Cs | MW | 120 | 1 | - | − | 4 | 33 (R = nPr) | 63 |
| 8 | nBuBr | Cs | Δ | 82 | 24 | MeCN | − | 3 | 90 (R = nBu) | 7 |
| 9 | nBuBr | K | MW | 120 | 2 | - | − | 20 | 65 (R = nBu) | 15 |

| Entry | nBuBr (equiv.) | TEBAC (%) | Composition (%) | ||||
|---|---|---|---|---|---|---|---|
| 22 | 24b | 25 | 26 | Other | |||
| 1 | 2.4 | 20 | 0 | 20 | 38 | 21 | 21 |
| 2 | 3.6 | 30 | 0 | 14 | 37 | 25 | 24 |
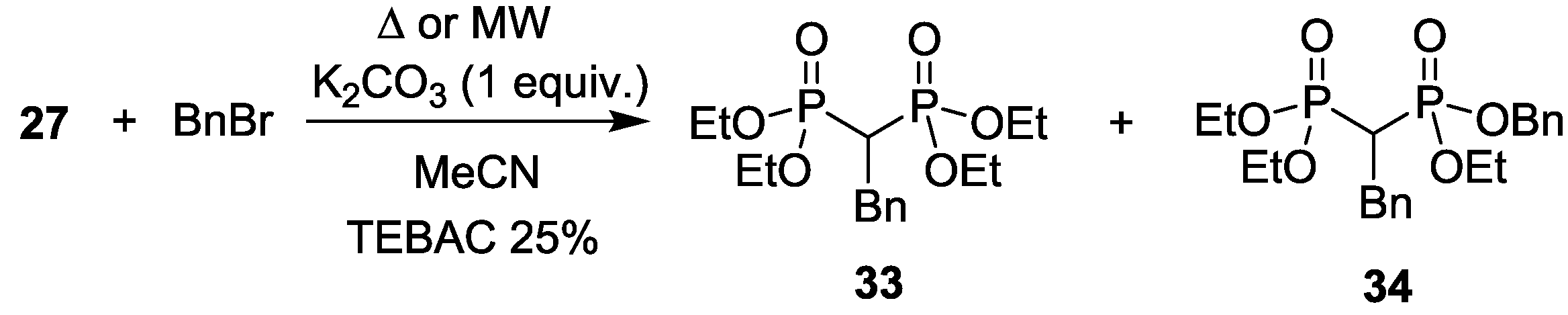
3.3. Alkylation of Tetraethyl Methylenebisphosphonate and Bis(diphenylphosphinoyl)methane under MW Conditions




| Entry | Mode of heating | T (°C) | t (h) | TEBAC (10%) | Conversion (%) | Yield of 36 R =Bn (%) |
|---|---|---|---|---|---|---|
| 1 | Δ | 82 | 24 | + | 60 | 44 |
| 2 | MW | 120 | 1.5 | − | 21 | - |
| 3 | MW | 120 | 3 | + | 57 | 45 |
| Entry | RX | Mode of heating | T (°C) | t (h) | TEBAC (10%) | Composition (%) | ||
|---|---|---|---|---|---|---|---|---|
| 35 | 36 | Other | ||||||
| 1 | nPrBr | MW | 180 | 4 | + | 36 | 47 (R = nPr) | 17 |
| 2 | nBuBr | MW | 180 | 4 | + | 26 | 52 (R = nBu) | 22 |
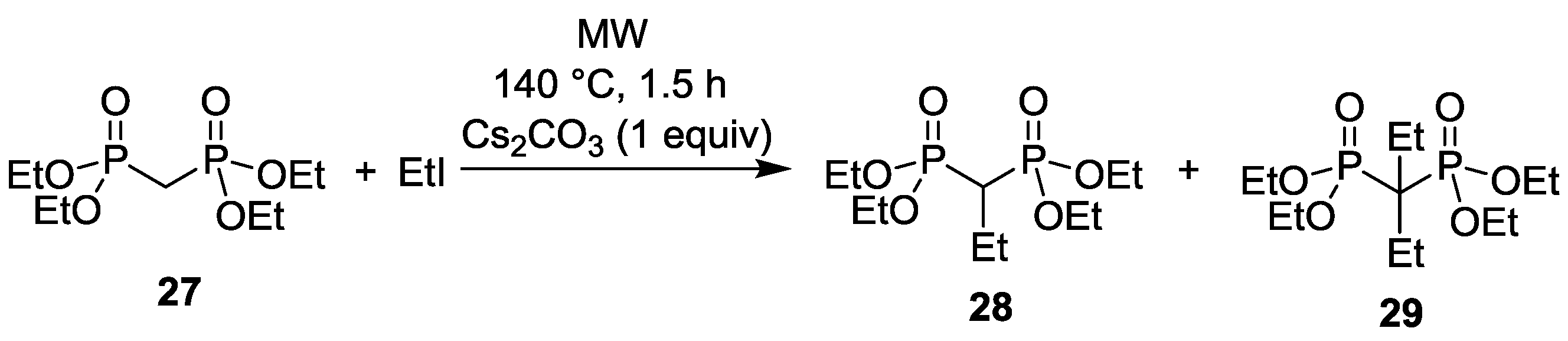
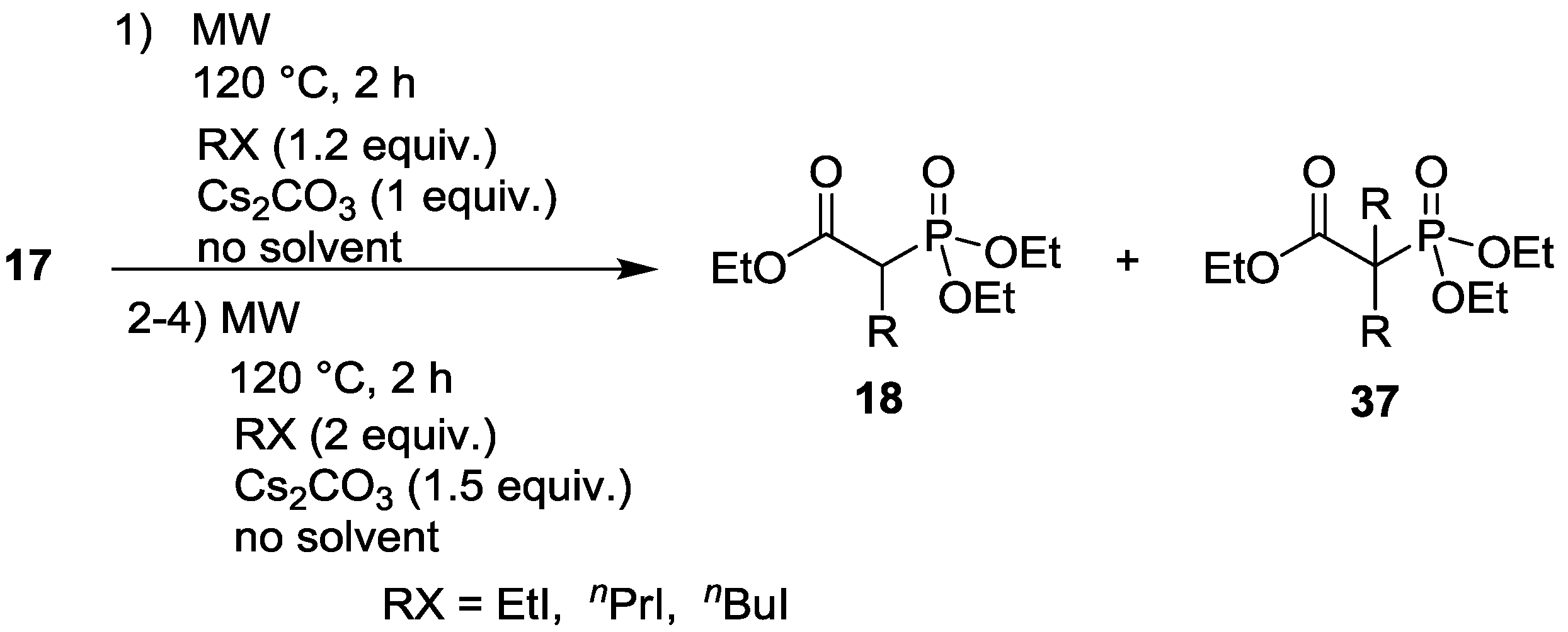
3.4. Dialkylation of Active Methylene Containing Compounds with P=O-function
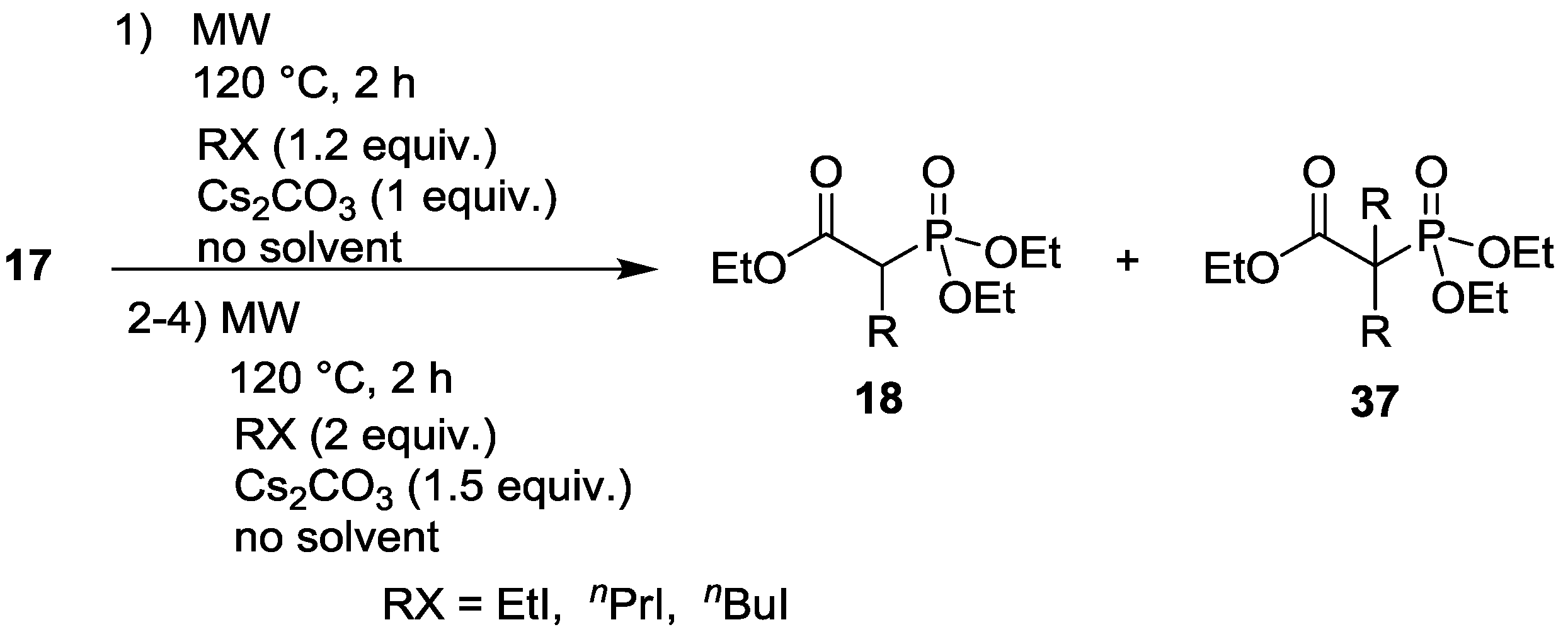
| Entry | Step | RX | Composition (%) | |||
|---|---|---|---|---|---|---|
| 17 | 18 | 37 | Other | |||
| 1 | 1. | EtI | 11 | 83 (R = Et) | 6 (R = Et) | - |
| 2. | EtI | - | 41 (R = Et) | 59 (R = Et) | - | |
| 3. | EtI | - | 9 (R = Et) | 91 (R = Et) | - | |
| 4. | EtI | - | 2 (R = Et) | 98 (R = Et) | - | |
| 2 | 1. | nPrI | 12 | 77 (R = nPr) | 3 (R = nPr) | 8 |
| 4. | nPrI | - | 16 (R = nPr) | 70 (R = nPr) | 14 | |
| 3 | 1. | nBuI | 14 | 77 (R = nBu) | 5 (R = nBu) | 4 |
| 4. | nBuI | - | 13 (R = nBu) | 76 (R = nBu) | 11 | |

| Entry | Step | Phase transfer catalyst | M2CO3 | Composition (%) | |||
|---|---|---|---|---|---|---|---|
| 17 | 18 (R = Et) | 37 (R = Et) | By-products | ||||
| 1 | 1. | TEBAC | Cs2CO3 | 8 | 75 | 7 | 10 |
| 2. | TEBAC | Cs2CO3 | - | 62 | 21 | 17 | |
| 2 | 1. | TBAB | Cs2CO3 | - | 75 | 25 | - |
| 2. | TBAB | Cs2CO3 | - | 57 | 43 | - | |

| Entry | Step | RX | Composition (%) | |||
|---|---|---|---|---|---|---|
| 17 | 18 | 38 | Other | |||
| 1 | 1. | PrI | 12 | 77 (R = nPr) | - | 11 |
| 2. | EtI | - | 37 (R = nPr) | 43 (R = nPr) | 20 | |
| 3. | EtI | - | 16 (R = nPr) | 59 (R = nPr) | 25 | |
| 4. | EtI | - | 1 (R = nPr) | 74 (R = nPr) | 25 | |
| 2 | 1. | BuBr | 14 | 79 (R = nBu) | - | 7 |
| 2. | EtI | - | 51 (R = nBu) | 26 (R = nBu) | 23 | |
| 3. | EtI | - | 25 (R = nBu) | 50 (R = nBu) | 25 | |
| 4. | EtI | - | 6 (R = nBu) | 71 (R = nBu) | 23 | |
| 3 | 1. | BnBr | - | 75 (R = Bn) | - | 25 |
| 2. | EtI | - | 21 (R = Bn) | 53 (R = Bn) | 26 | |
| 3. | EtI | - | - | 74 (R = Bn) | 26 | |
4. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Loupy, A. Microwaves in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Deshayes, S.; Liagre, M.; Loupy, A.; Luche, J.L.; Petit, A. Microwave activation in phase transfer catalysis. Tetrahedron 1999, 55, 10851–10870. [Google Scholar]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar]
- Keglevich, G.; Kiss, N.Z.; Mucsi, Z.; Körtvélyesi, T. Insights into a surprising reaction: The microwave-assisted direct esterification of phosphinic acids. Org. Biomol. Chem. 2012, 10, 2011–2018. [Google Scholar]
- Mucsi, Z.; Kiss, N.Z.; Keglevich, G. A quantum chemical study on the mechanism and energetics of the direct esterification, thioesterification and amidation of 1-hydroxy-3-methyl-3-phospholene 1-oxide. RSC Advances 2014, 4, 11948–11954. [Google Scholar]
- Glasnov, T.N.; Kappe, C.O. Microwave-assisted synthesis under continuous flow conditions. Macromol. Rapid Commun. 2007, 28, 395–410. [Google Scholar]
- Strauss, C.R. On scale up of organic reactions in closed vessel microwave systems. Org. Process Res. Dev. 2009, 13, 915–923. [Google Scholar]
- Cocagne, P.; Elguero, J.; Gallo, R. The present use and the possibilities of phase-transfer catalysis in drug synthesis. Heterocycles 1983, 20, 1379–1406. [Google Scholar]
- Starks, C.M.; Liotta, C.L.; Halpern, M. Phase Transfer Catalysis—Fundamentals, Applications and Industrial Perspectives; Chapman & Hall: New York, NY, USA; London, UK, 1994. [Google Scholar]
- Makosza, M. Phase-transfer catalysis. A general green methodology in organic synthesis. Pure Appl. Chem. 2000, 27, 1399–1403. [Google Scholar]
- Fedorynski, M.; Jezierska-Zieba, M.; Kakol, B. Phase transfer catalysis in pharmaceutical industry—where are we? Acta Pol. Pharm. 2008, 65, 647–654. [Google Scholar]
- Wang, Y.; Deng, R.; Mi, A.; Jiang, Y. Solid–liquid phase transfer catalytic synthesis. XII. Microwave irradiated alkylation of diethyl malonate. Synth. Commun. 1995, 25, 1761–1764. [Google Scholar]
- Deng, R.; Wang, Y.; Jiang, Y. Solid–liquid phase transfer catalytic synthesis. X. The rapid alkylation of ethyl acetoacetate under microwave irradiation. Synth. Commun. 1994, 24, 111–115. [Google Scholar]
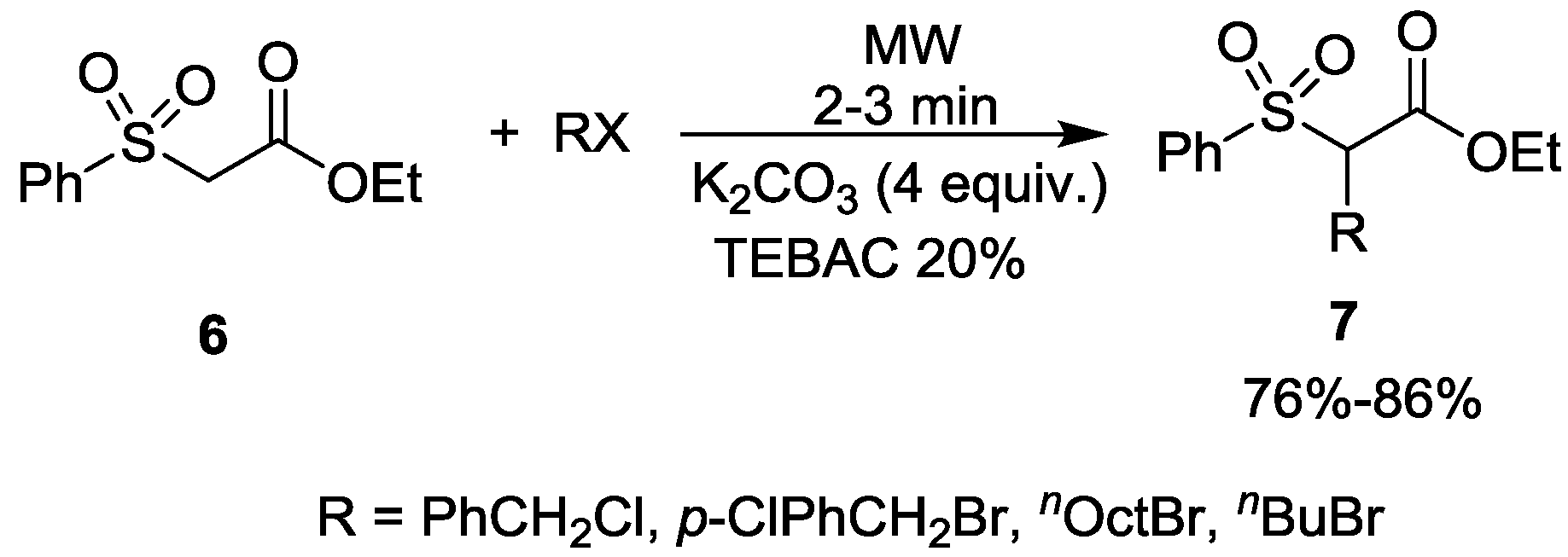
- Wang, Y.; Jiang, Y. Solid–liquid phase transfer catalytic synthesis. VIII. The rapid alkylation of ethyl phenylsulfonylacetate under microwave irradiation. Synth. Commun. 1992, 22, 2287–2291. [Google Scholar]
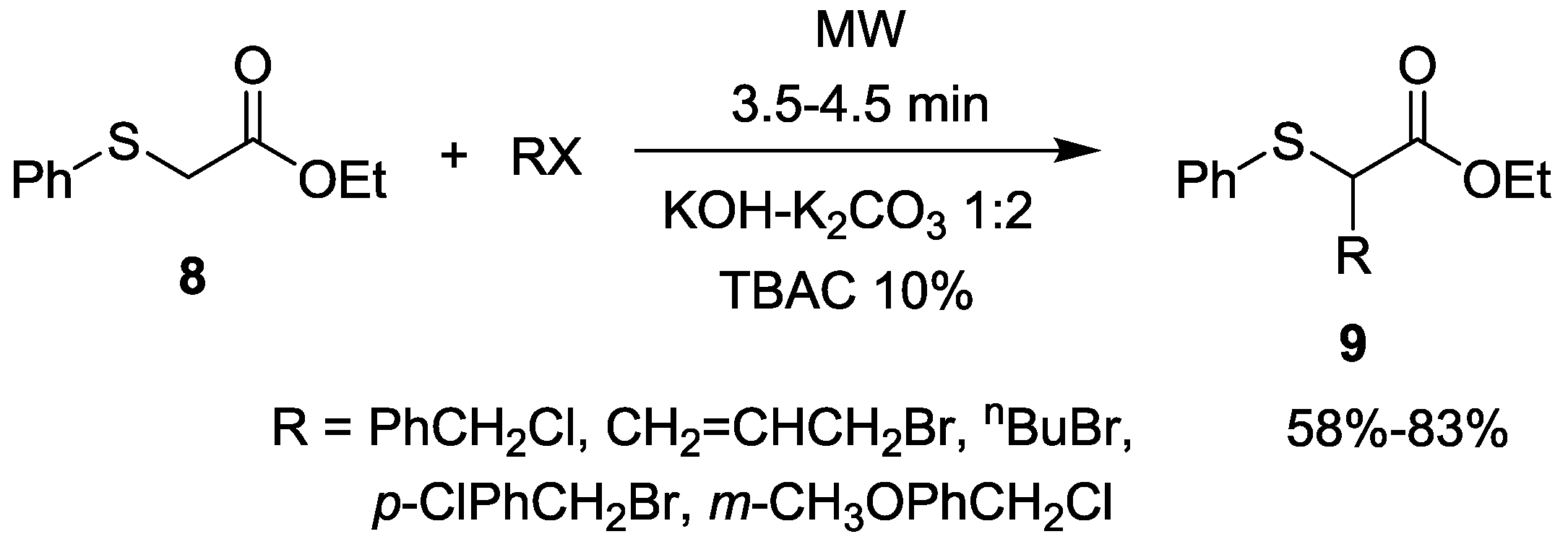
- Deng, R.; Wang, Y.; Jiang, Y. Solid–liquid phase transfer catalytic synthesis. XI. The convenient and efficient alkylation of ethyl phenylmercapto-acetate in the presence of quaternary ammonium salts under microwave irradiation. Synth. Commun. 1994, 24, 1917–1921. [Google Scholar]
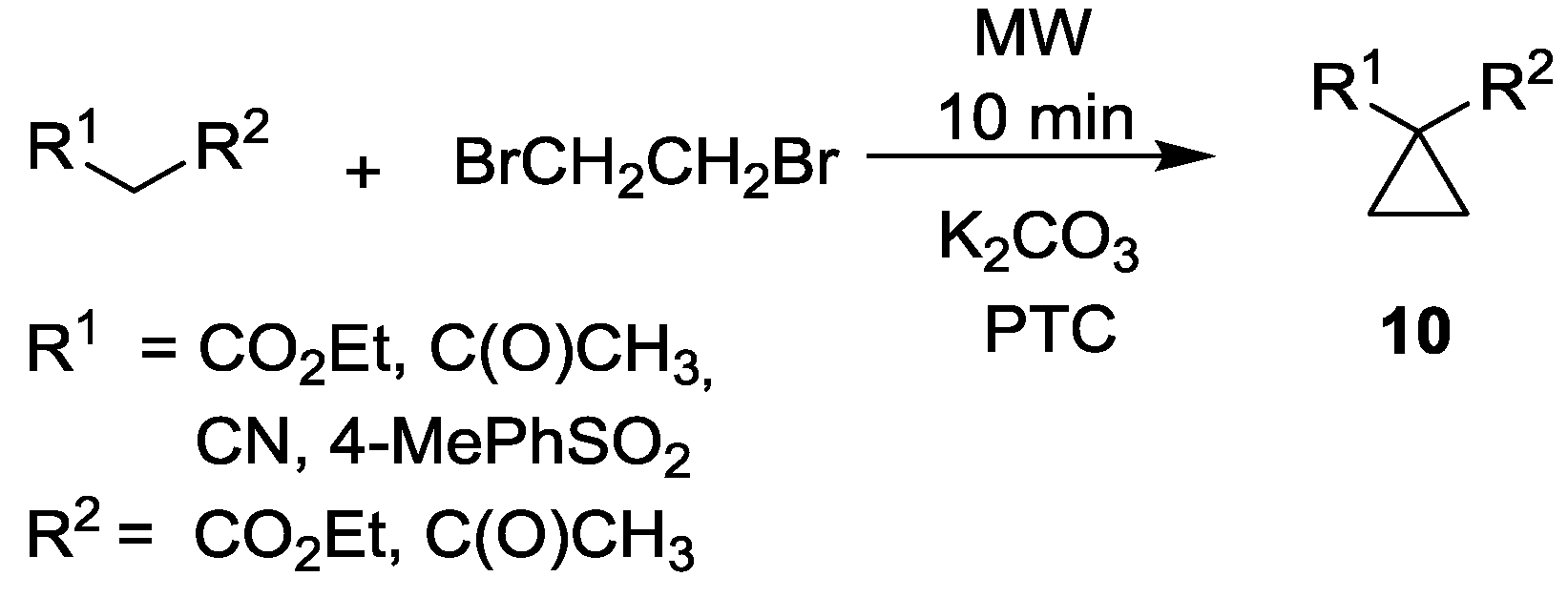
- Gumaste, V.K.; Khan, A.J.; Bhawal, B.M.; Deshmukh, A.R.A.S. Microwave assisted phase transfer catalysis: An efficient solvent free method for the synthesis of cyclopropane derivatives. Indian J. Chem. 2004, 43B, 420–423. [Google Scholar]
- Keglevich, G.; Novák, T.; Vida, L.; Greiner, I. Microwave irradiation as an alternative to phase transfer catalysis in the liquid–solid phase, solvent-free C-alkylation of active methylene containing substrates. Green Chem. 2006, 8, 1073–1075. [Google Scholar]
- Keglevich, G.; Majrik, K.; Vida, L.; Greiner, I. Microwave irradiation as a green alternative to phase transfer catalysis: solid–liquid phase alkylation of active methylene containing substrates under solvent-free conditions. Lett. Org. Chem. 2008, 5, 224–228. [Google Scholar]
- Greiner, I.; Grün, A.; Ludányi, K.; Keglevich, G. Solid–liquid two-phase alkylation of tetraethyl methylenebisphosphonate under microwave irradiation. Heteroatom Chem. 2011, 22, 11–14. [Google Scholar]
- Nakazato, A.; Kumagai, T.; Ohta, K.; Chaki, S.; Okuyama, S.; Tomisawa, K. Synthesis and SAR of 1-alkyl-2-phenylethylamine derivatives designed from N,N-dipropyl-4-methoxy-3-(2-phenylethoxy)phenylethylamine to discover σ1 ligands. J. Med. Chem. 1999, 42, 3965–3970. [Google Scholar]
- Kuroda, C.; Kimura, Y.; Nogami, H. Intramolecular cyclization of 2-(alkoxycarbonyl)allylsilanes with ynones. Nucleophilic and electrophilic aspects of the 2-(alkoxycarbonyl)allylsilane moiety. J. Chem. Res. (S) 1998, 174–175. [Google Scholar]
- Kuroda, C.; Tang, C.Y.; Tanabe, M.; Funakoshi, M. Proto- and iodo-lactonization reaction of substituted α,β:γ,δ-unsaturated carboxylic acid. Bull. Chem. Soc. Jap. 1999, 72, 1583–1587. [Google Scholar]
- Kuroda, C.; Koshio, H.; Koito, A.; Sumiya, H.; Murase, A.; Hirono, Y. Nazarov cyclization of 4-cycloalkylidene-5-(trimethylsilyl)pent-1-en-3-one derivatives. Synthesis of spiro[4.5]decane, spiro[4.4]nonane, and their derivatives. Tetrahedron 2000, 56, 6441–6455. [Google Scholar]
- Kuroda, C.; Honda, S.; Nogara, Y.; Koshio, H.; Shibue, T.; Takeshita, T. Synthesis of spiro[4.5]decane and bicyclo[4.3.0]nonane ring systems by self-cyclization of (Z)- and (E)-2-(trimethylsilylmethyl)pentadienal derivative. Tetrahedron 2004, 60, 319–331. [Google Scholar]
- Nakano, M.; Atsuumi, S.; Koike, Y.; Tanaka, S.; Funabashi, H.; Hashimoto, J.; Ohkubo, M.; Morishima, H. Synthesis of a homostatine-containing renin inhibitor which incorporates a sulfonemethylene isostere at its N-terminus. Bull. Chem. Soc. Jap. 1990, 63, 2224–2232. [Google Scholar]
- Gomez-Monterrey, I.; Turcaud, S.; Lucas, E.; Bruetschy, L.; Roques, B.P.; Fournib–Zaluski, M.-C. Exploration of neutral endopeptidase active site by a series of new thiol-containing inhibitors. J. Med. Chem. 1993, 36, 87–94. [Google Scholar]
- Chen, H.; Noble, F.; Mothe, A.; Meudal, H.; Coric, P.; Danascimento, S.; Roques, B. P.; George, P.; Fournib–Zaluski, M.-C. Phosphinic derivatives as new dual enkephalin-degrading enzyme inhibitors: Synthesis, biological properties, and antinociceptive activities. J. Med. Chem. 2000, 43, 1398–1408. [Google Scholar]
- Dhokte, U.P.; Rao, A.S. Synthesis of the pheromones, (E)-3,7-dimethyl-2,7-octadienyl propionate, (E)-3,7-dimethyl-2-octene-1,8-diol and frontalin from a common intermediate. Synth. Commun. 1988, 18, 811–822. [Google Scholar]
- Zhang, J.; Loh, T.-P. Ruthenium- and rhodium-catalyzed cross-coupling reaction of acrylamides with alkenes: efficient access to (Z,E)-dienamides. Chem. Commun. 2012, 48, 11232–11234. [Google Scholar]
- Doran, R.; Duggan, L.; Singh, S.; Duffy, C.D.; Guiry, P.J. Asymmetric synthesis of (+)-tanikolide and the β-methyl-substituted analogues of (+)-tanikolide and (–)-malyngolide. Eur. J. Org. Chem. 2011, 35, 7097–7106. [Google Scholar]
- Stritzke, K.; Schulz, S.; Nishida, R. Absolute configuration and synthesis of β- and δ-lactones present in the pheromone system of the giant white butterfly Idea leuconoe. Eur. J. Org. Chem. 2002, 22, 3884–3892. [Google Scholar]
- Senter, T.J.; Fadeyi, O.O.; Lindsley, C.W. Enantioselective total synthesis of (+)-amabiline. Org. Lett. 2012, 14, 1869–1871. [Google Scholar]
- Ferguson, A.C.; Adlington, R.M.; Martyres, D.H.; Rutledge, P.J.; Cowley, A.; Baldwin, J.E. Total synthesis of a novel 2-thiabicyclo[3.2.0]heptan-6-one analogue of penicillin N. Tetrahedron 2003, 59, 8233–8243. [Google Scholar]
- Wuts, P.G.M.; Putt, S.R.; Ritter, A.R. Synthesis of the dipeptide hyroxyethylene isostere of Leu-Val, a transition state mimic for the control of enzyme function. J. Org. Chem. 1988, 53, 4503–4508. [Google Scholar]
- Liao, C.-C.; Zhu, J.-L. Investigation on Lewis acid mediated Diels-Alder reactions of 2-phosphono-2-alkenoates. Application to total synthesis of (+/−)-α-Alasken-8-one via reductive alkylation of resulting adduct. J. Org. Chem. 2009, 74, 7873–7884. [Google Scholar]
- Pelotier, B.; Holmes, T.; Piva, O. Synthesis of anti-Alzheimer (R)-arundic acid. Tetrahedron Asymmetry 2005, 16, 1513–1520. [Google Scholar]
- Kirschleger, B.; Queignec, R. Heterogeneous mediated alkylation of ethyl diethylphosphonoacetate. A “one pot” access to α-alkylated acrylic esters. Synthesis 1986, 926–928. [Google Scholar]
- Nieminen, S.; Payne, T.G.; Senn, P.; Tamm, C. Biosynthesis of the rubratoxins. Helv. Chim. Acta 1981, 64, 2162–2174. [Google Scholar]
- Kosolapoff, G.M.; Powell, J.S. Alkylation of triethyl phosphonoacetate and related esters. J. Am. Chem. Soc. 1950, 72, 4198–4200. [Google Scholar]
- Grün, A.; Blastik, Z.; Drahos, L.; Keglevich, G. Microwave-assisted alkylation of diethyl ethoxycarbonylmethylphosphonate under solventless conditions. Heteroatom Chem. 2012, 23, 241–246. [Google Scholar]
- Simon, J.R.; Neidlein, R. Syntheses and chemical reactions of 1-cyano-1-isocyanoalkylphosphonic acid esters. Synthesis 2000, 1101–1108. [Google Scholar]
- Compagnone, R.S.; Suarez, A.I.; Zambrano, J.L.; Pina, I.C.; Dominguez, J.N. A short and versatile synthesis of 3-substituted 2-aminoquinolines. Synth. Commun. 1997, 27, 1631–1641. [Google Scholar]
- Bailey, P.D.; Morgan, K.M. A total asymmetric synthesis of (–)-suaveoline. Chem. Commun. 1996, 1479–1480. [Google Scholar]
- Bailey, P.D.; Morgan, K.M. The total synthesis of (−)-suaveoline. J. Chem. Soc. Perkin Trans. 1 2000, 3578–3583. [Google Scholar]
- Pudovik, A.N.; Lebedeva, N.M. Synthesis of esters of phosphonic and thiophosphonic acids. XXIV. Addition of phosphonoacetonitrile and its homologs to esters and nitriles of unsaturated carboxylic acids. Zh. Obshch Khim. 1955, 25, 2235–2240. [Google Scholar]
- Defacqz, N.; Touillaux, R.; Marchand-Brynaert, J. [4+2] Cycloaddition of N-buta-1,3-dienylsuccinimide to gem-substituted vinyl phosphonates. J. Chem. Res. (S) 1998, 512–513. [Google Scholar]
- Keglevich, G.; Grün, A.; Blastik, Z.; Greiner, I. Solid–liquid phase alkylation of P=O-functionalized CH acidic compounds utilizing phase transfer catalysis and microwave irradiation. Heteroatom Chem. 2011, 22, 174–179. [Google Scholar]
- Hays, H.R.; Logan, T.J. gem-Diphosphinoalkanes. Preparation and characterization. J. Org. Chem. 1966, 31, 3391–3394. [Google Scholar]
- Nguyen, L.M.; Niesor, E.; Bentzen, C.L. gem-Diphosphonate and gem-phosphonate-phosphate compounds with specific high density lipoprotein inducing activity. J. Med. Chem. 1987, 80, 1426–1433. [Google Scholar]
- Roth, A.G.; Drescher, D.; Reamer, S.; Arenz, C.; Yang, Y.; Uhlig, S. Potent and selective inhibition of acid sphingomyelinase by bisphosphonates. Angewandte Chem. Int. Ed. 2009, 48, 7560–7563. [Google Scholar]
- Kosolopoff, G.M. The chemistry of aliphatic phosphonic acids. I. Alkylation of methanediphosphonic acid. J. Am. Chem. Soc. 1953, 75, 1500–1501. [Google Scholar]
- Goebel, R.; Richte, F.; Weichmann, H. Synthesis and reactivity of methylene bridged diphosphoryl compounds. Phosphorous, Sulfur 1992, 73, 67–80. [Google Scholar]
- Cotton, F.A.; Schunn, R.A. Metal salts and complexes of dialkoxyphosphonylacetylmethanide ions. J. Am. Chem. Soc. 1963, 85, 2394–2402. [Google Scholar]
- Henecka, H. Methoden Der Organischen Chemie (Houben–Weyl); Müller, E., Ed.; Thieme Verlag: Stuttgart, Germany, 1976; Part II; pp. 1435–1447. [Google Scholar]
- Sustmann, R.; Korth, H.-G. Methoden Der Organischen Chemie (Houben-Weyl); Müller, E., Ed.; Thieme Verlag: Stuttgart, Germany, 1985; Part E5, Ch. 3.2.1.1.1.1.3; pp. 370–373. [Google Scholar]
- Davis, B.R.; Hinds, M.G. Synthetic, Structural and vibrational spectroscopic studies in bismuth(III) halide/N,N′-aromatic bidentate base systems. IV. Bismuth(III) halide/N,N′-bidentate ligand (1:1) systems. Aust. J. Chem. 1997, 50, 309–320. [Google Scholar]
- English, A.R.; Girard, D.; Jasys, V.J.; Martingano, R.J.; Kellogg, M.S. Orally effective acid prodrugs of the β-lactamase inhibitor sulbactam. J. Med. Chem. 1990, 33, 344–347. [Google Scholar]
- Baumstark, A.L.; Choudhary, A.; Vasquez, P.C.; Dotrong, M. Synthesis of 4,4-dimethyl-3,4-dihydro-3,3,5-trisubstituted-2H-pyrazoles and N-benzoyl derivatives: Method for “Hydrolysis” of unreactive amides and carbamates. J. Heterocycl. Chem. 1990, 27, 291–294. [Google Scholar]
- Quici, S.; Manfredi, A.; Raimondi, L.; Sironi, A. Synthesis and properties of new lipophilic macrotricyclic cylindrical cryptands. J. Org. Chem. 1995, 60, 6379–6388. [Google Scholar]
- Viera, I.; Manta, E.; Gonzalez, L.; Mahler, G. Synthesis of enantiomerically enriched α,α-disubstituted β,γ-epoxy esters using hydrolytic kinetic resolution catalyzed by salenCo(III). Tetrahedron:Asymmetry 2010, 21, 631–635. [Google Scholar]
- Bodnarchuk, N.D.; Malovik, V.V.; Derkach, G.I. Phosphono carboxylic acid derivatives. Zh. Obshch. Khim. 1970, 40, 1210–1217. [Google Scholar]
- Khachik, F.; Beecher, G.R.; Li, B.W.; Englert, G.J. Synthesis of 13C-labelled (all-E,3R,3′R)-β,β-carotene-3,3′-diol (zeaxanthin) at C(12), C(13), C(12′), and C(13′) via all-E-2,7-dimethylocta-2,4,6-triene-1,8-dial-13C4. Labelled Compd. Radiopharm. 1995, 36, 1157–1172. [Google Scholar]
- Noguchi, H.; Aoyama, T.; Shioiri, T. Total synthesis of analogs of topostin B, A DNA topoisomerase I inhibitor. Part 1. Synthesis of fragments of topostin B-1 analogs. Tetrahedron 1995, 51, 10531–10544. [Google Scholar]
- Singh, R.J. Alkylation Studies; Part II: Bis-alkylation of diethyl cyanomethanephosphonate. Synthesis 1986, 762–763. [Google Scholar]
- Grün, A.; Blastik, Z.; Drahos, L.; Keglevich, G. Dialkylation of diethyl ethoxycarbonylmethylphosphonate under microwave and solventless conditions. Heteroatom Chem. 2014, 25, 107–113. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grün, A.; Bálint, E.; Keglevich, G. Solid-Liquid Phase C-Alkylation of Active Methylene Containing Compounds under Microwave Conditions. Catalysts 2015, 5, 634-652. https://doi.org/10.3390/catal5020634
Grün A, Bálint E, Keglevich G. Solid-Liquid Phase C-Alkylation of Active Methylene Containing Compounds under Microwave Conditions. Catalysts. 2015; 5(2):634-652. https://doi.org/10.3390/catal5020634
Chicago/Turabian StyleGrün, Alajos, Erika Bálint, and György Keglevich. 2015. "Solid-Liquid Phase C-Alkylation of Active Methylene Containing Compounds under Microwave Conditions" Catalysts 5, no. 2: 634-652. https://doi.org/10.3390/catal5020634