
Utilization of Volatile Organic Compounds as an Alternative for Destructive Abatement


 ,
,
Abstract
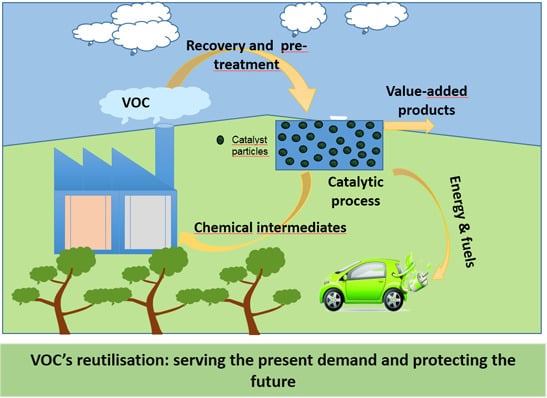
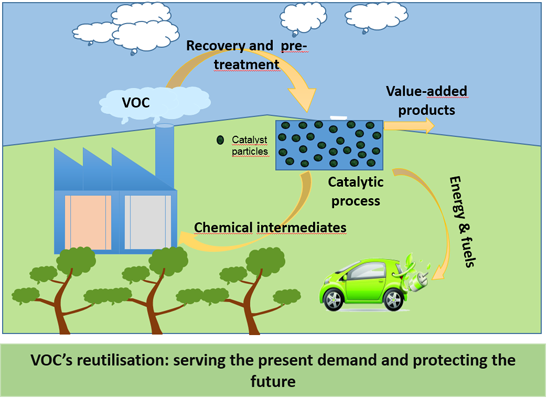
:
1. Introduction
2. Pre- and Post-Processing of the Emissions to Be Utilized
2.1. Recovery of the Raw Emissions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Recovery technique | Efficiency (%) | Indication of applied flow (m3 h−1) | Critical parameters |
|---|---|---|---|
| Adsorption: | |||
| Activated carbon | 80–98 | 100–100,000 | Fluid percentage, VOC concentration |
| Zeolite | 80–99 | <100,000 | Fluid percentage, dust in inlet gas |
| Polymeric | 95–98 | - | Dust in inlet gas |
| Membrane separation | 99.9 | <3000 | - |
| Wet scrubbing | 30–99 | 50–500,000 | Temperature |
| Condensation Cryocondensation | 60–90 >99 | 100–100,000 <5000 | Saturation of inlet gas Fluid percentage in inlet gas |
2.1.1. Adsorption
2.1.2. Membrane Separation
2.1.3. Wet Gas Scrubbing
2.1.4. Condensation and Cryocondensation
2.2. Purification of the Raw Emissions and the Products
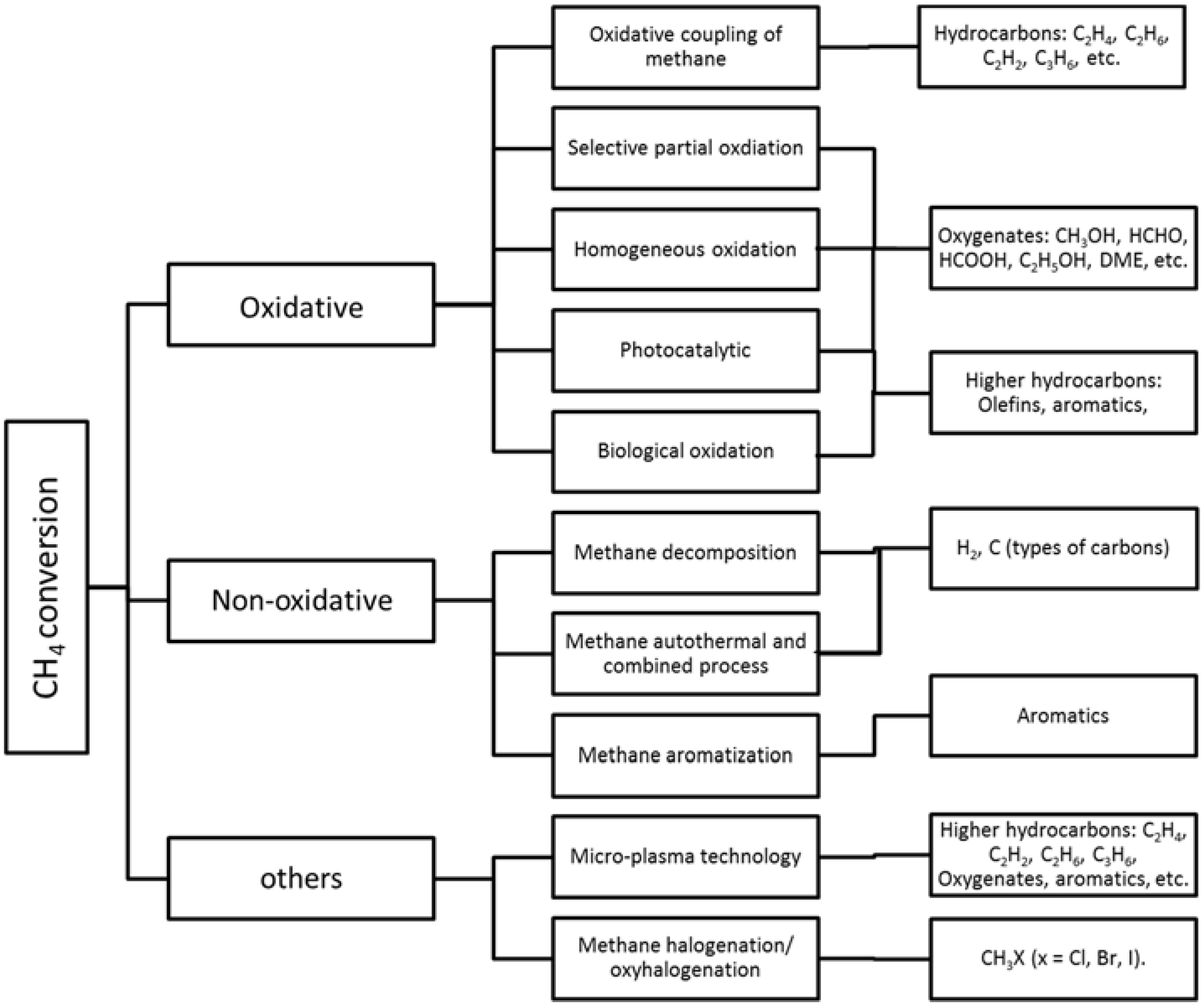
3. Catalytic Utilization of Methane Emissions
3.1. From Methane to Syngas and Hydrogen
3.1.1. Methane Dry Reforming and Partial Oxidation to Syngas

- (E1) Methane dry reforming (MDR)CH4 + CO2 ↔ 2H2 + 2CO ∆H°298 = 247 kJ mol−1
- (E2) Methane partial oxidation (POM)CH4 + 0.5O2 ↔ 2H2 + CO ΔH°298 = −36 kJ mol−1
- (E3) Methane steam reforming (MSR)CH4 + H2O ↔ 3H2 + CO ∆H°298 = 206 kJ mol−1
- (Insufficient steam)(E4) Methane steam reformingCH4 + 2H2O ↔ 4H2 + CO2 ∆H°298 = 165 kJ mol−1
- (Sufficient steam)(E5) Methane decompositionCH4 ↔ C + 2H2 ∆H°298 = 75 kJ mol−1
| Catalyst | Prepa-ration method | Reaction conditions | Conversion (%) | H2/CO | Comments | Ref. |
|---|---|---|---|---|---|---|
| LaNiO3@SiO2 | Stöber | RT Qtot = 50 mL min−1 CH4/CO2 = 1 | XCH4 = 67 XCO2 = 57 | 0.9 | NT-plasma, DBD reactor, core shell | [49] |
| Ni4.7Pt1 | ALD | T = 600 °C Qtot = 40 mL min−1 CH4/CO2 = 1 | XCH4 = 30 | na | small Ni particles ∼8 nm with ALD | [52] |
| Ni10Pt0.5/Al2O3 | WI | T = 750 °C, Qtot = 20 mL min−1 CH4/CO2 = 1 | XCH4 = 78 XCO2 = 95 | 0.63 | Pt addition reduce the carbon deposition and improve the geometric effects | [56] |
| Ni5Mn95O | CP | T = 750 °C, Qtot = 30 mL min−1 CH4/CO2 = 1 | XCH4 = 75, XCO2 = 75 | na | rapid deactivation due to Mn phase change leads to metal sintering | [59] |
| Ni5/diatomites | two solvents and DI | T = 650 °C, GHSV = 21,120 mL g−1 h−1 CH4/CO2 = 1 | XCH4 = 39 XCO2 = 49 | 0.88 | deactivation of Ni0 by re-oxidation, only C-β (reactive C-nanotubes) | [62] |
| Ni5/SiO2 | two solvents and DI | T = 650 °C, GHSV = 21,120 mL.g−1 h−1 CH4/CO2 = 1 | XCH4 = 61, XCO2 = 70 | 0.9 | deactivation of Ni0 by re-oxidation, two types of carbon forms: C-β and C-γ (less reactive sp3 C-graphite) | [62] |
| (NiCo)2.5/Ce77Zr20 | Glycothermal synthesis + DP + FD calcination | T = 750 °C p =1.2 bar | XCH4 = 90 XCO2 = 90 | 0.85 | TOS 20 h, calcination effect | [53] |
| Ni5-Ce6/SBA-15 | two solvents and IWI | T = 600 °C, GHSV = 264 Lg−1h−1 | XCH4 = 95, XCO2 = 90 | 0.96 | high dispersion inside pores, smaller NiO particles, mode of NiO addition after Ce | [51] |
| Ce2.5-Co5-Ni10/Al2O3 | IWI | T = 750 °C CH4:CO2 = 1 | XCH4 = 60 | TOS 5 h, effect of forced periodic cycling | [63] | |
| Ni15CeMgAl | refluxed co-precipitation | T = 750 °C GHSV = 48,000 h−1 CO2/CH4 = 1.04 | XCH4 = 96.5 XCO2 = 92 | 0.8 | bimodal pore, Ni sites in NiAl2O4 spinel structure had longer stability | [58] |
| Ni5/La2O3-ZrO2 | IWI | T = 400 °C, 12 mL min−1 | XCH4 = 7.5 XCO2 = 12 | na | effect of calcination temperature relation with Ni particle size | [64] |
| NiFe2O4/SiO2 | sol–gel | T = 800 °C Qtot = 30 mL min−1 CH4/CO2 = 2 | XCH4 = 66 XCO2 = 93 | 1.1 | surface acidity is crucial in limiting RWGSR | [65] |
| Rh0.2-Ni/SBA-15 | DI | T = 650 °C GHSV = 48,000 mL h−1 g−1 | - | 0.82 | Rh promoted the Ni reducibility and decrease the Ni particle size and enhances the stability | [54] |
| Ni/CNT | IWI | T = 750 °C W/F = 1 g.h.mol−1 | XCH4 = 66 XCO2 = 79 | 0.9 | TOS 8 h, Ni particles inside the CNTs are more active than outside, tube confinement effect | [66] |
| Ni/Al2O3 | IWI | T = 850 °C GHSV = 24 L·g−1·h−1 | XCH4 = 90 XCO2 = 94 | 1 | preparation of nano catalyst with high SA, good dispersion, | [61] |
| Ni15/TiO2 | sonication | T = 700 °C, GHSV=95,500 h−1 H2O/CH4 = 1.2 | XCH4 = 86 XCO2 = 84 | 0.9 | sonication is more efficient than impregnation, fine dispersion of Ni NPs, | [67] |
| Rh1/NiO20-Al2O3 | sol-gel/IWI | T = 700 °C Qtot = 37 L min−1 CH4:CO2 = 1:1 | XCH4 = 89 XCO2 = 91 | 1.22 | 210 min TOS, deactivation of Rh avoided by NiAl2O4 spinel phase, Ni enhance the Rh dispersion on the surface | [68] |
| Catalyst | Prepa-ration Method | Reaction conditions | Conversion and selectivity (%) | H2/CO | comments | ref. |
|---|---|---|---|---|---|---|
| Rh0.005/γ-Al2O3 | IWI | T = 900 °C GHSV = 24,640 h−1 CH4/O2 = 2 | XCH4 = ~88 SCO = 90 SH2 = 90 | 2 | TOS 160 h, CH4 conversion and CO selectivity decrease with Rh NP size | [70] |
| Rh5-doped CeO2 | solution-based hydrothermal | T = 700 °C Qtot = 100 mL·min−1 CH4/O2 = 2 | XCH4 = 95 SH2 = 93 SCO = 86 | 2.2 | surface chemistries with in situ studies are important to correlate the catalysts performance | [69] |
| Rh-honeycombs in microreactor | micro-structuring Rh foils | T = 1100 °C, p = 0.15 Mpa, GHSV = 195·103 h−1 CH4/O2 = 2 | XCH4 = 90 SH2 = 88 SCO = 87 | 2 | t > 10 ms; high pressure used in Rh honeycomb catalyst; minimizing pressure drop | [71] |
| Ni-Th-O | controlled oxidation | T = 750 °C CH4/N2O = 1 GHSV = 8500 mL·gcat−1·h−1 | XCH4 = 50 SCO = 90 SH2 = 90 | 2 | using N2O as oxidant, bimetallic Ni thorium used, accessibility and acidity | [72] |
| Ni-Pt/La0.2Zr0.4Ce0.4O x | wash coating and IWI | T = 800 °C GHSV = 820,000 h−1 Qtot = 4.0 mL·s−1 τ = 4.4 ms CH4/O2 = 2 | XCH4 = 82 SCO = 60 | 1.5 | microchannel reactor, very short residence time, high flow rates | [73] |
| Ce7-Fe3-O mixed oxide | CP | T = 900 °C | XCH4 = 95 SH2 = 99 SCO = 98.8 | 2 | oxygen carrier showed good, lattice oxygen is crucial and high O mobility | [62] |
| Ni30/MgO | IWI | T = 800 °C P = 0.1 MPa GHSV = 1200 h−1 CH4/O2 = 2 | XCH4 = 94 SCO = 94 SH2 = 94 | 2 | higher conversion with increasing calcination temperature, highly Ni dispersed | [74] |
| Rh1.5/CeO2/ monolith cordierite | solution combustion synthesis | T = 800 °C GHSV = 400,000 mL gcat−1 h−1 S/C = 1.2 O/C = 0.55 | XCH4 = 100 | 3 | Structured catalysts cordierite honeycombs, high performance at high WSV, process intensification | [75] |
| (CeO2)5/Pt2-SiC | micro-emulsion | T = 805 °C CH4/O2 = 2 GHSV = 1500 h−1 | XCH4 = 93 | 1 | composites with optimal Ce and Pt content to achieve high activity, | [76] |
| (NiO)48/CeO2 | one-step template | T = 850 °C GHSV = 9.5 × 106 h−1 CH4/O2 = 2 | XCH4 = 90 SCO = 84 SH2 = 90 | 2 | Fibrous nanocatalyst, highly active at very short contact time of 98 μs, | [77] |
| Ni10/CeO2-SiO2 | sol-gel/DI | T = 750 °C CH4/O2 = 2 Qtot = 30 mL·min−1 | XCH4 = ~84% | 2 | calcined at 700 °C had a weak surface acidity with high surface area and low carbon formation, atomic Ce/Si = 1, 15 h TOS | [78] |
| Pt10Ce0.5Zr0.5O2/Al2 O3 | IWI | T = 900 °C Qtot = 100 mL·min−1 CH4/O2 = 2 | XCH4 = 70 XCO = 80 SH2 = 96 | high reducibility and O2 mobility reduce the carbon formation | [79] |
3.1.2. Methane Steam Reforming and Decomposition to H2
| Catalyst | Prepa-ration method | Reaction conditions # | Conversion and H2 selectivity (%), yield (% or mole) | Comments | Ref. |
|---|---|---|---|---|---|
| Ni10/ZrO2-CeO2-La2O3 | WI | T = 500 °C GHSV = 70,000 h−1 S/C = 3 | XCH4 = 25 YH2 = 30 | Low temperature steam reforming | [85] |
| Ni/K2TixOy-Al2O3 | WI | T = 750 °C GHSV = 15,000 h−1 S/C = 2.5 TOS = 10 h | XCH4 = 97 YH2 = 3 mol | high activity due to weak interactions between Ni and support, addition of secondary support K2TiO2 improved the resistance to deactivation | [86] |
| NiO/SiO2 | sol-gel | T = 700 °C ST = 11.31 kgcat h.kmol−1 S/C = 3.5 | XCH4 = 96 YH2 = 3.8 mol | crystallite size of NiO catalysts can be controlled by calcination | [81] |
| Ni/Ce0.65Hf0.25Pr0.1 O2 | EDTA-citrate | T = 700 °C S/C = 2 | XCH4 = 85 | Dopants redox property crucial in enhancing the OSC in rare earth metals-doped | [87] |
| Cu5/Co6Al2 | precipitation and IWI | T = 650 °C S/C = 3 Qtot = 50 mL·min−1 | XCH4 = 96 YH2 = 2.60 mol | Cu favors the WGS reaction | [88] |
| Ni0.5Mg2.5AlO9 | co-precipitation | T = 900 °C S/C = 3.1, Qtot = 50 mL·min−1 | XCH4 = 100 | 150 h, 20 ms, high WHSV, higher dispersion of Ni particles, small particles | [89] |
| Ru/Co6Al2 | IWI | T = 600 °C, S/C = 3, Qtot = 20 mL·min−1, | XCH4 = 95 YH2 = 24 | 100 h, well dispersed at the surface and higher Co loading | [90] |
| Ni0.15Al0.85 | Solution-combustion/ IWI | T = 850 °C Qtot = 100 mL·min−1 S/C = 4 | XCH4 = 97.8 YH2 = 2.9 mol | high surface area and strong interaction between Ni and Al | [91] |
| Ir5/MgAl2O4 | IWI | T = 850°C GHSV = 284,000 h−1 S/C = 3 | XCH4 = 55 | Ir particles bind stronglyMgAl2O4 surface via redox process leading to a strong metal–support interaction and activate facile water dissociation | [92] |
| Au/Ni5-LaAl | anionic exchange /IWI | T = 700 °C S/C = 1.24 GHSV = 135,000 mL·g−1.h−1 | XCH4 = 34 | Au addition enhance the stability and decreases the carbon growth rate | [93] |
3.2. From Methane to Chemicals

- (E6) Methane partial oxidationCH4 + 0.5O2 ↔ CH3OH ΔH°298 = 127 kJ.mol−1CH4 + O2 ↔ HCHO + H2O ΔH°298 = −276 kJ.mol−1
- (E7) Methane combustion or total oxidationCH4 + 2O2 ↔ 2H2O + CO2 ΔH°298 = −802 kJ.mol−1
- (E8) Oxidative coupling of Methane (OCM)2CH4 + 0.5O2 ↔ C2H6 + H2O ΔH°298 = −174.2 kJ.mol−1C2H6 + 0.5O2 ↔ C2H4 + H2O ΔH°298 = −104 kJ.mol−1
- (E9) Methane dehydroaromatization (MDA)6CH4 ↔ 9H2 + C6H6 ΔH°298K = 88.7 kJ.mol−1
- (E10) Methane halogenation and oxyhalogenationCH4 + HCl + 2O2 ↔ CH3Cl + HClCH4 + HCl + 0.5O2 ↔ CH3Cl + H2O
3.2.1. Catalysts for Selective Partial Oxidation of Methane to Oxygenates
| Catalyst | Preparation method | Reaction conditions | Conversion, selectivity and yield (%) | Comments | Ref. |
|---|---|---|---|---|---|
| nano-Au/SiO2 | HAuCl4 + IL in SiO2 sol | T = 90 °C p = 20 atm | XCH4 = 25 SCH3OH = 72 YCH3OH = 18 | [Bmim]Cl ionic liquid as dissolution solvent, Liquid phase oxidation, 97% Au is recovered | [104] |
| CuFe2(P2O7)2 | IWI | T = 630 °C pCH4 = 65 kPa pN2O = 26 kPa Qtot = 3.6 L/h, CH4/N2O = 1.2 | XCH4 = 6.4 SHCHO = 26.6 SCH3OH = 1.8 Y*CH3OH+HCHO = 1.8 | interactions and synergistic effects of Fe and Cu leads to enhanced activity, crystalline phase | [110] |
| Fe2O3-CuO/γ-Al2O3 | IWI | T = 300 °C | XCH4 = 43 YCH3OH = 1.5 | Plasma-catalysis, inside packed catalyst configuration is more effective than post catalysis | [105] |
| Y3/Cu6-Zn2.5-Al1.5 | co-precipitation | CH4/O2 = 4 | XCH4 = 25 SCH3OH = 27 | Plasma-Catalysis, dielectric barrier discharge addition of Yttrium, also Pt and Fe, found to enhance performance | [106] |
| (V2O5)0.03/SiO2 | IWI | T = 650 °C GHSV = 6.6 × 104 h−1 Qtot = 71 mL·min−1 CH4/O2 = 2 | XCH4 = 34 YCH3OH+HCHO = 16 | 1% NO in feed enhanced the conversion and selectivity | [111] |
| PPFe+3OH/AlSiMg Fe/protoporphyrin | Activation | T = 180 °C H2O2/CH4 = 1.4 | XCH4 =50 YCH3OH = 60 SCH3OH = 97 | Biomimetic enzyme catalysis, using H2O2 oxidant, highest hydroxylating activity due to active O2 carriers, deactivates in 5 h | [112] |
| MoO3/SiO2 | sol-gel | T = 500 | XCH4 = 2.9 SHCHO = 52 SCH3OH = 8 YCH3OH+HCHO = 0.86 | catalyst preparation method influences the products selectivity, formation of silicomolybdic acid on the surface reduce the successive oxidation | [113] |
3.2.2. Oxidative Coupling of Methane (OCM) to Ethylene
3.2.3. Non-oxidative or Pyrolysis of Methane to Aromatics
4. Catalytic Utilization of Volatile Organic Compounds
4.1. From Volatile Organic Compounds to Syngas and Hydrogen
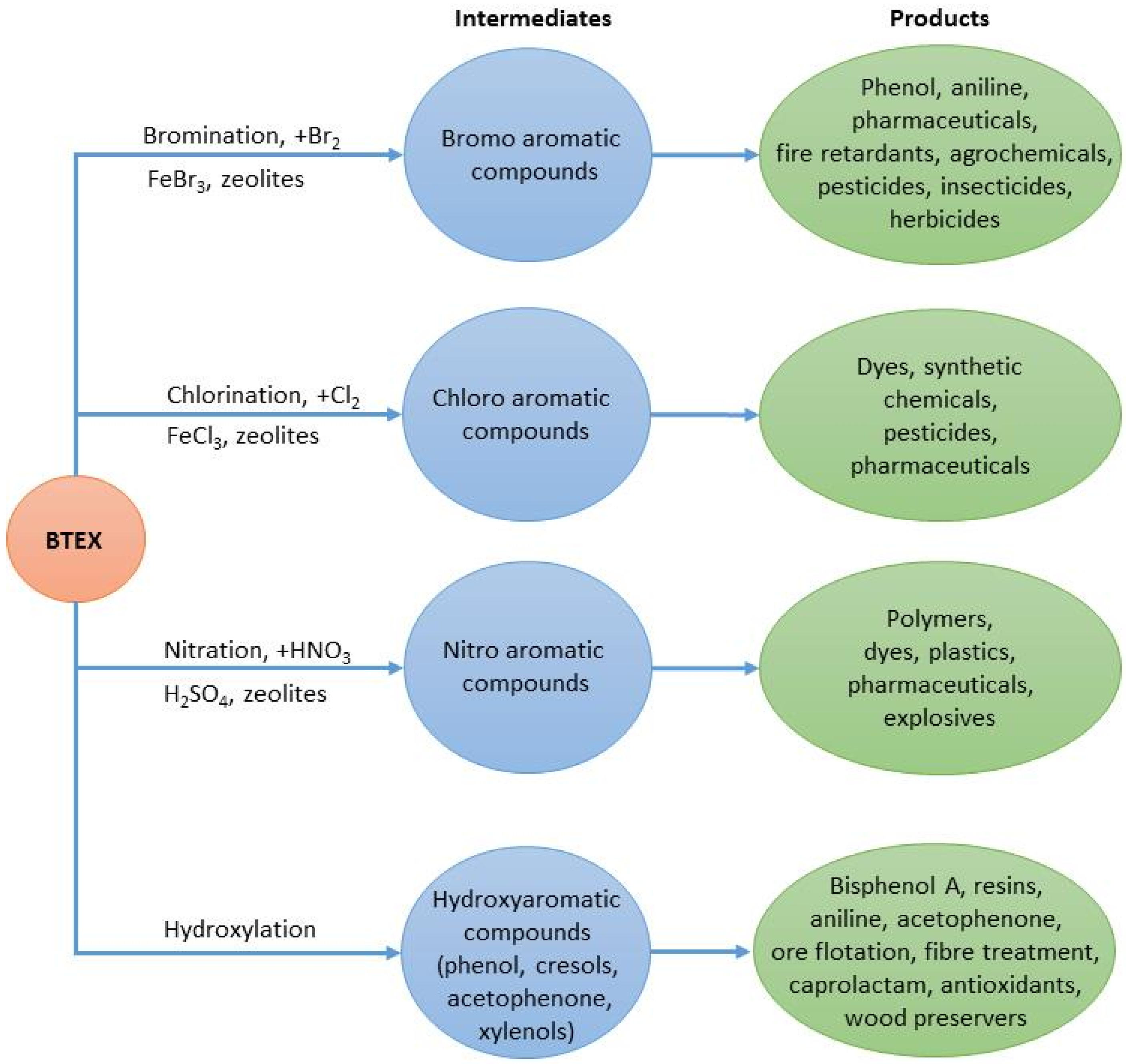
4.2. From Volatile Organic Compounds to Chemicals
| Sources of the Emission | Major Aromatic Compounds Emissions | Ref. |
|---|---|---|
| Vehicles exhaust (gasoline, diesel) | BTEX, trimethylbenzenes, ethylene, propene, 1-butene, ethane, acetylene, oxygenates | [182,183,200,201202] |
| Transportation | 1,3-butadiene, benzene, toluene, xylene, formaldehyde, acraldehyde | [207] |
| Fuel evaporation (gasoline, diesel) | Benzene, toluene, iso-pentane, pentenes, n-heptane | [182] |
| Biomass burning | BTEX, Benzene (major aromatic emission), acetylene, ethylene, propene, ethane, methylchloride, methanol, formaldehyde | [182,183,207] |
| Coal burning | BTEX, naphthalene, acetylene, ethylene, propylene, propane, ethane | [182,183] |
| Petrochemical industry | Styrene, benzene, hexane, methylcyclohexane, trichloroethylene, TRS compounds | [172,182,203,206] |
| Oil-refinery | Benzene, ethylene, hexane, cyclopentane, cyclohexane, methylcyclohexane, TRS compounds | [172,177,178,182,203] |
| Painting (Coating for building) | BTEX, styrene, n-butane | [182] |
| Electronics manufacturing (printed circuie board) | Toluene, 2-ethyl-1,3-dimethylbenzene, 1,2,4,5-tetramethylbenzene, ethanol, acetic acid, iso-propyl alcohol | [186] |
| Vehicle manufacturing | Toluene, ethylbenzene, p, o-xylene, trimethylbenzenes, ethyl acetate, 2-butanone, acetic acid | [186] |
| Flexographic Printing | o-xylene, acetic acid, ethyl acetate | [186] |
| Metal and plastic surface spraying | Ethylbenzene, p, o-xylene, ethyl acetate, 2-butanone, butyl acetate, ethanol | [186] |
| Furniture manufacturing | Toluene, ethylbenzene, p, o-xylene, trimethylbenzenes, trimethylbenzenes, acetic acid | [186] |
| Plastic waste recycling plants | BTEX, styrene, 1-butene, 2-hexene, pinenes, hexane, octane, 3-methylnonane | [194] |
| Iron and steel industry (cokemaking, sintering, hot forming, cold forming) | BTEX, trimethylbenzenes, isopentane, n-pentane, n-butane, methylhexanes, n-heptane, butenes, trichloroethylene, TRS compunds | [191,203] |
| Industrial solvent use (e.g., solvent production, paint and adhesive use) | Toluene, xylenes, n-hexane, oxygenates, substituted compounds | [200,201,202,205,207] |
| Pulp and paper production | oxygenates (mainly methanol), TRS compounds | 204 |
| Pharmaceutical industry | substituted compounds (alkyl halides) | 207 |
4.2.1. Potential Existing Technologies and New Considerations for VOC utilization in Chemicals’ Production
| Catalyst | Source compound | Oxidant | Conditions (°C) | Conversion | Product selectivity | Ref. |
|---|---|---|---|---|---|---|
| Zeolites | Benzene | N2O | 400–450 | N2O: 100% | Phenol: 80%–100% | [255,259] |
| Palladium membrane | Benzene | O2 | <250 | Benzene: 2%–16% | Phenol: 80%–97% | [259] |
| VO(acac)2 | Benzene | H2O2 | 65 | Benzene: 11% | Phenol: 100% | [264] |
| VO(acac)2 | Toluene | H2O2 | 65 | Toluene: 5% | o-cresol 37% m,p-cresol: 56% | [264] |
| VO(acac)2 | Anisole | H2O2 | 65 | Anisole:2% | p-hydroxyanisole: 26% | [264] |
| VOPc a | Benzene | H2O2 | 65 | Benzene: 22.4% | Phenol: 100% | [264] |
| VOPc a | Toluene | H2O2 | 65 | Toluene: 18.74% | o-cresol: 38% m,p-cresol: 57% | [264] |
| VOPc a | Anisole | H2O2 | 65 | Anisole: 6% | p-hydroxyanisole: 47% | [264] |
| Multi-walled carbon nanotubes | Benzene | - | 50–70 | Benzene: 2%–6% | Phenol: ~98% | [273] |
| Multi-walled carbon nanotubes | Toluene | - | 50–70 | - | o,m,p-cresol: <80% | [274] |
| H-[Al]ZSM-5 zeolites b | Benzene | N2O | 350 | Benzene: 22% | Phenol: ~98% | [267] |
| H-[Al]ZSM-5 zeolites b | Toluene | N2O | 350 | Toluene: 24% | - | [267] |
| H-[Al]ZSM-5 zeolites b | Anisole | N2O | 350 | Anisole:53% | - | [267] |

5. Considerations on Catalyst Durability in Utilization of Organic Emissions
6. Economic Issues Related to Utilization of Organic Emissions
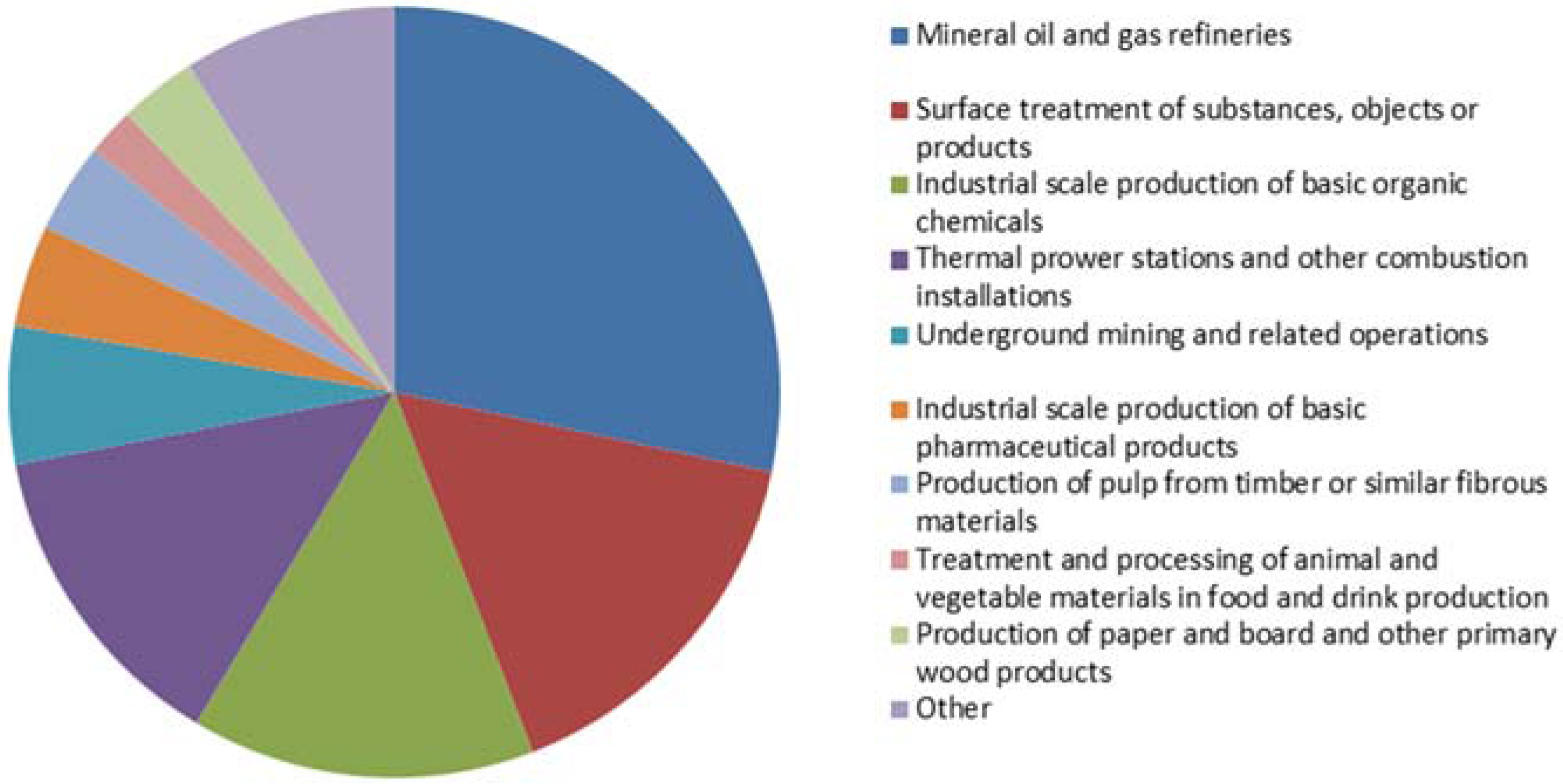
- (1)
- Energy sector
- (2)
- Production and processing of metals
- (3)
- Mineral industry
- (4)
- Chemical industry
- (5)
- Waste and waste water management
- (6)
- Paper and wood production processing
- (7)
- Intensive livestock production and aquaculture
- (8)
- Animal and vegetable products from the food and beverage sector
- (9)
- Other activities


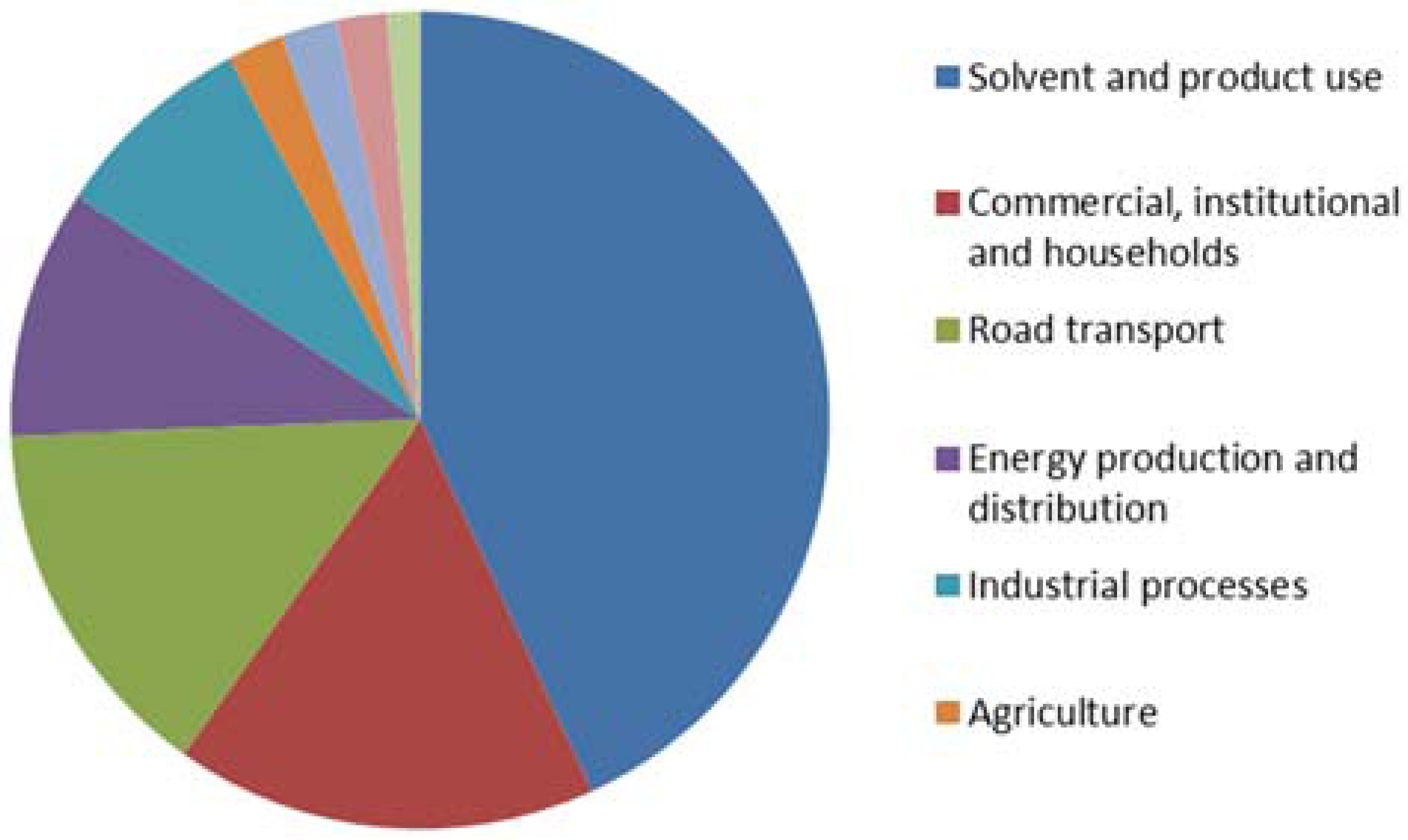
| substance class | Solvent use (%) | Transport (%) | Stationary combustion (%) | Production processes (%) |
|---|---|---|---|---|
| alkanes | 28 | 41 | 29 | 31 |
| alkenes | 1.3 | 16 | 19 | 15 |
| alkynes | - | 3 | 4 | 0.03 |
| aromatics | 22 | 27 | 37 | 5 |
| alcohols | 24 | - | - | 29 |
| aldehydes | - | 9 | 8 | 4 |
| esters | 9 | - | - | - |
| ethers | 1.3 | - | - | 2.4 |
| ketones | 6 | 1 | 2 | 0.2 |
| glycol derivates | 7 | - | - | - |
| halogenated hydrocarbons | 1.3 | - | - | 2.7 |
| carbonic acids | 0.1 | - | - | - |
| not allocated | - | 3 | 1 | 11 |
- (1)
- Stationary fuel combustion
- (2)
- Industrial processes
- (3)
- Highway vehicles
- (4)
- Non-road mobile
- (5)
- Miscellaneous excluding wildfires
7. Utilization of VOC Emissions and Sustainability
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ojala, S.; Pitkäaho, S.; Laitinen, T.; Niskala, N.; Brahmi, R.; Gaálová, J.; Matejova, L.; Kucherov, A.; Päivärinta, S.; Hirschmann, C.; et al. Catalysis in VOC abatement–A review. Top. Catal. 2011, 54, 1224–1256. [Google Scholar] [CrossRef]
- Council Directive 1999/13/EC of 11 March 1999 on the limitation of emissions of volatile organic compounds due to the use of organic solvents in certain activities and installations. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:31999L0013 (accessed on 28 February 2015).
- Directive 2010/75/EU of the European Parliament and of the Council of 24 November 2010 on industrial emissions (integrated pollution prevention and control). Available online: http://europa.eu/legislation_summaries/environment/soil_protection/ev0027_en.htm (accessed on 28 February 2015).
- Moretti, E.C. Reduce VOC and HAP Emissions. CEP Magazine. 2002, pp. 30–40. Available online: http://www.cepmagazine.org (accessed on 29 June 2015).
- Smith, N. Energy, Resources & Public Policy, Paper #2. 2003. Available online: http://www.brynmawr.edu/geology/206/smith2.htm (accessed on 29 June 2015).
- Su, S.; Beath, A.; Huo, H.; Mallett, C. An assessment of mine methane mitigation and utilization technologies. Prog. Energy Combust. Sci. 2005, 31, 123–170. [Google Scholar] [CrossRef]
- Belis-Bergouignan, M.-C.; Oltra, V.; Saint Jean, M. Trajectories towards clean technology: Example of volatile organic compound emission reductions. Ecol. Econ. 2004, 48, 201–220. [Google Scholar] [CrossRef]
- Melhus, Ö. Utilization of VOC in Diesel Engines. Ph.D. Thesis, Department of Marine Engineering, Norwegian University of Science and Technology, Trondheim, Norway, March 2002. [Google Scholar]
- US Department of Energy. Mid-Atlantic green energy application center, Penn State University, Clean energy–opportunity fuels–industrial by-products. Available online: http://www.maceac.psu.edu/clean_energy_opportunity_fuels_industrial.htm (accessed on 1 March 2015).
- Natural Resources Defense Council, NFDC. Renewable Energy for America, Harvesting the benefits of homegrown, renewable energy. Biogas Energy. Available online: http://www.nrdc.org/energy/renewables/biogas.asp (accessed on 29 June 2015).
- Nunez, C. VOCs: Sources, Definitions, and Considerations for Recovery, US EPA Seminar, EPA625/R-99/005, presented in 16 September 1998. Available online: http://nepis.epa.gov/ (accessed on 27 February 2015).
- BAT reference document for Common Waste Water and Waste Gas Treatment/Management Systems in the Chemical Sector. Available online: http://eippcb.jrc.ec.europa.eu/reference/BREF/CWW_Final_Draft_07_2014.pdf (accessed on 20 February 2015).
- Schenk, E.; Mieog, J.; Evers, D. Fact Sheets on Air Emission Abatement Techniques; DHV B.V.: Amersfoort, The Netherlands, 2009. [Google Scholar]
- Wu, J. Modeling Adsorption of Organic Compounds on Activated Carbon. Ph.D. Thesis, University of Umeå, Umeå, Sweden, 24 September 2004. [Google Scholar]
- Cruz, G.; Pirilä, M.; Huuhtanen, M.; Carrión, L.; Alvarenga, E.; Keiski, R.L. Production of Activated Carbon from Cocoa (Theobroma cacao) Pod Husk. J. Civ. Environ. Eng. 2012, 2, 109. [Google Scholar] [CrossRef]
- Girgis, B.S.; El-Hendawy, A.N.A. Porosity development in activated carbons obtained from date pits under chemical activation with phosphoric acid. Microporous Mesoporous Mater. 2002, 52, 105–117. [Google Scholar] [CrossRef]
- Olivares-Marín, M.; Fernández-González, C.; Macías-García, A.; Gómez-Serrano, V. Preparation of activated carbon from cherry stones by chemical activation with ZnCl2. Appl. Surf. Sci. 2006, 252, 5967–5971. [Google Scholar] [CrossRef]
- Okman, I.; Karagöz, S.; Tay, T.; Erdem, M. Activated Carbons from Grape Seeds by Chemical Activation with Potassium Carbonate and Potassium Hydroxide. Appl. Surf. Sci. 2014, 293, 138–142. [Google Scholar] [CrossRef]
- Subrenat, A.S.; le Cloirec, P.A. Volatile organic compound (VOC) removal by adsorption onto activated carbon fiber cloth and electrothermal desorption: An industrial application. Chem. Eng. Commun. 2006, 193, 478–486. [Google Scholar] [CrossRef]
- Khan, F.I.; Ghoshal, A.K. Removal of volatile organic compounds from polluted air. J. Loss Prev. Process Ind. 2000, 13, 527–545. [Google Scholar] [CrossRef]
- Anonymous. Hydrocarbon Processing’s Environmental Processes’ 98. Hydrocarbon Processing 1998, 77, 69–112. [Google Scholar]
- Engleman, V.S. Updates on choices of appropriate technology for control of VOC emissions. Met. Finish. 2000, 98, 433–445. [Google Scholar] [CrossRef]
- Reference Document on Best Available Techniques in the Large Volume Organic Chemical Industry. Available online: http://eippcb.jrc.ec.europa.eu/reference/BREF/lvo_bref_0203.pdf (accessed on 25 February 2015).
- Kruger, D.; Schultz, K.A. Guide for Methane Mitigation Projects: Gas-to-Energy at Coal Mines; US Environmental Protection Agency, Office of Air and Radiation 6202J: Washington, DC, USA, 1996.
- Xebec Adsorption Inc. Treatment solutions for landfill gas fuel applications. 2007. Available online: http://www.xebecinc.com/pdf/e_white_paper.pdf (accessed on 26 February 2015).
- Caballero, A.; Perez, P.J. Methane as raw material in synthetic chemistry: The final frontier. Chem. Soc. Rev. 2013, 42, 8809. [Google Scholar] [CrossRef] [PubMed]
- Kerr, R.A. Arctic Armageddon needs More Science, Less Hype. Science 2010, 329, 620–621. [Google Scholar] [CrossRef] [PubMed]
- US EPA. Summary Report: Global Anthropogenic Non-CO2 Greenhouse Gas Emissions: 1990–2030; EPA 430-S-12-002; EPA: Washington, DC, USA, 2012.
- Bousquet, P.; Tyler, S.C.; Peylin, P.; van der Werf, G.R.; Prigent, C.; Hauglustaine, D.A.; Dlugokencky, E.J.; Miller, J.B.; Ciais, P.; White, J.; et al. Contribution of anthropogenic and natural sources to atmospheric methane variability. Nature 2006, 443, 439–443. [Google Scholar] [CrossRef] [PubMed]
- McFarland, E. Unconventional Chemistry for Unconventional Natural Gas. Science 2012, 338, 340–342. [Google Scholar] [CrossRef] [PubMed]
- GGFR. Available online: http://www.flaringreductionforum.org/agenda.html (accessed on 1 March 2015).
- Ibitoye, F.I. Ending Natural Gas Flaring in Nigeria’s Oil Fields. J. Sustain. Dev. 2014, 7, 3. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, J.; Zhang, L.; Zheng, S.; Guo, M.; Yana, Y. Catalytic combustion of low-concentration coal bed methane over CuO/γ-Al2O3 catalyst: Effect of SO2. RSC Adv. 2014, 4, 39394–39399. [Google Scholar] [CrossRef]
- Horn, R.; Schlögl, R. Methane activation by heterogeneous catalysis. Catal. Lett. 2014, 145, 23–39. [Google Scholar] [CrossRef]
- Alvarez-Galvan, M.C.; Mota, N.; Ojeda, M.; Rojas, S.; Navarro, R.M.; Fierro, J.L.G. Direct methane conversion routes to chemicals and fuels. Catal. Today 2011, 171, 15–23. [Google Scholar] [CrossRef]
- Iglesia, E. Challenges and progress in the conversion of natural gas to fuels and chemicals. Fuel Chem. Div. Prepr. 2002, 47, 128. [Google Scholar]
- Lunsford, J.H. Catalytic conversion of methane to more useful chemicals and fuels: A challenge for the 21st century. Catal. Today 2000, 63, 165–174. [Google Scholar] [CrossRef]
- Tang, P.; Zhu, Q.; Wu, Z.; Ma, D. Methane activation: The past and future. Energy Environ. Sci. 2014, 7, 2580–2591. [Google Scholar] [CrossRef]
- Eliasson, B.; Liu, C.; Kogelschatz, U. Direct conversion of methane and carbon dioxide to higher hydrocarbons using catalytic dielectric-barrier discharges with zeolites. Ind. Eng. Chem. Res. 2000, 39, 1221–1227. [Google Scholar] [CrossRef]
- Lavoie, J.-M. Review on dry reforming of methane, a potentially more environmentally-friendly approach to the increasing natural gas exploitation. Front. Chem. 2014, 2, 81. [Google Scholar] [CrossRef] [PubMed]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; La Parola, V.; Pantaleo, G.; Puleo, F.; Venezia, A.M.; Liotta, L.F. Ni-Based Catalysts for Low Temperature Methane Steam Reforming: Recent Results on Ni-Au and Comparison with Other Bi-Metallic Systems. Catalysts 2013, 3, 563–583. [Google Scholar] [CrossRef]
- Budiman, A.W.; Song, S.H.; Chang, T.S.; Shin, C.H.; Choi, M.J. Dry reforming of methane over cobalt catalysts: A literature review of catalyst development. Catal. Surv. Asia 2012, 16, 183–197. [Google Scholar] [CrossRef]
- Fan, M.S.; Abdullah, A.Z.; Bhatia, S. Catalytic technology for carbon dioxide reforming of methane to synthesis gas. ChemCatChem 2009, 1, 192–208. [Google Scholar] [CrossRef]
- Ding, R.G.; Yan, Z.F.; Song, L.H.; Liu, X.M. A review of dry reforming of methane over various catalysts. J. Nat. Gas Chem. 2001, 10, 237–255. [Google Scholar]
- Al-Sayari, S.A. Recent Developments in the Partial Oxidation of Methane to Syngas. Open Catal. J. 2013, 6, 17–28. [Google Scholar] [CrossRef]
- Enger, B.C.; Lødeng, R.; Holmen, A. A review of catalytic partial oxidation of methane to synthesis gas with emphasis on reaction mechanisms over transition metal catalysts. Appl. Catal. A 2008, 346, 1–27. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhao, X.; Deng, Y. Advances in the partial oxidation of methane to synthesis gas. J. Nat. Gas Chem. 2004, 13, 191–203. [Google Scholar]
- Zheng, X.; Tan, S.; Dong, L.; Li, S.; Chen, H. Silica-coated LaNiO3 nanoparticles for non-thermal plasma assisted dry reforming of methane: Experimental and kinetic studies. Chem. Eng. J. 2015, 265, 147–156. [Google Scholar] [CrossRef]
- Ananth, A.; Gandhi, M.S.; Mok, Y.S. A dielectric barrier discharge (DBD) plasma reactor: An efficient tool to prepare novel RuO2 nanorods. J. Phys. D 2013, 46, 155–202. [Google Scholar] [CrossRef]
- Kaydouh, M.N.; El Hassan, N.; Davidson, A.; Casale, S.; El Zakhem, H.; Massiani, P. Effect of the order of Ni and Ce addition in SBA-15 on the activity in dry reforming of methane. Comptes Rendus Chim. 2015, 18, 293–301. [Google Scholar] [CrossRef]
- Gould, T.D.; Montemore, M.M.; Lubers, A.M.; Ellis, L.D.; Weimer, A.W.; Falconer, J.L.; Medlin, J.W. Enhanced dry reforming of methane on Ni and Ni-Pt catalysts synthesized by atomic layer deposition. Appl. Catal. A 2015, 492, 107–116. [Google Scholar] [CrossRef]
- Aw, M.S.; Črnivec, I.G.O.; Djinović, P.; Pintar, A. Strategies to enhance dry reforming of methane: Synthesis of ceria-zirconia/nickel-cobalt catalysts by freeze-drying and NO calcination. Int. J. Hydrogen Energy 2014, 39, 12636–12647. [Google Scholar] [CrossRef]
- Cai, W.-J.; Qian, L.-P.; Yue, B.; He, H.-Y. Rh doping effect on coking resistance of Ni/SBA-15 catalysts in dry reforming of methane. Chin. Chem. Lett. 2014, 25, 1411–1415. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- De Miguel, S.R.; Vilella, I.M.J.; Maina, S.P.; San José-Alonso, D.; Roman-Martinez, M.C.; Illan-Gomez, M.J. Influence of Pt addition to Ni catalysts on the catalytic performance for long term dry reforming of methane. Appl. Catal. A 2012, 435, 10–18. [Google Scholar] [CrossRef]
- Silva, P.P.; Silva, F.A.; Portela, L.S.; Mattos, L.V.; Noronha, F.B.; Hori, C.E. Effect of Ce/Zr ratio on the performance of Pt/CeZrO2/Al2O3 catalysts for methane partial oxidation. Catal. Today 2005, 108, 734–740. [Google Scholar] [CrossRef]
- Bao, Z.; Lu, Y.; Han, J.; Li, Y.; Yu, F. Highly active and stable Ni-based bimodal pore catalyst for dry reforming of methane. Appl. Catal. A 2015, 491, 116–126. [Google Scholar] [CrossRef]
- Littlewood, P.; Xie, X.; Bernicke, M.; Thomas, A.; Schomäcker, R. Ni0.05Mn0.95O catalysts for the dry reforming of methane. Catal. Today 2015, 242, 111–118. [Google Scholar] [CrossRef]
- Yonggang, W.; Hua, W.; Kongzhai, L. Ce-Fe-O mixed oxide as oxygen carrier for the direct partial oxidation of methane to syngas. J. Rare Earths 2010, 28, 560. [Google Scholar]
- Talkhoncheh, S.K.; Haghighi, M. Syngas production via dry reforming of methane over Ni-based nanocatalyst over various supports of clinoptilolite, ceria and alumina. J. Nat. Gas Sci. Eng. 2015, 23, 16–25. [Google Scholar] [CrossRef]
- Jabbour, K.; El Hassan, N.; Davidson, A.; Massiani, P.; Casale, S. Characterizations and performances of Ni/diatomite catalysts for dry reforming of methane. Chem. Eng. J. 2015, 264, 351–358. [Google Scholar] [CrossRef]
- Alenazey, F.S. Utilizing carbon dioxide as a regenerative agent in methane dry reforming to improve hydrogen production and catalyst activity and longevity. Int. J. Hydrogen Energy 2014, 39, 18632–18641. [Google Scholar] [CrossRef]
- Sokolov, S.; Kondratenko, E.V.; Pohl, M.M.; Rodemerck, U. Effect of calcination conditions on time on-stream performance of Ni/La2O3-ZrO2 in low-temperature dry reforming of methane. Int. J. Hydrogen Energy 2013, 38, 16121–16132. [Google Scholar] [CrossRef]
- Benrabaa, R.; Löfberg, A.; Caballero, J.G.; Bordes-Richard, E.; Rubbens, A.; Vannier, R.N.; Barama, A. Sol-gel synthesis and characterization of silica supported nickel ferrite catalysts for dry reforming of methane. Catal. Commun. 2015, 58, 127–131. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, D.; Wu, M.; Zhao, T.; Yoneyama, Y.; Tsubaki, N. Effect of catalytic site position: Nickel nanocatalyst selectively loaded inside or outside carbon nanotubes for methane dry reforming. Fuel 2013, 108, 430–438. [Google Scholar] [CrossRef]
- Shinde, V.M.; Madras, G. Catalytic performance of highly dispersed Ni/TiO2 for dry and steam reforming of methane. RSC Adv. 2014, 4, 4817–4826. [Google Scholar] [CrossRef]
- Drif, A.; Bion, N.; Brahmi, R.; Ojala, S.; Turpeinen, E.; Seelam, P.K.; Pirault-Roy, L.; Keiski, R.; Epron, F. Study of the dry reforming of methane and ethanol using Rh catalysts supported on doped alumina. Appl. Catal. A 2015, in press. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, S.; Shan, J.J.; Nguyen, L.; Zhan, S.; Gu, X.; Tao, F. In situ surface chemistries and catalytic performances of ceria doped with palladium, platinum, and rhodium in methane partial oxidation for the production of syngas. ACS Catal. 2013, 3, 2627–2639. [Google Scholar] [CrossRef]
- Kondratenko, V.A.; Berger-Karin, C.; Kondratenko, E.V. Partial Oxidation of Methane to Syngas Over γ-Al2O3-Supported Rh Nanoparticles: Kinetic and Mechanistic Origins of Size Effect on Selectivity and Activity. ACS Catal. 2014, 4, 3136–3144. [Google Scholar] [CrossRef]
- Fichtner, M.; Mayer, J.; Wolf, D.; Schubert, K. Microstructured rhodium catalysts for the partial oxidation of methane to syngas under pressure. Ind. Eng. Chem. Res. 2001, 40, 3475–3483. [Google Scholar] [CrossRef]
- Branco, J.B.; Ferreira, A.C.; Botelho do Rego, A.M.; Ferraria, A.M.; Almeida-Gasche, T. Conversion of Methane over Bimetallic Copper and Nickel Actinide Oxides (Th, U) Using Nitrous Oxide As Oxidant. ACS Catal. 2012, 2, 2482–2489. [Google Scholar] [CrossRef]
- Makarshin, L.L.; Sadykov, V.A.; Andreev, D.V.; Gribovskii, A.G.; Privezentsev, V.V.; Parmon, V.N. Syngas production by partial oxidation of methane in a microchannel reactor over a Ni-Pt/La0.2Zr0.4Ce0.4Ox catalyst. Fuel Process. Technol. 2015, 131, 21–28. [Google Scholar] [CrossRef]
- Requies, J.; Cabrero, M.A.; Barrio, V.L.; Güemez, M.B.; Cambra, J.F.; Arias, P.L.; Pérez-Alonso, F.J.; Ojeda, M.; Peña, M.A.; Fierro, J.L.G. Partial oxidation of methane to syngas over Ni/MgO and Ni/La2O3 catalysts. Appl. Catal. A 2005, 289, 214–223. [Google Scholar] [CrossRef]
- Vita, A.; Cristiano, G.; Italiano, C.; Specchia, S.; Cipitì, F.; Specchia, V. Methane oxy-steam reforming reaction: Performances of Ru/γ-Al2O3 catalysts loaded on structured cordierite monoliths. Int. J. Hydrogen Energy 2014, 32, 18592–18603. [Google Scholar] [CrossRef]
- Frind, R.; Borchardt, L.; Kockrick, E.; Mammitzsch, L.; Petasch, U.; Herrmann, M.; Kaskel, S. Complete and partial oxidation of methane on ceria/platinum silicon carbide nanocomposites. Catal. Sci. Technol. 2012, 2, 139–146. [Google Scholar] [CrossRef]
- Dong, D.; Shao, X.; Wang, Z.; Lievens, C.; Yao, J.; Wang, H.; Li, C.-Z. Fibrous NiO/CeO2 nanocatalysts for the partial oxidation of methane at microsecond contact times. RSC Adv. 2013, 3, 1341–1345. [Google Scholar] [CrossRef]
- Hu, J.; Yu, C.; Bi, Y.; Wei, L.; Chen, J.; Chen, X. Preparation and characterization of Ni/CeO2-SiO2 catalysts and their performance in catalytic partial oxidation of methane to syngas. Chin. J. Catal. 2014, 35, 8–20. [Google Scholar] [CrossRef]
- Silva, F.A.; Resende, K.A.; da Silva, A.M.; de Souza, K.R.; Mattos, L.V.; Montes, M.; Souza-Aguiar, E.F.; Noronha, F.B.; Hori, C.E. Syngas production by partial oxidation of methane over Pt/CeZrO2/Al2O3 catalysts. Catal. Today 2012, 180, 111–116. [Google Scholar] [CrossRef]
- Holladay, J.D.; Hu, J.; King, D.L.; Wang, Y. An overview of hydrogen production technologies. Catal. Today 2009, 139, 244–260. [Google Scholar] [CrossRef]
- Bej, B.; Pradhan, N.C.; Neogi, S. Production of hydrogen by steam reforming of methane over alumina supported nano-NiO/SiO2 catalyst. Catal. Today 2013, 207, 28–35. [Google Scholar] [CrossRef]
- Seelam, P.K.; Liguori, S.; Iulianelli, A.; Pinacci, P.; Calabrò, V.; Huuhtanen, M.; Keiski, R.; Piemonte, V.; Tosti, S.; Falco, M.; et al. Hydrogen production from bio-ethanol steam reforming reaction in a Pd/PSS membrane reactor. Catal. Today 2012, 193, 42–48. [Google Scholar] [CrossRef]
- Iulianelli, A.; Seelam, P.K.; Liguori, S.; Longo, T.; Keiski, R.; Calabrò, V.; Basile, A. Hydrogen production for PEM fuel cell by gas phase reforming of glycerol as byproduct of bio-diesel. The use of a Pd-Ag membrane reactor at middle reaction temperature. Int. J. Hydrogen Energy 2011, 36, 3827–3834. [Google Scholar] [CrossRef]
- Saraswat, S.K.; Pant, K.K. NieCueZn/MCM-22 catalysts for simultaneous production of hydrogen and multiwall carbon nanotubes via thermo-catalytic decomposition of methane. Int. J. Hydrogen Energy 2011, 36, 13352–13360. [Google Scholar] [CrossRef]
- Angeli, S.D.; Pilitsis, F.G.; Lemonidou, A.A. Methane steam reforming at low temperature: Effect of light alkanes’ presence on coke formation. Catal. Today 2015, 242, 119–128. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lim, H.; Woo, H.C. Catalytic activity and characterizations of Ni/K2TixOy-Al2O3 catalyst for steam methane reforming. Int. J. Hydrogen Energy 2014, 39, 17645–17655. [Google Scholar] [CrossRef]
- Harshini, D.; Hyung, D.; Lee, J.; Jeong, Y.; Kim, S.; Woo Nam, H.C.; Ham, J.H.H.; Lim, T.-H.; Yoon, C.W. Enhanced oxygen storage capacity of Ce0.65Hf0.25M0.1O2δ (M = rare earth elements): Applications to methane steam reforming with high coking resistance. Appl. Catal. B 2014, 148–149, 415–423. [Google Scholar] [CrossRef]
- Homsi, D.; Aouad, S.; Gennequin, C.; El Nakat, J.; Aboukaïs, A.; Abi-Aad, E. The effect of copper content on the reactivity of Cu/Co6Al2 solids in the catalytic steam reforming of methane reaction. Comptes Rendus Chim. 2014, 17, 454–458. [Google Scholar] [CrossRef]
- Zhai, X.; Ding, S.; Liu, Z.; Jin, Y.; Cheng, Y. Catalytic performance of Ni catalysts for steam reforming of methane at high space velocity. Int. J. Hydrogen Energy 2011, 36, 482–489. [Google Scholar] [CrossRef]
- Homsi, D.; Aouad, S.; Gennequin, C.; Aboukaïs, A.; Abi-Aad, E. A highly reactive and stable Ru/Co6−xMgxAl2 catalyst for hydrogen production via methane steam reforming. Int. J. Hydrogen Energy 2014b, 39, 10101–10107. [Google Scholar] [CrossRef]
- Lim, M.-W.; Yong, S.-T.; Chai, S.-P. Combustion-synthesized Nickel-based Catalysts for the Production of hydrogen from steam reforming of methane. Energy Procedia 2014, 61, 910–913. [Google Scholar] [CrossRef]
- Mei, D.; Glezakou, V.-A.; Lebarbier, V.; Kovarik, L.; Wan, H.; Albrecht, K.O.; Gerber, M.; Rousseau, R.; Dagle, R.A. Highly active and stable MgAl2O4-supported Rh and Ir catalysts for methane steam reforming: A combined experimental and theoretical study. J. Catal. 2014, 316, 11–23. [Google Scholar] [CrossRef]
- Palma, S.; Bobadilla, L.F.; Corrales, A.; Ivanova, S.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. Effect of gold on a NiLaO3 perovskite catalyst for methane steam reforming. Appl. Catal. B 2014, 144, 846–854. [Google Scholar] [CrossRef]
- Kondratenko, V.A. Mechanistic aspects of the Andrussow process over Pt-Rh gauzes. Pathways of formation and consumption of HCN. Appl. Catal. A 2010, 381, 74–82. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, B.; Zhang, Q.; Deng, W.; Wang, Y.; Yang, Y. Recent advances in heterogeneous selective oxidation catalysis for sustainable chemistry. Chem. Soc. Rev. 2014, 43, 3480–3524. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, W.R.; Ciuparu, D.; Pfefferle, L.D. Combustion of Methane over palladium-based catalysts: Catalytic deactivation and Role of the Support. J. Phys. Chem. C 2012, 116, 8587–8593. [Google Scholar] [CrossRef]
- Cargnello, M.; Delgado Jaén, J.J.; Hernández Garrido, J.C.; Bakhmutsky, K.; Montini, T.; Calvino Gámez, J.J.; Gorte, R.J.; Fornasiero, P. Exceptional Activity for Methane Combustion over Modular Pd@CeO2 Subunits on Functionalized Al2O3. Science 2012, 337, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Nozakia, T.; Agıral, A.; Yuzawa, S.; Han Gardeniers, J.G.E.; Okazaki, K. A single step methane conversion into synthetic fuels using microplasma reactor. Chem. Eng. J. 2011, 166, 288–293. [Google Scholar] [CrossRef]
- Holmen, A. Direct conversion of methane to fuels and chemicals. Catal. Today 2009, 142, 2–8. [Google Scholar] [CrossRef]
- Oshima, K.; Tanaka, K.; Yabe, T.; Kikuchi, E.; Sekine, Y. Oxidative coupling of methane using carbon dioxide in an electric field over La–ZrO2 catalyst at low external temperature. Fuel 2013, 107, 879–881. [Google Scholar] [CrossRef]
- Alayon, E.M.C. Copper Cores for the Conversion of Methane to Chemicals. Ph.D. Thesis, ETH Zurich for the degree of Doctor of Sciences, Zürich, Switzerland, 2012. [Google Scholar]
- Wei, X.; Ye, L.; Yuan, Y. Low temperature catalytic conversion of methane to formic acid by simple vanadium compound with use of H2O2. J. Nat. Gas Chem. 2009, 18, 295–299. [Google Scholar] [CrossRef]
- Villa, K.; Murcia-López, S.; Andreu, T.; Ramón Morante, J. Mesoporous WO3 photocatalyst for the partial oxidation of methane to methanol using electron scavengers. Appl. Catal. B 2015, 163, 150–155. [Google Scholar] [CrossRef]
- Li, T.; Wang, S.J.; Yu, C.S.; Ma, Y.C.; Li, K.L.; Lin, L.W. Direct conversion of methane to methanol over nano-[Au/SiO2] in [Bmim]Cl ionic liquid. Appl. Catal. A 2011, 398, 150–154. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.-W.; Huang, L.; Lei, L.-C. Partial oxidation of methane with air for methanol production in a post-plasma catalytic system. Chem. Eng. Process. 2009, 48, 1333–1340. [Google Scholar] [CrossRef]
- Indarto, A.; Ryook Yang, D.; Palgunadi, J.; Choi, J.-W.; Lee, H.; Song, H.K. Partial oxidation of methane with Cu-Zn-Al catalyst in a dielectric barrier discharge. Chem. Eng. Process. 2008, 47, 780–786. [Google Scholar] [CrossRef]
- Adebajo, M.O.; Frost, R.L. Recent Advances in Catalytic/Biocatalytic Conversion of Greenhouse Methane and Carbon Dioxide to Methanol and Other Oxygenates. In Greenhouse Gases-Capturing, Utilization and Reduction; Liu, G., Ed.; InTech Europe: Rijeka, Croatia, 2012; pp. 31–56. [Google Scholar]
- Hameed, A.; Ismail, I.M.I.; Aslam, M.; Gondal, M.A. Photocatalytic conversion of methane into methanol: Performance of silver impregnated WO3. Appl. Catal. A 2014, 470, 327–335. [Google Scholar] [CrossRef]
- Yuan, Q.; Deng, W.; Zhang, Q.; Wang, Y. Osmium-Catalyzed Selective Oxidations of Methane and Ethane with Hydrogen Peroxide in Aqueous Medium. Adv. Synth. Catal. 2007, 349, 1199–1209. [Google Scholar] [CrossRef]
- Polnišer, R.; Štolcová, M.; Hronec, M.; Mikula, M. Structure and reactivity of copper iron pyrophosphate catalysts for selective oxidation of methane to formaldehyde and methanol. Appl. Catal. A 2011, 400, 122–130. [Google Scholar] [CrossRef]
- Barbero, J.A.; Alvarez, M.C.; Bañares, M.A.; Peña, M.A.; Fierro, J.L.G. Breakthrough in the direct conversion of methane into C1-oxygenates. Chem. Commun. 2002, 1184–1185. [Google Scholar] [CrossRef]
- Nagiev, T.M.; Abbasova, M.T. Oxidation of methane to methanol by hydrogen peroxide on a supported hematin catalyst. Stud. Surf. Sci. Catal. 2000, 130, 3837–3842. [Google Scholar]
- Aoki, K.; Ohmae, M.; Nanba, T.; Takeishi, K.; Azuma, N.; Ueno, A.; Ohfune, H.; Hayashi, H.; Udagawa, Y. Direct conversion of methane into methanol over MoO3/SiO2 catalyst in an excess amount of water vapor. Catal. Today 1998, 45, 29–33. [Google Scholar] [CrossRef]
- Hiyoshi, N.; Ikeda, T. Oxidative coupling of methane over alkali chloride-Mn-Na2WO4/SiO2 catalysts: Promoting effect of molten alkali chloride. Fuel Process. Technol. 2015, 133, 29–34. [Google Scholar] [CrossRef]
- Vatani, A.; Jabbari, E.; Askarieh, M.; Torangi, A.M. Kinetic modeling of oxidative coupling of methane over Li/MgO catalyst by genetic algorithm. J. Nat. Gas Sci. Eng. 2014, 20, 347–356. [Google Scholar] [CrossRef]
- Ivanov, D.V.; Isupova, L.A.; Gerasimov, E.Y.; Dovlitova, L.S.; Glazneva, T.S.; Prosvirin, I.P. Oxidative methane coupling over Mg, Al, Ca, Ba, Pb-promoted SrTiO3 and Sr2TiO4: Influence of surface composition and microstructure. Appl. Catal. A 2014, 485, 10–19. [Google Scholar] [CrossRef]
- Ferreira, V.J.; Tavares, P.; Figueiredo, J.L.; Faria, J.L. Ce-Doped La2O3 based catalyst for the oxidative coupling of methane. Catal. Commun. 2013, 42, 50–53. [Google Scholar] [CrossRef]
- Wang, Z.; Zou, G.; Luo, X.; Liu, H.; Gao, R.; Chou, L.; Wang, X. Oxidative coupling of methane over BaCl2-TiO2-SnO2 catalyst. J. Nat. Gas Chem. 2012, 21, 49–55. [Google Scholar] [CrossRef]
- Farsi, A.; Ghader, S.; Moradi, A.; Mansouri, S.S.; Shadravan, V. A simple kinetic model for oxidative coupling of methane over La0.6Sr0.4Co0.8Fe0.2O3−δ nanocatalyst. J. Nat. Gas Chem. 2011, 20, 325–333. [Google Scholar] [CrossRef]
- Godini, H.R.; Gili, A.; Görke, O.; Arndt, S.; Simon, U.; Thomas, A.; Schomäcker, R.; Wozny, G. Sol–gel method for synthesis of Mn-Na2WO4/SiO2 catalyst for methane oxidative coupling. Catal. Today 2014, 236, 12–22. [Google Scholar] [CrossRef]
- Majhi, S.; Dalai, A.K.; Pant, K.K. Methanol assisted methane conversion for higher hydrocarbon over bifunctional Zn-modified Mo/HZSM-5 catalyst. J. Mol. Catal. A 2015, 398, 368–375. [Google Scholar] [CrossRef]
- Abdelsayed, V.; Shekhawat, D.; Smith, M.W. Effect of Fe and Zn promoters on Mo/HZSM-5 catalyst for methane dehydroaromatization. Fuel 2015, 139, 401–410. [Google Scholar] [CrossRef]
- Kenarsari, S.D.; Jiang, D.Y.G.; Zhang, S.; Wang, J.; Russell, A.G.; Wei, Q.; Fan, M. Review of recent advances in carbon dioxide separation and capture. RSC Adv. 2013, 3, 22739–22773. [Google Scholar] [CrossRef]
- Cao, Z.; Jiang, H.; Luo, H.; Baumann, S.; Meulenberg, W.A.; Assmann, J.; Mleczko, L.; Liu, Y.; Caro, J. Natural Gas to Fuels and Chemicals: Improved Methane Aromatization in an Oxygen-Permeable Membrane Reactor. Angew. Chem. Int. Ed. 2013, 52, 13794–13797. [Google Scholar] [CrossRef] [PubMed]
- Schädel, B.T.; Duisberg, M.; Deutschmann, O. Steam reforming of methane, ethane, propane, butane, and natural gas over a rhodium-based catalyst. Catal. Today 2009, 142, 42–51. [Google Scholar] [CrossRef]
- Huang, X.; Reimert, R. Kinetics of steam reforming of ethane on Ni/YSZ (yttria-stabilised zirconia) catalyst. Fuel 2013, 106, 380–387. [Google Scholar] [CrossRef]
- Olafsen, A.; Slagtern, Å.; Dahl, I.M.; Olsbye, U.; Schuurman, Y.; Mirodatos, C. Mechanistic features for propane reforming by carbon dioxide over a Ni/Mg (Al) O hydrotalcite-derived catalyst. J. Catal. 2005, 229, 163–175. [Google Scholar] [CrossRef]
- Resini, C.; Delgado, M.C.H.; Arrighi, L.; Alemany, L.J.; Marazza, R.; Busca, G. Propene versus propane steam reforming for hydrogen production over Pd-based and Ni-based catalysts. Catal. Commun. 2005, 6, 441–445. [Google Scholar] [CrossRef]
- Faria, W.L.; Dieguez, L.C.; Schmal, M. Autothermal reforming of propane for hydrogen production over Pd/CeO2/Al2O3 catalysts. Appl. Catal. B 2008, 85, 77–85. [Google Scholar] [CrossRef]
- Jensen, M.B.; Råberg, L.B.; Sjåstad, A.O.; Olsbye, U. Mechanistic study of the dry reforming of propane to synthesis gas over a Ni/Mg (Al) O catalyst. Catal. Today 2009, 145, 114–120. [Google Scholar] [CrossRef]
- Igarashi, A.; Ohtaka, T.; Motoki, S. Low-temperature steam reforming ofn-butane over Rh and Ru catalysts supported on ZrO2. Catal. Lett. 1992, 13, 189–194. [Google Scholar] [CrossRef]
- Wang, X.; Gorte, R.J. Steam reforming of n-butane on Pd/ceria. Catal. Lett. 2001, 73, 15–19. [Google Scholar] [CrossRef]
- Avcı, A.K.; Trimm, D.L.; Aksoylu, A.E.; Önsan, Z.I. Hydrogen production by steam reforming of n-butane over supported Ni and Pt-Ni catalysts. Appl. Catal. A 2004, 258, 235–240. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Sangtongkitcharoen, W.; Assabumrungrat, S. Catalytic steam reforming of ethane and propane over CeO 2-doped Ni/Al2O3 at SOFC temperature: Improvement of resistance toward carbon formation by the redox property of doping CeO2. Fuel 2006, 85, 323–332. [Google Scholar] [CrossRef]
- Nagaoka, K.; Sato, K.; Nishiguchi, H.; Takita, Y. Highly active Ni/MgO in oxidative steam pre-reforming of n-butane for fuel cell application. Catal. Commun. 2007, 8, 1807–1810. [Google Scholar] [CrossRef]
- Osipovs, S. Sampling of benzene in tar matrices from biomass gasification using two different solid-phase sorbents. Anal. Bioanal. Chem. 2008, 391, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.T.; Williams, E.A. Recycling plastic waste by pyrolysis. J. Inst. Energy 1998, 71, 81–93. [Google Scholar]
- Liu, Y.; Qian, J.; Wang, J. Pyrolysis of polystyrene waste in a fluidized-bed reactor to obtain styrene monomer and gasoline fraction. Fuel Process. Technol. 2000, 63, 45–55. [Google Scholar] [CrossRef]
- Li, B.; Chen, H.P.; Yang, H.P.; Yang, G.L.; Wang, X.H.; Zhang, S.H. Ni/γ-Al2O3 Catalyst for CO2 Reforming of Benzene as a Model Compound of Biomass Gasification Tar: Promotional Effect of Ultrasonic Treatment on Catalytic Performance. In Proceedings of the 20th International Conference on Fluidized Bed Combustion; Springer Berlin Heidelberg: Berlin, Germany, 2010; pp. 576–582. [Google Scholar]
- Park, H.J.; Park, S.H.; Sohn, J.M.; Park, J.; Jeon, J.K.; Kim, S.S.; Park, Y.K. Steam reforming of biomass gasification tar using benzene as a model compound over various Ni supported metal oxide catalysts. Bioresour. Technol. 2010, 101, S101–S103. [Google Scholar] [CrossRef] [PubMed]
- Sarvaramini, A.; Larachi, F. Mossbauer spectroscopy and catalytic reaction studies of chrysotile-catalyzed steam reforming of benzene. J. Phys. Chem. C 2011, 115, 6841–6848. [Google Scholar] [CrossRef]
- Swierczynski, D.; Courson, C.; Kiennemann, A. Study of steam reforming of toluene used as model compound of tar produced by biomass gasification. Chem. Eng. Process. 2008, 47, 508–513. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, X.; Chen, L.; Qu, R.; Meng, G.; Yi, X.; Sun, L. Steam reforming of toluene as model compound of biomass pyrolysis tar for hydrogen. Biomass Bioenergy 2010, 34, 140–144. [Google Scholar] [CrossRef]
- Grenoble, D.C. The chemistry and catalysis of the water/toluene reaction: 2. The role of support and kinetic analysis. J. Catal. 1978, 51, 212–220. [Google Scholar] [CrossRef]
- Duprez, D.; Miloudi, A.; Little, J.; Bousquet, J. The role of the metal/support interface in toluene steam reforming over rhodium-alumina catalysts. Appl. Catal. 1983, 5, 219–226. [Google Scholar] [CrossRef]
- Colby, J.L.; Wang, T.; Schmidt, L.D. Steam reforming of benzene as a model for biomass-derived syngas tars over Rh-based catalysts. Energy Fuels 2009, 24, 1341–1346. [Google Scholar] [CrossRef]
- Mei, D.; Lebarbier, V.M.; Rousseau, R.; Glezakou, V.A.; Albrecht, K.O.; Kovarik, L.; Dagle, R.A. Comparative investigation of benzene steam reforming over spinel supported Rh and Ir catalysts. ACS Catal. 2013, 3, 1133–1143. [Google Scholar] [CrossRef]
- Quitete, C.P.; Bittencourt, R.C. P.; Souza, M.M. Steam reforming of tar using toluene as a model compound with nickel catalysts supported on hexaaluminates. Appl. Catal. A 2014, 478, 234–240. [Google Scholar] [CrossRef]
- Jackson, S.D.; Thomson, S.J.; Webb, G. Carbonaceous deposition associated with the catalytic steam-reforming of hydrocarbons over nickel alumina catalysts. J. Catal. 1981, 70, 249–263. [Google Scholar] [CrossRef]
- Asadullah, M.; Ito, S.I.; Kunimori, K.; Yamada, M.; Tomishige, K. Energy efficient production of hydrogen and syngas from biomass: Development of low-temperature catalytic process for cellulose gasification. Environ. Sci. Technol. 2002, 36, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Świerczyński, D.; Libs, S.; Courson, C.; Kiennemann, A. Steam reforming of tar from a biomass gasification process over Ni/olivine catalyst using toluene as a model compound. Appl. Catal. B 2007, 74, 211–222. [Google Scholar] [CrossRef]
- Li, C.; Hirabayashi, D.; Suzuki, K. Development of new nickel based catalyst for biomass tar steam reforming producing H2-rich syngas. Fuel Process. Technol. 2009, 90, 790–796. [Google Scholar] [CrossRef]
- Li, C.; Hirabayashi, D.; Suzuki, K. Steam reforming of biomass tar producing H2-rich gases over Ni/MgOx/CaO1−x catalyst. Bioresour. Technol. 2010, 101, S97–S100. [Google Scholar] [CrossRef] [PubMed]
- Mukai, D.; Tochiya, S.; Murai, Y.; Imori, M.; Hashimoto, T.; Sugiura, Y.; Sekine, Y. Role of support lattice oxygen on steam reforming of toluene for hydrogen production over Ni/La0.7Sr0.3AlO3−δ catalyst. Appl. Catal. A 2013, 453, 60–70. [Google Scholar] [CrossRef]
- Oemar, U.; Ang, P.S.; Hidajat, K.; Kawi, S. Promotional effect of Fe on perovskite LaNixFe1−xO3 catalyst for hydrogen production via steam reforming of toluene. Int. J. Hydrogen Energy 2013, 38, 5525–5534. [Google Scholar] [CrossRef]
- Oemar, U.; Ang, M.L.; Hee, W.F.; Hidajat, K.; Kawi, S. Perovskite LaxM1−xNi0.8Fe0.2O3 catalyst for steam reforming of toluene: Crucial role of alkaline earth metal at low steam condition. Appl. Catal. B 2014, 148, 231–242. [Google Scholar] [CrossRef]
- Coll, R.; Salvado, J.; Farriol, X.; Montane, D. Steam reforming model compounds of biomass gasification tars: Conversion at different operating conditions and tendency towards coke formation. Fuel Process. Technol. 2001, 74, 19–31. [Google Scholar] [CrossRef]
- Bradford, M.C.; Vannice, M.A. Catalytic reforming of methane with carbon dioxide over nickel catalysts I. Catalyst characterization and activity. Appl. Catal. A 1996, 142, 73–96. [Google Scholar] [CrossRef]
- Vagia, E.C.; Lemonidou, A.A. Thermodynamic analysis of hydrogen production via steam reforming of selected components of aqueous bio-oil fraction. Int. J. Hydrogen Energy 2007, 32, 212–223. [Google Scholar] [CrossRef]
- Palmeri, N.; Chiodo, V.; Freni, S.; Frusteri, F.; Bart, J.C.J.; Cavallaro, S. Hydrogen from oxygenated solvents by steam reforming on Ni/Al2O3 catalyst. Int. J. Hydrogen Energy 2008, 33, 6627–6634. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, L.; Lu, G. Pruning of the surface species on Ni/Al2O3 catalyst to selective production of hydrogen via acetone and acetic acid steam reforming. Appl. Catal. A 2012, 427, 49–57. [Google Scholar] [CrossRef]
- Trane-Restrup, R.; Resasco, D.E.; Jensen, A.D. Steam reforming of light oxygenates. Catal. Sci. Technol. 2013, 3, 3292–3302. [Google Scholar] [CrossRef]
- Navarro, R.M.; Guil-Lopez, R.; Gonzalez-Carballo, J.M.; Cubero, A.; Ismail, A.A.; Al-Sayari, S.A.; Fierro, J.L.G. Bimetallic MNi/Al2O3-La catalysts (M = Pt, Cu) for acetone steam reforming: Role of M on catalyst structure and activity. Appl. Catal. A 2014, 474, 168–177. [Google Scholar] [CrossRef]
- Navarro, R.M.; Guil-Lopez, R.; Ismail, A.A.; Al-Sayari, S.A.; Fierro, J.L.G. Ni- and PtNi-catalysts supported on Al2O3 for acetone steam reforming: Effect of the modification of support with Ce, La and Mg. Catal. Today 2015, 242, 60–70. [Google Scholar] [CrossRef]
- Güell, B.M.; Babich, I.; Nichols, K.P.; Gardeniers, J.G.E.; Lefferts, L.; Seshan, K. Design of a stable steam reforming catalyst—A promising route to sustainable hydrogen from biomass oxygenates. Appl. Catal. B 2009, 90, 38–44. [Google Scholar] [CrossRef]
- Devianto, H.; Han, J.; Yoon, S.P.; Nam, S.W.; Lim, T.-H.; Oh, I.-H.; Hong, S.-A.; Lee, H.-I. The effect of impurities on the performance of bioethanol-used internal reforming molten carbonate fuel cell. Int. J. Hydrogen Energy 2011, 36, 10346–10354. [Google Scholar] [CrossRef]
- Rass-Hansen, J.; Johansson, R.; Moller, M.; Christensen, C.H. Steam reforming of technical bioethanol for hydrogen production. Int. J. Hydrogen Energy 2008, 33, 4547–4554. [Google Scholar] [CrossRef]
- Medrano, J.A.; Oliva, M.; Ruiz, J.; García, L.; Arauzo, J. Catalytic steam reforming of butanol in a fluidized bed and comparison with other oxygenated compounds. Fuel Process. Technol. 2014, 124, 123–133. [Google Scholar] [CrossRef]
- Nahar, G.A.; Madhani, S.S. Thermodynamics of hydrogen production by the steam reforming of butanol: Analysis of inorganic gases and light hydrocarbons. Int. J. Hydrogen Energy 2010, 35, 98–109. [Google Scholar] [CrossRef]
- Piccot, D.S.; Watson, J.J.; Jones, W.J. A Global inventory of organic compounds emissions from anthropogenic sources. J. Geophys. Res. 1992, 97, 9897–9912. [Google Scholar] [CrossRef]
- Wei, W.; Wang, S.; Hao, J.; Cheng, S. Trends of chemical speciation profiles of anthropogenic volatile organic compounds emission in China, 2005–2020. Front. Environ. Sci. Eng. 2014, 8, 27–41. [Google Scholar] [CrossRef]
- Katzenstein, A.S.; Doezema, L.A.; Simpson, I.J.; Blake, D.R.; Rowland, F.S. Extensive regional atmospheric hydrocarbon pollution in the southwestern United States. Proc. Natl. Acad. Sci. 2003, 100, 11975–11979. [Google Scholar] [CrossRef] [PubMed]
- Birch, L.; Bachofen, R. Microbial production of hydrocarbons. In Biotechnology; Rehm, H.-J., Ed.; VCH: Weinheim, Germany, 1988; Volume 6b, pp. 71–99. [Google Scholar]
- Tissot, B.; Welte, D. Petroluem Formation and Occurrence; Springer Verlag: Berlin, Germany, 1984. [Google Scholar]
- Van Zyl, R.L.; Seatlholo, S.T.; van Vuuren, S.F.; Viljoen, A.M. The biological activities of 20 nature identical essential oil constituents. J. Essent. Oil Res. 2006, 18, 129–133. [Google Scholar]
- Ferris, S.W. Handbook of Hydrocarbons; Academic Press: Yew York, NY, USA, 1955; p. 324. [Google Scholar]
- Wert, B.P.; Trainer, M.; Fried, A.; Ryerson, T.B.; Henry, B.; Potter, W.; Wisthaler, A. (2003). Signatures of terminal alkene oxidation in airborne formaldehyde measurements during TexAQS. J. Geophys. Res.: Atmos. 2000, 108. [Google Scholar] [CrossRef]
- Johansson, J.K.; Mellqvist, J.; Samuelsson, J.; Offerle, B.; Lefer, B.; Rappenglück, B.; Yarwood, G. Emission measurements of alkenes, alkanes, SO2, and NO2 from stationary sources in Southeast Texas over a 5 year period using SOF and mobile DOAS. J. Geophys. Res. 2014, 119, 1973–1991. [Google Scholar]
- Bruice, P.Y. Organic Chemistry, 5th ed.; Pearson Education: Upper Saddle River, NJ, USA, 2007; pp. 258–286. [Google Scholar]
- Schobert, H. Production of Acetylene and Acetylene-based Chemicals from Coal. Chem. Rev. 2014, 114, 1743–1760. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Smith, D.B. The combustion and oxidation of acetylene. Chem. Rev. 1970, 70, 267–293. [Google Scholar] [CrossRef]
- Liu, Y.; Shao, M.; Fu, L.; Lu, S.; Zeng, L.; Tang, D. Source profiles of volatile organic compounds (VOCs) measured in China: Part I. Atmos. Environ. 2008, 42, 6247–6260. [Google Scholar] [CrossRef]
- Parrish, D.D.; Kuster, W.C.; Shao, M.; Yokouchi, Y.; Kondo, Y.; Goldan, P.D.; de Gouw, J.A.; Koike, M.; Shirai, T. Comparison of air pollutant emissions among mega-cities. Atmos. Environ. 2009, 43, 6435–6441. [Google Scholar] [CrossRef]
- Lin, T.J.; Meng, X.; Shi, L. Catalytic hydrocarboxylation of acetylene to acrylic acid using Ni2O3 and cupric bromide as combined catalysts. J. Mol. Catal. A: Chem. 2015, 396, 77–83. [Google Scholar] [CrossRef]
- Matar, S.; Hatch, L.F. Chemistry of Petrochemical Processes, 2nd ed.; Gulf Professional Publishing: Boston, MA, USA, 2001; pp. 262–300. [Google Scholar]
- Wang, H.; Lie, N.; Li, L.; Wang, Y.; Wang, G.; Wang, J.; Hao, Z. Characterization and assessment of volatile organic compounds (VOCs) emissions from typical industries. Chin. Sci. Bull. 2013, 58, 724–730. [Google Scholar] [CrossRef]
- El-Naas, M.; Acio, J.A.; El Telib, A.E. Aerobic biodegradation of BTEX: Progresses and Prospects. J. Environ. Chem. Eng. 2014, 2, 1104–1122. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency (EPA). Available online: http://www.epa.gov/reg3hwmd/bf-lr/regional/analytical/semi-volatile.htm (accessed on 30 January 2015).
- Yassaa, N.; Brancaleoni, E.; Frattoni, M.; Ciccioli, P. Isomeric analysis of BTEXs in the atmosphere using b-cyclodextrin capillary chromatography coupled with thermal desorption and mass spectrometry. Chemosphere 2006, 63, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Durmusoglu, E.; Taspinar, F.; Karademir, A. Health risk assessment of BTEX emissions in the landfill environment. J. Hazard. Mater. 2010, 176, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.-H.; Lin, K.-H.; Chen, C.-Y.; Lai, N.; Ma, S.-Y.; Chiang, H.-L. Volatile organic compound constituents from an integrated iron and steel facility. J. Hazard. Mater. 2008, 157, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Geng, F.; Tie, X.; Yu, Q.; An, J. Characteristics and source apportionment of VOCs measured in Shanghai, China. Atmos. Environ. 2010, 44, 5005–5014. [Google Scholar] [CrossRef]
- Atkinson, R. Gas phase tropospheric chemistry of organic compounds. In Volatile Organic Compounds in the Atmosphere, Issues in Environmental Science and Technology 4; Hester, R.E., Harrison, R.M., Eds.; The Royal Society of Chemistry: Letchworth, UK, 1995; pp. 65–89. [Google Scholar]
- Huang, D.-Y.; Zhou, S.-G.; Hong, W.; Feng, W.-F.; Tao, L. Pollution characteristics of volatile organic compounds, polycyclic aromatic hydrocarbons and phthalate esters emitted from plastic wastes recycling granulation plants in Xingtan Town, South China. Atmos. Environ. 2013, 71, 327–334. [Google Scholar] [CrossRef]
- Huang, H.; Ye, X.; Huang, W.; Chen, J.; Xu, Y.; Wu, M.; Shao, Q.; Peng, Z.; Ou, G.; Shi, J.; et al. Ozone-catalytic oxidation of gaseous benzene over MnO2/ZSM-5 at ambient temperature: Catalytic deactivation and its suppression. Chem. Eng. J. 2015, 264, 24–31. [Google Scholar] [CrossRef]
- Scirè, S.; Liotta, L.F. Supported gold catalysts for the total oxidation of volatile organic compounds. Appl. Catal. B 2012, 125, 222–246. [Google Scholar] [CrossRef]
- Einaga, H.; Teraoka, Y.; Ogata, A. Catalytic oxidation of benzene by ozone over manganese oxides supported on USY zeolite. J. Catal. 2013, 305, 227–237. [Google Scholar] [CrossRef]
- Ren, C.; Liu, X.; Wang, G.; Miao, S.; Chen, Y. Thermo-photocatalytic degradation of benzene on Pt-loaded TiO2/ZrO2. J. Mol. Catal. A 2012, 358, 31–37. [Google Scholar] [CrossRef]
- Rezaei, E.; Soltan, J. EXAFS and kinetic study of MnOχ/γ-alumina in gas phase catalytic oxidation of toluene by ozone. Appl. Catal. B 2014, 148–149, 70–79. [Google Scholar] [CrossRef]
- Legreid, G.; Lööv, J.B.; Staehelin, J.; Hueglinh, C.; Hill, M.; Buchmann, B.; Prevot, A.S.H.; Reimann, S. Oxygenated volatile organic compounds (OVOCs) at an urban background site in Zürich (Europe): Seasonal variation and source allocation. Atmos. Environ. 2007, 41, 8409–8423. [Google Scholar] [CrossRef]
- Placet, M.; Mann, C.O.; Gilbert, R.O.; Niefer, M.J. Emissions of ozone precursors from stationary sources: A critical review. Atmos. Environ. 2000, 34, 2183–2204. [Google Scholar] [CrossRef]
- Sawyer, R.F.; Harley, R.A.M.; Cadle, S.H.; Norbeck, J.M.; Slott, R.; Bravo, H.A. Mobile sources critical review: 1998 NARSTO assessment. Atmos. Environ. 2000, 34, 2161–2181. [Google Scholar] [CrossRef]
- AMEC Earth & Environmental Limited and University of Calgary. Assessment report on reduced sulphur compounds for developing ambient air quality objectives, Alberta environment. 2004. Available online: http://environment.gov.ab.ca/info/library/6664.pdf (accessed on 3 March 2015). [Google Scholar]
- Kulkarni, S. Alkyl Halides. In Encyclopedia of Toxicology; Elsevier: Berlin, Germany, 2014; pp. 144–145. [Google Scholar]
- Cram101 Textbook Reviews. In Study Guide for Introductory Chemistry: Atoms First by Russo, Steve, 5th ed.; Cram101: Bellevue, WA, USA, 2014.
- Huang, B.; Lei, C.; Wei, C.; Zeng, G. Chlorinated volatile organic compounds (Cl-VOCs) in environment—Sources, potential human health impacts, and current remediation technologies. Environ. Int. 2014, 71, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wang, S.; Chatani, S.; Klimont, Z.; Cofala, J.; Hao, J. Emission and speciation of non-methane volatile organic compounds from anthropogenic sources in China. Atmos. Environ. 2008, 42, 4976–4988. [Google Scholar] [CrossRef]
- US EPA. EPA Coalbed Methane Outreach Program Technical Options Series; United States Environmental Protection Agency, Office of Air and Radiation 6202J: Washington, DC, USA, 1998.
- Rojo, F. Enzymes for aerobic degradation of alkanes. In Handbook of Hydrocarbon and Lipid Microbiology; Springer: Berlin, Germany, 2010; pp. 781–797. [Google Scholar]
- Singh, S.N.; Kumari, B.; Mishra, S. Microbial degradation of alkanes. In Microbial Degradation of Xenobiotics; Springer: Berlin, Germany, 2012; pp. 439–469. [Google Scholar]
- Wentzel, A.; Ellingsen, T.E.; Kotlar, H.K. Bacterial metabolism of long-chain n-alkanes. Appl. Microbial. Biotechnol. 2007, 76, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Heider, J.; Spormann, A.M.; Beller, H.R. Anaerobic bacterial metabolism of hydrocarbons. FEMS Microbiol. Rev. 1998, 22, 459–473. [Google Scholar] [CrossRef]
- Rojo, F. Degradation of alkanes by bacteria. Environ. Microbial. 2009, 11, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T.; Daligault, F.; Brunner, B.; Hinrichs, H.; Deege, A. Directed evolution of cyclohexanone monooxygenases: Enantioselective biocatalysts for the oxidation of prochiral thioethers. Angew. Chem. 2004, 116, 4170–4173. [Google Scholar] [CrossRef]
- De Gonzalo, G.; Mihovilovic, M.D.; Fraaije, M.W. Recent developments in the application of Baeyer–Villiger monooxygenases as biocatalysts. ChemBioChem 2010, 11, 2208–2231. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Dalton, H. Biocatalysis by methane monooxygenase and its implications for the petroleum industry. Stud. Surf. Sci. Catal. 2004, 151, 177–192. [Google Scholar]
- Akella, S.; Pothini, S.; Veerashekhar, G.E. Recent progress in oxidation of n-alkanes by heterogeneous catalysis. Res. Rev. Mater. Sci. Chem. 2012, 1, 75–103. [Google Scholar]
- Kirillov, A.M.; Kirillova, M.V.; Pombeiro, A.J. Multicopper complexes and coordination polymers for mild oxidative functionalization of alkanes. Coord. Chem. Rev. 2012, 256, 2741–2759. [Google Scholar] [CrossRef]
- Chepaikin, E.G. Oxidative functionalization of alkanes under dioxygen in the presence of homogeneous noble metal catalysts. J. Mol. Catal. A 2014, 385, 160–174. [Google Scholar] [CrossRef]
- Jia, C.; Kitamura, T.; Fujiwara, Y. Catalytic functionalization of arenes and alkanes via CH bond activation. Acc. Chem. Res. 2001, 34, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Maihom, R.; Khongpracha, P.; Sirijaraensre, J.; Limytakul, J. Mechanistic studies on the transformation of ethanol into ethene over Fe-ZSM-5 zeolite. ChemPhysChem 2013, 14, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, G.S.; Rychlicki, G.; Terzyk, A.P. Catalytic conversion of ethanol on carbon catalysts. Carbon 1994, 32, 265–271. [Google Scholar] [CrossRef]
- Eastham, G.R.; Heaton, B.T.; Iggom, J.A.M.; Tooze, R.P.; Whyman, R.; Zacchini, S. Synthesis and spectroscopic characterization of all the intermediates in the Pd-catalysed methoxycarbonylation of ethene. Chem. Commun. 2000, 7, 609–910. [Google Scholar] [CrossRef]
- Samel, U.-R.; Kohler, W.; Gamer, A.O.; Keuser, U. Propionic acid and derivatives. Ullman’s Encycl. Ind. Chem. 2000. [Google Scholar] [CrossRef]
- OECD SIDS–Ethylene, UNEP Publications. Available online: http://en.wikipedia.org/wiki/Ethylene (accessed on 2 March 2015).
- Material Safety Data Sheets, 1-Pentene, Acros Organics. Available online: http://www.fishersci.com (accessed on 30 June 2015).
- Hashimi, S.K.; Sehwarz, L. Switch from Palladium-Catalyzed Cycloisomerization/Dimerization of Terminal Allenyl Ketones to Preferential Formation of Monomers by a 5-Palladatricyclo[4.1.0.02,4]heptane Catalyst: Synthesis of Furans from Substrates Incompatible with the Commonly Used Silver Catalysts. Eur. J. Inorg. Chem. 2006. [Google Scholar] [CrossRef]
- Davarpanah, J.; Kiasat, A.R. Synthesis and characterization of SBA-polyperoxyacid: An efficient heterogeneous solid peroxyacid catalyst for epoxidation of alkenes. Catal. Commun. 2014, 46, 75–80. [Google Scholar] [CrossRef]
- Crosthwaite, J.M.; Farmer, V.A.; Hallett, J.P.; Welton, T. Epoxidation of alkenes by Oxone™ using 2-alkyl-3,4-dihydroisoquinolium salts as catalysts in ionic liquids. J. Mol. Catal. A 2008, 279, 148–152. [Google Scholar] [CrossRef]
- Pescarmona, P.P.; Jacobs, P.A. A high-throughput experimentation study of the epoxidation of alkenes with transition-metal-free heterogeneous catalysts. Catal. Today 2008, 137, 52–60. [Google Scholar] [CrossRef]
- Salmi, T.; Carucci, J.H.; Roche, M.; Eränen, K.; Wärnå, J.; Murzin, D. Microreactors as tools in kinetic investigations: Ethylene oxide formation on silver catalyst. Chem. Eng. Sci. 2013, 87, 306–314. [Google Scholar] [CrossRef]
- Hernández Carucci, J.R.; Halonen, V.; Eränen, K.; Wärnå, J.; Ojala, S.; Huuhtanen, M.; Keiski, R.; Salmi, T. Ethylene Oxide Formation in a Microreactor: From Qualitative Kinetics to Detailed Modelling. Ind. Eng. Chem. Res. 2010, 49, 10897–10907. [Google Scholar] [CrossRef]
- Smidt, J.; Hafner, W.; Jira, R.; Sedlmeier, J.; Siener, R.; Rüttinger, R.; Kojer, H. Catalytic reactions of olefins on compounds of the platinum group. Angew. Chem. 1959, 71, 176–182. [Google Scholar] [CrossRef]
- Cornell, C.N.; Sigman, M.S. Recent progress in Wacker oxidations: Moving toward molecular oxygen as the sole oxidant. Inorg. Chem. 2007, 46, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Che, C.-M. A Practical and mild method for the highly selective conversion of terminal alkanes into aldehydes through epoxidation–isomerization with ruthenium (IV)-phorphyrin catalysts. Angew. Chem. 2004, 116, 5058–5062. [Google Scholar] [CrossRef]
- Conte, M.; Carley, A.F.; Heirene, C.; Willock, D.J.; Johnston, P.; Herzing, A.; Kiely, C.J.; Hutching, G.J. Hydrochlorination of acetylene using a supported gold catalyst: A study of the reaction mechanism. J. Catal. 2007, 250, 231–239. [Google Scholar] [CrossRef]
- Huang, C.; Zhu, M.; Kang, L.; Dai, B. Active carbon supported TiO2-AuCl3/AC catalyst with excellent stability for acetylene hydrochlorination reaction. Chem. Eng. J. 2014, 242, 69–75. [Google Scholar] [CrossRef]
- Lin, T.J.; Meng, X.; Shi, L. Ni-exchanged Y-zeolite: An efficient heterogeneous catalyst for acetylene hydrocarboxylation. Appl. Catal. A 2014, 485, 163–171. [Google Scholar] [CrossRef]
- Tang, C.-M.; Zeng, Y.; Yang, X.-G.; Lei, Y.-C.; Wang, G.-Y. The palladium catalyzed hydrocarboxylation of acetylene with carbon monoxide to acrylic acid under mild conditions. J. Mol. Catal. A 2009, 314, 15–20. [Google Scholar] [CrossRef]
- Beesu, M.; Periasamy, M. Stereoselective synthesis of α,β-unsaturated carboxylic acids from alkynes using the Fe(CO)5/t-BuOK/AcOH/CH2Cl2 reagent system. J. Organomet. Chem. 2012, 705, 30–33. [Google Scholar] [CrossRef]
- Zhang, Y.; Riduan, S.N. Catalytic Hydrocarboxylation of Alkenes and Alkynes with CO2. Angew. Chem. Int. Ed. 2011, 50, 6210–6212. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Radhakrishnan, U.; Rameshkumar, C.; Brumet, J.-J. A New method for the regio and stereoselective hydrocarboxylation of aIkynes using NaHFe(CO)4/CH2CI2 System. Tetrahedron Lett. 1997, 38, 1623–1626. [Google Scholar] [CrossRef]
- Fujihara, T.; Xu, T.; Semba, K.; Terao, J.; Tsuji, Y. Copper catalyzed hydrocarboxylation of alkynes using carbon dioxide and hydrosilanes. Angew. Chem. Int. Ed. 2011, 50, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Dérien, S.; Duñach, E.; Périchon, J. From stoichiometry to catalysis: Electroreductive coupling of alkynes and carbon dioxide with nickel-bipyridine complexes. Magnesium ions as the key for catalysis. J. Am. Chem. Soc. 1991, 113, 8447–8454. [Google Scholar] [CrossRef]
- Zhang, H.; Dai, B.; Wang, X.; Xu, L.; Zhu, M. Hydrochlorination of acetylene to vinyl chloride monomer over bimetallic Au-La/SAC catalysts. J. Ind. Eng. Chem. 2012, 18, 49–54. [Google Scholar] [CrossRef]
- Li, X.; Pan, X.; Bao, X. Nitrogen doped carbon catalyzing acetylene conversion to vinyl chloride. J. Energy Chem. 2014, 23, 131–135. [Google Scholar] [CrossRef]
- Gulyaeva, Y.K.; Kaicher, V.V.; Zaikovskii, V.I.; Kovalyov, E.V.; Suknev, A.P.; Bal’zhinimaev, B.S. Selective hydrogenation of acetylene over novel Pd/fiberglass catalyst. Catal. Today 2015, 245, 139–146. [Google Scholar] [CrossRef]
- Yan, X.; Wheeler, J.; Jang, B.; Lin, W.-Y.; Zhao, B. Stable Au catalyst for selective hydrogenation of acetylene in ethylene. Appl. Catal. A 2014, 487, 36–44. [Google Scholar] [CrossRef]
- Azizi, Y.; Petit, C.; Pitchon, V. Formation of polymer-grade ethylene by selective hydrogenation of acetylene over Au/CeO2 catalyst. J. Catal. 2008, 256, 338–344. [Google Scholar] [CrossRef]
- Teschner, D.; Vass, E.; Hävecker, M.; Zafeiratos, S.; Schnörch, P.; Sauer, H.; Knop-Gericke, A.; Schlögl, R.; Chamam, M.; Wootsch, A.; et al. Alkyne hydrogenation over Pd catalysts: A new paradigm. J. Catal. 2006, 242, 26–37. [Google Scholar] [CrossRef]
- McCue, A.J.; McRitchie, C.J.; Shepherd, A.M.; Anderson, J.A. Cu/Al2O3 catalysts modified with Pd for selective acetylene hydrogenation. J. Catal. 2014, 319, 127–135. [Google Scholar] [CrossRef]
- Liptáková, B.; Báhidský, M.; Hronec, M. Preparation of phenol from benzene by one-step reaction. Appl. Catal. A 2004, 263, 33–38. [Google Scholar] [CrossRef]
- Molinary, R.; Argurio, P.; Poerio, T. Vanadyl acetylacetonate filled PVDF membranes as the core of a liquid phase continuous process for pure phenol production from benzene. J. Membr. Sci. 2015, 476, 490–499. [Google Scholar] [CrossRef]
- Busca, G.; Berardinelli, S.; Resini, C.; Arrighi, L. Technologies for the removal of phenol from fluid streams: A short review of recent developments. J. Hazard. Mater. 2008, 160, 265–288. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R. Industrial catalytic processes—Phenol production. Appl. Catal. A 2005, 280, 89–103. [Google Scholar] [CrossRef]
- Maity, D.; Jagtap, R.; Kaistha, N. Systematic top-down economic plantwide control of thecumene process. J. Process Control 2013, 23, 1426–1440. [Google Scholar] [CrossRef]
- Sayyar, M.H.; Wakeman, R.J. Comparing two new routes for benzene hydroxylation. Chem. Eng. Res. Des. 2008, 86, 517–526. [Google Scholar] [CrossRef]
- INEOS group AG. Available online: http://www.ineos.com/ (accessed on 1 March 2015).
- Molinary, R.; Argurio, P.; Lavorato, C. Review on reduction and partial oxidation of organic in photocatalytic (membrane) reactors. Curr. Org. Chem. 2013, 17, 2516–2537. [Google Scholar] [CrossRef]
- Baykan, D.; Oztas, N.A. Synthesis of iron orthophosphate catalysts by solution and solution combustion methods for the hydroxylation of benzene to phenol. Mater. Res. Bull. 2015, 64, 294–300. [Google Scholar] [CrossRef]
- Vulpescu, G.D.; Ruitenbeek, M.; van Lieshout, L.L; Correia, L.A.; Meyer, D.; Pex, P.P.A.C. One-step selective oxidation over a Pd-based catalytic membrane; evaluation of the oxidation of benzene to phenol as a model reaction. Catal. Commun. 2004, 5, 347–351. [Google Scholar] [CrossRef]
- Bahidsky, M.; Hronec, M. Direct gas phase hydroxylation of benzene over phosphate catalysts. Catal. Today 2004, 91–92, 13–16. [Google Scholar] [CrossRef]
- Lemke, K.; Ehrich, H.; Lohse, U.; Berndt, H.; Jähnisch, K. Selective hydroxylation of benzene to phenol over supported vanadium oxide catalysts. Appl. Catal. A 2003, 243, 41–51. [Google Scholar] [CrossRef]
- Joseph, J.; Singhal, S.; Jain, S.L.; Sivakumaran, R.; Kumar, B.; Sain, B. Studies on vanadium catalyzed direct hydroxylation of aromatic hydrocarbons using hydrogen peroxide as oxidant. Catal. Today 2009, 141, 211–214. [Google Scholar] [CrossRef]
- Callanan, L.H.; Burton, R.M.; Mullineux, J.; Engelbrecht, J.M.M.; Rau, U. Effect of semi-batch reactor configuration on aromatic hydroxylation reactions. Chem. Eng. J. 2012, 180, 255–262. [Google Scholar] [CrossRef]
- Yashima, T.; Kobayashi, Y.; Komatsu, T.; Namba, S. Hydroxylation of toluene with hydrogen peroxide on HY zeolites with various Si/Al ratios. Stud. Surf. Sci. Catal. 1993, 75, 1689–1692. [Google Scholar]
- Motz, J.L.; Heinichen, H.; Hölderich, W.L. Direct hydroxylation of aromatics to their corresponding phenols catalysed by H-[Al]ZSM-5 zeolite. J. Mol. Catal. A 1998, 136, 175–184. [Google Scholar] [CrossRef]
- Bartoli, J.-F.; Lambert, F.; Morgenstern-Badarau, I.; Battioni, P.; Mansuy, D. Unusual efficiency of a non-heme iron complex as catalyst for the hydroxylation of aromatic compounds by hydrogen peroxide: Comparison with iron porphyrins. C.R. Chim. 2002, 5, 263–266. [Google Scholar] [CrossRef]
- Balland, V.; Mathieu, D.; Pons-Y-Moll, N.; Bartoli, J.F.; Banse, F.; Battioni, P.; Girerd, J.J.; Daniel Mansuyb, D. Non-heme iron polyazadentate complexes as catalysts for oxidations by H2O2: Particular efficiency in aromatic hydroxylations and beneficial effects of a reducing agent. J. Mol. Catal. A 2004, 215, 81–87. [Google Scholar] [CrossRef]
- Bianchi, D.; Bertoli, M.; Tassinari, R.; Ricci, M.; Vignola, R. Direct synthesis of phenols by iron-catalyzed biphasic oxidation of aromatic hydrocarbons with hydrogen peroxide. J. Mol. Catal. A 2003, 200, 111–116. [Google Scholar] [CrossRef]
- Yashima, T.; Nagase, S.; Komatsu, T.; Namba, S. Liquid-phase hydroxylation of xylene with hydrogen peroxide over zeolite catalysts. Stud. Surf. Sci. Catal. 1993, 77, 417–420. [Google Scholar]
- Kang, Z.; Wang, E.; Mao, B.; Su, Z.; Gao, L.; Niu, L.; Shan, H.; Xu, L. Heterogeneous hydroxylation catalyzed by multi-walled carbon nanotubes at low temperature. Appl. Catal. A 2006, 299, 212–217. [Google Scholar] [CrossRef]
- Singh, A.P. Selective chlorination of various aromatics over zeolite catalysts. Catal. Stud. Surf. Sci. Catal. 1998, 113, 419–423. [Google Scholar]
- Singh, A.P.; Mirajkar, S.P.; Sharma, S. Liquid phase bromination of aromatics over zeolite H-beta catalyst. J. Mol. Catal. A 1999, 150, 241–250. [Google Scholar] [CrossRef]
- Daou, T.J.; Boltz, M.; Tzanis, L.; Michelin, L.; Louis, B. Gas-phase chlorination of aromatics over FAU- and EMT-type zeolites. Catal. Commun. 2013, 39, 10–13. [Google Scholar] [CrossRef]
- Ju, J.; Li, Y.J.; Gao, J.R.; Jia, R.H.; Han, L.; Sheng, W.J.; Jia, Y.X. High selectively oxidative bromination of toluene derivatives by the H2O2–HBr system. Chin. Chem. Lett. 2011, 22, 382–384. [Google Scholar] [CrossRef]
- Olah, G.A.; Kuhn, S.J.; Flood, S.H.; Hardie, B.A. Aromatic Substitution. XIV. Ferric chloride catalyzed bromination of benzene and alkylbenzenes with bromine in nitromethane solution. J. Am. Chem. Soc. 1964, 86, 1039–1044. [Google Scholar] [CrossRef]
- Ratnasamy, P.; Singh, A.P.; Sharma, S. Halogenation over zeolite catalysts. Appl. Catal. A 1996, 135, 25–55. [Google Scholar] [CrossRef]
- Wang, P.-C.; Lu, M.; Zhu, J.; Song, Y.-M.; Xiong, X.-F. Regioselective nitration of aromatics under phase-transfer catalysis conditions. Catal. Commun. 2011, 14, 42–47. [Google Scholar] [CrossRef]
- Ma, X.M.; Li, B.D.; Chen, L.; Lu, M.; Lv, C.X. Selective nitration of aromatic compounds catalysed by Hβ zeolite using N2O5. Chin. Chem. Lett. 2012, 23, 809–812. [Google Scholar] [CrossRef]
- Nowrouzi, N.; Mehranpour, A.M.; Bashiri, E.; Shayan, Z. Aromatic nitration under neutral conditions using N-bromosuccinimide/silver(I) nitrate. Tetrahedron Lett. 2012, 53, 4841–4842. [Google Scholar] [CrossRef]
- Ott, J.; Gronemann, V.; Pontzen, F.; Fiedler, E.; Grossmann, G.; Kersebohm, D.B.; Weiss, G.; Witte, C. Methanol. In Ullmann’s Encyclopedia of Industrial Chemistry; Bellussi, G., Bohnet, M., Bus, J., Drauz, K., Faulhammer, H., Greim, H., Jäckel, K.-P., Karst, U., Kleemann, A., Kutscher, B., et al., Eds.; John Wiley and Sons: Weinheim, Germany, 2012; pp. 1–27. [Google Scholar]
- Kung, H.H.; Smith, K.J. Methanol to chemical. In Methanol Production and Use; Cheng, W.H., Kung, H.H., Eds.; Marcel Dekker: New York, NY, USA, 1994; pp. 175–214. [Google Scholar]
- Hordeski, M.F. Alternative Fuels: The Future of the Hydrogen, 2nd ed.; The Fairmont Press: Lilburn, GA, USA, 2008; pp. 1–32. [Google Scholar]
- Kosaric, N.; Duvnjak, Z.; Farkas, A.; Sahm, H.; Bringer-Meyer, S.; Goebel, O.; Mayer, D. Ethanol. In Ullmann’s Encyclopedia of Industrial Chemistry; Bellussi, G., Bohnet, M., Bus, J., Drauz, K., Faulhammer, H., Greim, H., Jäckel, K.-P., Karst, U., Kleemann, A., Kutscher, B., et al., Eds.; John Wiley and Sons: Weinheim, Germany, 2011; pp. 333–403. [Google Scholar]
- Hahn, H.D.; Dämbkes, G.; Rupprich, N.; Bahl, H.; Frey, G.D. Buthanols. In Ullmann’s Encyclopedia of Industrial Chemistry; Bellussi, G., Bohnet, M., Bus, J., Drauz, K., Faulhammer, H., Greim, H., Jäckel, K.-P., Karst, U., Kleemann, A., Kutscher, B., et al., Eds.; John Wiley and Sons: Weinheim, Germany, 2013; pp. 1–13. [Google Scholar]
- McGrath, K.M.; Prakash, G.K.S.; Olah, G.A. Direct methanol fuel cells. J. Ind. Eng. Chem. 2004, 10, 1063–1080. [Google Scholar]
- Kamarudin, S.K.; Achmad, F.; Daud, W.R.W. Overview on the application of direct methanol fuel cell (DMFC) for portable electronic devices. Int. J. Hydrogen Energy 2009, 34, 6902–6916. [Google Scholar] [CrossRef]
- Kundu, A.; Jang, J.H.; Gil, J.H.; Jung, C.R.; Lee, H.R.; Kim, S.-H.; Ku, B.; Oh, Y.S. Micro-fuel cells—Current development and applications. J. Power Sources 2007, 170, 67–78. [Google Scholar] [CrossRef]
- Ojala, S.; Laakso, I.; Maunula, T.; Silvonen, R.; Ylönen, R.; Lassi, U.; Keiski, R.-L. Abatement of malodorous pulp mill emissions by catalytic oxidation–Pilot experiments in Stora Enso Pulp Mill Oulu, Finland. TAPPI J. 2005, 4, 9–13. [Google Scholar]
- Koivikko, N.; Laitinen, T.; Ojala, S.; Pitkäaho, S.; Kucherov, A.; Keiski, R.L. Formaldehyde production from methanol and methyl mercaptan over titania and vanadia based catalysts. Appl. Catal. B 2011, 103, 72–78. [Google Scholar] [CrossRef]
- Henze, M.; van Loosdrecht, M.; Ekama, G.; Brdjanovic, D. Biological Wastewater Treatment: Principles, Modeling and Design; IWA Publishing: London, UK, 2008; pp. 87–154. [Google Scholar]
- Sakuth, M.; Mensing, T.; Schuler, J.; Heitmann, W.; Strehlke, G.; Mayer, D. Ethers, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Bellussi, G., Bohnet, M., Bus, J., Drauz, K., Faulhammer, H., Greim, H., Jäckel, K.-P., Karst, U., Kleemann, A., Kutscher, B., et al., Eds.; John Wiley and Sons: Weinheim, Germany, 2010; pp. 433–449. [Google Scholar]
- Lewis, R.A. Lewis’ Dictionary of Toxicology; CRC Press LLC: Boca Raton, MA, USA, 1998. [Google Scholar]
- Leisch, H.; Omori, A.T.; Finn, K.J.; Gilmet, J.; Bissett, T.; Ilceski, D.; Hudlicky, T. Chemoenzymatic enantiodivergent total synthesis of (+)- and (+)-cdeine. Tetrahedron 2009, 65, 9862–9875. [Google Scholar] [CrossRef]
- Nadim, F.; Zack, P.; hoag, G.E.; Liu, S. United States experience with gasoline additives. Energy Policy 2001, 29, 1–5. [Google Scholar] [CrossRef]
- Wang, S.Z.; Ishihara, T.; Takita, Y. Partial oxidation of dimethyl ether over various supported metal catalysts. Appl. Catal. A 2002, 228, 167–176. [Google Scholar] [CrossRef]
- Yu, L.; Xu, J.; Sun, M.; Wang, X. Catalytic oxidation of dimethyl ether to hydrocarbons over SnO2/MgO and SnO2/CaO catalysts. J. Nat. Gas Chem. 2007, 16, 200–203. [Google Scholar] [CrossRef]
- Sun, M.; Yu, L.; Ye, F.; Diao, G.; Yu, Q.; Hao, Z.; Zheng, Y.; Yuan, L. Transition metal doped cryptomelane-type manganese oxide for low temperature catalytic combustion of dimethyl ether. Chem. Eng. J. 2013, 220, 320–327. [Google Scholar] [CrossRef]
- Park, S.; Watanabe, Y.; Nishita, Y.; Fukuoka, T.; Inagaki, S.; Kubota, Y. Catalytic conversion of dimethyl ether into propylene over MCM-68 zeolite. J. Catal. 2014, 319, 265–273. [Google Scholar] [CrossRef]
- Siegel, H.; Eggersdorfer, M. Ketones. In Ullmann’s Encyclopedia of Industrial Chemistry; Bellussi, G., Bohnet, M., Bus, J., Drauz, K., Faulhammer, H., Greim, H., Jäckel, K.-P., Karst, U., Kleemann, A., Kutscher, B., et al., Eds.; John Wiley and Sons: Weinheim, Germany, 2000; pp. 187–207. [Google Scholar]
- Weber, M.; Pompetzki, W.; Bonmann, R.; Weber, M. Acetone. In Ullmann’s Encyclopedia of Industrial Chemistry; Bellussi, G., Bohnet, M., Bus, J., Drauz, K., Faulhammer, H., Greim, H., Jäckel, K.-P., Karst, U., Kleemann, A., Kutscher, B., et al., Eds.; John Wiley and Sons: Weinheim, Germany, 2014; pp. 1–19. [Google Scholar]
- Suzuki, T.; Yamagiwa, N.; Matsuo, Y.; Sakamoto, S.; Yamaguchi, K.; Shibasaki, M.; Noyori, R. Catalytic asymmetric aldol reaction of ketones and aldehydes using chiral alkoxides. Tetrahedron Lett. 2001, 42, 4669–4671. [Google Scholar] [CrossRef]
- Vetere, V.; Merlo, A.B.; Casella, M.L. Chemoselective hydrogenation of aromatic ketones with Pt-based heterogeneous catalysts. Substituent effects. Appl. Catal. A 2015, 491, 70–77. [Google Scholar] [CrossRef]
- Malyala, R.V.; Rode, C.V.; Arai, M.; Hegde, S.G.; Chaudhari, R.V. Activity, selectivity and stability of Ni and bimetallic Ni-Pt supported on zeolite Y catalysts for hydrogenation of acetophenone and its substituted derivatives. Appl. Catal. A 2000, 193, 76–86. [Google Scholar] [CrossRef]
- Trasarti, A.F.; Bertero, N.M.; Apesteguia, C.R.; Marchi, A.J. Liquid-phase hydrogenation of acetophenone over silica-supported Ni, Co and Cu catalysts: Influence of metal and solvent. Appl. Catal. A 2014, 475, 282–291. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Hajek, J.; Salmi, T.; Murzin, D.Y. Chemoselective hydrogenation of carbonyl compounds over heterogeneous catalysts. Appl. Catal. A 2005, 292, 1–49. [Google Scholar] [CrossRef]
- Bergault, I.; Fouilloux, P.; Joly-Vuillemin, C.; Delmas, H. Kinetics and intraparticle diffusion modelling of a complex multistep reaction: Hydrogenation of acetophenone over Rhodium catalyst. J. Catal. 1998, 175, 328–337. [Google Scholar] [CrossRef]
- Deshmukh, A.; Kinage, A.; Kumar, R.; Meijboom, R. An efficient heterogeneous catalytic system for chemoselective hydrogenation of unsaturated ketones in aqueous medium. Polyhedron 2010, 29, 3262–3268. [Google Scholar] [CrossRef]
- Madduri, A.V.R.; Harutyunyan, S.R.; Minnaard, A.J. Catalytic asymmetric alkylation of ketones using organometallic reagents. Drug Discov. Today: Technol. 2013, 10, e21–e27. [Google Scholar] [CrossRef] [PubMed]
- Anneken, D.J.; Both, S.; Christoph, R.; Fieg, G.; Steinberger, U.; Westfechtel, A. Fatty acids. In Ullmann’s Encyclopedia of Industrial Chemistry; Bellussi, G., Bohnet, M., Bus, J., Drauz, K., Faulhammer, H., Greim, H., Jäckel, K.-P., Karst, U., Kleemann, A., Kutscher, B., et al., Eds.; John Wiley and Sons: Weinheim, Germany, 2006; pp. 73–116. [Google Scholar]
- Kubitschke, J.; Lange, H.; Strutz. Carboxylic acids, aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Bellussi, G., Bohnet, M., Bus, J., Drauz, K., Faulhammer, H., Greim, H., Jäckel, K.-P., Karst, U., Kleemann, A., Kutscher, B., et al., Eds.; John Wiley and Sons: Weinheim, Germany, 2014; pp. 1–18. [Google Scholar]
- Mérel, D.S.; Do, M.L.T.; Gaillard, S.; Dupau, P.; Renaud, J.-L. Iron-catalyzed reduction of carboxylic and carbonic acid derivatives. Coord. Chem. Rev. 2015, 288, 50–68. [Google Scholar] [CrossRef]
- Misal Castro, L.C.; Li, H.; Sortais, J.-B.; Darcel, C. Selective switchable iron-catalyzed hydrosilylation of carboxylic acids. Chem. Commun. 2012, 48, 10514–10516. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Q.; Liu, J.; Zhang, Y.-H.; Shao, C.D.; Yu, J.-X. Copper-catalyzed amide bond formation from formamides and carboxylic acids. Chin. Chem. Lett. 2015, 26, 11–14. [Google Scholar] [CrossRef]
- Riemenschneider, W.; Bolt, H. Esters, Organic. In Ullmann’s Encyclopedia of Industrial Chemistry; Bellussi, G., Bohnet, M., Bus, J., Drauz, K., Faulhammer, H., Greim, H., Jäckel, K.-P., Karst, U., Kleemann, A., Kutscher, B., et al., Eds.; John Wiley and Sons: Weinheim, Germany, 2005; pp. 245–266. [Google Scholar]
- Koritala, S. Hydrogenation of unsaturated fatty esters with copper-chromite catalyst: Kinetics, mechanism and isomerization. J. Am. Oil Chem. Soc. 1970, 47, 463–466. [Google Scholar] [CrossRef]
- Niczke, L.; Czechowski, F.; Gawel, I. Oxidized rapseed oil methyl ester as a bitumen flux: Structural changes in the ester during catalytic oxidation. Prog. Org. Coat. 2007, 59, 304–311. [Google Scholar] [CrossRef]
- Nguyen, B.N.T.; Leclerc, C.A. Catalytic partial oxidation of methyl acetate as a model to investigate the conversion of methyl esters to hydrogen. Int. J. Hydrog. Energy 2008, 33, 1295–1303. [Google Scholar] [CrossRef]
- Wachs, I.E. Production of Formaldehyde from Methyl Mercaptans. US Patent N: 5969191, 6 September 1999. [Google Scholar]
- Mouammine, A.; Ojala, S.; Pirault-Roy, L.; Bensitel, M.; Keiski, R.; Brahmi, R. Catalytic Partial Oxidation of Methanol and Methyl Mercaptan: Studies on the Selectivity of TiO2 and CeO2 Supported V2O5 Catalysts. Top. Catal. 2013, 56, 650–657. [Google Scholar] [CrossRef]
- Krause, E.; Rroka, K. Making Formaldahyde from Methylene Chloride. US Patent N: 1616533, 7 August 1927. [Google Scholar]
- Stauffer, J.E. Formaldehyde process. Separation. US patent N: 6822123 B2, 02 April 2003. [Google Scholar]
- Gong, H.; Chen, Z.; Zhang, M.; Wu, W.; Wang, W.; Wang, W. Study on the deactivation of the deoxygen catalyst during the landfill gas upgrading process. Fuel 2015, 144, 43–49. [Google Scholar] [CrossRef]
- Karacan, Ö.; Ruiz, F.A.; Cotè, M.; Phipps, S. Coal mine methane: A review of capture and utilization practices with benefits to mining safety and to greenhouse gas reduction. Int. J. Coal Geol. 2011, 86, 121–156. [Google Scholar] [CrossRef]
- Stasiñska, B.; Machocki, A. Catalysts for the utilization of methane from the coal mine ventilation air. Pol. J. Chem. Technol. 2007, 9, 29–32. [Google Scholar] [CrossRef]
- McMinn, E.; Moates, F.C.; Richardson, J.T. Catalytic Steam Reforming of Chlorocarbons: Catalyst Deactivation. Appl. Catal. B 2001, 31, 93–105. [Google Scholar] [CrossRef]
- Zhang, Q.-H.; Li, Y.; Xu, B.-Q. Reforming of Methane and Coalbed Methane over Nanocomposite Ni/ZrO2. Prepr. Pap.-Am. Chem. Soc. Div. Fuel Chem. 2004, 49, 137. [Google Scholar] [CrossRef]
- Ojeifo, E.; Abaa, K.; Orsulak, M.; Pamidimukkala, P.K.; Sircar, S.; Sharma, S.; Vatsa, T. Coalbed Methane: Recovery & Utilization in North Western San Juan; Colorado EME 580: Integrative Design; Department of Energy and Mineral Engineering, Penn State University: State College, PA, USA, 2010. [Google Scholar]
- Wachs, I.E. Treating Methanol-Containing Waste Gas Streams. U.S. Patent 5907066, 25 May 1999. [Google Scholar]
- Burgess, T.L.; Gibson, A.G.; Furstein, S.J.; Wachs, I.E. Converting waste gases from pulp mills into value-added chemicals. Environ. Prog. 2002, 21, 137–141. [Google Scholar] [CrossRef]
- Kalantar Neyestanaki, A.; Klingstedt, F.; Salmi, T.; Murzin, D.Y. Deactivation of post combustion catalysts, a review. Fuel 2004, 83, 395–408. [Google Scholar] [CrossRef]
- Spivey, J.J.; Butt, J.B. Deactivation of catalysts in the oxidation of volatile organic compounds. Catal. Today 1992, 11, 465–500. [Google Scholar] [CrossRef]
- Spivey, J.J. Deactivation of reforming Catalysts. In Fuel Cells: Technologies for fuel processing; Shekhawat, D, Spivey, J.J., Berry, D.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; pp. 285–315. [Google Scholar]
- Hulteberg, C. Sulphur-tolerant catalysts in small-scale hydrogen production, a review. Int. J. Hydrogen Energy 2012, 37, 3978–3992. [Google Scholar] [CrossRef]
- Pitkäaho, S.; Ojala, S.; Maunula, T.; Savimäki, A.; Kinnunen, T.; Keiski, R.L. Oxidation of dichloromethane and perchloroethylene as single compounds and in mixtures. Appl. Catal. B 2011, 102, 395–403. [Google Scholar] [CrossRef]
- Pitkäaho, S.; Matejova, L.; Jiratova, K.; Ojala, S.; Keiski, R.L. Oxidation of perchloroethylene–Activity and selectivity of Pt, Pd, Rh and V2O5 catalysts supported on Al2O3, Al2O3-TiO2, Al2O3-CeO2, Part 2. Appl. Catal. B 2012, 126, 215–224. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A 2001, 212, 17–60. [Google Scholar] [CrossRef]
- De Rivas, B.; López-Fonseca, R.; Gutiérrez-Ortiz, M.A.; Gutiérrez-Ortiz, J.I. Impact of induced chlorine-poisoning on the catalytic behavior of Ce0.5Zr0.5O2 and Ce0.15Zr0.85O2 in the gas-phase oxidation of chlorinated VOCs. Appl. Catal. B 2011, 104, 373–381. [Google Scholar] [CrossRef]
- EEA. Non-methane volatile organic compounds (NMVOC) emissions (Ape 004). European Environment Agency, 2014. Available online: http://www.eea.europa.eu/data-and-maps/indicators/eea-32-non-methane-volatile-1/assessment-4 (accessed on 31 January 2015).
- E-PRTR (The European Pollutant Release and Transfer Register) 2012 data. Available online: http://prtr.ec.europa.eu (accessed on 28 January 2015).
- Theloke, J.; Friedrich, R. Compilation of a database on the composition of anthropogenic VOC emissions for atmospheric modeling in Europe. Atmos. Environ. 2007, 41, 4148–4160. [Google Scholar] [CrossRef]
- U.S. EPA. Profile of the 2011 National Air emissions inventory U.S. EPA 2011 NEI version1.0. Office of Air Quality Planning&Standards, Emissions inventory&Analysis group, United States Environmental Protection Agency, April 2014. Available online: http://www.epa.gov/ttn/chief/net/lite_finalversion_ver10.pdf (accessed on 31 January 2015). [Google Scholar]
- Wei, W.; Wang, S.; Hao, J.; Cheng, S. Projection of anthropogenic volatile organic compounds (VOCs) emissions in China for the period 2010–2020. Atmos. Environ. 2011, 45, 6863–6871. [Google Scholar] [CrossRef]
- EEA. Costs of air pollution from European industrial facilities 2008–2012–an updated assessment. European Environment Agency, 2014. Available online: http://www.eea.europa.eu/publications/costs-of-air-pllution-2008–2012 (accessed on 31 January 2015).
- Centi, G.; Ciambelli, P.; Perathoner, S.; Russo, P. Environmental catalysis: Trends and outlook. Catal. Today 2002, 75, 3–15. [Google Scholar] [CrossRef]
- Centi, G.; van Santen, R. Catalysis for Renewables; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; p. 448. [Google Scholar]
- Ballivet-Tkatchenko, D.; Chambrey, S.; Keiski, R.; Ligabue, R.; Plasseraud, L.; Richard, P.; Turunen, H. Direct synthesis of dimethyl carbonate with supercritical carbon dioxide: Characterization of a key organotin oxide intermediate. Catal. Today 2006, 115, 80–87. [Google Scholar] [CrossRef]
- Jenssen, F.J.J.G.; van Santen, R.A. Environmental Catalysis. NIOK Catal. Sci. Ser. 1999, 1, 369. [Google Scholar]
- Lowell Center for Sustainable Production. Available online: http://sustainableproduction.org/ (accessed on 2 May 2009).
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Anastas, P.T.; Heine, G.L.; Williamson, T.C. Green Engineering; Oxford University Press: Washington, DC, USA, 2001. [Google Scholar]
- Anastas, P.T.; Williamson, T.C. Green Chemistry, Frontiers in Benign Chemical Syntheses and Processes; Bookcraft Ltd.: Oxford, UK, 1998. [Google Scholar]
- Saavalainen, P.; Majala, A.; Pongrácz, E.; Keiski, R.L. Designing for sustainability: Developing a tool for sustainable chemical process design. Manuscript submitted in 2011.
- Manley, J.B.; Anastas, P.T.; Cue, B.W. Frontiers in Green Chemistry: Meeting the grand challenges for sustainability in R&D and manufacturing. J. Clean. Prod. 2008, 16, 743–750. [Google Scholar]
- Quinn, M.; Kriebel, D.; Geiser, K.; Moure-Eraso, R. Sustainable production. A proposed strategy for the work environment. In Approaches to Sustainable Development; Forrant, R., Pyle, J.L., Lazonick, W., Levenstein, C., Eds.; University of Massachusetts Press: Amherst, MA, USA, 2001. [Google Scholar]
- Grunes, J.; Zhu, J.; Somorjai, G.A. Catalysis and nanoscience. Chem. Commun. 2003, 2257–2260. [Google Scholar] [CrossRef]
- Somorjai, G.A. On the move. Nature 2004, 430, 730. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Hermans, S.; Somorjai, G.A. Nanotechnology in Catalysis; Springer: New York, NY, USA, 2004; Volumes 1–3, p. 555,333. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojala, S.; Koivikko, N.; Laitinen, T.; Mouammine, A.; Seelam, P.K.; Laassiri, S.; Ainassaari, K.; Brahmi, R.; Keiski, R.L. Utilization of Volatile Organic Compounds as an Alternative for Destructive Abatement. Catalysts 2015, 5, 1092-1151. https://doi.org/10.3390/catal5031092
Ojala S, Koivikko N, Laitinen T, Mouammine A, Seelam PK, Laassiri S, Ainassaari K, Brahmi R, Keiski RL. Utilization of Volatile Organic Compounds as an Alternative for Destructive Abatement. Catalysts. 2015; 5(3):1092-1151. https://doi.org/10.3390/catal5031092
Chicago/Turabian StyleOjala, Satu, Niina Koivikko, Tiina Laitinen, Anass Mouammine, Prem K. Seelam, Said Laassiri, Kaisu Ainassaari, Rachid Brahmi, and Riitta L. Keiski. 2015. "Utilization of Volatile Organic Compounds as an Alternative for Destructive Abatement" Catalysts 5, no. 3: 1092-1151. https://doi.org/10.3390/catal5031092