
Spatial Concentration Profiles for the Catalytic Partial Oxidation of Jet Fuel Surrogates in a Rh/Al2O3 Coated Monolith

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
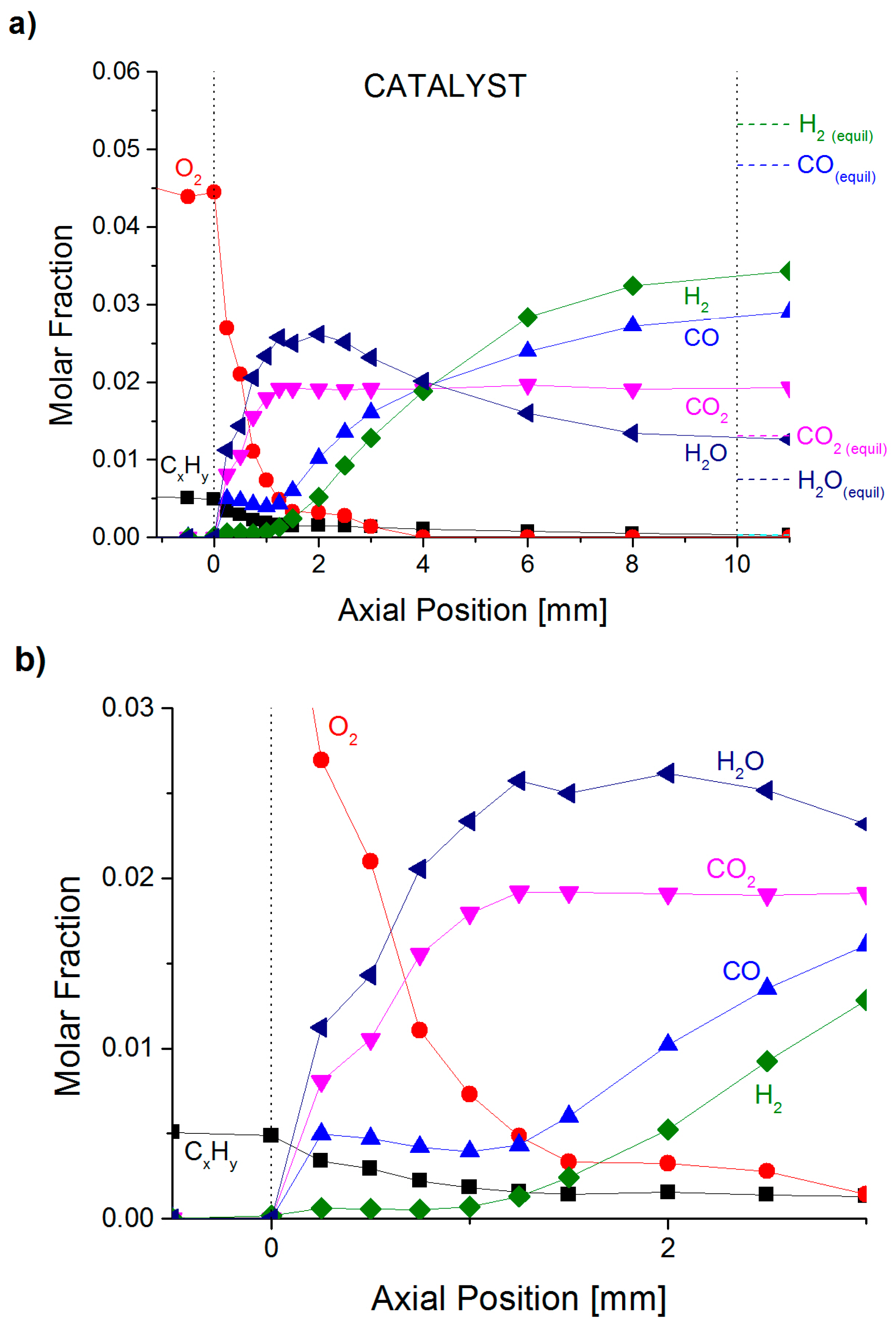
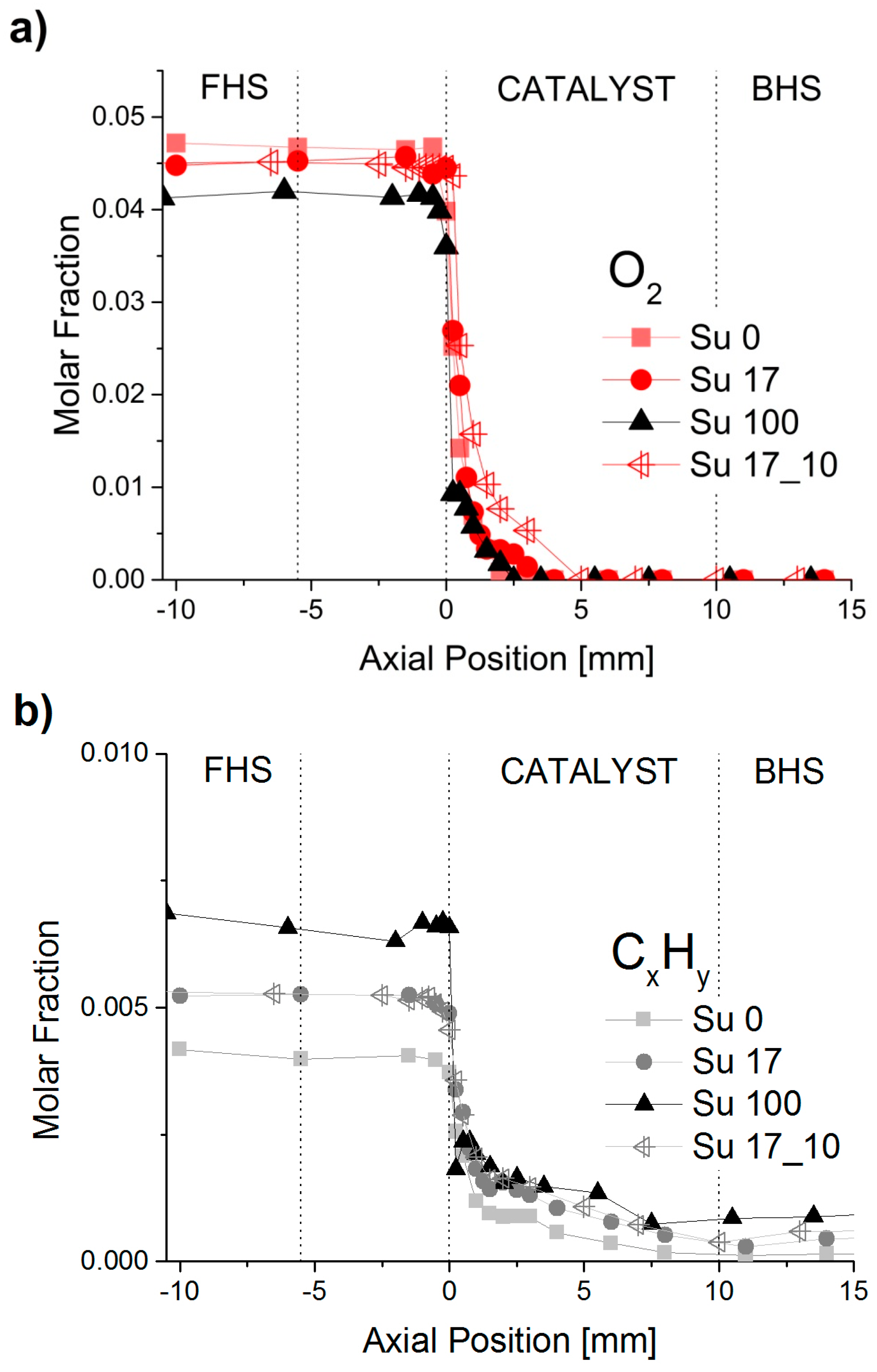
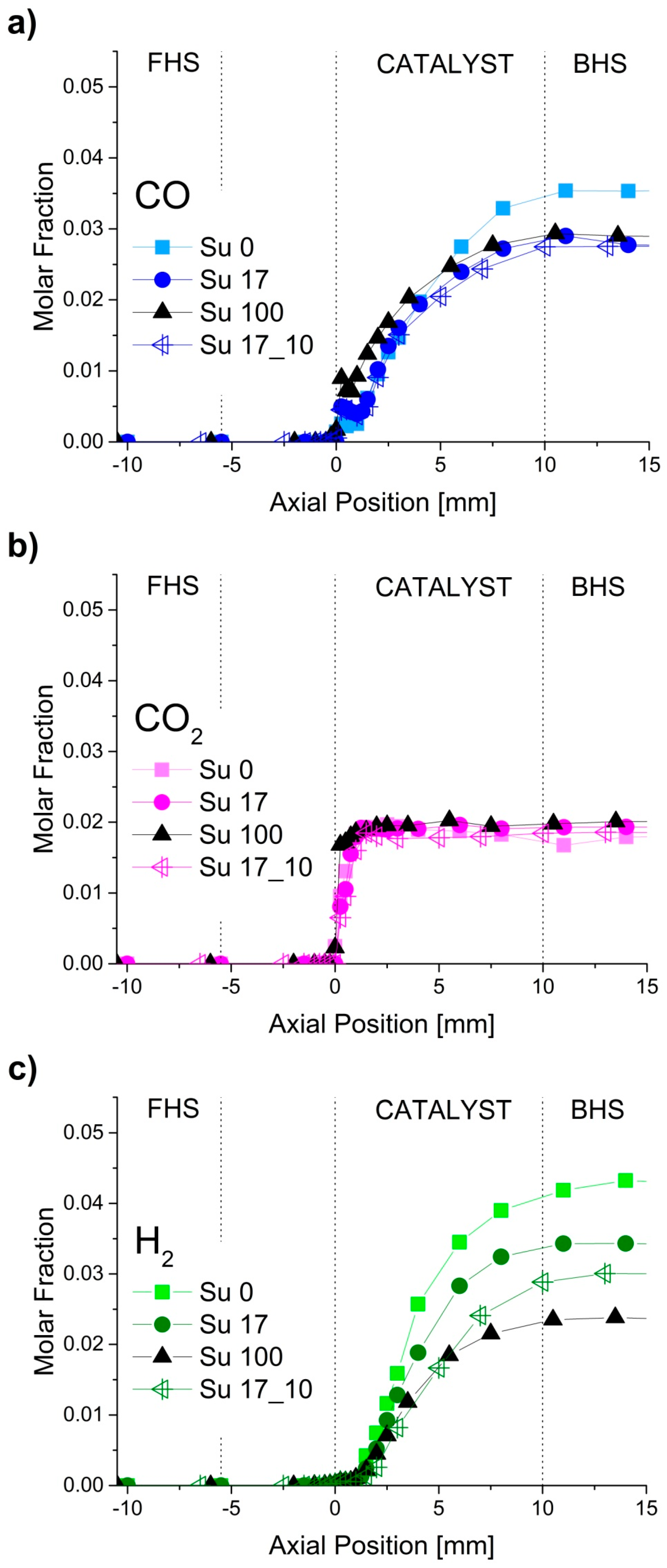
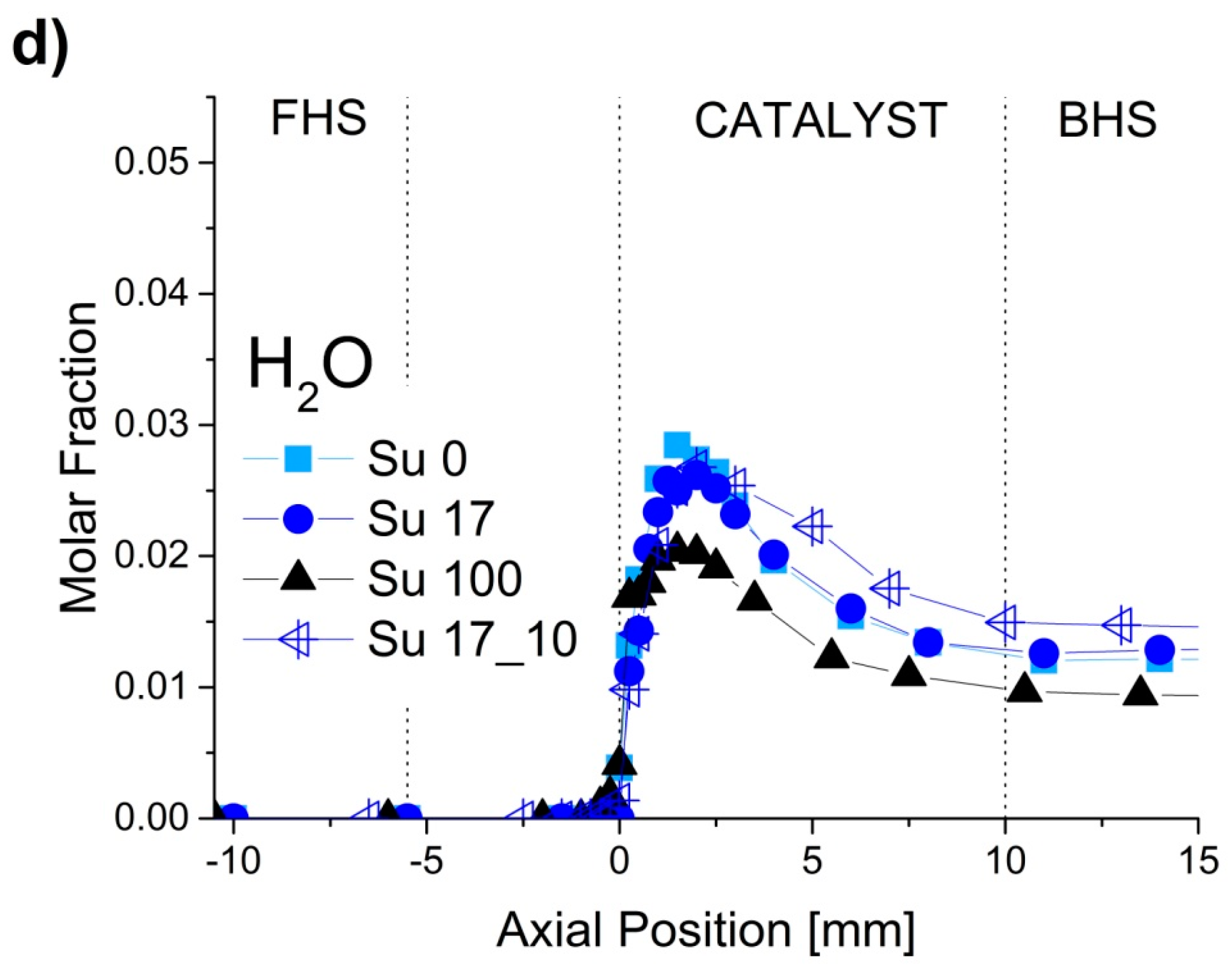
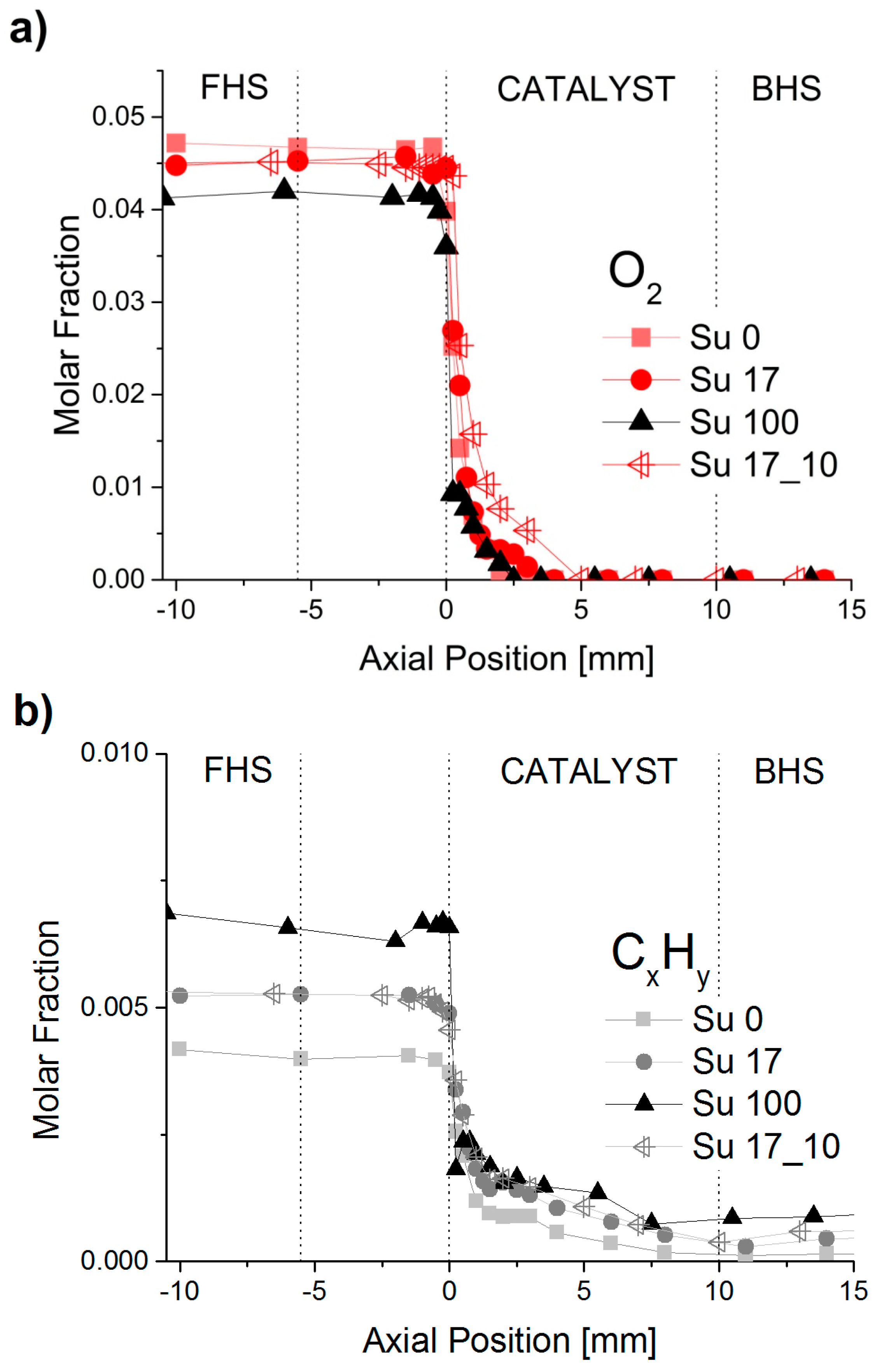
2.1. Spatially-Resolved Concentration and Temperature Profiles
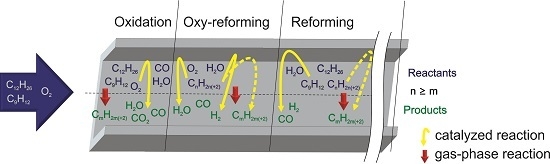
2.2. Reaction Zones in CPOX of Higher Hydrocarbons
2.3. Reaction Zones in Methane CPOX
2.4. Formation of by-Products for CPOX of Higher Hydrocarbons
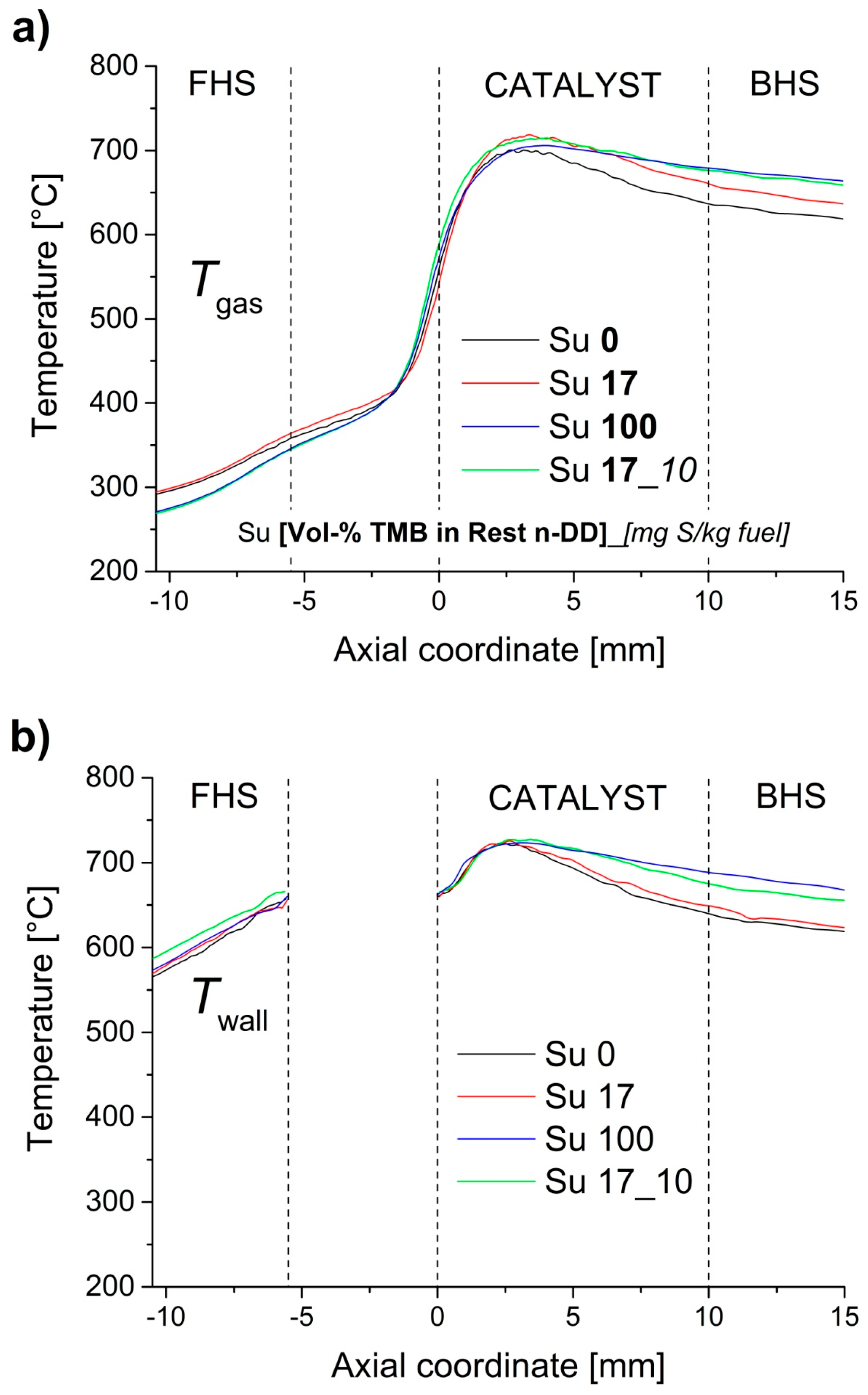
2.5. Influence of Aromatic and Sulfur Content
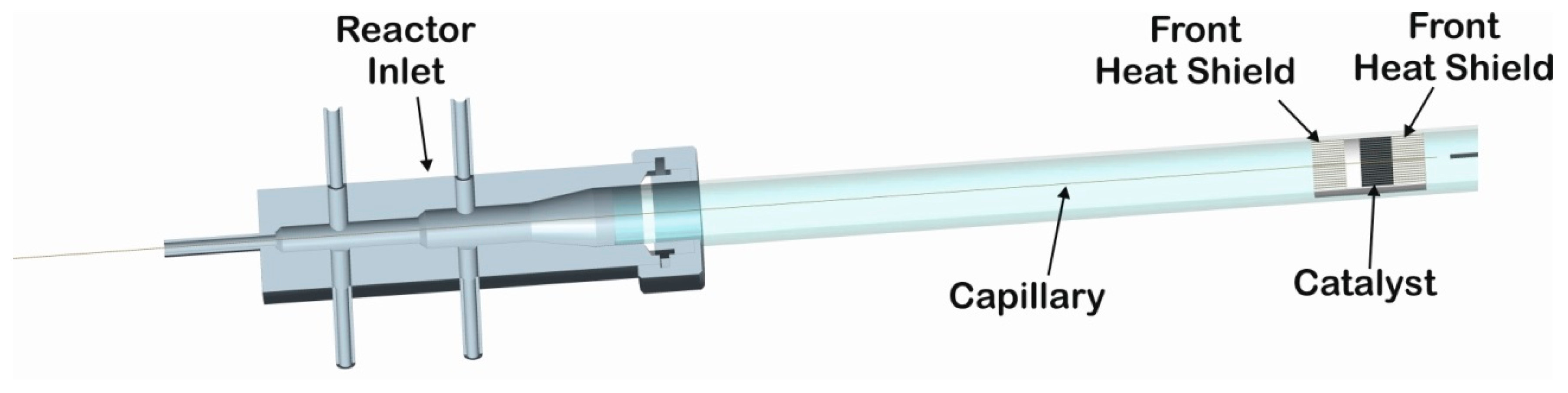
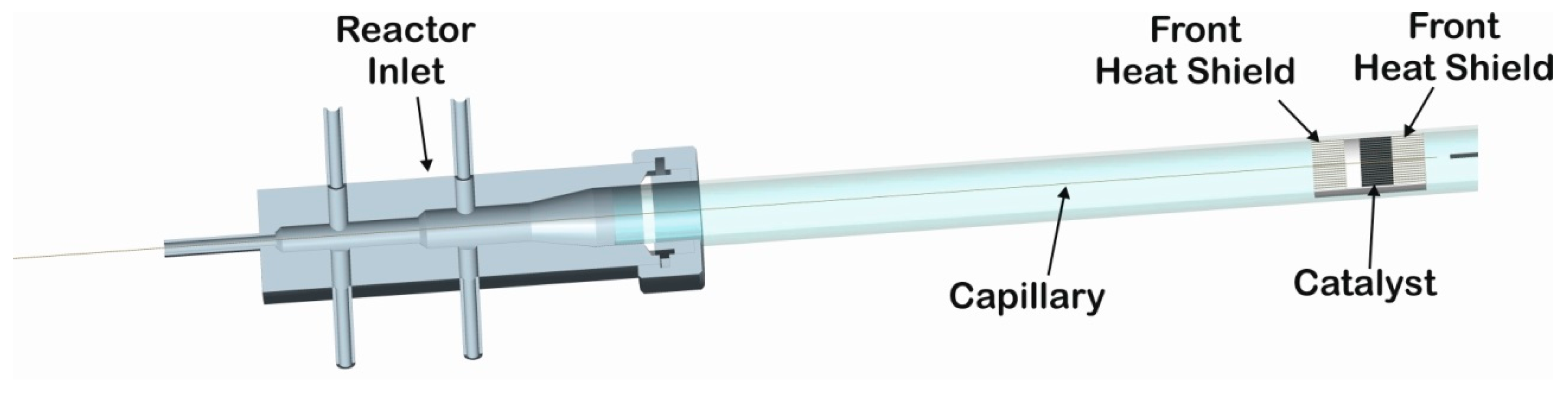
3. Materials and Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aicher, T.; Lenz, B.; Gschnell, F.; Groos, U.; Federici, F.; Caprile, L.; Parodi, L. Fuel processors for fuel cell APU applications. J. Power Sources 2006, 154, 503–508. [Google Scholar] [CrossRef]
- Cheekatamarla, P.K.; Finnerty, C.M. Synthesis gas production via catalytic partial oxidation reforming of liquid fuels. Int. J. Hydrogen Energy 2008, 33, 5012–5019. [Google Scholar] [CrossRef]
- Lindström, B.; Karlsson, J.A.J.; Ekdunge, P.; De Verdier, L.; Häggendal, B.; Dawody, J.; Nilsson, M.; Pettersson, L.J. Diesel fuel reformer for automotive fuel cell applications. Int. J. Hydrogen Energy 2009, 34, 3367–3381. [Google Scholar] [CrossRef]
- Kaltschmitt, T.; Deutschmann, O. Fuel processing for fuel cells. Adv. Chem. Eng. 2012, 41, 1–64. [Google Scholar]
- Pasel, J.; Samsun, R.C.; Peters, R.; Stolten, D. Fuel processing of diesel and kerosene for auxiliary power unit applications. Energy Fuels 2013, 27, 4386–4394. [Google Scholar] [CrossRef]
- Xu, X.; Li, P.; Shen, Y. Small-scale reforming of diesel and jet fuels to make hydrogen and syngas for fuel cells: A review. Appl. Energy 2013, 108, 202–217. [Google Scholar] [CrossRef]
- Hickman, D.A.; Schmidt, L.D. Production of syngas by direct catalytic oxidation of methane. Science 1993, 259, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Schwiedernoch, R.; Tischer, S.; Correa, C.; Deutschmann, O. Experimental and numerical study on the transient behavior of partial oxidation of methane in a catalytic monolith. Chem. Eng. Sci. 2003, 58, 633–642. [Google Scholar] [CrossRef]
- Eriksson, S.; Nilsson, M.; Boutonnet, M.; Järås, S. Partial oxidation of methane over rhodium catalysts for power generation applications. Catal. Today 2005, 100, 447–451. [Google Scholar] [CrossRef]
- Horn, R.; Williams, K.A.; Degenstein, N.J.; Schmidt, L.D. Syngas by catalytic partial oxidation of methane on rhodium: Mechanistic conclusions from spatially resolved measurements and numerical simulations. J. Catal. 2006, 242, 92–102. [Google Scholar] [CrossRef]
- Bitsch-Larsen, A.; Degenstein, N.J.; Schmidt, L.D. Effect of sulfur in catalytic partial oxidation of methane over Rh–Ce coated foam monoliths. Appl. Catal. B 2008, 78, 364–370. [Google Scholar] [CrossRef]
- Bitsch-Larsen, A.; Horn, R.; Schmidt, L.D. Catalytic partial oxidation of methane on rhodium and platinum: Spatial profiles at elevated pressure. Appl. Catal. A 2008, 348, 165–172. [Google Scholar] [CrossRef]
- Christian Enger, B.; Lødeng, R.; Holmen, A. A review of catalytic partial oxidation of methane to synthesis gas with emphasis on reaction mechanisms over transition metal catalysts. Appl. Catal. A 2008, 346, 1–27. [Google Scholar] [CrossRef]
- Donazzi, A.; Livio, D.; Maestri, M.; Beretta, A.; Groppi, G.; Tronconi, E.; Forzatti, P. Synergy of homogeneous and heterogeneous chemistry probed by in situ spatially resolved measurements of temperature and composition. Angew. Chem. Int. Ed. 2011, 50, 3943–3946. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Kruger, J.S.; Hermann, R.J.; Blass, S.D.; Schmidt, L.D. Spatial profiles in partial oxidation of methane and dimethyl ether in an autothermal reactor over rhodium catalysts. Appl. Catal. A 2014, 483, 97–102. [Google Scholar] [CrossRef]
- Maestri, M.; Vlachos, D.; Beretta, A.; Forzatti, P.; Groppi, G.; Tronconi, E. Dominant reaction pathways in the catalytic partial oxidation of CH4 on Rh. Top. Catal. 2009, 52, 1983–1988. [Google Scholar] [CrossRef]
- Nogare, D.D.; Degenstein, N.J.; Horn, R.; Canu, P.; Schmidt, L.D. Modeling spatially resolved data of methane catalytic partial oxidation on Rh foam catalyst at different inlet compositions and flowrates. J. Catal. 2011, 277, 134–148. [Google Scholar] [CrossRef]
- Diehm, C.; Deutschmann, O. Hydrogen production by catalytic partial oxidation of methane over staged Pd/Rh coated monoliths: Spatially resolved concentration and temperature profiles. Int. J. Hydrogen Energy 2014, 39, 17998–18004. [Google Scholar] [CrossRef]
- Wang, D.; Dewaele, O.; Groote, A.M.D.; Froment, G.F. Reaction mechanism and role of the support in the partial oxidation of methane on Rh/Al2O3. J. Catal. 1996, 159, 418–426. [Google Scholar] [CrossRef]
- Mallens, E.P.J.; Hoebink, J.H.B.J.; Marin, G.B. The reaction mechanism of the partial oxidation of methane to synthesis gas: A transient kinetic study over rhodium and a comparison with platinum. J. Catal. 1997, 167, 43–56. [Google Scholar] [CrossRef]
- Livio, D.; Diehm, C.; Donazzi, A.; Beretta, A.; Deutschmann, O. Catalytic partial oxidation of ethanol over Rh/Al2O3: Spatially resolved temperature and concentration profiles. Appl. Catal. A 2013, 467, 530–541. [Google Scholar] [CrossRef]
- Hartmann, M.; Maier, L.; Minh, H.D.; Deutschmann, O. Catalytic partial oxidation of iso-octane over rhodium catalysts: An experimental, modeling, and simulation study. Combust. Flame 2010, 157, 1771–1782. [Google Scholar] [CrossRef]
- Curran, H.J.; Gaffuri, P.; Pitz, W.J.; Westbrook, C.K. A comprehensive modeling study of iso-octane oxidation. Combust. Flame 2002, 129, 253–280. [Google Scholar] [CrossRef]
- Krummenacher, J.J.; West, K.N.; Schmidt, L.D. Catalytic partial oxidation of higher hydrocarbons at millisecond contact times: decane, hexadecane, and diesel fuel. J. Catal. 2003, 215, 332–343. [Google Scholar] [CrossRef]
- Cheekatamarla, P.K.; Lane, A.M. Catalytic autothermal reforming of diesel fuel for hydrogen generation in fuel cells: I. Activity tests and sulfur poisoning. J. Power Sources 2005, 152, 256–263. [Google Scholar] [CrossRef]
- Shekhawat, D.; Gardner, T.H.; Berry, D.A.; Salazar, M.; Haynes, D.J.; Spivey, J.J. Catalytic partial oxidation of n-tetradecane in the presence of sulfur or polynuclear aromatics: Effects of support and metal. Appl. Catal. A 2006, 311, 8–16. [Google Scholar] [CrossRef]
- Hartmann, M.; Kaltschmitt, T.; Deutschmann, O. Catalytic partial oxidation of higher hydrocarbon fuel components on Rh/Al2O3 coated honeycomb monoliths. Catal. Today 2009, 147, S204–S209. [Google Scholar] [CrossRef]
- Haynes, D.J.; Berry, D.A.; Shekhawat, D.; Spivey, J.J. Catalytic partial oxidation of n-tetradecane using Rh and Sr substituted pyrochlores: Effects of sulfur. Catal. Today 2009, 145, 121–126. [Google Scholar] [CrossRef]
- Shekhawat, D.; Berry, D.A.; Haynes, D.J.; Spivey, J.J. Fuel constituent effects on fuel reforming properties for fuel cell applications. Fuel 2009, 88, 817–825. [Google Scholar] [CrossRef]
- DuBois, T.G.; Nieh, S. Selection and performance comparison of jet fuel surrogates for autothermal reforming. Fuel 2011, 90, 1439–1448. [Google Scholar] [CrossRef]
- Diehm, C.; Kaltschmitt, T.; Deutschmann, O. Hydrogen production by partial oxidation of ethanol/gasoline blends over Rh/Al2O3. Catal. Today 2012, 197, 90–100. [Google Scholar] [CrossRef]
- Granlund, M.Z.; Jansson, K.; Nilsson, M.; Dawody, J.; Pettersson, L.J. Evaluation of Co, La, and Mn promoted Rh catalysts for autothermal reforming of commercial diesel. Appl. Catal. B 2014, 154–155, 386–394. [Google Scholar] [CrossRef]
- Granlund, M.Z.; Jansson, K.; Nilsson, M.; Dawody, J.; Pettersson, L.J. Evaluation of Co, La, and Mn promoted Rh catalysts for autothermal reforming of commercial diesel: Aging and characterization. Appl. Catal. B 2015, 172–173, 145–153. [Google Scholar] [CrossRef]
- Bär, J.N.; Rocha, M.I.; Oliviera, E.J.; Deutschmann, O. Impact of sulfur on catalytic partial oxidation of jet fuel surrogates over Rh/Al2O3. Int. J. Hydrogen Energy 2015, 41, 3701–3711. [Google Scholar] [CrossRef]
- Livio, D.; Donazzi, A.; Beretta, A.; Groppi, G.; Forzatti, P. Experimental and modeling analysis of the thermal behavior of an autothermal C3H8 catalytic partial oxidation reformer. Ind. Eng. Chem. Res. 2012, 51, 7573–7583. [Google Scholar] [CrossRef]
- Beretta, A.; Donazzi, A.; Groppi, G.; Maestri, M.; Tronconi, E.; Forzatti, P. Gaining insight into the kinetics of partial oxidation of light hydrocarbons on Rh, through a multiscale methodology based on advanced experimental and modeling techniques. Catalysis 2013, 25, 1–49. [Google Scholar]
- Donazzi, A.; Livio, D.; Beretta, A.; Groppi, G.; Forzatti, P. Surface temperature profiles in CH4 CPO over honeycomb supported Rh catalyst probed with in situ optical pyrometer. Appl. Catal. A 2011, 402, 41–49. [Google Scholar] [CrossRef]
- Hettel, M.; Diehm, C.; Torkashvand, B.; Deutschmann, O. Critical evaluation of in situ probe techniques for catalytic honeycomb monoliths. Catal. Today 2013, 216, 2–10. [Google Scholar] [CrossRef]
- Hickman, D.A.; Schmidt, L.D. Synthesis gas formation by direct oxidation of methane over Pt monoliths. J. Catal. 1992, 138, 267–282. [Google Scholar] [CrossRef]
- Fathi, M.; Monnet, F.; Schuurman, Y.; Holmen, A.; Mirodatos, C. Reactive oxygen species on platinum gauzes during partial oxidation of methane into synthesis gas. J. Catal. 2000, 190, 439–445. [Google Scholar] [CrossRef]
- Subramanian, R.; Panuccio, G.J.; Krummenacher, J.J.; Lee, I.C.; Schmidt, L.D. Catalytic partial oxidation of higher hydrocarbons: reactivities and selectivities of mixtures. Chem. Eng. Sci. 2004, 59, 5501–5507. [Google Scholar] [CrossRef]
- Hartmann, M.; Maier, L.; Deutschmann, O. Hydrogen production by catalytic partial oxidation of iso-octane at varying flow rate and fuel/oxygen ratio: From detailed kinetics to reactor behavior. Appl. Catal. A 2011, 391, 144–152. [Google Scholar] [CrossRef]
- Kaltschmitt, T.; Maier, L.; Hartmann, M.; Hauck, C.; Deutschmann, O. Influence of gas-phase reactions on catalytic reforming of isooctane. Proc. Combust. Inst. 2011, 33, 3177–3183. [Google Scholar] [CrossRef]
- Aartun, I.; Silberova, B.; Venvik, H.; Pfeifer, P.; Görke, O.; Schubert, K.; Holmen, A. Hydrogen production from propane in Rh-impregnated metallic microchannel reactors and alumina foams. Catal. Today 2005, 105, 469–478. [Google Scholar] [CrossRef]
- Shinjoh, H.; Muraki, H.; Fujitani, Y. Periodic operation effects in propane and propylene oxidation over noble metal catalysts. Appl. Catal. 1989, 49, 195–204. [Google Scholar] [CrossRef]
- Calo, J.M.; Perkins, M.T. A heterogeneous surface model for the “steady-state” kinetics of the Boudouard reaction. Carbon 1987, 25, 395–407. [Google Scholar] [CrossRef]
- Lim, J.Y.; McGregor, J.; Sederman, A.J.; Dennis, J.S. The role of the Boudouard and water–gas shift reactions in the methanation of CO or CO2 over Ni/γ-Al2O3 catalyst. Chem. Eng. Sci. 2016, 152, 754–766. [Google Scholar] [CrossRef]
- Antonio, G.F.M.; Franco, F.; Batalha, N.; Pereira, M.M. Coupling CH4 pyrolysis with CO2 activation via reverse Boudouard reaction in the presence of O2 trhough a multifunctional catalyst Ni-V-Li/Al2O3. J. CO2 Util. 2016, 16, 458–465. [Google Scholar] [CrossRef]
- Deutschmann, O.; Grunwaldt, J.-D. Exhaust gas aftertreatment in mobile systems: Status, challenges, and perspectives. Chem. Ing. Tech. 2013, 85, 595–617. [Google Scholar] [CrossRef]
- Pryor, W. Frontiers of Free Radical Chemistry; Elsevier Science: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Hazlett, R.N. Thermal Oxidation Stability of Aviation Turbine Fuels; ASTM: West Conshohocken, PA, USA, 1991. [Google Scholar]
- Yoon, E.M.; Selvaraj, L.; Song, C.; Stallman, J.B.; Coleman, M.M. High-temperature stabilizers for jet fuels and similar hydrocarbon mixtures. 1. Comparative studies of hydrogen donors. Energy Fuels 1996, 10, 806–811. [Google Scholar] [CrossRef]
- Yoon, E.M.; Selvaraj, L.; Eser, S.; Coleman, M.M. High-temperature stabilizers for jet fuels and similar hydrocarbon mixtures. 2. Kinetic studies. Energy Fuels 1996, 10, 812–815. [Google Scholar] [CrossRef]
- Partridge, W.P.; Storey, J.M.; Lewis, S.; Smithwick, R.; DeVault, G.L.; Cunningham, M.J.; Currier, N.; Yonushonis, T.M. Time-resolved measurements of emission transients by mass spectrometry. SAE Tech. Paper 2000. 2000-01-2952. [Google Scholar] [CrossRef]
- Deutschmann, O.; Tischer, S.; Correa, C.; Chatterjee, D.; Kleditzsch, S.; Janardhanan, V.M.; Mladenov, N.; Minh, H.D.; Karadeniz, H.; Hettel, M. DETCHEMTM Software Package, Version 2.5; DETCHEM: Karlsruhe, Germany, 2014.




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bär, J.N.; Antinori, C.; Maier, L.; Deutschmann, O. Spatial Concentration Profiles for the Catalytic Partial Oxidation of Jet Fuel Surrogates in a Rh/Al2O3 Coated Monolith. Catalysts 2016, 6, 207. https://doi.org/10.3390/catal6120207
Bär JN, Antinori C, Maier L, Deutschmann O. Spatial Concentration Profiles for the Catalytic Partial Oxidation of Jet Fuel Surrogates in a Rh/Al2O3 Coated Monolith. Catalysts. 2016; 6(12):207. https://doi.org/10.3390/catal6120207
Chicago/Turabian StyleBär, Julian N., Claudia Antinori, Lubow Maier, and Olaf Deutschmann. 2016. "Spatial Concentration Profiles for the Catalytic Partial Oxidation of Jet Fuel Surrogates in a Rh/Al2O3 Coated Monolith" Catalysts 6, no. 12: 207. https://doi.org/10.3390/catal6120207