
Nanostructured Inorganic Materials at Work in Electrochemical Sensing and Biofuel Cells

 ,
, 

Abstract
:
1. Introduction
2. From Enzymatically to Abiotically Assisted Sensing
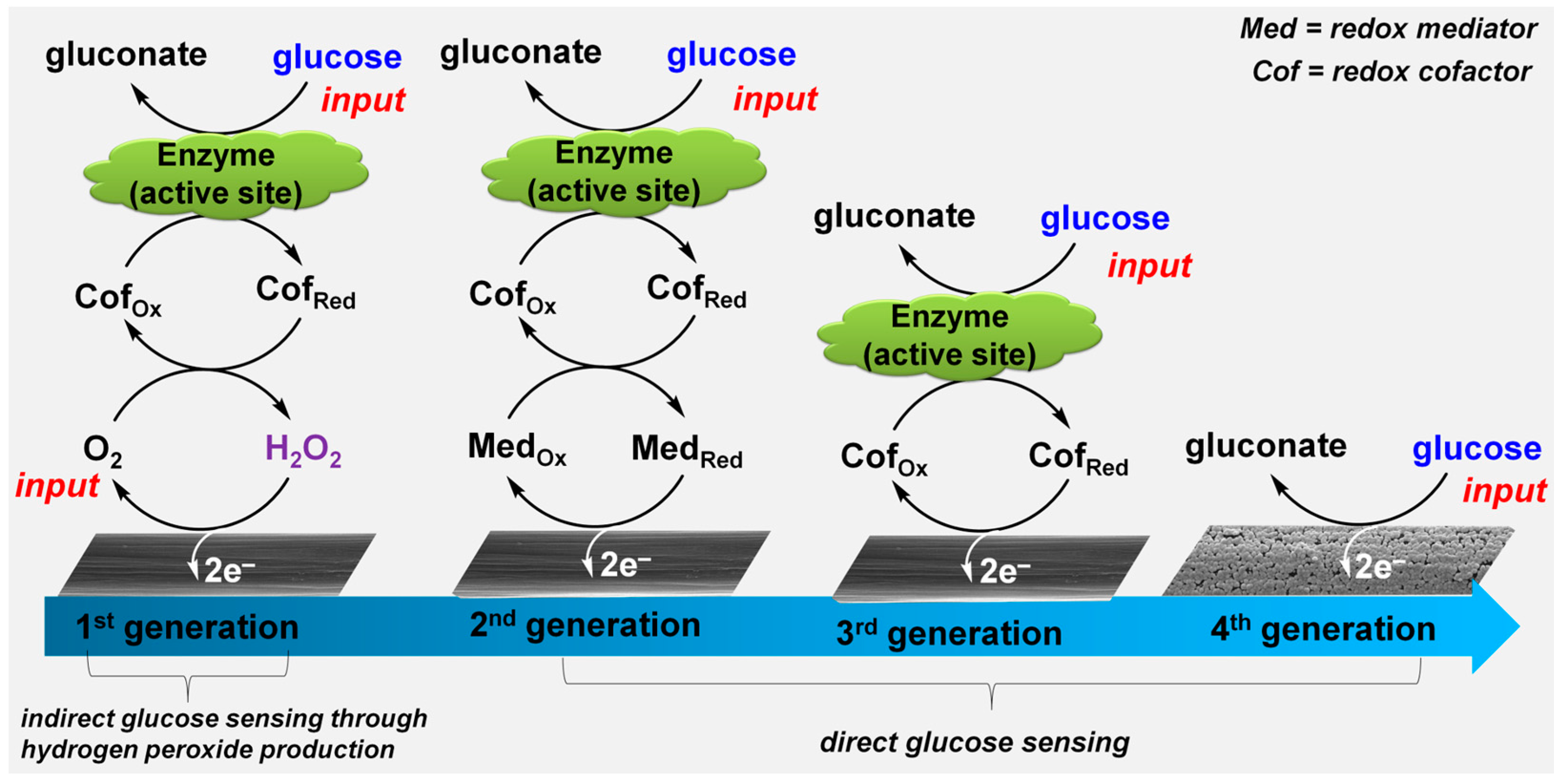
2.1. First-Generation Glucose-Sensing Bioelectrocatalysis
2.2. Second-Generation Glucose-Sensing Bioelectrocatalysis
2.3. Third-Generation Glucose-Sensing Bioelectrocatalysis
2.4. Towards a Fourth-Generation Glucose-Sensing Electrocatalysis
3. Non-Enzymatic Electrochemical Sensors for Biomedical and Non-Biomedical Applications
3.1. Clarifying the Requirements of Electrochemical Sensors for Biomedical and Non-Physiological Situations
3.1.1. Which Type of Electrochemical Sensor?
3.1.2. Which Precision for Which Goal?
3.1.3. Reliability of Tests: Addressing our Experimental Needs
3.1.4. Current Methods: Running the Electrochemical Assays Experimentally
3.1.5. Screening Electroactive Interferences and Electrode Fouling in the Case of Glucose Sensors
3.2. Nanomaterials for Sensors Based on Glucose Monitoring for Biomedical and Food Industry Targets
3.2.1. Nanostructured Metals: Porous and Non-Porous Materials
3.2.2. Noble Metal (Gold and Platinum)–Based Electrodes
3.2.3. Platinum and Gold-Free Electrodes: Earth-Abundant Transition Metals
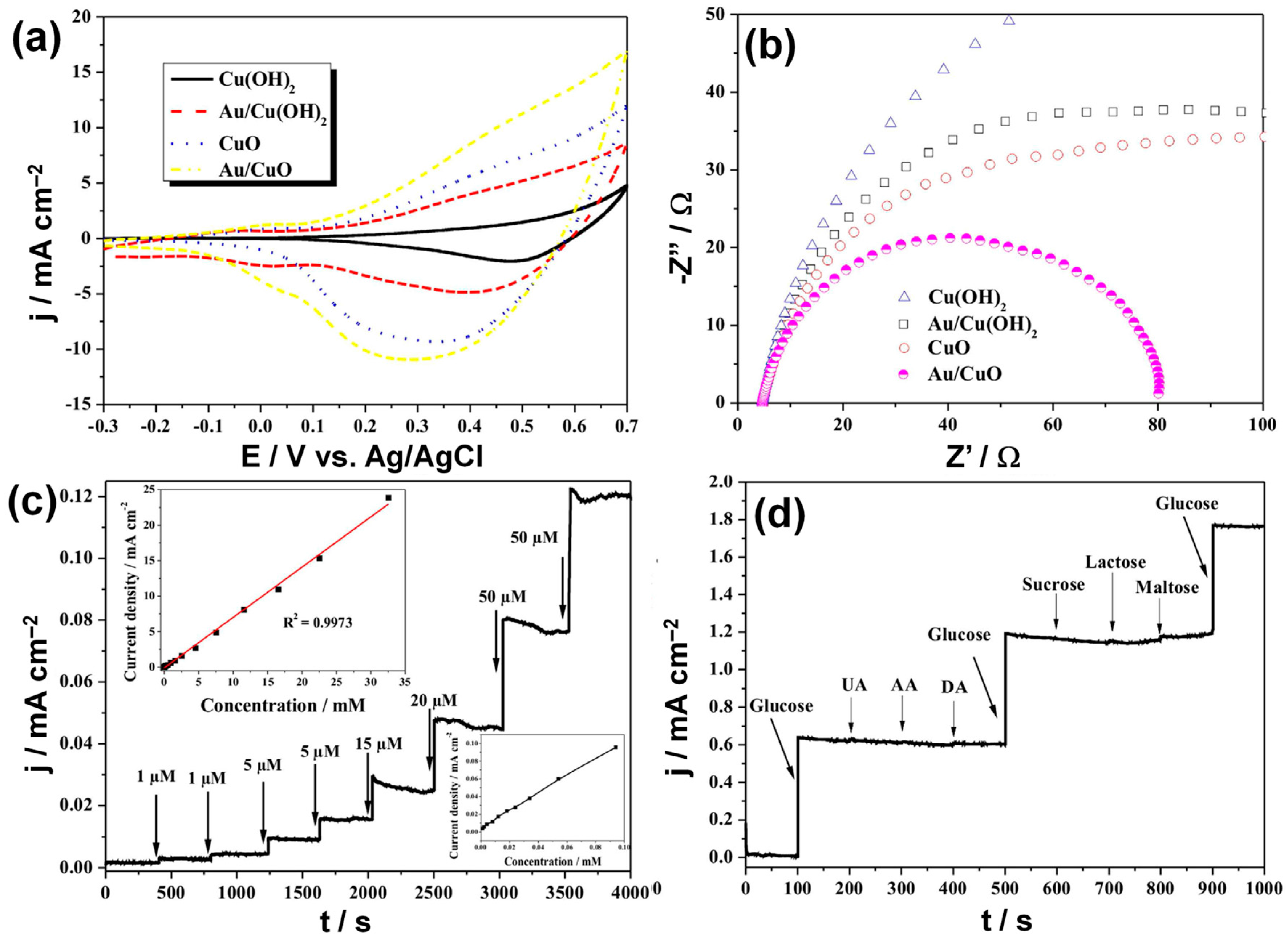
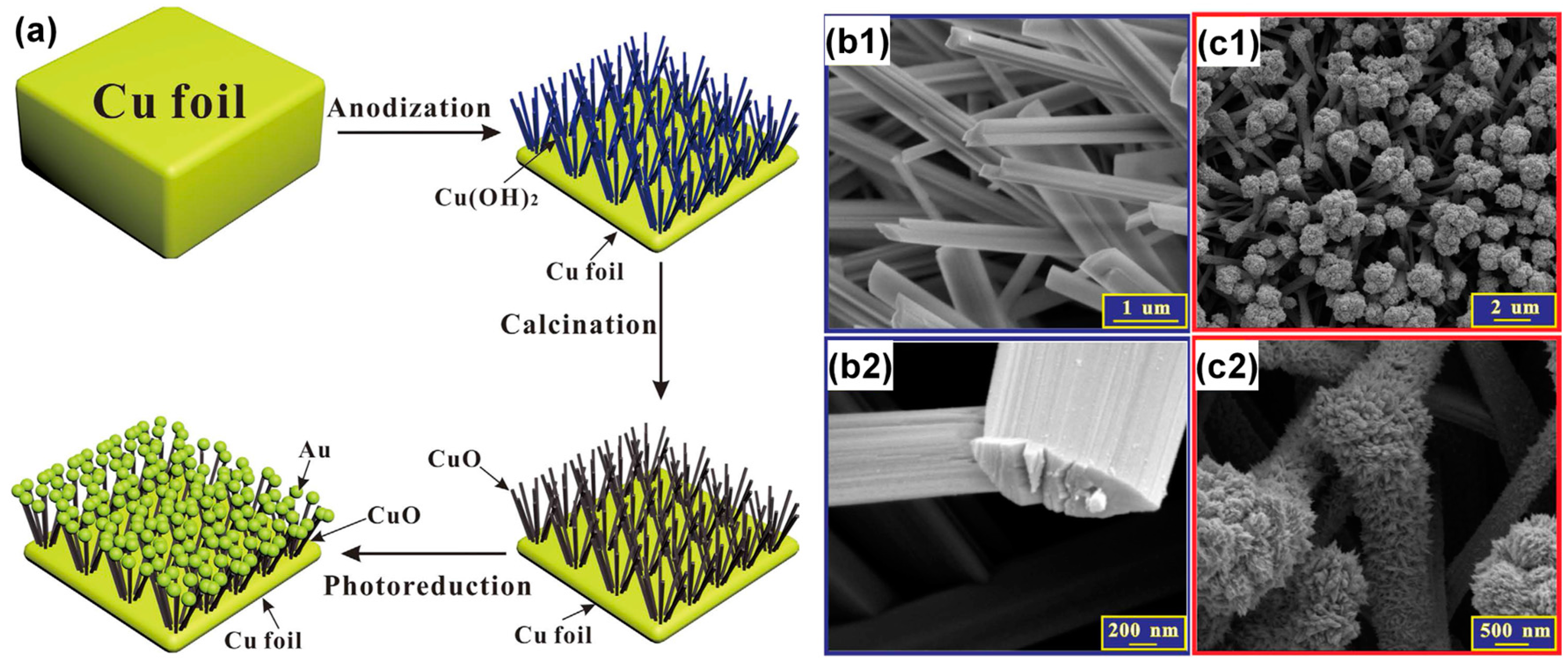
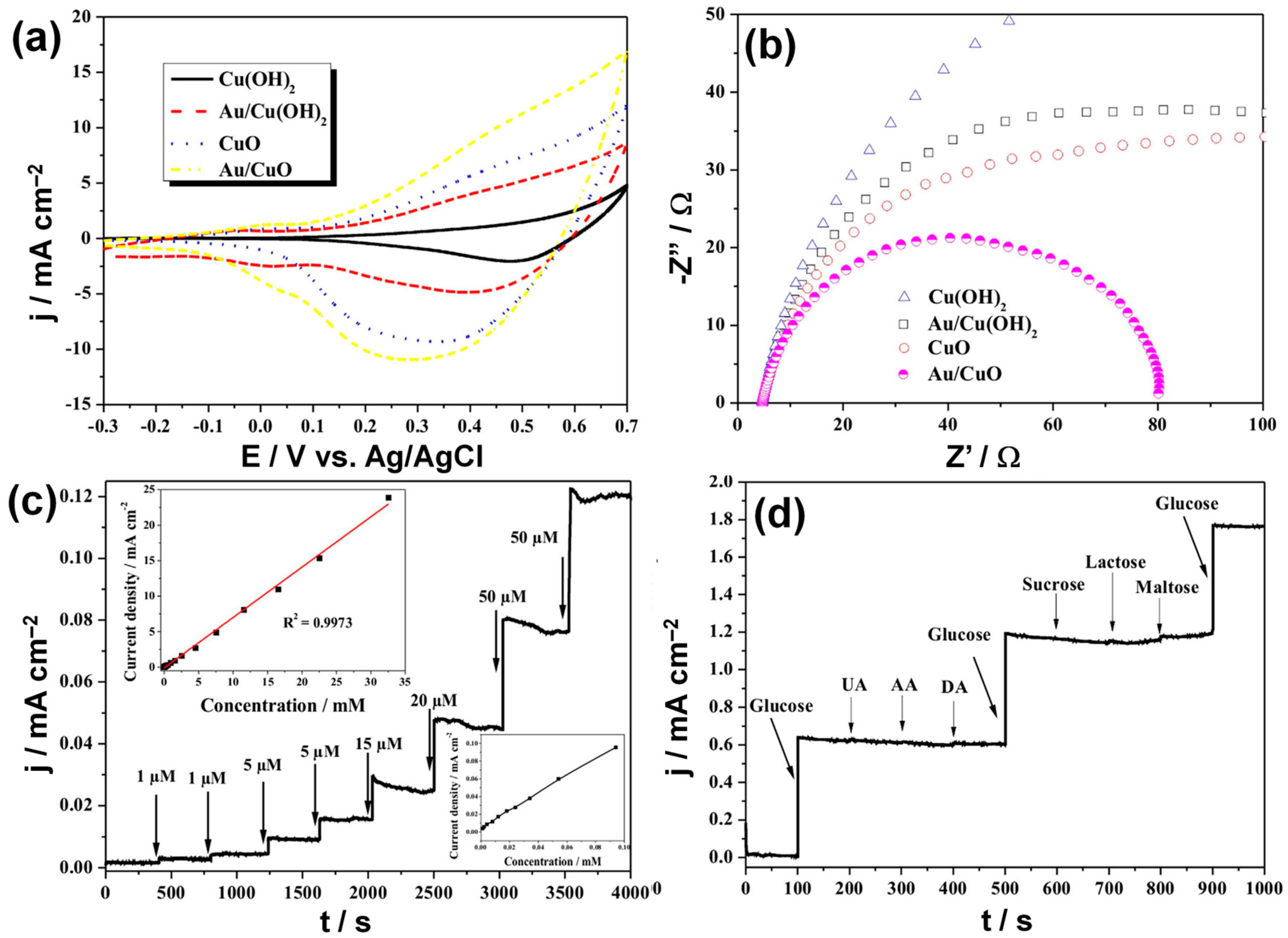
3.2.4. Heterogeneous Electrode Materials: Au- and Cu-Based Hydroxide or Oxide Interfaces
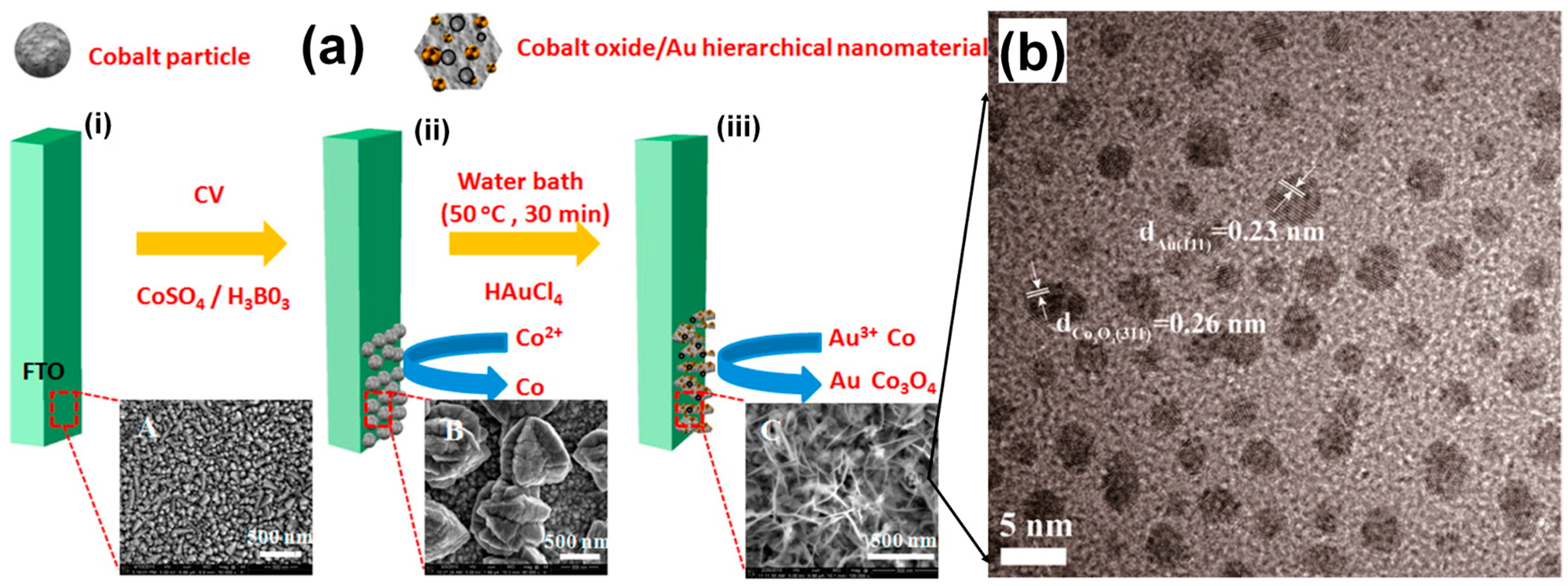
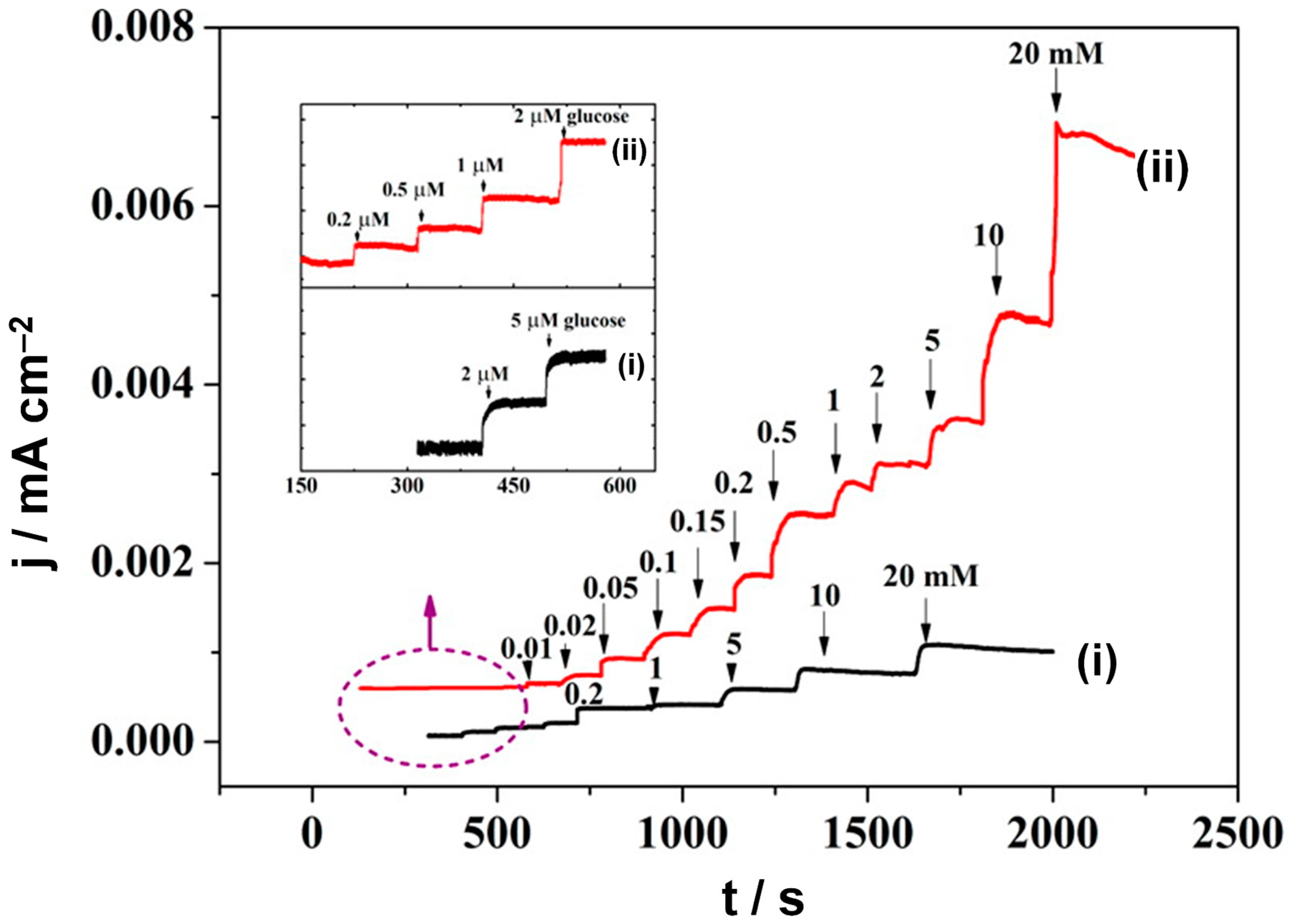
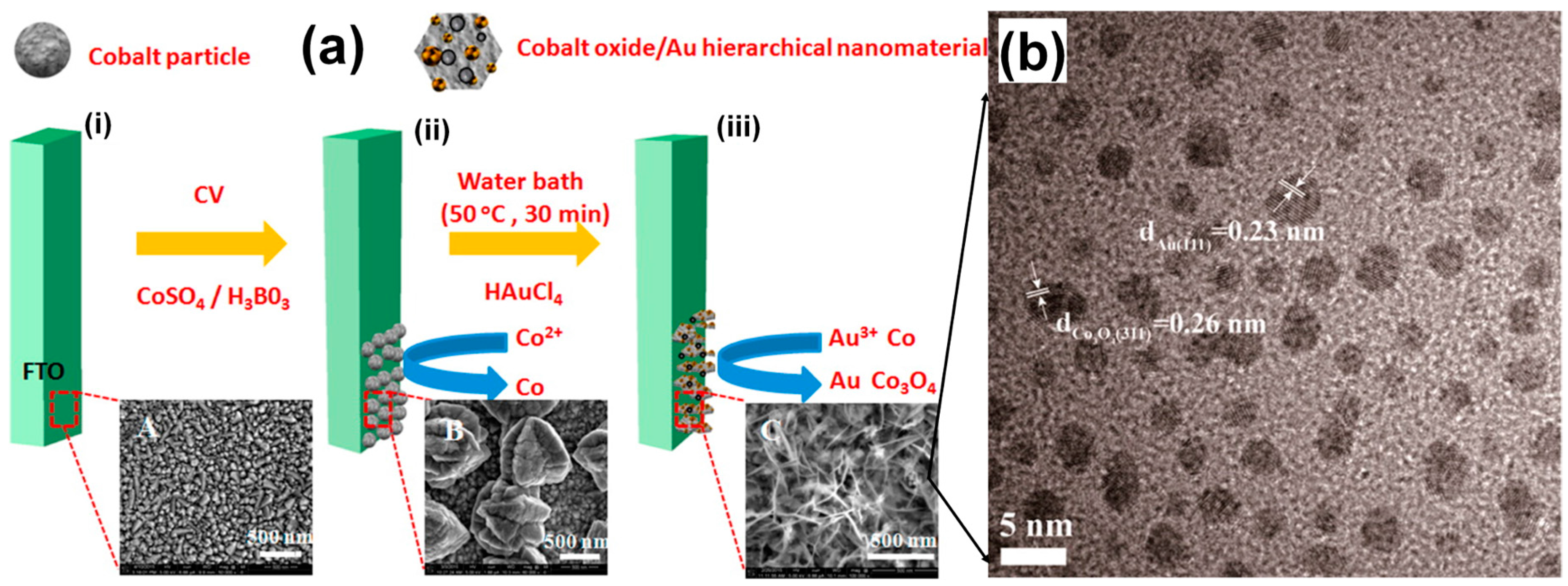
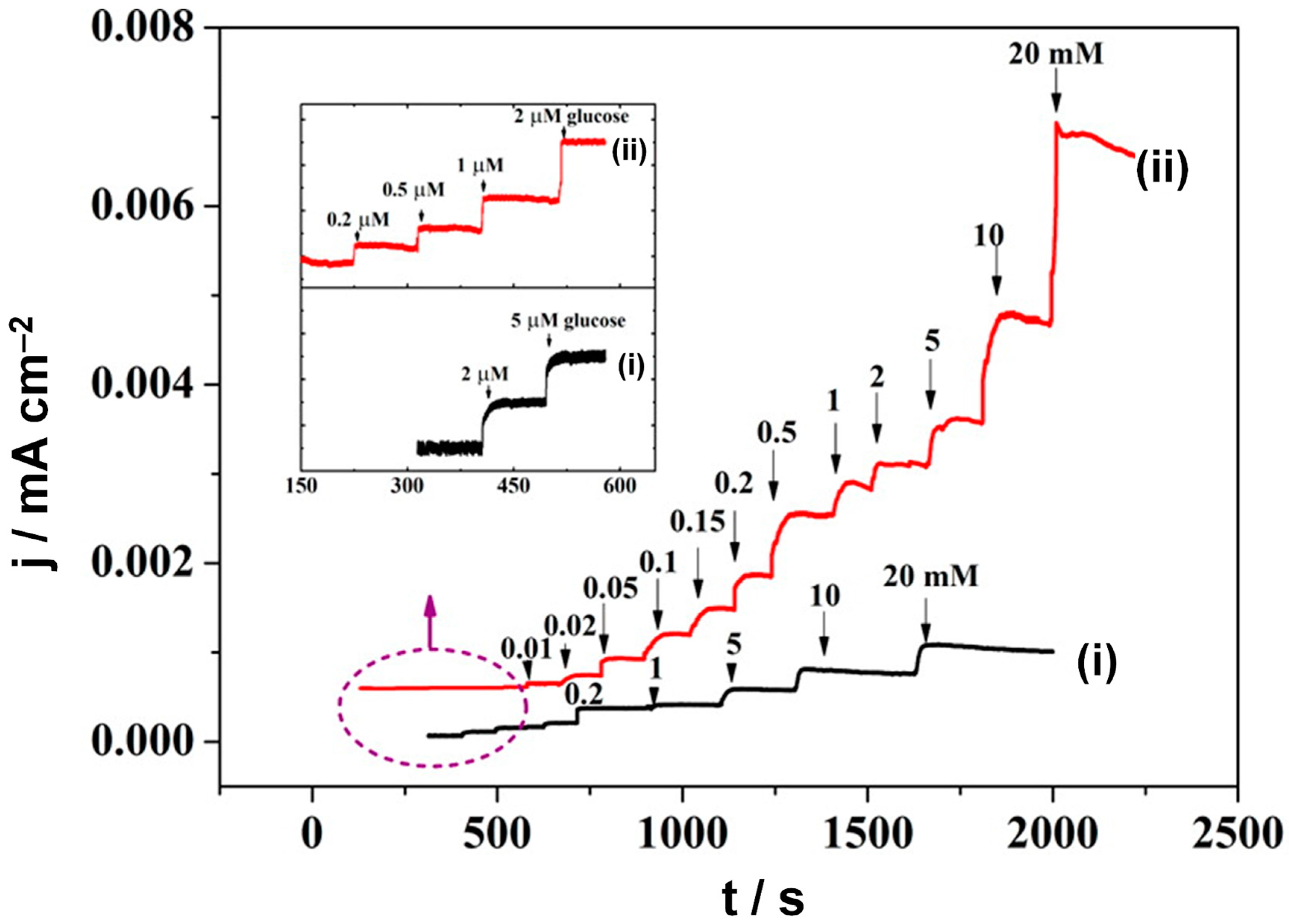
3.2.5. Heterogeneous Electrode Materials: Au- and Co-Based Hydroxide or Oxide Interfaces
3.2.6. Heterogeneous Electrode Materials: Metallic Ni– and Ni-Based Hydroxide or Oxide Interfaces
3.2.7. Carbon-Based Nanomaterials
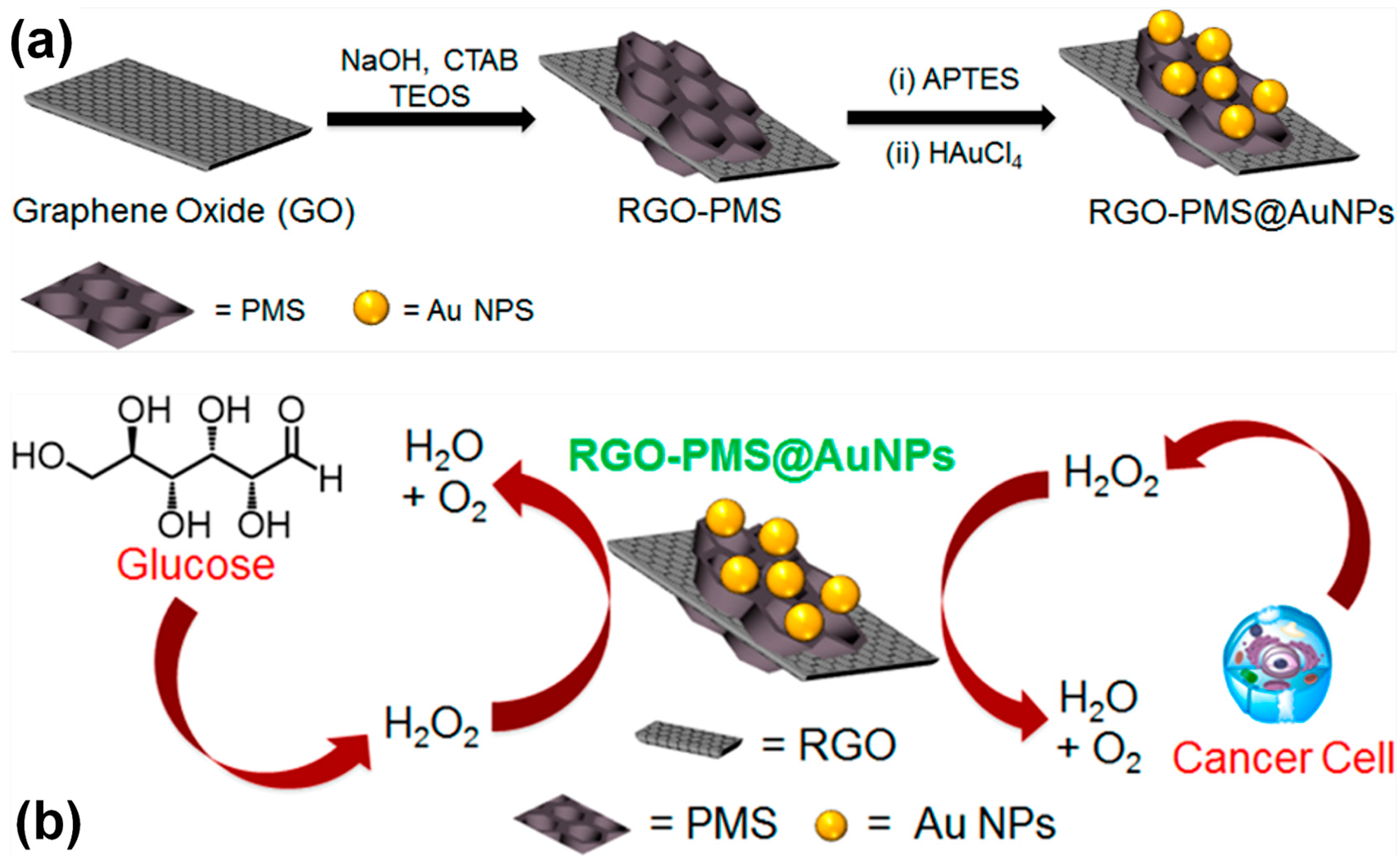
3.3. Nanomaterials for the Electroanalysis of H2O2: Tracing the Progress of Biochemical Reactions
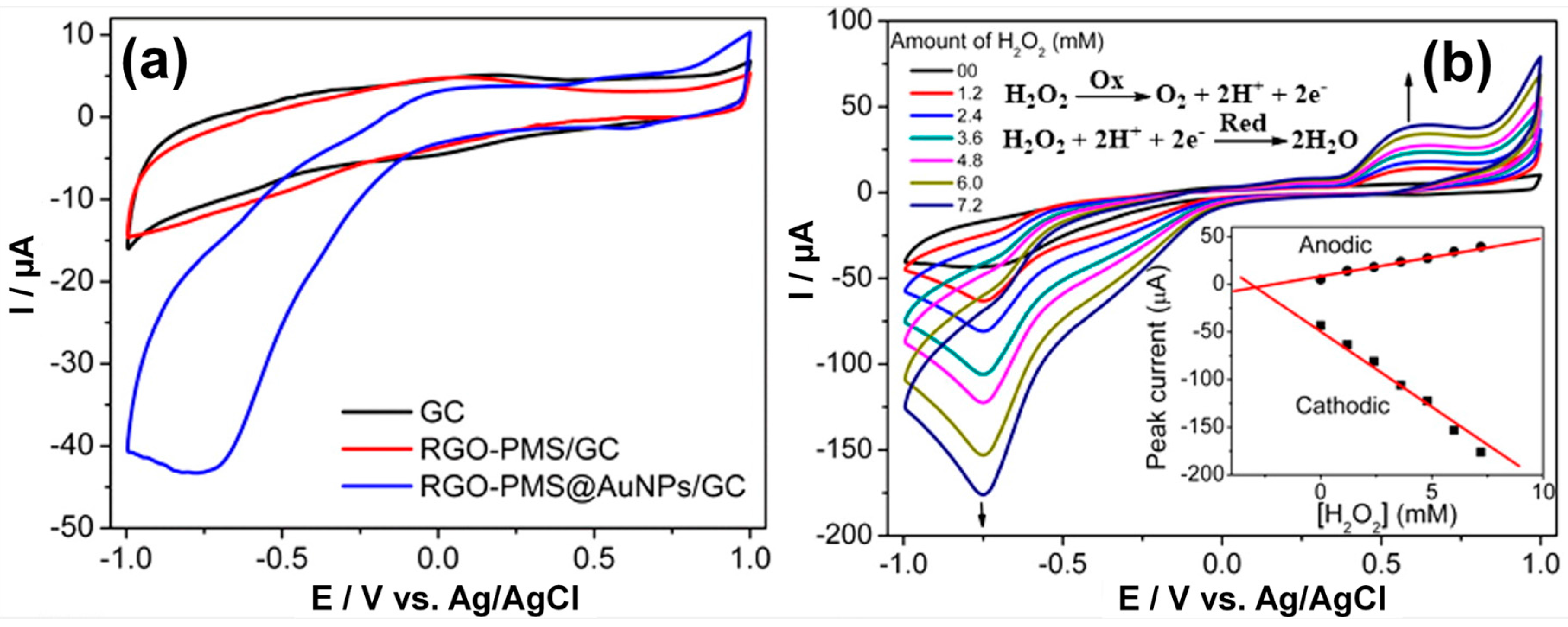
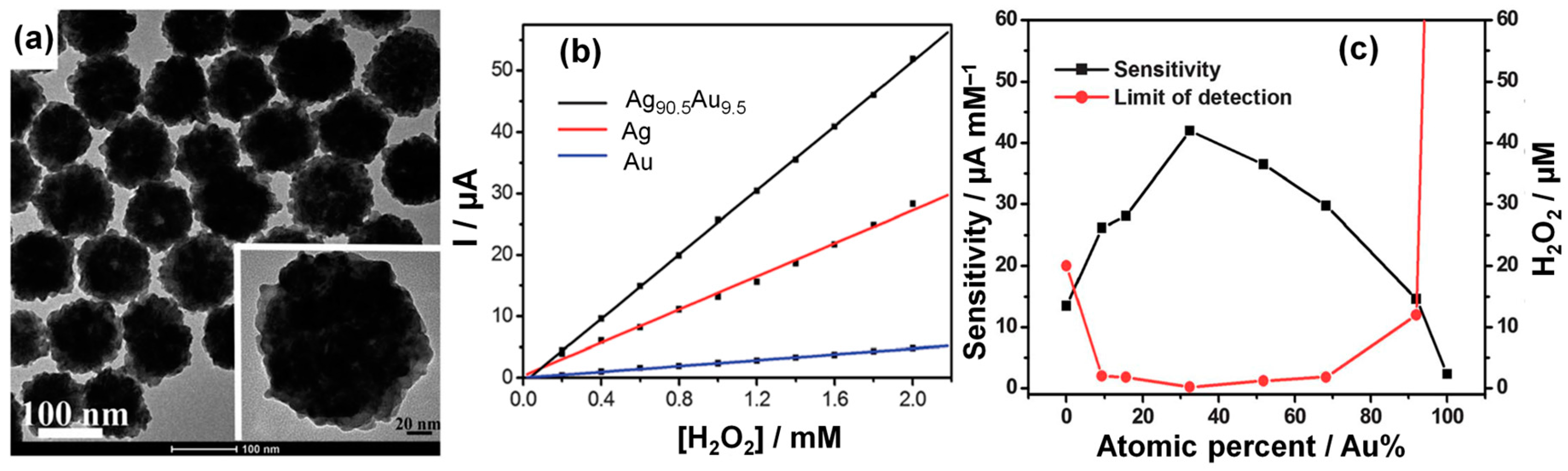
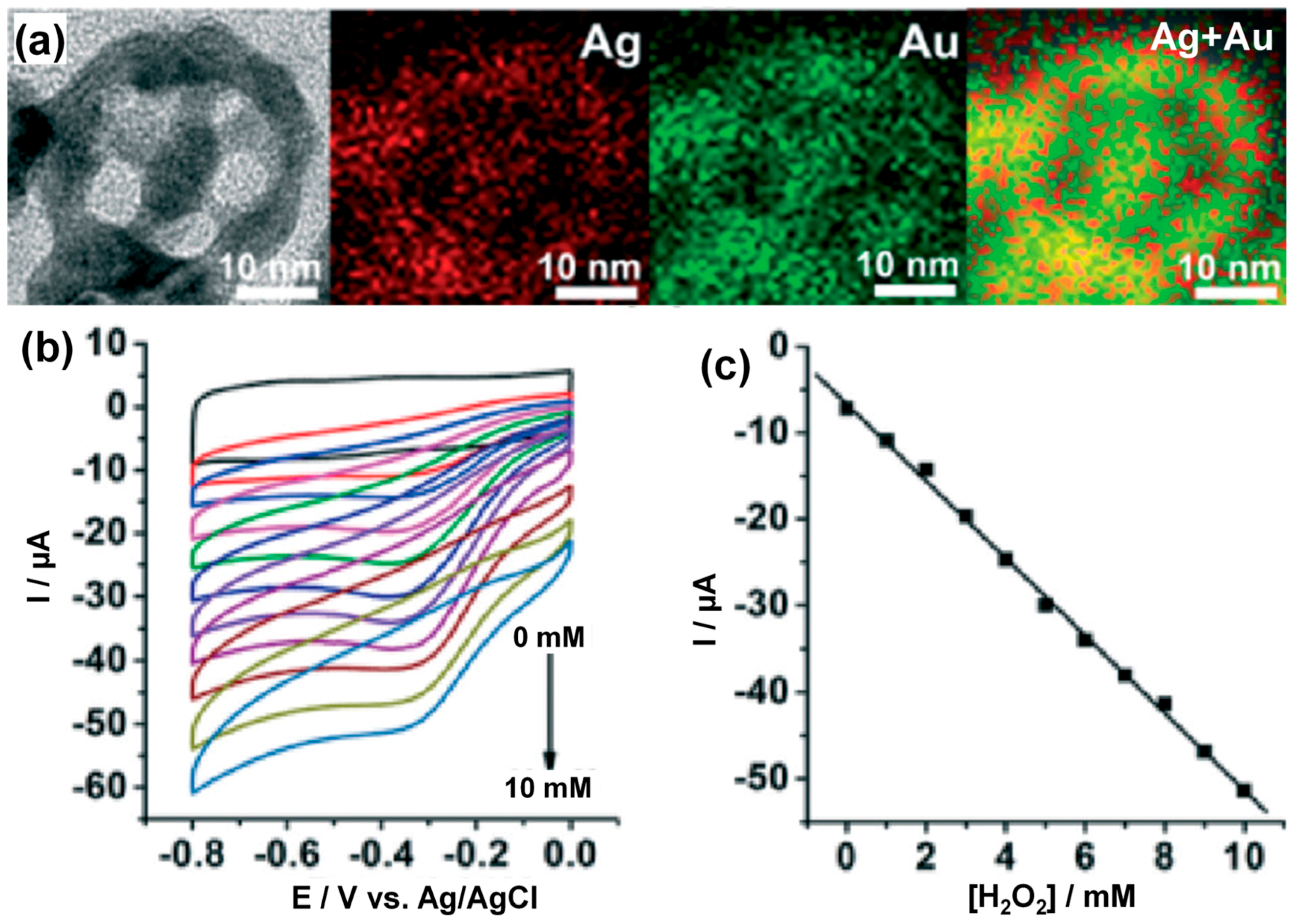
3.3.1. Noble Metal (Gold)-Based Electrodes
3.3.2. Heterogeneous Electrode Materials: Metal- and Carbon-Based Oxide Interfaces
3.3.3. Noble Metal–Based Electrodes
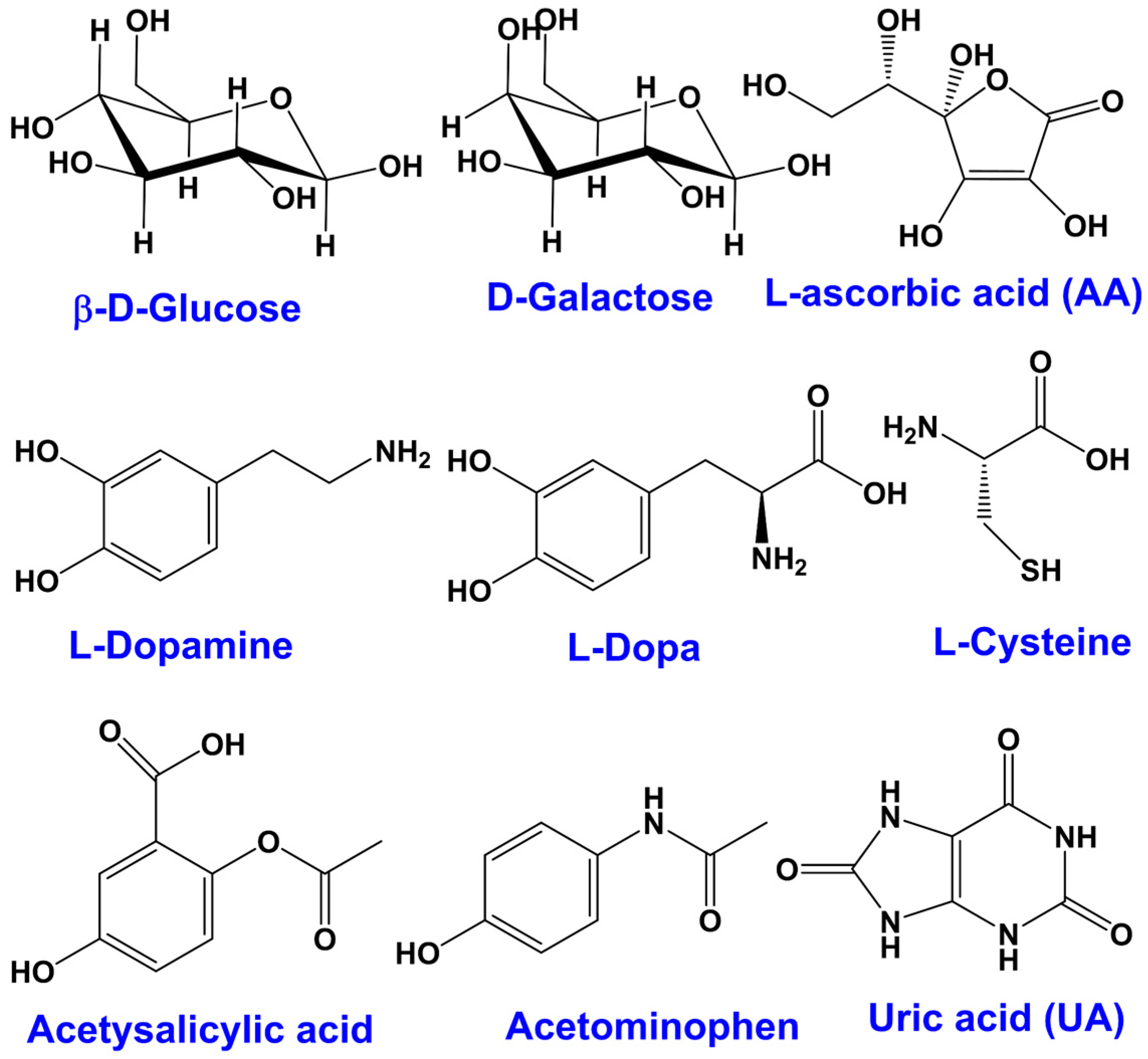
3.4. Nanomaterials for the Electroanalysis of Species of Biological Interest: DNA, Ascorbic Acid, Dopamine
4. Nanomaterials as Abiotic Catalysts at Work in Fuel Cells for the Activation of Bio-Devices
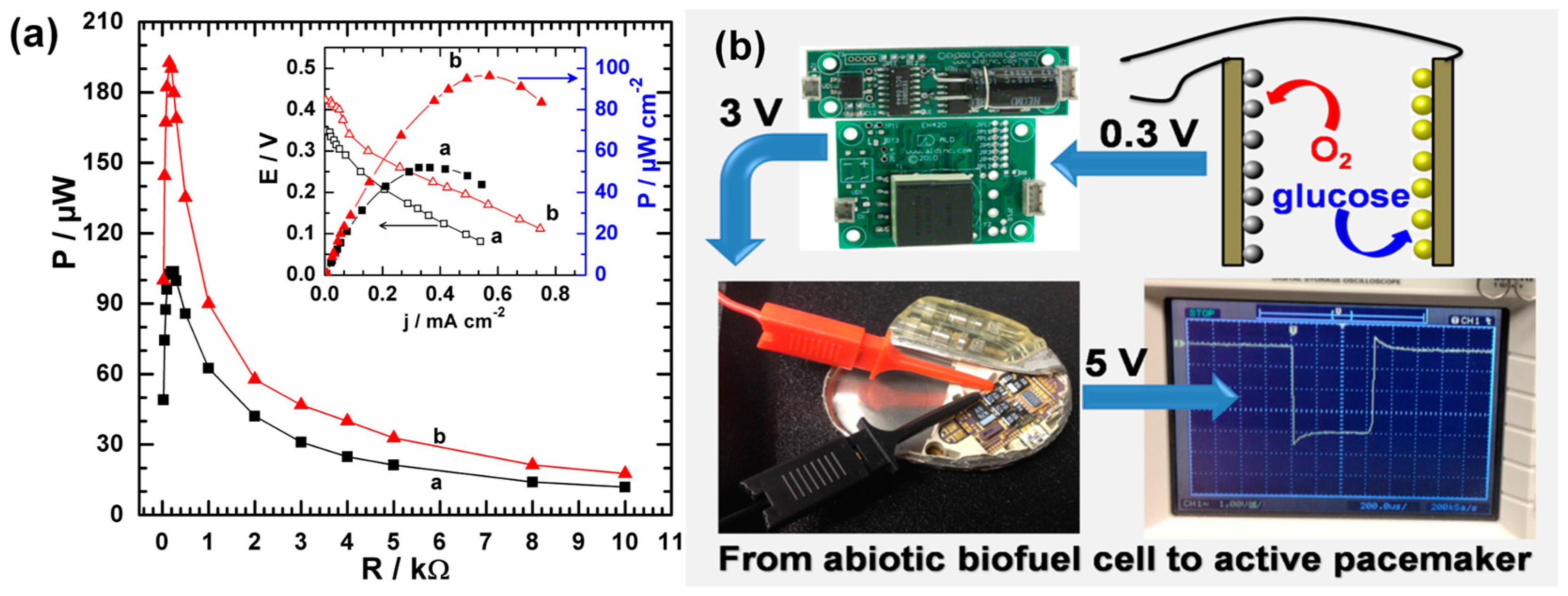
4.1. From Enzymatically to Abiotically Catalyzed Glucose Fuel Cell for Implantable Targets
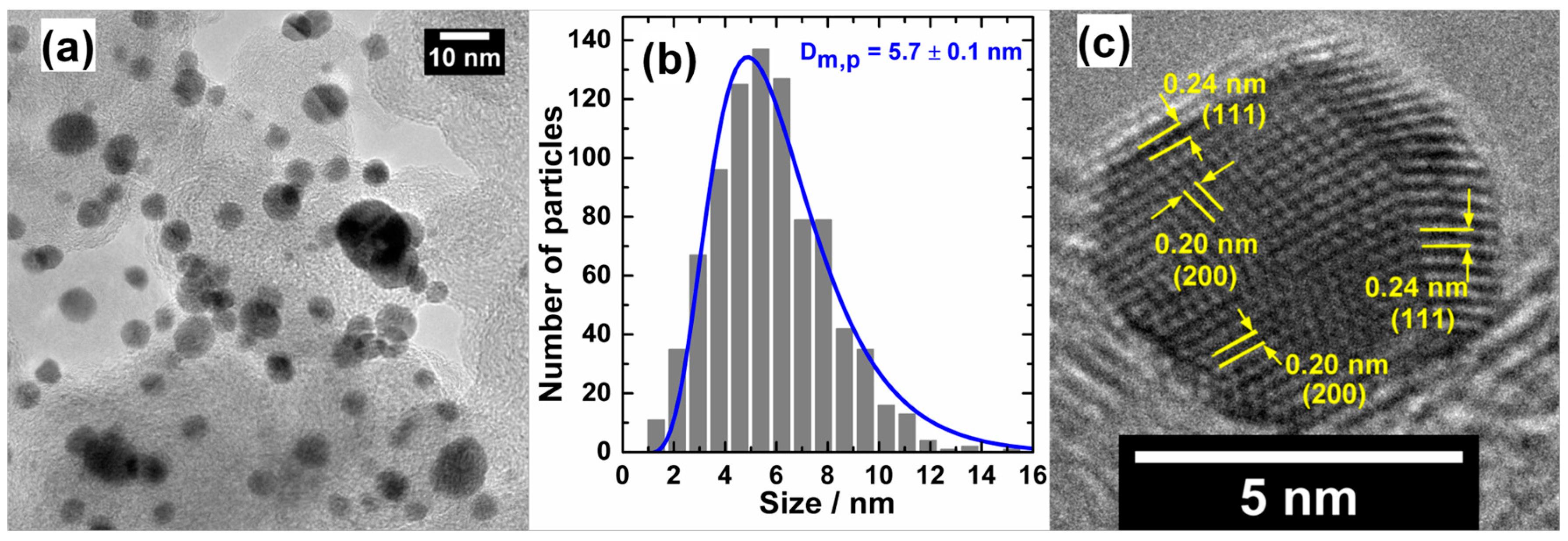
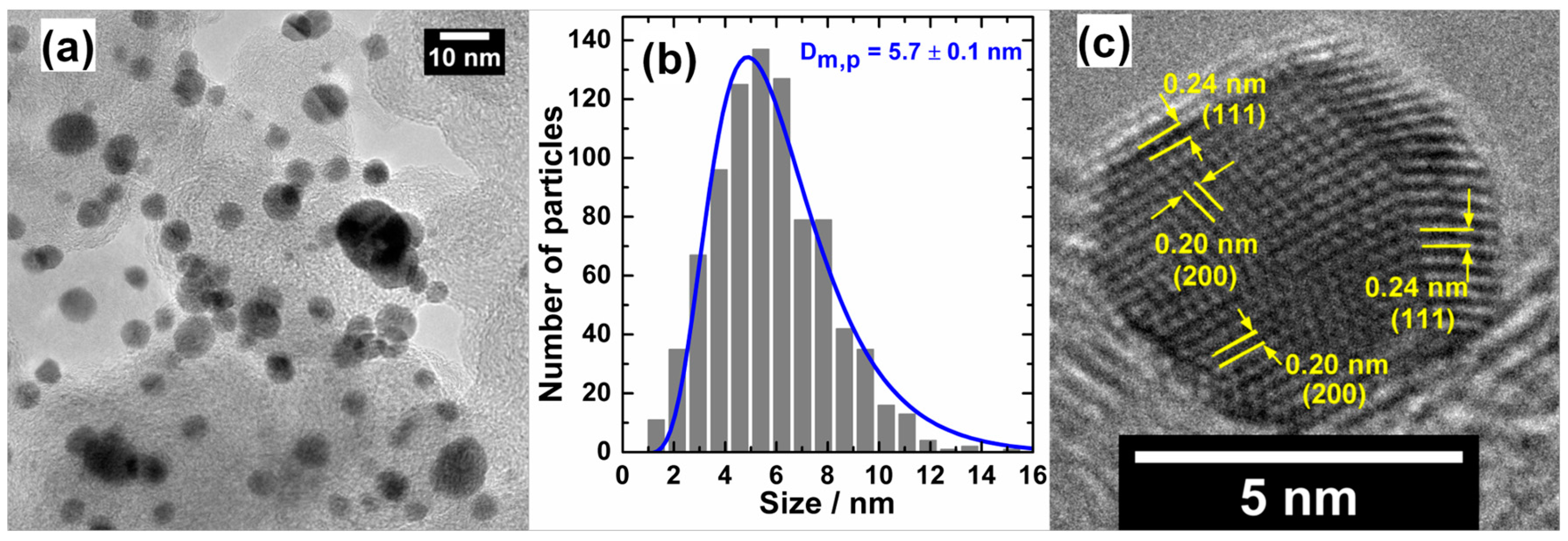
4.2. Target-Directed Development of Gold-Based Nanostructures by “Bromide Anion Exchange” Method
4.2.1. Preparation of Nanoelectrocatalysts Scheduled to Operate in Membraneless Cells
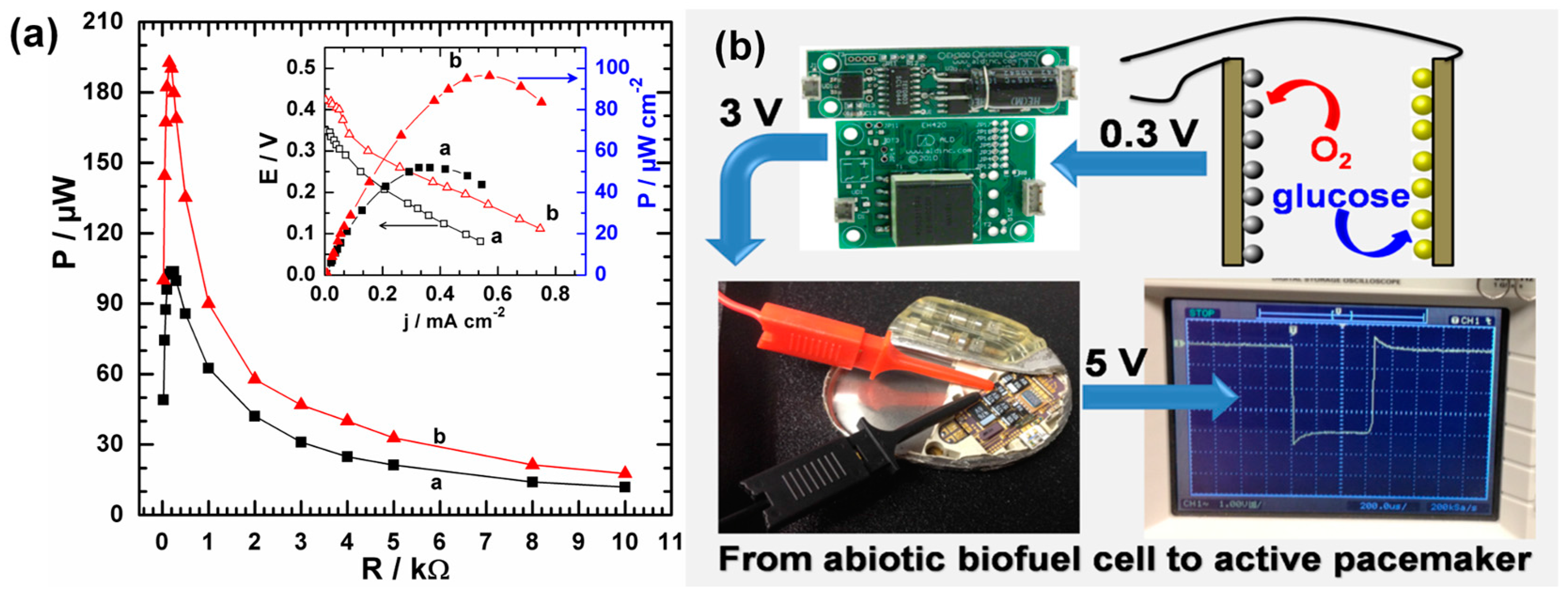
4.2.2. Design and Fabrication of Electrodes with Improved Selectivity in Human Serum Solution
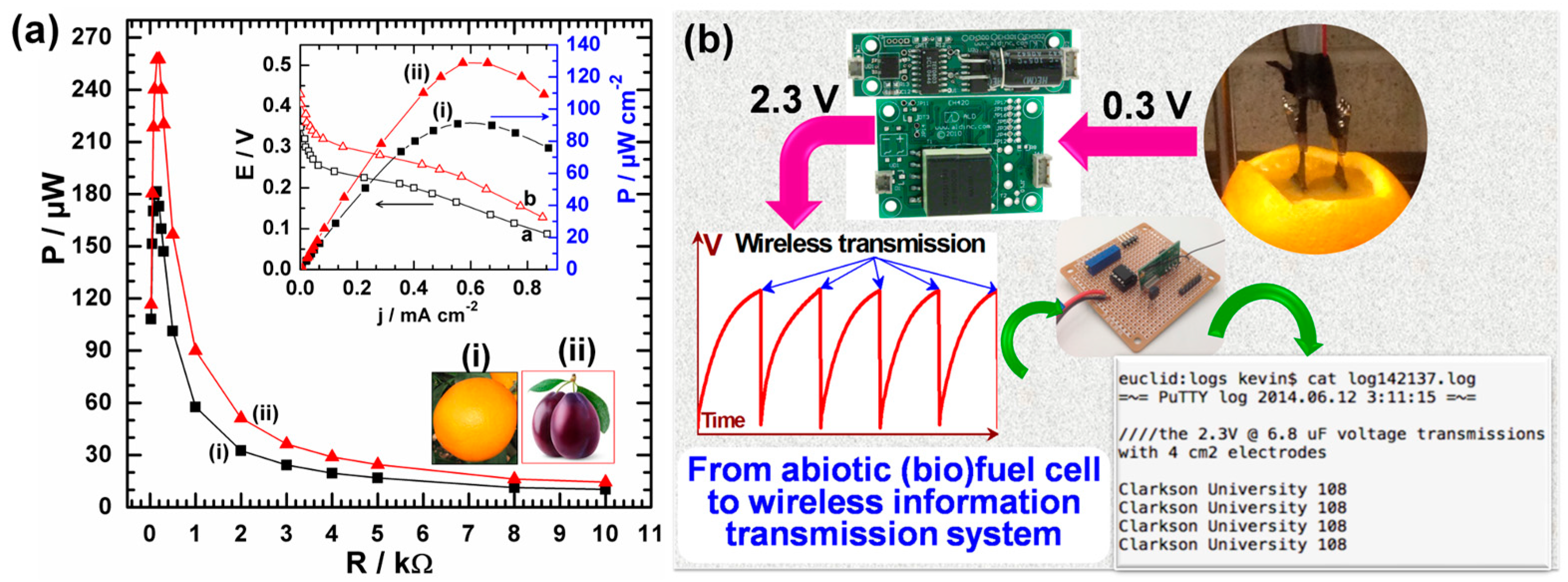
4.2.3. Design and Fabrication of Electrodes to Activate Wireless Information Transmission Systems
5. Concluding Remarks and Further Outlook
Acknowledgments
Conflicts of Interest
References
- Willner, I.; Katz, E. Bioelectronics: From Theory to Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; p. 492. [Google Scholar]
- Katz, E. Implantable Bioelectronics; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; p. 450. [Google Scholar]
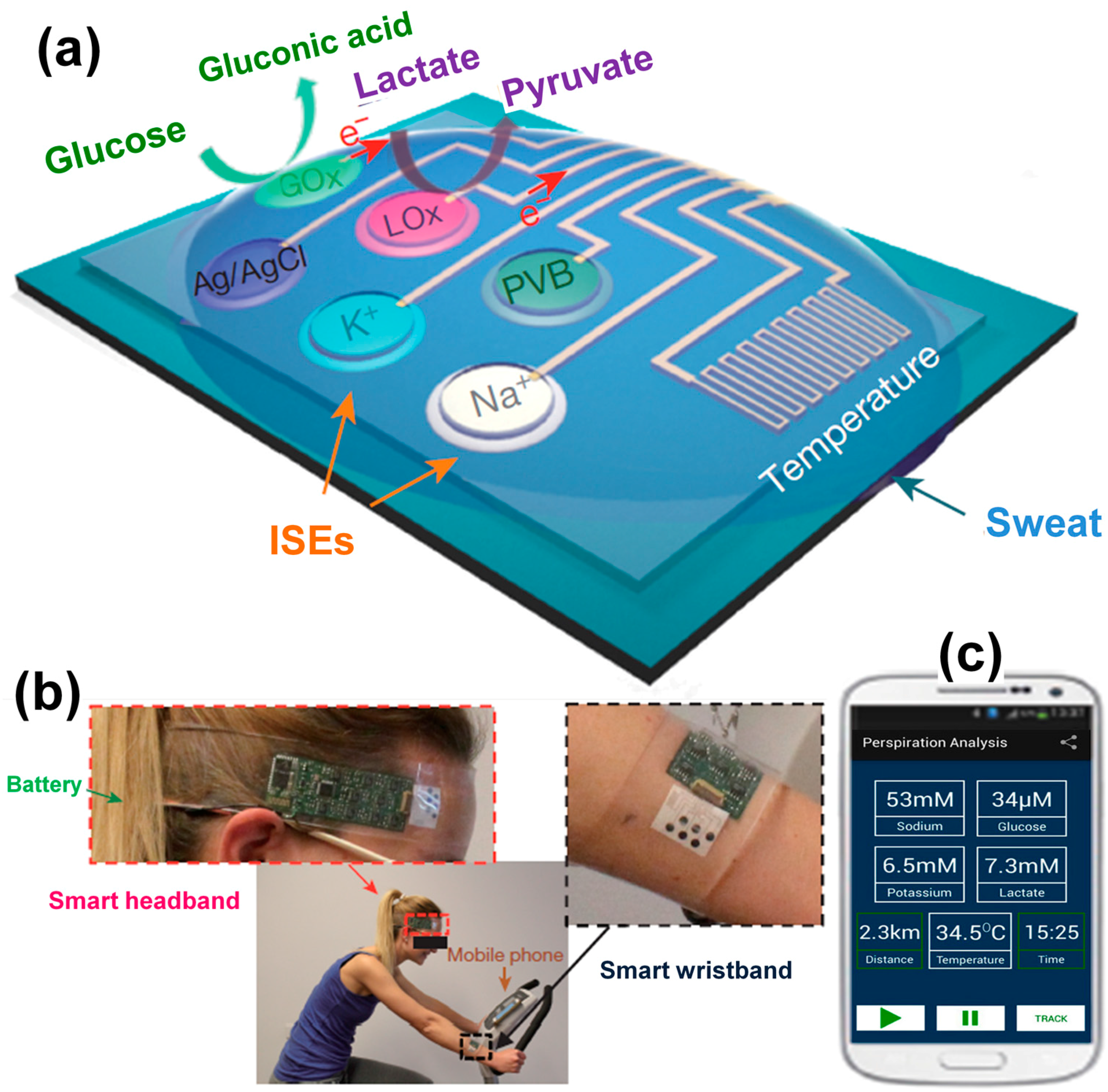
- Gao, W.; Emaminejad, S.; Nyein, H.Y.Y.; Challa, S.; Chen, K.; Peck, A.; Fahad, H.M.; Ota, H.; Shiraki, H.; Kiriya, D.; et al. Fully integrated wearable sensor arrays for multiplexed in situ perspiration analysis. Nature 2016, 529, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Nagel, B.; Dellweg, H.; Gierasch, L. Glossary for chemists of terms used in biotechnology (IUPAC recommendations 1992). Pure Appl. Chem. 1992, 64, 143–168. [Google Scholar] [CrossRef]
- International Union of Pure and Applied Chemistry. Compendium of Chemical Terminology, 2nd ed.; the “Gold Book”; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Wang, L.; Ma, W.; Xu, L.; Chen, W.; Zhu, Y.; Xu, C.; Kotov, N.A. Nanoparticle-based environmental sensors. Mater. Sci. Eng. R Rep. 2010, 70, 265–274. [Google Scholar] [CrossRef]
- Zhu, C.; Yang, G.; Li, H.; Du, D.; Lin, Y. Electrochemical sensors and biosensors based on nanomaterials and nanostructures. Anal. Chem. 2015, 87, 230–249. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Sumana, G.; Nagarajan, R.; Malhotra, B.D. Application of nanostructured ZnO films for electrochemical DNA biosensor. Thin Solid Films 2010, 519, 1196–1201. [Google Scholar] [CrossRef]
- Kang, X.; Wang, J.; Wu, H.; Liu, J.; Aksay, I.A.; Lin, Y. A graphene-based electrochemical sensor for sensitive detection of paracetamol. Talanta 2010, 81, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liu, J.-H.; Lu, H.-T.; Zhang, Q. Electrochemical behavior and voltammetric determination of paracetamol on Nafion/TiO2-graphene modified glassy carbon electrode. Colloids Surf. B 2011, 85, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Chen, Y.; Xu, H.; Tan, Y.; Wang, S. Detection of dopamine based on tyrosinase-Fe3O4 nanoparticles-chitosan nanocomposite biosensor. Am. J. Biomed. Sci. 2010, 2, 209–216. [Google Scholar] [CrossRef]
- Puangjan, A.; Chaiyasith, S.; Wichitpanya, S.; Daengduang, S.; Puttota, S. Electrochemical sensor based on PANI/MnO2-Sb2O3 nanocomposite for selective simultaneous voltammetric determination of ascorbic acid and acetylsalicylic acid. J. Electroanal. Chem. 2016, 782, 192–201. [Google Scholar] [CrossRef]
- Van Nguyen, K.; Minteer, S.D. DNA-functionalized Pt nanoparticles as catalysts for chemically powered micromotors: Toward signal-on motion-based DNA biosensor. Chem. Commun. 2015, 51, 4782–4784. [Google Scholar] [CrossRef] [PubMed]
- Hickey, D.P.; Milton, R.D.; Rasmussen, M.; Abdellaoui, S.; Nguyen, K.; Minteer, S.D. Fundamentals and applications of bioelectrocatalysis. In Electrochemistry; The Royal Society of Chemistry: London, UK, 2016; Volume 13, pp. 97–132. [Google Scholar]
- Clark, L.C.; Wolf, R.; Granger, D.; Taylor, Z. Continuous recording of blood oxygen tensions by polarography. J. Appl. Physiol. 1953, 6, 189–193. [Google Scholar] [PubMed]
- Clark, L.C.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Leech, D.; Kavanagh, P.; Schuhmann, W. Enzymatic fuel cells: Recent progress. Electrochim. Acta 2012, 84, 223–234. [Google Scholar] [CrossRef]
- Ivnitski, D.; Branch, B.; Atanassov, P.; Apblett, C. Glucose oxidase anode for biofuel cell based on direct electron transfer. Electrochem. Commun. 2006, 8, 1204–1210. [Google Scholar] [CrossRef]
- Degani, Y.; Heller, A. Direct electrical communication between chemically modified enzymes and metal electrodes. I. Electron transfer from glucose oxidase to metal electrodes via electron relays, bound covalently to the enzyme. J. Phys. Chem. 1987, 91, 1285–1289. [Google Scholar] [CrossRef]
- Heller, A. Electrical wiring of redox enzymes. Acc. Chem. Res. 1990, 23, 128–134. [Google Scholar] [CrossRef]
- Wilson, R.; Turner, A.P.F. Glucose oxidase: An ideal enzyme. Biosens. Bioelectron. 1992, 7, 165–185. [Google Scholar] [CrossRef]
- Wohlfahrt, G.; Witt, S.; Hendle, J.; Schomburg, D.; Kalisz, H.M.; Hecht, H.-J. 1.8 and 1.9 Å resolution structures of the Penicillium amagasakiense and Aspergillus niger glucose oxidases as a basis for modelling substrate complexes. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Bankar, S.B.; Bule, M.V.; Singhal, R.S.; Ananthanarayan, L. Glucose oxidase—An overview. Biotechnol. Adv. 2009, 27, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, J.; Chen, L.; Lu, F. Highly selective membrane-free, mediator-free glucose biosensor. Anal. Chem. 1994, 66, 3600–3603. [Google Scholar] [CrossRef]
- Wang, J.; Romero, E.G.; Reviejo, A.J. Improved alcohol biosensor based on ruthenium-dispersed carbon paste enzyme electrodes. J. Electroanal. Chem. 1993, 353, 113–120. [Google Scholar] [CrossRef]
- Weizmann, Y.; Patolsky, F.; Katz, E.; Willner, I. Amplified telomerase analysis by using rotating magnetic particles: The rapid and sensitive detection of cancer cells. ChemBioChem 2004, 5, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Lioubashevski, O.; Katz, E.; Niazov, T.; Willner, I. Two coupled enzymes perform in parallel the ‘AND’ and ‘inhibAND’ logic gate operations. Org. Biomol. Chem. 2006, 4, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Elouarzaki, K.; Bourourou, M.; Holzinger, M.; Le Goff, A.; Marks, R.S.; Cosnier, S. Freestanding HRP-GOx redox buckypaper as an oxygen-reducing biocathode for biofuel cell applications. Energy Environ. Sci. 2015, 8, 2069–2074. [Google Scholar] [CrossRef]
- Kavanagh, P.; Leech, D. Mediated electron transfer in glucose oxidising enzyme electrodes for application to biofuel cells: Recent progress and perspectives. Phys. Chem. Chem. Phys. 2013, 15, 4859–4869. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Willner, I.; Kotlyar, A.B. A non-compartmentalized glucose/O2 biofuel cell by bioengineered electrode surfaces. J. Electroanal. Chem. 1999, 479, 64–68. [Google Scholar] [CrossRef]
- Schlesinger, M. Applications of Electrochemistry in Medicine; Springer: New York, NY, USA, 2013; Volume 56, p. 452. [Google Scholar]
- Toghill, K.E.; Compton, R.G. Electrochemical non-enzymatic glucose sensors: A perspective and an evaluation. Int. J. Electrochem. Sci. 2010, 5, 1246–1301. [Google Scholar]
- Meredith, M.T.; Kao, D.-Y.; Hickey, D.; Schmidtke, D.W.; Glatzhofer, D.T. High current density ferrocene-modified linear poly(ethylenimine) bioanodes and their use in biofuel cells. J. Electrochem. Soc. 2011, 158, B166–B174. [Google Scholar] [CrossRef]
- Meredith, M.T.; Minson, M.; Hickey, D.; Artyushkova, K.; Glatzhofer, D.T.; Minteer, S.D. Anthracene-modified multi-walled carbon nanotubes as direct electron transfer scaffolds for enzymatic oxygen reduction. ACS Catal. 2011, 1, 1683–1690. [Google Scholar] [CrossRef]
- Reuillard, B.; Le Goff, A.; Agnes, C.; Holzinger, M.; Zebda, A.; Gondran, C.; Elouarzaki, K.; Cosnier, S. High power enzymatic biofuel cell based on naphthoquinone-mediated oxidation of glucose by glucose oxidase in a carbon nanotube 3D matrix. Phys. Chem. Chem. Phys. 2013, 15, 4892–4896. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, P.; Tuurala, S.; Vaari, A.; Valkiainen, M.; Smolander, M.; Leech, D. A comparison of glucose oxidase and aldose dehydrogenase as mediated anodes in printed glucose/oxygen enzymatic fuel cells using ABTS/laccase cathodes. Bioelectrochemistry 2012, 87, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Mano, N.; Heller, A. Long tethers binding redox centers to polymer backbones enhance electron transport in enzyme “wiring” hydrogels. J. Am. Chem. Soc. 2003, 125, 4951–4957. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Mao, F.; Heller, A. Characteristics of a miniature compartment-less glucose-O2 biofuel cell and its operation in a living plant. J. Am. Chem. Soc. 2003, 125, 6588–6594. [Google Scholar] [CrossRef]
- Milton, R.D.; Hickey, D.P.; Abdellaoui, S.; Lim, K.; Wu, F.; Tan, B.; Minteer, S.D. Rational design of quinones for high power density biofuel cells. Chem. Sci. 2015, 6, 4867–4875. [Google Scholar] [CrossRef]
- Milton, R.D.; Lim, K.; Hickey, D.P.; Minteer, S.D. Employing FAD-dependent glucose dehydrogenase within a glucose/oxygen enzymatic fuel cell operating in human serum. Bioelectrochemistry 2015, 106 Pt A, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Zakeeruddin, S.M.; Fraser, D.M.; Nazeeruddin, M.K.; Grätzel, M. Towards mediator design: Characterization of tris-(4,4′-substituted-2,2′-bipyridine) complexes of iron(II), ruthenium(II) and osmium(II) as mediators for glucose oxidase of aspergillus niger and other redox proteins. J. Electroanal. Chem. 1992, 337, 253–283. [Google Scholar] [CrossRef]
- Gallaway, J.W.; Calabrese Barton, S.A. Kinetics of redox polymer-mediated enzyme electrodes. J. Am. Chem. Soc. 2008, 130, 8527–8536. [Google Scholar] [CrossRef] [PubMed]
- Takagi, K.; Kano, K.; Ikeda, T. Mediated bioelectrocatalysis based on NAD-related enzymes with reversible characteristics. J. Electroanal. Chem. 1998, 445, 211–219. [Google Scholar] [CrossRef]
- Yan, Y.-M.; Tel-Vered, R.; Yehezkeli, O.; Cheglakov, Z.; Willner, I. Biocatalytic growth of Au nanoparticles immobilized on glucose oxidase enhances the ferrocene-mediated bioelectrocatalytic oxidation of glucose. Adv. Mater. 2008, 20, 2365–2370. [Google Scholar] [CrossRef]
- Willner, I.; Heleg-Shabtai, V.; Blonder, R.; Katz, E.; Tao, G.; Bückmann, A.F.; Heller, A. Electrical wiring of glucose oxidase by reconstitution of FAD-modified monolayers assembled onto Au-electrodes. J. Am. Chem. Soc. 1996, 118, 10321–10322. [Google Scholar] [CrossRef]
- Riklin, A.; Katz, E.; Willner, I.; Stocker, A.; Buckmann, A.F. Improving enzyme–electrode contacts by redox modification of cofactors. Nature 1995, 376, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta (BBA) Rev. Bioenerg. 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Lojou, E. Hydrogenases as catalysts for fuel cells: Strategies for efficient immobilization at electrode interfaces. Electrochim. Acta 2011, 56, 10385–10397. [Google Scholar] [CrossRef]
- Vassilyev, Y.B.; Khazova, O.A.; Nikolaeva, N.N. Kinetics and mechanism of glucose electrooxidation on different electrode-catalysts: Part I. Adsorption and oxidation on platinum. J. Electroanal. Chem. Interfacial Electrochem. 1985, 196, 105–125. [Google Scholar] [CrossRef]
- Vassilyev, Y.B.; Khazova, O.A.; Nikolaeva, N.N. Kinetics and mechanism of glucose electrooxidation on different electrode-catalysts: Part II. Effect of the nature of the electrode and the electrooxidation mechanism. J. Electroanal. Chem. Interfacial Electrochem. 1985, 196, 127–144. [Google Scholar] [CrossRef]
- Zhao, C.; Shao, C.; Li, M.; Jiao, K. Flow-injection analysis of glucose without enzyme based on electrocatalytic oxidation of glucose at a nickel electrode. Talanta 2007, 71, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Mehmet, S.; Quan, G.; Thomas, S.; Goldsmith, D. Important causes of hypoglycaemia in patients with diabetes on peritoneal dialysis. Diabet. Med. 2001, 18, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.J.; Rajagopalan, R.; Heller, A. “Wired” enzyme electrodes for amperometric determination of glucose or lactate in the presence of interfering substances. Anal. Chem. 1994, 66, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.-W.; Wu, B.; Cha, C.-S.; Kita, H. Electro-oxidation of glucose on platinum in alkaline solution and selective oxidation in the presence of additives. J. Electroanal. Chem. 1995, 382, 103–110. [Google Scholar] [CrossRef]
- Guerrieri, A.; Ciriello, R.; Centonze, D. Permselective and enzyme-entrapping behaviours of an electropolymerized, non-conducting, poly(o-aminophenol) thin film-modified electrode: A critical study. Biosens. Bioelectron. 2009, 24, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Ndamanisha, J.C.; Guo, L. Nonenzymatic glucose detection at ordered mesoporous carbon modified electrode. Bioelectrochemistry 2009, 77, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Yeo, I.-H.; Johnson, D.C. Electrochemical response of small organic molecules at nickel–copper alloy electrodes. J. Electroanal. Chem. 2001, 495, 110–119. [Google Scholar] [CrossRef]
- Holade, Y.; Hickey, D.P.; Minteer, S.D. Halide-regulated growth of electrocatalytic metal nanoparticles directly onto a carbon paper electrode. J. Mater. Chem. A 2016, 4, 17154–17162. [Google Scholar] [CrossRef]
- Alloyeau, D.; Mottet, C.; Ricolleau, C. Nanoalloys: Synthesis, Structure and Properties; Springer: London, UK, 2012; p. 420. [Google Scholar]
- Wieckowski, A.; Savinova, E.R.; Vayenas, C.G. Catalysis and Electrocatalysis at Nanoparticle Surfaces; Marcel Dekker, Inc.: New York, NY, USA, 2003; p. 970. [Google Scholar]
- Lahmani, M.; Bréchignac, C.; Houdy, P. Les Nanosciences: 2. Nanomatériaux et Nanochimie, 2nd ed.; Belin: Paris, France, 2012; p. 732. [Google Scholar]
- Doherty, R.P.; Krafft, J.-M.; Methivier, C.; Casale, S.; Remita, H.; Louis, C.; Thomas, C. On the promoting effect of Au on CO oxidation kinetics of Au-Pt bimetallic nanoparticles supported on SiO2: An electronic effect? J. Catal. 2012, 287, 102–113. [Google Scholar] [CrossRef]
- Pedersen, M.Ø.; Helveg, S.; Ruban, A.; Stensgaard, I.; Lægsgaard, E.; Nørskov, J.K.; Besenbacher, F. How a gold substrate can increase the reactivity of a Pt overlayer. Surf. Sci. 1999, 426, 395–409. [Google Scholar] [CrossRef]
- Holade, Y.; Servat, K.; Rousseau, J.; Canaff, C.; Poulin, S.; Napporn, T.W.; Kokoh, K.B. Electrochemical and physicochemical characterizations of gold-based nanomaterials: Correlation between surface composition and electrocatalytic activity. J. Electrochem. Soc. 2015, 162, H929–H937. [Google Scholar] [CrossRef]
- Sing, K.; Everett, D.; Haul, R.; Moscou, L.; Pierotti, R.; Rouquerol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems. Pure Appl. Chem. 1982, 54, 2201. [Google Scholar]
- Sing, K.; Everett, D.; Haul, R.; Moscou, L.; Pierotti, R.; Rouquerol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Aoun, S.B.; Bang, G.S.; Koga, T.; Nonaka, Y.; Sotomura, T.; Taniguchi, I. Electrocatalytic oxidation of sugars on silver-UPD single crystal gold electrodes in alkaline solutions. Electrochem. Commun. 2003, 5, 317–320. [Google Scholar] [CrossRef]
- Aoun, S.B.; Dursun, Z.; Koga, T.; Bang, G.S.; Sotomura, T.; Taniguchi, I. Effect of metal ad-layers on Au(111) electrodes on electrocatalytic oxidation of glucose in an alkaline solution. J. Electroanal. Chem. 2004, 567, 175–183. [Google Scholar] [CrossRef]
- Holade, Y.; Servat, K.; Napporn, T.W.; Morais, C.; Berjeaud, J.-M.; Kokoh, K.B. Highly selective oxidation of carbohydrates in an efficient electrochemical energy converter: Cogenerating organic electrosynthesis. ChemSusChem 2016, 9, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Salinas, D.; Mayer, C.; Schmidt, E.; Staikov, G.; Lorenz, W.J. Ag UPD on Au(100) and Au(111). Electrochim. Acta 1998, 43, 3007–3019. [Google Scholar] [CrossRef]
- Hebié, S.; Napporn, T.W.; Morais, C.; Kokoh, K.B. Size-dependent electrocatalytic activity of free gold nanoparticles for the glucose oxidation reaction. ChemPhysChem 2016, 17, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, J.; Xiong, Y.; Yang, J.; Gao, Y.; Liu, Y.; Lu, X.; Tang, Z. Shape-dependent electrocatalytic activity of monodispersed gold nanocrystals toward glucose oxidation. Chem. Commun. 2011, 47, 6894–6896. [Google Scholar] [CrossRef] [PubMed]
- Hebié, S.; Kokoh, K.B.; Servat, K.; Napporn, T. Shape-dependent electrocatalytic activity of free gold nanoparticles toward glucose oxidation. Gold Bull. 2013, 46, 311–318. [Google Scholar] [CrossRef]
- Hebié, S.; Cornu, L.; Napporn, T.W.; Rousseau, J.; Kokoh, B.K. Insight on the surface structure effect of free gold nanorods on glucose electrooxidation. J. Phys. Chem. C 2013, 117, 9872–9880. [Google Scholar] [CrossRef]
- Hebié, S.; Holade, Y.; Maximova, K.; Sentis, M.; Delaporte, P.; Kokoh, K.B.; Napporn, T.W.; Kabashin, A.V. Advanced electrocatalysts on the basis of bare Au nanomaterials for biofuel cell applications. ACS Catal. 2015, 5, 6489–6496. [Google Scholar] [CrossRef]
- Wang, J.; Thomas, D.F.; Chen, A. Nonenzymatic electrochemical glucose sensor based on nanoporous PtPb networks. Anal. Chem. 2008, 80, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, Y.; Parisi, J.; Zhang, L.; Lei, Y. A novel Ni–Au hybrid nanobelts based sensor for sensitive and selective glucose detection. Biosens. Bioelectron. 2011, 28, 393–398. [Google Scholar] [CrossRef] [PubMed]
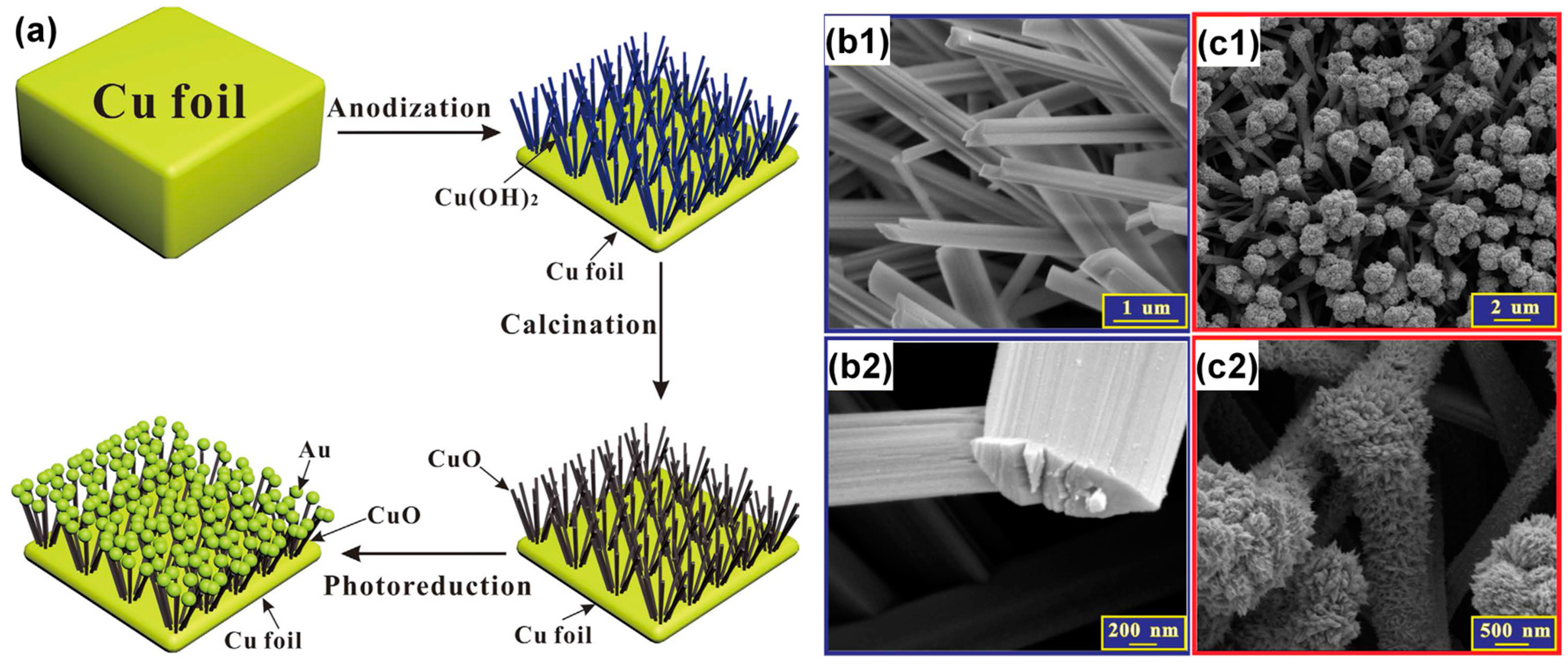
- Xiao, X.; Wang, M.; Li, H.; Pan, Y.; Si, P. Non-enzymatic glucose sensors based on controllable nanoporous gold/copper oxide nanohybrids. Talanta 2014, 125, 366–371. [Google Scholar] [CrossRef] [PubMed]
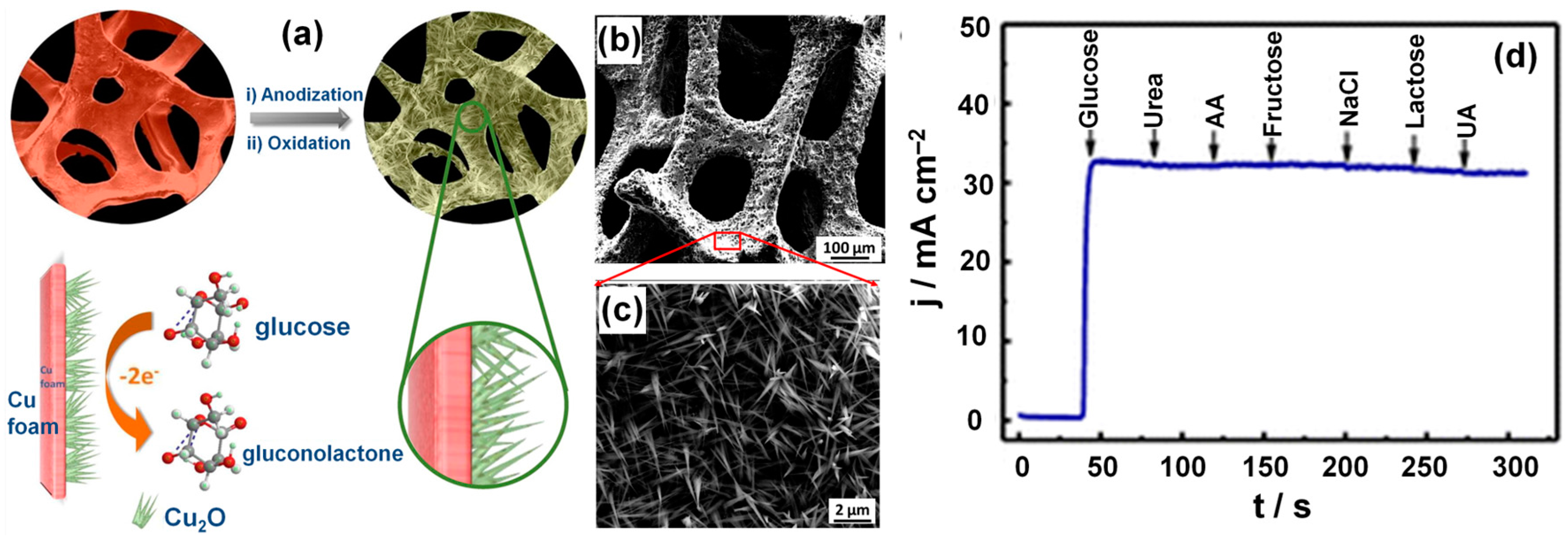
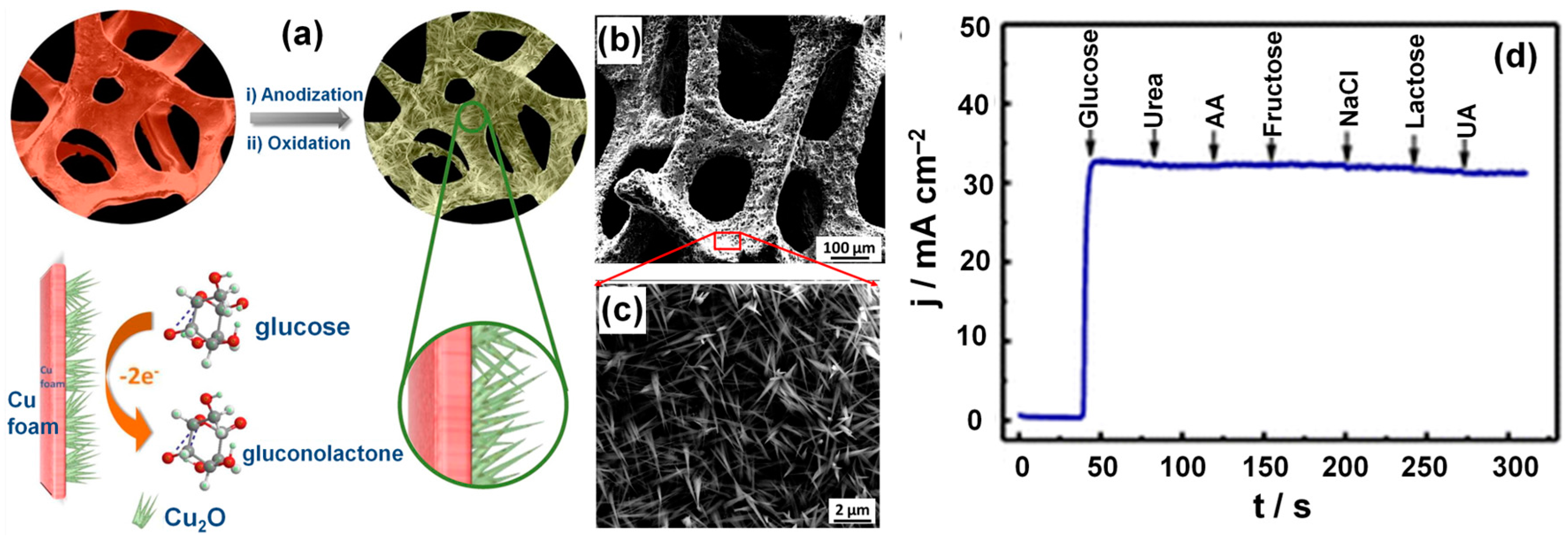
- Dong, C.; Zhong, H.; Kou, T.; Frenzel, J.; Eggeler, G.; Zhang, Z. Three-dimensional cu foam-supported single crystalline mesoporous Cu2O nanothorn arrays for ultra-highly sensitive and efficient nonenzymatic detection of glucose. ACS Appl. Mater. Interfaces 2015, 7, 20215–20223. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, Y.; Shi, G.; Xian, Y.; Jin, L. Facile synthesis of leaf-like CuO nanoparticles and their application on glucose biosensor. Electroanalysis 2011, 23, 497–502. [Google Scholar] [CrossRef]
- Dong, J.; Ren, L.; Zhang, Y.; Cui, X.; Hu, P.; Xu, J. Direct electrodeposition of cable-like Cu@Cu nanowires array for non-enzymatic sensing. Talanta 2015, 132, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Tang, L.; Zhang, X.; Sun, S.; Yang, S.; Song, X.; Yang, Z. Templating synthesis of hollow CuO polyhedron and its application for nonenzymatic glucose detection. J. Mater. Chem. A 2014, 2, 7306–7312. [Google Scholar] [CrossRef]
- Song, M.-J.; Lee, S.-K.; Kim, J.-H.; Lim, D.-S. Non-enzymatic glucose sensor based on cu electrode modified with CuO nanoflowers. J. Electrochem. Soc. 2013, 160, B43–B46. [Google Scholar] [CrossRef]
- Zhuang, Z.; Su, X.; Yuan, H.; Sun, Q.; Xiao, D.; Choi, M.M.F. An improved sensitivity non-enzymatic glucose sensor based on a CuO nanowire modified Cu electrode. Analyst 2008, 133, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, G.; Wang, M.; Chatrathi, M.P. Carbon-nanotube/copper composite electrodes for capillary electrophoresis microchip detection of carbohydrates. Analyst 2004, 129, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhao, Y.; Xu, J.Z.; Zhu, J.-J. Preparation of functionalized copper nanoparticles and fabrication of a glucose sensor. Sens. Actuators B 2006, 114, 379–386. [Google Scholar] [CrossRef]
- Burke, L.D.; Bruton, G.M.; Collins, J.A. The redox properties of active sites and the importance of the latter in electrocatalysis at copper in base. Electrochim. Acta 1998, 44, 1467–1479. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, Z.; Liu, L. Electrochemical studies of a Cu(II)-Cu(III) couple: Cyclic voltammetry and chronoamperometry in a strong alkaline medium and in the presence of periodate anions. Electrochim. Acta 1997, 42, 2719–2723. [Google Scholar] [CrossRef]
- Ogorevc, B.; Tavčar, G.; Hudnik, V.; Pejovnik, S. Electrochemical behaviour of a Cu(II)-Cu(III) couple: Cyclic voltammetry and kinetic parameters at a platinum electrode in a strong alkaline medium and in the presence of tellurate anions. J. Electroanal. Chem. 1993, 351, 81–90. [Google Scholar] [CrossRef]
- Kang, X.; Mai, Z.; Zou, X.; Cai, P.; Mo, J. A sensitive nonenzymatic glucose sensor in alkaline media with a copper nanocluster/multiwall carbon nanotube-modified glassy carbon electrode. Anal. Biochem. 2007, 363, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-X.; Cao, W.-M.; Li, Y.; Liu, G.; Wen, Y.; Yang, H.-F.; Yang, S.-P. In situ growth of copper nanoparticles on multiwalled carbon nanotubes and their application as non-enzymatic glucose sensor materials. Electrochim. Acta 2010, 55, 3734–3740. [Google Scholar] [CrossRef]
- Li, Z.; Xin, Y.; Zhang, Z.; Wu, H.; Wang, P. Rational design of binder-free noble metal/metal oxide arrays with nanocauliflower structure for wide linear range nonenzymatic glucose detection. Sci. Rep. 2015, 5, 10617. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Luo, B.; Zhang, J.Z. Controllable cobalt oxide/Au hierarchically nanostructured electrode for nonenzymatic glucose sensing. Anal. Chem. 2016, 88, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Mayers, B.T.; Xia, Y. Template-engaged replacement reaction: A one-step approach to the large-scale synthesis of metal nanostructures with hollow interiors. Nano Lett. 2002, 2, 481–485. [Google Scholar] [CrossRef]
- Lu, X.; Au, L.; McLellan, J.; Li, Z.-Y.; Marquez, M.; Xia, Y. Fabrication of cubic nanocages and nanoframes by dealloying Au/Ag alloy nanoboxes with an aqueous etchant based on Fe(NO3)3 or NH4OH. Nano Lett. 2007, 7, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Xia, X.; Peng, H.-C. Shape-controlled synthesis of colloidal metal nanocrystals: Thermodynamic versus kinetic products. J. Am. Chem. Soc. 2015, 137, 7947–7966. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.-P.; Wan, L.-J.; Bai, C.-L.; Jiang, L. Gold hollow nanospheres: Tunable surface plasmon resonance controlled by interior-cavity sizes. J. Phys. Chem. B 2005, 109, 7795–7800. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yang, Z.; Cao, X.; Liu, P.; Chen, S.; Cao, Z. Hierarchical macro-mesoporous Ni(OH)2 for nonenzymatic electrochemical sensing of glucose. J. Electrochem. Soc. 2014, 161, B201–B206. [Google Scholar] [CrossRef]
- Zhan, B.; Liu, C.; Chen, H.; Shi, H.; Wang, L.; Chen, P.; Huang, W.; Dong, X. Free-standing electrochemical electrode based on Ni(OH)2/3D graphene foam for nonenzymatic glucose detection. Nanoscale 2014, 6, 7424–7429. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Huo, H.; Xu, C. Ni0.31Co0.69S2 nanoparticles uniformly anchored on a porous reduced graphene oxide framework for a high-performance non-enzymatic glucose sensor. J. Mater. Chem. A 2015, 3, 4922–4930. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, T.; Jiang, L.; Zhang, K.; Yuen, M.M.F.; Xu, J.-B.; Fu, X.-Z.; Sun, R.; Wong, C.-P. NiO mesoporous nanowalls grown on RGO coated nickel foam as high performance electrodes for supercapacitors and biosensors. Electrochim. Acta 2016, 192, 205–215. [Google Scholar] [CrossRef]
- Rengaraj, A.; Haldorai, Y.; Kwak, C.H.; Ahn, S.; Jeon, K.-J.; Park, S.H.; Han, Y.-K.; Huh, Y.S. Electrodeposition of flower-like nickel oxide on CVD-grown graphene to develop an electrochemical non-enzymatic biosensor. J. Mater. Chem. B 2015, 3, 6301–6309. [Google Scholar] [CrossRef]
- Marimuthu, T.; Mohamad, S.; Alias, Y. Needle-like polypyrrole–NiO composite for non-enzymatic detection of glucose. Synth. Met. 2015, 207, 35–41. [Google Scholar] [CrossRef]
- Zhao, X.; Muench, F.; Schaefer, S.; Brötz, J.; Duerrschnabel, M.; Molina-Luna, L.; Kleebe, H.-J.; Liu, S.; Tan, J.; Ensinger, W. Electroless decoration of macroscale foam with nickel nano-spikes: A scalable route toward efficient catalyst electrodes. Electrochem. Commun. 2016, 65, 39–43. [Google Scholar] [CrossRef]
- Holade, Y.; Morais, C.; Arrii-Clacens, S.; Servat, K.; Napporn, T.W.; Kokoh, K.B. New preparation of PdNi/C and PdAg/C nanocatalysts for glycerol electrooxidation in alkaline medium. Electrocatalysis 2013, 4, 167–178. [Google Scholar] [CrossRef]
- Oliveira, V.L.; Morais, C.; Servat, K.; Napporn, T.W.; Tremiliosi-Filho, G.; Kokoh, K.B. Glycerol oxidation on nickel based nanocatalysts in alkaline medium—Identification of the reaction products. J. Electroanal. Chem. 2013, 703, 56–62. [Google Scholar] [CrossRef]
- Ye, J.-S.; Wen, Y.; De Zhang, W.; Ming Gan, L.; Xu, G.Q.; Sheu, F.-S. Nonenzymatic glucose detection using multi-walled carbon nanotube electrodes. Electrochem. Commun. 2004, 6, 66–70. [Google Scholar] [CrossRef]
- Wang, S.G.; Zhang, Q.; Wang, R.; Yoon, S.F. A novel multi-walled carbon nanotube-based biosensor for glucose detection. Biochem. Biophys. Res. Commun. 2003, 311, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Premlatha, S.; Sivasakthi, P.; Ramesh Bapu, G.N.K. Electrodeposition of a 3D hierarchical porous flower-like cobalt-MWCNT nanocomposite electrode for non-enzymatic glucose sensing. RSC Adv. 2015, 5, 74374–74380. [Google Scholar] [CrossRef]
- Ci, S.; Mao, S.; Huang, T.; Wen, Z.; Steeber, D.A.; Chen, J. Enzymeless glucose detection based on CoO/graphene microsphere hybrids. Electroanalysis 2014, 26, 1326–1334. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Bai, W.; Zheng, J. Layer-by-layer assembly of copper nanoparticles and manganese dioxide-multiwalled carbon nanotubes film: A new nonenzymatic electrochemical sensor for glucose. Talanta 2016, 149, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, K.; Raj kumar, T.; Babu, K.J.; Gnana kumar, G. Ni-Co bimetal nanowires filled multiwalled carbon nanotubes for the highly sensitive and selective non-enzymatic glucose sensor applications. Sci. Rep. 2016, 6, 36583. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Wu, W.; Wu, H.; Wang, S.; Feng, C.; Ding, Y. Synthesis of PtxSn/MWCNTS and their application in non-enzymatic glucose and hydrogen peroxide sensors. Electroanalysis 2016. [Google Scholar] [CrossRef]
- Guo, C.; Li, H.; Zhang, X.; Huo, H.; Xu, C. 3D porous CNT/MnO2 composite electrode for high-performance enzymeless glucose detection and supercapacitor application. Sens. Actuators B 2015, 206, 407–414. [Google Scholar] [CrossRef]
- Masoomi-Godarzi, S.; Khodadadi, A.A.; Vesali-Naseh, M.; Mortazavi, Y. Highly stable and selective non-enzymatic glucose biosensor using carbon nanotubes decorated by Fe3O4 nanoparticles. J. Electrochem. Soc. 2014, 161, B19–B25. [Google Scholar] [CrossRef]
- Rong, L.-Q.; Yang, C.; Qian, Q.-Y.; Xia, X.-H. Study of the nonenzymatic glucose sensor based on highly dispersed Pt nanoparticles supported on carbon nanotubes. Talanta 2007, 72, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Nana, C.; Hongjuan, W.; Xiaomeng, L.; Liande, Z. Amperometric glucose biosensor based on integration of glucose oxidase with palladium nanoparticles/reduced graphene oxide nanocomposite. Am. J. Anal. Chem. 2012, 3, 312–319. [Google Scholar]
- Xu, C.; Liu, Y.; Su, F.; Liu, A.; Qiu, H. Nanoporous ptag and PtCu alloys with hollow ligaments for enhanced electrocatalysis and glucose biosensing. Biosens. Bioelectron. 2011, 27, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.F.; Park, J.Y. Amperometric glucose biosensor based on Pt-Pd nanoparticles supported by reduced graphene oxide and integrated with glucose oxidase. Electroanalysis 2014, 26, 940–951. [Google Scholar] [CrossRef]
- Putzbach, W.; Ronkainen, N. Immobilization techniques in the fabrication of nanomaterial-based electrochemical biosensors: A review. Sensors 2013, 13, 4811–4840. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Viry, L.; Maugey, M.; Poulin, P.; Mano, N. Engineering hybrid nanotube wires for high-power biofuel cells. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Giroud, F.; Milton, R.D.; Tan, B.-X.; Minteer, S.D. Simplifying enzymatic biofuel cells: Immobilized naphthoquinone as a biocathodic orientational moiety and bioanodic electron mediator. ACS Catal. 2015, 5, 1240–1244. [Google Scholar] [CrossRef]
- Narváez Villarrubia, C.W.; Artyushkova, K.; Garcia, S.O.; Atanassov, P. NAD+/NADH tethering on mwnts-bucky papers for glucose dehydrogenase-based anodes. J. Electrochem. Soc. 2014, 161, H3020–H3028. [Google Scholar] [CrossRef]
- MacVittie, K.; Halamek, J.; Halamkova, L.; Southcott, M.; Jemison, W.D.; Lobel, R.; Katz, E. From “cyborg” lobsters to a pacemaker powered by implantable biofuel cells. Energy Environ. Sci. 2013, 6, 81–86. [Google Scholar] [CrossRef]
- Halámková, L.; Halámek, J.; Bocharova, V.; Szczupak, A.; Alfonta, L.; Katz, E. Implanted biofuel cell operating in a living snail. J. Am. Chem. Soc. 2012, 134, 5040–5043. [Google Scholar] [CrossRef] [PubMed]
- Ciaccafava, A.; De Poulpiquet, A.; Techer, V.; Giudici-Orticoni, M.T.; Tingry, S.; Innocent, C.; Lojou, E. An innovative powerful and mediatorless H2/O2 biofuel cell based on an outstanding bioanode. Electrochem. Commun. 2012, 23, 25–28. [Google Scholar] [CrossRef]
- Poulpiquet, A.d.; Marques-Knopf, H.; Wernert, V.; Giudici-Orticoni, M.T.; Gadiou, R.; Lojou, E. Carbon nanofiber mesoporous films: Efficient platforms for bio-hydrogen oxidation in biofuel cells. Phys. Chem. Chem. Phys. 2014, 16, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Cai, Z.; Chen, J.; Zhang, H.; Cai, C. Electrochemical measurement of the flux of hydrogen peroxide releasing from RAW 264.7 macrophage cells based on enzyme-attapulgite clay nanohybrids. Biosens. Bioelectron. 2011, 26, 4012–4017. [Google Scholar] [CrossRef] [PubMed]
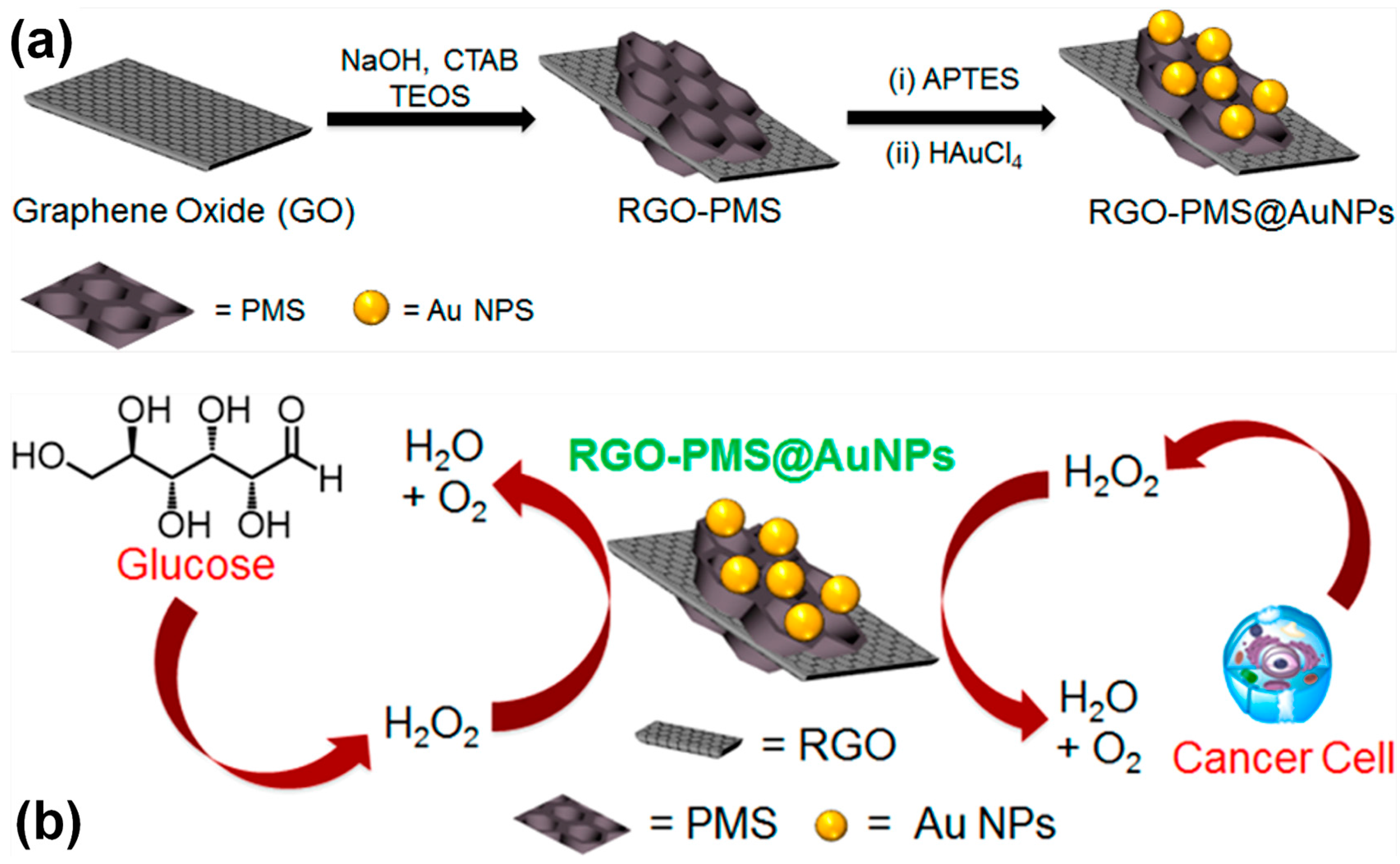
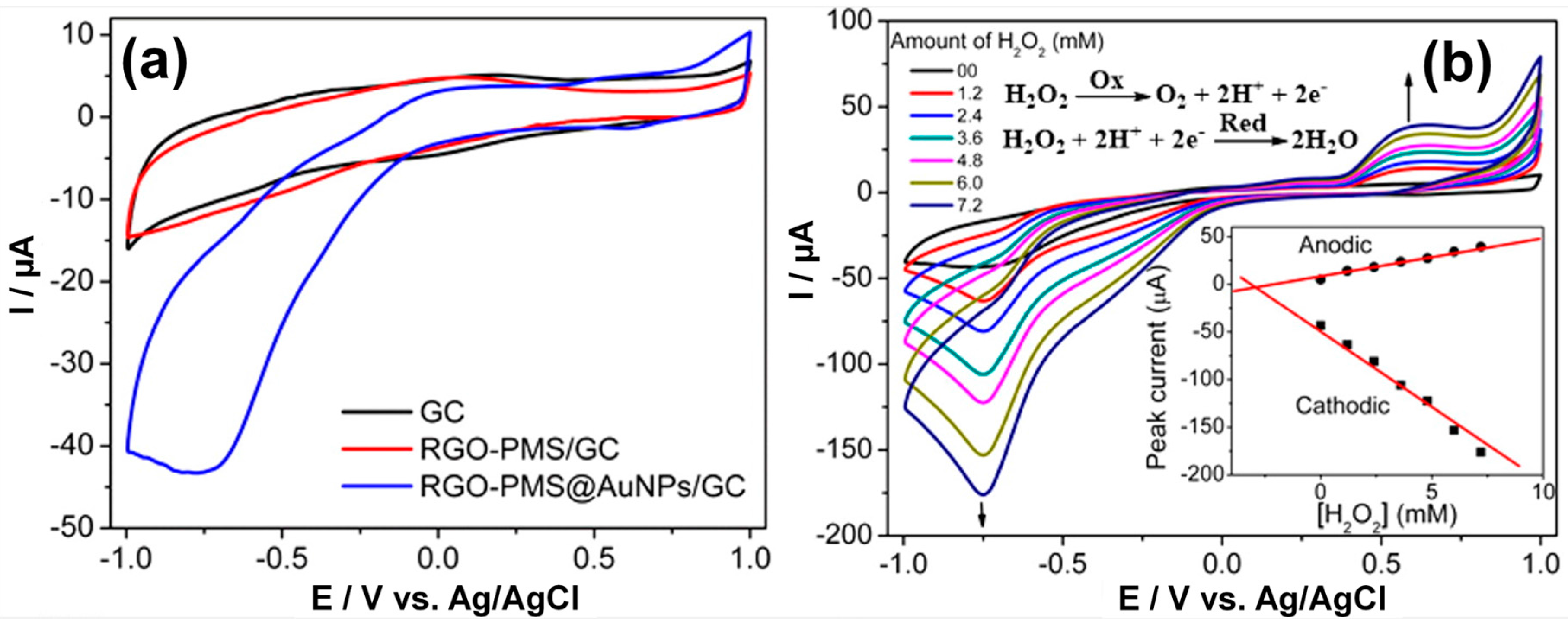
- Maji, S.K.; Sreejith, S.; Mandal, A.K.; Ma, X.; Zhao, Y. Immobilizing gold nanoparticles in mesoporous silica covered reduced graphene oxide: A hybrid material for cancer cell detection through hydrogen peroxide sensing. ACS Appl. Mater. Interfaces. 2014, 6, 13648–13656. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.W.; Albers, A.E.; Pralle, A.; Isacoff, E.Y.; Chang, C.J. Boronate-based fluorescent probes for imaging cellular hydrogen peroxide. J. Am. Chem. Soc. 2005, 127, 16652–16659. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, W.; Dostert, P.; Matsubara, K.; Naoi, M. N-methyl(r)salsolinol produces hydroxyl radicals: Involvement to neurotoxicity. Free Radic. Biol. Med. 1995, 19, 67–75. [Google Scholar] [CrossRef]
- Chang, M.C.Y.; Pralle, A.; Isacoff, E.Y.; Chang, C.J. A selective, cell-permeable optical probe for hydrogen peroxide in living cells. J. Am. Chem. Soc. 2004, 126, 15392–15393. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, H.; Tatemichi, M.; Sawa, T. Chemical basis of inflammation-induced carcinogenesis. Arch. Biochem. Biophys. 2003, 417, 3–11. [Google Scholar] [CrossRef]
- Devadoss, A.; Han, H.; Song, T.; Kim, Y.-P.; Paik, U. Gold nanoparticle-composite nanofibers for enzymatic electrochemical sensing of hydrogen peroxide. Analyst 2013, 138, 5025–5030. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhuang, J.; Nie, L.; Zhang, J.; Zhang, Y.; Gu, N.; Wang, T.; Feng, J.; Yang, D.; Perrett, S.; et al. Intrinsic peroxidase-like activity of ferromagnetic nanoparticles. Nat. Nano 2007, 2, 577–583. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wu, X.; Liu, J.; Hu, X.; Zhang, K.; Hou, S.; Zhou, W.; Xie, S. Design of AgM bimetallic alloy nanostructures (M = Au, Pd, Pt) with tunable morphology and peroxidase-like activity. Chem. Mater. 2010, 22, 2988–2994. [Google Scholar] [CrossRef]
- Li, W.; Kuai, L.; Qin, Q.; Geng, B. Ag-Au bimetallic nanostructures: Co-reduction synthesis and their component-dependent performance for enzyme-free H2O2 sensing. J. Mater. Chem. A 2013, 1, 7111–7117. [Google Scholar] [CrossRef]
- Lee, Y.; Loew, A.; Sun, S. Surface- and structure-dependent catalytic activity of Au nanoparticles for oxygen reduction reaction. Chem. Mater. 2010, 22, 755–761. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Wang, S. Water-soluble Au nanocages for enzyme-free H2O2 sensor and 4-nitrophenol reduction. CrystEngComm 2015, 17, 2368–2375. [Google Scholar] [CrossRef]
- Sun, Y.; Xia, Y. Shape-controlled synthesis of gold and silver nanoparticles. Science 2002, 298, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xia, Y. Mechanistic study on the replacement reaction between silver nanostructures and chloroauric acid in aqueous medium. J. Am. Chem. Soc. 2004, 126, 3892–3901. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, M.S.; Cheng, Y.; Chen, J.; Cobley, C.M.; Zhang, Q.; Rycenga, M.; Xie, J.; Kim, C.; Song, K.H.; Schwartz, A.G.; et al. Gold nanocages covered by smart polymers for controlled release with near-infrared light. Nat. Mater. 2009, 8, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, M.; Zhang, Q.; Cho, E.C.; Cobley, C.M.; Kim, C.; Glaus, C.; Wang, L.V.; Welch, M.J.; Xia, Y. Gold nanocages: A novel class of multifunctional nanomaterials for theranostic applications. Adv. Funct. Mater. 2010, 20, 3684–3694. [Google Scholar] [CrossRef]
- Zhang, L.; Roling, L.T.; Wang, X.; Vara, M.; Chi, M.; Liu, J.; Choi, S.-I.; Park, J.; Herron, J.A.; Xie, Z.; et al. Platinum-based nanocages with subnanometer-thick walls and well-defined, controllable facets. Science 2015, 349, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Skrabalak, S.E.; Au, L.; Li, X.; Xia, Y. Facile synthesis of Ag nanocubes and Au nanocages. Nat. Protoc. 2007, 2, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Cobley, C.M.; Campbell, D.J.; Xia, Y. Tailoring the optical and catalytic properties of gold-silver nanoboxes and nanocages by introducing palladium. Adv. Mater. 2008, 20, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-C.; Lai, Y.-S.; Tsai, C.-M.; Kong, Y.-T.; Lee, C.-I.; Huang, C.-L. One-pot synthesis of monodispersed silver nanodecahedra with optimal sers activities using seedless photo-assisted citrate reduction method. J. Phys. Chem. C 2012, 116, 24292–24300. [Google Scholar] [CrossRef]
- Hu, K.; Lan, D.; Li, X.; Zhang, S. Electrochemical DNA biosensor based on nanoporous gold electrode and multifunctional encoded DNA-Au bio bar codes. Anal. Chem. 2008, 80, 9124–9130. [Google Scholar] [CrossRef] [PubMed]
- Daggumati, P.; Matharu, Z.; Seker, E. Effect of nanoporous gold thin film morphology on electrochemical DNA sensing. Anal. Chem. 2015, 87, 8149–8156. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhang, Z.; Zhuang, Z.; Meng, Z.; Li, C.; Shen, Y. PdPt bimetallic alloy nanowires-based electrochemical sensor for sensitive detection of ascorbic acid. RSC Adv. 2016, 6, 42008–42013. [Google Scholar] [CrossRef]
- Satheesh Babu, T.G.; Varadarajan, D.; Murugan, G.; Ramachandran, T.; Nair, B.G. Gold nanoparticle-polypyrrole composite modified TiO2 nanotube array electrode for the amperometric sensing of ascorbic acid. J. Appl. Electrochem. 2012, 42, 427–434. [Google Scholar] [CrossRef]
- Nancy, T.E.M.; Kumary, V.A. Synergistic electrocatalytic effect of graphene/nickel hydroxide composite for the simultaneous electrochemical determination of ascorbic acid, dopamine and uric acid. Electrochim. Acta 2014, 133, 233–240. [Google Scholar] [CrossRef]
- Sanghavi, B.J.; Srivastava, A.K. Simultaneous voltammetric determination of acetaminophen, aspirin and caffeine using an in situ surfactant-modified multiwalled carbon nanotube paste electrode. Electrochim. Acta 2010, 55, 8638–8648. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; p. 850. [Google Scholar]
- Chen, A.; Chatterjee, S. Nanomaterials based electrochemical sensors for biomedical applications. Chem. Soc. Rev. 2013, 42, 5425–5438. [Google Scholar] [CrossRef] [PubMed]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, M.S. Electrical energy storage and intercalation chemistry. Science 1976, 192, 1126–1127. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Brodd, R.J. What are batteries, fuel cells, and supercapacitors? Chem. Rev. 2004, 104, 4245–4270. [Google Scholar] [CrossRef] [PubMed]
- Steele, B.C.H.; Heinzel, A. Materials for fuel-cell technologies. Nature 2001, 414, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Grove, W.R. On voltaic series and the combination of gases by platinum. Philos. Mag. 1839, 14, 127–130. [Google Scholar] [CrossRef]
- Grove, W.R. On a small voltaic battery of great energy; some observations on voltaic combinations and forms of arrangement; and on the inactivity of a copper positive electrode in nitro-sulphuric acid. Philos. Mag. 1839, 15, 287–293. [Google Scholar] [CrossRef]
- Grove, W.R. On a gaseous voltaic battery. Philos. Mag. 1842, 21, 417–420. [Google Scholar]
- Yahiro, A.T.; Lee, S.M.; Kimble, D.O. Bioelectrochemistry: I. Enzyme utilizing bio-fuel cell studies. Biochim. Biophys. Acta 1964, 88, 375–383. [Google Scholar] [CrossRef]
- Young, T.G.; Hadjipetrou, L.; Lilly, M.D. The theoretical aspects of biochemical fuel cells. Biotechnol. Bioeng. 1966, 8, 581–593. [Google Scholar] [CrossRef]
- Zhao, F.; Slade, R.C.T.; Varcoe, J.R. Techniques for the study and development of microbial fuel cells: An electrochemical perspective. Chem. Soc. Rev. 2009, 38, 1926–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.B.; Yarbrough, H.F. Preliminary experiments on a microbial fuel cell. Science 1962, 137, 615–616. [Google Scholar] [CrossRef]
- Higgins, S.R.; Lau, C.; Atanassov, P.; Minteer, S.D.; Cooney, M.J. Standardized characterization of a flow through microbial fuel cell. Electroanalysis 2011, 23, 2174–2181. [Google Scholar] [CrossRef]
- Liu, H.; Ramnarayanan, R.; Logan, B.E. Production of electricity during wastewater treatment using a single chamber microbial fuel cell. Environ. Sci. Technol. 2004, 38, 2281–2285. [Google Scholar] [CrossRef] [PubMed]
- Logan, B.E.; Hamelers, B.; Rozendal, R.; Schröder, U.; Keller, J.; Freguia, S.; Aelterman, P.; Verstraete, W.; Rabaey, K. Microbial fuel cells: Methodology and technology. Environ. Sci. Technol. 2006, 40, 5181–5192. [Google Scholar] [CrossRef] [PubMed]
- Heller, A.; Feldman, B. Electrochemical glucose sensors and their application in diabetes management. In Applications of Electrochemistry in Medicine; Schlesinger, M., Ed.; Springer: New York, NY, USA, 2013; Volume 56, pp. 121–187. [Google Scholar]
- Heller, A. Integrated medical feedback systems for drug delivery. AIChE J. 2005, 51, 1054–1066. [Google Scholar] [CrossRef]
- Holmes, C.F. Electrochemical power sources and the treatment of human illness. Electrochem. Soc. Interface 2003, 12, 26–29. [Google Scholar]
- Heller, A. Potentially implantable miniature batteries. Anal. Bioanal. Chem. 2006, 385, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Kerzenmacher, S.; Ducrée, J.; Zengerle, R.; von Stetten, F. An abiotically catalyzed glucose fuel cell for powering medical implants: Reconstructed manufacturing protocol and analysis of performance. J. Power Sources 2008, 182, 66–75. [Google Scholar] [CrossRef]
- Kwon, C.H.; Lee, S.-H.; Choi, Y.-B.; Lee, J.A.; Kim, S.H.; Kim, H.-H.; Spinks, G.M.; Wallace, G.G.; Lima, M.D.; Kozlov, M.E.; et al. High-power biofuel cell textiles from woven biscrolled carbon nanotube yarns. Nat. Commun. 2014, 5, 3928. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Filanovsky, B.; Willner, I. A biofuel cell based on two immiscible solvents and glucose oxidase and microperoxidase-11 monolayer-functionalized electrodes. New J. Chem. 1999, 23, 481–487. [Google Scholar] [CrossRef]
- Katz, E.; Willner, I. A biofuel cell with electrochemically switchable and tunable power output. J. Am. Chem. Soc. 2003, 125, 6803–6813. [Google Scholar] [CrossRef]
- Zebda, A.; Gondran, C.; Le Goff, A.; Holzinger, M.; Cinquin, P.; Cosnier, S. Mediatorless high-power glucose biofuel cells based on compressed carbon nanotube-enzyme electrodes. Nat. Commun. 2011, 2, 370. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Minteer, S.D. Enzymatic biofuel cell for oxidation of glucose to CO2. ACS Catal. 2012, 2, 91–94. [Google Scholar] [CrossRef]
- Holade, Y.; Both Engel, A.; Tingry, S.; Cherifi, A.; Cornu, D.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Insights on hybrid glucose biofuel cell based on bilirubin oxidase cathode and gold-based nanomaterials anode. ChemElectroChem 2014, 1, 1976–1987. [Google Scholar] [CrossRef]
- Holade, Y.; MacVittie, K.; Conlon, T.; Guz, N.; Servat, K.; Napporn, T.W.; Kokoh, K.B.; Katz, E. Pacemaker activated by an abiotic biofuel cell operated in human serum solution. Electroanalysis 2014, 26, 2445–2457. [Google Scholar] [CrossRef]
- Cinquin, P.; Gondran, C.; Giroud, F.; Mazabrard, S.; Pellissier, A.; Boucher, F.; Alcaraz, J.-P.; Gorgy, K.; Lenouvel, F.; Mathé, S.; et al. A glucose biofuel cell implanted in rats. PLoS ONE 2010, 5, e10476. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Ritzmann, R.E.; Lee, I.; Pollack, A.J.; Scherson, D. An implantable biofuel cell for a live insect. J. Am. Chem. Soc. 2012, 134, 1458–1460. [Google Scholar] [CrossRef] [PubMed]
- Szczupak, A.; Halamek, J.; Halamkova, L.; Bocharova, V.; Alfonta, L.; Katz, E. Living battery—Biofuel cells operating in vivo in clams. Energy Environ. Sci. 2012, 5, 8891–8895. [Google Scholar] [CrossRef]
- Andoralov, V.; Falk, M.; Suyatin, D.B.; Granmo, M.; Sotres, J.; Ludwig, R.; Popov, V.O.; Schouenborg, J.; Blum, Z.; Shleev, S. Biofuel cell based on microscale nanostructured electrodes with inductive coupling to rat brain neurons. Sci. Rep. 2013, 3, 3270. [Google Scholar] [CrossRef] [PubMed]
- Castorena-Gonzalez, J.A.; Foote, C.; MacVittie, K.; Halámek, J.; Halámková, L.; Martinez-Lemus, L.A.; Katz, E. Biofuel cell operating in vivo in rat. Electroanalysis 2013, 25, 1579–1584. [Google Scholar] [CrossRef]
- Katz, E.; MacVittie, K. Implanted biofuel cells operating in vivo—Methods, applications and perspectives—Feature article. Energy Environ. Sci. 2013, 6, 2791–2803. [Google Scholar] [CrossRef]
- Zebda, A.; Cosnier, S.; Alcaraz, J.P.; Holzinger, M.; Le Goff, A.; Gondran, C.; Boucher, F.; Giroud, F.; Gorgy, K.; Lamraoui, H.; et al. Single glucose biofuel cells implanted in rats power electronic devices. Sci. Rep. 2013, 3, 1516. [Google Scholar] [CrossRef] [PubMed]
- Habrioux, A.; Servat, K.; Tingry, S.; Kokoh, K.B. Enhancement of the performances of a single concentric glucose/O2 biofuel cell by combination of bilirubin oxidase/Nafion cathode and Au-Pt anode. Electrochem. Commun. 2009, 11, 111–113. [Google Scholar] [CrossRef]
- Habrioux, A.; Sibert, E.; Servat, K.; Vogel, W.; Kokoh, K.B.; Alonso-Vante, N. Activity of platinum-gold alloys for glucose electrooxidation in biofuel cells. J. Phys. Chem. B 2007, 111, 10329–10333. [Google Scholar] [CrossRef] [PubMed]
- Both Engel, A.; Bechelany, M.; Fontaine, O.; Cherifi, A.; Cornu, D.; Tingry, S. One-pot route to gold nanoparticles embedded in electrospun carbon fibers as an efficient catalyst material for hybrid alkaline glucose biofuel cells. ChemElectroChem 2016, 3, 629–637. [Google Scholar] [CrossRef]
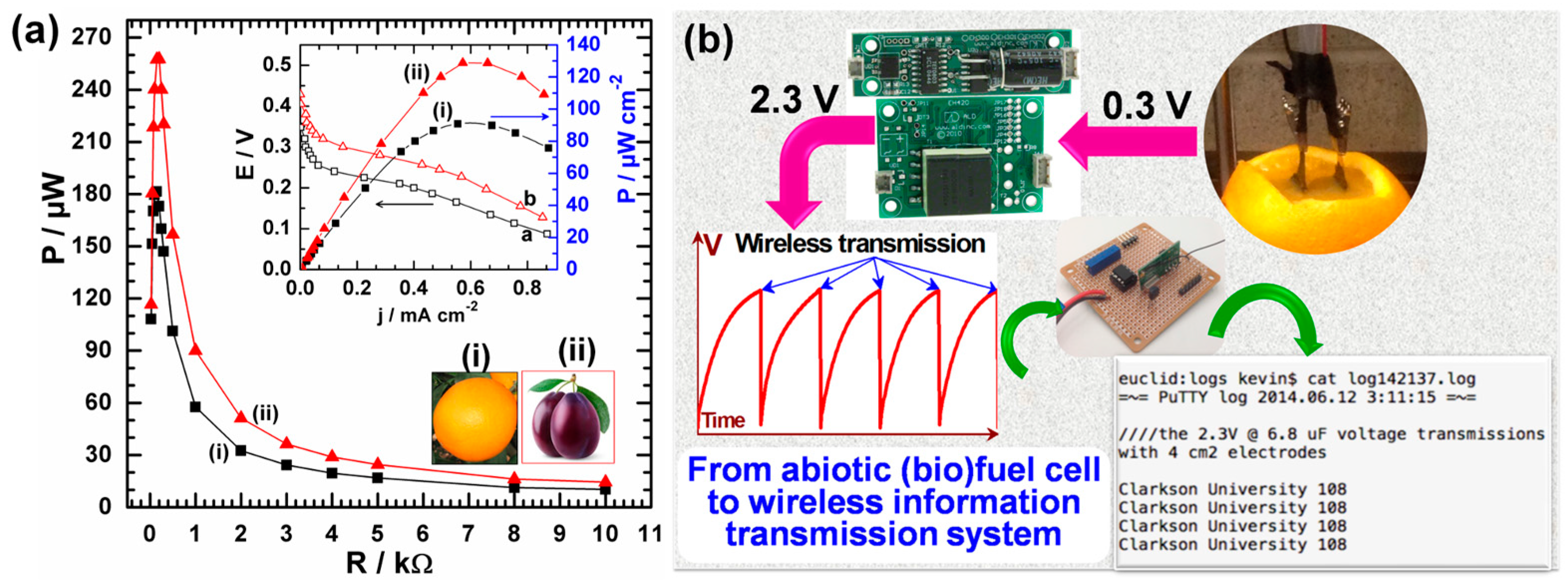
- Holade, Y.; MacVittie, K.; Conlon, T.; Guz, N.; Servat, K.; Napporn, T.W.; Kokoh, K.B.; Katz, E. Wireless information transmission system powered by an abiotic biofuel cell implanted in an orange. Electroanalysis 2015, 27, 276–280. [Google Scholar] [CrossRef]
- Kerzenmacher, S.; Ducrée, J.; Zengerle, R.; von Stetten, F. Energy harvesting by implantable abiotically catalyzed glucose fuel cells. J. Power Sources 2008, 182, 1–17. [Google Scholar] [CrossRef]
- Kloke, A.; Biller, B.; Kräling, U.; Kerzenmacher, S.; Zengerle, R.; von Stetten, F. A single layer glucose fuel cell intended as power supplying coating for medical implants. Fuel Cells 2011, 11, 316–326. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Chu, M.; Deng, W.; Tan, Y.; Ma, M.; Su, X.; Xie, Q.; Yao, S. Facile fabrication of network film electrodes with ultrathin Au nanowires for nonenzymatic glucose sensing and glucose/O2 fuel cell. Biosens. Bioelectron. 2014, 52, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fan, L.; Hong, B.; Zhang, Y.; Zhang, M.; Que, Q.; Ji, J. Three-dimensional porous palladium foam-like nanostructures as electrocatalysts for glucose biofuel cells. Energy Technol. 2016, 4, 249–255. [Google Scholar] [CrossRef]
- Werner, H.; Robinson, B.W. A glucose cell. In Proceedings of the Digest of the 7th International Conference on Medical and Biological Engineering, Stockholm, Sweden, 14–19 August 1967; Jacobson, B., Ed.; The Royal Academy of Engineering Sciences: Stockholm, Sweden, 1967; p. 520. [Google Scholar]
- Kloke, A.; Köhler, C.; Zengerle, R.; Kerzenmacher, S. Porous platinum electrodes fabricated by cyclic electrodeposition of PtCu alloy: Application to implantable glucose fuel cells. J. Phys. Chem. C 2012, 116, 19689–19698. [Google Scholar] [CrossRef]
- Slaughter, G.; Sunday, J. A membraneless single compartment abiotic glucose fuel cell. J. Power Sources 2014, 261, 332–336. [Google Scholar] [CrossRef]
- Oncescu, V.; Erickson, D. A microfabricated low cost enzyme-free glucose fuel cell for powering low-power implantable devices. J. Power Sources 2011, 196, 9169–9175. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, Y.; Chen, C.; Yang, X.; Li, C. Carbon nanotubes coated with platinum nanoparticles as anode of biofuel cell. Particuology 2012, 10, 450–455. [Google Scholar] [CrossRef]
- Wen, D.; Liu, W.; Herrmann, A.-K.; Eychmüller, A. A membraneless glucose/O2 biofuel cell based on Pd aerogels. Chem. Eur. J. 2014, 20, 4380–4385. [Google Scholar] [CrossRef] [PubMed]
- Santiago, Ó.; Navarro, E.; Raso, M.A.; Leo, T.J. Review of implantable and external abiotically catalysed glucose fuel cells and the differences between their membranes and catalysts. Appl. Energy 2016, 179, 497–522. [Google Scholar] [CrossRef]
- Kerzenmacher, S.; Zehnle, S.; Volk, T.; Jansen, D.; von Stetten, F.; Zengerle, R. An efficient low-power DC-DC converter enables operation of a cardiac pacemaker by an integrated glucose fuel cell. In Proceedings of the PowerMEMS 2008, Sendai, Japan, 9–12 November 2008; pp. 189–192.
- Holade, Y.; Napporn, T.W.; Kokoh, B.K. Advanced surfactant-free nanomaterials for electrochemical energy conversion systems: From electrocatalysis to bionanotechnology. In Advanced Electrode Materials; Tiwari, A., Kuralay, F., Uzun, L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 103–146. [Google Scholar]
- Both Engel, A.; Holade, Y.; Tingry, S.; Cherifi, A.; Cornu, D.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Electrospun carbon fibers: Promising electrode material for abiotic and enzymatic catalysis. J. Phys. Chem. C 2015, 119, 16724–16733. [Google Scholar] [CrossRef]
- Holade, Y.; Sahin, N.; Servat, K.; Napporn, T.; Kokoh, K. Recent advances in carbon supported metal nanoparticles preparation for oxygen reduction reaction in low temperature fuel cells. Catalysts 2015, 5, 310–348. [Google Scholar] [CrossRef]
- Holade, Y.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Electrocatalytic properties of nanomaterials synthesized from “bromide anion exchange” method—Investigations of glucose and glycerol oxidation. Electrochim. Acta 2015, 162, 205–214. [Google Scholar] [CrossRef]
- Holade, Y. Powering implantable micro-devices: Towards a new joker? L’Act. Chim. 2016, 412, 23–31. [Google Scholar]
- Southcott, M.; MacVittie, K.; Halamek, J.; Halamkova, L.; Jemison, W.D.; Lobel, R.; Katz, E. A pacemaker powered by an implantable biofuel cell operating under conditions mimicking the human blood circulatory system—Battery not included. Phys. Chem. Chem. Phys. 2013, 15, 6278–6283. [Google Scholar] [CrossRef] [PubMed]
- Pistoia, G. Batteries for Portable Devices; Elsevier Science B.V.: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Szczesny, S.; Jetzki, S.; Leonhardt, S. Review of Current Actuator Suitability for Use in Medical Implants. In Proceedings of the 28th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBS ’06), New York City, NY, USA, 30 August 2006–3 September 2006; pp. 5956–5959.
- Hebié, S.; Holade, Y.; Servat, K.; Kokoh, B.K.; Napporn, T.W. Electrochemical reactivity at free and supported gold nanocatalysts surface. In Catalytic Application of Nano-Gold Catalysts; Mishra, N.K., Ed.; Intech: Rijeka, Croatia, 2016; pp. 101–130. [Google Scholar]
- Gellett, W.; Kesmez, M.; Schumacher, J.; Akers, N.; Minteer, S.D. Biofuel cells for portable power. Electroanalysis 2010, 22, 727–731. [Google Scholar] [CrossRef]
- Katz, E. Implantable biofuel cells operating in vivo—Potential power sources for bioelectronic devices. Bioelectron. Med. 2015, 2, 1–12. [Google Scholar]
- MacVittie, K.; Conlon, T.; Katz, E. A wireless transmission system powered by an enzyme biofuel cell implanted in an orange. Bioelectrochemistry 2015, 106, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Schröder, U. From in vitro to in vivo—Biofuel cells are maturing. Angew. Chem. Int. Ed. 2012, 51, 7370–7372. [Google Scholar] [CrossRef] [PubMed]
- Schwefel, J.; Ritzmann, R.E.; Lee, I.N.; Pollack, A.; Weeman, W.; Garverick, S.; Willis, M.; Rasmussen, M.; Scherson, D. Wireless communication by an autonomous self-powered cyborg insect. J. Electrochem. Soc. 2015, 161, H3113–H3116. [Google Scholar] [CrossRef]
- Falk, M.; Alcalde, M.; Bartlett, P.N.; De Lacey, A.L.; Gorton, L.; Gutierrez-Sanchez, C.; Haddad, R.; Kilburn, J.; Leech, D.; Ludwig, R.; et al. Self-powered wireless carbohydrate/oxygen sensitive biodevice based on radio signal transmission. PLoS ONE 2014, 9, e109104. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Shleev, S.; Narváez Villarrubia, C.W.; Babanova, S.; Atanassov, P. Biological fuel cells for biomedical applications. In Enzymatic Fuel Cells: From Fundamentals to Applications; Luckarift, H.R., Atanassov, P., Johnson, G.R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 422–450. [Google Scholar]
- Jia, W.; Valdés-Ramírez, G.; Bandodkar, A.J.; Windmiller, J.R.; Wang, J. Epidermal biofuel cells: Energy harvesting from human perspiration. Angew. Chem. Int. Ed. 2013, 52, 7233–7236. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Bandodkar, A.J.; Valdés-Ramírez, G.; Windmiller, J.R.; Yang, Z.; Ramírez, J.; Chan, G.; Wang, J. Electrochemical tattoo biosensors for real-time noninvasive lactate monitoring in human perspiration. Anal. Chem. 2013, 85, 6553–6560. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.; Coyle, S.; Scarmagnani, S.; Hayes, J. Wireless sensor networks and chemo-/biosensing. Chem. Rev. 2008, 108, 652–679. [Google Scholar] [CrossRef] [PubMed]
- Windmiller, J.R.; Wang, J. Wearable electrochemical sensors and biosensors: A review. Electroanalysis 2013, 25, 29–46. [Google Scholar] [CrossRef]
- Luckarift, H.R.; Atanassov, P.; Johnson, G.R. Enzymatic Fuel Cells: From Fundamentals to Applications, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; p. 496. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Status | Chemicals | Comments |
|---|---|---|
| Known sugar interferences | Maltose | food, immune globulins, peritoneal dialysis |
| Galactose | food | |
| Xylose | malabsorption testing | |
| Icodextrin | peritoneal dialysis | |
| Possible sugar interferences | Mannose | food |
| Lactose | ||
| Ribose | ||
| Arabinose | ||
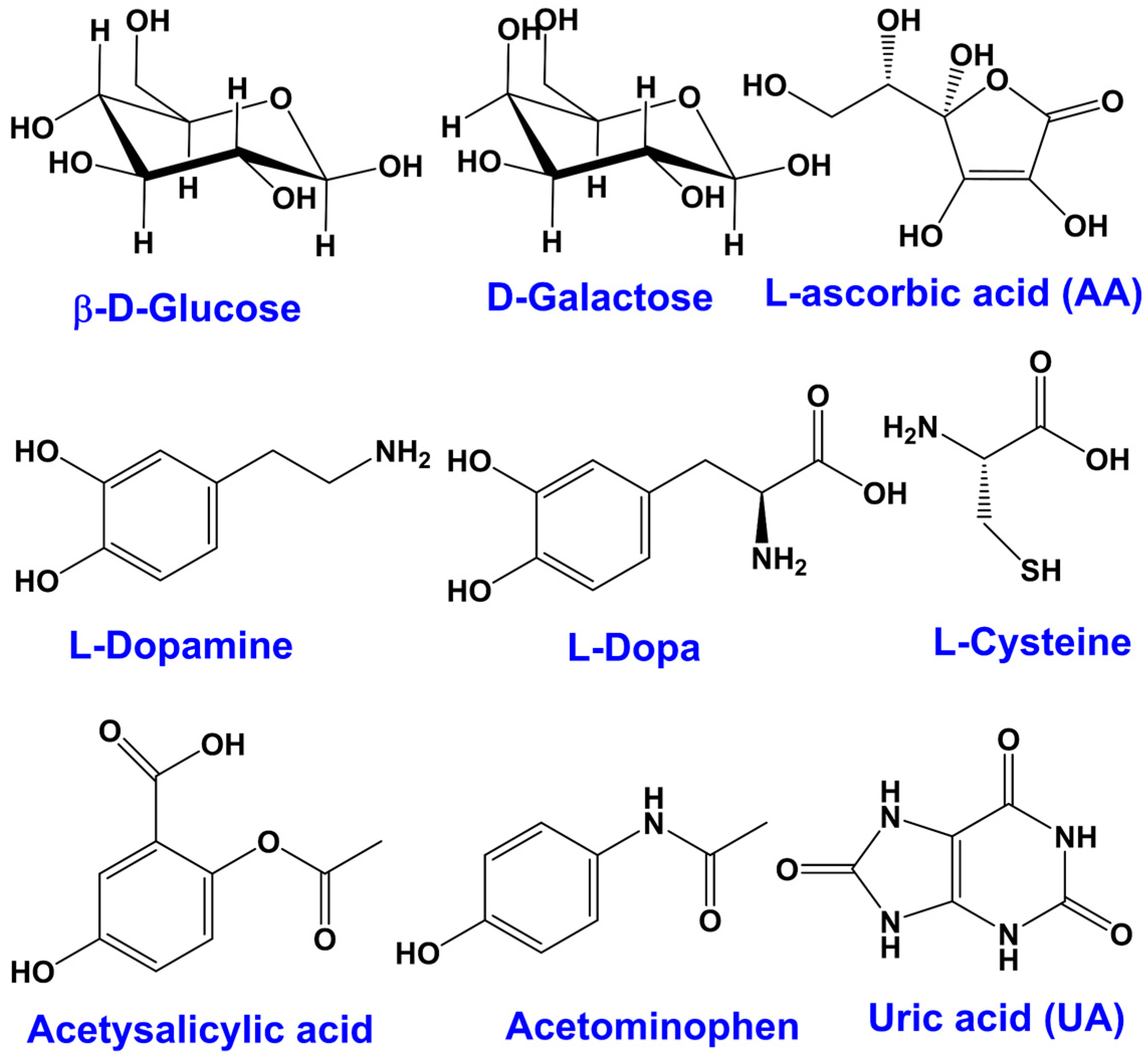
| Naturally occurring interferences | Ascorbic acid (AA) | Like glucose and commonly known as vitamin C, it is a strong reducing agent, concentrations in blood vary on dietary intake, normal blood: 23–85 µM |
| Uric acid (UA) | A strong reducing agent like glucose, present as the final metabolite of purine and in the blood as an antioxidant at ca. 0.18 to 0.42 mM | |
| Cysteine | Present in the blood at very low concentrations of 3 to 15 μM | |
| Dopamine | Present at low levels naturally, is known to be oxidized at a similar potential window to glucose | |
| Pharmacological interferences (chemicals from drugs) | Acetaminophen (AP) | A constituent of paracetamol 1 that is administered as a drug: therefore commonly used and of a variable concentration in the blood, with therapeutic levels of up to 0.2 mM |
| Salicylic acid | A component of aspirin administered as a drug, thus may also be present in the blood (up to 2.2 mM) at an interfering level via salicylates | |
| Dopamine | Naturally occurring and also administered as a drug in the form of its precursor L-Dopa | |
| Proteins and chloride anions | Proteins | In the blood, proteins (6–8.4 g·dL‒1) readily adsorb and cause inhibitory electrode fouling, mostly on Pt and Au electrodes. Triglycerides in blood can have a similar effect, competing and lowering the glucose oxidation signal |
| Chloride | Having a high concentration in the blood, 98–106 mM, Cl‒ anions have an important poisoning rate: they greatly interfere with glucose electrooxidation signals, as they strongly chemisorb irreversibly onto the metal surface (inhibiting the electrocatalysis) | |
| Alcohol | Ethanol | Present in blood, up to 65 mM. Can be readily oxidized at metallic electrodes, thus interferes |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holade, Y.; Tingry, S.; Servat, K.; Napporn, T.W.; Cornu, D.; Kokoh, K.B. Nanostructured Inorganic Materials at Work in Electrochemical Sensing and Biofuel Cells. Catalysts 2017, 7, 31. https://doi.org/10.3390/catal7010031
Holade Y, Tingry S, Servat K, Napporn TW, Cornu D, Kokoh KB. Nanostructured Inorganic Materials at Work in Electrochemical Sensing and Biofuel Cells. Catalysts. 2017; 7(1):31. https://doi.org/10.3390/catal7010031
Chicago/Turabian StyleHolade, Yaovi, Sophie Tingry, Karine Servat, Teko W. Napporn, David Cornu, and Kouakou Boniface Kokoh. 2017. "Nanostructured Inorganic Materials at Work in Electrochemical Sensing and Biofuel Cells" Catalysts 7, no. 1: 31. https://doi.org/10.3390/catal7010031