Enzymatic Systems for Cellulose Acetate Degradation
by
 , ,
, ,
Oskar Haske-Cornelius
1, 
Alessandro Pellis
1,
Gregor Tegl
1,
Stefan Wurz
1,
Bodo Saake
2,
Roland Ludwig
3,
Andries Sebastian
4,
Gibson S. Nyanhongo
1,* and
Georg M. Guebitz
1 1
Institute of Environmental Biotechnology, University of Natural Resources and Life Sciences Vienna, Konrad-Lorenz-Strasse 20, 3430 Tulln an der Donau, Austria
2
Chemical Wood Technology, Department of Wood Science, University of Hamburg, Leuschnerstrasse 91, 21031 Hamburg, Germany
3
Department of Food Science and Technology, University of Natural Resources and Life Sciences Vienna, Muthgasse 18, 1190 Vienna, Austria
4
R. J. Reynolds Tobacco Company, 401 North Main Street, Winston-Salem, NC 27101, USA
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(10), 287; https://doi.org/10.3390/catal7100287
Submission received: 13 August 2017
/
Revised: 25 September 2017
/
Accepted: 25 September 2017
/
Published: 27 September 2017
(This article belongs to the Special Issue Biocatalysis and Biotransformations)
Abstract
:Cellulose acetate (CA)-based materials, like cigarette filters, contribute to landscape pollution challenging municipal authorities and manufacturers. This study investigates the potential of enzymes to degrade CA and to be potentially incorporated into the respective materials, enhancing biodegradation. Deacetylation studies based on Liquid Chromatography-Mass Spectrometry-Time of Flight (LC-MS-TOF), High Performance Liquid Chromatography (HPLC), and spectrophotometric analysis showed that the tested esterases were able to deacetylate the plasticizer triacetin (glycerol triacetate) and glucose pentaacetate (cellulose acetate model compound). The most effective esterases for deacetylation belong to the enzyme family 2 (AXE55, AXE 53, GAE), they deacetylated CA with a degree of acetylation of up to 1.8. A combination of esterases and cellulases showed synergistic effects, the absolute glucose recovery for CA 1.8 was increased from 15% to 28% when an enzymatic deacetylation was performed. Lytic polysaccharide monooxygenase (LPMO), and cellobiohydrolase were able to cleave cellulose acetates with a degree of acetylation of up to 1.4, whereas chitinase showed no activity. In general, the degree of substitution, chain length, and acetyl group distribution were found to affect CA degradation. This study shows that, for a successful enzyme-based deacetylation system, a cocktail of enzymes, which will randomly cleave and generate shorter CA fragments, is the most suitable.
1. Introduction
Cellulose acetate (CA), produced from high-quality cellulose materials obtained from cotton or wood dissolving pulp, is used in many consumer products. As blend material with different water soluble and insoluble polymers, like poly(-lactic acid) or starch, CA can be found in several plastics and coating materials [1,2], which, if handled improperly, can have a severe environmental impact. Other sources of CA pollution are cigarette filters and textiles, which end up as litter, causing multiple problems in the environment [3,4]. This is particularly true for cigarette filters, which are inappropriately disposed by smokers. An investigation in the USA showed that 25–50% of all litter items collected on streets and roadways were cigarette butts [5]. Thus, developing biodegradable cigarette filters may help to overcome pollution and reduce effort in litter cleanup activities. For some regions these litter issues already influenced legislation: in 2012 the state of New York took into consideration a law that will promote sales of biodegradable cigarette filters [6].
CA-based filters are produced by chemical acetylation of cellulose (95% α-cellulose), achieved in a two-step process using an excess of acetic anhydride in the presence of sulfuric acid or perchloric acid as catalysts. This first step is followed by a partial deacetylation, which enables the control of acetylation across the molecule.
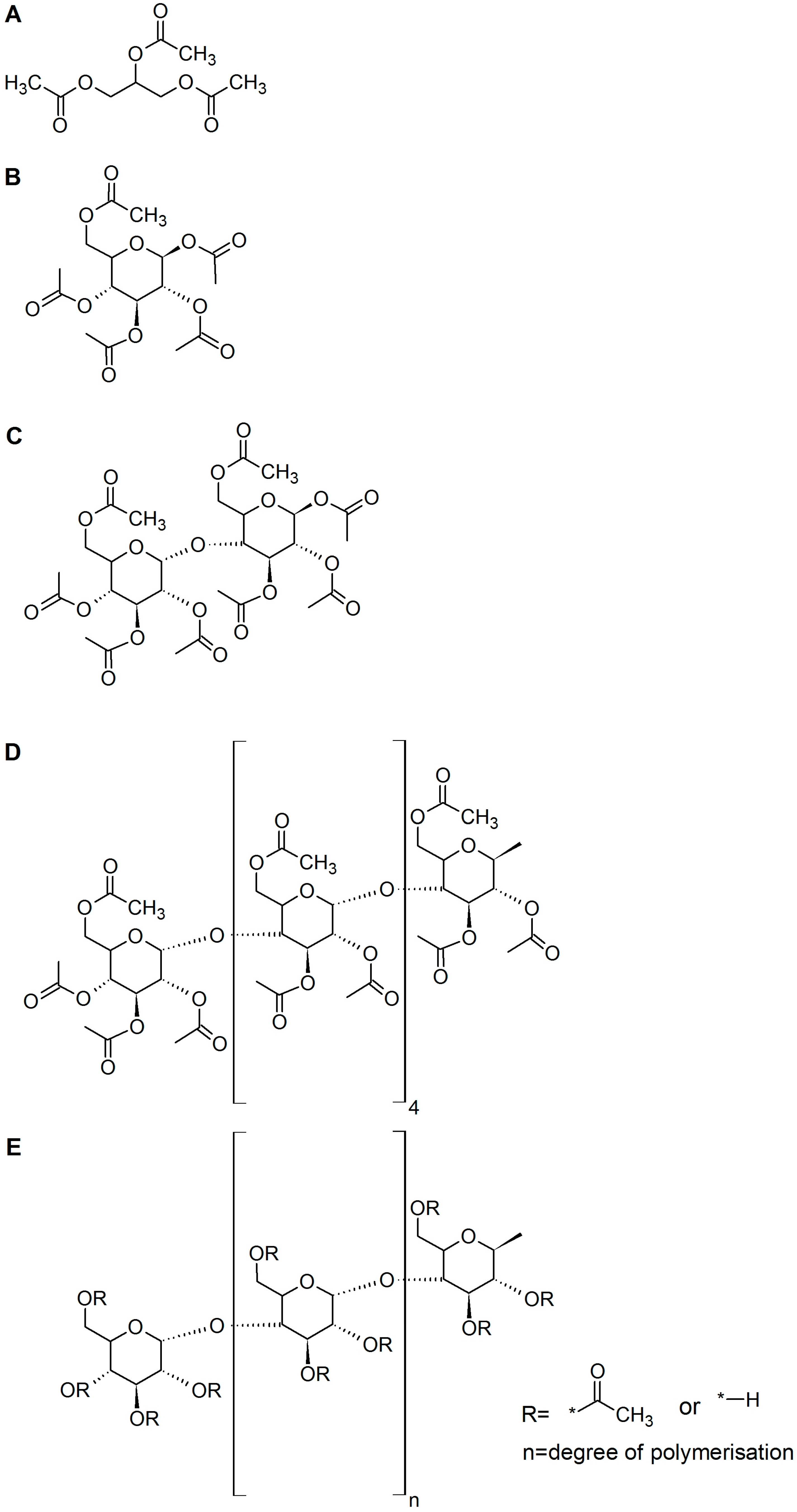
For CA degradation, several natural mechanisms are known. Microbial-caused reduction in the degree of substitution and photodegradation of CA products, like fibers and films were previously reported. Depending on multiple factors, like product configuration (film or fiber), DS and light exposure degradation times vary from days to weeks [7,8]. Here it might be useful to introduce an enzymatic systems supporting the natural degradation of CA materials. Thereby, enzyme incorporation strategies into CA fibrous materials [9] and cellulose [10] are known. An important aspect in enzymatic degradation is to monitor the influences of different substrate properties on the enzymatic degradation. CA can be characterized according to its chain length, which is described by the degree of polymerization (amount of acetylated glucose subunits in a chain) and the number of acetyl groups per monomer, which is given as degree of substitution (DS). A DS of 1.9 means that on ten glucose subunits 19 of 30 potential positions are acetylated. An overview about all used substrates is given Figure 1.
Cigarette filters, for example, have a degree of acetylation of 2.5 [3,4]. This high degree of acetylation makes tobacco CA filters difficult to degrade by microorganisms [11]. In addition, high molecular weight, crystallinity, and the physical form influence the microbial degradation efficacy [12,13]. However, the degradation ability of highly-acetylated CA is still under investigation in the scientific community [4]. Saki et al. reported the ability of Neisseria sicca to produce extracellular enzymes able to degrade CA with a 2.3 degree of acetylation. The authors suggested a cooperative system of esterases deacetylating CA, followed by cellulases breaking the backbone into smaller fragments, which can be quickly taken up by the cells [13]. An approach using single enzymes or a combination of biocatalysts may lead to an industrial application for planned litter depletion. For cellulases, a decrease in degradation for acetylated pulps was reported compared to non-acetylated pulps [14]. Data published by Olaru et al. show a reduced action of cellulases, based on the viscometry of CA, with an increasing degree of substitution (DS) [15]. Apart from mechanistic insights, identification of more efficient CA degrading enzyme systems could also allow the incorporation in CA materials enhancing biodegradation upon contact with water. Thereby it could be sufficient to incorporate one of the functions into the material whereby the second step can be carried out by the natural biome in the surrounding of the litter item.
However, only a few representatives of esterases have been assessed so far for CA deacetylation. While the effect of oxidoreductases like lytic polysaccharide monooxygenase (LPMO), has not yet been investigated on CA as a substrate. Therefore, in this study CA cleavage was systematically investigated using a variety of enzymes belonging to different classes. All enzymes were tested on different substrates displaying different properties of CA materials.
In this study we evaluate the eligibility of different enzymes from different classes, which are involved either in the deacetylation of CA or in backbone cleavage of the polymer. Enzymes were tested extensively on different substrates (Figure 1) and in combination to enhance the performance on CA.
2. Results and Discussion
2.1. Deacetylation
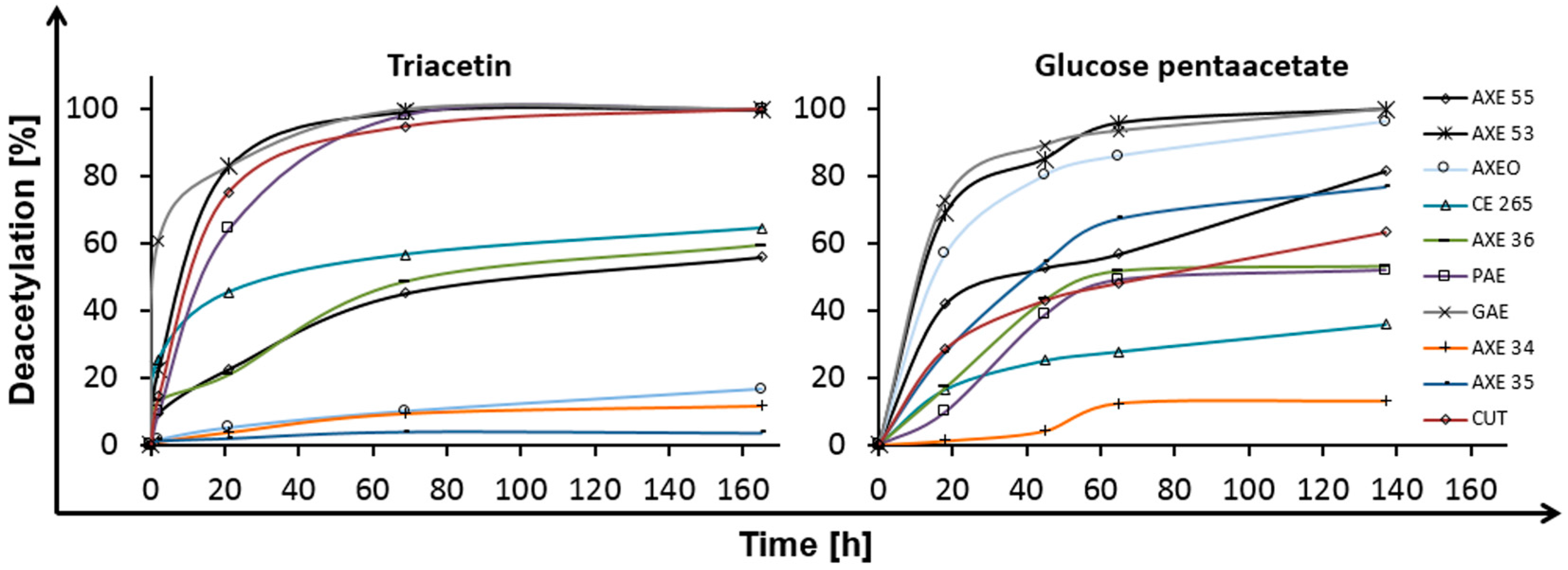
In a first step, deacetylation of glucose pentaacetate (GPA) and triacetin (TA) by different esterases was investigated. Hydrolysis of triacetin was studied since this compound is used as plasticizer in many CA materials, while hydrolysis may have an impact on biodegradation of CA [16]. Although several enzymes were able to completely deacetylate both compounds, there were significant differences between the individual enzymes (Figure 2). Major differences were seen for acetyl xylan esterases (AXE) 35 and AXE O), which deacetylated GPA to 80% and 90%, respectively, but had only minor effects on triacetin. Classified as AXE, these enzymes clearly prefer sugar-bound esters compared to aliphatic esters, like in triacetin. On the other hand, cutinase (CUT) showed similar activities on both substrates and was previously described to cleave ester bonds in hydrophobic aromatic and aliphatic polyesters [17]. Moreover, different applications of cutinase are known, e.g., a wild-type cutinase was reported to esterify the hydroxyl groups of cellulose [18]. This makes cutinase a promising enzyme in cellulose acetate treatment.
AXE 53 and glucomannan acetyl esterase (GAE) showed high capacities to deacetylate both substrates. Especially for GAE, 60% of triacetin were deacetylated in 2 min. For AXE’s deacetylation of different model compounds, representing acetylated polysaccharides and non-polysaccharide compounds, was reported before for chitosan, chitin [19], and cephalosporin-C [20].
On the other hand, AXE 34 only weakly deacetylated both substrates. Altaner et al. found evidence for regioselectivity of esterases derived from different families. They claim that enzymes of the carbohydrate esterase (CE) family 1 exclusively deacetylated CA in the C2- and C3-carbon positions, without cleaving C6 in the sugar. Furthermore, they postulate differences in cleavability for different substitution positions on the polymer chain [21]. Complete deacetylation was observed for pectin acetyl esterase PAE, GAE, AXE O, and AXE 53. For some enzymes, e.g., AXE 34 only one acetyl group of GPA was attacked. For triacetin, the degrees of deacetylation in Figure 2 are arranged into three groups corresponding to the three acetyl groups of the molecule. This indicates cleavage of only one ester bond for AXE O, AXE 34, and AXE 35. Two bonds were cleaved for CE 265, CUT, and AXE 36. All other enzymes deacetylated triacetin completely. An influence of the enzyme family for this behavior is not visible. Apart from GPA, enzymatic hydrolysis of acetylated oligomers, namely cellobiose octaacetate and cellohexose eicosaacetate, was investigated. Within 68 h only GAE (45%) AXE 55 (14%), and CUT 1 (13.5%) showed significant deacetylation of cellohexaose eicosaacetate. All other tested enzymes reached deacetylation degrees lower than 4%. Additionally, for GPA, an influence of the enzyme family cannot be stated.
In nature, glucomannan acetyl esterase is part of the wood degradation process. O-Acetylgalactoglucomannans (AcGGM) are the principal hemicellulose components in softwoods. They are mainly water insoluble, but their acetylation pattern influences their behavior in water [22]. The structure of AcGGM is a linear backbone of (1→4)-linked β-d-mannopyranosyl and (1→4)-linked β-d-glucopyranosyl units, with (1→6)-linked α-d-galactopyranosyl units. Naturally, the mannose subunits can be acetylated at C-2 and C-3, however, via chemical acetylation galactose and glucose subunits were also acetylated [23]. It was shown that linear oligosaccharides from AcGGM could be obtained when an acetyl mannan esterases and a α-galactosidases were used in combination [22]. It was also suggested that galactomannan deacetylation is an inherent property of some AXEs [11]. Due to catalytic similarities between acetyl xylan esterases and glucomannan esterases, the latter enzyme is of interest for degradation processes of acetylcellulose.
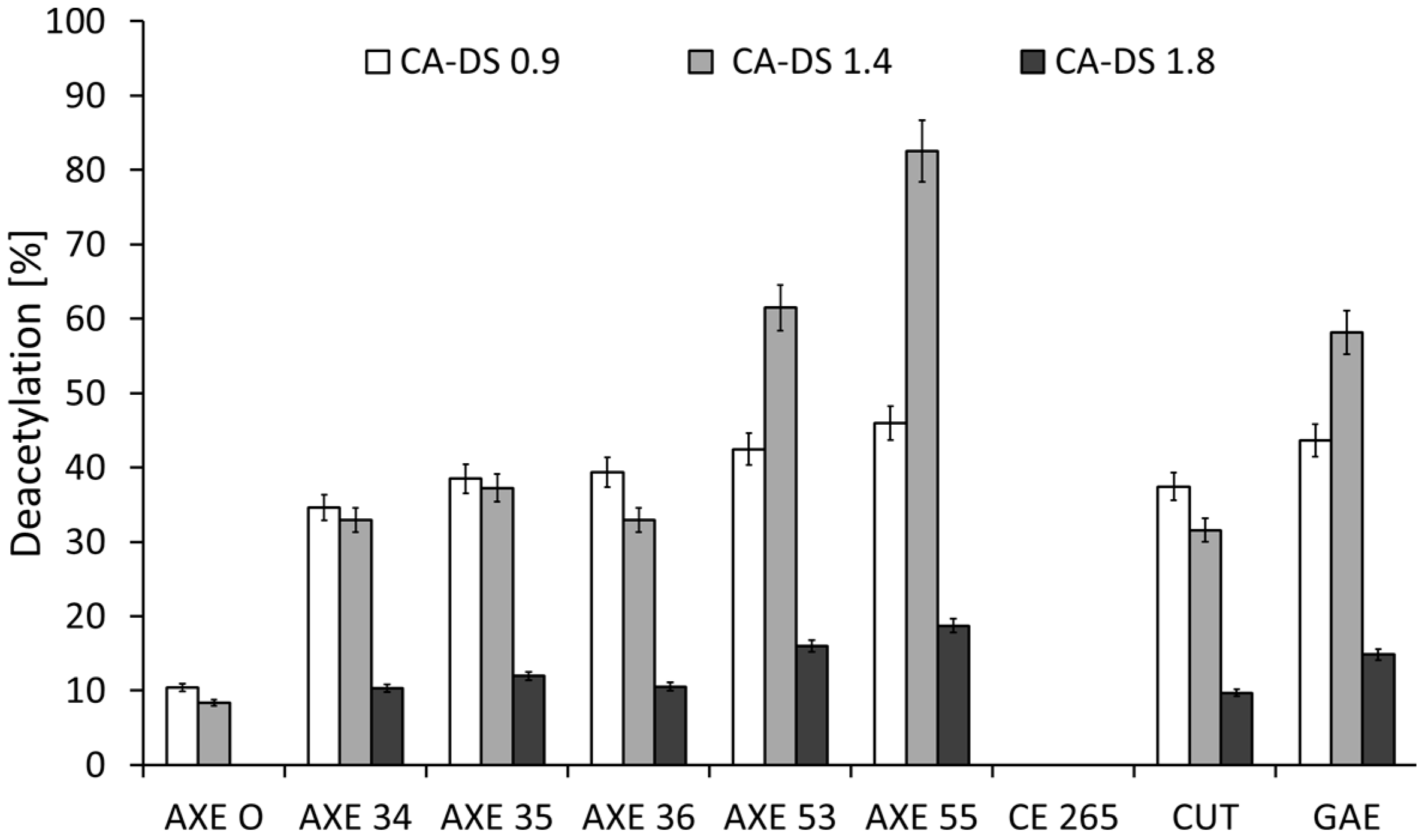
In a next step, enzymatic hydrolysis of CA with different degrees of acetylation was investigated (Figure 3). In general, the activity of the enzymes increased with decreasing degree of acetylation confirming the “protective” function of acetyl groups as reported before [11]. Only for AXE 55, AXE 53, and GAE was deacetylation weaker for CA-DS 0.9 (degree of substitution) than for CA-DS 1.4, which belongs to the same family (Table 1). Poutanen et al. reported the behavior of acetyl xylan esterases on their natural substrates. AXE prefers polymeric substrates, whereas acetyl esterase showed high affinity on acetyl xylobiose. Out of this, they claim a high deacetylation specificity, which depends on the specific position of the acetyl groups and not on the degree of polymerization. Using acetyl xylan esterase in combination with other enzymes they obtained a complete degradation of polymeric substrates to acetic acid and xylose using xylanase and β-xylosidase [24]. Regioselectivity claimed for AXE’s natural model substrates explains the non-complete deacetylation of CA materials. Due to the higher number of acetyl groups per monomer, regioselectivity plays a more important role for CAs with a high degree of substitution. The probability to find a critical position blocked is, therefore, larger for highly-acetylated substrates.
Interestingly, most of the enzymes not belonging to family II showed almost slightly lower activity on CA-DS 1.4 compared to CA-DS 0.9, while the activity dramatically decreased for CA-DS 1.8. As mentioned before, family II enzymes showed regioselectivity for deacetylation. These three enzymes (AXE 55, AXE 53, GAE) had the highest deacetylation activity. A possible explanation might be given by the distribution of the acetyl groups across the polymer. Hence, for enzymes, cellulose acetates with different degrees of substitutions pose to be substrates with different properties. Based on the conserved motive, AXE 55, AXE 53, and GAE are representatives of the so-called GDSL-family or family II, and all esterases within lipolytic enzymes can be classified into the thirteen families [25]. This family shares five highly-conserved homology blocks, which are important for their classification. GDSL hydrolases have a flexible active site and they can change conformation in the presence of different substrates [26]. This flexibility might be an explanation for their good deacetylation efficiency over a broad range of substrates. Enzymes of this family were also reported to show broad regioselectivity, which probably makes them suitable for degradation of CAs with a heterogeneous acetylation pattern [27].
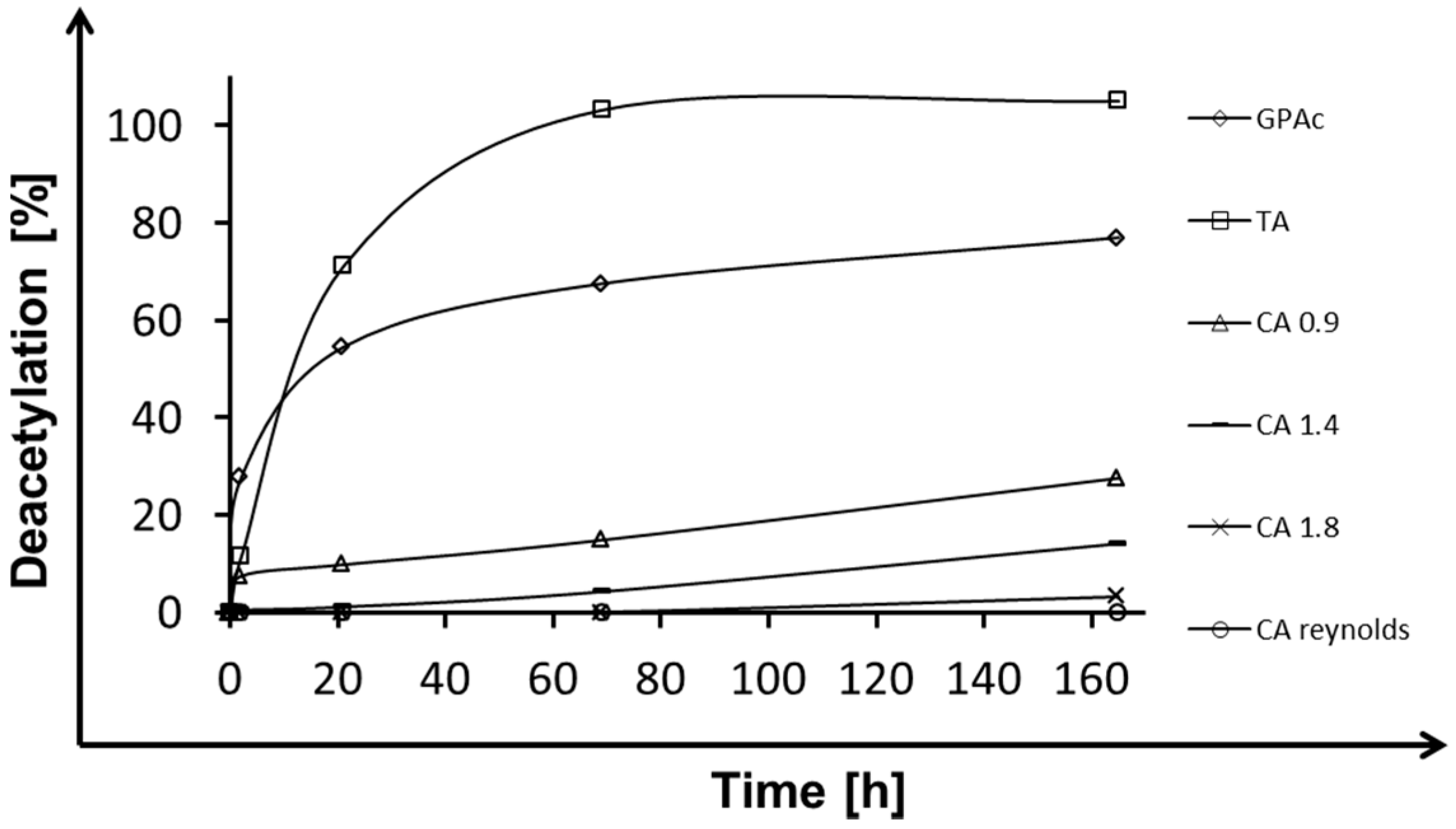
Despite investigations for triacetin and glucose pentaacetate (Figure 2), for PAE deacetylation was examined over time by using different cellulose acetate model compounds varying in degree of substitution (CA-DS 0.9, CA-DS 1.4, CA-DS 1.8) and real cigarette filter material (R. J. Reynolds Tobacco Company, Winston-Salem, NC, USA). Figure 4 compares deacetylation efficiencies for cellulose acetates over time. For the used model compounds, deacetylation decreased from 20% for low-acetylated substrate (CA 0.9) to 5% for highly-acetylated material (CA 1.8) after 165 h of incubation. Real filter material with a degree of substitution of 2.5 revealed a high resistance against enzymatic degradation with PAE, resulting in no detectable release of acetic acid. As shown in Figure 2 for glucose pentaacetate and triacetin, PAE exhibited high and medium ability to degrade small acetylated substrates, whereas larger substrates, like different cellulose acetates (Figure 4), were less deacetylated even when the DS was lower. For the substrates reported in this study, the polymer size influenced the deacetylation efficiency of PAE to a great extent. The highest deacetylation efficiency was visible for CA-DS 0.9 within the first 2 h. This can be explained by auto-degradation of the polymer in combination with the enzyme action. For all other conditions, deacetylation was linear for the whole reaction time course.
The results of enzymatic deacetylation of a variety of substrates with different degrees of acetylation are summarized in Table 2. None of the enzymes were able to deacetylate CA with a degree of substitution higher than 1.8. The most promising enzymes for the degradation of large and highly substituted polymers were of family II (AXE 55, AXE 53, GAE). Small- and highly-acetylated molecules, such as cellobiose octaacetate and cellohexose eicosaacetate, were less deacetylated than larger molecules with less acetyl groups per monomer. An increase in the number of glucose subunits from one (glucose pentaacetate) to six (cellohexaose eicosaacetate) strongly decreased deacetylation efficiency for all enzymes except for PAE and GAE (compare Figure 4 and Table 2). For example, cellohexaose eicosaacetate was less deacetylated than glucose pentaacetate with a higher degree of substitution. Hence, it seems that for shorter molecules the degree of acetylation has a lower impact on deacetylation efficiency than the chain length.
2.2. Combined CA Degradation with Esterases and Cellulases
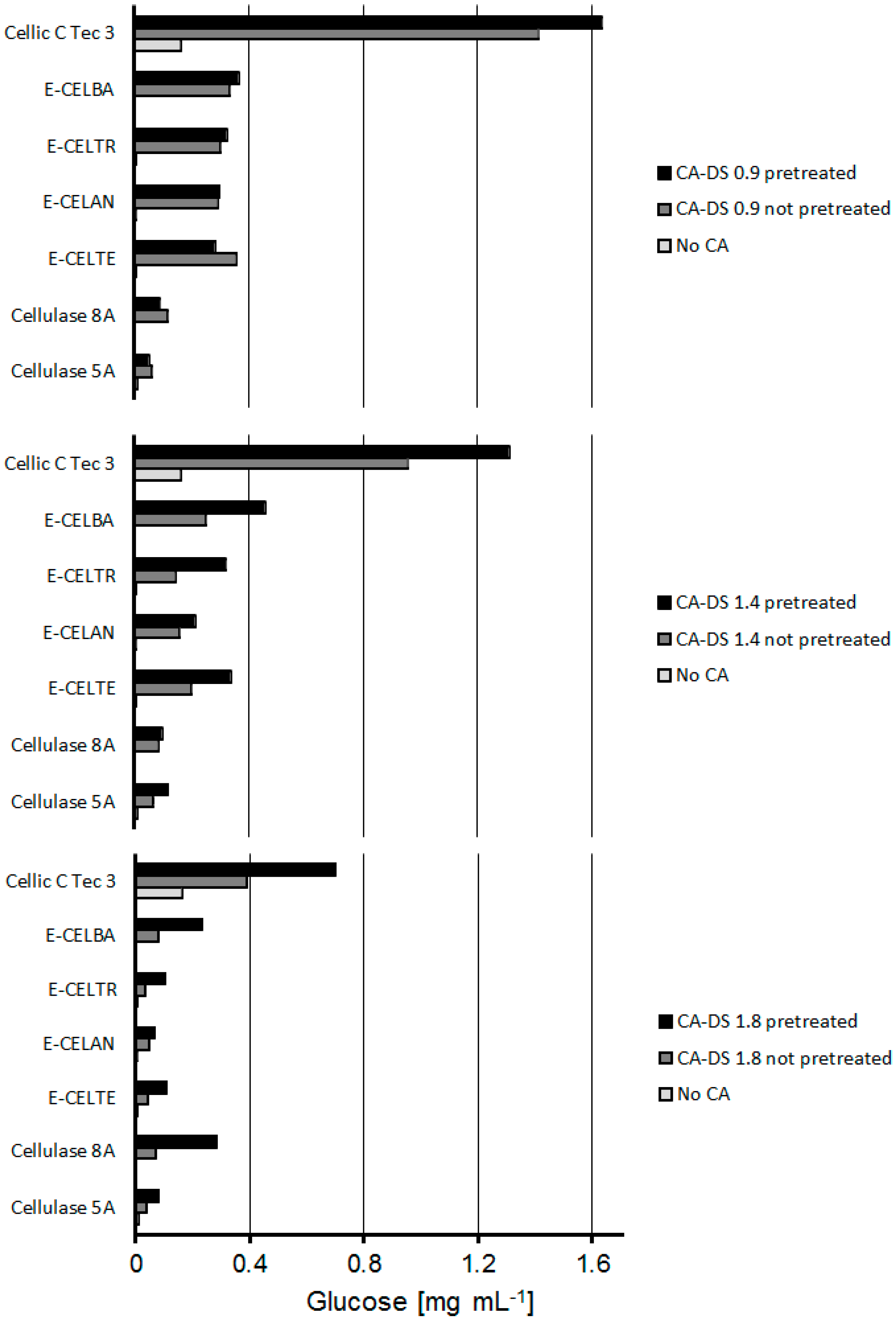
To achieve full cellulose acetate degradation, the ability of different cellulases to hydrolyze esterase-pretreated CA was investigated. The synergistic mechanism of esterases and cellulases has previously been reported for cellulose acetate degradation in microbial systems [13]. Moriyoshi et al. isolated different enzymes out of Neisseria sicca performing a synergistic reaction of deacetylation and degradation [28]. Figure 5 shows the differences in glucose release for a wide spectrum of cellulases preparations applied on pretreated and not-pretreated cellulose acetates with different degrees of substitution (DS). Only cellulase 8A and E-CELBA did not release glucose. For all cellulases, the glucose release increased upon prior deacetylation with esterases. The best working enzyme approach for pretreated and non-pretreated samples was Cellic C Tec 3. Multiple enzyme activities like LPMO, endoglucanases, exoglucanases, and cellobiohydrolases are involved in the cellulose degradation in nature and, hence, also contained in commercial cellulase preparations [29,30]. This makes a broad screening of enzymes for a possible application necessary. The most pronounced difference between pretreatment and no pretreatment was seen for CA-DS 1.8. Here, deacetylation increased the glucose liberation by almost 50%, confirming the “protective” effect of acetyl groups towards enzymatic hydrolysis of cellulosic materials. Irrespective of the pretreatment, the glucose release decreased with increasing acetylation for all enzymes (Table 2).
The glucose recovery halved when the degree of acetylation increases from 0.9 to 1.8. Table 2 indicates that the protective function of acetyl groups is not linear with the degree of substitution. Increasing the acetyl content from 0.9 to 1.4 had fewer effects than an increment from 1.4 to 1.8.
2.3. Lytic Polysaccharide Monooxygenase (LPMO) Hydrolysis of CA
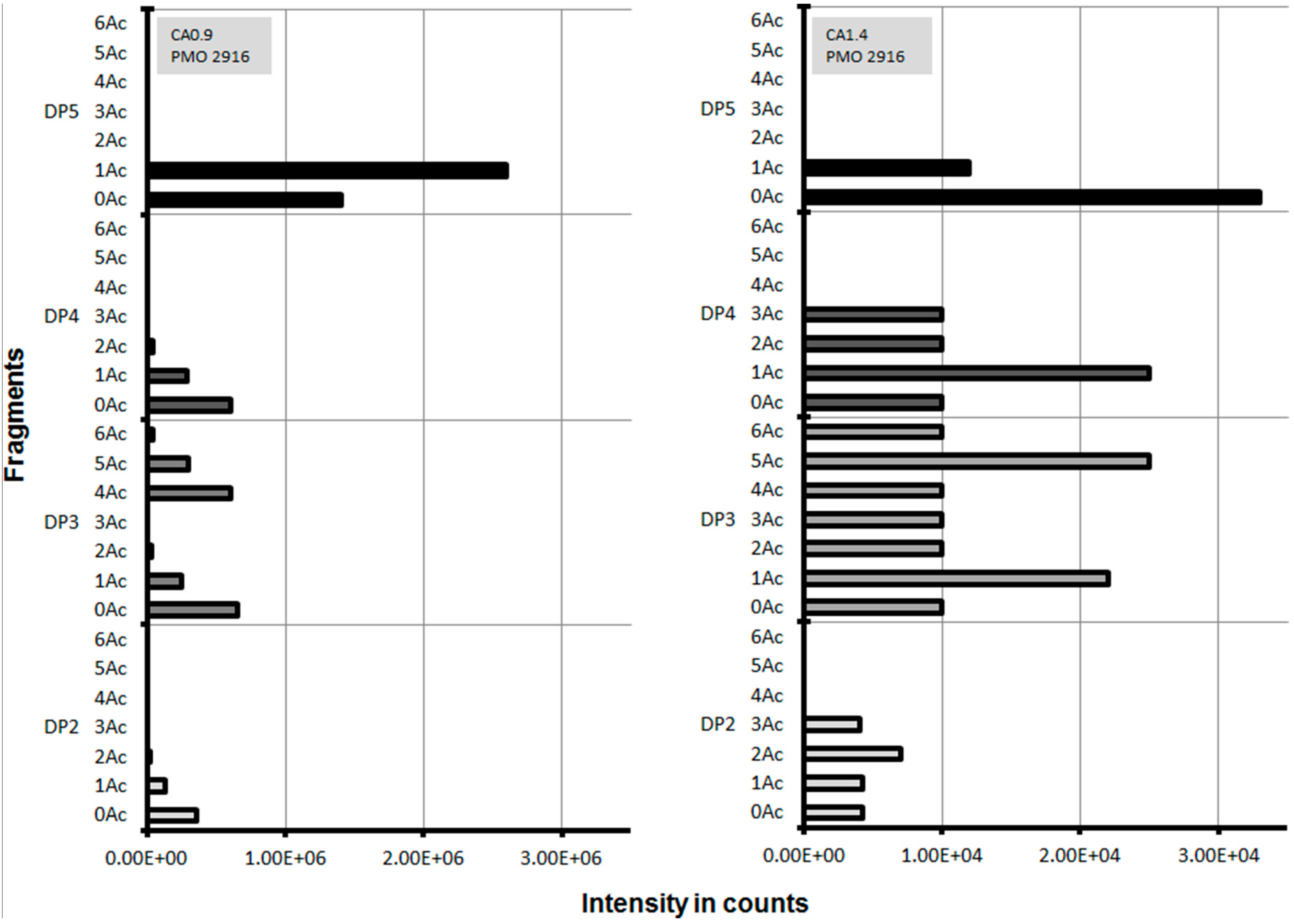
Since the degree of enzymatic deacetylation decreased with increasing polymer chain length, it was speculated that cleavage of the polymer into smaller fragments prior to deacetylation increases CA degradation. Lytic polysaccharide monooxygenases (LPMOs) are known to attack cellulose by an oxidative mechanism, which cleaves the glycoside bond at either the C1 or the C4 [29]. An interesting feature of LPMO is its flat substrate binding surface, which fits onto the cellulose surfaces and brings the active-site copper with an activated oxygen species into close contact with the species [30]. This enzyme can theoretically bind at any position to cellulose to perform cleavage. Some LPMOs like LPMO-02916 (also known as LPMO 9C) from Neurospora crassa can also act on the less-structured substrates, hemicellulose and cellooligosaccharides [31,32], which suggests that LPMO-02916 might also be suitable to act on CA. In experiments where LPMO-02916 was incubated with a reductant and cellulose (PASC, Avicel, or steam-exploded spruce), fragments between two and five monomers were detected [31]. CA with a DS of 0.9 and 1.4 was incubated separately with LPMO-02916 and cleavage fragments were analyzed. For CA with a DS of 0.9 after incubation with LPMO no fragments larger than five monomers and more than one acetyl group were detected. For CA 0.9, the number of different fragments was decreasing for increasing amount of acetyl groups. LPMO liberated fragments for both CA with DS 0.9 and 1.4 (Figure 6), while no fragments were detected for CA with higher DS. Preferentially, fragments with a low DS were released. This indicates that acetyl groups interfere with LPMO’s substrate binding site or disturb the catalytic reaction. It also indicates that CA may not be uniformly acetylated, allowing the LPMO to cleave in those regions with a lower DS. In this context synergies of LPMOs in combination with other cellulose degrading enzymes were observed and are worth of further investigations [33]. Cleavage of CA with DS 0.9 and 1.4 by LPMO was investigated by analyzing the released fragments (Figure 6).
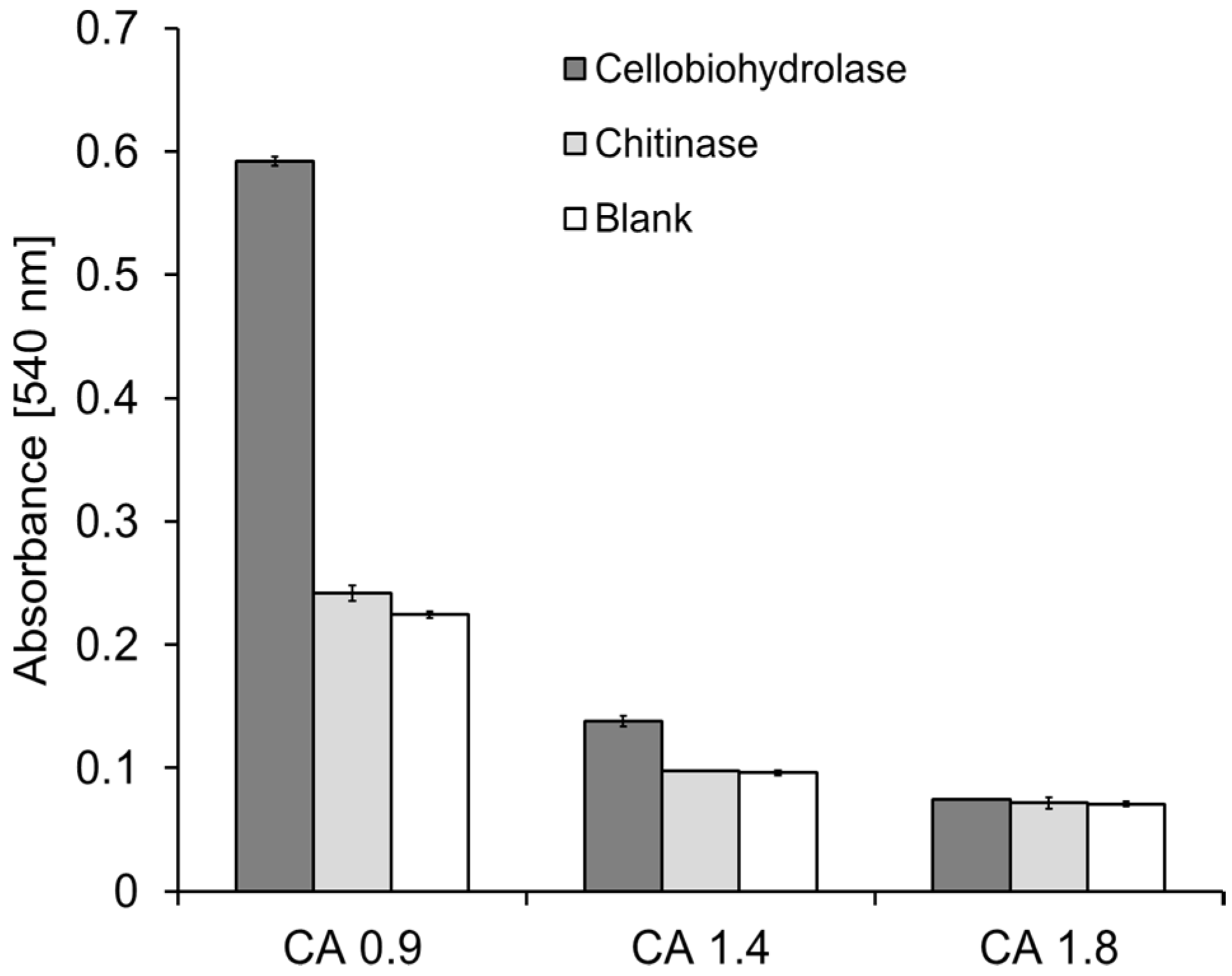
Figure 7 shows backbone cleavage by cellobiohydrolase I and chitinase after 145 h of reaction. Cleavage activity was monitored photometrically by detection of the reducing sugar ends using the dinitrosalicylic acid (DNS) method [32]. For cellobiohydrolase I, the amount of reducing ends is increased by a factor of three for low-substituted material (CA-DS 0.9). Minor increases are visible for medium-substituted substrates (DS 1.4). No cleavage activity was measured for highly-substituted cellulose acetate (CA-DS 1.8). As visible for the deacetylation with esterases (Figure 3), the decrease in activity, for highly-acetylated substrates, is also not linear for glycosidic-acting enzymes in backbone cleavage, with the substrates used here. Cellobiohydrolase I is an enzyme with broad product specificity, reported to bind only the hydrophobic parts of the cellulose crystal structure [34]. Ike et al. reported chitinase activity for cellobiohydrolase I [35] and Textor et al. mentioned the cleavage of carboxymethyl cellulose by the enzyme. This presence of a cellulose binding module (CBM) and low end-product inhibition promise applicability of cellobiohydrolase I in industrial degradation processes [36]. The ability of cellobiohydrolases to decrease the degree of polymerization was reported before by Saake et al. for low- and medium-acetylated substrates, based on size exclusion chromatography of acetylated cellulose [37].
Due to the chemical similarities of the polymers chitin and cellulose acetate, chitinase was tested on its ability to cleave glycosidic bonds in different cellulose acetates. Chitinase only showed small changes for CA-DS 0.9 and no shift in absorbance for other substrates. The N-acetamide group seems to be essential for the ability of chitinase to detect glycosidic bonds. The binding ability for a chitinase on chitin and cellulose was determined to be equal for both polymers [38]. The effects of reduced binding affinity to substrates with at least one acetyl group per monomer might be the reason for reduced backbone cleavage of cellobiohydrolase for CA 1.4 and CA 1.8.
3. Materials and Methods
3.1. Enzymes, Substrates, and Other Chemicals
Enzymatic deacetylation and CA backbone cleavage was investigated using different model substrates, like triacetin, glucose pentaacetate, cellobiose octaacetate, cellohexaose eicosaacetate, CA with different DS (0.9, 1.4, 1.8, 2.3, and 2.5), and CA cigarette filters were from R. J. Reynolds Tobacco Company (Winston-Salem, NC, USA) with a DS of 2.5. CA model compounds with different DS (0.9, 1.4, 1.8, 2.3, and 2.5) and chain lengths were prepared by acid deacetylation of cellulose triacetate, as previously described [39]. All other substrates were purchased from Sigma-Aldrich (Vienna, Austria) and used as received if not otherwise specified. Esterases were obtained from Nzytech (Lisbon, Portugal) (GAE, PAE), Megazyme (Wicklow, Ireland) (AXE O), and Prozomix (Haltwhistle, UK), and used without further purification, whereas cutinase from Thermobifida cellulosilytica was produced and purified as previously described [40]. Additionally, chemical hydrolysis with 0.1 M NaOH was carried out for 24 h using 100 mg of each substrate. Table 3 shows all used esterases for the screening, including the optimal reaction conditions and enzyme family.
Cellulases used for combinatorial experiments with esterases were purchased from Megazyme, Ireland (E-CELAN, E-CELTR, E-CELTE, E-CELBA), Nzytech, Portugal (Cellulase 8A, Cellulase 5A) and Novozymes, Denmark (Cellic C Tec 3). Table 4 shows all used cellulases for the screening, including optimal reaction conditions and enzyme family.
Chitinase and Cellobiohydrolase were supplied by Sigma-Aldrich (Vienna, Austria) and Nzytech (Lisbon, Portugal), respectively, and were used without further purification. LPMO from Neurospora crassa PMO 2916 [41] was used for the direct CA backbone cleavage.
3.2. Deacetylation of CA and Acetylated Oligomer
For the investigation of enzymatic deacetylation, 100 mg of each substrate were used, except for CA with a DS of 0.9, 1.4, and 1.8, of which only 50 mg were weighted. The enzymatic hydrolysis was carried out in 0.1 M sodium phosphate buffer using the optimum pH for each enzyme, using 20 (with 50 mg substrate) or 50 mL (with 100 mg substrate) as reaction volume. The dosage of each enzyme was 0.1 U·mg−1 substrate (with 1 unit defined as the µmol of substrate converted in 1 min by the enzyme). The samples were incubated at their respective optimum temperature and samples were taken at certain time points.
3.3. Combined Deacetylation and Hydrolysis of CA
For deacetylation, CA was pretreated with the best performing esterase, which was AXE 55. CA concentration was 7.54 mg·mL−1 in 20 mM Tris-HCl buffer at pH 8.5 for the esterase treatment. Enzyme concentration for the pretreatment was 34 µg·mL−1. Blanks were performed without esterase. All samples were incubated for 165 h. After the pretreatment, the pH was changed for all cellulase treatments. Therefore, 100 mM buffer with the right pH was added. Change of pH to 4.5 and 5 was done by the addition of citrate buffer, and pH 6.5 and 7.5 were obtained using sodium phosphate buffer. The final CA concentration was 3.77 mg·mL−1. For the different cellulases, the protein content was determined and between 0.01 and 1 mg enzyme was added. All samples were incubated for 168 h at the optimal temperature.
3.4. Direct Enzymatic Hydrolysis of CA
For all experiments, 5 mg of the different substrates were used. Conversion of CA by LPMO was carried out in 975 µL sodium phosphate buffer (50 mM, pH 6) with 20 µL of 10 mM gallic acid and 5 µL enzyme, (with an initial concentration of 60 mg·mL−1). Samples were incubated at 25 °C and 900 rpm in the dark. To ensure appropriate oxygen supply, samples were covered with an O2 permeable foil. Experiments containing chitinase (EC 3.2.1.14) and cellobiohydrolase (EC 3.2.1.91) were performed in a 1.5 mL total reaction volume containing buffer (50 mM citrate) and 10 µL of the enzyme solution. Optimal pH values were 6.5 for chitinase and 5.0 for cellobiohydrolase. Ten microliters (10 µL) of the enzyme were used (corresponding to 250 U for chitinase and 13 U for cellobiohydrolase). Samples were incubated for 145 h and 200 rpm at the temperature optimum (cellobiohydrolase: 50 °C, chitinase 60 °C).
3.5. Monitoring Deacetylation of CA and Oligomers
Deacetylation was monitored using a high-performance liquid chromatography (HPLC) system equipped with a transgenomic ION-300 column (New Haven, CT, USA). H2SO4 (0.01 N) was used as the mobile phase with a flow rate of 0.325 mL·min−1 at 45 °C. Forty microliters (40 µL) of the desired solution were injected at a runtime of 60 min. The method was calibrated using acetic acid standards within a 10–1000 mg·L−1 range. In order to render the deacetylation results for substrates with different degrees of substitution comparable, all values were converted into percentage values.
3.6. Monitoring Enzymatic Cleavage of CA
Backbone cleavage resulting in the formation of reducing sugars was monitored using the DNS method as previously reported by Ghose [33] with slight modifications. Furthermore, released CA fragments were analyzed by liquid chromatography electronspray ionization time-of-flight mass spectroscopy (LC-ESI-TOF MS), injecting 30 µL samples, using a Poroshell 120 EC-C18 (Agilent, Santa Clara, CA, USA) column at 40 °C with a flow rate of 0.4 mL·min−1. The system was operated with a linear gradient of 100% water containing 0.1% formic acid to 100% acetonitrile with 0.1% formic acid in 45 min. For mass spectrometry, a Dual ESI G6230B TOF (Agilent) was used. The sample was ionized with a nebulizer at 40 psig and positive ion mode with a gas temperature of 200 °C at 8 L·min−1 gas flow. The fragmentor voltage in the system was set to 200 V and the skimmer voltage was 65 V for mass correction the masses 121.0509 m/z and 922.0098 m/z. Mass range for the analysis was from 50 to 3000 m/z. The acquired data were analyzed with the Agilent MassHunter software (Version B07.00).
Samples treated with a combination of esterases and cellulases were analyzed by HPLC monitoring the glucose release for pretreated and non-pretreated samples. Forty microliters (40 µL) of sample were injected and the mobile phase was 0.01 N sulfuric acid with a flow rate of 0.325 mL·min−1. The column temperature was 45 °C using a transgenomic ION-300 column (New Haven, CT, USA). Signal recording was achieved by refractive index measurement using an Agilent 1260 Infinity II detector. The method was calibrated using glucose standards between 10 and 1000 mg·L−1.
4. Conclusions
Here we show an extensive screening of many representatives of different classes of enzymes, which are all involved in the cellulase acetate degradation, either on the acetyl group of the polymer or on the backbone of the chain. The behavior of different substrates was investigated. To our knowledge the synergistic performance of esterases and cellulases in combination, outside of a microbial system, was described for the first time. Additionally we introduced a new enzyme, which is called lytic polysscharide monooxygenase, to the collection of promising biocatalysts for CA degradation.
Most of the investigated esterases were able to completely deacetylate fully-acetylated glucose pentaacetate and triacetin. However, the ability of the deacetylation by esterases decreased with increasing DS. CAs with a DS up to 1.8 were deacetylated to various extents. Esterases were not able to deacetylate cellulose acetates with higher DS than 1.8. Experiments with small model compounds showed that increasing the chain length and degree of acetylation negatively affected the ability of esterases to deacetylate. A combination of esterases with cellulases increased the glucose recovery from cellulose acetate. Furthermore, a hydrolytic enzyme (LPMO) randomly cleaved low-acetylated substrates into short fragments with at least one non-acetylated monomer. In summary, combinations of backbone cleaving and deacetylating enzymes can enhance CA degradation up to a maximum DS of 1.8.
Acknowledgments
This work has been supported by the Federal Ministry of Science, Research, and Economy (BMWFW), the Federal Ministry of Traffic, Innovation, and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol, and the Government of Lower Austria and Business Agency Vienna through the COMET-Funding Program managed by the Austrian Research Promotion Agency FFG. The authors are grateful for the financial assistance and filter material provided by the R. J. Reynolds Tobacco Company (Winston-Salem, NC, USA).
Author Contributions
B.S. and A.S. conceived and designed the experiments; O.H.-C. and S.W. performed the experiments; O.H.-C., A.P., and G.T. analyzed the data; R.L. expressed and purified the CBH enzyme; O.H.C. and A.P. prepared and formatted the graphs and the manuscript; and O.H.-C., G.S.N., and G.M.G. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Quintana, R.; Persenaire, O.; Lemmouchi, Y.; Bonnaud, L.; Dubois, P. Compatibilization of Co-Plasticized Cellulose Acetate/water Soluble Polymers Blends by Reactive Extrusion. Polym. Degrad. Stabil. 2016, 126, 31–38. [Google Scholar] [CrossRef]
- Rosa, D.S.G.; Uedes, C.G.F.; Casarin, F.; Braganca, F.C. The Effect of the Mw of PEG in PCL/CA Blends. Polym. Test. 2005, 24, 542–548. [Google Scholar] [CrossRef]
- Daud, W.W.R.; Djuned, F.M. Cellulose Acetate from Oil Palm Empty Fruit Bunch via a One Step Heterogeneous Acetylation. Carbohydr. Polym. 2015, 132, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Puls, J.; Wilson, S.A.; Hölter, D. Degradation of Cellulose Acetate-Based Materials: A Review. J. Polym. Environ. 2011, 19, 152–165. [Google Scholar] [CrossRef]
- Novotny, T.E.; Lum, K.; Smith, E.; Wang, V.; Barnes, R. Cigarettes Butts and the Case for an Environmental Policy on Hazardous Cigarette Waste. Int. J. Envrion. Res. Public Health 2009, 6, 1691–1705. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.M.; Thomas, W.C.; Suthar, J.N.; Brown, D.M. Accelerated Degradation of Cellulose Acetate Cigarette Filters Using Controlled-Release Acid Catalysis. Green Chem. 2012, 14, 2266–2272. [Google Scholar] [CrossRef]
- Buchanan, C.M.; Gardner, R.M.; Komarek, R. Aerobic Biodegradation of Cellulose Acetate. Appl. Polym. 1972, 47, 1709–1719. [Google Scholar] [CrossRef]
- Hon, N. Photodegradation of Cellulose Acetate Fibers. Polym. Chem. 1977, 15, 725–744. [Google Scholar] [CrossRef]
- Sternberg, R.; Bindra, D.S.; Wilson, G.S.; Thévenot, D.R. Covalent Enzyme Coupling on Cellulose Acetate Membranes for Glucose Sensor Developmen. Anal. Chem. 1988, 12, 2781–2786. [Google Scholar] [CrossRef] [Green Version]
- Yudanova, T.N.; Skokova, I.F.; Gal, L.S. Fabrication of Biologically Active Fibre Materilas with Predetermined Properties. Fibre Chem. 2000, 6, 411–413. [Google Scholar] [CrossRef]
- Biely, P. Microbial Carbohydrate Esterases Deacetylating Plant Polysaccharides. Biotechnol. Adv. 2012, 30, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Abrusci, C.; Marquina, D.; Santos, A.; Del Amo, A.; Corrales, T.; Catalina, F. Biodeterioration of Cinematographic Cellulose Triacetate by Sphingomonas Paucimobilis Using Indirect Impedance and Chemiluminescence Techniques. Int. Biodeterior. Biodegrad. 2009, 63, 759–764. [Google Scholar] [CrossRef]
- Sakai, K.; Yamauchi, T.; Nakasu, F.; Ohe, T. Biodegradation of Cellulose Acetate by Neisseria sicca. Biosci. Biotechnol. Biochem. 1996, 60, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Gilkes, N.; Saddler, J.N. Effect of Acetyl Groups on Enzymatic Hydrolysis of Cellulosic Substrates. Int. J. Biol. Chem. Phys. Technol. Wood 2006, 60, 398–401. [Google Scholar] [CrossRef]
- Olaru, L.; Olaru, N.; Popa, V.I. On Enzymatic Degradation of Cellulose Acetate. Iran. Polym. J. 2004, 13, 235–240. [Google Scholar]
- Crawford, R.R.; Esmerian, O.K. Effect of Plasticizers on Some Physical Properties of Cellulose Acetate Phthalate Films. J. Pharm. Sci. 1971, 60, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Perz, V.; Bleymaier, K.; Sinkel, C.; Kueper, U.; Bonnekessel, M.; Ribitsch, D.; Guebitz, G.M. Substrate Specificities of Cutinases on Aliphatic-Aromatic Polyesters and on Their Model Substrates. N. Biotechnol. 2016, 33, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Matamá, T.; Casal, M.; Cavaco-Paulo, A. Direct Enzymatic Esterification of Cotton and Avicel with Wild-Type and Engineered Cutinases. Cellulose 2013, 20, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Morley, K.L.; Chauve, G.; Kazlauskas, R.; Dupont, C.; Shareck, F.; Marchessault, R.H. Acetyl Xylan Esterase-Catalyzed Deacetylation of Chitin and Chitosan. Carbohydr. Polym. 2006, 63, 310–315. [Google Scholar] [CrossRef]
- Krastanova, I.; Guarnaccia, C.; Zahariev, S.; Degrassi, G.; Lamba, D. Heterologous Expression, Purification, Crystallization, X-ray Analysis and Phasing of the Acetyl Xylan Esterase from Bacillus Pumilus. Biochim. Biophys. Acta 2005, 1748, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Altaner, C.; Saake, B.; Tenkanen, M.; Eyzaquirre, J.; Faulds, C.B.; Biely, P.; Viikari, L.; Siika-aho, M.; Puls, J. Regioselective Deacetylation of Cellulose Acetates by Acetyl Xylan Esterases of Different CE-Families. J. Biotechnol. 2003, 105, 95–104. [Google Scholar] [CrossRef]
- Willför, S.; Sundberg, K.; Tenkanen, M.; Holmbom, B. Spruce-Derived Mannans—A Potential Raw Material for Hydrocolloids and Novel Advanced Natural Materials. Carbohydr. Polym. 2008, 72, 197–210. [Google Scholar] [CrossRef]
- Xu, C.; Leppanen, A.-S.; Eklund, P.; Holmlund, P.; Sjoholm, R.; Sundberg, K.; Willfor, S. Acetylation and Characterization of Spruce (Picea abies) Galactoglucomannans. Carbohydr. Res. 2010, 345, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Poutanen, K.; Sundberg, M.; Korte, H.; Puls, J. Deacetylation of Xylans by Acetyl Esterases of Trichoderma Reesei. Appl. Microbiol. Biotechnol. 1990, 33, 506–510. [Google Scholar] [CrossRef]
- Rao, L.; Xue, Y.; Zhou, C.; Tao, J.; Li, G.; Lu, G.R.; Ma, Y. A Thermostable Esterase from Thermoanaerobacter Tengcongensis Opening up a New Family of Bacterial Lipolytic Enzymes. Biochim. Biophys. Acta 2011, 1814, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Chepyshko, H.; Lai, C.-P.; Huang, L.-M.; Liu, J.-H.; Shaw, J.-F. Multifunctionality and Diversity of GDSL Esterase/lipase Gene Family in Rice (Oryza sativa L. Japonica) Genome: New Insights from Bioinformatics Analysis. BMC Genom. 2012, 13, 309. [Google Scholar] [CrossRef] [PubMed]
- Akoh, C.C.; Lee, G.C.; Liaw, Y.C.; Huang, T.H.; Shaw, J.F. GDSL Family of Serine Esterases/lipases. Prog. Lipid Res. 2004, 43, 534–552. [Google Scholar] [CrossRef] [PubMed]
- Moriyoshi, K.; Koma, D.; Yamanaka, H.; Sakai, K.; Ohmoto, T. Expression and Characterization of a Thermostable Acetylxylan Esterase from Caldanaerobacter subterraneus Subsp. Tengcongensis Involved in the Degradation of Insoluble Cellulose Acetate. Biosci. Biotechnol. Biochem. 2013, 77, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Frandsen, K.E.H.; Simmons, T.J.; Dupree, P.; Poulsen, J.-C.N.; Hemsworth, J.-C.N.; Ciano, L.; Johnston, E.M.; Tovborg, M.; Johansen, K.S.; von Freiesleben, P.; et al. The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat. Chem. Biol. 2016, 12, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Dimarogona, M.; Topakas, E.; Christakopoulos, P. Cellulose degradation by oxidative enzymes. Comput. Struct. Biotechnol. J. 2012, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Isaksen, T.; Westereng, B.; Aachmann, F.L.; Agger, J.W.; Kracher, D.; Kittl, R.; Ludwig, R.; Haltrich, D.; Eijsink, V.G.H.; Horn, S.J. A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 2014, 289, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- Žifčáková, L.; Baldrian, P. Fungal polysaccharide monooxygenases: New players in the decomposition of cellulose. Fungal Ecol. 2012, 5, 481–489. [Google Scholar] [CrossRef]
- Ghose, T.K. International Union of Pure Commission on Biotechnology. Measurement of cellulase activity. Pure Appl. Chem. 1987, 59, 257–268. [Google Scholar] [CrossRef]
- Liu, Y.S.; Baker, J.O.; Zeng, Y.; Himmel, M.E.; Haas, T.; Ding, S.Y. Cellobiohydrolase Hydrolyzes Crystalline Cellulose on Hydrophobic Faces. J. Biol. Chem. 2011, 286, 11195–11201. [Google Scholar] [CrossRef] [PubMed]
- Ike, M.; Ko, Y.; Yokohama, K.; Sumitani, J.-I.; Kawaguchi, T.; Ogasawara, W.; Okada, H.; Morikawa, Y. Cellobiohydrolase I (Cel7A) from Trichoderma Reesei Has Chitosanase Activity. J. Mol. Catal. B Enzym. 2007, 47, 159–163. [Google Scholar] [CrossRef]
- Textor, L.C.; Colussi, F.; Silveira, R.L.; Serpa, V.; de Mello, B.L.; Muniz, J.R.; Squina, F.M.; Pereira, N., Jr.; Skaf, M.S.; Polikarpov, I. Joint X-ray Crystallographic and Molecular Dynamics Study of Cellobiohydrolase I from Trichoderma Harzianum: Deciphering the Structural Features of Cellobiohydrolase Catalytic Activity. FEBS J. 2013, 280, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Saake, B.; Horner, S.; Puls, J. Progress in the Enzymatic Hydrolysis of Cellulose Derivatives. Cellul. Deriv. 1998, 15, 201–216. [Google Scholar] [CrossRef]
- Kikkawa, Y.; Fukuda, M.; Kashiwada, A.; Matsuda, K.; Kanesato, M.; Wada, M.; Imanaka, T.; Tanaka, T. Binding Ability of Chitinase onto Cellulose: An Atomic Force Microscopy Study. Polym. J. 2011, 43, 742–744. [Google Scholar] [CrossRef]
- Lee, S.-J.; Altaner, C.; Puls, J.; Saake, B. Determination of the Substituent Distribution along Cellulose Acetate Chains as Revealed by Enzymatic and Chemical Methods. Carbohydr. Polym. 2003, 54, 353–362. [Google Scholar] [CrossRef]
- Pellis, A.; Haernvall, K.; Pichler, C.M.; Ghazaryan, G.; Breinbauer, R.; Guebitz, G.M. Enzymatic Hydrolysis of Poly(ethylene Furanoate). J. Biotechnol. 2015, 235, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Kittl, R.; Kracher, D.; Burgstaller, D.; Haltrich, D.; Ludwig, R. Production of Four Neurospora Crassa Lytic Polysaccharide Monooxygenases in Pichia Pastoris Monitored by a Fluorimetric Assay. Biotechnol. Biofuels 2012, 5, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Substrates for enzymatic CA degradation. Triacetin (A); glucose pentaacetate (B); cellobiose octaacetate (C); hellohexose eicosacetate (D); and CA (E). For CA: R refers for the degree of substitution and n + 2 is the degree of polymerization.
Figure 1.
Substrates for enzymatic CA degradation. Triacetin (A); glucose pentaacetate (B); cellobiose octaacetate (C); hellohexose eicosacetate (D); and CA (E). For CA: R refers for the degree of substitution and n + 2 is the degree of polymerization.
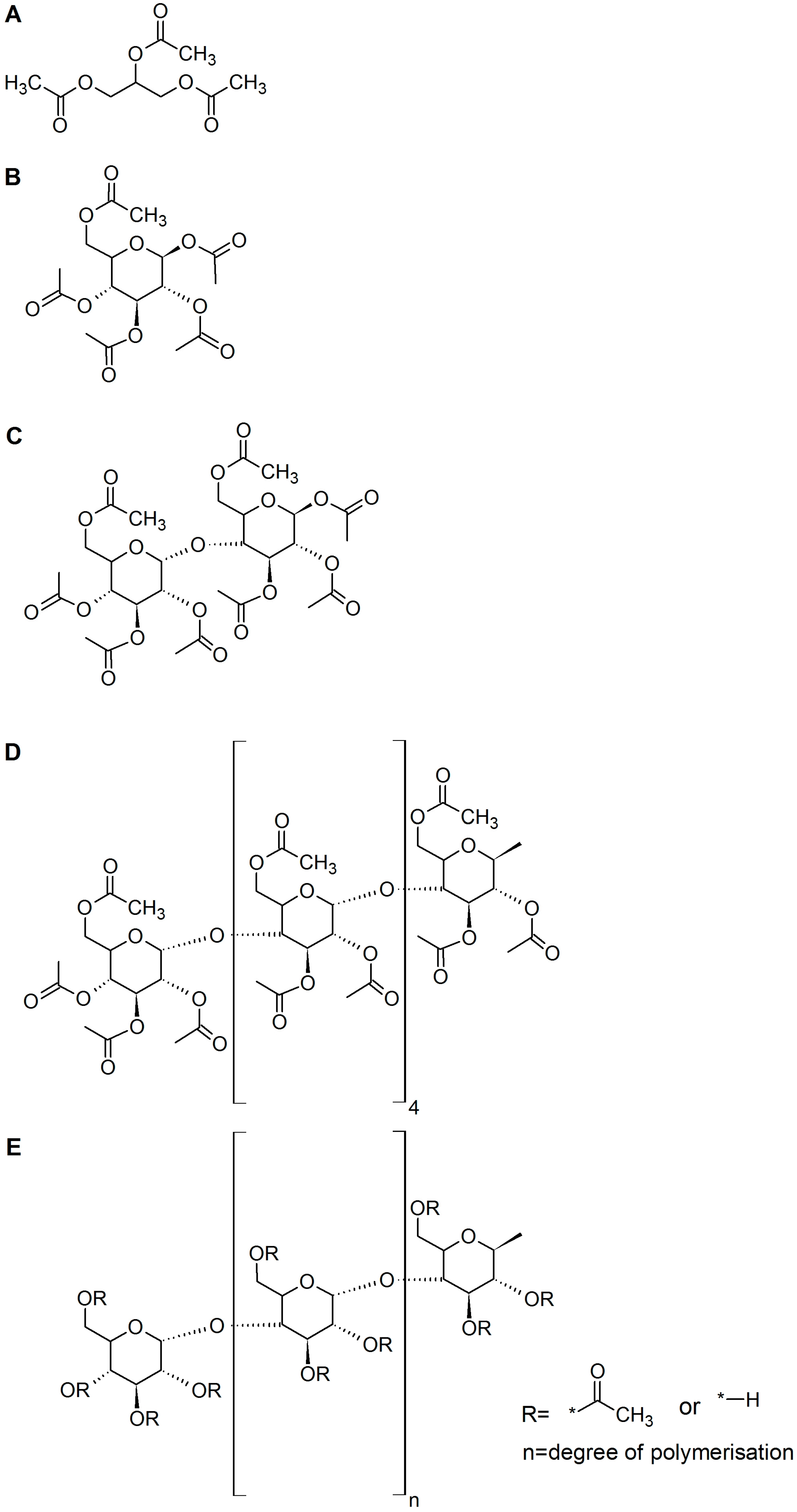
Figure 2.
Deacetylation of glucose pentaacetate and triacetin by different esterases. Triacetin is a plasticizer in cigarette filters and glucose pentaacetate serves as small model compound for CA. Axetyl xylan estarase (AXE); carbohydrate esterase (CE); cutinase (CUT); glucomannan acetyl esterase (GAE); and pectin acetyl esterase (PAE).
Figure 2.
Deacetylation of glucose pentaacetate and triacetin by different esterases. Triacetin is a plasticizer in cigarette filters and glucose pentaacetate serves as small model compound for CA. Axetyl xylan estarase (AXE); carbohydrate esterase (CE); cutinase (CUT); glucomannan acetyl esterase (GAE); and pectin acetyl esterase (PAE).
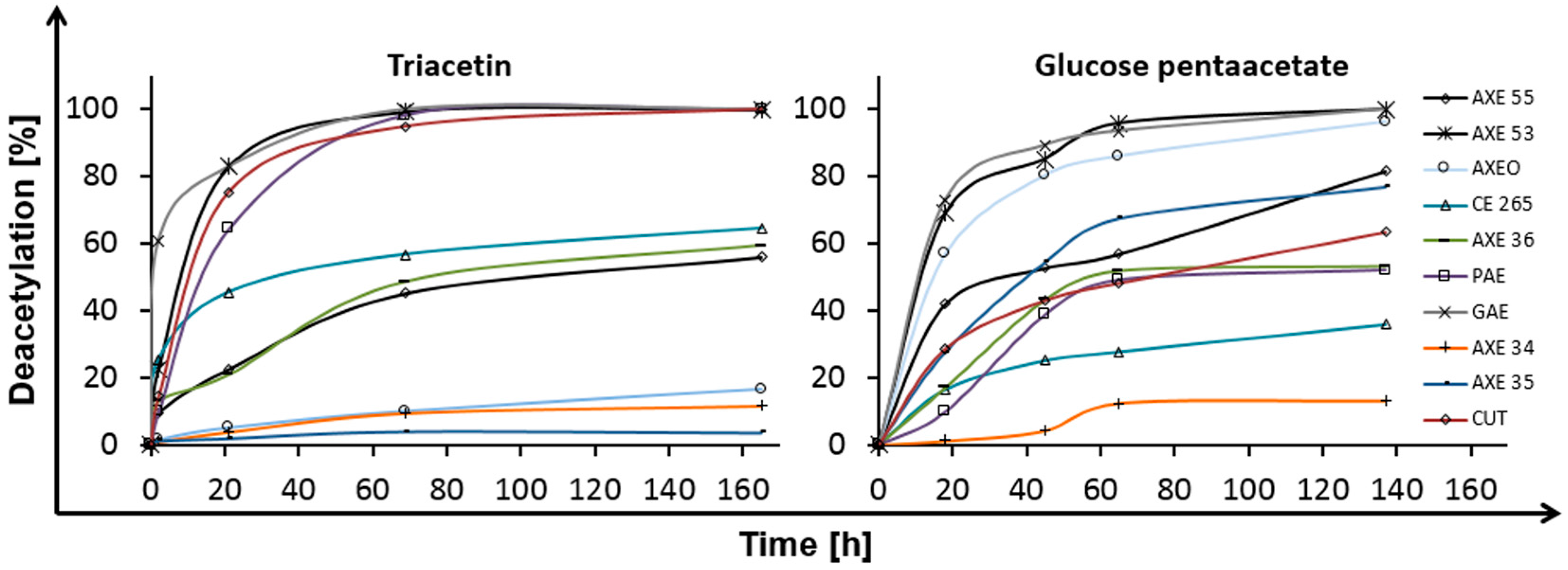
Figure 3.
Deacetylation of cellulose acetate with different degree of substitution (DS) and model compounds by pectin acetyl esterase (PAE) after 168 h of incubation. Glucose pentaacetate (GPA); Triacetin (TA)
Figure 3.
Deacetylation of cellulose acetate with different degree of substitution (DS) and model compounds by pectin acetyl esterase (PAE) after 168 h of incubation. Glucose pentaacetate (GPA); Triacetin (TA)

Figure 4.
Deacetylation of different cellulose acetates by esterases. Cellulose acetate with 0.9 acetyl groups per monomer (CA-DS 0.9), cellulose acetate with 1.4 acetyl groups per monomer (CA-DS 1.4), and cellulose acetate with 1.8 acetyl groups per monomer (CA-DS 1.8). Axetyl Xylan Estarase (AXE); carbohydrate esterase (CE); cutinase (CUT); glucomannan acetyl esterase (GAE); pectin acetyl esterase (PAE).
Figure 4.
Deacetylation of different cellulose acetates by esterases. Cellulose acetate with 0.9 acetyl groups per monomer (CA-DS 0.9), cellulose acetate with 1.4 acetyl groups per monomer (CA-DS 1.4), and cellulose acetate with 1.8 acetyl groups per monomer (CA-DS 1.8). Axetyl Xylan Estarase (AXE); carbohydrate esterase (CE); cutinase (CUT); glucomannan acetyl esterase (GAE); pectin acetyl esterase (PAE).

Figure 5.
HPLC results of synergistic cellulose acetate degradation by cellulases. Substrates were pretreated with esterase AXE 55. Samples without cellulose acetate were examined to estimate the glucose content of the used enzyme formulations.
Figure 5.
HPLC results of synergistic cellulose acetate degradation by cellulases. Substrates were pretreated with esterase AXE 55. Samples without cellulose acetate were examined to estimate the glucose content of the used enzyme formulations.
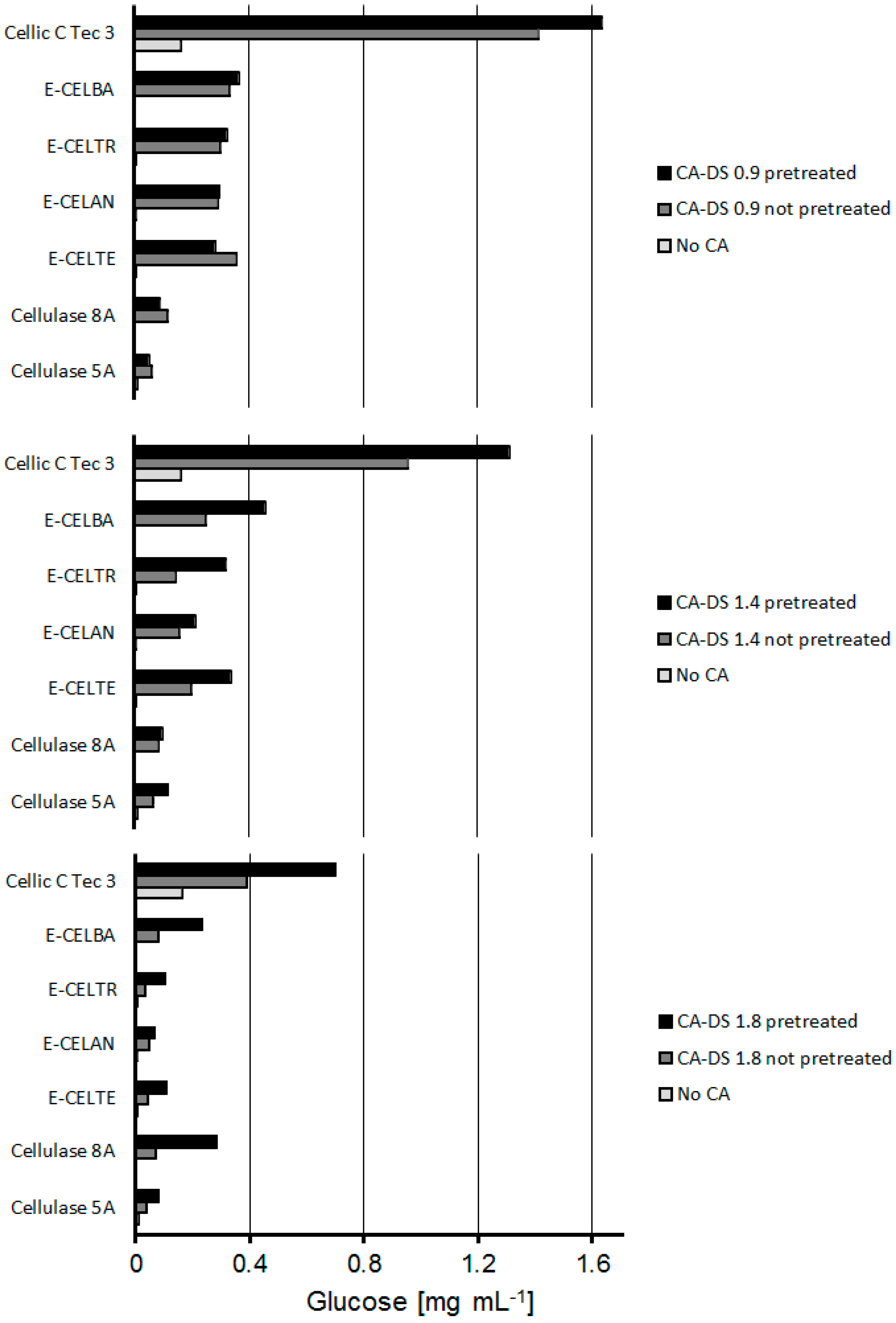
Figure 6.
Hydrolysis of cellulose acetates with different degree of acetylation (CA0.9, CA1.4) by lytic polysaccharide monooxygenases (LPMO 2916) monitored using LC-TOF-MS. Degree of polymerization (DP); number of acetyl groups per fragment (Ac).
Figure 6.
Hydrolysis of cellulose acetates with different degree of acetylation (CA0.9, CA1.4) by lytic polysaccharide monooxygenases (LPMO 2916) monitored using LC-TOF-MS. Degree of polymerization (DP); number of acetyl groups per fragment (Ac).
Figure 7.
Backbone cleavage of cellulose acetates with chitinase and cellobiohydrolase I after 145 h.
Figure 7.
Backbone cleavage of cellulose acetates with chitinase and cellobiohydrolase I after 145 h.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Esterase screening for several esterases substrates. Very strong deacetylation (++++), strong deacetylation (+++), moderate deacetylation (++), weak deacetylation (+), no deacetylation (-). Axetyl xylan estarase (AXE); carbohydrate esterase (CE); cutinase (CUT); glucomannan acetyl esterase (GAE); ND pectin acetyl esterase (PAE).
Table 1.
Esterase screening for several esterases substrates. Very strong deacetylation (++++), strong deacetylation (+++), moderate deacetylation (++), weak deacetylation (+), no deacetylation (-). Axetyl xylan estarase (AXE); carbohydrate esterase (CE); cutinase (CUT); glucomannan acetyl esterase (GAE); ND pectin acetyl esterase (PAE).
| Esterase | Triacetin | Glucose Pentaacetate | Cellobiose Octaacetate | Cellohexaose Eicosaacetate | CA-DS 0.9 | CA-DS 1.4 | CA-DS 1.8 | CA-DS 2.3 | CA-DS 2.5 |
|---|---|---|---|---|---|---|---|---|---|
| AXE O | +++ | +++ | + | + | + | - | - | - | - |
| AXE 34 | + | + | + | + | ++ | ++ | + | - | - |
| AXE 35 | + | ++ | + | - | ++ | ++ | + | - | - |
| AXE 36 | + | ++ | + | + | ++ | ++ | + | - | - |
| AXE 53 | ++++ | ++++ | + | + | +++ | +++ | ++ | - | - |
| AXE 55 | +++ | +++ | + | + | +++ | ++++ | + | - | - |
| CE 265 | + | + | - | - | - | - | - | - | - |
| CUT | ++++ | ++++ | + | + | +++ | +++ | + | - | - |
| GAE | ++++ | ++++ | +++ | ++ | +++ | +++ | ++ | - | - |
| PAE | ++++ | +++ | +++ | ++ | +++ | +++ | ++ | - | - |
Table 2.
Glucose recovery with Cellic C Tec 3 after esterase pretreatment and without pretreatment of CA with different degrees of substitution. Cellulose acetate with a degree of substitution of 0.9 (CA-DS 0.9). The degree of substitution refers to the number of acetyl groups per glucose unit in the molecule.
Table 2.
Glucose recovery with Cellic C Tec 3 after esterase pretreatment and without pretreatment of CA with different degrees of substitution. Cellulose acetate with a degree of substitution of 0.9 (CA-DS 0.9). The degree of substitution refers to the number of acetyl groups per glucose unit in the molecule.
| Substrate | % Glucose (w/w) | % Acetic Acid (w/w) | Glucose Recovery Pretreated (%) | Glucose Recovery not Pretreated (%) |
|---|---|---|---|---|
| CA-DS 0.9 | 80.6 | 19.4 | 53.8 | 46.5 |
| CA-DS 1.4 | 72.7 | 27.3 | 47.8 | 34.9 |
| CA-DS 1.8 | 67.4 | 32.6 | 27.6 | 15.3 |
Table 3.
Esterases and their optimal reaction conditions investigated for deacetylation of oligomeric model substrates and of CA. Family assignments correspond to the classification of carbohydrate esterases according to the Carbohydrate Active Enzymes database (www.cazy.org).
Table 3.
Esterases and their optimal reaction conditions investigated for deacetylation of oligomeric model substrates and of CA. Family assignments correspond to the classification of carbohydrate esterases according to the Carbohydrate Active Enzymes database (www.cazy.org).
| Enzyme | Origin | pH | Temp. (°C) | Family |
|---|---|---|---|---|
| Acetyl xylan esterase (AXE O) | Orpinomyces sp. | 6.7–7 | 40 | 6 |
| Acetyl xylan esterase (AXE 34) | Clostridium thermocellum | 7 | 50 | 3 |
| Acetyl xylan esterase (AXE 35) | Clostridium thermocellum | 6.5 | 50 | 4 |
| Acetyl xylan esterase (AXE 36) | Clostridium thermocellum | 6.5 | 50 | 4 |
| Acetyl xylan esterase (AXE 53) | Cellvibrio japonicus | 8.5 | 25 | 2 |
| Acetyl xylan esterase (AXE 55) | Cellvibrio japonicus | 8.5 | 25 | 2 |
| Carboxylesterase (CE 265) | Bacillus subtilis | 7 | 37 | N.A. |
| Cutinase (CUT) | Thermobifida cellulosilytica | 7 | 50 | N.A. |
| Glucomannan acetyl esterase (GAE) | Clostridium thermocellum | 7 | 50 | 2 |
| Pectin acetyl esterase (PAE) | Clostridium thermocellum | 6.5 | 50 | 12 |
Table 4.
Cellulases used for degradation of deacetylated cellulose acetate with their optimal conditions and origin. Family classification follows the Carbohydrate-Active Enzymes Database.
Table 4.
Cellulases used for degradation of deacetylated cellulose acetate with their optimal conditions and origin. Family classification follows the Carbohydrate-Active Enzymes Database.
| Enzyme | Origin | pH | Temp. (°C) | Family |
|---|---|---|---|---|
| Cellulase E-CELAN | Aspergillus niger | 4.5 | 55 | GH12 |
| Cellulase E-CELTR | Trichoderma longibrachiatum | 4.5 | 70 | GH7 |
| Cellulase E-CELTE | Talaromyces emersonii. | 4.5 | 70 | GH5 |
| Cellulase E-CELBA | Bacillus amyloliquefaciens | 6.5 | 55 | GH5 |
| Cellulase 8A | Escherichia coli | 7 | 40 | GH8 |
| Cellulase 5A | Bacillus subtilis | 7.5 | 55 | GH5 |
| Cellulase Cellic C Tec 3 | N.A. | 5 | 55 | N.A. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Haske-Cornelius, O.; Pellis, A.; Tegl, G.; Wurz, S.; Saake, B.; Ludwig, R.; Sebastian, A.; Nyanhongo, G.S.; Guebitz, G.M. Enzymatic Systems for Cellulose Acetate Degradation. Catalysts 2017, 7, 287. https://doi.org/10.3390/catal7100287
AMA Style
Haske-Cornelius O, Pellis A, Tegl G, Wurz S, Saake B, Ludwig R, Sebastian A, Nyanhongo GS, Guebitz GM. Enzymatic Systems for Cellulose Acetate Degradation. Catalysts. 2017; 7(10):287. https://doi.org/10.3390/catal7100287
Chicago/Turabian StyleHaske-Cornelius, Oskar, Alessandro Pellis, Gregor Tegl, Stefan Wurz, Bodo Saake, Roland Ludwig, Andries Sebastian, Gibson S. Nyanhongo, and Georg M. Guebitz. 2017. "Enzymatic Systems for Cellulose Acetate Degradation" Catalysts 7, no. 10: 287. https://doi.org/10.3390/catal7100287
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.