Co-Production of Ethanol and 1,2-Propanediol via Glycerol Hydrogenolysis Using Ni/Ce–Mg Catalysts: Effects of Catalyst Preparation and Reaction Conditions

Abstract
:1. Introduction
2. Results and Discussion
2.1. Catalyst Characterization
2.1.1. Textural Properties
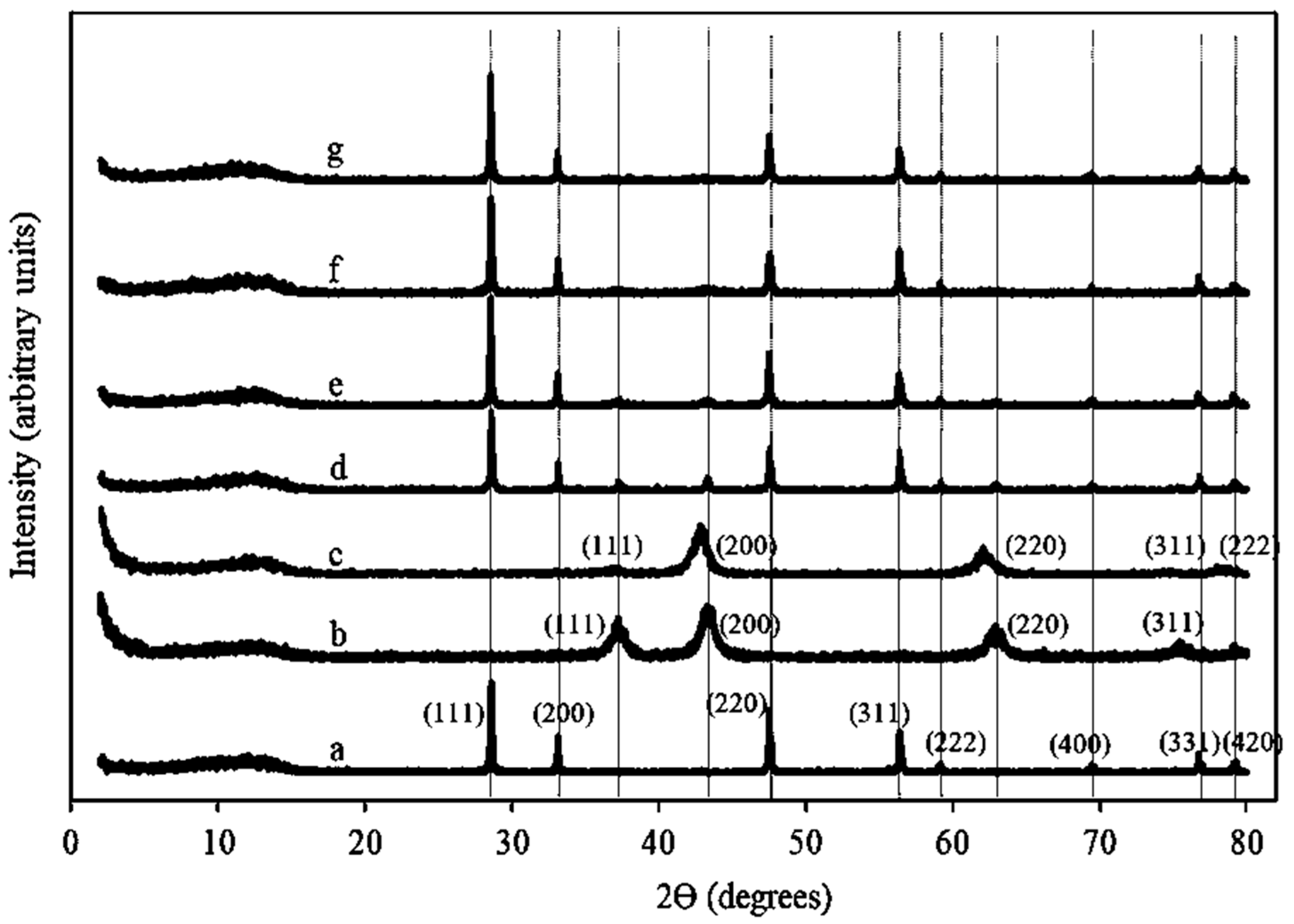
2.1.2. XRD Patterns of Ni/Ce–Mg Catalysts
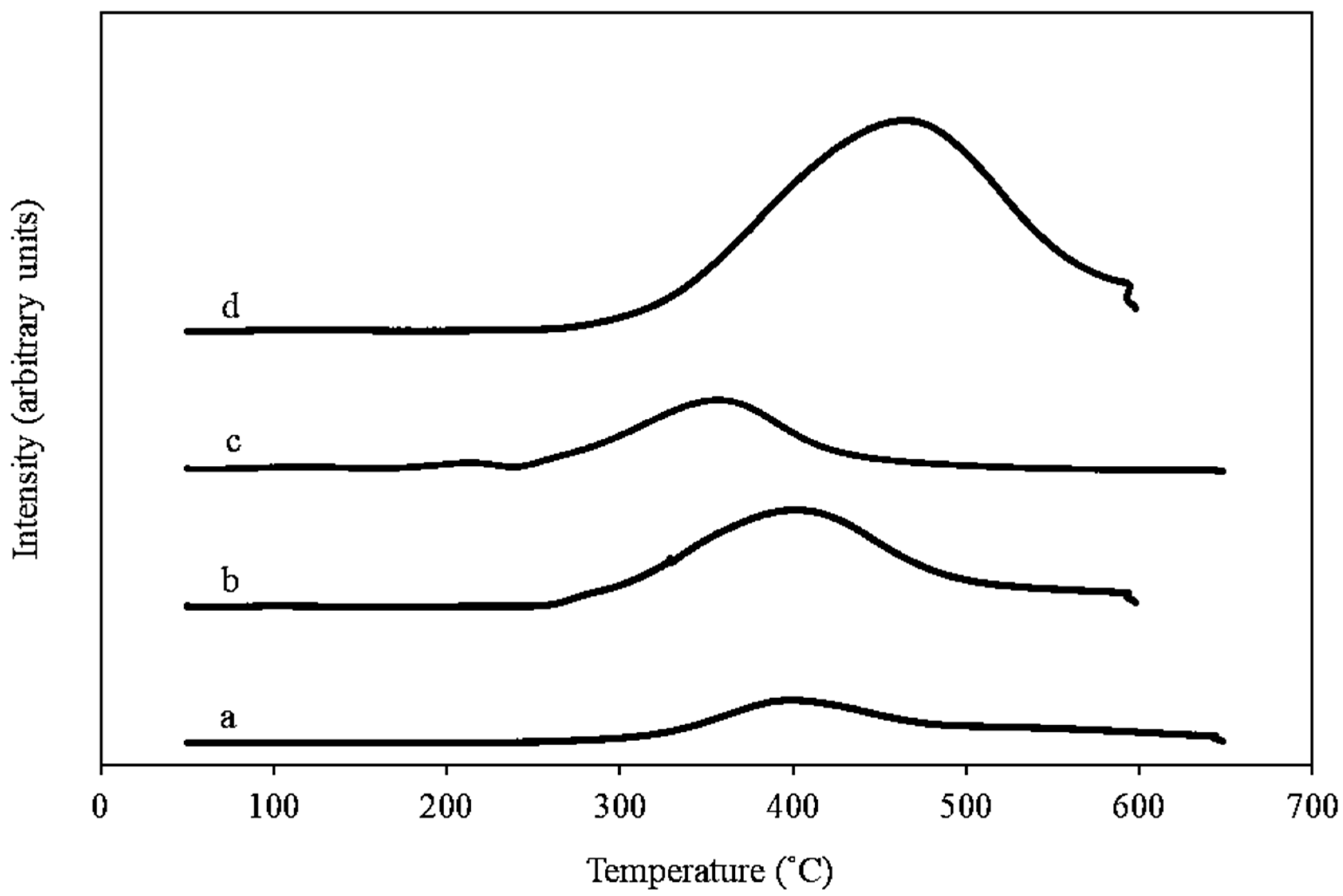
2.1.3. Temperature Programmed Reduction (H2-TPR)
2.2. Effect of Preparation Method
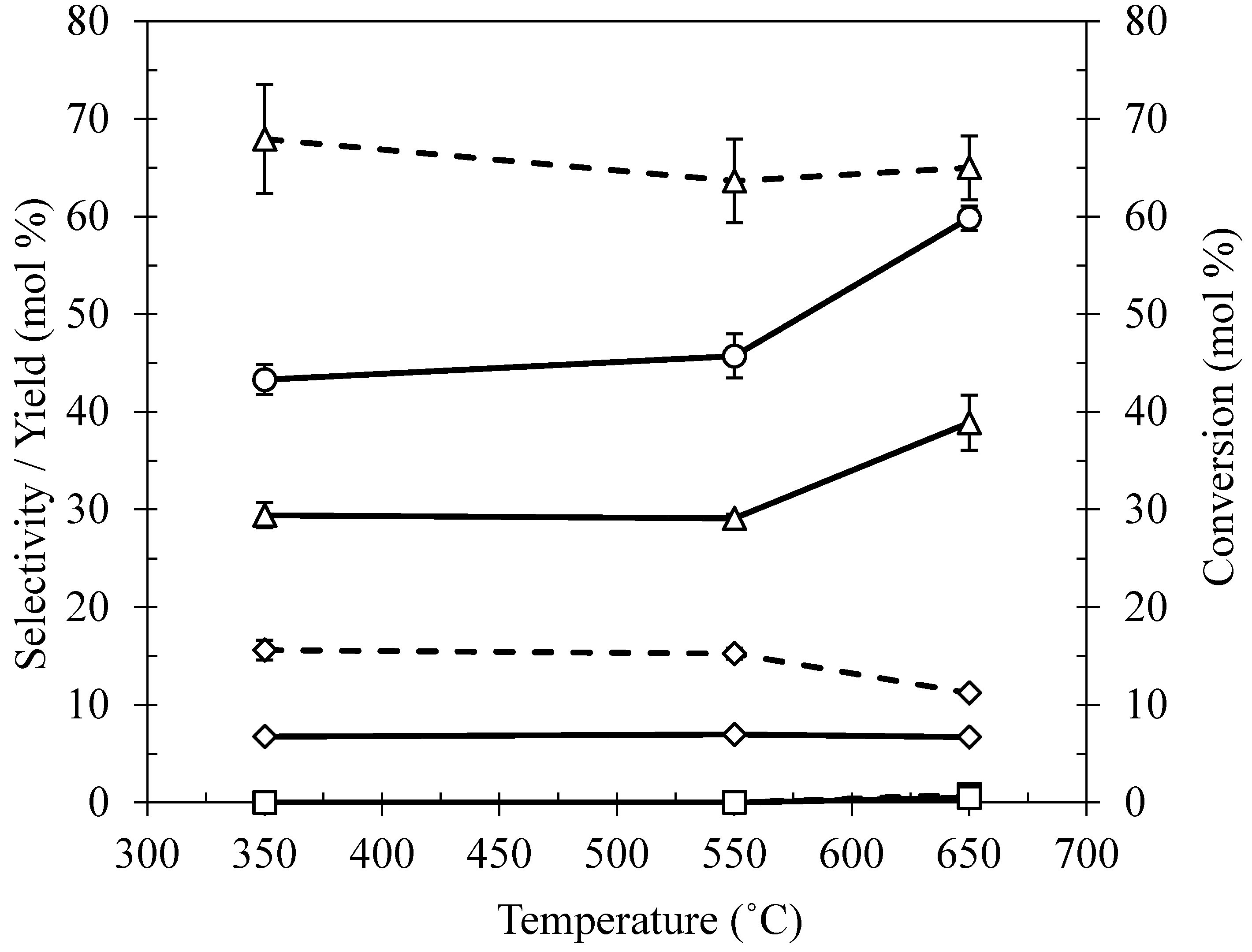
2.3. Effect of Calcination Temperature on Ni/Ce–Mg Catalysts Prepared by DP
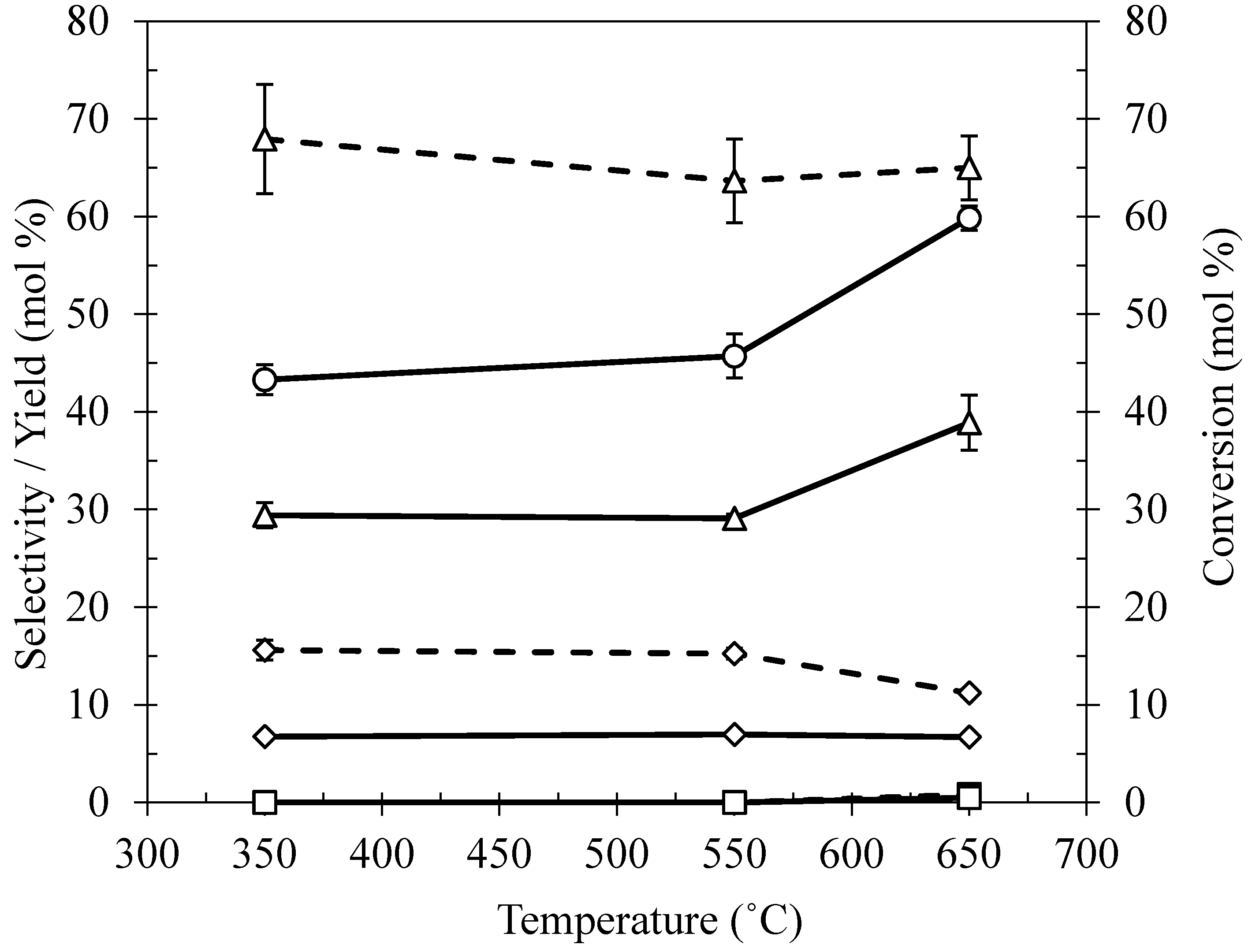
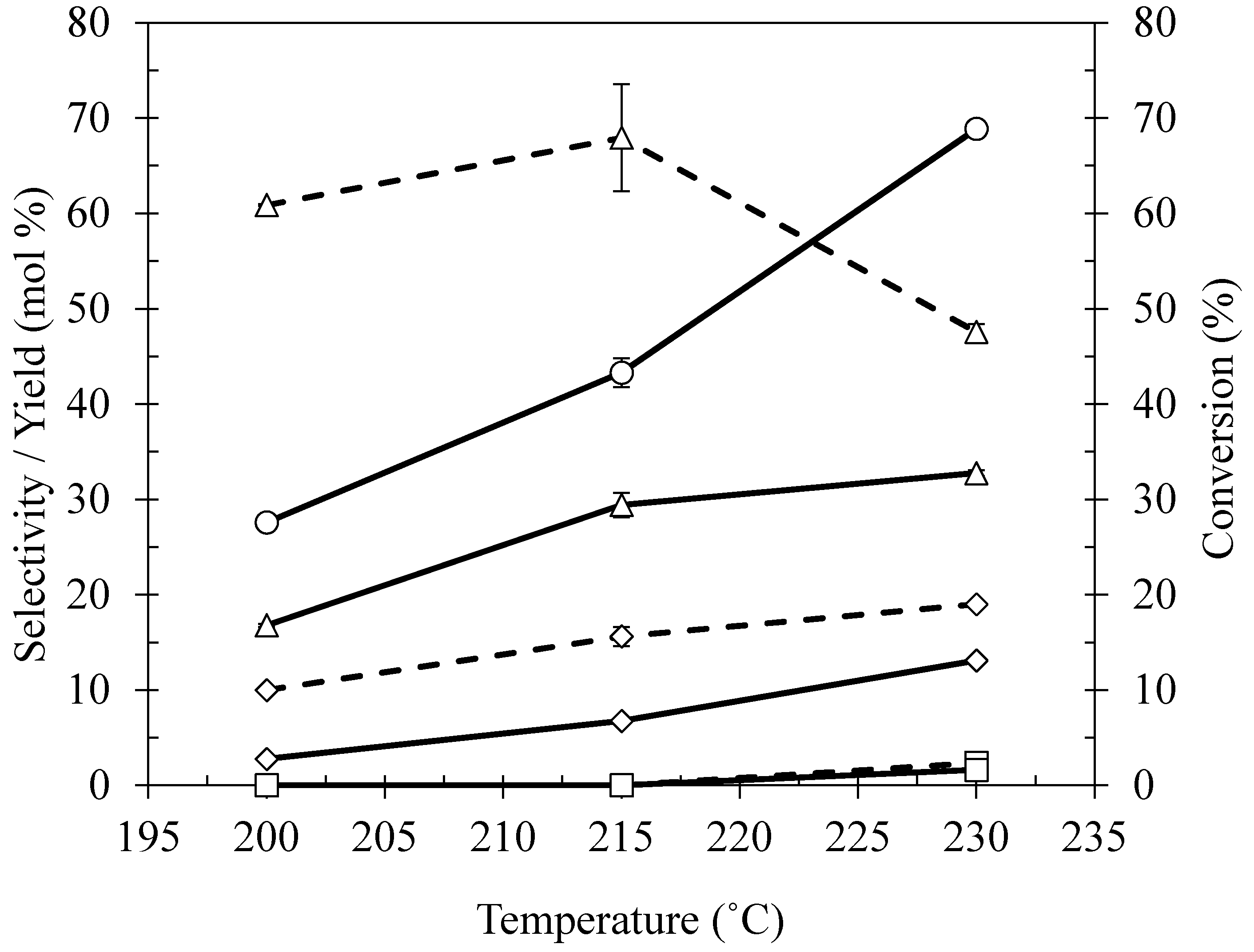
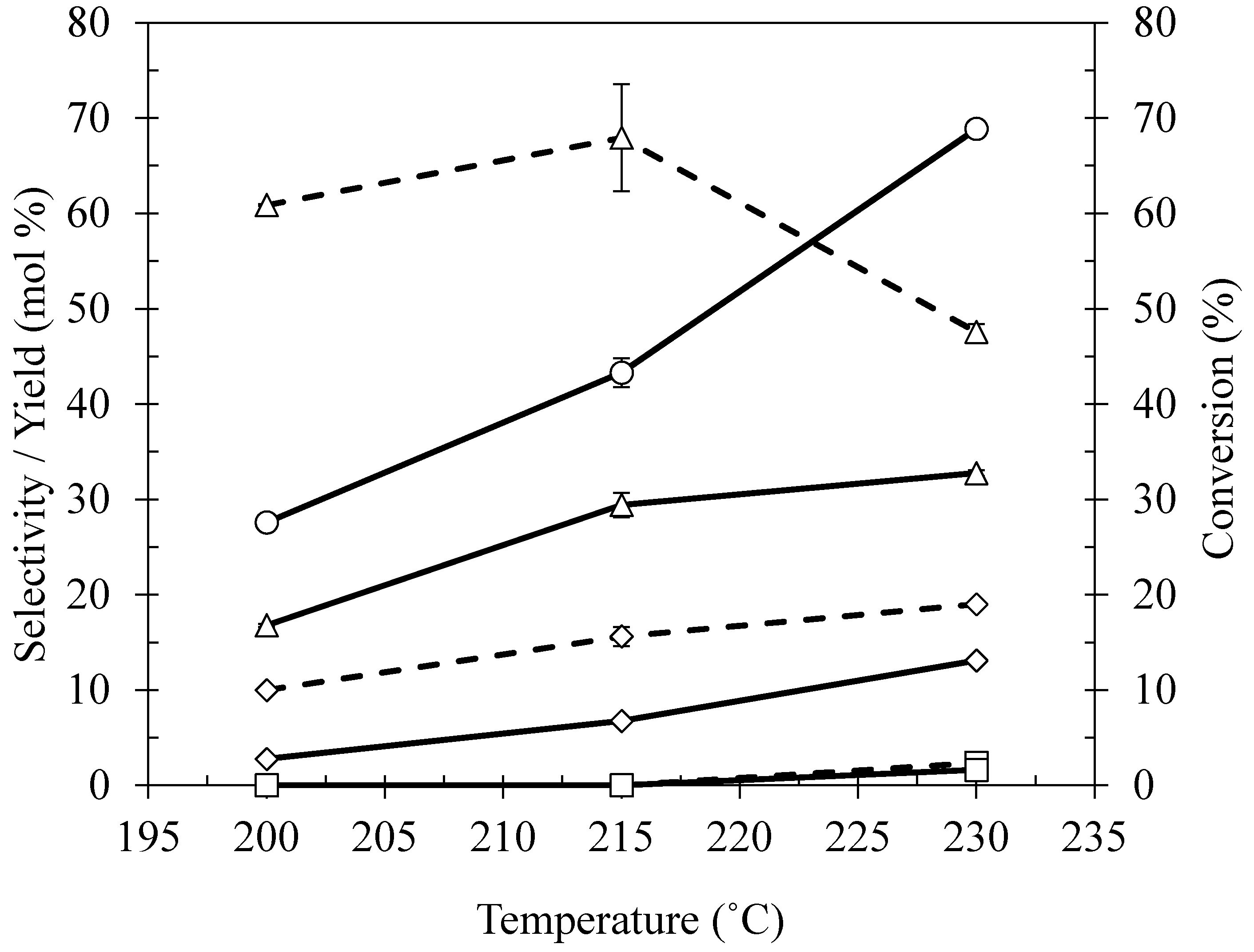
2.4. Effect of Reaction Temperature
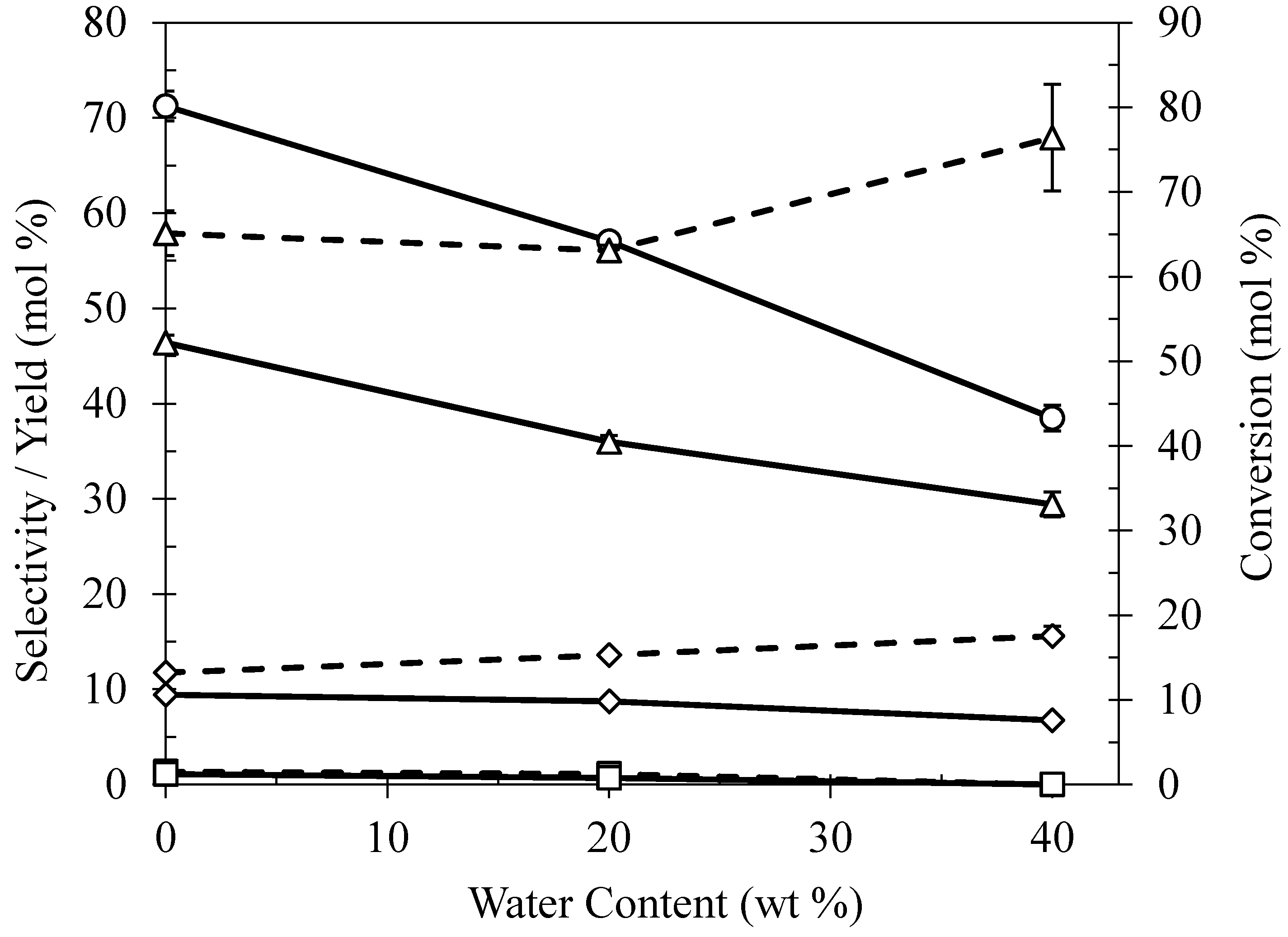
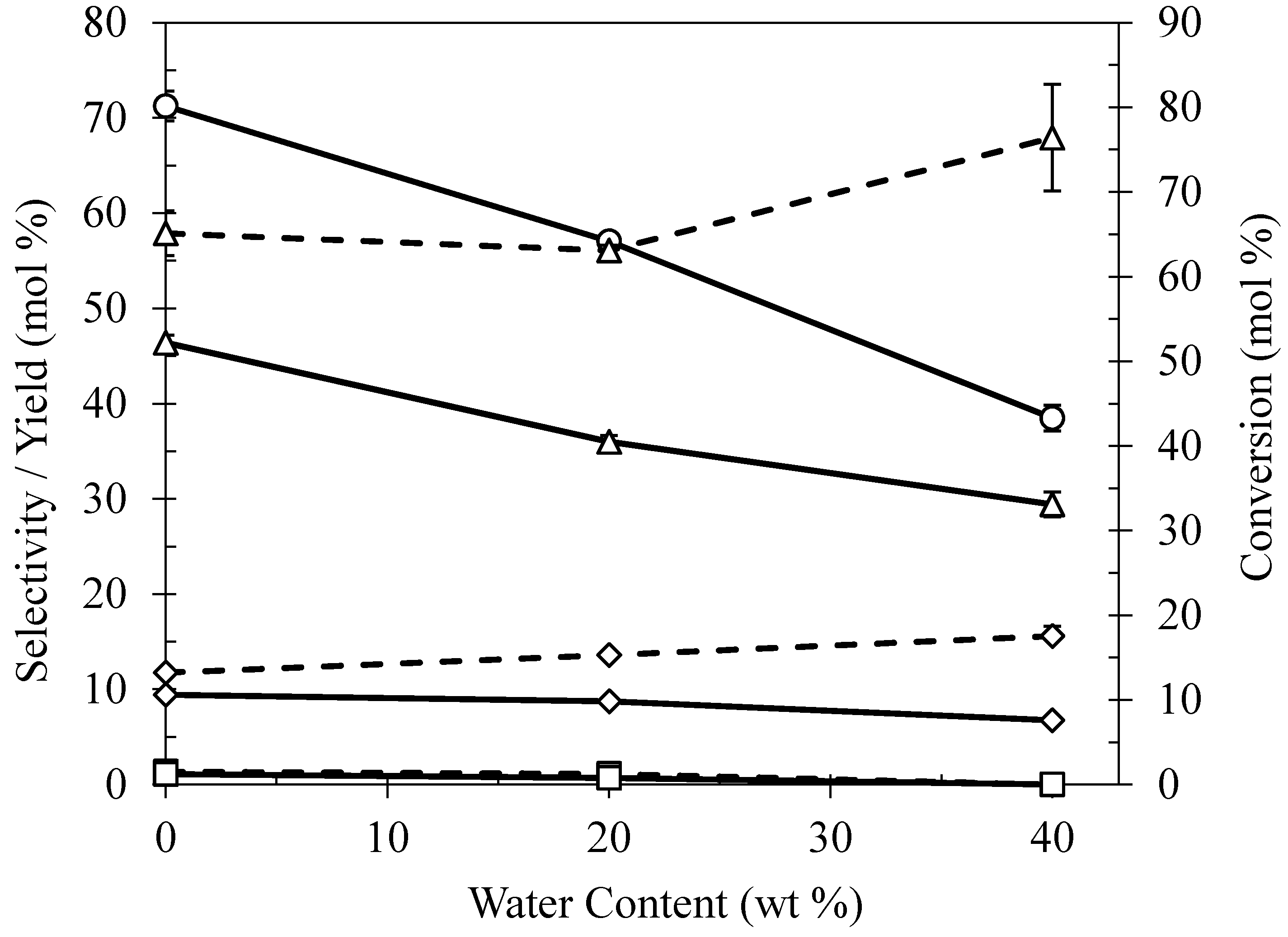
2.5. Effect of Water Content
2.6. Effect of Initial Hydrogen Pressure
2.7. Effect of Reaction Time
2.8. Comparison with Raney® Ni Catalyst
3. Experimental Section
3.1. Materials
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.4. Catalytic Activity Testing
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Barrett, D.; Delwiche, M.; Stroeve, P. Methods for pretreatment of lignocellulosic biomass for efficient hydrolysis and biofuel production. Ind. Eng. Chem. Res. 2009, 48, 3713–3729. [Google Scholar] [CrossRef]
- Sheldon, R.A. Utilisation of biomass for sustainable fuels and chemicals, molecules, methods and metrics. Catal. Today 2011, 167, 3–13. [Google Scholar] [CrossRef]
- Leung, D.Y.C.; Wu, X.; Leung, M.K.H. A review on biodiesel production using catalyzed transesterification. Appl. Energy 2010, 87, 1083–1095. [Google Scholar] [CrossRef]
- Silitonga, A.S.; Ong, H.C.; Mahlia, T.M.I.; Masjuki, H.H.; Chong, W.T. Biodiesel conversion from high FFA crude Jatropha curcas, Calophyllum inophyllum and Ceiba pentandra Oil. Energy Procedia 2014, 61, 480–483. [Google Scholar] [CrossRef]
- Shahid, E.M.; Jamal, Y. Production of biodiesel, a technical review. Renew. Sustain. Energy Rev. 2011, 15, 4732–4745. [Google Scholar] [CrossRef]
- US EIA. U.S. Biodiesel Production Capacity and Production. Available online: https //www.eia.gov/biofuels/biodiesel/production/table1.pdf (accessed on 15 August 2017).
- International Energy Agency. Medium-Term Renewable Energy Market Report. 2014. Available online: http://www.iea.org/Textbase/npsum/MTrenew2014SUM.pdf (accessed on 16 August 2017).
- Anand, P.; Saxena, R.K. A comparative study of solvent-assisted pretreatment of biodiesel derived crude glycerol on growth and 1,3-propanediol production from Citrobacter freundii. New Biotechnol. 2012, 29, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Hanna, M.A.; Sun, R. Value-added uses for crude glycerol—A byproduct of biodiesel production. Biotechnol. Biofuels 2012, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.T.; Taconi, K.A. The glycerin glut, options for the value-added conversion of crude glycerol resulting from biodiesel production. Environ. Prog. 2007, 26, 338–348. [Google Scholar] [CrossRef]
- Pagliaro, M.; Rossi, M. Glycerol, Properties and Production. In The Future of Glycerol, 2nd ed.; The Royal Society of Chemistry: London, UK, 2010; Chapter 1; pp. 1–28. [Google Scholar]
- Zhou, C.H.; Beltramini, J.; Fan, Y.X.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Balat, M. Production of bioethanol from lignocellulosic materials via the biochemical pathway: A review. Energy Convers. Manag. 2011, 52, 858–875. [Google Scholar] [CrossRef]
- Kosaric, N.; Duvnjak, Z.; Farkas, A.; Sahm, H.; Bringer-Meyer, S.; Goebel, O.; Mayer, D. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Nakagawa, Y.; Tomishige, K. Heterogeneous catalysis of the glycerol hydrogenolysis. Catal. Sci. Technol. 2011, 1, 179–190. [Google Scholar] [CrossRef]
- Perosa, A.; Tundo, P. Selective hydrogenolysis of glycerol with Raney nickel. Ind. Eng. Chem. Res. 2005, 44, 8535–8537. [Google Scholar] [CrossRef] [Green Version]
- Van Ryneveld, E.; Mahomed, A.S.; van Heerden, P.S.; Green, M.J.; Friedrich, H.B. A catalytic route to lower alcohols from glycerol using Ni-supported catalysts. Green Chem. 2011, 13, 1819–1827. [Google Scholar] [CrossRef]
- Huang, J.; Chen, J. Comparison of Ni2P/SiO2 and Ni/SiO2 for hydrogenolysis of glycerol, a consideration of factors influencing catalyst activity and product selectivity. Chin. J. Catal. 2012, 33, 790–796. [Google Scholar] [CrossRef]
- Menchavez, R.N.; Morra, J.M.; He, B.B. Glycerol hydrogenolysis using a Ni/Ce-Mg catalyst for improved ethanol and 1,2-propanediol selectivities. Can. J. Chem. Eng. 2017, 95, 1332–1339. [Google Scholar] [CrossRef]
- Wawrzetz, A.; Peng, B.; Hrabar, A.; Jentys, A.; Lemonidou, A.A.; Lercher, J.A. Towards understanding the bifunctional hydrodeoxygenation and aqueous phase reforming of glycerol. J. Catal. 2010, 269, 411–420. [Google Scholar] [CrossRef]
- Peng, B.; Zhao, C.; Mejía-Centeno, I.; Fuentes, G.A.; Jentys, A.; Lercher, J.A. Comparison of kinetics and reaction pathways for hydrodeoxygenation of C3 alcohols on Pt/Al2O3. Catal. Today 2012, 183, 3–9. [Google Scholar] [CrossRef]
- Mauriello, F.; Vinci, A.; Espro, C.; Gumina, B.; Musolino, M.G.; Pietropaolo, R. Hydrogenolysis vs. aqueous phase reforming (APR) of glycerol promoted by a heterogeneous Pd/Fe catalyst. Catal. Sci. Technol. 2015, 5, 4466–4473. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green chemistry, principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Werpy, T.; Petersen, G.R.; Aden, A.; Bozell, J.J.; Holladay, J.; White, J.; Manheim, A.; Eliot, D.; Lasure, L.; Jones, S. Top value added chemicals from biomass. In Results of Screening for Potential Candidates from Sugars and Synthesis Gas; National Renewable Energy Laboratory: Springfield, VA, USA, 2004; Volume 1. [Google Scholar]
- Liu, J.; Shi, J.; He, D.; Zhang, Q.; Wu, X.; Liang, Y.; Zhu, Q. Surface active structure of ultra-fine Cu/ZrO2 catalysts used for the CO2+H2 to methanol reaction. Appl. Catal. A Gen. 2001, 218, 113–119. [Google Scholar] [CrossRef]
- Yaakob, Z.; Bshish, A.; Ebshish, A.; Tasirin, S.; Alhasan, F. Hydrogen production by steam reforming of ethanol over nickel catalysts supported on sol gel made alumina, influence of calcination temperature on supports. Materials 2013, 6, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Daza, C.; Kiennemann, A.; Moreno, S.; Molina, R. Stability of Ni-Ce catalysts supported over Al-PVA modified mineral clay in dry reforming of methane. Energy Fuels 2009, 23, 3497–3509. [Google Scholar] [CrossRef]
- Hernández-Enríquez, J.M.; Silva-Rodrigo, R.; García-Alamilla, R.; García-Serrano, L.A.; Handy, B.E.; Cárdenas-Galindo, G.; Cueto-Hernández, A. Synthesis and physico-chemical characterization of CeO2/ZrO2-SO42- mixed oxides. J. Mex. Chem. Soc. 2012, 56, 115–120. [Google Scholar]
- Veranitisagul, C.; Kaewvilai, A.; Sangngern, S.; Wattanathana, W.; Suramitr, S.; Koonsaeng, N.; Laobuthee, A. Novel recovery of nano-structured Ceria (CeO2) from Ce(III)-benzoxazine dimer complexes via thermal decomposition. Int. J. Mol. Sci. 2011, 12, 4365–4377. [Google Scholar] [CrossRef] [PubMed]
- El-Kemary, M.; Nagy, N.; El-Mehasseb, I. Nickel oxide nanoparticles, synthesis and spectral studies of interactions with glucose. Mater. Sci. Semicond. Process. 2013, 16, 1747–1752. [Google Scholar] [CrossRef]
- Umaralikhan, L.; Jamal Mohamed Jaffar, M. Green synthesis of MgO nanoparticles and its antibacterial activity. Iran. J. Sci. Technol. Trans. A Sci. 2016. [CrossRef]
- Bindhu, M.R.; Umadevi, M.; Kavin Micheal, M.; Arasu, M.V.; Abdullah Al-Dhabi, N. Structural, morphological and optical properties of MgO nanoparticles for antibacterial applications. Mater. Lett. 2016, 166, 19–22. [Google Scholar] [CrossRef]
- Daza, C.E.; Parkhomenko, K.; Kiennemann, A. Ni-Ce/Mg Catalysts prepared by self-combustion for CO2 reforming of methane. Adv. Chem. Lett. 2013, 1, 321–329. [Google Scholar] [CrossRef]
- Wang, S.F.; Yeh, C.T.; Wang, Y.R.; Wu, Y.C. Characterization of samarium-doped ceria powders prepared by hydrothermal synthesis for use in solid state oxide fuel cells. J. Mater. Res. Technol. 2013, 2, 141–148. [Google Scholar] [CrossRef]
- Arena, F.; Frusteri, F.; Parmaliana, A.; Plyasova, L.; Shmakov, A.N. Effect of calcination on the structure of Ni/MgO catalyst, an X-ray diffraction study. J. Chem. Soc. Faraday Trans. 1996, 92, 469–471. [Google Scholar] [CrossRef]
- Li, L.; Zhang, L.; Shi, X.; Zhang, Y.; Li, J. Carbon dioxide reforming of methane over nickel catalysts supported on mesoporous MgO. J. Porous Mater. 2014, 21, 217–224. [Google Scholar] [CrossRef]
- Zangouei, M.; Moghaddam, A.Z.; Arasteh, M. The influence of nickel loading on reducibility of NiO/Al2O3 catalysts synthesized by sol-gel method. Chem. Eng. Res. Bull. 2010, 14, 97–102. [Google Scholar] [CrossRef]
- Taufiq-Yap, Y.H.; Sudarno, U.; Zainal, Z. CeO2–SiO2 supported nickel catalysts for dry reforming of methane toward syngas production. Appl. Catal. A 2013, 468, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Montoya, J.A.; Romero-Pascual, E.; Gimon, C.; Del Angel, P.; Monzón, A. Methane reforming with CO2 over Ni/ZrO2–CeO2 catalysts prepared by sol–gel. Catal. Today 2000, 63, 71–85. [Google Scholar] [CrossRef]
- Kim, N.D.; Oh, S.; Joo, J.B.; Jung, K.S.; Yi, J. Effect of preparation method on structure and catalytic activity of Cr-promoted Cu catalyst in glycerol hydrogenolysis. Korean J. Chem. Eng. 2010, 27, 431–434. [Google Scholar] [CrossRef]
- Sato, S.; Akiyama, M.; Inui, K.; Yokota, M. Selective conversion of glycerol into 1,2-propanediol at ambient hydrogen pressure. Chem. Lett. 2009, 38, 560–561. [Google Scholar] [CrossRef]
- Xiao, Y.; Xiao, G.; Varma, A. A universal procedure for crude glycerol purification from different Feedstocks in biodiesel production, experimental and simulation study. Ind. Eng. Chem. Res. 2013, 52, 14291–14296. [Google Scholar] [CrossRef]
- Maglinao, R.L.; He, B.B. Catalytic thermochemical conversion of glycerol to simple and polyhydric alcohols using Raney nickel catalyst. Ind. Eng. Chem. Res. 2011, 50, 6028–6033. [Google Scholar] [CrossRef]
- Xiao, Z.; Li, C.; Xiu, J.; Wang, X.; Williams, C.T.; Liang, C. Insights into the reaction pathways of glycerol hydrogenolysis over Cu–Cr catalysts. J. Mol. Catal. A Chem. 2012, 365, 24–31. [Google Scholar] [CrossRef]
- Guo, L.; Zhou, J.; Mao, J.; Guo, X.; Zhang, S. Supported Cu catalysts for the selective hydrogenolysis of glycerol to propanediols. Appl. Catal. A 2009, 367, 93–98. [Google Scholar] [CrossRef]
- Meher, L.C.; Gopinath, R.; Naik, S.N.; Dalai, A.K. Catalytic hydrogenolysis of glycerol to propylene glycol over mixed oxides derived from a hydrotalcite-type precursor. Ind. Eng. Chem. Res. 2009, 48, 1840–1846. [Google Scholar] [CrossRef]
- Zhao, B.; Li, C.; Xu, C. Insight into the catalytic mechanism of glycerol hydrogenolysis using basal spacing of hydrotalcite as a tool. Catal. Sci. Technol. 2012, 2, 1985–1994. [Google Scholar] [CrossRef]
- Dasari, M.A.; Kiatsimkul, P.P.; Sutterlin, W.R.; Suppes, G.J. Low-pressure hydrogenolysis of glycerol to propylene glycol. Appl. Catal. A Gen. 2005, 281, 225–231. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preparation Method a | Calcination Temp. (°C) | BET Surface (m2/g) | Pore Volume b (cm3/g) | Pore Diameter c (nm) |
|---|---|---|---|---|
| Impregnation (IM) | 550 | 8.08 | 0.0521 | 27.1 |
| Deposition Precipitation (DP) | 550 | 27.33 | 0.0879 | 10.2 |
| 650 | 27.78 | 0.0832 | 10.4 | |
| 350 | 70.78 | 0.0973 | 4.6 |
| Preparation Method | Conversion (mol %) | Ethanol | EG | n-Propanol | 1,2-PDO | Others | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| S | Y | S | Y | S | Y | S | Y | S | ||
| AE b | AE | AE | AE | AE | ||||||
| RE | RE | RE | RE | RE | ||||||
| Impregnation (IM) | 28.63 | 15.28 | 4.38 | 5.34 | 1.53 | ND | ND | 62.14 | 17.79 | 17.25 |
| 0.48 | 0.41 | 0.17 | - | 1.55 | ||||||
| 1.68 | 0.68 | 3.18 | - | 2.49 | ||||||
| Deposition Precipitation (DP) | 45.71 | 15.26 | 6.98 | 5.76 | 2.63 | ND | ND | 63.64 | 29.09 | 15.34 |
| 2.26 | 0.57 | 0.36 | - | 4.28 | ||||||
| 4.94 | 3.74 | 6.25 | - | 6.72 | ||||||
| Conditions b pH2 (MPa); Time (h) | Conversion (mol %) | Ethanol | EG | n-Propanol | 1,2-PDO | Others | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| S | Y | S | Y | S | Y | S | Y | S | ||
| AE | AE | AE | AE | AE | ||||||
| RE | RE | RE | RE | RE | ||||||
| 6.9; 24 | 43.29 | 15.61 | 6.75 | 6.80 | 2.94 | ND | ND | 67.93 | 29.41 | 9.66 |
| 1.53 | 1.02 | 0.54 | - | 5.6 | ||||||
| 3.53 | 6.53 | 7.94 | - | 8.24 | ||||||
| 8.6; 24 | 51.40 | 12.40 | 6.38 | 7.38 | 3.80 | 0.73 | 0.38 | 64.12 | 33 | 15.37 |
| 1.66 | 0.04 | 0.3 | 0.01 | 1.50 | ||||||
| 3.23 | 0.32 | 4.07 | 1.37 | 2.34 | ||||||
| 8.6; 48 | 75.64 | 15.43 | 11.68 | 7.30 | 5.53 | 1.34 | 1.02 | 60.81 | 46.0 | 15.12 |
| 1.93 | 0.65 | 0.08 | 0.07 | 0.36 | ||||||
| 2.55 | 4.21 | 1.10 | 5.22 | 0.59 | ||||||
| Catalyst | T (°C) | Conversion (mol %) | Ethanol | EG | n-Propanol | 1,2-PDO | Others | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S | Y | S | Y | S | Y | S | Y | S | |||
| SD | SD | SD | SD | SD | |||||||
| CV | CV | CV | CV | CV | |||||||
| Ni/Ce–Mg | 200 | 27.57 | 9.99 | 2.75 | 5.77 | 1.59 | ND | ND | 60.87 | 16.78 | 23.37 |
| 0.46 | 1.06 | 0.17 | - | 3.49 | |||||||
| 1.66 | 9.95 | 2.84 | - | 5.54 | |||||||
| 215 | 43.29 | 15.61 | 6.75 | 6.80 | 2.94 | ND | ND | 67.93 | 29.41 | 9.66 | |
| 2.58 | 1.76 | 0.85 | - | 5.41 | |||||||
| 5.80 | 10.70 | 11.72 | - | 7.93 | |||||||
| 230 | 68.87 | 19.02 | 13.10 | 6.80 | 2.94 | 2.36 | 1.63 | 47.57 | 32.76 | 22.62 | |
| 0.62 | 1.31 | 0.15 | 0.16 | 1.76 | |||||||
| 0.91 | 7.18 | 2.55 | 6.34 | 3.63 | |||||||
| Raney® Ni | 200 | 31.00 | 4.59 | 1.42 | 52.80 | 16.39 | ND | ND | 22.34 | 6.93 | 20.27 |
| 1.09 | 0.41 | 1.89 | - | 0.36 | |||||||
| 3.50 | 8.93 | 3.57 | - | 1.59 | |||||||
| 215 | 49.40 | 9.07 | 4.48 | 34.73 | 17.16 | 0.76 | 0.38 | 26.15 | 12.92 | 29.9 | |
| 1.34 | 0.26 | 1.33 | 0.01 | 0.60 | |||||||
| 2.72 | 2.82 | 5.73 | 1.29 | 2.29 | |||||||
| 230 | 67.46 | 11.43 | 7.71 | 18.63 | 12.57 | 1.19 | 0.80 | 26.70 | 18.01 | 42.05 | |
| 2.62 | 0.33 | 1.73 | 0.04 | 0.34 | |||||||
| 3.88 | 2.85 | 9.28 | 3.66 | 1.29 | |||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menchavez, R.N.; Morra, M.J.; He, B.B. Co-Production of Ethanol and 1,2-Propanediol via Glycerol Hydrogenolysis Using Ni/Ce–Mg Catalysts: Effects of Catalyst Preparation and Reaction Conditions. Catalysts 2017, 7, 290. https://doi.org/10.3390/catal7100290
Menchavez RN, Morra MJ, He BB. Co-Production of Ethanol and 1,2-Propanediol via Glycerol Hydrogenolysis Using Ni/Ce–Mg Catalysts: Effects of Catalyst Preparation and Reaction Conditions. Catalysts. 2017; 7(10):290. https://doi.org/10.3390/catal7100290
Chicago/Turabian StyleMenchavez, Russel N., Matthew J. Morra, and B. Brian He. 2017. "Co-Production of Ethanol and 1,2-Propanediol via Glycerol Hydrogenolysis Using Ni/Ce–Mg Catalysts: Effects of Catalyst Preparation and Reaction Conditions" Catalysts 7, no. 10: 290. https://doi.org/10.3390/catal7100290