Reactivity of Trapped and Accumulated Electrons in Titanium Dioxide Photocatalysis

Department of Pharmacy, School of Pharmacy, Hyogo University of Health Sciences, 1-3-6 Minatojima, Chuo-ku, Kobe 650-8530, Japan
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(10), 303; https://doi.org/10.3390/catal7100303
Submission received: 20 September 2017
/
Revised: 5 October 2017
/
Accepted: 8 October 2017
/
Published: 13 October 2017
(This article belongs to the Special Issue Titanium Dioxide Photocatalysis)
Abstract
:Electrons, photogenerated in conduction bands (CB) and trapped in electron trap defects (Tids) in titanium dioxide (TiO2), play crucial roles in characteristic reductive reactions. This review summarizes the recent progress in the research on electron transfer in photo-excited TiO2. Particularly, the reactivity of electrons accumulated in CB and trapped at Tids on TiO2 is highlighted in the reduction of molecular oxygen and molecular nitrogen, and the hydrogenation and dehalogenation of organic substrates. Finally, the prospects for developing highly active TiO2 photocatalysts are discussed.
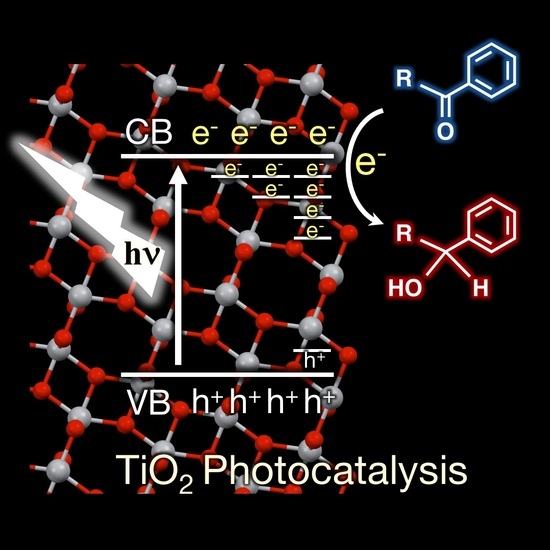
1. Introduction
Since Fujishima and Honda discovered photoelectrochemical water splitting on titanium dioxide (TiO2) photoelectrodes in the early 1970s [1], TiO2 photocatalysis has been applied in various fields, such as the storage of solar energy [2,3,4,5], environmental purification [6], organic synthesis [7,8,9,10,11], anti-bacterial applications [12], and anti-fogging treatments [12,13]. These characteristic photo-functionalities are induced by incident light, in which the behavior of photogenerated electrons and holes, as well as the roles of defects formed on surface and in lattice, are of particular importance. The defect sites are the recombination centers for the photogenerated electrons and holes, because photocatalytic activities decrease with increasing the amount of defects created [2,6,8]. However, Amano et al. reported that the introduction of defect states in TiO2 with H2 reduction treatment greatly enhanced the photocatalytic activity for the water oxidation reaction in aqueous solution [14,15]. Moreover, Kong and coworkers claimed that tuning the relative concentration ratio of bulk defects/surface defects in TiO2 nanocrystal improves the separation efficiencies of photogenerated electrons and holes, thereby enhancing the photocatalytic activity [16]. Thus, further understanding of the defects in TiO2 necessitates the development of highly active photocatalysts.
The properties of defects—such as energy levels, structures, and interactions with adsorbates—have been reviewed by Diebold [17], Henderson [18], and Nowotny [19,20] in detail, but many unanswered questions remain. Recent studies in this field have made the considerable progress during the last decade. This review summarizes the recent progress in the research on the defects in TiO2. Herein, we focus on the properties of electron trap defects formed within the bandgap of TiO2 associated with Ti defects, specifically the intra-bandgap Ti states (Tids). Firstly, the fate of photogenerated electron and holes in TiO2 are described with respect to Tids and hole trap sites in Section 2. Next, the origin of Tids and their energy distribution in TiO2 are considered in Section 3. In Section 4, the reactivity of electrons trapped at Tids and accumulated in the conduction band (CB) on the representative reductive reactions are highlighted. Finally, the prospect for developing a highly active TiO2 catalyst is discussed.
2. Fate of Photogenerated Electrons and Holes in TiO2
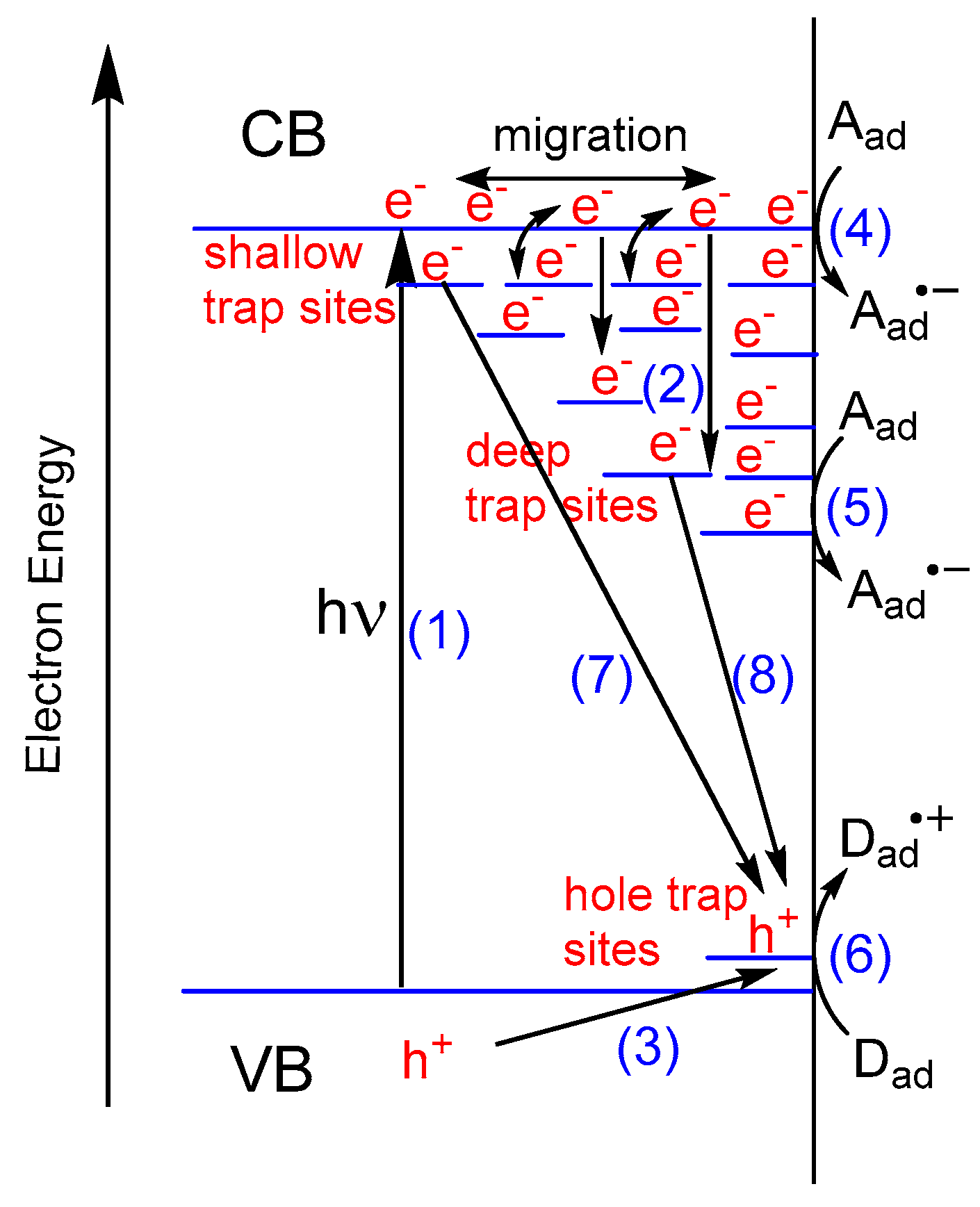
Although several models exist for the charge transport, trapping, and the reaction of photogenerated electrons and holes on photoexcited TiO2, we adopted a schematic model for the anatase TiO2 based on the recent selected reviews and reports as illustrated in Figure 1 [11,21,22,23,24,25].
This model consists of several steps:
- Step 1.
- Electron–hole pair generationTiO2 + hν → TiO2 (e− + h+)
- Step 2.
- Trapping CB electrons (ecb−) at defect Ti4+ sitesTids4+ + ecb− → Tids3+
- Step 3.
- Trapping valence band holes (hvb+) at terminal Ti–OH or surface Ti–O–Ti sitesTi–OsH or Ti–Os–Ti + hvb+ → Ti–OsH・+ or Ti–Os・+−Ti
- Step 4.
- Reduction of adsorbed electron acceptor (Aad) with ecb− at reduction sitesecb− + Aad → Aad・−
- Step 5.
- Reduction of Aad with electrons trapped at defect sites (Tids3+)Tids3+ + Aad → Tids4+ + Aad・−
- Step 6.
- Oxidation of adsorbed electron donor (Dad) by trapped holes at oxidation sitesTi–OsH・+ or Ti–Os・+–Ti + Dad → Ti–OsH or Ti–Os–Ti + Dad・+
- Step 7.
- Recombination of ecb− with trapped holesecb− + Ti–OsH・+ or Ti–Os・+–Ti → Ti–OsH or Ti–Os–Ti
- Step 8.
- Recombination of Tids3+ with trapped holesTids3+ + Ti–OsH・+ or Ti–Os・+–Ti → Tidt4+ + Ti–OsH or Ti–Os–Ti
where time scales for each step are described in brackets [21,22,23,24]. The time scales depend on the crystalline phases, crystallinity, specific surface area, and the presence of bulk and surface defect states in TiO2. The following assumptions were applied to this model: (a) CB electrons (ecb−) contain both electrons in CB and electrons trapped at shallow sites, located just below the CB edge of TiO2 within 0–0.05 eV. These electrons were assumed to be in thermal equilibrium in the bulk CB and at the shallow trap sites; (b) Valence band holes (hvb+) are rapidly transported to the surface hole trap sites (Ti–OsH or Ti–Os–Ti) (Step 3); (c) trapped holes (Ti–OsH・+ or Ti–Os・+–Ti) are the main oxidants for the adsorbed electron donor (Dad) (Step 6); and (d) charge carrier recombination occurs between ecb− and holes trapped at the surface trap sites (Step 7), as well as between electrons trapped at Ti defect states (Tids3+) and holes trapped at the surface trap sites (Step 8), whereas the interband electron–hole carrier recombination (e− + h+→hv or heat) is negligible.
These assumptions can be justified as follows. Tamaki and coworkers described the charge carrier dynamics under weak excitation conditions for nano-crystalline anatase TiO2 samples in femtosecond to microsecond time scales [22,23], which should be compatible with the actual photocatalytic reactions under the usual UV irradiation conditions. They observed the ecb− and hvb+ pair generation within 100 fs, and the ecb− migration between CB and shallow trap sites in equilibrium. These electrons then relaxed to deep trap sites (Tids) with an approximate 500 ps time constant. Meanwhile, hvb+ was rapidly trapped to the surface terminal Ti–OsH sites within 100 fs to create Ti–OsH・+ [22,23]. If the photoinduced event occurred in alcohols, the lifetime of the Ti–OsH・+ generated on the TiO2 surface would be in the nanosecond or sub-nanosecond time scale (approximately 0.1–3 ns in alcohols) due to the fast reaction of Ti–OsH・+ with the abundant alcohol adsorbed on the TiO2 surface [24]. Therefore, the free hvb+ rarely presents in the bulk or on the surface of TiO2, so that ecb− may recombine only with the trapped holes.
3. Origin and Energy Distribution of Electron Trap Defects (Tids)
The bulk and surface Tids are formed in reduced or doped TiO2 in both rutile and anatase phases [17,18,19,20]. As depicted in Diebold’s review [17], the bulk Tids are easily created in the rutile single crystal by thermal annealing in a vacuum, resulting in the formation of blue color centers, indicating high conductivity. Therefore, TiO2 is classified as an n-type semiconductor. The H2 reduction of TiO2 creates both oxygen vacancies and Ti3+ ions, which is an electron trapped in a Ti4+ lattice site, as described in Reaction (1) using Kröger–Vink notation [14,15,19,20]
where is an O2− ion in the oxygen lattice site, is an oxygen vacancy with a double positive charge, and Ti’Ti is a Ti3+ ion in the titanium lattice site. The two Ti’Ti that are created per have two excess electrons, which are responsible for the n-type conductivity, the blue-black colorization, and the enhancement of photocatalytic activity on TiO2. The H2 reduction on TiO2 can also induce a disordered structure in the surface layer of TiO2 nanocrystals, indicated by black TiO2 [26,27,28]. Black TiO2 exhibits high photocatalytic performance in decomposing organic pollutants and in generating hydrogen gas in an aqueous methanol solution under solar light irradiation. The other titanium oxides that have Tids are the F-doped or Nb-doped TiO2, in which oxygen atoms are substituted with fluorine atoms or Ti atoms are replaced with Nb atoms, respectively [29]. Another type of Ti defect in TiO2 is titanium interstitials () possessing excess Ti atoms or ions in the lattice or in the near-surface region on TiO2 surface [17,18,19,20,30].
Facile laser ablation and processing techniques have been developed to introduce the defects into TiO2 nanocrystals and colloids in liquid [31,32,33,34]. In a typical procedure, TiO2 suspensions are irradiated by a high-intensity pulsed laser with frequent repetition rates to produce the characteristic blue-black TiO2. The obtained TiO2 nanoparticles enhanced the photocatalytic activities in decomposing an organic dye [31] and in a water splitting reaction [32].
The electronic energy of Tids is located just below the CB edge in the band gap of TiO2 in a broad range of 0–1.8 eV [35]. Di Valentin and co-workers theoretically calculated the energy levels of point defects in bulk anatase TiO2, which are located at 0.3, 0.4, 0.7, and 0.8 eV below the CB edge, for six-fold-coordinated Ti introduced by F- or Nb-doping, Ti–OH species associated with hydrogen doping, five-coordinated Ti’Ti associated with the oxygen vacancy site, and titanium interstitials , respectively [29]. Deskins et al. calculated the relative energies of Tids formed in the {110}-terminated rutile TiO2 surface by means of the density functional theory (DFT), known as the DFT + U method [36,37]. They modeled the formation of Tids at various Ti sites, such as the five-coordinated Ti and oxygen vacancies [37]. The calculation for the five-coordinated Ti in the presence of surface hydroxyls indicated that deep Tids sites may exist in the second Ti layer from the surface or under the five-coordinated Ti rows [36].
The presence of these Tids species, such as and , can be experimentally confirmed by means of electron spin resonance (ESR) [38,39,40,41,42], infrared radiation (IR) [40,42,43,44,45,46,47,48], ultraviolet-visible absorption (UV–vis) [14,42,49,50,51], photoluminescence (PL) [52], photoacoustic [53,54,55,56], and photoelectron spectroscopies [17,18,30]. Scanning tunneling microscopy (STM) and atomic force microscopy (AFM) are powerful tools for the direct observation of surface Tids [17,18,30,57,58,59]. The oxygen vacancy (Ti’Ti accompanied by ) [57,58,59] and interstitial Ti () [30] sites were directly observed on the TiO2 surface.
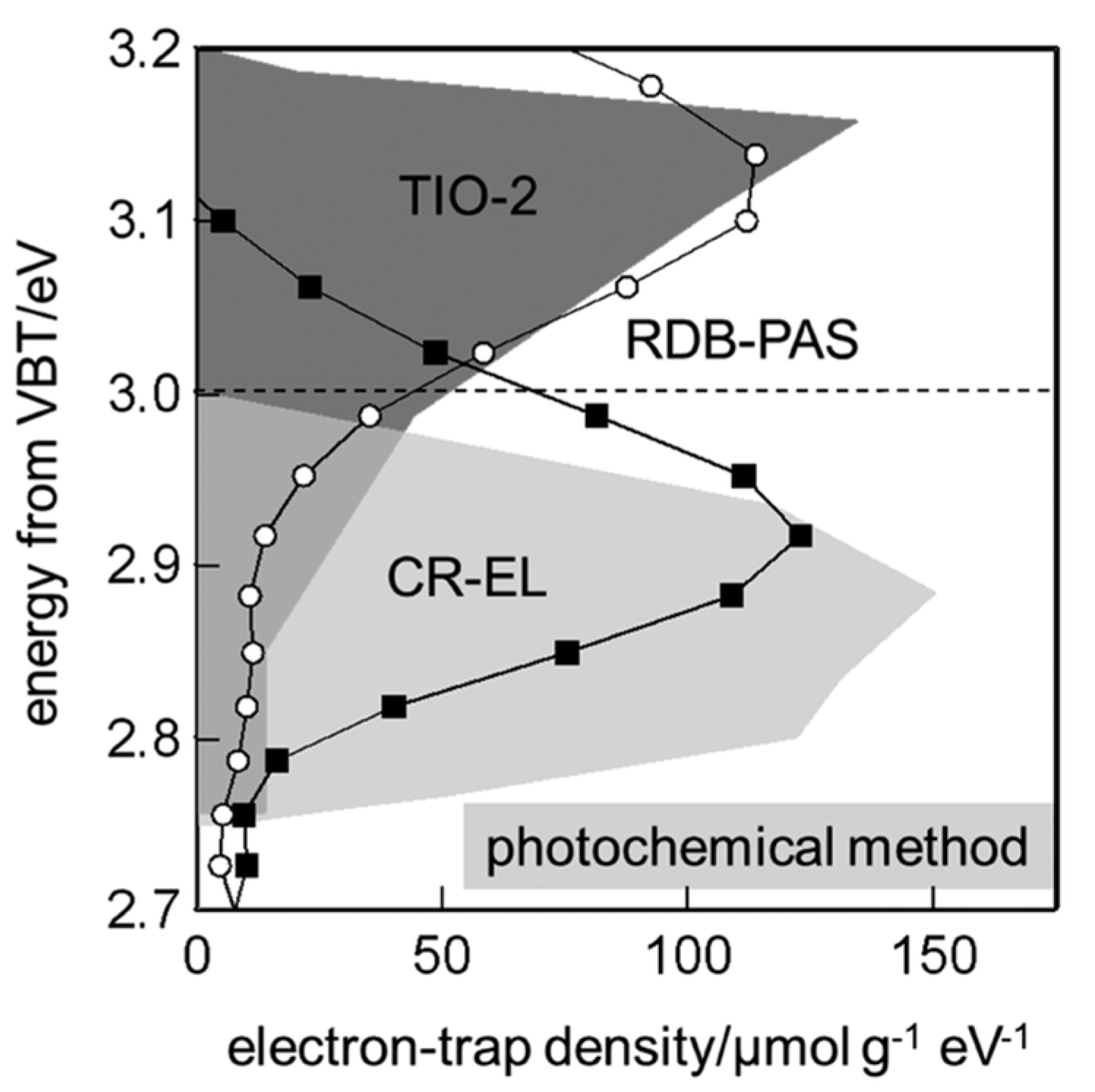
The Tids described above are not the only type of point defects. There are many types of lattice defects including step edges, line defects, grain boundaries, and impurities [17]. The Tids energies can be strongly affected by site heterogeneity due to the local structures. Furthermore, under actual photocatalytic conditions, the TiO2 surface is always covered by adsorbates, especially solvent molecules; thus, the energy of Tids should depend on the adsorbed species [19,20]. Therefore, the information from the theoretical calculations and the photoelectron spectroscopy, applied to the clean catalyst surfaces under ultra-high vacuum conditions, may be limited for actual photocatalytic systems. To address this issue, Ohtani et al. developed a powerful tool for measuring the energy-resolved distribution of electron trap states for many types of TiO2 powders, composed of anatase, rutile, and brookite in methanol-containing gas phase, by means of reversed double-beam photoacoustic spectroscopy (RDB-PAS) [56]. They showed that the electron energy of the trap states is distributed around the CB edge of TiO2 and the distribution range of anatase TiO2 is relatively broader than that of rutile TiO2 (Figure 2). The energy distribution patterns for both anatase and rutile TiO2 powders are similar to those obtained by the photochemical method, which uses the surface reaction of the trapped electrons with methyl viologen to release its cation radical in de-aerated aqueous solution containing methanol as a sacrificial reagent [60]. They also revealed that the total density of the traps is well-correlated to the specific surface area of TiO2 powders, suggesting that the electron trap sites are predominantly located on the surface of TiO2, and they do not depend on the type of crystallites in anatase, rutile, or their mixtures.
The energy distribution of Tids can also be obtained by an electrochemical method [61,62]. In this method, the potential variation in accumulated charge at the TiO2 electrode can be measured in aqueous solution. By calculating the derivative of the accumulated charge (Q) versus the applied potential (U), the energy density of Tids is directly proportional to dQ/dU and the plot of dQ/dU vs. U reflects the energy distribution of Tids. The maximum density of Tids is located around 0.25–0.4 eV below the CB edge for nanostructured anatase TiO2 and P25 TiO2 samples with a ratio of anatase to rutile of approximately 80:20. Thus, the distribution of Tids within 0–0.4 eV is predominant, so that electrons trapped in these trap states may participate in the reductive reaction on TiO2.
4. Reactivity of Trapped and Accumulated Electrons
This section highlights the reactivity of electrons trapped at Tids and accumulated in CB in the reductions of molecular oxygen O2 and molecular nitrogen N2, and the hydrogenation and dehalogenation of selected organic substrates occurring on TiO2. The reductive reactions associated with Tids have been extensively investigated under various experimental conditions and through theoretical calculation methods. Although many studies have been performed for clean surfaces on TiO2 under high-vacuum conditions [17,18,30,57,58,59], here we focus on the reactions occurring on powder or colloidal TiO2 under conventional gas or liquid phase conditions.
Here we define the terms ‘accumulated electrons’ (ecb−) and ‘trapped electrons’ (Tids3+) to distinguish them. Accumulated electrons contain both electrons in CB and electrons trapped at shallow Ti states, located just below the CB edge of TiO2, within 0–0.05 eV. These electrons can be in thermal equilibrium between the bulk CB and at the shallow trap sites, and easily migrate through these states. The accumulation of electrons in these states should occur after saturation of the intra-bandgap Ti states (Tids) during UV irradiation. These electrons are highly reactive at the TiO2 interface (Step 4 in Figure 1).The trapped electrons Tids3+ mean the electrons trapped at Tids are located in relatively deep energy from the CB edge. Therefore, the trapped electrons Tids3+ cannot be excited thermally to the CB or the shallow states, exhibiting either low or no reactivity at the TiO2 interface (Step 5 in Figure 1).
The trapped electrons Tids3+ and the accumulated electrons ecb− are quite stable in the presence of a good sacrificial hole scavenger, such as alcohols or amines, and in the absence of electron acceptors such as O2, the lifetime may exceed several hours [50,51,63]. This extremely long lifetime of Tids3+ and ecb− is attributable to the excellent hole scavenging ability of the sacrificial reagents on the TiO2 surface [24], which prevents the recombination of Tids3+ and ecb− with the surface trapped holes. In other words, the hole scavengers inhibit Steps 7 and 8 in Figure 1. The excess charges caused by electrons Tids3+ and ecb− on the irradiated TiO2 are balanced by the insertion (intercalation) of protons into the TiO2 lattice [47,62,64,65]. The electrons Tids3+ and ecb− show the unique blue-black coloration from the visible to the IR region [14,40,42,43,44,45,46,47,48,49,50,51], which enables the tracing of the lifetimes of the species generated on the irradiated TiO2. Interestingly, the electrons Tids3+ and ecb− are distinguishable by measuring IR spectra. The free electrons ecb− exhibit the typical exponential frequency-dependent spectrum that is attributed to the intra-CB transition [43,44,46,48], whereas the trapped electrons Tids3+ are characterized by a broad absorption in the mid-IR region that is ascribed to a direct optical transition [46,48].
4.1. Reduction of Molecular Oxygen and Hydrogen Peroxide
The interfacial electron transfer between molecular oxygen (O2) and electrons Tids3+ or ecb− on nanocrystalline TiO2 films was examined using a transient UV–vis absorption spectroscopy in gas phase [66]. In the presence of ethanol, as a sacrificial hole scavenger under ethanol-saturated conditions (5.8%), the half-life (t50%) of the electron-species Tids3+ and ecb− was approximately 0.5 s in the absence of O2. The t50% value drastically decreased with increasing O2 concentration, to approximately 12 μs in an oxygen concentration of 21% (air saturated conditions). Thus, the efficient electron transfer of molecular oxygen in a gaseous phase occurred on TiO2 films by using ethanol as the hole scavenger. The dynamics of the electron transfer between O2 and the nanosized TiO2 particles in a liquid phase were also investigated by employing a simple and facile stopped flow technique [50,51]. With methanol as the hole scavenger, the electrons on the TiO2 particles, in an argon-purged and de-aerated aqueous solution, were accumulated by pre-UV irradiation, causing blue colorization characterized by a broad absorption band in the visible light region of 400–800 nm. In the stopped flow experiment, the TiO2 solution containing the electron-species Tids3+ and ecb− was mixed with the aqueous solution containing O2 at pH2.3, and the change in absorbance of the electrons was recorded at 600 nm. The absorbance signal decreased slowly with a rate constant of 8.9 × 10−7 mol L−1 s−1 in the absence of O2, whereas the signal rapidly disappeared within a few seconds under O2-saturated conditions. Thus, efficient electron transfer from the accumulated TiO2 to O2 proceeded even in the aqueous solution. The reduction of hydrogen peroxide (H2O2) was also confirmed by the same stopped flow experiment [50,51]. The decay rate of the electron-species Tids3+ and ecb− in the H2O2 reduction was slower than in the O2 reduction under similar conditions.
The reduction of O2 on TiO2 results in the formation of reactive oxygen species (ROS), such as superoxide anion radicals (O2˙−), hydroperoxy radicals (˙OOH), hydrogen peroxide (H2O2), and hydroxyl radicals (˙OH), under both aqueous and aerated conditions [6,21]. These ROS play a crucial role in the photocatalysis on TiO2 for water purification, air cleaning, self-cleaning, self-sterilization, etc. The sequential ROS generation on photo-irradiated TiO2 under acidic conditions can be depicted as follows [6,21,50,51]:
| Photoreduction: | O2 + (Tids3+ and ecb−) → O2˙− |
| Protonation: | O2˙− + H+ →˙OOH |
| Disproportionation: | 2˙OOH → H2O2 + O2 |
| Photoreduction: | H2O2 + (Tids3+ and ecb−) →˙OH + OH− |
Though the CB edge of anatase TiO2 is located at −0.27 V vs. standard hydrogen electrode (SHE) at pH2.3 [67], anatase TiO2 produced the superoxide anion radial by photoexcitation; O2/O2˙− was approximately 0.33 V vs. SHE [68]. This could be due to the strong adsorption of O2 at the Tids sites, such as , which may lead to a positive shift of the redox potential (O2/O2˙−). In this electron transfer step, an electron seems to be transferred from the Ti 3d orbital to the π* orbital of the adsorbed O2.
4.2. Reduction of Molecular Nitrogen, Nitrate, and Nitrite Ions
Molecular nitrogen (N2) is chemically stable, so photocatalytic reduction of N2 to ammonia (NH3) under ambient temperature and pressure is challenging. Hirakawa and coworkers recently reported that the photocatalytic conversion of N2 to ammonia with water occurred on the bare TiO2 powders under ambient conditions [69]. They stated that the active sites for N2 reduction are the Tids with oxygen vacancies mainly formed on the rutile {110} surface. They investigated the photocatalytic reductions of nitrate (NO3−) and nitrite (NO2−) ions to ammonia and N2 on bare TiO2 under ambient conditions [70]. They proposed that the Tids sites selectively promoted the eight-electron reduction of NO3− to NH3 (NO3− + 9H+ + 8e− → NH3 + 3H2O), while the Lewis acid site promoted nonselective reduction, resulting in N2 and NH3 formation. Thus, TiO2 with many Tids and a small number of Lewis acid sites produced ammonia with very high selectivity. The use of artificial fertilizers in agriculture has caused a great deal of concern for water pollution caused by the production of NO3− and NO2− ions from fertilizers [71]. Therefore, a chemical process for the reduction of NO3− and NO2− ions on TiO2 may be useful for an environmental recycling process.
4.3. Hydrogenation of Carbonyl Compounds
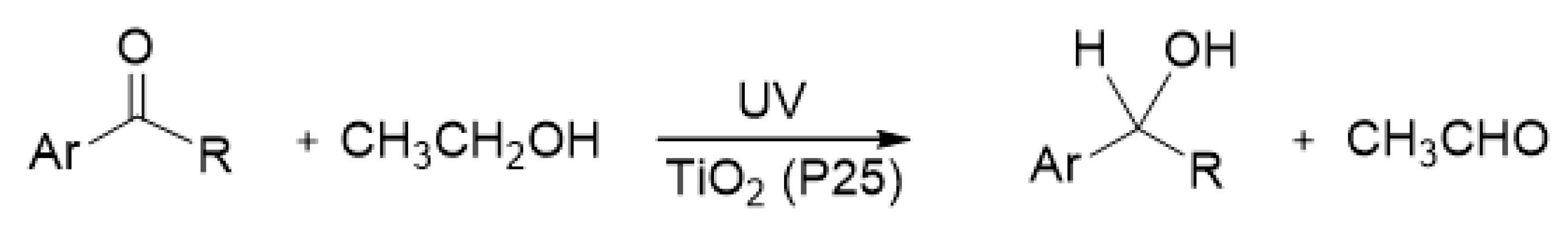
Kohtani et al. examined whether electrons Tids3+ and ecb− transfer to acetophenone (AP) derivatives adsorbed on TiO2 [63,72]. The photoreductive hydrogenation of several aromatic carbonyl compounds was confirmed to occur on UV-irradiated P25 TiO2 as illustrated in Scheme 1 [72,73]. They evaluated the number of transferred electrons in the injection experiment using a pre-irradiated TiO2 suspension. When the P25 TiO2 powder was dispersed in de-aerated ethanol as a hole scavenger and irradiated with UV light for 2 h, the white color of TiO2 powder changed to blue-gray. After the blue-gray color change was confirmed, a large amount of AP derivatives was injected into this TiO2 suspension in the dark. In this experiment, ethanol acted not only as a solvent but also as a hole scavenger.
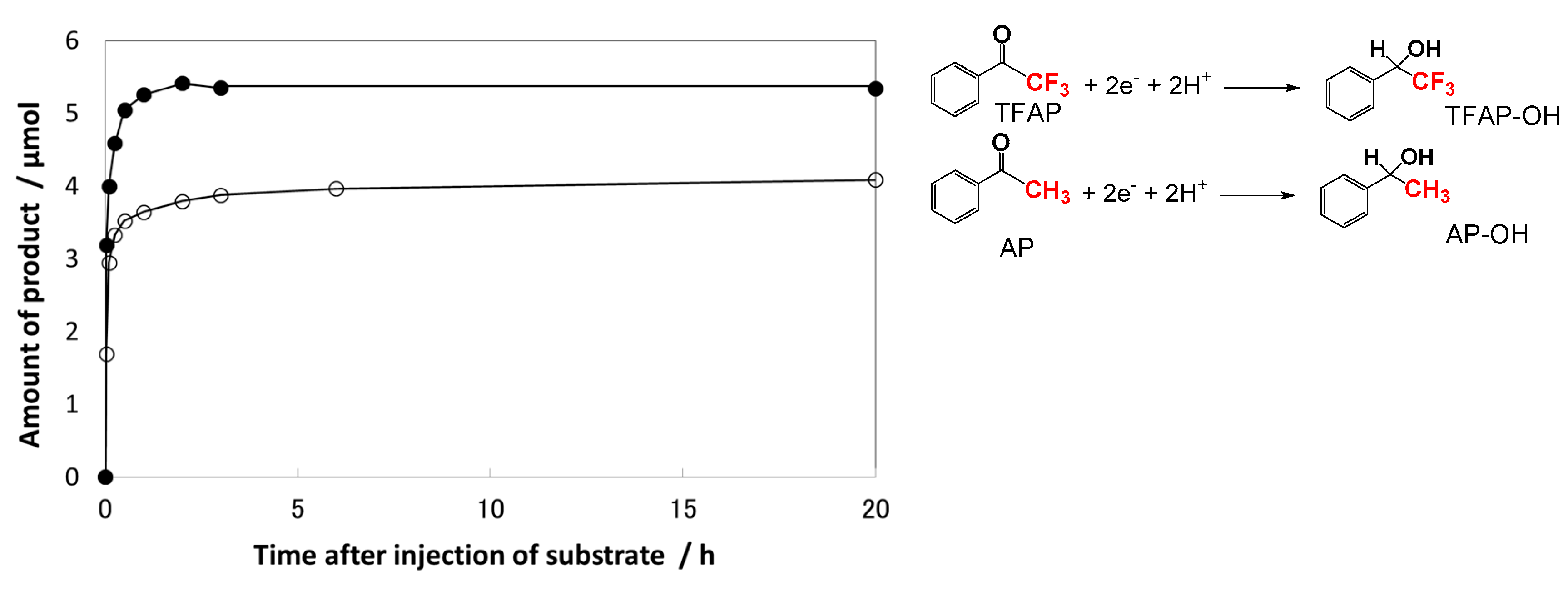
Figure 3a shows that the blue-gray color of the pre-irradiated P25 TiO2 suspension remained for a few days in the absence of AP derivatives [63], meaning that the electrons accumulated on the P25 TiO2 surface are quite stable in the de-aerated ethanol. Figure 3b,c show the color change induced by the addition of aromatic carbonyl compounds. The blue-gray color of TiO2 rapidly changed after the injection of 2,2,2-trifluoacetophenone (TFAP) where the aromatic ring (Ar) was C6H5 and the R was CF3 (Scheme 1). The change from blue-gray to white was completed within 3 h in the TFAP solution as shown in Figure 3c. This result indicates that all the trapped and accumulated electrons on TiO2 were consumed in the reduction of TFAP within 3 h. On the other hand, with AP, a part of the blue-gray species on TiO2 was remarkably stable even after 50 h, as shown in Figure 3b, which may be due to the remaining electrons trapped at the deep states of TiO2.
Figure 4 depicts the time evolution of the secondary alcohols 1-phenylethanol (AP-OH) or 1-phenyl-2,2,2-trifluoroethanol (TFAP-OH), as products after the injection of substrates AP or TFAP, into the sufficient pre-irradiated TiO2 suspension, respectively [63]. The amount of each product quickly grew within 0.5 h, which agrees with the observation of color change in the TiO2 suspension (Figure 3b,c). The amount of TFAP-OH product obtained from the reactive substrate TFAP rapidly increased, and reached 5.4 μmoL within 1 h. Assuming all Tids3+ and ecb− electrons were consumed in the reductive hydrogenation of TFAP, the total amount of Tids3+ and ecb− on the TiO2 powder was estimated to be about 100 μmol∙g−1. The time evolution of AP-OH from the less reactive substrate AP consisted of a fast component within 0.5 h, and a slow component after 0.5 h, which increased gradually to reach 4.1 μmol (Figure 4). The slow component represents the slow electron transfer event from middle Tids, between shallow and deep states, to AP adsorbed on TiO2. The total amount of AP-OH production was about 25% smaller than that of TFAP-OH production. In the reduction of the less reactive substrate AP, the deep Tids3+ species remained on the TiO2 surface for a long time (>20 h) after the injection of AP. The amount of deep Tids3+ that remained on TiO2 was roughly estimated to be 25% given the difference between the amounts of TFAP-OH (5.4 μmoL) and AP-OH (4.1 μmoL). Thus, the residual 25% electrons could not react with AP and remained at the deep trap sites.
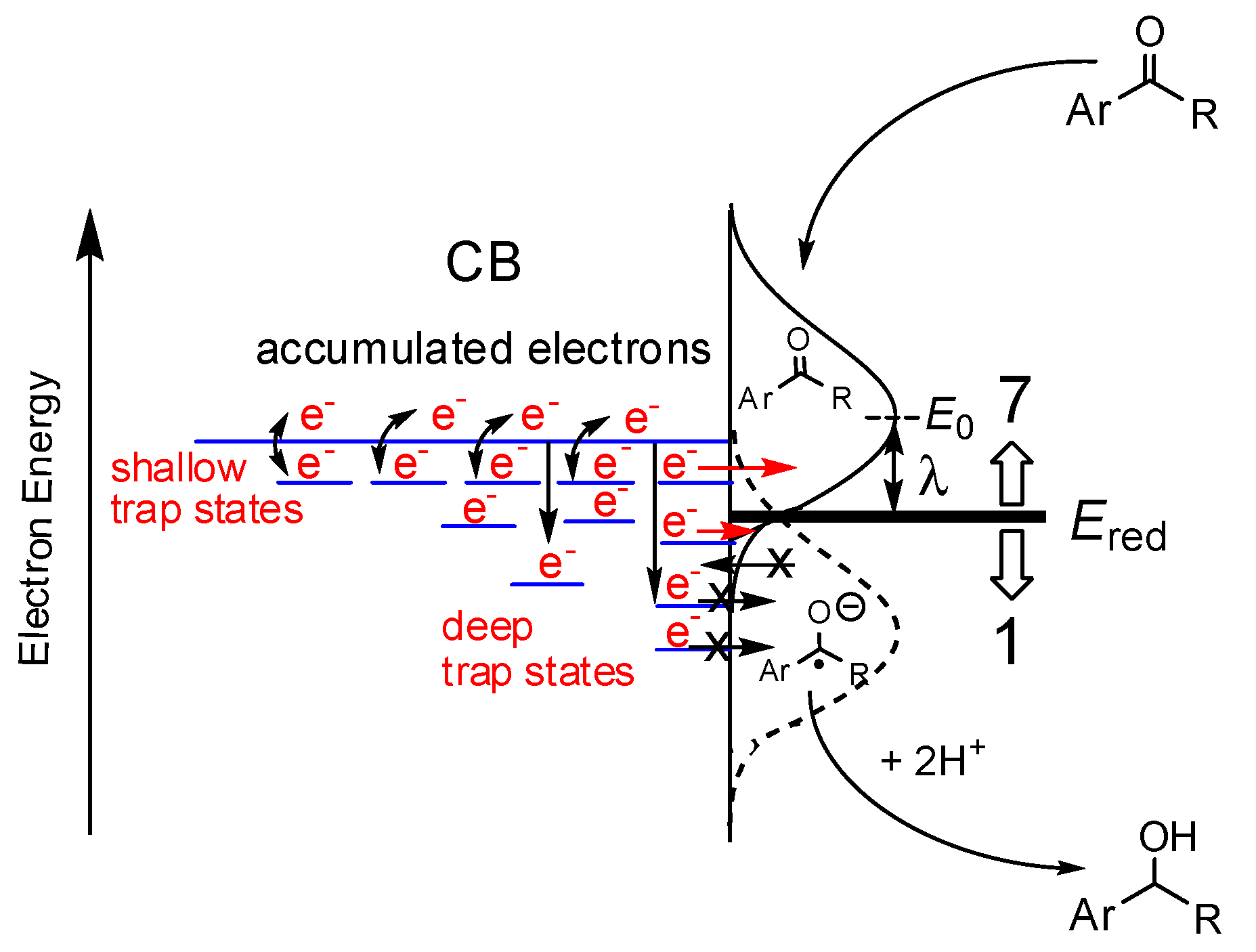
Assuming that all Tids3+ and ecb− electrons react with TFAP, the percentages of reacted electrons were estimated for other AP derivatives, as summarized in Table 1 [72]. The number of reacted electrons showed a tendency to decrease with decreasing the reduction potential (Ered) of the substrates according to the dependence on the actual reaction rates as listed in Table 1. This implies that the rates of photocatalytic hydrogenation of AP derivatives are governed by the electron transfer efficiency from the Tids sites to the adsorbed AP sites. Notably, the relative position between the Tids energy distributed within the bandgap and the acceptor level of the AP derivatives (the solid Gaussian curve) should be appropriate (Figure 5) [72]. The energy distribution of Tids in TiO2 powders and colloids can be obtained by photochemical [60], electrochemical [61,62], and RDB-PAS [56] methods. The acceptor levels of AP derivatives can be estimated by the Marcus theory [72,74,75,76].
Kohtani et al. also reported the photohydrogenation of AP derivatives on P25 TiO2, modified with metal-free organic dyes such as rhodamine B, fluorescein, and coumarin derivatives [77,78]. The use of these organic dyes successfully extended the UV response of TiO2 to the visible light region, though these reaction rates were much slower than the hydrogenation rate using UV excitation of non-modified TiO2. In this dye-sensitized system, the electron injection from dye into TiO2 can take place in two different ways: (1) injection via a lowest unoccupied molecular orbital (LUMO) level of the excited dye to the CB of TiO2, and (2) direct injection of TiO2 to CB on the excitation of the charge transfer complex (TiO2δ− dyeδ+) [77]. The injected electrons should then be distributed to the Tids sites on the P25 TiO2 surface. The accumulated electrons were observed with the blue-gray color for all dye-TiO2 powders during visible light irradiation.
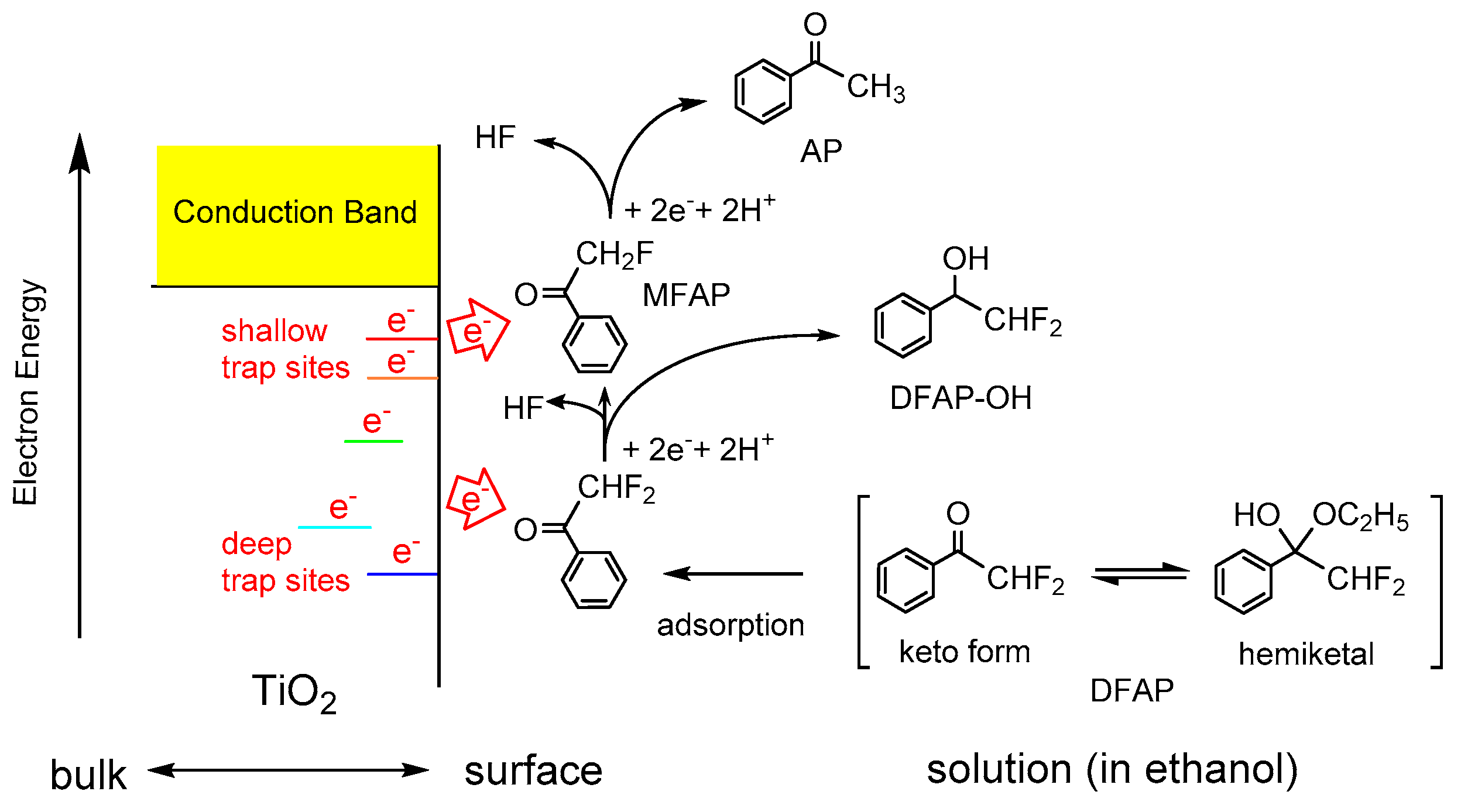
4.4. Defluorination of Fluorinated AP Derivatives
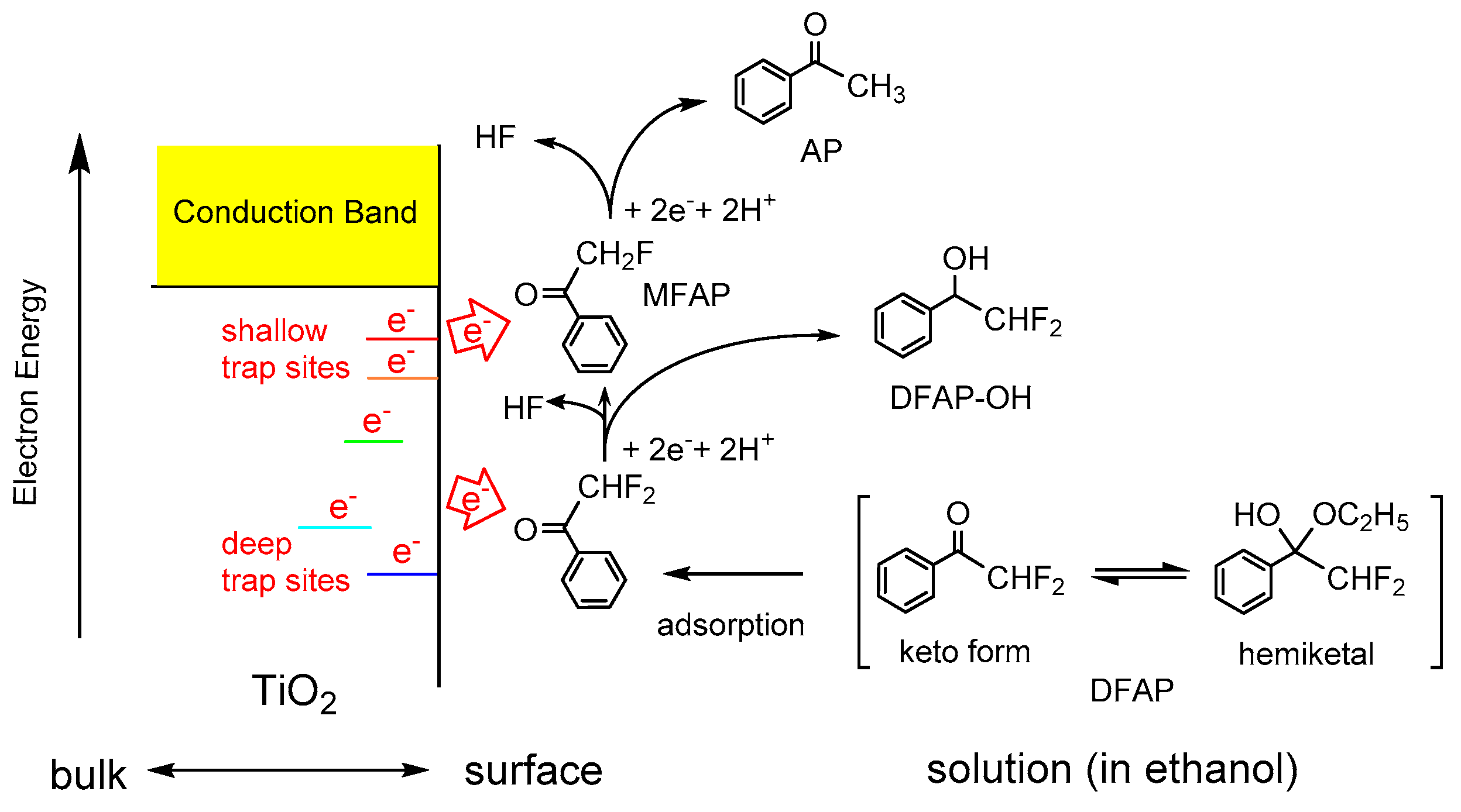
Compounds containing fluorine atoms are often used as pharmaceutical and agrochemical reagents. Since the C–F bond is one of the strongest bonds, C–F bond activation and cleavage is a field of current interest in organic chemistry [79], although less is known about the catalytic activation and cleavage methods. Photocatalytic reaction is one of the promising methods to promote the activation and cleavage of the C–F bond of fluorinated compounds under mild conditions. Therefore, an attempt was made to use trapped and accumulated electrons on TiO2 for the sequential multi-step electron transfer in the reduction of fluorinated AP derivatives (Figure 6) [80]. The reaction of fluorinated AP derivatives on TiO2 showed the two photocatalytic reductive transformations, i.e., the defluorination and reduction of the carbonyl group. The reduction of 2-fluoromethylacetophenone (MFAP) only provided the ketone AP because of the C–F bond cleavage, whereas the reaction of TFAP only provided the alcohol TFAP-OH as a result of the reduction of the carbonyl group. Interestingly, the reduction of 2,2-difluoromethyacetophenone (DFAP), possessing characteristics between those of MFAP and TFAP, gave the defluorinated ketones, MFAP and AP, as well as hydrogenated alcohol 1-phenyl-2,2-difluoroethanol (DFAP-OH), as shown in Figure 6. The defluorination reactions became unfavorable with increasing the number of fluorine atoms on the substrates. This mainly arises because of the increase in the bond dissociation energy of the C–F bond and the positive shift of the reduction potential of fluorinated AP derivatives with the increasing number of fluorine atoms [80].
4.5. Hydrogenation of Nitroaromatic Compounds
Several organic nitroaromatic compounds can be easily hydrogenated to create the corresponding amino compounds on the UV-irradiated TiO2 in the presence of 2-propanol as a sacrificial hole scavenger (Scheme 2).
Shiraishi et al. reported that some kinds of rutile TiO2 particles promote the highly efficient photocatalytic hydrogenation of nitroaromatic compounds [81,82]. They claimed that the oxygen vacancy sites on the rutile {110} behave as the adsorption sites for the nitroaromatic compounds and the electron trap sites, resulting in the formation of aniline derivatives with significantly high yields of greater than 25% at 370 nm [81]. They also found that the activity of rutile particles depends on the number of defects on the particles [82]. The inner (bulk) defects behave as the deactivation sites for the recombination of electrons and holes, whereas the surface defects behave as the active reaction sites as well as the deactivation sites. As a result, the reaction rate is proportional to the ratio of the amount of surface defects to that of total defects (Nsurface/Ntotal) in the rutile TiO2 particles, which aligns with the report of Kong and coworkers [16].
Molinari and coworkers examined the selective hydrogenation of NO2–C6H4–CHO, bearing the two reducible functional groups, –NO2 and –CHO [62]. They found that the nitro group was easily reduced by the trapped electrons at Tids within the bandgap, whereas the aldehyde group was reduced by electrons accumulated on CB. Therefore, the chemoselective reduction of functional groups can be controlled by the energy distribution patterns of Tids, which may depend on the type of TiO2 powders. The selective reduction of functional groups is difficult to achieve through conventional thermal catalysis. Therefore, this topic is an interesting issue for chemoselective photocatalysis.
5. Summary and Potential for the Development of Efficient Photocatalysis
According to the reports of Kong et al. [16] and Shiraishi et al. [82], the photocatalytic efficiencies increased with increasing the ratio of the amount of surface to bulk defects (Nsurface/Nbulk) or the amount of surface to total defects (Nsurface/Ntotal). For example, molecular oxygen (O2) in a gaseous phase would be easily adsorbed on the surface Tids and reduced by electrons Tids3+ and ecb−, resulting in the efficient formation of ROS, which oxidize benzene efficiently [16]. Further, the surface Tids behaves as the active reaction site in the efficient reduction of nitrobenzene [81]. Thus, the surface defects favorably act as adsorption sites as well as reaction sites.
In addition, the relative position between the energy distributions of Tids and the acceptor level (reduction potential) of substrates should be appropriate as indicated in Figure 5 [72]. The electrons accumulated in CB and trapped at shallow Tids can easily participate in the reaction, whereas those trapped at deep Tids cannot [63]. These unreacted electrons remain at the deep Tids sites and exhibit an extremely long lifetime in alcohols. Thus, the shallow traps enhance photocatalytic activity, while the deep traps cause a reduction. Furthermore, Amano et al. proposed that the creation of shallow Tids greatly enhances electrical conductivity, thereby facilitating the charge transport and separation caused by the formation of band bending in the space charge layer at the TiO2-liquid interface [14,15].
In conclusion, the development of highly active photocatalysts necessitates precise control of the structural properties; the density of surface shallow traps should be maximized and the density of deep traps as well as inner (bulk) traps minimized, as proposed by Ohtani [83]. One of the promising strategies for meeting these requirements is the use of highly uniform TiO2 nanocrystals with specific exposure of the reactive facets [42,84,85,86,87]. In particular, the anatase {101} and {001} facets have been reported to be favorable for the reductive and oxidative reactions in TiO2 photocatalysis, respectively [87,88,89,90,91]. If the reductive and oxidative facets could be selectively covered with a large amount of the active shallow Tids and the terminal Ti–OsH hole trapping sites, respectively, the photocatalytic activity on the TiO2 nanocrystals would be greatly enhanced by the effective electron-hole charge separation, followed by the subsequent charge transfer reactions at the specific reductive and oxidative sites. Therefore, special attention should be directed toward the development of TiO2 nanocrystals with precisely controlled facets.
Acknowledgments
Conflicts of Interest
The author declares no conflict of interest.
References
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Zhang, X.; Tryk, D. Heterogeneous photocatalysis: From water photolysis to applications in environmental cleanup. Int. J. Hydrog. Energy 2007, 32, 2664–2672. [Google Scholar] [CrossRef]
- Grätzel, M. Recent advances in sensitized mesoscopic solar cells. Acc. Chem. Res. 2009, 42, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Martin, S.; Choi, W.; Bahnemann, D. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Sakata, T. Photocatalysis of irradiated semiconductor surfaces: Its application to water splitting and some organic reactions. J. Photochem. 1985, 29, 205–215. [Google Scholar] [CrossRef]
- Kisch, H. Semiconductor photocatalysis for organic synthesis advances in photochemistry. Adv. Photochem. 2001, 26, 93–143. [Google Scholar]
- Palmisano, G.; Augugliaro, V.; Pagliaro, M.; Palmisano, L. Photocatalysis: A promising route for 21st century organic chemistry. Chem. Commun. 2007, 3425–3437. [Google Scholar] [CrossRef] [PubMed]
- Kohtani, S.; Yoshioka, E.; Miyabe, H. Photocatalytic hydrogenation on semiconductor particles. Hydrogenation 2012, 291–308. [Google Scholar] [CrossRef]
- Kisch, H. Semiconductor photocatalysis-mechanistic and synthetic aspects. Angew. Chem. Int. Ed. 2013, 52, 812–847. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Rao, T.; Tryk, D. Titanium dioxide photocatalysis. J. Photochem. Photobiol. C 2000, 1, 1–21. [Google Scholar] [CrossRef]
- Thompson, T.; Yates, J. Surface science studies of the photoactivation of TiO2 new photochemical processes. Chem. Rev. 2006, 106, 4428–4453. [Google Scholar] [CrossRef] [PubMed]
- Amano, F.; Nakata, M.; Yamamoto, A.; Tanaka, T. Effect of Ti3+ ions and conduction band electrons on photocatalytic and photoelectrochemical activity of rutile titania for water oxidation. J. Phys. Chem. C 2016, 120, 6467–6474. [Google Scholar] [CrossRef]
- Amano, F.; Nakata, M.; Yamamoto, A.; Tanaka, T. Rutile titanium dioxide prepared by hydrogen reduction of degussa P25 for highly efficient photocatalytic hydrogen evolution. Catal. Sci. Technol. 2016, 6, 5693–5699. [Google Scholar] [CrossRef]
- Kong, M.; Li, Y.; Chen, X.; Tian, T.; Fang, P.; Zheng, F.; Zhao, X. Tuning the relative concentration ratio of bulk defects to surface defects in TiO2 nanocrystals leads to high photocatalytic efficiency. J. Am. Chem. Soc. 2011, 133, 16414–16417. [Google Scholar] [CrossRef] [PubMed]
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Henderson, M. A Surface science perspective on TiO2 photocatalysis. Surf. Sci. Rep. 2011, 66, 185–297. [Google Scholar] [CrossRef]
- Nowotny, M.K.; Sheppard, L.R.; Bak, T.; Nowotny, J. Defect chemistry of titanium dioxide. Application of defect engineering in processing of TiO2-based photocatalysts. J. Phys. Chem. C 2008, 112, 5275–5300. [Google Scholar] [CrossRef]
- Nowotny, J.; Alim, M.; Bak, T.; Idris, M.; Ionescu, M.; Prince, K.; Sahdan, M.; Sopian, K.; Mat Teridi, M.; Sigmund, W. Defect chemistry and defect engineering of TiO2-based semiconductors for solar energy conversion. Chem. Soc. Rev. 2015, 44, 8424–8442. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Furube, A.; Murai, M.; Hara, K.; Katoh, R.; Tachiya, M. Dynamics of efficient electron-hole separation in TiO2 nanoparticles revealed by femtosecond transient absorption spectroscopy under the weak-excitation condition. Phys. Chem. Chem. Phys. 2007, 9, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Hara, K.; Katoh, R.; Tachiya, M.; Furube, A. Femtosecond visible-to-IR spectroscopy of TiO2 nanocrystalline films: Elucidation of the electron mobility before deep trapping. J. Phys. Chem. C 2009, 113, 11741–11746. [Google Scholar] [CrossRef]
- Tamaki, Y.; Furube, A.; Murai, M.; Hara, K.; Katoh, R.; Tachiya, M. Direct observation of reactive trapped holes in TiO2 undergoing photocatalytic oxidation of adsorbed alcohols: Evaluation of the reaction rates and yields. J. Am. Chem. Soc. 2006, 128, 416–417. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, X.; Jia, Y.; Chen, X.; Han, H.; Li, C. Titanium dioxide-based nanomaterials for photocatalytic fuel generations. Chem. Rev. 2014, 114, 9987–10043. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, L.; Yu, P.Y.; Mao, S.S. Increasing solar absorption for photocatalysis with black hydrogenated titanium dioxide nanocrystals. Science 2011, 331, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, L.; Huang, F. Black titanium dioxide (TiO2) nanomaterials. Chem. Soc. Rev. 2015, 44, 1861–1885. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Li, W.; Wang, J.-Q.; Qu, Y.; Yang, Y.; Xie, Y.; Zhang, K.; Wang, L.; Fu, H.; Zhao, D. Ordered mesoporous black TiO2 as highly efficient hydrogen evolution photocatalyst. J. Am. Chem. Soc. 2014, 136, 9280–9283. [Google Scholar] [CrossRef] [PubMed]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Reduced and n-type doped TiO2: Nature of Ti3+ species. J. Phys. Chem. C 2009, 113, 20543–20552. [Google Scholar] [CrossRef]
- Wendt, S.; Sprunger, P.T.; Lira, E.; Madsen, G.K.H.; Li, Z.; Hansen, J.O.; Matthiesen, J.; Blekinge Rasmussen, A.; Laegsgaard, E.; Hammer, B.; et al. The role of interstitial sites in the Ti3d defect state in the band gap of titania. Science 2008, 320, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, D.; Liu, K.; Wang, C.; Liu, L.; Li, B.; Zhang, Z.; Shen, D. Laser-modified black titanium oxide nanospheres and their photocatalytic activities under visible light. ACS Appl. Mater. Interfaces 2015, 7, 16070–16077. [Google Scholar] [CrossRef] [PubMed]
- Filice, S.; Compagnini, G.; Fiorenza, R.; Scirè, S.; D’Urso, L.; Fragalà, M.E.; Russo, P.; Fazio, E.; Scalese, S. Laser processing of TiO2 colloids for an enhanced photocatalytic water splitting activity. J. Colloid Interface Sci. 2017, 489, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Liang, R.; He, R.X.; Zhou, Y.N. Phase transformation of TiO2 nanoparticles by femtosecond laser ablation in aqueous solutions and deposition on conductive substrates. Nanoscale 2017, 9, 6167–6177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, J.; Li, P.; Tian, Z.; Liang, C. Recent advances in surfactant-free, surface-charged, and defect-rich catalysts developed by laser ablation and processing in liquids. ChemNanoMat 2017, 3, 512–533. [Google Scholar] [CrossRef]
- Weiler, B.; Gagliardi, A.; Lugli, P. Kinetic monte carlo simulations of defects in anatase titanium dioxide. J. Phys. Chem. C 2016, 120, 10062–10077. [Google Scholar] [CrossRef]
- Deskins, N.A.; Rousseau, R.; Dupuis, M. Localized electronic states from surface hydroxyls and polarons in TiO2 (110). J. Phys. Chem. C 2009, 113, 14583–14586. [Google Scholar] [CrossRef]
- Deskins, N.A.; Rousseau, R.; Dupuis, M. Distribution of Ti3+ surface sites in reduced TiO2. J. Phys. Chem. C 2011, 115, 7562–7572. [Google Scholar] [CrossRef]
- Howe, R.; Grätzel, M. EPR observation of trapped electrons in colloidal titanium dioxide. J. Phys. Chem. 1985, 89, 4495–4499. [Google Scholar] [CrossRef]
- Hurum, D.; Agrios, A.; Gray, K.; Rajh, T.; Thurnauer, M. Explaining the enhanced photocatalytic activity of degussa P25 mixed-phase TiO2 using EPR. J. Phys. Chem. B 2003, 107, 4545–4549. [Google Scholar] [CrossRef]
- Berger, T.; Sterrer, M.; Diwald, O.; Knözinger, E.; Panayotov, D.; Thompson, T.L.; Yates, J.T. Light-induced charge separation in anatase TiO2 particles. J. Phys. Chem. B 2005, 109, 6061–6068. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Dimitrijevic, N.; Chen, L.; Nichols, J.; Rajh, T.; Gray, K. The important role of tetrahedral Ti4+ sites in the phase transformation and photocatalytic activity of TiO2 nanocomposites. J. Am. Chem. Soc. 2008, 130, 5402–5403. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Cargnello, M.; Paik, T.; Mangolini, F.; Weber, R.; Fornasiero, P.; Murray, C. Nonaqueous synthesis of TiO2 nanocrystals using TiF4 to engineer morphology, oxygen vacancy concentration, and photocatalytic activity. J. Am. Chem. Soc. 2012, 134, 6751–6761. [Google Scholar] [CrossRef] [PubMed]
- Szczepankiewicz, S.; Colussi, A.J.; Hoffmann, M. Infrared spectra of photoinduced species on hydroxylated titania surfaces. J. Phys. Chem. B 2000, 104, 9842–9850. [Google Scholar] [CrossRef]
- Szczepankiewicz, S.; Moss, J.; Hoffmann, M. Slow surface charge trapping kinetics on irradiated TiO2. J. Phys. Chem. B 2002, 106, 2922–2927. [Google Scholar] [CrossRef]
- Takeuchi, M.; Martra, G.; Coluccia, S.; Anpo, M. Verification of the photoadsorption of H2O molecules on TiO2 semiconductor surfaces by vibrational absorption spectroscopy. J. Phys. Chem. C 2007, 111, 9811–9817. [Google Scholar] [CrossRef]
- Panayotov, D.; Burrows, S.; Morris, J. Infrared spectroscopic studies of conduction band and trapped electrons in UV-photoexcited, H-atom n-doped, and thermally reduced TiO2. J. Phys. Chem. C 2012, 116, 4535–4544. [Google Scholar] [CrossRef]
- Savory, D.; McQuillan, A.J. Influence of formate adsorption and protons on shallow trap infrared absorption (STIRA) of anatase TiO2 during photocatalysis. J. Phys. Chem. C 2013, 117, 23645–23656. [Google Scholar] [CrossRef]
- Litke, A.; Hensen, E.J.M.; Hofmann, J. Role of dissociatively adsorbed water on the formation of shallow trapped electrons in TiO2 photocatalysts. J. Phys. Chem. C 2017, 121, 10153–10162. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Deguchi, J.; Sakai, S.; Anpo, M. Effect of H2O vapor addition on the photocatalytic oxidation of ethanol, acetaldehyde and acetic acid in the gas phase on TiO2 semiconductor powders. Appl. Catal. B Environ. 2010, 96, 218–223. [Google Scholar] [CrossRef]
- Mohamed, H.; Dillert, R.; Bahnemann, D. Reaction dynamics of the transfer of stored electrons on TiO2 nanoparticles: A stopped flow study. J. Photochem. Photobiol. A Chem. 2011, 217, 271–274. [Google Scholar] [CrossRef]
- Mohamed, H.; Mendive, C.; Dillert, R.; Bahnemann, D. Kinetic and mechanistic investigations of multielectron transfer reactions induced by stored electrons in TiO2 nanoparticles: A stopped flow study. J. Phys. Chem. A 2011, 115, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Knorr, F.; Mercado, C.; McHale, J. Trap-state distributions and carrier transport in pure and mixed-phase TiO2: Influence of contacting solvent and interphasial electron transfer. J. Phys. Chem. C 2008, 112, 12786–12794. [Google Scholar] [CrossRef]
- Leytner, S.; Hupp, J. Evaluation of the energetics of electron trap states at the nanocrystalline titanium dioxide/aqueous solution interface via time-resolved photoacoustic spectroscopy. Chem. Phys. Lett. 2000, 330, 231–236. [Google Scholar] [CrossRef]
- Murakami, N.; Prieto Mahaney, O.; Torimoto, T.; Ohtani, B. Photoacoustic spectroscopic analysis of photoinduced change in absorption of titanium(IV) oxide photocatalyst powders: A novel feasible technique for measurement of defect density. Chem. Phys. Lett. 2006, 426, 204–208. [Google Scholar] [CrossRef]
- Murakami, N.; Prieto Mahaney, O.; Abe, R.; Torimoto, T.; Ohtani, B. Double-beam photoacoustic spectroscopic studies on transient absorption of titanium(IV) oxide photocatalyst powders. J. Phys. Chem. C 2007, 111, 11927–11935. [Google Scholar] [CrossRef]
- Nitta, A.; Takase, M.; Takashima, M.; Murakami, N.; Ohtani, B. A fingerprint of metal-oxide powders: Energy-resolved distribution of electron traps. Chem. Commun. 2016, 52, 12096–12099. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.; Lun Pang, C.; Lindsay, R.; Thornton, G. Chemical reactions on rutile TiO2 (110). Chem. Soc. Rev. 2008, 37, 2328–2353. [Google Scholar] [CrossRef] [PubMed]
- Papageorigiou, A.; Papageorgiou, A.C.; Beglitis, N.S.; Pang, C.L.; Teobaldi, G.; Cabailh, G.; Chen, Q.; Fisher, A.J.; Hofer, W.A.; Thornton, G. Electron traps and their effect on the surface chemistry of TiO2 (110). Proc. Natl. Acad. Sci. USA 2010, 107, 2391–2396. [Google Scholar] [CrossRef] [PubMed]
- Setvin, M.; Aschauer, U.; Scheiber, P.; Li, Y.F.; Hou, W.; Schmid, M.; Selloni, A.; Diebold, U. Reaction of O2 with subsurface oxygen vacancies on TiO2 anatase (101). Science 2013, 341, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Sugiyama, N.; Murakami, S.; Kominami, H.; Kera, Y.; Noguchi, H.; Uosaki, K.; Torimoto, T.; Ohtani, B. Quantitative analysis of defective sites in titanium(IV) oxide photocatalyst powders. Phys. Chem. Chem. Phys. 2003, 5, 778–783. [Google Scholar] [CrossRef]
- Wang, H.; He, J.; Boschloo, G.; Lindström, H.; Hagfeldt, A.; Lindquist, S.-E. Electrochemical investigation of traps in a nanostructured TiO2 film. J. Phys. Chem. B 2001, 105, 2529–2533. [Google Scholar] [CrossRef]
- Molinari, A.; Maldotti, A.; Amadelli, R. Probing the role of surface energetics of electrons and their accumulation in photoreduction processes on TiO2. Chem. Eur. J. 2014, 20, 7759–7765. [Google Scholar] [CrossRef] [PubMed]
- Kohtani, S.; Yoshioka, E.; Saito, K.; Kudo, A.; Miyabe, H. Adsorptive and kinetic properties on photocatalytic hydrogenation of aromatic ketones upon UV irradiated polycrystalline titanium dioxide: Differences between acetophenone and its trifluoromethylated derivative. J. Phys. Chem. C 2012, 116, 17705–17713. [Google Scholar] [CrossRef]
- Lemon, B.; Hupp, J. Photochemical quartz crystal microbalance study of the nanocrystalline titanium dioxide semiconductor electrode/water interface: Simultaneous photoaccumulation of electrons and protons. J. Phys. Chem. 1996, 100, 14578–14580. [Google Scholar] [CrossRef]
- Jimenez, J.; Jiménez, J.; Bourret, G.; Berger, T.; McKenna, K. Modification of charge trapping at particle/particle interfaces by electrochemical hydrogen doping of nanocrystalline TiO2. J. Am. Chem. Soc. 2016, 138, 15956–15964. [Google Scholar] [CrossRef] [PubMed]
- Peiró, A.; Colombo, C.; Doyle, G.; Nelson, J.; Mills, A.; Durrant, J. Photochemical reduction of oxygen adsorbed to nanocrystalline TiO2 films: A transient absorption and oxygen scavenging study of different TiO2 preparations. J. Phys. Chem. B 2006, 110, 23255–23263. [Google Scholar] [CrossRef] [PubMed]
- Dung, D.; Ramsden, J.; Grätzel, M. Dynamics of interfacial electron-transfer processes in colloidal semiconductor systems. J. Am. Chem. Soc. 1982, 104, 2977–2985. [Google Scholar] [CrossRef]
- Ilan, Y.; Meisel, D.; Czapski, G. The redox potential of the O2/O2− system in aqueous media. Isr. J. Chem. 1974, 12, 891–895. [Google Scholar] [CrossRef]
- Hirakawa, H.; Hashimoto, M.; Shiraishi, Y.; Hirai, T. Photocatalytic conversion of nitrogen to ammonia with water on surface oxygen vacancies of titanium dioxide. J. Am. Chem. Soc. 2017, 139, 10929–10936. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, H.; Hashimoto, M.; Shiraishi, Y.; Hirai, T. Selective nitrate-to-ammonia transformation on surface defects of titanium dioxide photocatalysts. ACS Catal. 2017, 7, 3713–3720. [Google Scholar] [CrossRef]
- Burt, T.P.; Howden, N.J.K.; Worrall, F.; Whelan, M.J. Long-term monitoring of river water nitrate: How much data do we need? J. Environ. Monit. 2010, 12, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kohtani, S.; Kamoi, Y.; Yoshioka, E.; Miyabe, H. Kinetic study on photocatalytic hydrogenation of acetophenone derivatives on titanium dioxide. Catal. Sci. Technol. 2014, 4, 1084–1091. [Google Scholar] [CrossRef]
- Kohtani, S.; Yoshioka, E.; Saito, K.; Kudo, A.; Miyabe, H. Photocatalytic hydrogenation of acetophenone derivatives and diaryl ketones on polycrystalline titanium dioxide. Catal. Commun. 2010, 11, 1049–1053. [Google Scholar] [CrossRef]
- Lewis, N. Progress in understanding electron-transfer reactions at semiconductor/liquid interfaces. J. Phys. Chem. B 1998, 102, 4843–4855. [Google Scholar] [CrossRef]
- Hamann, T.; Gstrein, F.; Brunschwig, B.; Lewis, N. Measurement of the free-energy dependence of interfacial charge-transfer rate constants using ZnO/H2O semiconductor/liquid contacts. J. Am. Chem. Soc. 2005, 127, 7815–7824. [Google Scholar] [CrossRef] [PubMed]
- Ondersma, J.; Hamann, T. Measurements and modeling of recombination from nanoparticle TiO2 electrodes. J. Am. Chem. Soc. 2011, 133, 8264–8271. [Google Scholar] [CrossRef] [PubMed]
- Kohtani, S.; Nishioka, S.; Yoshioka, E.; Miyabe, H. Dye-sensitized photo-hydrogenation of aromatic ketones on titanium dioxide under visible light irradiation. Catal. Commun. 2014, 43, 61–65. [Google Scholar] [CrossRef]
- Kohtani, S.; Mori, M.; Yoshioka, E.; Miyabe, H. Photohydrogenation of acetophenone using coumarin dye-sensitized titanium dioxide under visible light irradiation. Catalysts 2015, 5, 1417–1424. [Google Scholar] [CrossRef]
- Beier, P.; Alexandrova, A.; Zibinsky, M.; Surya Prakash, G.K. Nucleophilic difluoromethylation and difluoromethylenation of aldehydes and ketones using diethyl difluoromethylphosphonate. Tetrahedron 2008, 64, 10977–10985. [Google Scholar] [CrossRef] [PubMed]
- Kohtani, S.; Kurokawa, T.; Yoshioka, E.; Miyabe, H. Photoreductive transformation of fluorinated acetophenone derivatives on titanium dioxide: Defluorination vs. reduction of carbonyl group. Appl. Catal. A Genel. 2016, 521, 68–74. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Togawa, Y.; Tsukamoto, D.; Tanaka, S.; Hirai, T. Highly efficient and selective hydrogenation of nitroaromatics on photoactivated rutile titanium dioxide. ACS Catal. 2012, 2, 2475–2481. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Hirakawa, H.; Togawa, Y.; Sugano, Y.; Ichikawa, S.; Hirai, T. Rutile crystallites isolated from Degussa (Evonik) P25 TiO2: Highly efficient photocatalyst for chemoselective hydrogenation of nitroaromatics. ACS Catal. 2013, 3, 2318–2326. [Google Scholar] [CrossRef]
- Ohtani, B. Titania photocatalysis beyond recombination: A critical review. Catalysts 2013, 3, 942–953. [Google Scholar] [CrossRef]
- Yang, H.; Sun, C.; Qiao, S.; Zou, J.; Liu, G.; Smith, S.; Cheng, H.; Lu, G. Anatase TiO2 single crystals with a large percentage of reactive facets. Nature 2008, 453, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Amano, F.; Yasumoto, T.; Prieto Mahaney, O.-O.; Uchida, S.; Shibayama, T.; Ohtani, B. Photocatalytic activity of octahedral single-crystalline mesoparticles of anatase titanium(IV) oxide. Chem. Commun. 2009, 2311–2313. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yang, H.; Pan, J.; Yang, Y.; Lu, G.Q.; Cheng, H.-M. Titanium dioxide crystals with tailored facets. Chem. Rev. 2014, 114, 9559–9612. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Olds, D.; Peng, R.; Yu, L.; Foo, G.; Qian, S.; Keum, J.; Guiton, B.; Wu, Z.; Page, K. Quantitative analysis of the morphology of {101} and {001} faceted anatase TiO2 nanocrystals and its implication on photocatalytic activity. Chem. Mater. 2017, 29, 5591–5604. [Google Scholar] [CrossRef]
- Roy, N.; Sohn, Y.; Pradhan, D. Synergy of low-energy {101} and high-energy {001} TiO2 crystal facets for enhanced photocatalysis. ACS Nano 2013, 7, 2532–2540. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Koenigsmann, C.; Ding, W.; Rudshteyn, B.; Yang, K.; Regan, K.; Konezny, S.; Batista, V.; Brudvig, G.; Schmuttenmaer, C.; Kim, J.-H. Facet-dependent photoelectrochemical performance of TiO2 nanostructures: An experimental and computational study. J. Am. Chem. Soc. 2015, 137, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zhang, H.; Ji, H.; Ma, W.; Chen, C.; Zhao, J. Modulating the photocatalytic redox preferences between anatase TiO2 {001} and {101} surfaces. Chem. Commun. 2017, 53, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Chamtouri, M.; Kenens, B.; Aubert, R.; Lu, G.; Inose, T.; Fujita, Y.; Masuhara, A.; Hofkens, J.; Uji-i, H. Facet-dependent diol-induced density of states of anatase TiO2 crystal surface. ACS Omega 2017, 2, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic model of the earlier stage of photocatalysis in the anatase titanium dioxide (TiO2). CB: conduction band; VB: valence band; Aad: adsorbed electron acceptor; Dad: adsorbed electron donor.
Figure 1.
Schematic model of the earlier stage of photocatalysis in the anatase titanium dioxide (TiO2). CB: conduction band; VB: valence band; Aad: adsorbed electron acceptor; Dad: adsorbed electron donor.

Figure 2.
Comparison of energy-resolved distribution of electron traps (ERDT) patterns measured using the photochemical method shown by the grey patterns [60], and reversed double-beam photoacoustic spectroscopy (RDB-PAS), shown by the plots [56], for representative anatase (TIO−2; 18 m2g−1) and rutile (7 m2g−1) samples. The top line, at 3.2 eV, and the dashed line show the conduction bands (CB) edge positions of anatase and rutile estimated by the reported bandgaps at 3.2 and 3.0 eV, respectively. Reprinted from reference [56], an open access article under conditions of the Creative Commons Attribution (CC BY) license.
Figure 2.
Comparison of energy-resolved distribution of electron traps (ERDT) patterns measured using the photochemical method shown by the grey patterns [60], and reversed double-beam photoacoustic spectroscopy (RDB-PAS), shown by the plots [56], for representative anatase (TIO−2; 18 m2g−1) and rutile (7 m2g−1) samples. The top line, at 3.2 eV, and the dashed line show the conduction bands (CB) edge positions of anatase and rutile estimated by the reported bandgaps at 3.2 and 3.0 eV, respectively. Reprinted from reference [56], an open access article under conditions of the Creative Commons Attribution (CC BY) license.

Scheme 1.
Photocatalytic hydrogenation of aromatic carbonyl compounds: Ar = aromatic ring, R = H, Me, Et, i-Pr, or CF3. Reprinted with permission from reference [72]. Copyright (2014) The Royal Society of Chemistry.
Scheme 1.
Photocatalytic hydrogenation of aromatic carbonyl compounds: Ar = aromatic ring, R = H, Me, Et, i-Pr, or CF3. Reprinted with permission from reference [72]. Copyright (2014) The Royal Society of Chemistry.
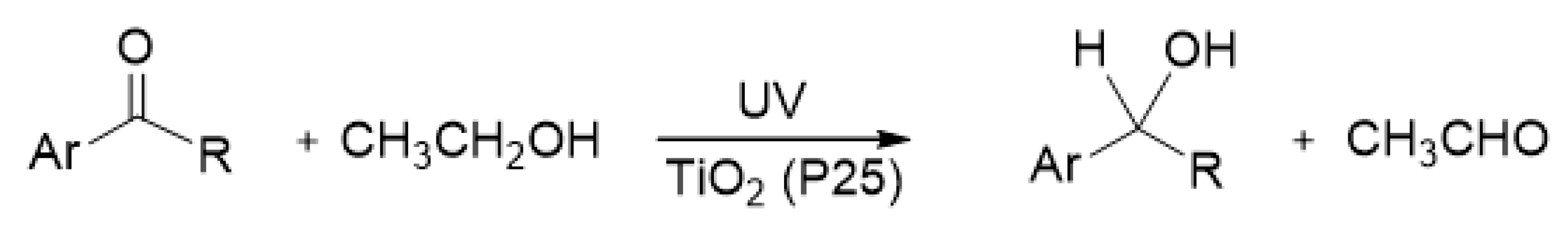
Figure 3.
Color changes of the pre-irradiated P25 TiO2 powder dispersed in de-aerated ethanol (a) in the absence of substrates, and after injection of (b) 300 μmol acetophenone (AP), or (c) 2,2,2-trifluoacetophenone (TFAP). Adapted with permission from reference [63]. Copyright 2012 American Chemical Society. UV irrad.: UV irraditaion.
Figure 3.
Color changes of the pre-irradiated P25 TiO2 powder dispersed in de-aerated ethanol (a) in the absence of substrates, and after injection of (b) 300 μmol acetophenone (AP), or (c) 2,2,2-trifluoacetophenone (TFAP). Adapted with permission from reference [63]. Copyright 2012 American Chemical Society. UV irrad.: UV irraditaion.

Figure 4.
Time evolutions of the amount of AP-OH (○) and TFAP-OH (●) after the 300 μmol injection of substrate (AP or TFAP) into the 2 h pre-irradiated TiO2 suspension at 32 °C Adapted with permission from reference [63]. Copyright (2012) American Chemical Society.
Figure 4.
Time evolutions of the amount of AP-OH (○) and TFAP-OH (●) after the 300 μmol injection of substrate (AP or TFAP) into the 2 h pre-irradiated TiO2 suspension at 32 °C Adapted with permission from reference [63]. Copyright (2012) American Chemical Society.
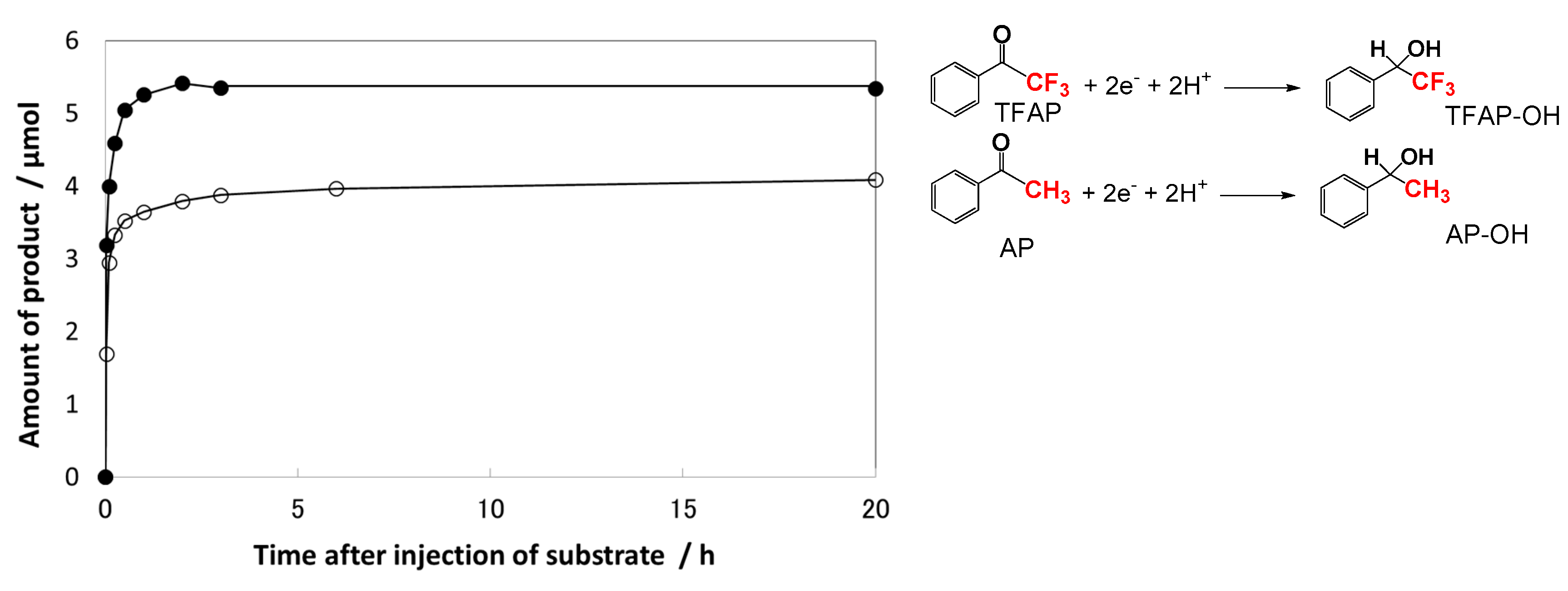
Figure 5.
Schematic illustration of the electron transfer reaction from the Tids states to the adsorbed AP derivatives, where Ered is the reduction potential of AP derivatives, λ is the reorganization energy (approximately 0.7 eV for AP), and E0 is the energy at the top of curve for the acceptor level, calculated by qEred – λ and shown by the solid line. The dotted line indicates the donor energy level for anionic species. Reprinted with permission from reference [72]. Copyright (2014) The Royal Society of Chemistry.
Figure 5.
Schematic illustration of the electron transfer reaction from the Tids states to the adsorbed AP derivatives, where Ered is the reduction potential of AP derivatives, λ is the reorganization energy (approximately 0.7 eV for AP), and E0 is the energy at the top of curve for the acceptor level, calculated by qEred – λ and shown by the solid line. The dotted line indicates the donor energy level for anionic species. Reprinted with permission from reference [72]. Copyright (2014) The Royal Society of Chemistry.

Figure 6.
Schematic illustration on the photocatalytic reduction of 2,2-difluoromethyacetophenone (DFAP) on the UV irradiated TiO2. Reprinted with permission from reference [80]. Copyright (2016) Elsevier B.V.
Figure 6.
Schematic illustration on the photocatalytic reduction of 2,2-difluoromethyacetophenone (DFAP) on the UV irradiated TiO2. Reprinted with permission from reference [80]. Copyright (2016) Elsevier B.V.
Scheme 2.
Photocatalytic reduction of nitroaromatic compounds and oxidation of 2-propanol as a sacrificial hole scavenger.
Scheme 2.
Photocatalytic reduction of nitroaromatic compounds and oxidation of 2-propanol as a sacrificial hole scavenger.
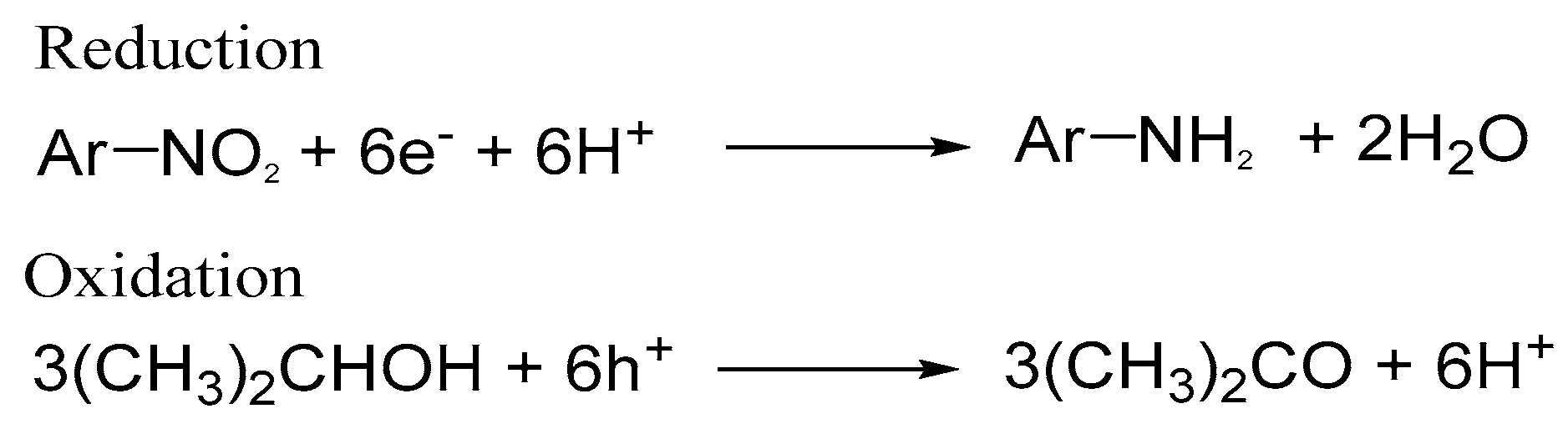

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
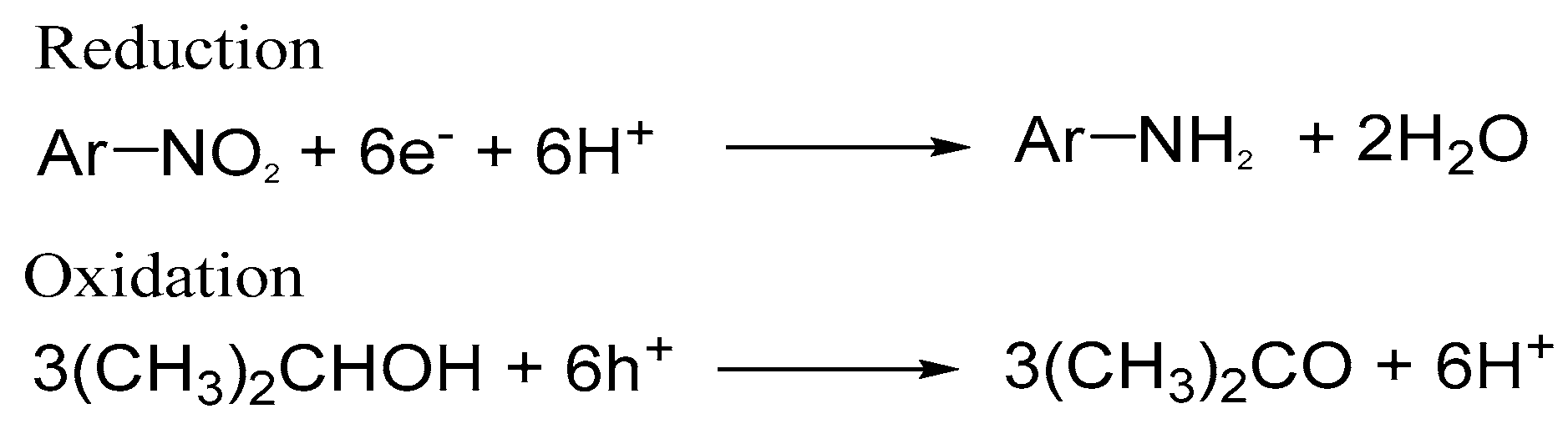{kind=link}
Table 1.
Reduction potentials, amount of reacted electrons, percentages of reacted electrons, and reaction rates at maximum concentration of substrates [72]. Adapted with permission from reference [72]. Copyright (2014) The Royal Society of Chemistry.
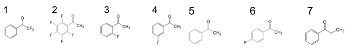
| Substrate | Ered/V a | Amount of Reacted Electrons b/μmoL | Percentage c/% | Reaction Rate d/mMh−1 |
|---|---|---|---|---|
| 1 (TFAP) | –1.35 | 10.2 | 100 | – |
| 2 | –1.59 | 8.22 | 81 | 3.4 ± 0.2 |
| 3 | –1.62 | 6.32 | 62 | 2.2 ± 0.2 |
| 4 | –1.80 | 6.09 | 60 | 2.0 ± 0.1 |
| 5 (AP) | –1.89 | 7.38 | 72 | 1.9 ± 0.1 |
| 6 | –0.92 | 5.70 | 56 | 1.2 ± 0.1 |
| 7 | –1.94 | 4.76 | 47 | 0.75 ± 0.05 |
a Reduction potentials vs. SHE (standard hydrogen electrode). b Molar number of reacted electrons estimated by the injection experiment using a pre-irradiated TiO2 suspension. c Percentage of the reacted electrons per the total amount of Tist3+ and ecb− (10.2 μmol) generated on 0.10 g TiO2. d Reaction rates at the maximum concentration of substrates.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kohtani, S.; Kawashima, A.; Miyabe, H. Reactivity of Trapped and Accumulated Electrons in Titanium Dioxide Photocatalysis. Catalysts 2017, 7, 303. https://doi.org/10.3390/catal7100303
AMA Style
Kohtani S, Kawashima A, Miyabe H. Reactivity of Trapped and Accumulated Electrons in Titanium Dioxide Photocatalysis. Catalysts. 2017; 7(10):303. https://doi.org/10.3390/catal7100303
Chicago/Turabian StyleKohtani, Shigeru, Akira Kawashima, and Hideto Miyabe. 2017. "Reactivity of Trapped and Accumulated Electrons in Titanium Dioxide Photocatalysis" Catalysts 7, no. 10: 303. https://doi.org/10.3390/catal7100303
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.