Application of Artificial Neural Networks for Catalysis: A Review


1
College of Chemistry, Sichuan University, Chengdu 610064, China
2
School of Chemistry and Chemical Engineering, Chongqing University of Technology, Chongqing 400054, China
3
Department of Power Engineering, School of Energy, Power and Mechanical Engineering, North China Electric Power University, Baoding 071003, China
*
Authors to whom correspondence should be addressed.
†
Present Address: Department of Chemistry and Institute for Computational and Engineering Sciences, The University of Texas at Austin, 105 E. 24th Street, Stop A5300, Austin, TX 78712, USA.
‡
These authors contributed equally to this work.
Catalysts 2017, 7(10), 306; https://doi.org/10.3390/catal7100306
Submission received: 28 September 2017
/
Revised: 14 October 2017
/
Accepted: 16 October 2017
/
Published: 18 October 2017
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Machine learning has proven to be a powerful technique during the past decades. Artificial neural network (ANN), as one of the most popular machine learning algorithms, has been widely applied to various areas. However, their applications for catalysis were not well-studied until recent decades. In this review, we aim to summarize the applications of ANNs for catalysis research reported in the literature. We show how this powerful technique helps people address the highly complicated problems and accelerate the progress of the catalysis community. From the perspectives of both experiment and theory, this review shows how ANNs can be effectively applied for catalysis prediction, the design of new catalysts, and the understanding of catalytic structures.
Keywords:
machine learning; artificial neural network (ANN); catalyst; catalysis; experiment; theory1. Introduction
Machine learning, a powerful technique of artificial intelligence (AI), has been widely used for numerical prediction [1,2], classification [3,4], and pattern recognition [5,6]. As its name implies, machine learning is able to “learn” the highly complicated relationships between the independent and dependent variables via non-linear “black box” data processing. During the past decades, it has been widely used in many scientific and industrial areas, such as biology [7,8,9], medicine [10,11,12], energy [13,14,15,16,17,18,19], environment [20,21,22], engineering [23,24,25], and information technology (IT) [26,27]. These application studies indicate that machine learning techniques have dramatically boosted the development of many different areas. With the developing concept of big data and data-mining [1], there is an increasing demand for knowledge-based machine learning models in practical applications. Artificial neural network (ANN) [28], as a non-linear fitting algorithm, has become one of the most popular machine learning techniques due to its advantages of easy-training, adaptive structure, and tunable training parameters [13]. With the development of algorithms, there are currently a large number of ANN methods, such as the back-propagation neural network (BPNN) [29], general regression neural network (GRNN) [30], and extreme learning machine (ELM) [31]. More recently, the deep neural network (DNN) has raised broad interests due to its strong learning capacity and the popular concept of deep learning techniques [32,33]. Previous studies have shown that different neural network algorithms have different advantages for practical applications [17,18,34,35].
However, compared to other areas, machine learning has not been well-studied in the field of catalysis during the past decades, due to the following two reasons: (i) Lacking of a sufficiently large database: both experimental and theoretical studies on catalytic reactions require a large amount of time and resources; (ii) Too many input variables: to predict the reaction properties, there are usually a large number of independent variables that correlate (or partially correlate) with the predicted properties (e.g., experimental conditions, reaction coordinates, electronic structures, and orbital properties). These problems significantly hinder the development and application of machine learning for the catalysis community. For the catalysis using nano-size catalysts, things become even more complicated: experimental characterizations of their catalysts are complicated and there are many nano-size effects that are difficult to observe. Theoretically, though there are some state-of-the-art computational techniques (e.g., quantum mechanical calculations [36]) that can precisely calculate the electronic structure and energy properties of a catalytic system (e.g., nanoparticles and nanorods), the high computational cost is still a huge challenge. Fortunately, during the recent decade, with the rapid development of both experimental and computational techniques, scientists have been able to more effectively and precisely acquire a large number of properties with a shorter timescale. Meanwhile, the quick developments of computer and machine learning algorithms also ensure that even with lots of independent variables, some machine learning techniques can still perform fast training with a sufficiently large database.
Nevertheless, to the best of our knowledge, there are very few review studies that summarize the machine learning applications for catalysis research. Therefore, detailed discussions on the potential applications of ANNs for both experimental and theoretical catalysis are also necessary. Here, we aim to summarize the recent progress of ANN applications for catalysis, from the perspectives of both experiments and theories. The content of this review consists of three parts: (i) Algorithmic introduction of an ANN; (ii) Applications of ANNs for experimental catalysis; and (iii) Applications of ANNs for theoretical catalysis.
2. Principle of ANN
2.1. Schematic Structure of an ANN
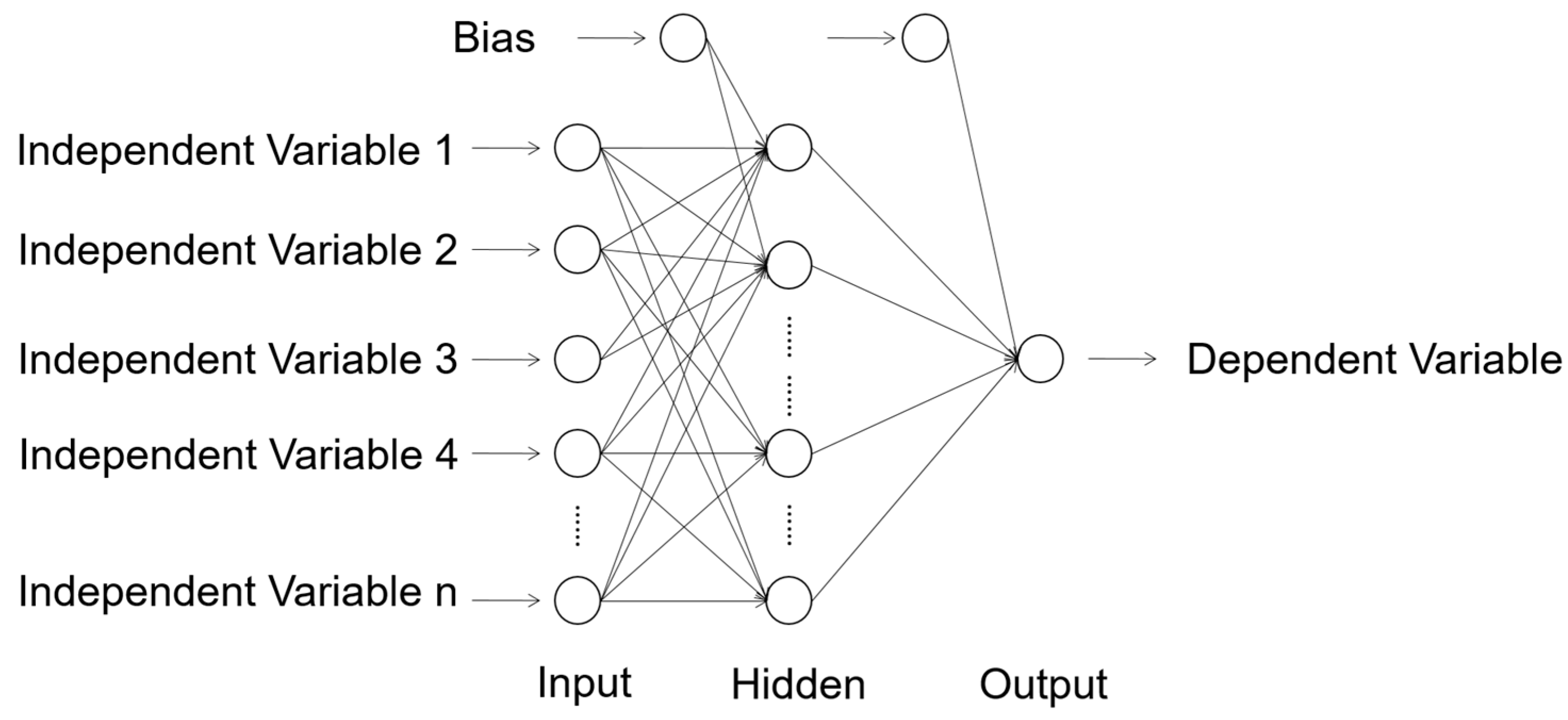
A complete algorithmic structure of a conventional ANN consists of at least three different layers: the input, hidden, and output layers (Figure 1) [37]. Each layer consists of a certain number of neurons. Each neuron inter-connects with all the neurons in the following layer. Each connection represents a weight that contributes to the fitting. With a proper activation function, a combination of optimized weights can generate the prediction of the dependent variable:
where represents the weight value of a connection, represents an inputted independent variable, and represents a bias. For the activation function (), the sigmoid function is one of the most popular forms that can introduce a smooth non-linear fitting to the training of an ANN (Equation (2)). The training of an ANN is essentially the optimization of each weight contribution based on the data groups in the training set. The most commonly used weight optimization method is the back-propagation algorithm, which iteratively analyzes the errors and optimizes each weight value based on the errors generated by the next layer. As we have mentioned above, there are also some other types of networks like GRNN and ELM. Though there are some differences in the weight training and algorithmic structures, the basic principles, as well as the training and testing processes, are very similar. More details about their principles can be found in References [28,30,38].
2.2. Model Development
The rational development of a knowledge-based ANN model consists of two parts: (i) training and (ii) testing. The training process is the so-called “learning” process from the database, while the testing process is the validation of the trained model using the data groups that are not previously involved in the training process. Detailed discussions are shown in Section 2.2.1 and Section 2.2.2. It should be noted that the training and testing processes are not only suitable for the topic discussed in this review, but are also suitable for ANN model development in many other areas.
2.2.1. Model Training
The training of an ANN consists of database preparation and the selection of variables. The size of the database should be sufficiently large to avoid over-fitting. For each variable (especially the dependent variable), the data range should be wide enough to ensure good training. If the data range is too narrow, the trained model might only have a good prediction capacity in a very local region. For the numerical prediction, the dependent variables are usually the properties that are hard to acquire from regular measurements or calculations. The independent variables, on the other hand, should be easily-measured and have potential relationships with the selected dependent variable. More details about the training criterion can be found in Reference [37].
2.2.2. Model Testing
To validate the trained ANN, a testing process is necessary. The testing of a model should use the data groups that are not used for the training process. With the inputs of the testing set, the outputted data can be compared with the actual data in the testing set, with the root mean square error (RMSE) calculated by Equation (3):
where represents the predicted value outputted by the ANN, is the actual value, and represents the total number of samples. If the calculated RMSE from the testing set is relatively small, it means that the ANN is well-trained. It should be noted that for the training and testing of an ANN, a cross-validation [39] process should be performed using different components of the training and testing datasets. If the database is relatively large, a sensitivity test can be performed to replace the cross-validation, in order to avoid a high computational cost.
It should be noted that, for a typical ANN algorithm (e.g., BPNN), it is necessary to optimize the overall ANN structure before deciding the final numbers of the hidden layer and hidden nodes. Repeated training and testing should be performed with different ANN structures. On the one hand, if the numbers of hidden layers and/or hidden neurons are too high, there is a risk of over-fitting [40]; on the other hand, if their numbers are too low, this leads to under-fitting. Usually, the best ANN algorithmic configuration can be defined by comparing the average RMSEs from the testing sets during a cross-validation or sensitivity test.
3. Applications of ANN for Catalysis: Experiment
3.1. Prediction of Catalytic Activity
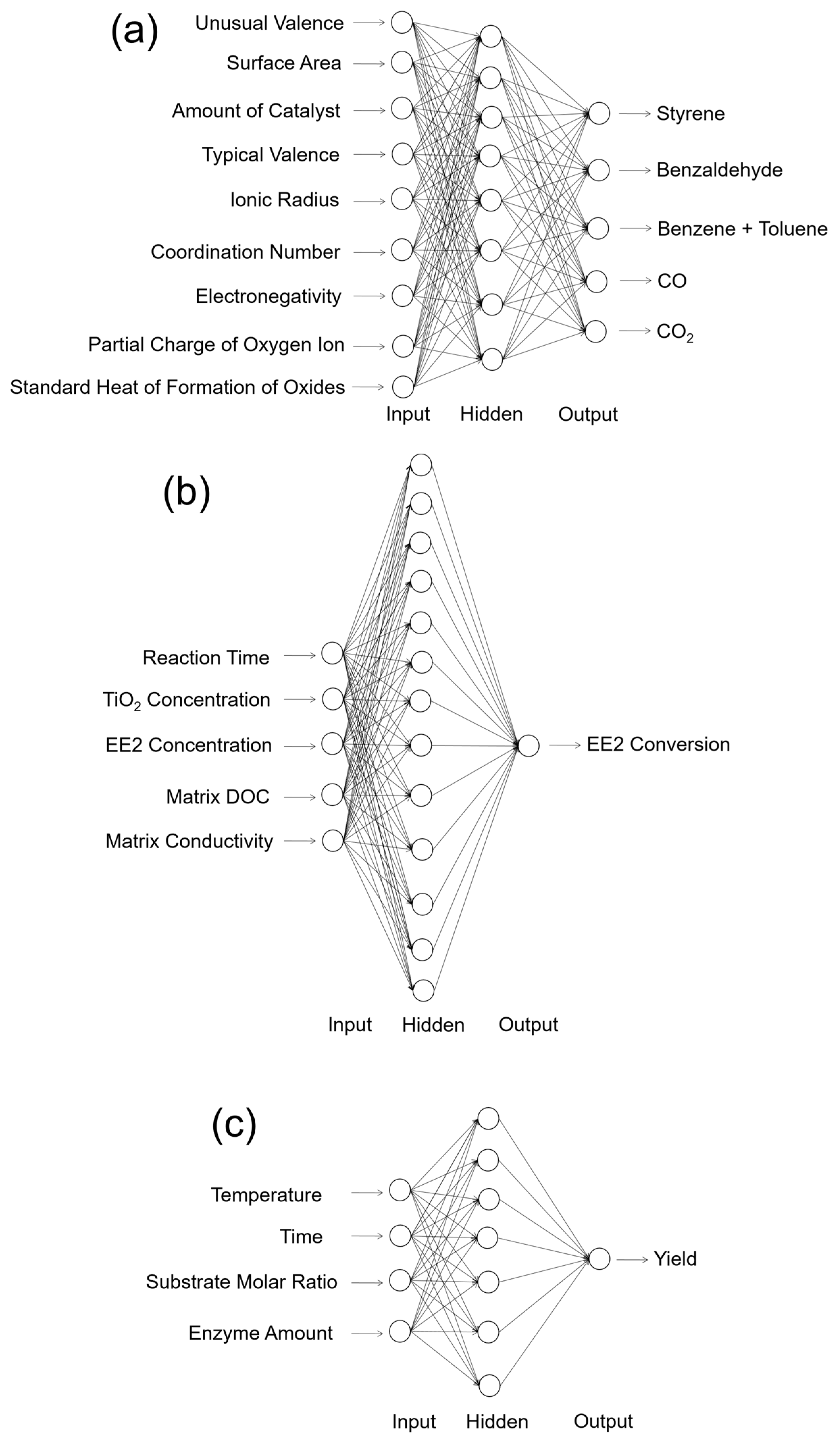
The most straightforward application of ANN is the numerical prediction. One of the earliest works for the catalytic application was done by Kito et al. in 1994 [41]. In this work, they predicted the product distribution of ethylbenzene oxidative hydrogenation, with the product components of styrene, benzaldehyde, benzene + toluene, CO, and CO2 as the outputs of a network. In terms of the inputs of the ANN, they used nine different independent variables that had potential relationships with the productivity and selectivity of the catalytic reaction, including: unusual valence, surface area of the catalyst, amount of catalyst, typical valence, ionic radius, coordination number, electronegativity, partial charge of oxygen ion, and standard heat of formation of oxides. Their results found that with a good experimental database, a single hidden layer ANN could perform precise predictions for the product selectivity. The schematic structure of the ANN they used for the prediction is reconstructed in Figure 2a. In the work done by Sasaki et al. [42], they first proposed that ANNs could be used for catalytic activity predictions, as well as experimental condition optimizations. Setting the experimental conditions such as compositional quantity, temperature, and pressure, they showed that the yield and byproducts of NO decomposition over the Cu/ZSM-5 zeolite catalyst could be precisely predicted by a well-trained ANN [42]. For other more complicated reactions, such as 1-hexene epoxidation catalyzed by polymer supported Mo(VI) complexes, Mohammeda et al. showed that the ANN had a powerful predictive capacity for forecasting its catalytic activities, in excellent agreement with their experimental conclusions [43]. For photocatalysis, the ANN also shows its strong predictive capacity: Frontistis et al. studied the photocatalytic degradation of 17-ethynylestradiol (EE2) using TiO2 catalysts with varying concentrations [44]. With the inputs of reaction time, TiO2 concentration, EE2 initial concentration, matrix dissolved organic carbon (DOC), and matrix conductivity as the inputs, they found that a single hidden layer ANN could perform the minimized average RMSE during the testing processes (Figure 2b) [44]. In terms of the biotechnical catalysis, Rahman et al. also found that using the temperature, reaction time, substrate molar ratio, and enzyme amount as the inputs, an optimized ANN structure could be used for the yield prediction of lipase-catalyzed dioctyl adipate synthesis (Figure 2c) [45]. Recently, with the developing concept of data mining, Günay and Yildirim successfully used 1337 data points from 20 studies of selective CO oxidation over Cu-based catalysts. They concluded that ANN modeling could be used to extract valuable experimental results from previous literature data and provides powerful guidance for future experimental designs [46]. In addition to catalysis, Raccuglia et al. further found that a similar concept could even help assist the materials discovery from failed experimental data [47]. From these typical studies, it can be seen that with different types of reaction systems, catalysis, and datasets, the optimal ANN structures for prediction are significantly different. As can be seen in Figure 2, different reaction types have very different input variables and output indicators. This means that each prediction task should be predicted by a specific model with an optimal weight contribution and network structure.
3.2. Optimization of Catalysis
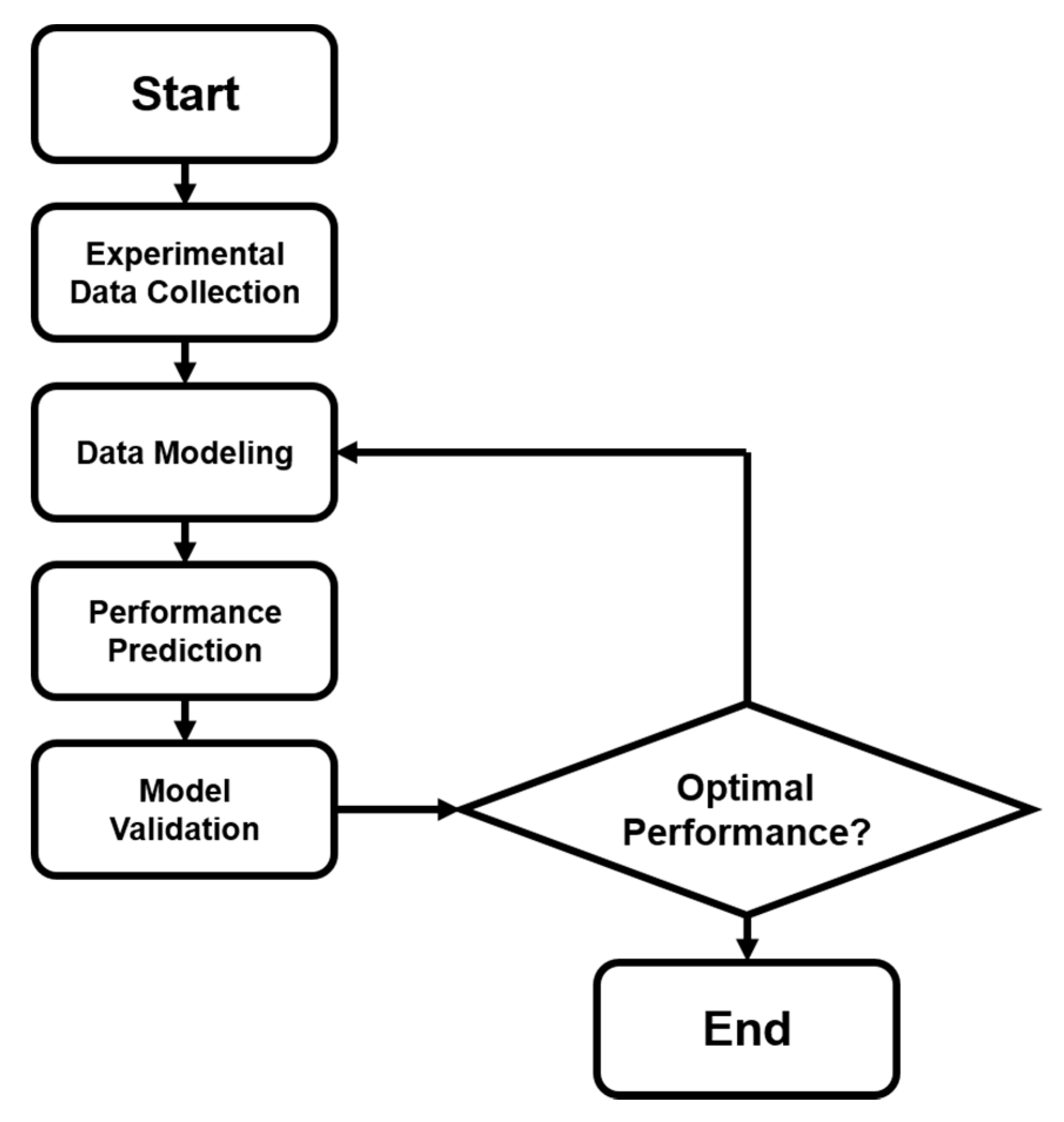
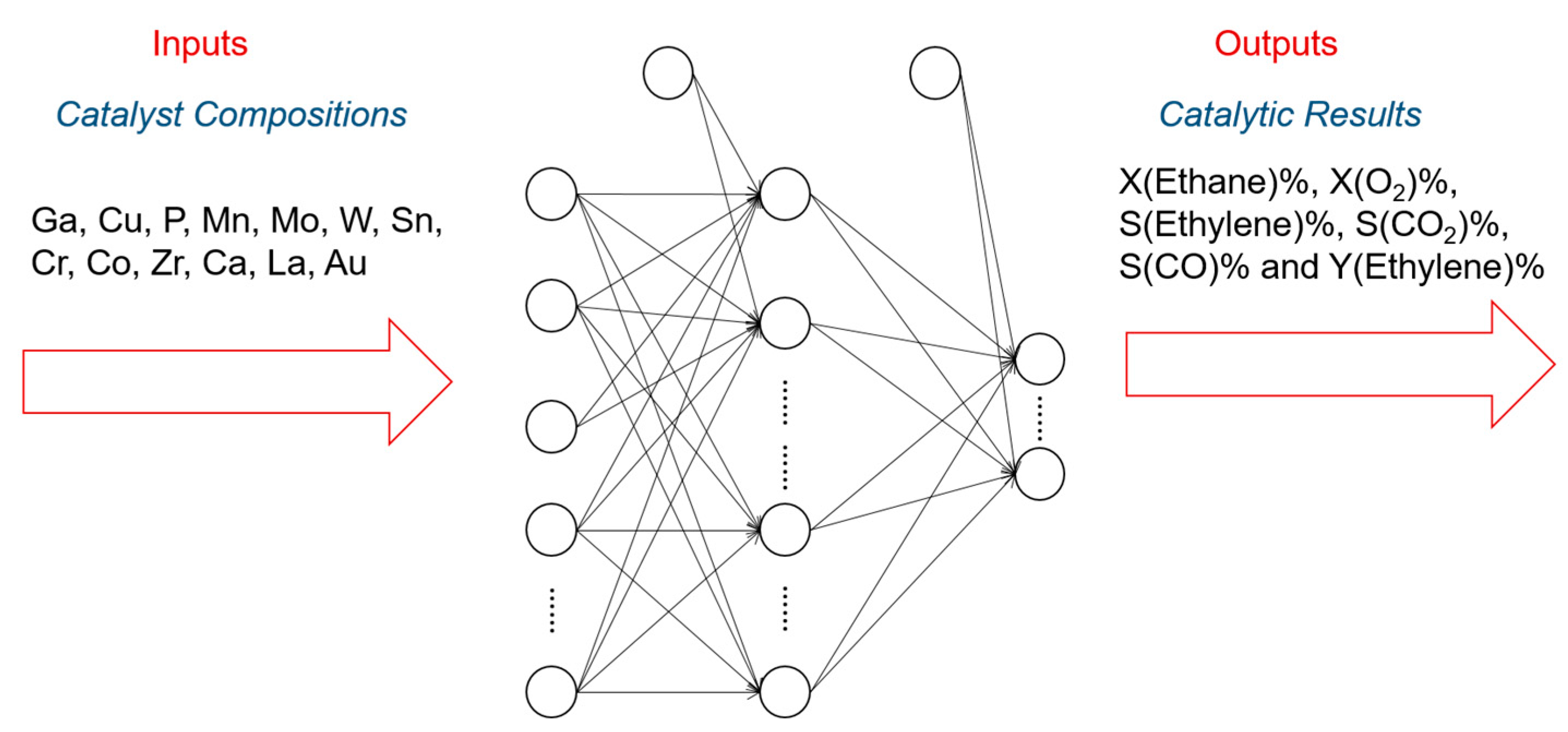
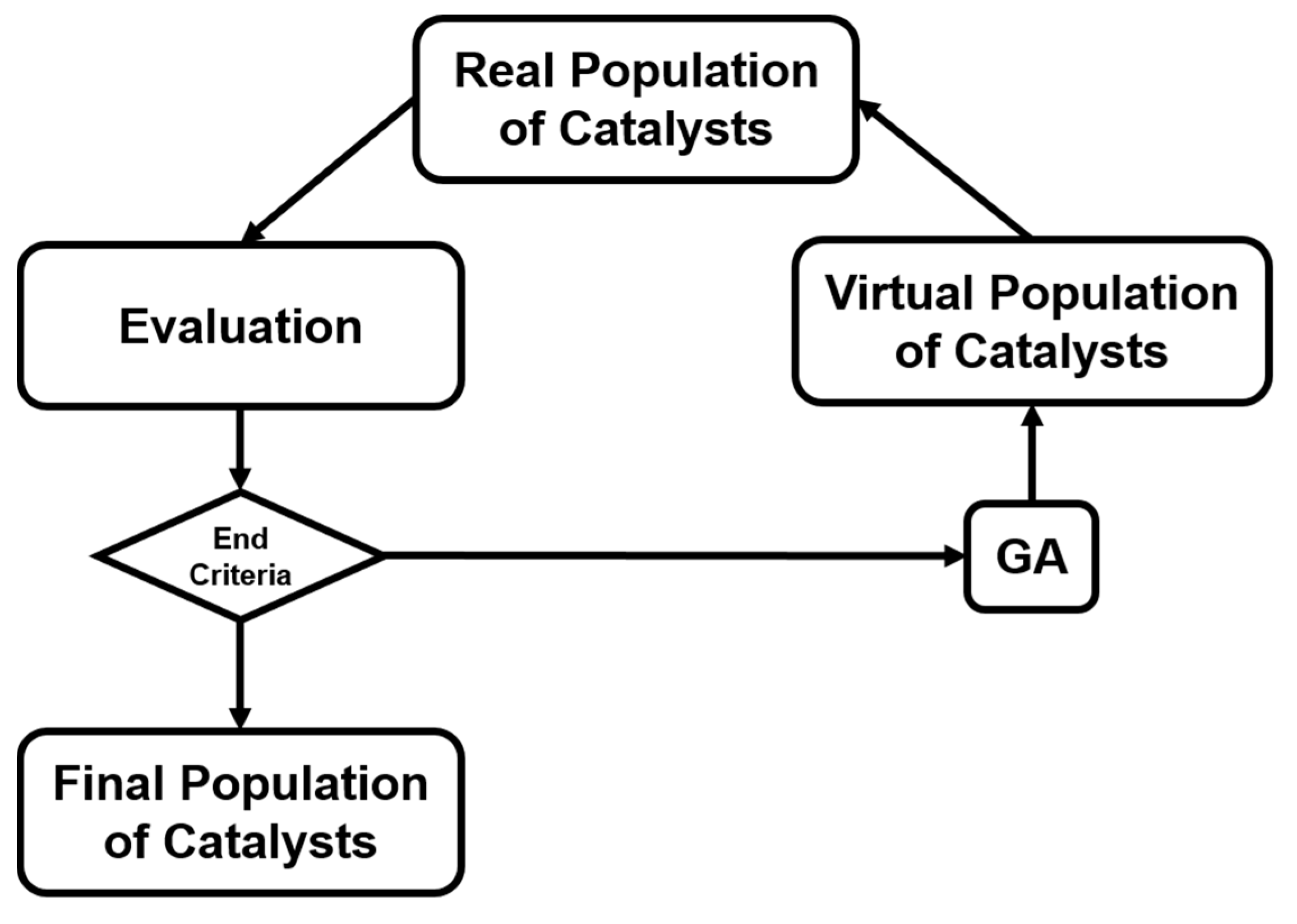
In addition to the activity prediction, scientists started to think about a more practical question: how can we cost-effectively design novel catalysts using the predictive power of ANN? Now that we know that ANNs can precisely predict the catalytic performances of various catalytic systems, we may want to design and generate new inputs of new expected catalysts, and acquire their predicted performances from a well-trained ANN. A general algorithmic flow chart of catalyst optimization summarized by Maldonado and Rothenberg is reconstructed in Figure 3 [48]. This is actually more challenging than a straightforward prediction. For modeling and optimizing the catalysis, one of the primary works was done by Corma et al., who first applied ANNs for the optimization of potential catalyst compositions for the oxidative dehydrogenation of ethane (ODHE) [49]. The scheme of the optimization strategy in their study is shown in Figure 4, with the inputs of various combinatorial compositions and the output of catalytic results. It should be noted that in this catalysis optimization, a genetic algorithm (GA) was introduced as the promoter of design generation [50]. Omata and Yamada developed an ANN to predict the effective additives to a Ni/active carbon (AC) catalyst for methanol vapor-phase carbonylation [51]. Using a well-trained network, they found that Sn was an effective element that could optimize this catalytic process. Hou et al. first proposed an ANN-based computer-aided framework for catalyst design [52]. They found that such a method could be effective for the design of promising propane ammoxidation catalysts. In a similar catalytic system for propane ammoxidation, Cundari et al. further combined the ANN with a GA method for a quick catalyst selection [53]. With the use of the GA method, people can more rationally design the catalysts by optimizing the inputs of ANNs. Similarly, Umegaki et al. combined the GA and ANN with a parallel activity test for optimizing a Cu−Zn−Al−Sc oxide catalyst for methanol synthesis [54]. Rodemerck et al. generalized the GA-assisted ANN method and proposed a general framework for new solid catalytic materials screening, in good agreement with their experimental data [55]. Based on the previous developments of the GA-assisted ANN methods, Baumes et al. further developed an “ANN fliter” for the high-throughput screening (HTS) of heterogeneous catalysis discovery [56]. Using the water-gas shift (WGS) reaction as an example, they showed that though the optimization method previously developed by Corma et al. was successful for ODHE (as mentioned at the beginning of this Subsection) [49], it failed to precisely estimate the WGS reaction activities. However, with a well-trained ANN classifier as a filter that could help define the “good” and “bad” catalysts, WGS catalysts could be rationally designed with a GA-assisted HTS method. Being similar to Reference [56], their framework is summarized and reconstructed in Figure 5.
In addition to the GA-assisted ANN method, there are other strategies that could also help speed up the design and/or screening of catalysts. Kasiri et al. modeled and optimized the heterogeneous photo-Fenton process using both response surface methodology (RSM) and ANN [57], with the experimental measured H2O2 concentration, catalyst concentration, initial pH, and initial dye concentration as the inputs of the models. They found that for a catalyst design process, ANN is as powerful as RSM. Basri et al. also found that, for optimizing the lipase-catalyzed synthesis of a palm-based wax ester, ANN superiorly outperformed RSM [58]. More comparative studies using RSM and ANN can be found in References [59,60,61,62,63,64,65].
4. Applications of ANN for Catalysis: Theory
4.1. Prediction of Reaction Descriptors
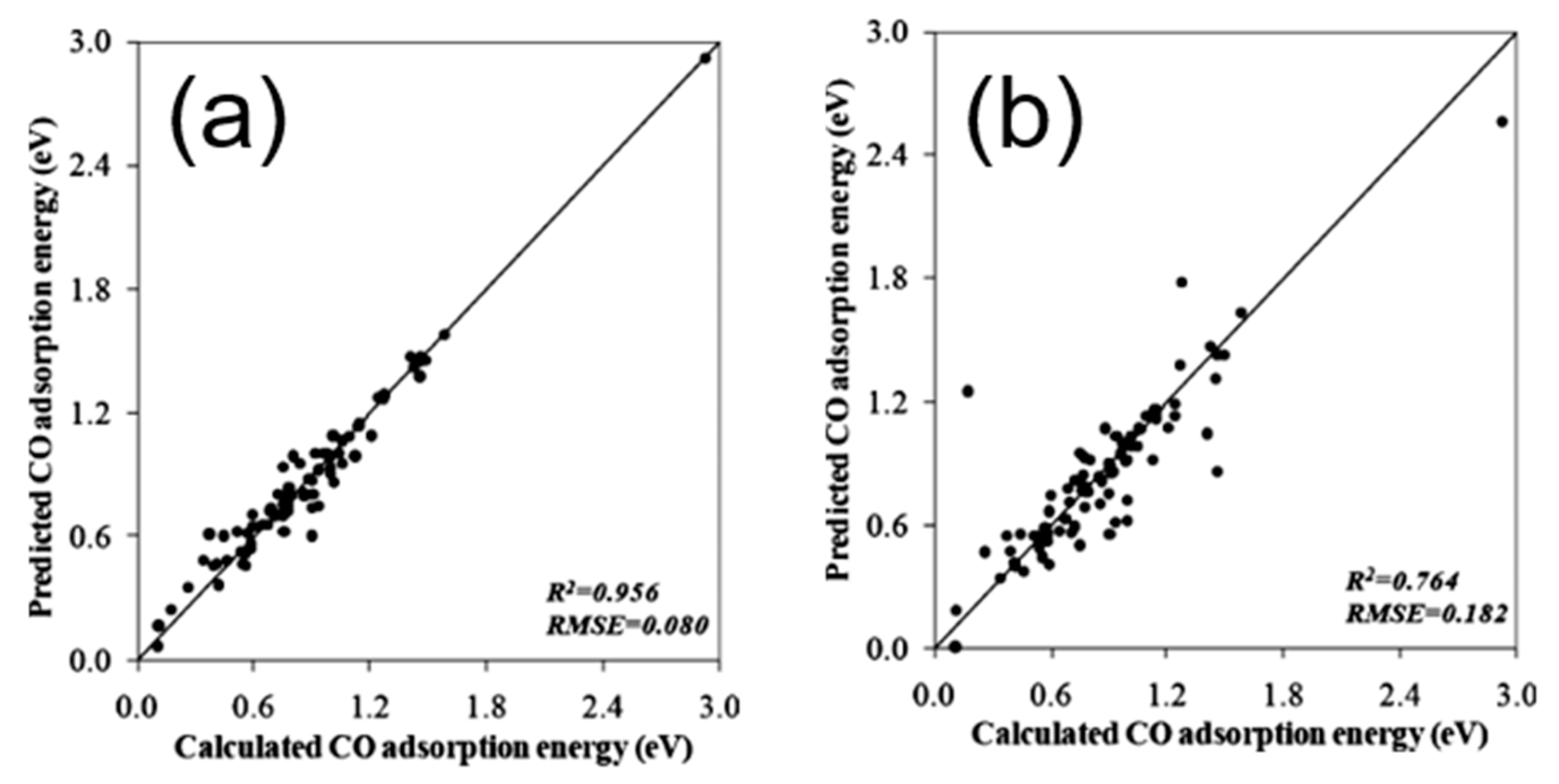
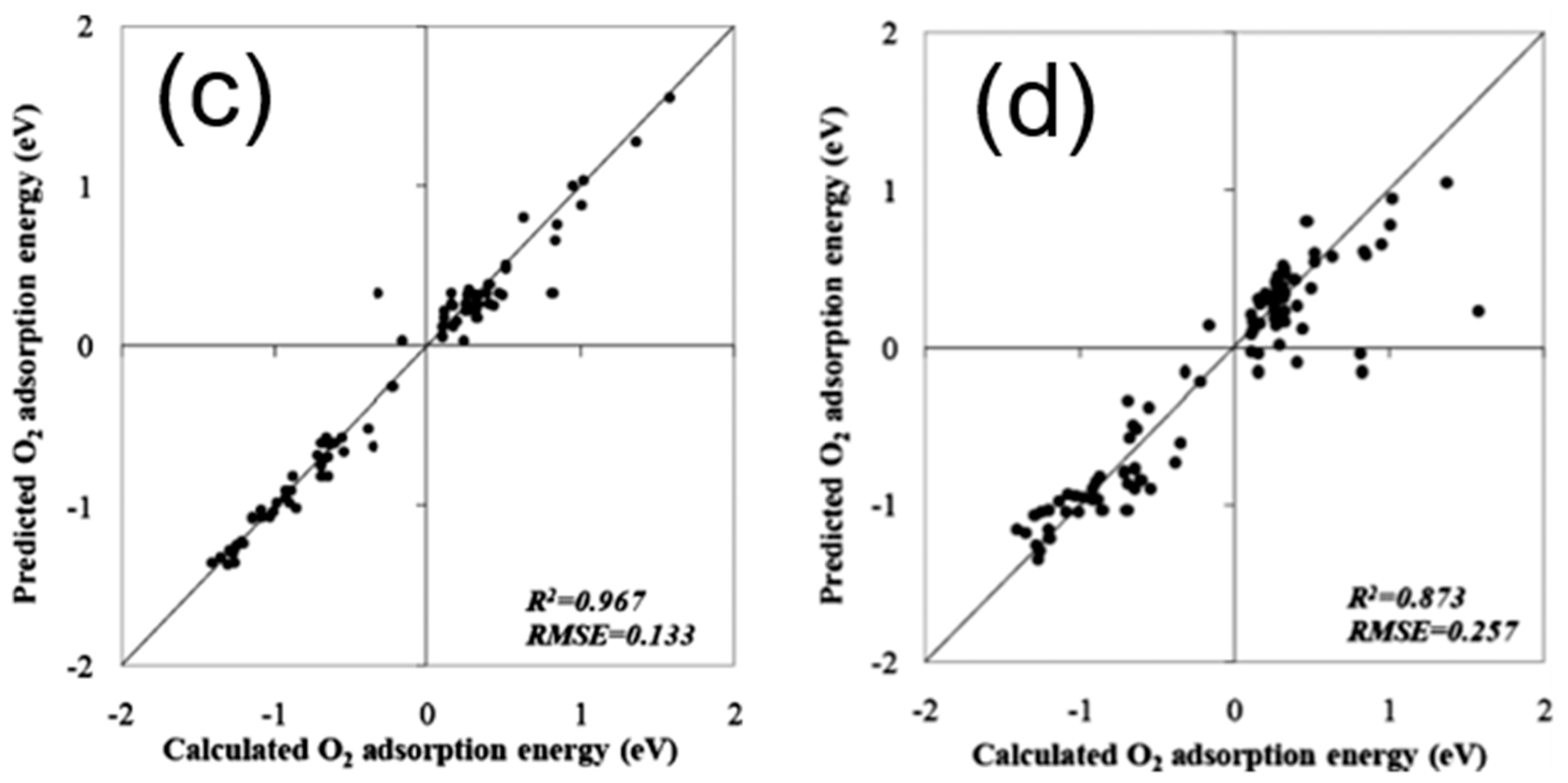
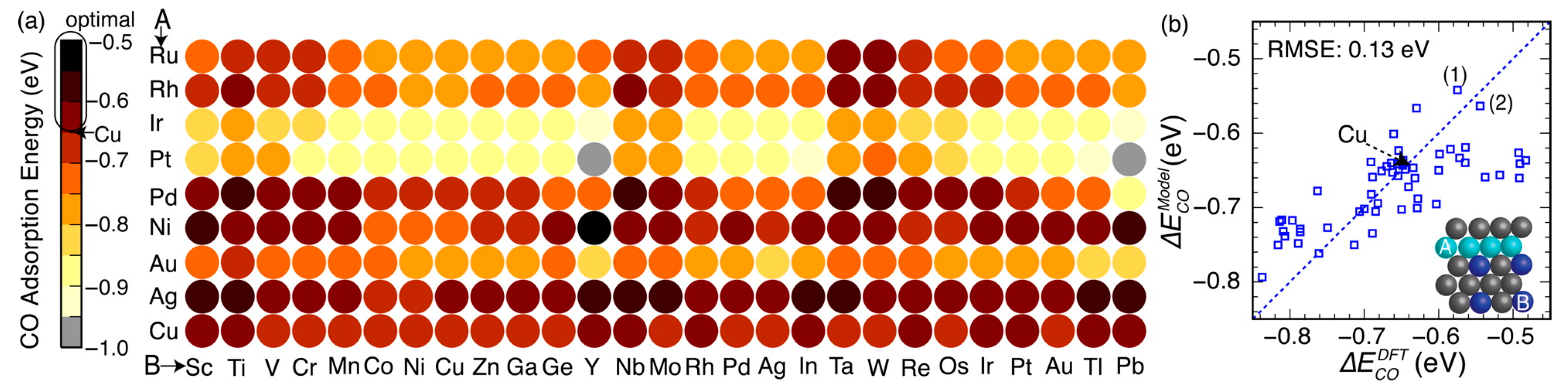
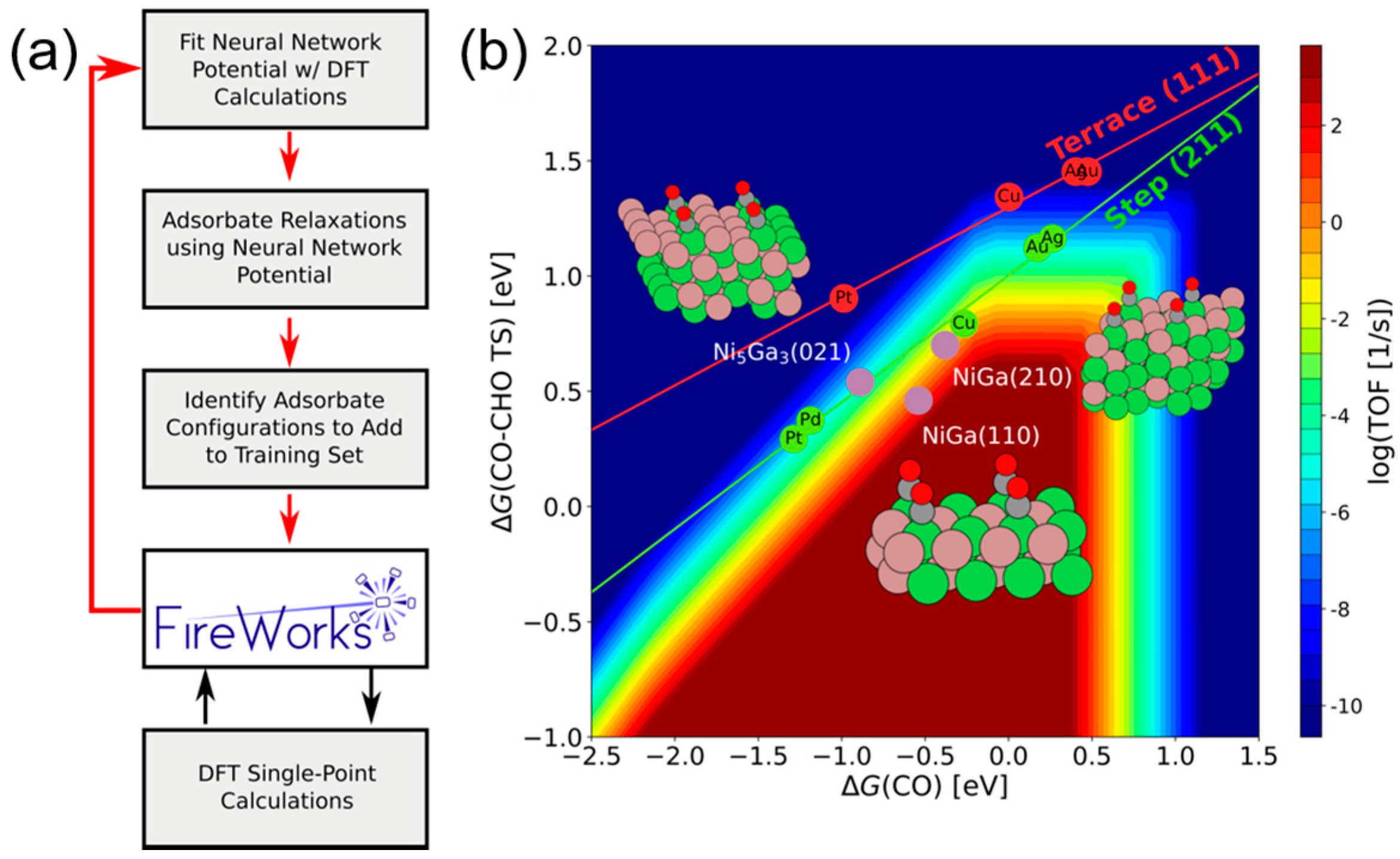
To theoretically assess the catalytic activity of a reaction, density functional theory (DFT) [66,67] calculations and molecular dynamic (MD) [68,69] simulations have been widely used to calculate the energy barriers of a catalytic reaction. With the applications of the transition state theory (TST), people can calculate the theoretical reaction rates with the knowledge of an energy barrier or reaction energy [70]. However, both DFT and MD are limited by the high computational cost with a relatively large catalytic system, which dramatically hinders the further developments of the computer-aided catalyst design and its relevant mechanism study. To address this problem, Nørskov and his co-workers successfully developed a “volcano activity plot” method to estimate the theoretical activities of monometallic heterogeneous catalysts [71,72,73]. By estimating the energy barriers using the Brønsted–Evans–Polanyi (BEP) relationship (the linear or nearly-linear relationships between the energy barrier and reaction energy under the same reaction mechanism [74]) [75,76], and predicting the binding energies of reaction intermediate species using the scaling relationship [77,78], they modeled the general trends of the catalytic activities of heterogeneous catalysts, which significantly boosted the subsequent industrial community for new catalyst design. Interestingly, they found that the heterogeneous catalytic activities always correlate well with one or two binding energies of the reaction species (e.g., CO and O for CO oxidation [73], CO for CO2 electroduction [79], O and OH for oxygen reduction [71], and H for hydrogen evolution [72]). Using these binding energies as the reaction descriptors, a large number of new mono- and multimetallic catalysts have been discovered [80,81]. However, with the increase of industrial demand, using DFT calculations for a screening of the binding energies still requires a large number of computational resources. Especially for the design of alloy catalysts, the computational cost would further increase due to the atomic ensemble effects (specific arrangement of group of atoms [82]). To develop a knowledge-based alternative, scientists started to use ANNs to help acquire the binding energies of adsorbates on the catalytic surface. Davran-Candan et al. employed an ANN to predict the CO and O2 binding energies over Au2 to Au10, with excellent modeling results [83]. Their training and testing results for CO and O2 binding predictions are shown in Figure 6. To predict the more complicated system, the alloy catalysts, Ma et al. used an ANN model to screen the CO binding energies on various bimetallic models. Based on the ANN-predicted results, they found that Cu3Y−Ni@Cu and Cu3Sc−Ni@Cu had the desired CO binding energies for CO2 electroreduction [79]. Their screening results are shown in Figure 7. As a further step for the CO2 electroreduction catalyst search, Ulissi and Nørskov et al. developed a framework that combined ANN potential energy fitting and the binding energy prediction for bimetallic alloy design [84]. They concluded that a machine learning technique can enable an exhaustive screening on both bimetallic facets and active atomic ensembles for a given catalysis. Figure 8 illustrates their machine learning-assisted screening framework and their screening results shown in a volcano activity plot of CO2 electroreduction. With the increasing demand of catalyst design and screening, DFT calculations alone may no longer fulfill the experimental requirements. With these machine learning-assisted methods, it has been shown that some state-of-the-art data mining techniques (e.g., ANNs) are able to perform ultra-fast and precise predictions of catalytic descriptors based on a sufficiently large DFT-calculated database. The research done by Ulissi and Nørskov et al. [84] can be a good milestone that illustrates how DFT can work with machine learning and perform a quick and exhaustive catalysts screening and optimization.
4.2. Prediction of Potential Energy Surface
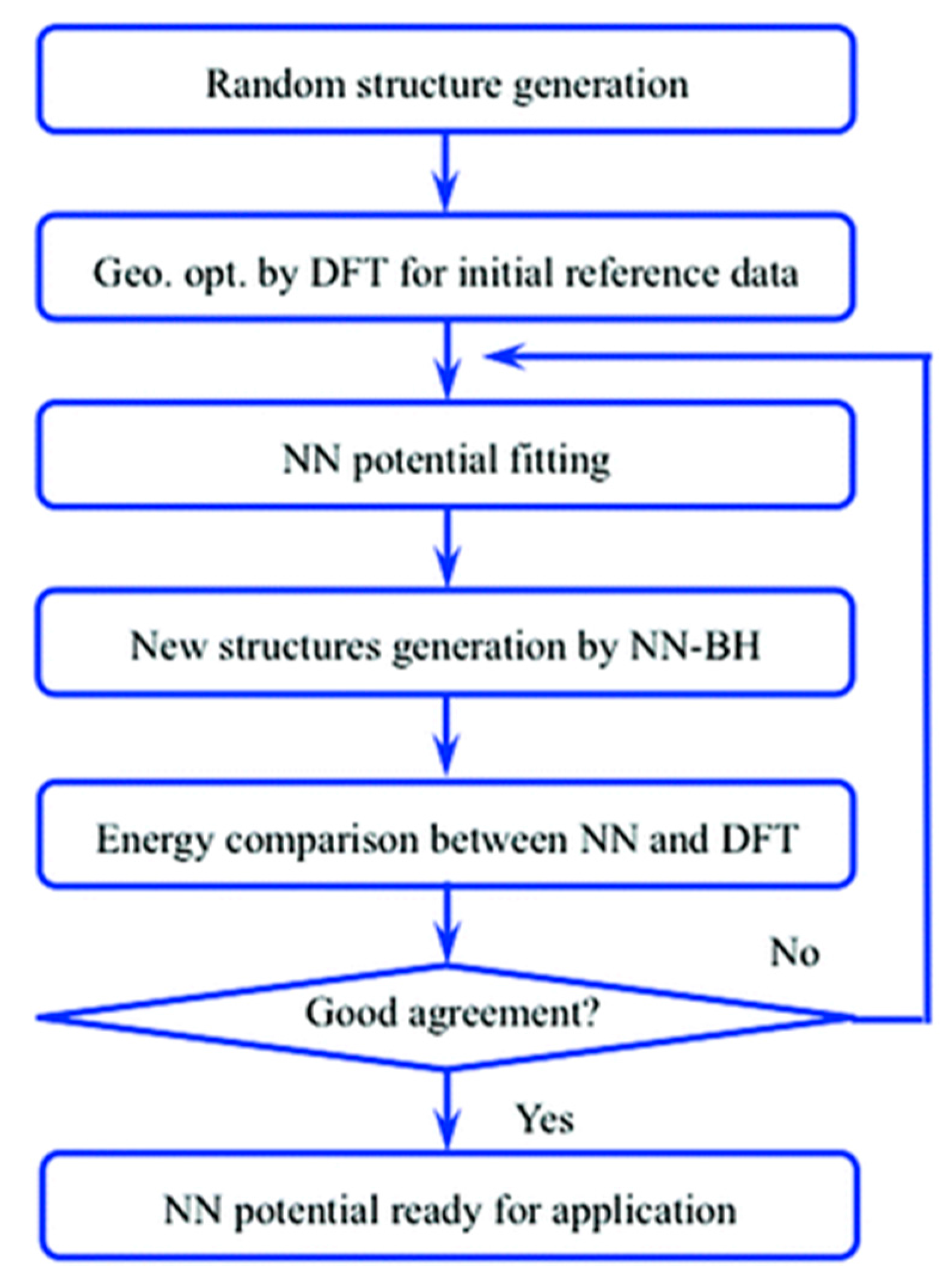
In addition to the study of catalytic descriptors, knowledge about the configurations of catalytic systems is also vital for catalysis study. Usually, even with some state-of-the-art experimental techniques, it is still hard to detect the most favorable configurations of a catalyst system, especially for nano catalytic systems. Fortunately, Monte Carlo (MC) [86] methods are able to provide simulations that perform global optimizations for a specific catalytic system (e.g., looking for the global minima of a specific metallic cluster catalysts). However, these methods strongly rely on the energy potential for acquiring the total energy. To use an alternative potential energy, Sumpter and Noid first systemically showed that neural networks are able to map the vibrational motion determined from spectra onto a fully coupled potential energy surface (PES) with a high accuracy [87]. Subsequently, people started to use ANN and other machine learning techniques for mapping the PES of some simple chemical reactions (e.g., H2 dissociation [88]) and molecular interactions (e.g., water dimer [89]). In terms of the global optimization of catalytic systems, Ouyang et al. used an ANN with 56 independent variables in the input layer to fit the PES of Au58 clusters [90]. With a well-trained ANN, they performed a basin-hopping (BH) [91] configuration search using the network as the “black-box” potential (Figure 9). Validated by DFT calculations, they found a new global minima that was more energetically stable than all the previously studied Au58 clusters.
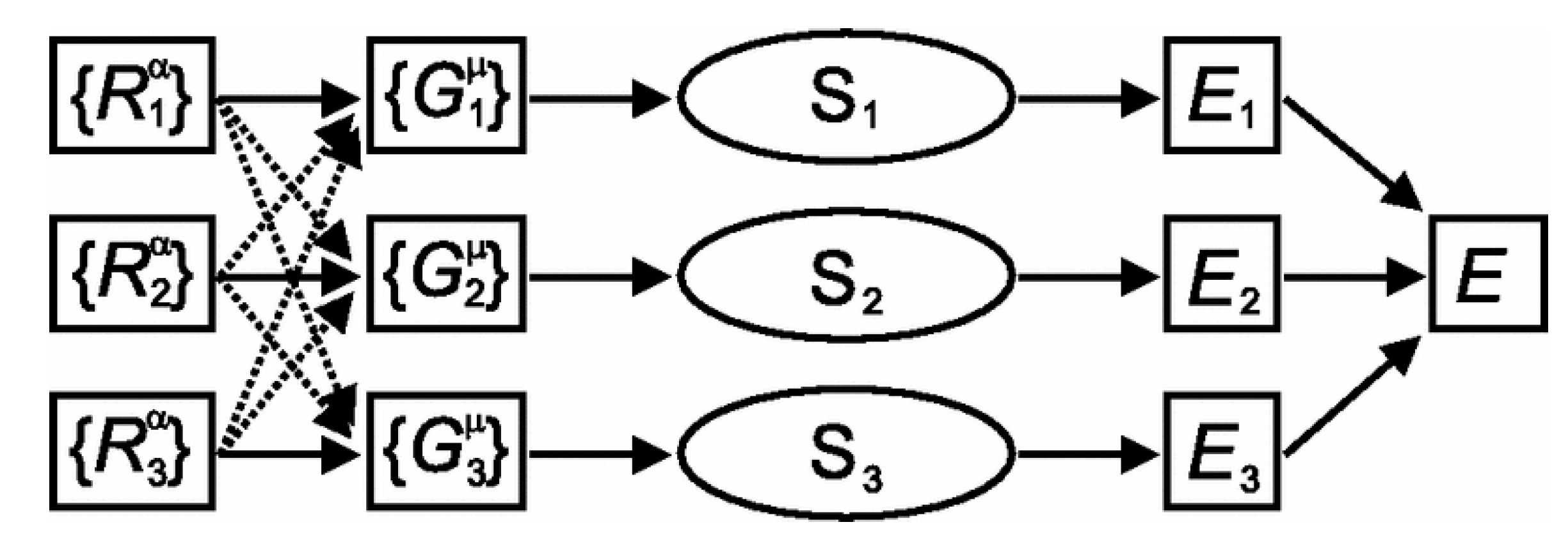
However, most of the previous ANN-potential constructions used molecular coordinates or bond lengths as the inputs, which is seriously limited by the size of the system. If a cluster is large, there will be a large number of input variables. This would lead to an extremely long time-consumption for model training. To address this problem, reducing the number of inputs for each single network training is particularly important. To propose a good solution, Behler and Parrinello first suggested a generalized neural-network representation of high-dimensional PES (Figure 10) [92]. Using the radial symmetry function, the atomic environment of each atom can be described by the set of , which only includes the independent variables of the pair interaction (Equation (4)) and angular terms (Equation (5)):
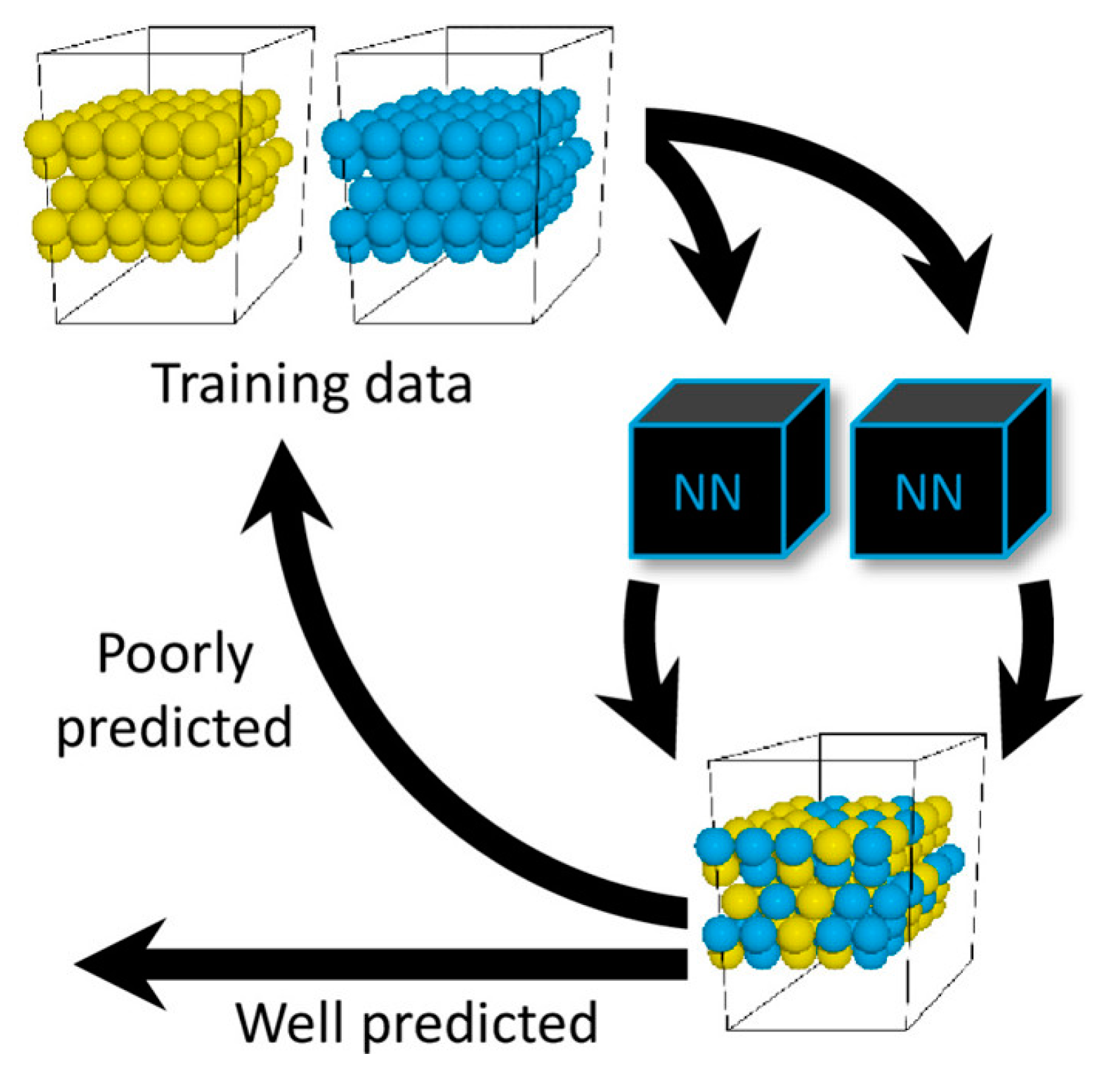
where and are tunable parameters and is the cutoff function of the interatomic distance (Equation (6)) with a defined cutoff distance . With this representation, ANN training of the PES can be described as the training of separate ANN models with only two inputs. Many subsequent applications of this representation method have shown that it is effective to precisely acquire the PES with an acceptable modeling time [93,94,95,96,97]. Chiriki et al. studied the dynamics and thermodynamics of Aun nanocatalysts () using ANN potentials [98]. Similar studies on varying Au configurations were also reported by Boes et al. [99]. With a similar method, Zhai and Alexandrova developed a GPU-accelerated deep neural network (DNN) [32] for the global optimization of Pt13 clusters [100]. To provide a general package for PES fitting, Khorshidi and Peterson developed an atomistic machine-learning package (Amp) combined with the new neural network representation shown above [101]. Based on this package, some catalytic structural information studies were published (e.g., oxygen interaction on Pd [102]). To expand this method to multi-element systems, Boes and Kitchin studied the surface segregation effects of PdAu random alloy models. They found that together with the network representation proposed by Behler and Parrinello, using two separate models, they were able to acquire a quick and precise potential for the atomic segregation studies of PdAu (Figure 11) [103]. Combined with the MC method, they discovered a moderate surface segregation of Pd under high temperature, which might lead to different catalytic activities.
5. Remarks and Prospects
In this review, we have shown that as a powerful predictive tool, ANN has been widely used in catalytic studies for both experiment and theory. It can be clearly seen that though the applications of ANN for experimental studies began in the 1990s, its application for theoretical catalytic studies was not popular until the last decade. The remarks can be summarized as follows:
- (1)
- As the most straightforward application, ANN has been widely used for the prediction of catalytic performance during the past two decades. Though there are various relevant studies, the motifs are quite similar: setting the experimental conditions and/or the properties of the catalytic system as the inputs, and the catalytic activities as the output of the model. Figure 2 summarizes three typical examples of such an application. It shows that the number of output variables can be more than one. That means an ANN with a sufficiently large database is able to perform multiple outputs to predict the product distribution and reaction selectivity.
- (2)
- In the catalysis community, the optimization and design of catalysts are usually more important. In addition to predicting the catalytic activities, some studies generated new input combinations for a well-trained ANN model, and acquired the predicted output activities. For the generations of new input combinations, GA is the most popular and (so far) the most successful strategy for input generation with less time-consumption. It is expected that in addition to the GA method, a machine learning-assisted HTS can be more sufficient for the generation of inputs in future study [37,104].
- (3)
- In terms of the theoretical catalysis study, ANN has proven to be a good tool for catalytic descriptor prediction (e.g., binding energy of adsorbate on a catalytic surface). Based on a DFT-calculated database with proper independent variables as the inputs, ANN is able to “learn” the highly-complicated intrinsic properties via a non-linear fitting process. Several successful studies such as the research done by Ulissi and Nørskov et al. [84] have shown that machine learning can be a good choice to reduce the computational cost of theoretical catalysis study, and meanwhile, provides precise and ultra-fast screening for new catalyst discovery.
- (4)
- To theoretically study the PES for a catalytic system and perform global optimizations, ANN has shown its capacity for mapping the PES for a specific reaction and/or a specific type of catalyst, combined with the MC methods. To reduce the inputs, Behler and Parrinello [92] developed a new ANN representation for atomic systems, which dramatically reduces the number of inputs for network training and saves much time and manpower. It is expected that this method would be more widely used for discovering the favorable structures of the catalytic systems (especially the metallic catalytic systems) and screening good potential catalysts.
- (5)
- It should be noted that though machine learning development has boosted the ANN applications to the catalysis study of both experiment and theory, many previous studies failed to use an appropriate training-and-testing method. Many of the studies did not perform optimization on ANN structures before they used the model for further applications. This is clearly not doing it in the correct way and could be risky for real applications. To define the optimal structure of ANN, different numbers of hidden neurons and (even) hidden layers should be tried for multiple training and testing. Failing to do this would lead to the severe potential risks of under- or over-fitting.
Though there is a huge success of ANNs that could facilitate the progress of the catalysis community, there are still some common challenges that should be addressed in future studies:
- (1)
- So far, most of the relevant studies have been done by a conventional ANN (e.g., BPNN). However, with the development of machine learning, conventional ANNs are sometimes no longer the best choice. For example, for fitting a PES of a metallic cluster, a BPNN with regular activation functions sometimes cannot provide smooth fitting, leading to incorrect forces. Also, the required training time of a conventional ANN is another challenge: with a larger database and higher number of hidden neurons and layers, the required training time would become much longer. At this moment, finding out the optimal ANN structure would become harder. Actually, with the algorithm developments, there are many machine learning methods that are sometimes more precise and much faster than the conventional ANN (e.g., GRNN, support vector machine (SVM) [105], and ELM [31]). Several comparative studies have been performed to compare the speed and accuracy of different algorithms [18,34,35]. It is expected that an increasing number of machine learning algorithms will be applied to catalysis studies in the future.
- (2)
- Similarly, with the rapid development of big data analysis and deep learning techniques [32], it is expected that they could be widely applied for catalytic activity predictions and global structural optimizations of catalytic systems. Though, so far, only a few relevant studies have focused on deep learning techniques (e.g., Zhai et al. [100]), more applicable studies should emerge in the near future. It is also expected that some of the current challenges, such as CO2 electroreduction selectivity and machine learning-assisted MD simulations, could be well-addressed and understood by state-of-the-art data-mining analysis and deep learning techniques.
- (3)
- Compared to other research areas, the applications of machine learning for the catalysis community are still not popular and not well-studied. The main reasons include: (i) acquiring the original database for model training is expensive; (ii) too many input variables have to be considered for modeling training; and (iii) there is a lack of user-friendly platforms. The first two points can be addressed by the developments of experimental and computational techniques and devices. The development of good atomic representations can also help reduce the input variables. In terms of the third point, despite there being some chemical packages that could help speed up the machine learning fitting, due to the complexity of chemical systems, very few of them are effective and user-friendly enough. It is expected that future inter-disciplinary study would help address this issue and more well-developed software platforms can be provided for more complicated catalytic studies.
Acknowledgments
We are grateful to Tian-Yi Ma (Guest Editor of Catalysts) and Shelly Liu (from the MDPI Office) for inviting us to submit this review paper. We also acknowledge all the publishers, journals, and authors who provided their permission for us to reproduce their figure results in this review. We would like to acknowledge the financial support from the Open Fund of Key Laboratory of Low-grade Energy Utilization Technologies and Systems, Ministry of Education of China (No. LLEUTS-201708), and Scientific and Technological Research Program of Chongqing Municipal Education Commission (No. KJ1709193).
Author Contributions
Hao Li provided the algorithmic discussions. Zhien Zhang provided most of the catalytic insights in both experiments and theories. This review paper was mainly done when Hao Li was in Sichuan University. All the authors participated in the discussion, writing, and revision of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Nomenclature
| AI | artificial intelligence |
| IT | information technology |
| ANN | artificial neural network |
| BPNN | back-propagation neural network |
| GRNN | general regression neural network |
| ELM | extreme learning machine |
| DNN | deep neural network |
| RMSE | root mean square error |
| EE2 | 17-ethynylestradiol |
| DOC | dissolved organic carbon |
| ODHE | oxidative dehydrogenation of ethane |
| GA | genetic algorithm |
| AC | active carbon |
| HTS | high-throughput screening |
| WGS | water-gas shift |
| RSM | response surface methodology |
| DFT | density functional theory |
| MD | molecular dynamics |
| TST | transition state theory |
| MC | Monte Carlo |
| PES | potential energy surface |
| BH | basin hopping |
| Amp | atomistic machine-learning package |
| SVM | support vector machine |
References
- Witten, I.H.; Frank, E. Data Mining: Practical Machine Learning Tools and Techniques; Morgan Kaufman: Burlington, MA, USA, 2005. [Google Scholar]
- Mair, C.; Kadoda, G.; Le, M.; Phalp, K.; Scho, C.; Shepperd, M.; Webster, S. An investigation of machine learning based prediction systems. J. Syst. Softw. 2000, 53, 23–29. [Google Scholar] [CrossRef]
- Kotsiantis, S.B. Supervised Machine Learning: A Review of Classification Techniques. Informatica 2007, 31, 249–268. [Google Scholar]
- Carpenter, G.A.; Grossberg, S.; Reynolds, J.H. ARTMAP: Supervised real-time learning and classification of nonstationary data by a self-organizing neural network. Neural Netw. 1991, 4, 565–588. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recognition and Machine Learning; Springer: Berlin, Germany, 2006; Volume 4. [Google Scholar]
- Raudys, S.J.; Jain, A.K. Small sample size effects in statistical pattern recognition: Recommendations for practitioners. IEEE Trans. Pattern Anal. Mach. Intell. 1991, 13, 252–264. [Google Scholar] [CrossRef]
- Chen, F.; Li, H.; Xu, Z.; Hou, S.; Yang, D. User-friendly optimization approach of fed-batch fermentation conditions for the production of iturin A using artificial neural networks and support vector machine. Electron. J. Biotechnol. 2015, 18. [Google Scholar] [CrossRef]
- Sommer, C.; Gerlich, D.W. Machine learning in cell biology—Teaching computers to recognize phenotypes. J. Cell Sci. 2013, 126, 5529–5539. [Google Scholar] [CrossRef] [PubMed]
- Tarca, A.L.; Carey, V.J.; Chen, X.; Romero, R.; Drăghici, S. Machine Learning and Its Applications to Biology. PLoS Comput. Biol. 2007, 3, e116. [Google Scholar] [CrossRef] [PubMed]
- Wernick, M.; Yang, Y.; Brankov, J.; Yourganov, G.; Strother, S. Machine learning in medical imaging. IEEE Signal Proc. Mag. 2010, 27, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Wei, J.S.; Ringnér, M.; Saal, L.H.; Ladanyi, M.; Westermann, F.; Berthold, F.; Schwab, M.; Antonescu, C.R.; Peterson, C.; et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med. 2001, 7, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Deo, R.C. Machine learning in medicine. Circulation 2015, 132, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Kalogirou, S. Applications of artificial neural networks in energy systems. Energy Convers. Manag. 1999, 40, 1073–1087. [Google Scholar] [CrossRef]
- Sanaye, S.; Asgari, H. Thermal modeling of gas engine driven air to water heat pump systems in heating mode using genetic algorithm and Artificial Neural Network methods. Int. J. Refrig. 2013, 36, 2262–2277. [Google Scholar] [CrossRef]
- Kalogirou, S.A.; Panteliou, S.; Dentsoras, A. Artificial neural networks used for the performance prediction of a thermosiphon solar water heater. Renew. Energy 1999, 18, 87–99. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, K.; Li, H.; Zhang, X.; Jin, G.; Cheng, K. Artificial Neural Networks-Based Software for Measuring Heat Collection Rate and Heat Loss Coefficient of Water-in-Glass Evacuated Tube Solar Water Heaters. PLoS ONE 2015, 10, e0143624. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, H.; Tang, X.; Zhang, X.; Lin, F.; Cheng, K. Extreme learning machine: A new alternative for measuring heat collection rate and heat loss coefficient of water-in-glass evacuated tube solar water heaters. Springerplus 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, H.; Zhang, X.; Jin, G.; Cheng, K. Novel method for measuring the heat collection rate and heat loss coefficient of water-in-glass evacuated tube solar water heaters based on artificial neural networks and support vector machine. Energies 2015, 8, 8814–8834. [Google Scholar] [CrossRef]
- Jung, H.C.; Kim, J.S.; Heo, H. Prediction of building energy consumption using an improved real coded genetic algorithm based least squares support vector machine approach. Energy Build. 2015, 90, 76–84. [Google Scholar] [CrossRef]
- Gardner, M.; Dorling, S. Artificial neural networks (the multilayer perceptron)—A review of applications in the atmospheric sciences. Atmos. Environ. 1998, 32, 2627–2636. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Cao, G. Quick Estimation Model for the Concentration of Indoor Airborne Culturable Bacteria: An Application of Machine Learning. Int. J. Environ. Res. Public Health 2017, 14, 857. [Google Scholar] [CrossRef] [PubMed]
- Recknagel, F. Applications of machine learning to ecological modelling. Ecol. Modell. 2001, 146, 303–310. [Google Scholar] [CrossRef]
- Peng, H.; Ling, X. Optimal design approach for the plate-fin heat exchangers using neural networks cooperated with genetic algorithms. Appl. Therm. Eng. 2008, 28, 642–650. [Google Scholar] [CrossRef]
- Kalogirou, S.A. Designing and Modeling Solar Energy Systems. In Solar Energy Engineering; Academic Press: Cambridge, MA, USA, 2014; pp. 583–699. [Google Scholar]
- Liu, Z.; Li, H.; Liu, K.; Yu, H.; Cheng, K. Design of high-performance water-in-glass evacuated tube solar water heaters by a high-throughput screening based on machine learning: A combined modeling and experimental study. Sol. Energy 2017. [Google Scholar] [CrossRef]
- Gao, J.; Jamidar, R. Machine Learning Applications for Data Center Optimization. Google White Pap. 2014, 1–13. [Google Scholar]
- Li, M. Scaling Distributed Machine Learning with the Parameter Server. In Proceedings of the 2014 International Conference on Big Data Science and Computing—BigDataScience’14, Broomfield, CO, USA, 6–8 October 2014. [Google Scholar]
- Hopfield, J.J. Artificial neural networks. IEEE Circuits Devices Mag. 1988, 4, 3–10. [Google Scholar] [CrossRef]
- Nawi, N.M.; Khan, A.; Rehman, M.Z. A New Back-Propagation Neural Network Optimized. ICCSA 2013, 2013, 413–426. [Google Scholar]
- Specht, D.F. A general regression neural network. IEEE Trans. Neural Netw. 1991, 2, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Huang, G.B.; Song, S.; You, K. Trends in extreme learning machines: A review. Neural Netw. 2015, 61, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Schmidhuber, J. Deep Learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef] [PubMed]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet Classification with Deep Convolutional Neural Networks. Adv. Neural Inf. Process. Syst. 2012, 1106–1114. [Google Scholar]
- Li, H.; Tang, X.; Wang, R.; Lin, F.; Liu, Z.; Cheng, K. Comparative Study on Theoretical and Machine Learning Methods for Acquiring Compressed Liquid Densities of 1,1,1,2,3,3,3-Heptafluoropropane (R227ea) via Song and Mason Equation, Support Vector Machine, and Artificial Neural Networks. Appl. Sci. 2016, 6, 25. [Google Scholar] [CrossRef]
- Li, H.; Chen, F.; Cheng, K.; Zhao, Z.; Yang, D. Prediction of Zeta Potential of Decomposed Peat via Machine Learning : Comparative Study of Support Vector Machine and Artificial Neural Networks. Int. J. Electrochem. Sci. 2015, 10, 6044–6056. [Google Scholar]
- Paxton, A.T.; Gumbsch, P.; Methfessel, M. A quantum mechanical calculation of the theoretical strength of metals. Philos. Mag. Lett. 1991, 63, 267–274. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.; Liu, K.; Zhang, Z. Predictive Power of Machine Learning for Optimizing Solar Water Heater Performance: The Potential Application of High-Throughput Screening. Int. J. Photoenergy 2017. [Google Scholar] [CrossRef]
- Huang, G.-B.; Zhu, Q.-Y.; Siew, C.-K. Extreme learning machine: Theory and applications. Neurocomputing 2006, 70, 489–501. [Google Scholar] [CrossRef]
- Browne, M.W. Cross-Validation Methods. J. Math. Psychol. 2000, 44, 108–132. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Livingstone, D.J.; Luik, A.I. Neural Network Studies. 1. Comparison of Overfitting and Overtraining. J. Chem. Inf. Comput. Sci. 1995, 35, 826–833. [Google Scholar] [CrossRef]
- Kito, S.; Hattori, T.; Murakami, Y. Estimation of catalytic performance by neural network—Product distribution in oxidative dehydrogenation of ethylbenzene. Appl. Catal. A Gen. 1994, 114. [Google Scholar] [CrossRef]
- Sasaki, M.; Hamada, H.; Kintaichi, Y.; Ito, T. Application of a neural network to the analysis of catalytic reactions Analysis of NO decomposition over Cu/ZSM-5 zeolite. Appl. Catal. A Gen. 1995, 132, 261–270. [Google Scholar] [CrossRef]
- Mohammed, M.L.; Patel, D.; Mbeleck, R.; Niyogi, D.; Sherrington, D.C.; Saha, B. Optimisation of alkene epoxidation catalysed by polymer supported Mo(VI) complexes and application of artificial neural network for the prediction of catalytic performances. Appl. Catal. A Gen. 2013, 466, 142–152. [Google Scholar] [CrossRef]
- Frontistis, Z.; Daskalaki, V.M.; Hapeshi, E.; Drosou, C.; Fatta-Kassinos, D.; Xekoukoulotakis, N.P.; Mantzavinos, D. Photocatalytic (UV-A/TiO2) degradation of 17α-ethynylestradiol in environmental matrices: Experimental studies and artificial neural network modeling. J. Photochem. Photobiol. A Chem. 2012, 240, 33–41. [Google Scholar] [CrossRef]
- Abdul Rahman, M.B.; Chaibakhsh, N.; Basri, M.; Salleh, A.B.; Abdul Rahman, R.N.Z.R. Application of artificial neural network for yield prediction of lipase-catalyzed synthesis of dioctyl adipate. Appl. Biochem. Biotechnol. 2009, 158, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Günay, M.E.; Yildirim, R. Neural network analysis of selective CO oxidation over copper-based catalysts for knowledge extraction from published data in the literature. Ind. Eng. Chem. Res. 2011, 50. [Google Scholar] [CrossRef]
- Raccuglia, P.; Elbert, K.C.; Adler, P.D.F.; Falk, C.; Wenny, M.B.; Mollo, A.; Zeller, M.; Friedler, S.A.; Schrier, J.; Norquist, A.J. Machine-learning-assisted materials discovery using failed experiments. Nature 2016, 533, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, A.G.; Rothenberg, G. Predictive modeling in catalysis—From dream to reality. Chem. Eng. Prog. 2009, 105, 26–32. [Google Scholar]
- Corma, A.; Serra, J.M.; Argente, E.; Botti, V.; Valero, S. Application of artificial neural networks to combinatorial catalysis: Modeling and predicting ODHE catalysts. ChemPhysChem 2002, 3, 939–945. [Google Scholar] [CrossRef]
- Kumar, M.; Husian, M.; Upreti, N.; Gupta, D. Genetic Algorithm: Review and Application. Int. J. Inf. Technol. Knowl. Manag. 2010, 2, 451–454. [Google Scholar]
- Yamada, M.; Omata, K. Prediction of Effective Additives to a Ni/Active Carbon Catalyst for Vapor-Phase Carbonylation of Methanol by an Artificial Neural Network. Ind. Eng. Chem. Res. 2004, 43, 6622–6625. [Google Scholar]
- Hou, Z.-Y.; Dai, Q.; Wu, X.-Q.; Chen, G.-T. Artificial neural network aided design of catalyst for propane ammoxidation. Appl. Catal. A Gen. 1997, 161, 183–190. [Google Scholar] [CrossRef]
- Zhao, Y.; Cundari, T.; Deng, J. Design of a Propane Ammoxidation Catalyst Using Artificial Neural Networks and Genetic Algorithms. Ind. Eng. Chem. Res. 2001, 40, 5475–5480. [Google Scholar]
- Umegaki, T.; Watanabe, Y.; Nukui, N.; Omata, K.; Yamada, M. Optimization of catalyst for methanol synthesis by a combinatorial approach using a parallel activity test and genetic algorithm assisted by a neural network. Energy Fuels 2003, 17, 850–856. [Google Scholar] [CrossRef]
- Rodemerck, U.; Baerns, M.; Holena, M.; Wolf, D. Application of a genetic algorithm and a neural network for the discovery and optimization of new solid catalytic materials. Appl. Surf. Sci. 2004, 223, 168–174. [Google Scholar] [CrossRef]
- Baumes, L.; Farrusseng, D.; Lengliz, M.; Mirodatos, C. Using artificial neural networks to boost high-throughput discovery in heterogeneous catalysis. QSAR Comb. Sci. 2004, 23, 767–778. [Google Scholar] [CrossRef]
- Kasiri, M.B.; Aleboyeh, H.; Aleboyeh, A. Modeling and optimization of heterogeneous photo-fenton process with response surface methodology and artificial neural networks. Environ. Sci. Technol. 2008, 42, 7970–7975. [Google Scholar] [CrossRef] [PubMed]
- Basri, M.; Rahman, R.N.Z.R.A.; Ebrahimpour, A.; Salleh, A.B.; Gunawan, E.R.; Rahman, M.B.A. Comparison of estimation capabilities of response surface methodology (RSM) with artificial neural network (ANN) in lipase-catalyzed synthesis of palm-based wax ester. BMC Biotechnol. 2007, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Prakash Maran, J.; Priya, B. Comparison of response surface methodology and artificial neural network approach towards efficient ultrasound-assisted biodiesel production from muskmelon oil. Ultrason. Sonochem. 2015, 23, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Betiku, E.; Taiwo, A.E. Modeling and optimization of bioethanol production from breadfruit starch hydrolyzate vis-a-vis response surface methodology and artificial neural network. Renew. Energy 2015, 74, 87–94. [Google Scholar] [CrossRef]
- Talebian-Kiakalaieh, A.; Amin, N.A.S.; Zarei, A.; Noshadi, I. Transesterification of waste cooking oil by heteropoly acid (HPA) catalyst: Optimization and kinetic model. Appl. Energy 2013, 102, 283–292. [Google Scholar] [CrossRef]
- Rajković, K.M.; Avramović, J.M.; Milić, P.S.; Stamenković, O.S.; Veljković, V.B. Optimization of ultrasound-assisted base-catalyzed methanolysis of sunflower oil using response surface and artifical neural network methodologies. Chem. Eng. J. 2013, 215–216, 82–89. [Google Scholar] [CrossRef]
- Mohammad Fauzi, A.H.; Saidina Amin, N.A. Optimization of oleic acid esterification catalyzed by ionic liquid for green biodiesel synthesis. Energy Convers. Manag. 2013, 76, 818–827. [Google Scholar] [CrossRef]
- Stamenković, O.S.; Rajković, K.; Veličković, A.V.; Milić, P.S.; Veljković, V.B. Optimization of base-catalyzed ethanolysis of sunflower oil by regression and artificial neural network models. Fuel Process. Technol. 2013, 114, 101–108. [Google Scholar] [CrossRef]
- Ayodele, O.B.; Auta, H.S.; Md Nor, N. Artificial neural networks, optimization and kinetic modeling of amoxicillin degradation in photo-fenton process using aluminum pillared montmorillonite-supported ferrioxalate catalyst. Ind. Eng. Chem. Res. 2012, 51, 16311–16319. [Google Scholar] [CrossRef]
- Pople, J.A.; Gill, P.M.W.; Johnson, B.G. Kohn—Sham density-functional theory within a finite basis set. Chem. Phys. Lett. 1992, 199, 557–560. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Berendsen, H.J.C. Computer Simulation of Molecular Dynamics: Methodology, Applications, and Perspectives in Chemistry. Angew. Chem. Int. Ed. Engl. 1990, 29, 992–1023. [Google Scholar] [CrossRef]
- Rapaport, D.C. Molecular dynamics simulation. Comput. Sci. Eng. 1999, 1, 537–542. [Google Scholar] [CrossRef]
- Pechukas, P. Transition State Theory. Annu. Rev. Phys. Chem. 1981, 32, 159–177. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef] [Green Version]
- Falsig, H.; Hvolbæk, B.; Kristensen, I.S.; Jiang, T.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. Trends in the catalytic CO oxidation activity of nanoparticles. Angew. Chem. Int. Ed. 2008, 47, 4835–4839. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.G.; Polanyi, M. Inertia and driving force of chemical reactions. Trans. Faraday Soc. 1938, 34, 11. [Google Scholar] [CrossRef]
- Logadottir, A.; Rod, T.; Nørskov, J.; Hammer, B.; Dahl, S.; Jacobsen, C.J. The Brønsted–Evans–Polanyi Relation and the Volcano Plot for Ammonia Synthesis over Transition Metal Catalysts. J. Catal. 2001, 197, 229–231. [Google Scholar] [CrossRef]
- Loffreda, D.; Delbecq, F.; Vigné, F.; Sautet, P. Fast prediction of selectivity in heterogeneous catalysis from extended brønsted-evans-polanyi relations: A theoretical insight. Angew. Chem. Int. Ed. 2009, 48, 8978–8980. [Google Scholar] [CrossRef] [PubMed]
- Fernndez, E.M.; Moses, P.G.; Toftelund, A.; Hansen, H.A.; Martínez, J.I.; Abild-Pedersen, F.; Kleis, J.; Hinnemann, B.; Rossmeisl, J.; Bligaard, T.; et al. Scaling relationships for adsorption energies on transition metal oxide, sulfide, and nitride surfaces. Angew. Chem. Int. Ed. 2008, 47, 4683–4686. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Tsai, C.; Chen, L.D.; Abild-Pedersen, F.; Nørskov, J.K.; Chan, K. Scaling Relations for Adsorption Energies on Doped Molybdenum Phosphide Surfaces. ACS Catal. 2017, 7, 2528–2534. [Google Scholar] [CrossRef]
- Ma, X.; Li, Z.; Achenie, L.E.K.; Xin, H. Machine-Learning-Augmented Chemisorption Model for CO2 Electroreduction Catalyst Screening. J. Phys. Chem. Lett. 2015, 6, 3528–3533. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.B.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Nørskov, J.K. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.Y.; Mullen, G.M.; Flaherty, D.W.; Mullins, C.B. Selective hydrogen production from formic acid decomposition on Pd-Au bimetallic surfaces. J. Am. Chem. Soc. 2014, 136, 11070–11078. [Google Scholar] [CrossRef] [PubMed]
- Davran-Candan, T.; Günay, M.E.; Yildirim, R. Structure and activity relationship for CO and O2 adsorption over gold nanoparticles using density functional theory and artificial neural networks. J. Chem. Phys. 2010, 132, 174113. [Google Scholar] [CrossRef] [PubMed]
- Ulissi, Z.W.; Tang, M.T.; Xiao, J.; Liu, X.; Torelli, D.A.; Karamad, M.; Cummins, K.; Hahn, C.; Lewis, N.S.; Jaramillo, T.F.; et al. Machine-Learning Methods Enable Exhaustive Searches for Active Bimetallic Facets and Reveal Active Site Motifs for CO2 Reduction. ACS Catal. 2017, 7b01648. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Chen, W.; Medasani, B.; Qu, X.; Kocher, M.; Brafman, M.; Petretto, G.; Rignanese, G.M.; Hautier, G.; et al. FireWorks: A dynamic workflow system designed for high-throughput applications. Concurr. Comput. 2015, 27, 5037–5059. [Google Scholar] [CrossRef]
- Wolf, U.; Arkhipov, V.I.; Bässler, H. Current injection from a metal to a disordered hopping system. I. Monte Carlo simulation. Phys. Rev. B 1999, 59, 7507–7513. [Google Scholar] [CrossRef]
- Sumpter, B.G.; Noid, D.W. Potential energy surfaces for macromolecules. A neural network technique. Chem. Phys. Lett. 1992, 192, 455–462. [Google Scholar] [CrossRef]
- Lorenz, S.; Scheffler, M.; Gross, A. Descriptions of surface chemical reactions using a neural network representation of the potential-energy surface. Phys. Rev. B 2006, 73, 115431. [Google Scholar] [CrossRef]
- Tai No, K. Description of the potential energy surface of the water dimer with an artificial neural network. Chem. Phys. Lett. 1997, 271, 152–156. [Google Scholar] [CrossRef]
- Ouyang, R.; Xie, Y.; Jiang, D. Global minimization of gold clusters by combining neural network potentials and the basin-hopping method. Nanoscale 2015, 7, 14817–14821. [Google Scholar] [CrossRef] [PubMed]
- Wales, D.J. Global Optimization of Clusters, Crystals, and Biomolecules. Science 1999, 285, 1368–1372. [Google Scholar] [CrossRef] [PubMed]
- Behler, J.; Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 2007, 98. [Google Scholar] [CrossRef] [PubMed]
- Behler, J. Neural network potential-energy surfaces in chemistry: A tool for large-scale simulations. Phys. Chem. Chem. Phys. 2011, 13, 17930. [Google Scholar] [CrossRef] [PubMed]
- Behler, J. Constructing high-dimensional neural network potentials: A tutorial review. Int. J. Q. Chem. 2015, 115, 1032–1050. [Google Scholar] [CrossRef]
- Morawietz, T.; Behler, J. A density-functional theory-based neural network potential for water clusters including van der waals corrections. J. Phys. Chem. A 2013, 117, 7356–7366. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Behler, J. Neural network molecular dynamics simulations of solid–liquid interfaces: Water at low-index copper surfaces. Phys. Chem. Chem. Phys. 2016, 18, 28704–28725. [Google Scholar] [CrossRef] [PubMed]
- Artrith, N.; Behler, J. High-dimensional neural network potentials for metal surfaces: A prototype study for copper. Phys. Rev. B—Condens. Matter Mater. Phys. 2012, 85. [Google Scholar] [CrossRef]
- Chiriki, S.; Jindal, S.; Bulusu, S.S. Neural network potentials for dynamics and thermodynamics of gold nanoparticles. J. Chem. Phys. 2017, 146. [Google Scholar] [CrossRef] [PubMed]
- Boes, J.R.; Groenenboom, M.C.; Keith, J.A.; Kitchin, J.R. Neural network and ReaxFF comparison for Au properties. Int. J. Q. Chem. 2016, 116, 979–987. [Google Scholar] [CrossRef]
- Zhai, H.; Alexandrova, A.N. Ensemble-Average Representation of Pt Clusters in Conditions of Catalysis Accessed through GPU Accelerated Deep Neural Network Fitting Global Optimization. J. Chem. Theory Comput. 2016, 12, 6213–6226. [Google Scholar] [CrossRef] [PubMed]
- Khorshidi, A.; Peterson, A.A. Amp: A modular approach to machine learning in atomistic simulations. Comput. Phys. Commun. 2016, 207, 310–324. [Google Scholar] [CrossRef]
- Boes, J.R.; Kitchin, J.R. Neural network predictions of oxygen interactions on a dynamic Pd surface. Mol. Simul. 2017, 43, 346–354. [Google Scholar] [CrossRef]
- Boes, J.R.; Kitchin, J.R. Modeling Segregation on AuPd(111) Surfaces with Density Functional Theory and Monte Carlo Simulations. J. Phys. Chem. C 2017, 121, 3479–3487. [Google Scholar] [CrossRef]
- Hautier, G.; Fischer, C.C.; Jain, A.; Mueller, T.; Ceder, G. Finding natures missing ternary oxide compounds using machine learning and density functional theory. Chem. Mater. 2010, 22, 3762–3767. [Google Scholar] [CrossRef]
- Suykens, J.A.; Vandewalle, J. Least Squares Support Vector Machine Classifiers. Neural Process. Lett. 1999, 9, 293–300. [Google Scholar] [CrossRef]
Figure 1.
Algorithmic structure of a typical artificial neural network (ANN). Each circle represents a neuron in the ANN algorithm. All the variables only pass to the same direction (from left to right). Reproduced with permission from Reference [37].
Figure 1.
Algorithmic structure of a typical artificial neural network (ANN). Each circle represents a neuron in the ANN algorithm. All the variables only pass to the same direction (from left to right). Reproduced with permission from Reference [37].

Figure 2.
Schematic structure of the ANNs modeled for the activity prediction of thermal catalysis, photocatalysis, and enzymatic catalysis. (a) Prediction of the catalytic activity of ethylbenzene oxidative hydrogenation reported in Reference [41]. The network contains nine input neurons, eight hidden neurons, and five output neurons (9-8-5). (b) Prediction of the photocatalytic activity of the 17-ethynylestradiol (EE2) conversion reported in [44]. The network contains five input neurons, thirteen hidden neurons, and one output neuron (5-13-1). (c) Prediction of lipase-catalyzed dioctyl adipate synthesis reported in [45]. The network contains four input neurons, seven hidden neurons, and one output neuron (4-7-1). The biases of the ANNs are not shown in these structures.
Figure 2.
Schematic structure of the ANNs modeled for the activity prediction of thermal catalysis, photocatalysis, and enzymatic catalysis. (a) Prediction of the catalytic activity of ethylbenzene oxidative hydrogenation reported in Reference [41]. The network contains nine input neurons, eight hidden neurons, and five output neurons (9-8-5). (b) Prediction of the photocatalytic activity of the 17-ethynylestradiol (EE2) conversion reported in [44]. The network contains five input neurons, thirteen hidden neurons, and one output neuron (5-13-1). (c) Prediction of lipase-catalyzed dioctyl adipate synthesis reported in [45]. The network contains four input neurons, seven hidden neurons, and one output neuron (4-7-1). The biases of the ANNs are not shown in these structures.
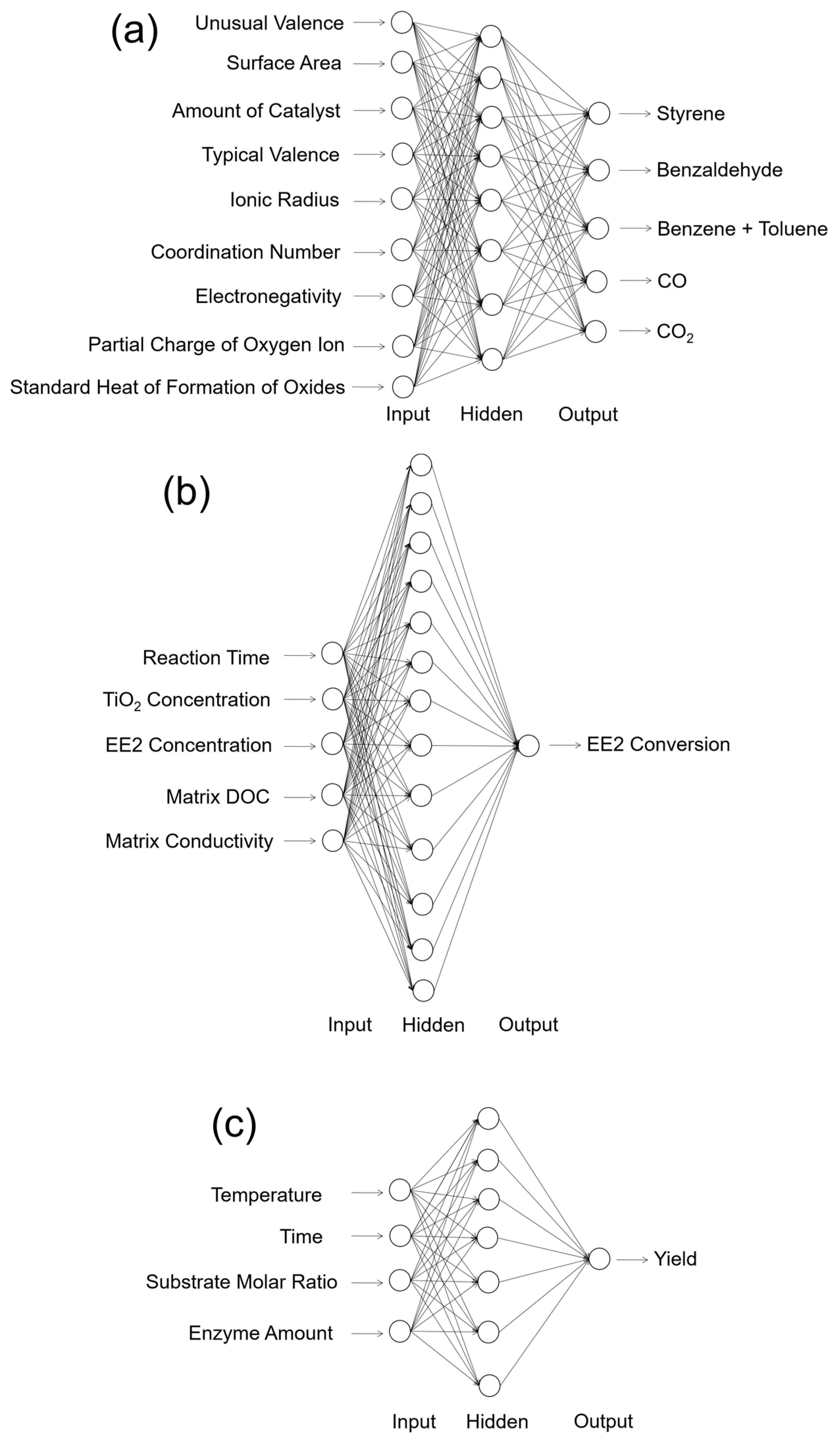
Figure 3.
Optimization modeling that combines both experimental and computational steps. Flow chart summarized by Maldonado and Rothenberg [48].
Figure 3.
Optimization modeling that combines both experimental and computational steps. Flow chart summarized by Maldonado and Rothenberg [48].
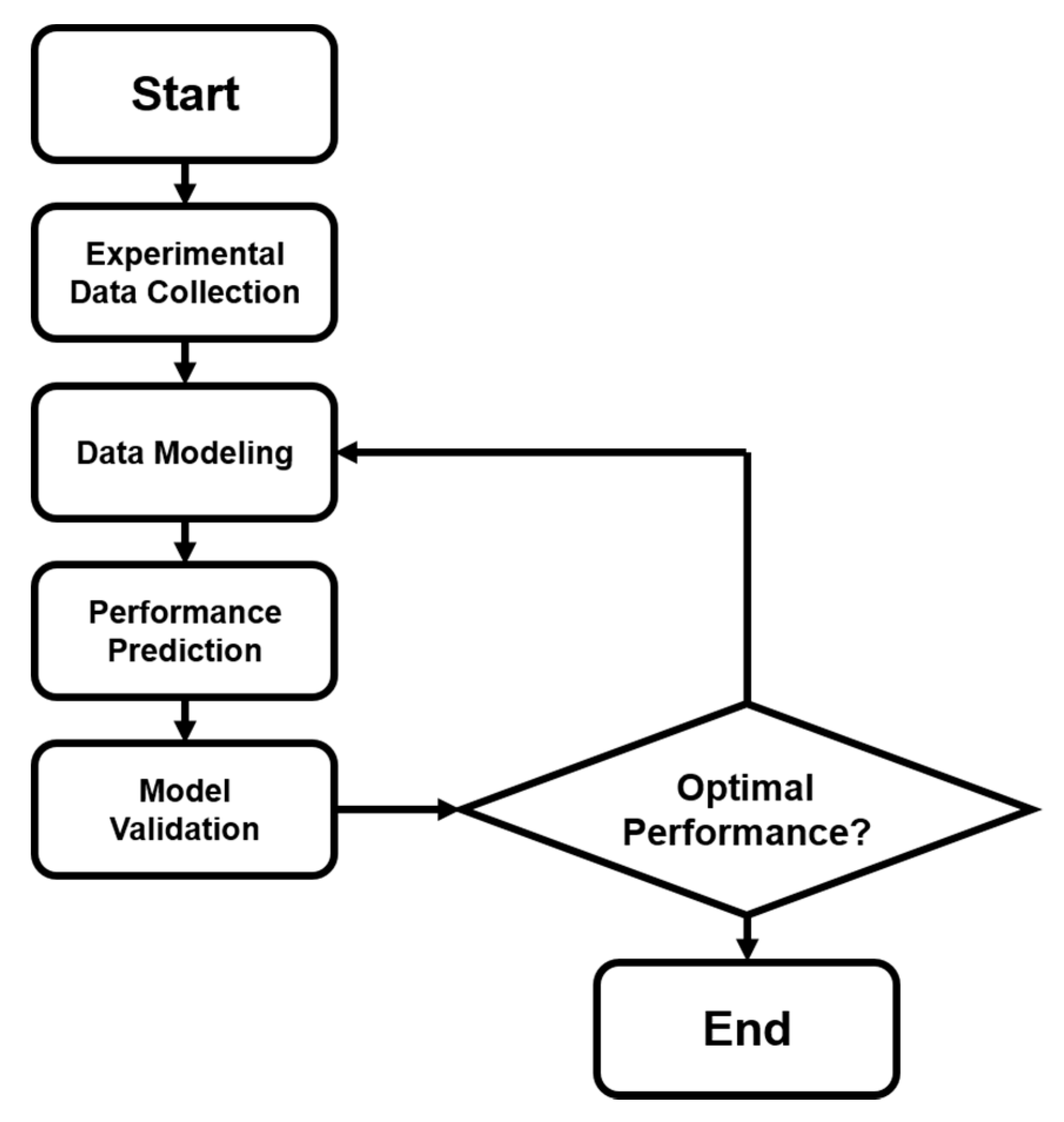
Figure 4.
Scheme of the ANN applications for predicting the combinatorial catalysts for the oxidative dehydrogenation of ethane (ODHE). Research reported by Corma et al. [49].
Figure 4.
Scheme of the ANN applications for predicting the combinatorial catalysts for the oxidative dehydrogenation of ethane (ODHE). Research reported by Corma et al. [49].
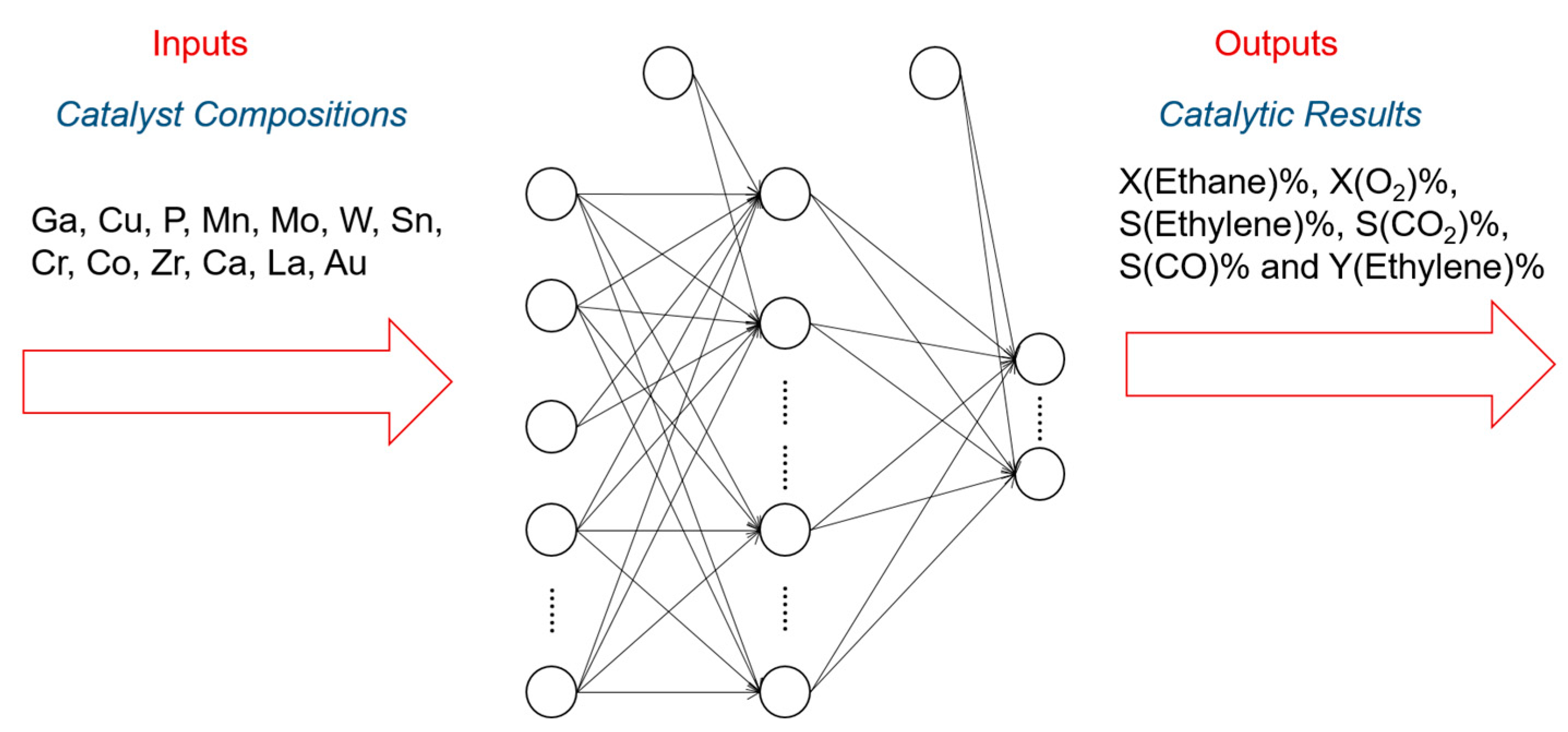
Figure 5.
A framework of the methodology proposed for boosting primary screening in heterogeneous catalysis [56]. GA: genetic algorithm.
Figure 5.
A framework of the methodology proposed for boosting primary screening in heterogeneous catalysis [56]. GA: genetic algorithm.

Figure 6.
(a) Training and (b) testing results for CO binding energy prediction using an ANN. (c) Training and (d) testing results for O2 binding energy predictions using an ANN. Reproduced with permission from Reference [83].
Figure 6.
(a) Training and (b) testing results for CO binding energy prediction using an ANN. (c) Training and (d) testing results for O2 binding energy predictions using an ANN. Reproduced with permission from Reference [83].
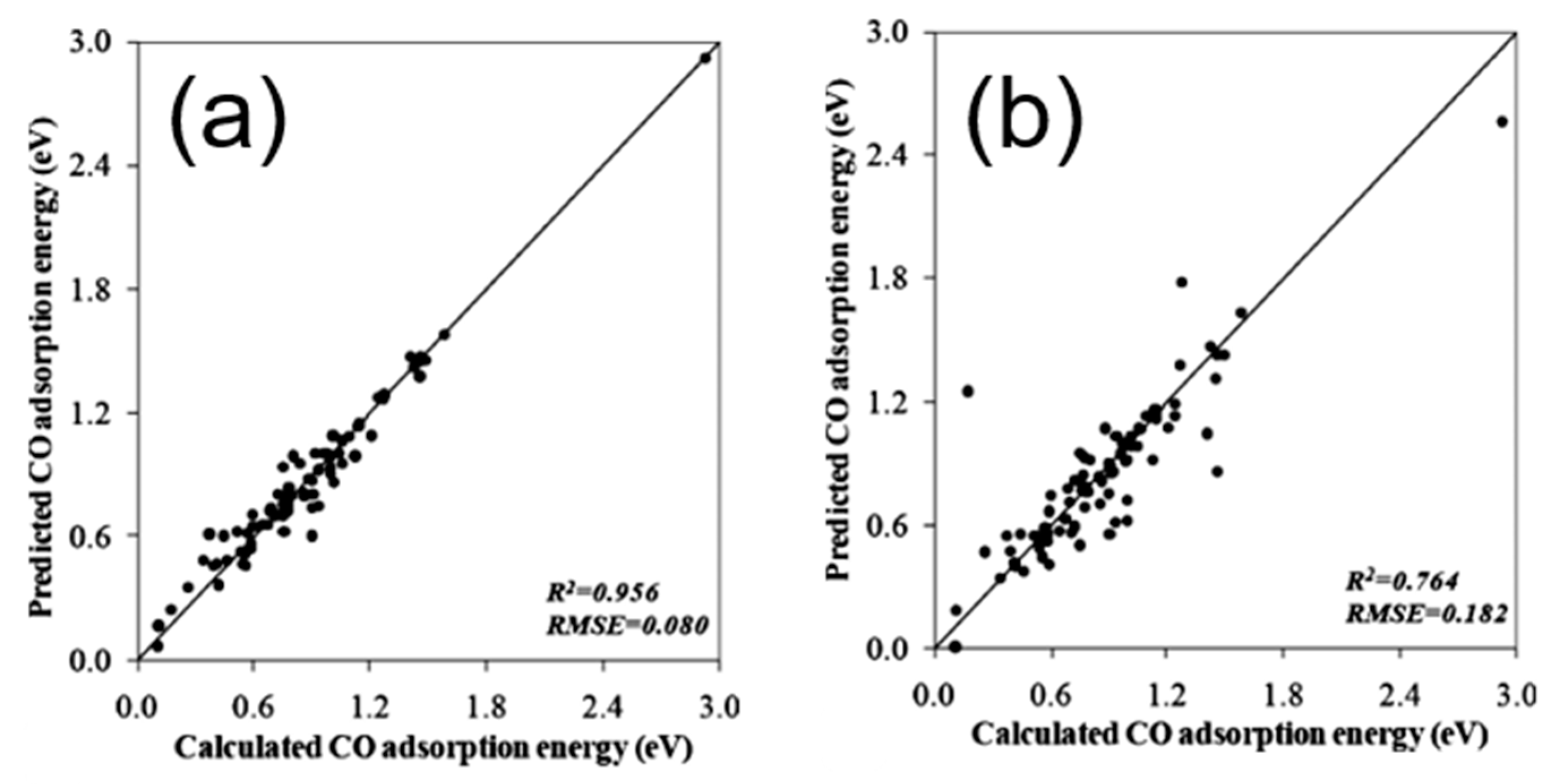
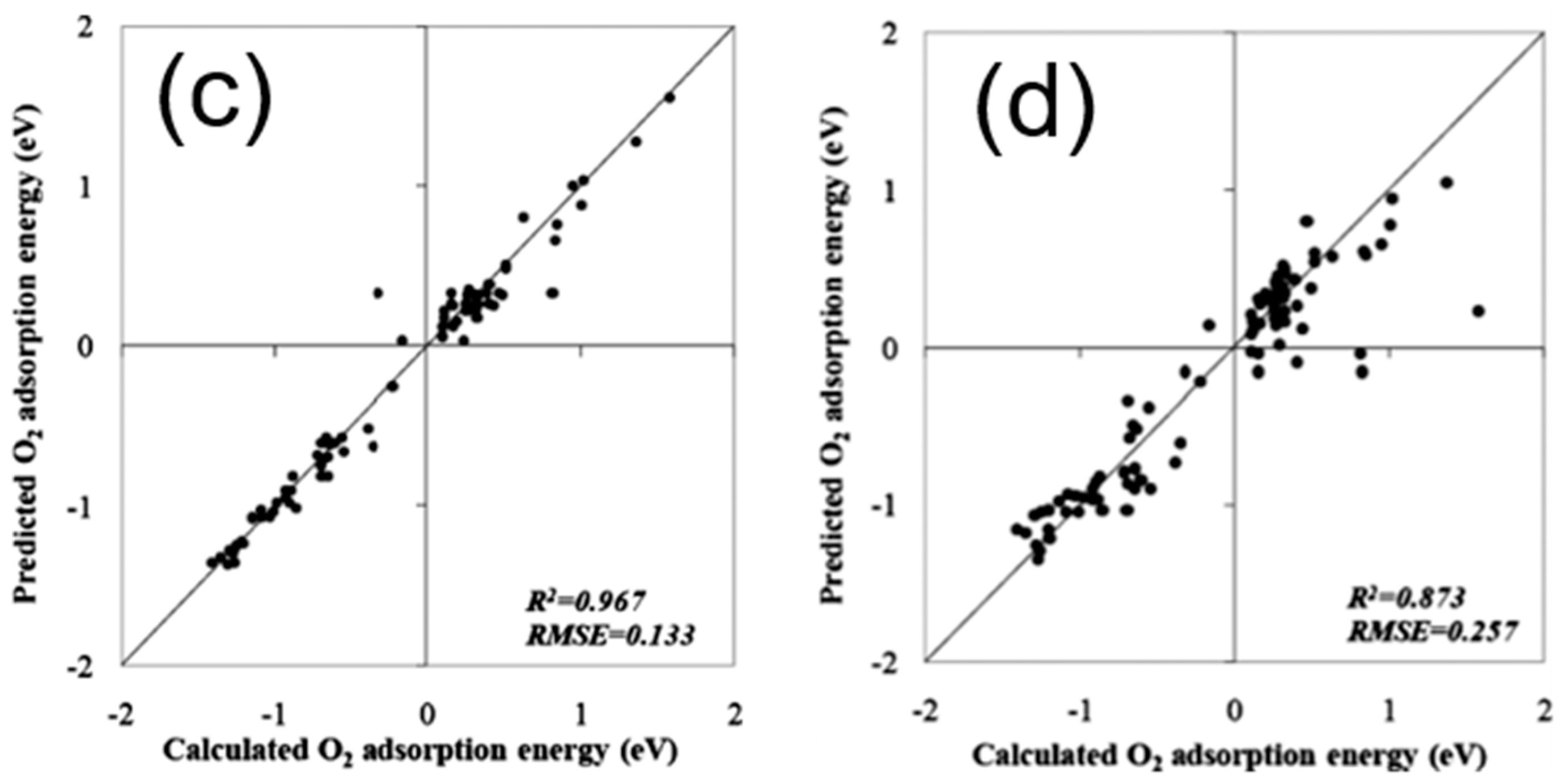
Figure 7.
(a) Screened results of CO binding energy on the second-generation core–shell alloy surfaces (Cu3B-A@Cu) via a trained ANN model. (b) Comparison of the CO binding energies on selected Cu monolayer alloys calculated using both DFT and knowledge-based ANN. Reproduced with permission from Reference [79].
Figure 7.
(a) Screened results of CO binding energy on the second-generation core–shell alloy surfaces (Cu3B-A@Cu) via a trained ANN model. (b) Comparison of the CO binding energies on selected Cu monolayer alloys calculated using both DFT and knowledge-based ANN. Reproduced with permission from Reference [79].
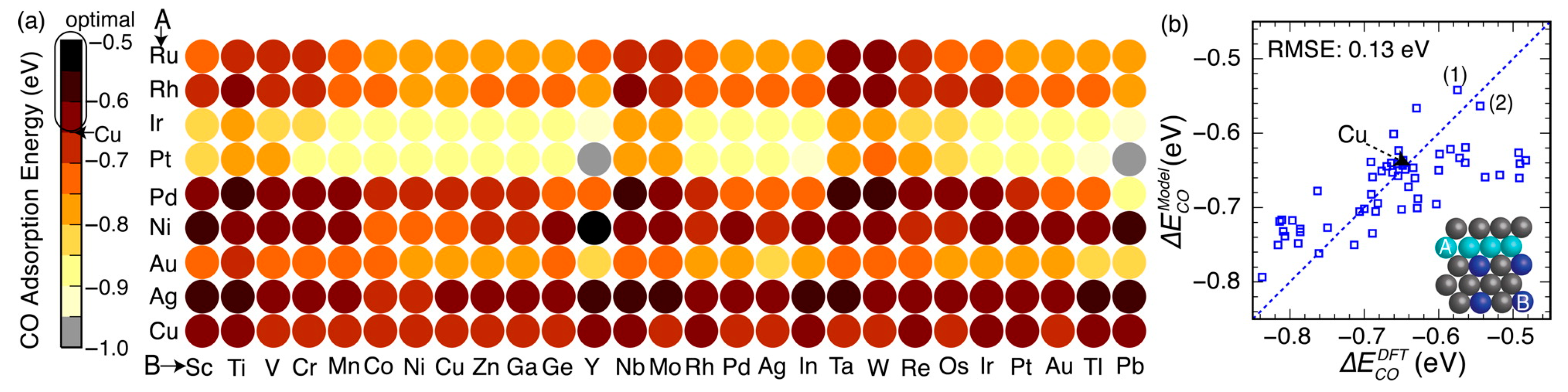
Figure 8.
(a) Framework of ANN model development and its application of the catalyst screening. The acquisition of new training data via DFT single-point calculations was performed by a FireWorks workflow developed in Reference [85]. (b) Volcano activity plot of CO2 electroreduction. Green and red circles are the DFT calculated transition state energies of single-metal step or terrace surfaces. Purple circles are the three Ni-Ga facets. Reproduced with permission from Reference [84].
Figure 8.
(a) Framework of ANN model development and its application of the catalyst screening. The acquisition of new training data via DFT single-point calculations was performed by a FireWorks workflow developed in Reference [85]. (b) Volcano activity plot of CO2 electroreduction. Green and red circles are the DFT calculated transition state energies of single-metal step or terrace surfaces. Purple circles are the three Ni-Ga facets. Reproduced with permission from Reference [84].
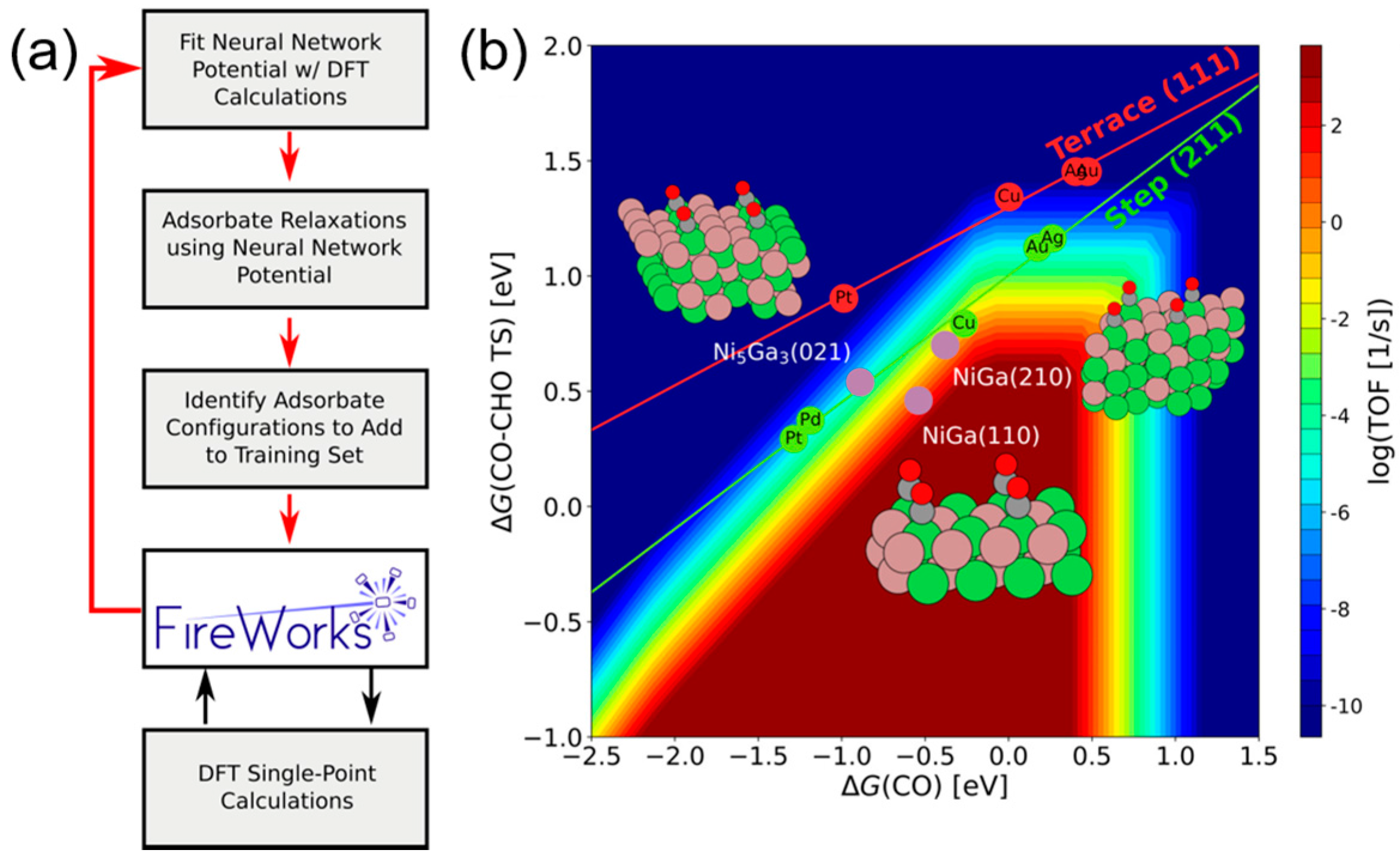
Figure 9.
Flow chart of the ANN potential fitting for Au58 clusters studied by Ouyang et al. [90], an Open Access Article licensed under a Creative Commons Attribution 3.0 Unported License.
Figure 9.
Flow chart of the ANN potential fitting for Au58 clusters studied by Ouyang et al. [90], an Open Access Article licensed under a Creative Commons Attribution 3.0 Unported License.
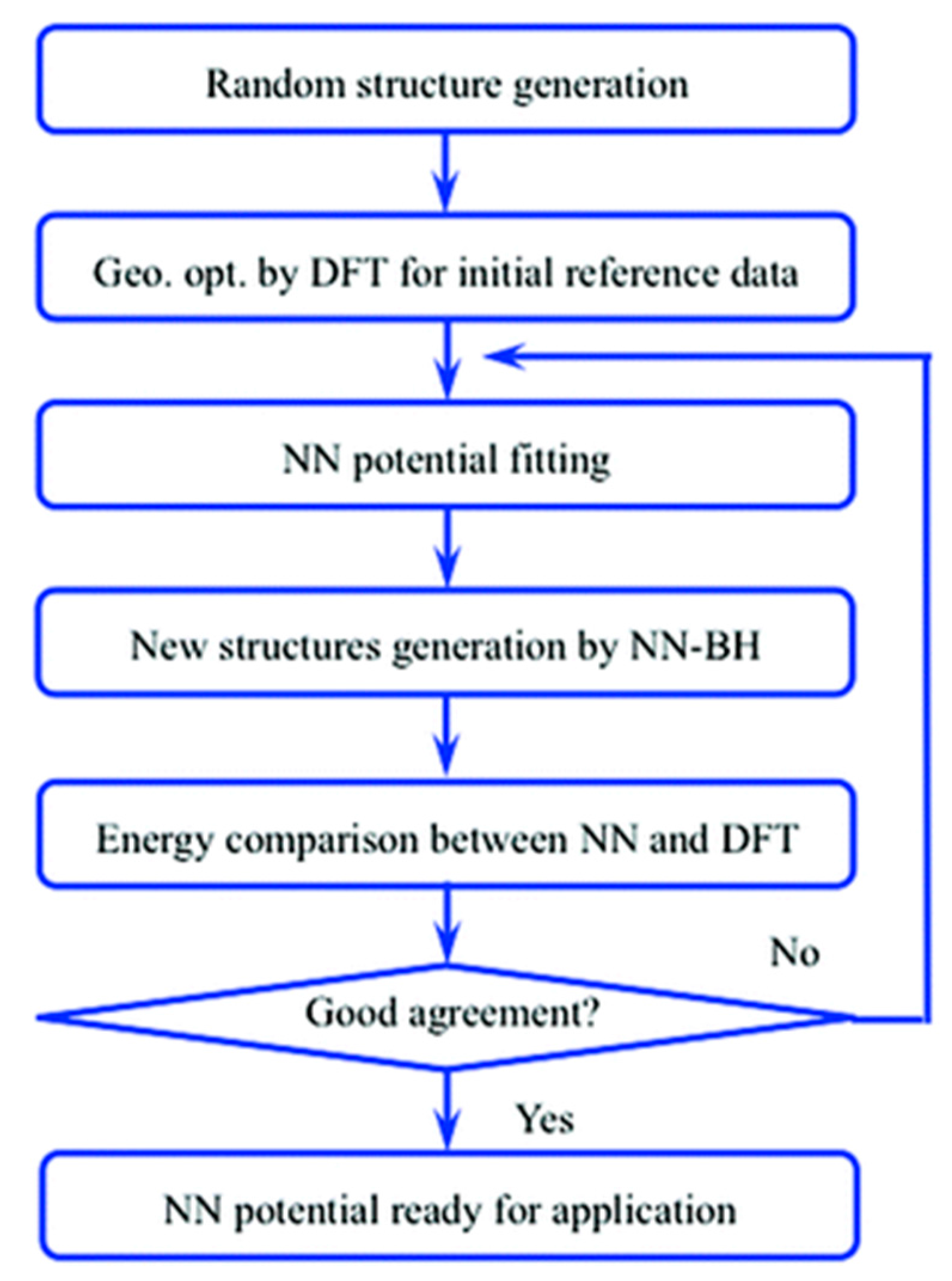
Figure 10.
A generalized ANN representation of high-dimensional potential energy surface (PES) proposed by Behler and Parrinello [92]. represents the Cartesian coordinates of the ith atom. They are transformed into the local geometric environment (assigned as ). Each consists of the pair interaction and angular terms. Each represents a single ANN model with the inputs of and the output of the energy contribution of the ith atom. Total energy is represented as the sum of . Reproduced with permission from Reference [92].
Figure 10.
A generalized ANN representation of high-dimensional potential energy surface (PES) proposed by Behler and Parrinello [92]. represents the Cartesian coordinates of the ith atom. They are transformed into the local geometric environment (assigned as ). Each consists of the pair interaction and angular terms. Each represents a single ANN model with the inputs of and the output of the energy contribution of the ith atom. Total energy is represented as the sum of . Reproduced with permission from Reference [92].
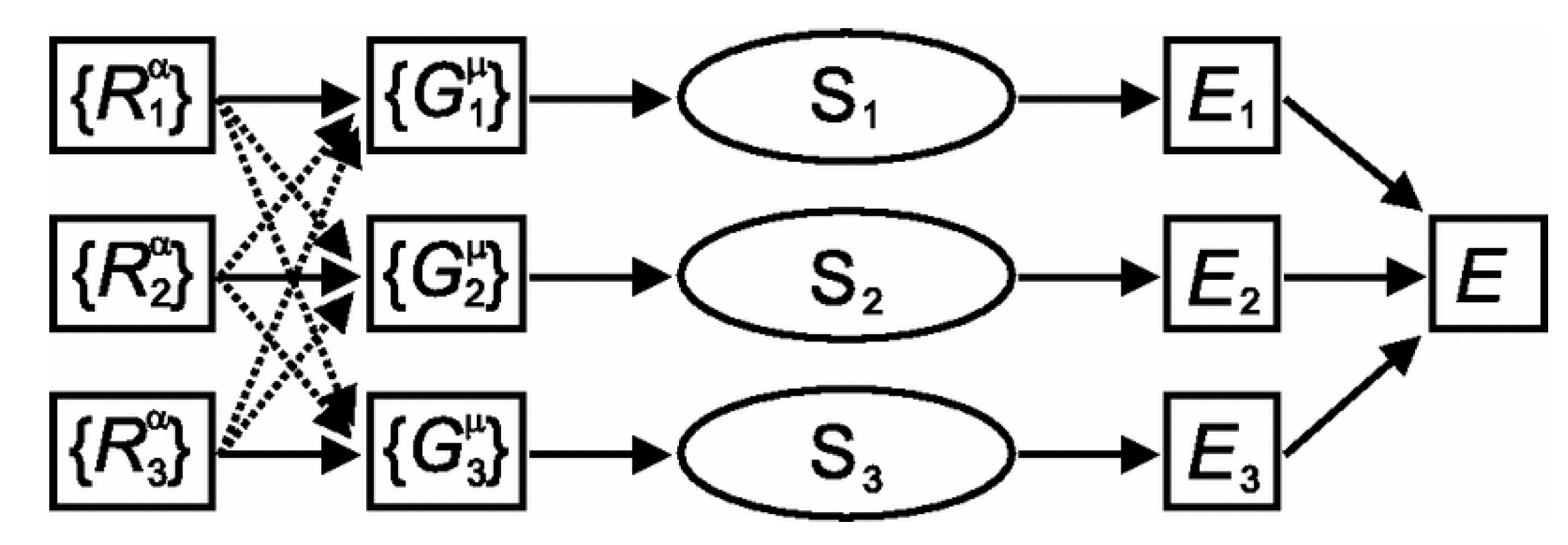
Figure 11.
Diagram of the PES training of a PdAu alloy system. Yellow spheres represent Au atoms. Blue spheres represent Pd atoms. The two black boxes represent the two ANN models for the training of Pd and Au, respectively. Reproduced with permission from Reference [103].
Figure 11.
Diagram of the PES training of a PdAu alloy system. Yellow spheres represent Au atoms. Blue spheres represent Pd atoms. The two black boxes represent the two ANN models for the training of Pd and Au, respectively. Reproduced with permission from Reference [103].

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, H.; Zhang, Z.; Liu, Z. Application of Artificial Neural Networks for Catalysis: A Review. Catalysts 2017, 7, 306. https://doi.org/10.3390/catal7100306
AMA Style
Li H, Zhang Z, Liu Z. Application of Artificial Neural Networks for Catalysis: A Review. Catalysts. 2017; 7(10):306. https://doi.org/10.3390/catal7100306
Chicago/Turabian StyleLi, Hao, Zhien Zhang, and Zhijian Liu. 2017. "Application of Artificial Neural Networks for Catalysis: A Review" Catalysts 7, no. 10: 306. https://doi.org/10.3390/catal7100306
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.