Metal-Catalyzed Intra- and Intermolecular Addition of Carboxylic Acids to Alkynes in Aqueous Media: A Review



Laboratorio de Compuestos Organometálicos y Catálisis (Unidad Asociada al CSIC), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Departamento de Química Orgánica e Inorgánica, IUQOEM, Facultad de Química, Universidad de Oviedo, Julián Clavería 8, E-33006 Oviedo, Spain
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(11), 328; https://doi.org/10.3390/catal7110328
Submission received: 19 October 2017
/
Revised: 1 November 2017
/
Accepted: 2 November 2017
/
Published: 6 November 2017
(This article belongs to the Special Issue Homogeneous Catalysis and Mechanisms in Water and Biphasic Media)
Abstract
:The metal-catalyzed addition of carboxylic acids to alkynes is a very effective tool for the synthesis of carboxylate-functionalized olefinic compounds in an atom-economical manner. Thus, a large variety of synthetically useful lactones and enol-esters can be accessed through the intra- or intermolecular versions of this process. In order to reduce the environmental impact of these reactions, considerable efforts have been devoted in recent years to the development of catalytic systems able to operate in aqueous media, which represent a real challenge taking into account the tendency of alkynes to undergo hydration in the presence of transition metals. Despite this, different Pd, Pt, Au, Cu and Ru catalysts capable of promoting the intra- and intermolecular addition of carboxylic acids to alkynes in a selective manner in aqueous environments have appeared in the literature. In this review article, an overview of this chemistry is provided. The synthesis of β-oxo esters by catalytic addition of carboxylic acids to terminal propargylic alcohols in water is also discussed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
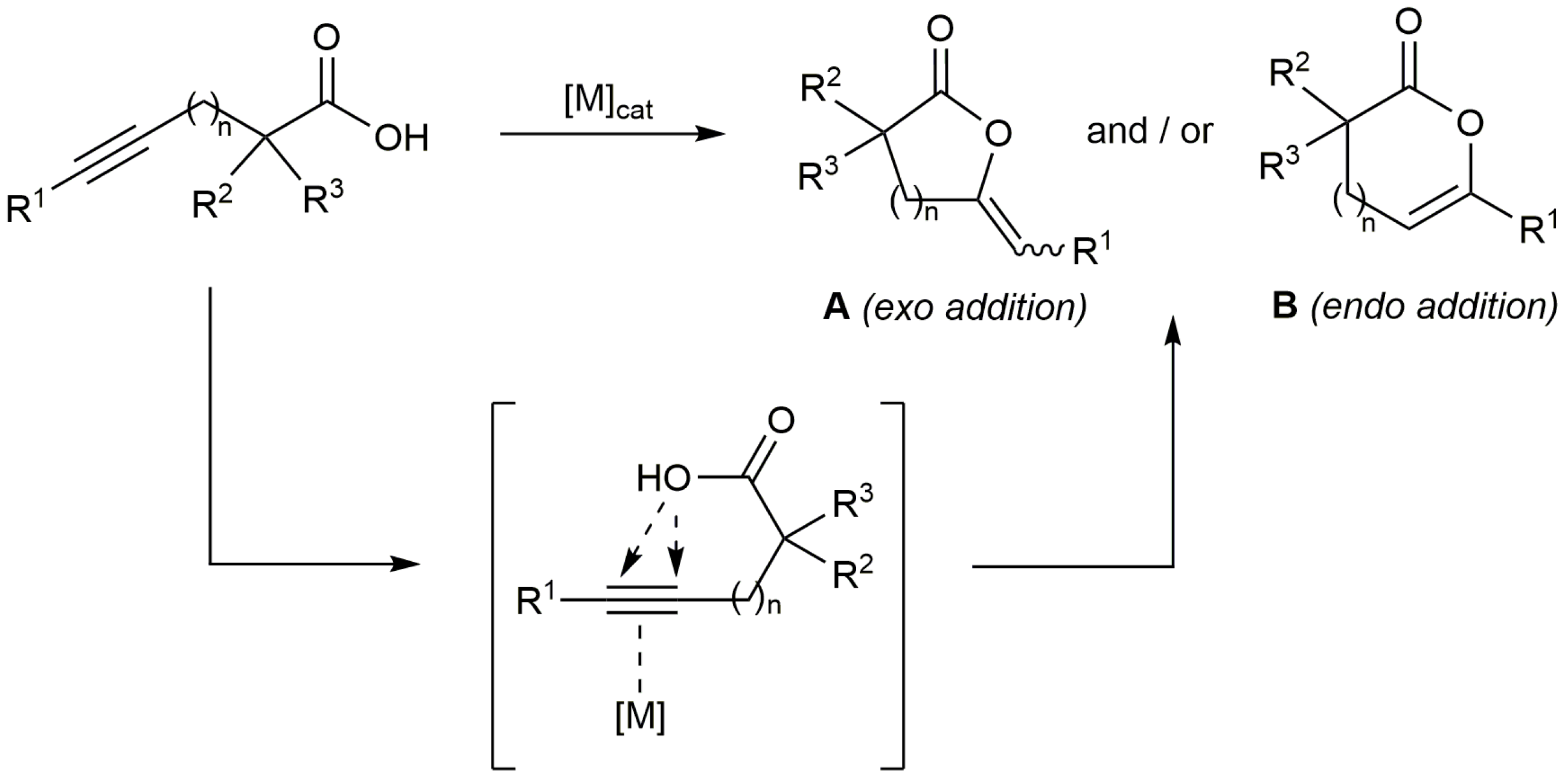
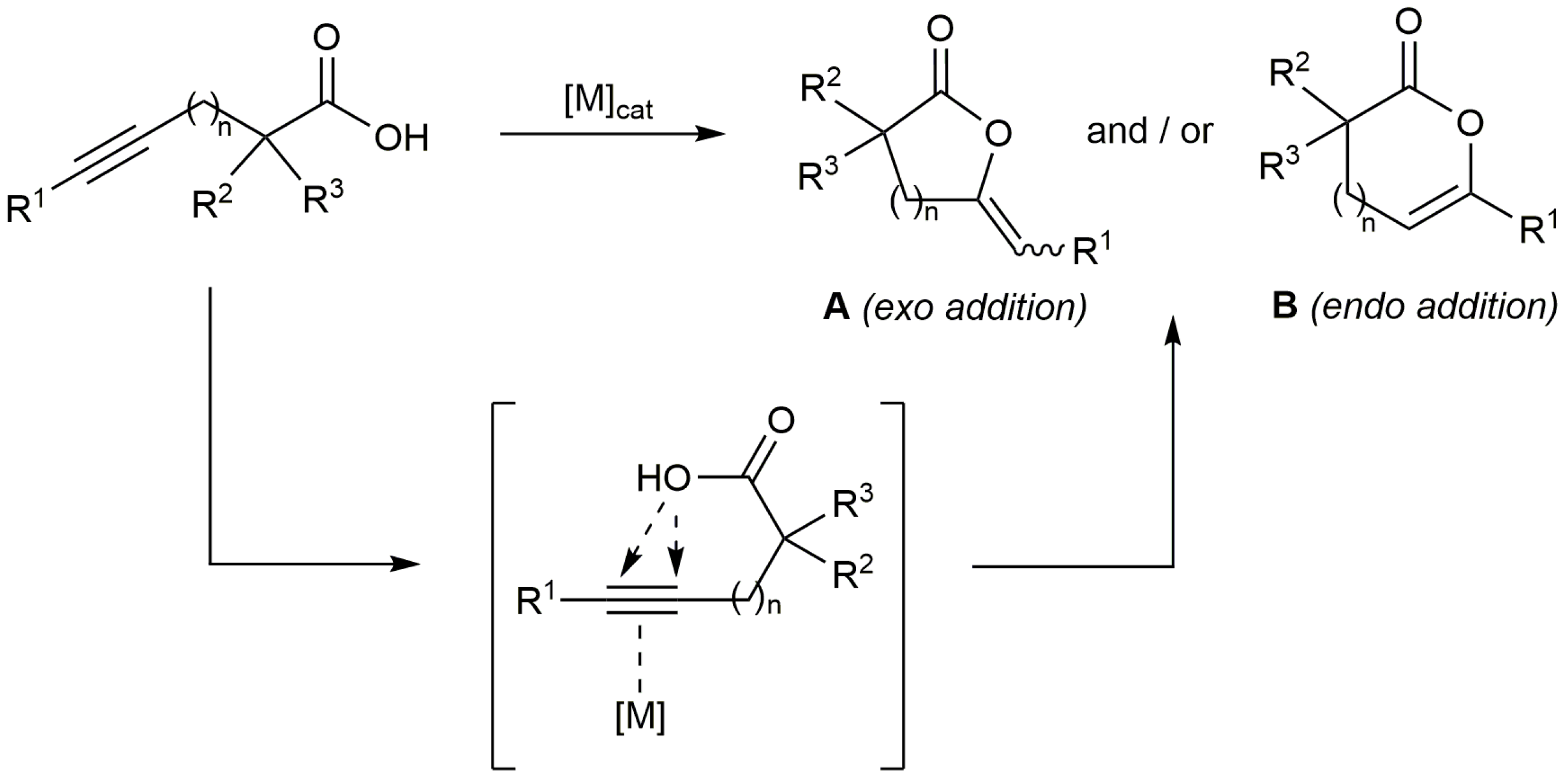
The transition metal-catalyzed heterofunctionalization of alkynes by addition of nucleophiles to the C≡C bond has emerged in recent years as a versatile synthetic tool in organic chemistry [1,2,3]. In particular, the addition of carboxylic acids to alkynes, reaction also referred in the literature as hydro-oxycarbonylation or hydrocarboxylation of alkynes, represents a straightforward way to obtain different types of olefinic esters with atom economy [4,5,6]. Thus, the intramolecular version of the process, i.e., the cycloisomerization of alkynoic acids, produces unsaturated lactones (Scheme 1) which are common structural motifs found in natural products and biologically active molecules, as well as valuable synthetic intermediates [7,8,9,10]. A large variety of transition metal complexes have been used to catalyze these reactions through the π-activation of the carbon-carbon triple bond of the alkynoic acid, the regioselectivity (exo or endo addition of the carboxylate to the C≡C bond leading to lactones A or B, respectively) and stereoselectivity of the process being dependent on the metal employed, the length of the hydrocarbon chain connecting the acid and alkyne units, and the terminal or internal nature of the alkyne functionality [1,2,3,4,5,6]. Among the most commonly employed metals are Pd, Ag, Au and Rh, including examples of heterogeneous systems [11], which have shown a high efficacy for the selective preparation of 5-, 6-, and to a lesser extent, 7-membered ring lactones [1,2,3,4,5,6].
The intermolecular addition of carboxylic acids to alkynes also presents enormous interest since the reaction products, i.e., enol esters (C or D in Scheme 2), are versatile building blocks in organic synthesis and material science. For example, to name just a few of their myriad applications, they are widely employed as mild acylating reagents [12,13], as monomers in diverse polymerization reactions [14,15,16], as substrates in asymmetric hydrogenation for the generation of enantioenriched alcohols [17,18,19], or as starting materials for different cross-coupling processes [20,21,22]. As for the cycloisomerization of alkynoic acids, a huge number of catalytic systems involving late transition metals have been described, with those based on ruthenium playing a prominent role [1,2,3,4,5]. However, for a long time, this reaction was limited to terminal alkynes, since most catalysts failed to enable the hydrocarboxylation of internal alkynes as a consequence of their greater steric hindrance, which disfavours their coordination to the metal catalyst. At this point, it should be noted that a reduced reactivity of internal vs. terminal C≡C bonds is also commonly observed in the cyclization reactions of alkynoic acids, although in this case the problem is not so marked as the intramolecular process is much more favoured from a thermodynamic point of view. Fortunately, recent works focused in the design of increasingly active catalysts have made possible to overcome this limitation and some Ru-, Pd-, Ag- and Au-based systems active with internal alkynes are now available [23].
On the other hand, the increasing awareness of environmental concerns has stimulated the development of metal-catalyzed reactions in aqueous media, since water is cheaper, safer and benigner compared to the traditional petroleum-derived organic solvents [24,25,26]. In addition, the use of water (or aqueous biphasic mixtures) allows in many cases an easy catalyst/product separation, thus allowing the effective recycling of the catalytically active species, which is another key aspect in the Green Chemistry context [27]. However, despite the growing interest in aqueous catalysis, the use of this environmentally benign solvent in the hydrocarboxylation of alkynes been for long time neglected, probably due to the concerns of a competing hydration of the alkyne substrates to form carbonyl compounds, a process that is also catalyzed by transition metals [1,2,3,4,5,6,28]. In fact, in a general review on the “catalytic reactions of alkynes in water”, published by Chen and Li in 2006, no examples of hydrocarboxylation reactions were collected [28]. Catalytic systems able to operate selectively in aqueous environments have only appeared in the literature in recent years, and are the subject of the present review article. Intra- and intermolecular processes are covered, including examples of sequential transformations of synthetic interest. The access to β-oxo esters by the catalytic addition of carboxylic acids to terminal propargylic alcohols in water is also discussed.
2. Intramolecular Processes
2.1. Cycloisomerization of Preformed or In Situ Generated Alkynoic Acids
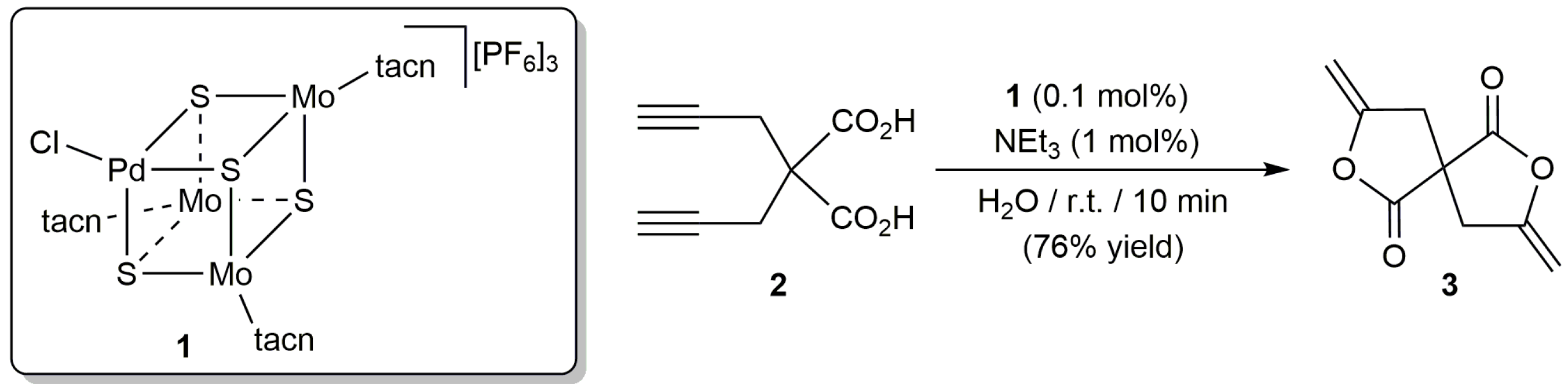
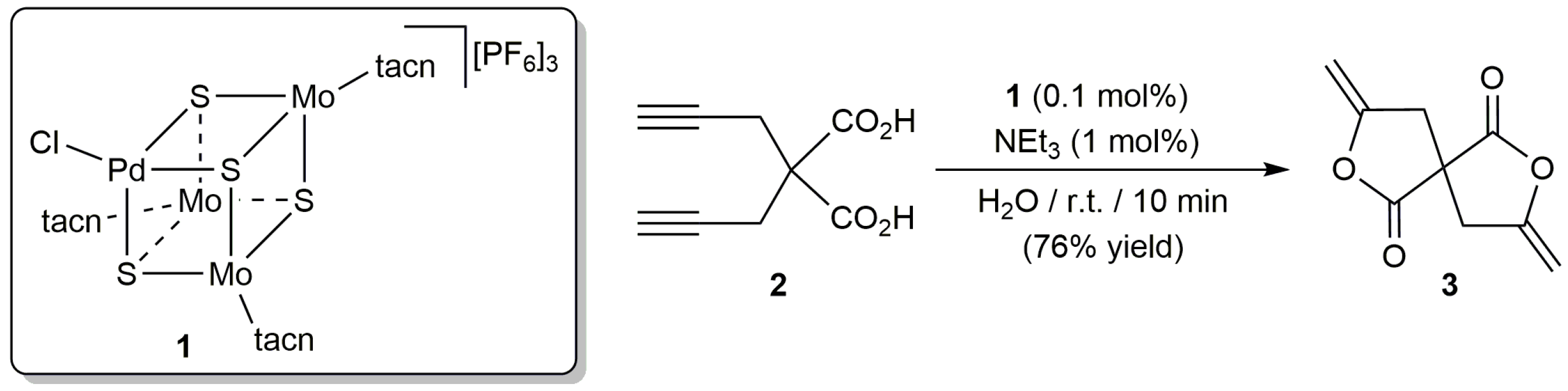
The first example of such reactions in an aqueous environment was described by Hidai and co-workers in 1996 employing the mixed-metal cubane-type cluster complex [PdMo3S4(tacn)3Cl][PF6]3 (1; tacn = 1,4,7-triazacyclononane) as catalyst [29]. Thus, they found that, in combination with NEt3, 1 was able to transform dipropargylmalonic acid 2 into the enol lactone 3 in water at room temperature (Scheme 3). Unfortunately, although the rate and yield of the reaction were very similar to those observed in acetonitrile, this latter solvent was selected by the authors to study the scope of the process. In this regard, different 5-, 6- and 7-membered ring lactones could be obtained from the corresponding alkynoic acids with complex 1, which showed an enhanced reactivity in comparison with classical mononuclear Pd(II) sources, such as [PdCl2(PhCN)2] or Na2[PdCl4]. In line with this, despite the presence of three molybdenum atoms in the structure of 1, the catalytic reactions were assumed to proceed only at the palladium center.
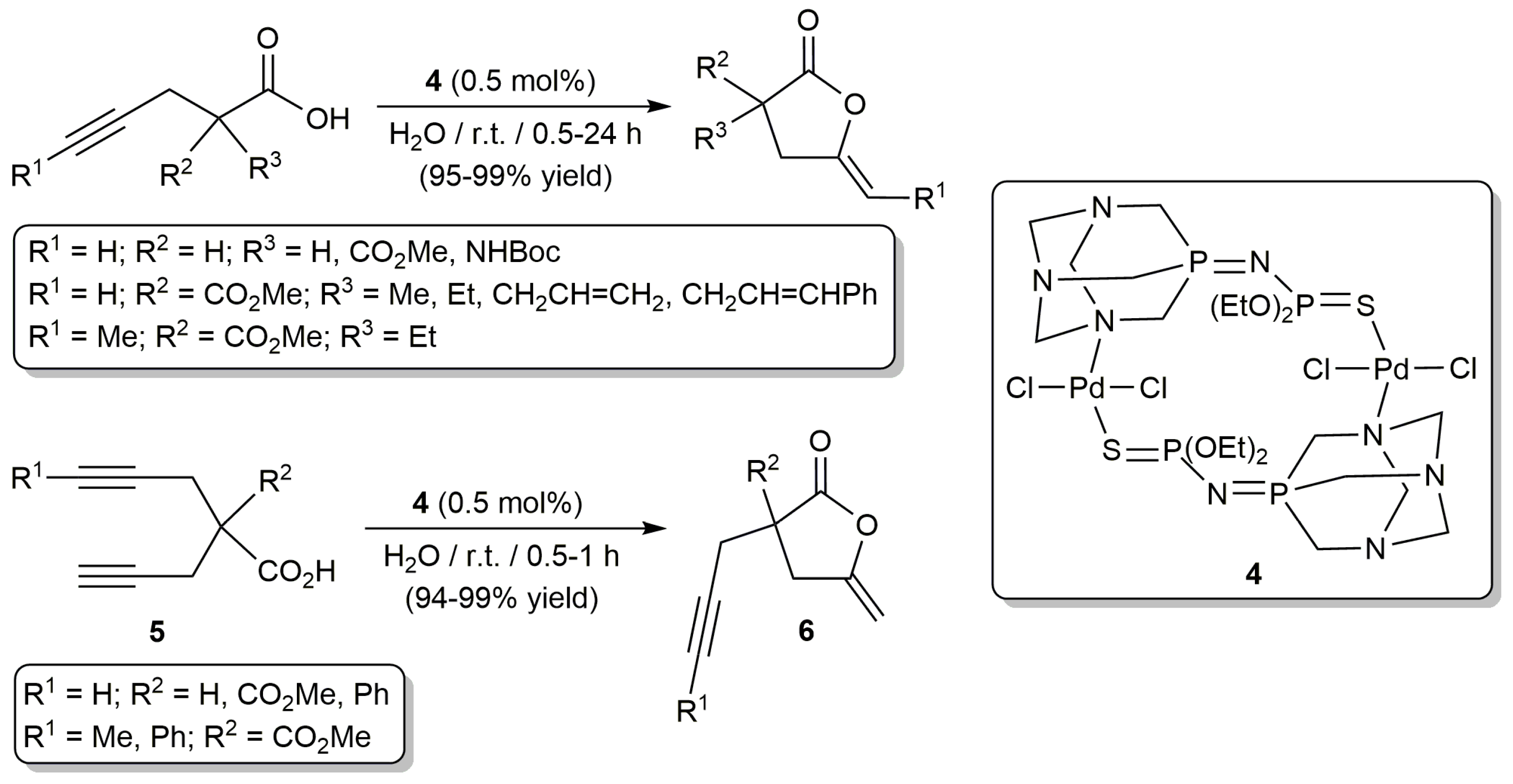
More recently, other palladium-based catalysts for the cyclization of alkynoic acids in aqueous media have been described. In particular, García-Álvarez and co-workers disclosed that transformation of γ-alkynoic acids into five-membered ring enol-lactones (5-exo-dig cyclization) can be conveniently and selectively achieved in pure water, and under aerobic conditions, by using catalytic amounts of the dinuclear Pd(II) derivative trans-[PdCl2{µ2-N,S-(PTA)=NP(=S)(OEt)2}]2 (4) (Scheme 4) [30]. This catalyst features a bridging iminophosphorane-type ligand derived from the well-known hydrophilic phosphine PTA (1,3,5-triaza-7-phosphaadamantane) [31,32], which facilitates its solubility in water.
As shown in Scheme 4, the reactions proceeded cleanly at r.t. (room temperature) without formation of any by-product derived from the hydration of the C≡C bond of the substrates or from the hydrolysis of the (Z)-γ-alkylidene butyrolactone products. The process was operative with γ-alkynoic acids containing both terminal and internal C≡C bonds, although for the latter a much longer reaction time was needed (24 instead of 0.5–2 h). It is also worth noting that (i) this catalytic system could be recycled up to 10 consecutive runs for the cyclization of the model 4-pentynoic acid (cumulative TON (turnover number) of 982), and (ii) it could be applied in the desymmetrization of bispropargylic carboxylic acids 5, affording enol-lactones 6 containing an intact propargylic side arm in excellent yields.
Although in a limited number of examples (only six substrates were explored), the group of Nakamura also demonstrated the capacity of the NCN-pincer Pd(II) derivative 7 (Figure 1) to promote the high-yield formation of five-membered ring lactones from both γ- and β-alkynoic acids (5-exo-dig and 5-endo-dig cyclization, respetively) in pure water [33]. Complex 7 easily generates hydrogen-bonding-based supramolecular gels, with well-organized Pd-arrays, in organic solvents. For the catalytic reactions, which were performed at 70 °C and in the presence of Et3N (3 mol %), a water-insoluble xerogel prepared from 7 in toluene was employed (0.5 mol % Pd content). Interestingly, the Pd-gel catalyst could be recovered by filtration and reused three times in the cyclization of 4-pentynoic acid without loss of catalytic activity. However, it should also be mentioned that, contrary to the case of the dinuclear Pd(II) complex 4, when a bispropargylic carboxylic acid was employed as substrate, partial hydration of propargylic side arm of the enol-lactone product was observed.
Selective 5-exo-dig cyclization of pentynoic acids RC≡CCH2CH2CO2H (R = aryl or heteroaryl group) to afford the corresponding (Z)-γ-alkylidene butyrolactones could also be achieved, in water at 50 °C, using the related amphiphilic NCN-pincer Pd(II) complex 8 (2 mol %) (Figure 1) in combination with Et3N (6 mol %) (yields in the range 38–94% after 1–6 h) [34]. Complex 8 self-assembles in aqueous solution forming bilayered vesicles which were found to be essential for the promotion of the catalytic process (markedly lower yields were obtained when the same reactions were performed in organic solvents or employing the amorphous complex 8 in water).
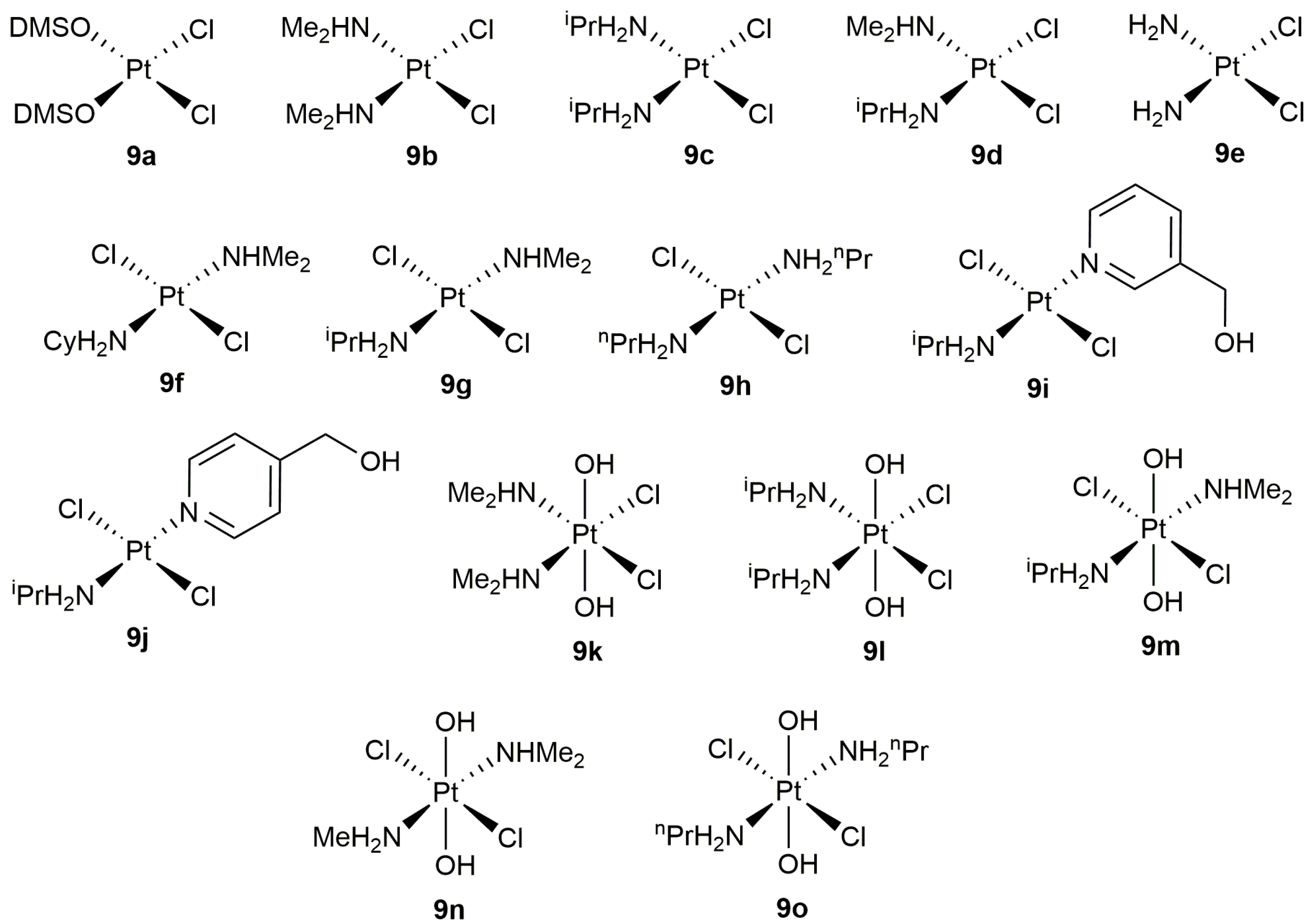
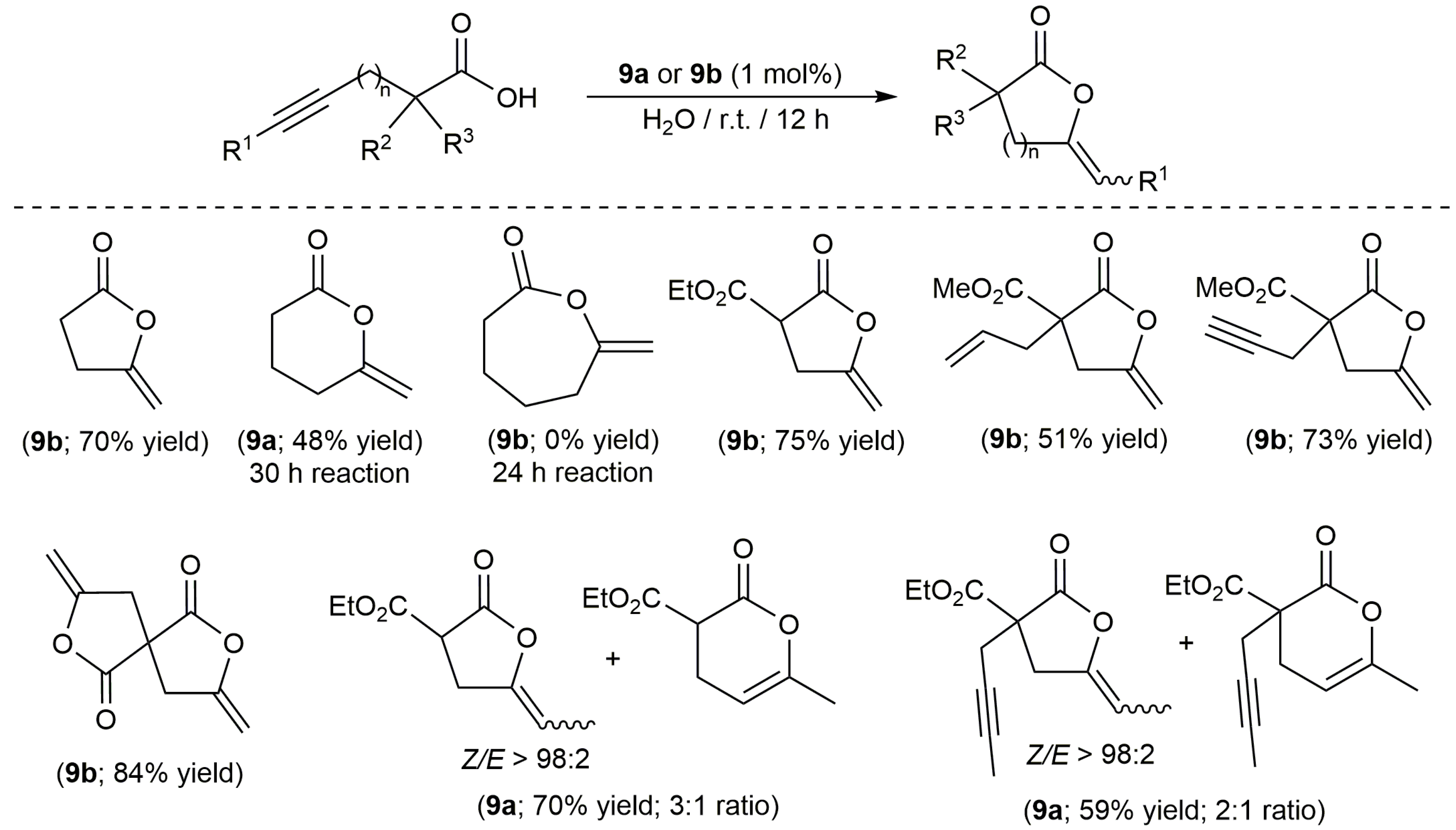
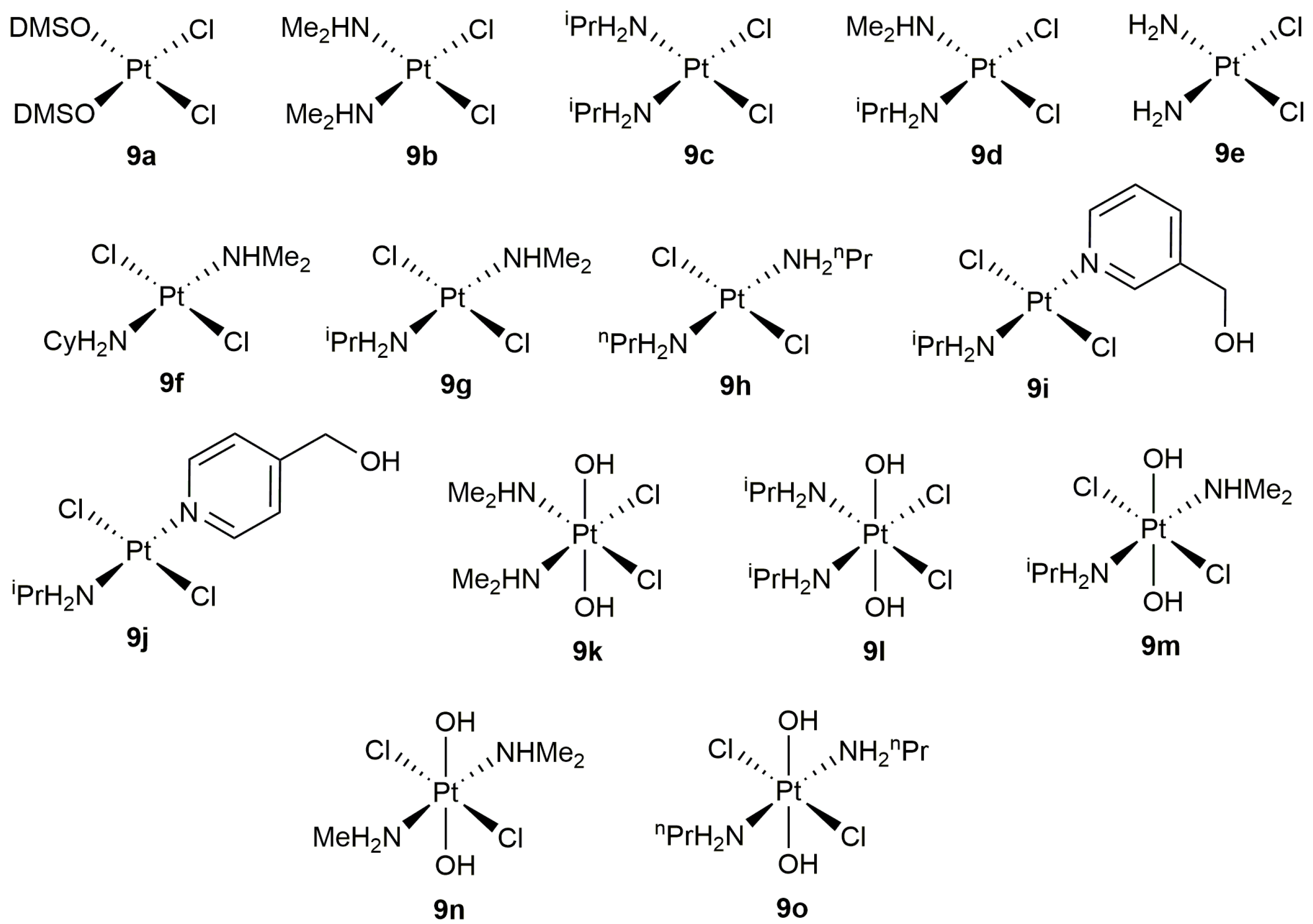
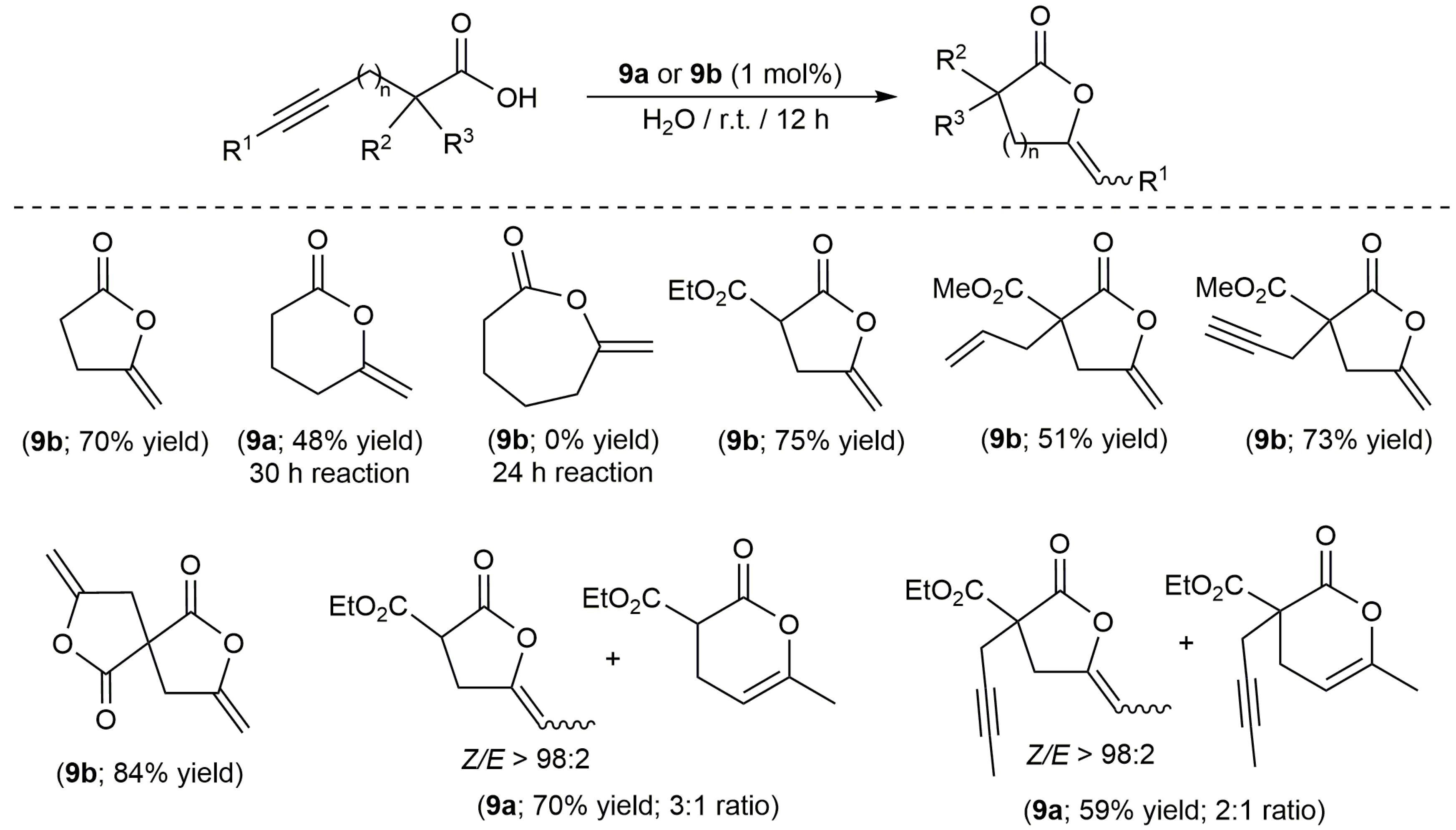
On the other hand, despite the well-known ability of platinum compounds to promote the cycloisomerization of polyunsaturated organic molecules [35,36], this metal has been much less used than palladium to catalyze the cyclization of alkynoic acids [37]. In this regard, the work published by Alemán, Navarro-Ranninger and co-workers merits highlighting [38]. These authors evaluated the catalytic behaviour of a broad family of anticancer platinum(II) and platinum(IV) amino-complexes (9a–j and 9k–o, respectively; see Figure 2) in the cycloisomerization of 4-pentynoic acid into 5-methylene-dihydrofuran-2-one (5-exo-dig cyclization), finding that both the oxidation state of the metal and the stereochemistry of the complex are key factors for the reaction to proceed efficiently. Thus, while the cis-platinum(II) derivatives 9a–e showed a high reactivity in acetone at r.t., their trans-platinum(II) counterparts 9f–j and the Pt(IV) species 9k–o (regardless of their cis or trans configuration) turned out to be practically inactive.
In addition, they also demonstrated the possibility of using water (or even blood plasma) as solvent. In fact, the scope of the catalytic reaction was explored in water using complexes 9a,b [38,39]. Thus, as shown in Scheme 5, different 5- and 6-membered ring lactones could be synthesized with these complexes, although mixtures of regioisomers were systematically formed starting from internal alkynes. On the other hand, it is also worth noting that the attempt made to generate a seven-membered ring lactone with this type of catalysts failed.
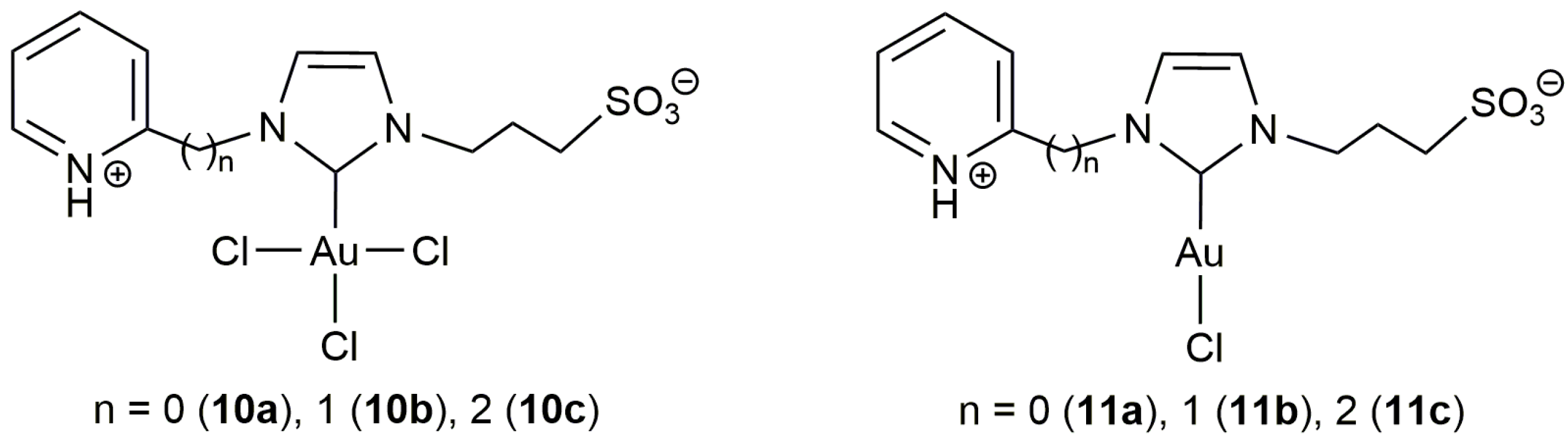
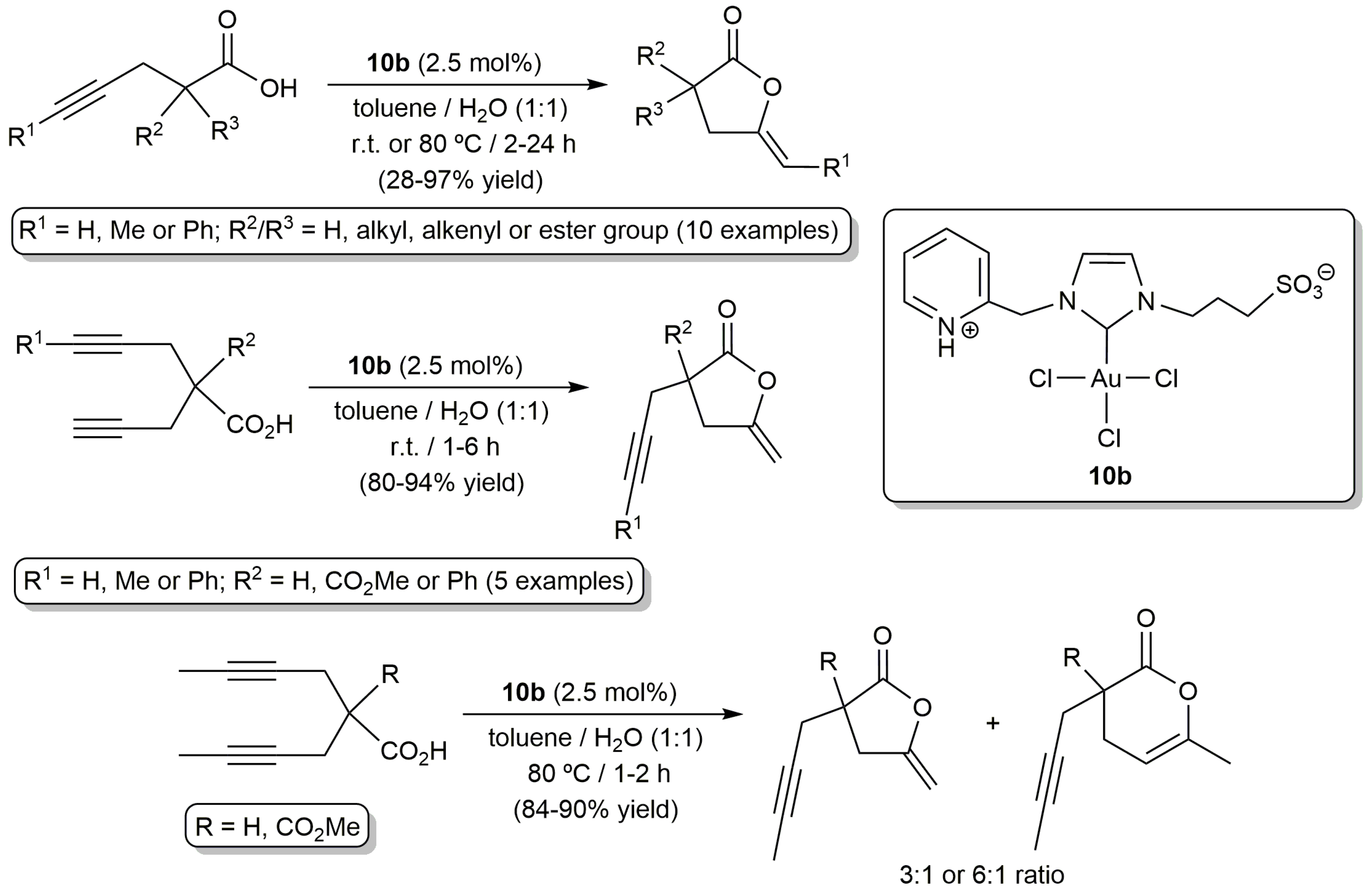
Gold compounds are currently recognized as the most effective systems for the electrophilic π-activation of unsaturated carbon–carbon bonds [40,41], and, in 2006, Michelet and co-workers demonstrated for the first time their usefulness in the cycloisomerization of acetylenic acids [42]. The same group was also a pioneer in the use of gold catalysts in aqueous environments. In particular, in 2008, they reported on the tolerance of the heterogeneous system Au2O3 towards the presence of water during the cycloisomerization of 2-phenyl-4-pentynoic acid into 5-methylene-3-phenyl-dihydrofuran-2-one (92% yield performing the reaction in MeCN-H2O (6:1) at r.t. with 2.5 mol % of Au2O3 for 3 h; 95% yield when acetonitrile was employed alone under identical reaction conditions) [43]. Since then, a number of gold-based catalysts capable of operating in pure water or in aqueous biphasic mixtures have been described, most involving functionalized N-heterocyclic carbenes (NHC) as auxiliary ligands. In this regard, the groups of Michelet, Cadierno and Conejero developed the zwitterionic Au(III)-NHC complex 10b which proved to be active in the cycloisomerization of a broad range of γ-alkynoic acids under biphasic toluene/water conditions (Scheme 6) [44]. Remarkably, the participation of a silver(I) co-catalyst, usually employed in catalytic gold chemistry to generate vacant coordination sites on the metal through chloride ligands abstraction, was not required. Also of note is the fact that, despite the well-known ability of gold complexes to promote the hydration of alkynes, competitive hydration processes were not observed under the biphasic conditions employed, even during the cycloisomerization of bispropargylic substrates. However, it should be noted that, if pure water is used as solvent, partial hydrolysis of the lactone products takes place, making the use of biphasic conditions more advantageous. In general, the reactions proceeded in air under very mild temperature conditions (r.t.), except with those substrates containing internal C≡C bonds which showed a markedly lower reactivity and required of heating at 80 °C. Concerning the regioselectivity of the process, 5-membered ring enol-lactones were selectively formed when terminal C≡C bonds were involved in the cyclization process. On the other hand, although the 5-exo-dig cyclization was also the preferred reaction pathway with internal alkynes, mixtures containing the corresponding 5- and 6-membered ring lactones were in some cases obtained.
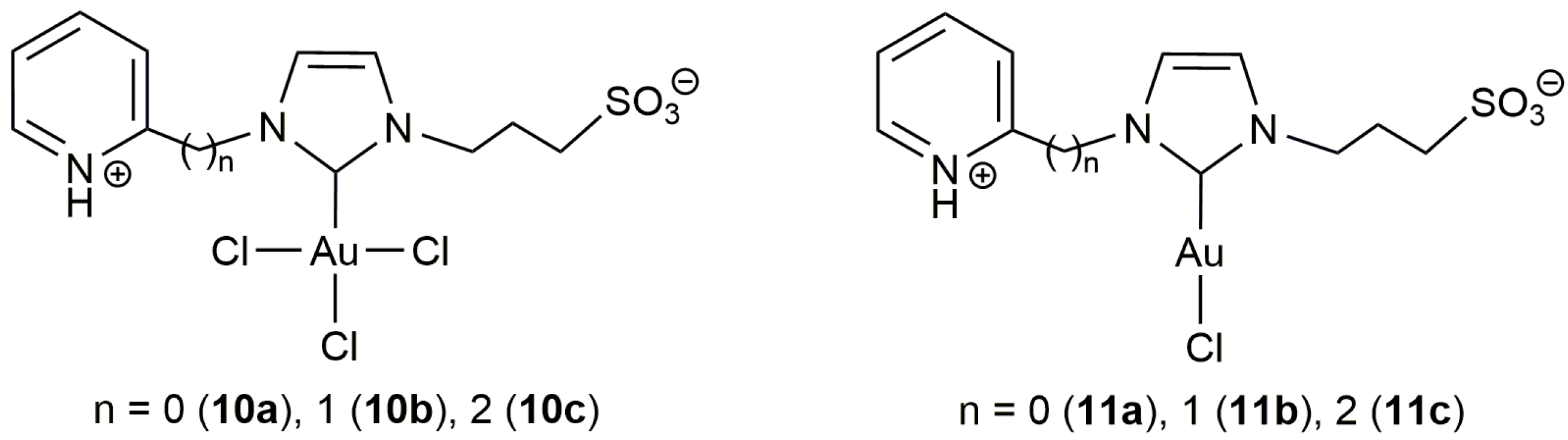
This initial work with 10b was subsequently extended to other Au(III) and Au(I) complexes containing related zwitterionic NHC ligands bearing 3-sulfonatopropyl, and 2-pyridyl, 2-pycolyl or pyridylethyl substituents (10a,c and 11a–c in Figure 3) [45]. All of them proved to be catalytically active, even with Au loadings of only 0.1 mol %, showing in general performances similar to that of 10b. Interestingly, all these gold-carbenes showed a very high recyclability (up to 10 consecutive runs) by simple phase separation (the gold catalyst remained in the aqueous phase while the lactone product was completely dissolved in the toluene one). In this regard, some differences were observed between the Au(I) and Au(III) species, the recyclability of the latter being much more effective due to their higher stability in the aqueous medium (the Au(I) derivatives 11a–c undergo with time partial decomposition into catalytically inactive Au(0) nanoparticles; a process also observed with 10a–c but which takes place much more slowly).
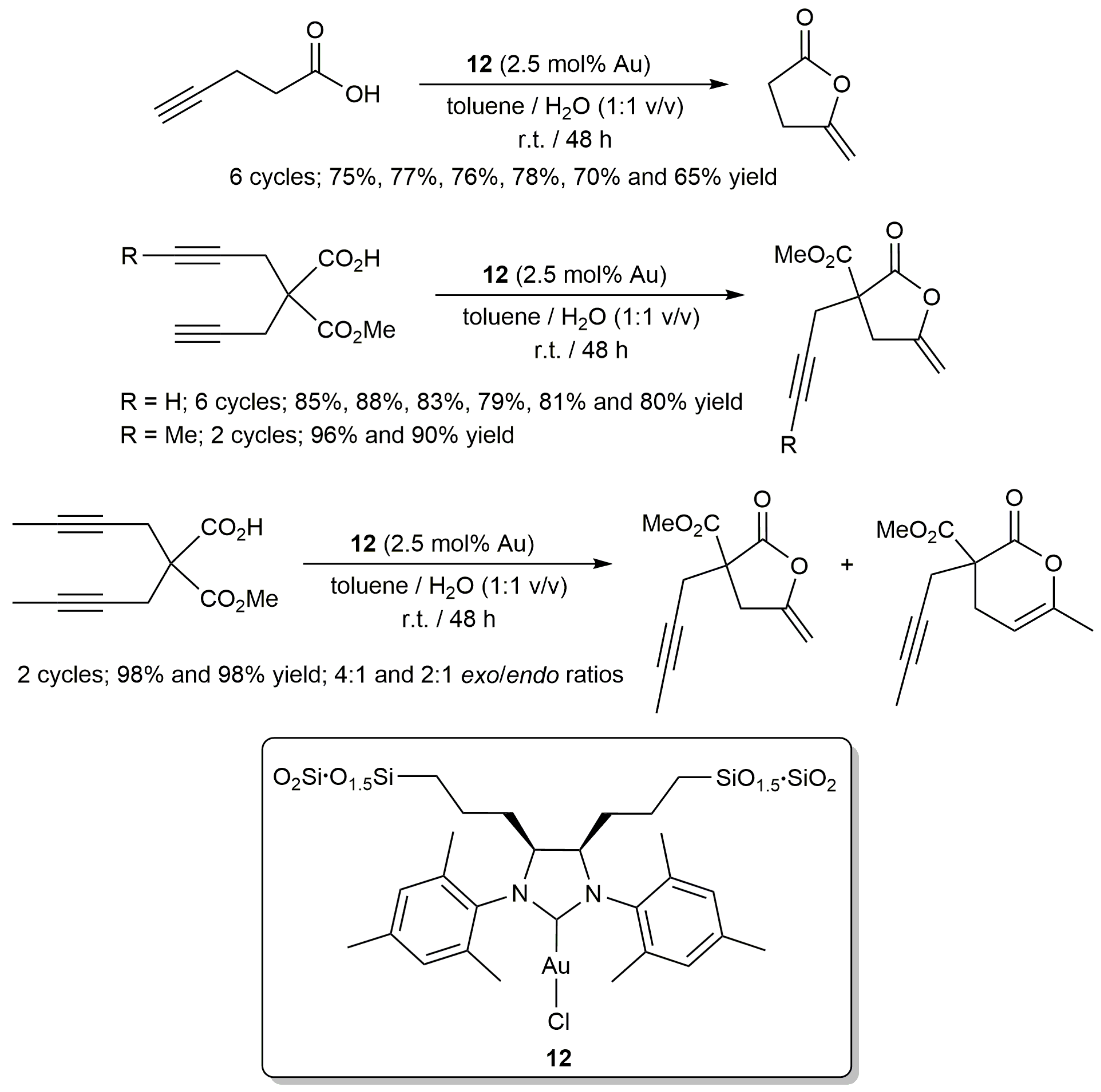
A good recyclability was also reported by Pleixats and co-workers for the sol-gel immobilized Au(I)-NHC complex 12 (Scheme 7) [46]. This organometallic hybrid silica material proved to be active in the cycloisomerization of different γ-alkynoic acids at room temperature using, as in the previous example, a toluene/water biphasic system and in the absence of silver salts. However, due to the heterogeneous nature of 12, longer reaction times and a wrist-type shaker stirring (which allows a good mixing of the immiscible layers and the insoluble catalyst) were in this case required to obtain the enol-lactone products in high yields (no reaction was observed using conventional magnetic stirring). Concerning the regioselectivity of the process, five-membered enol-lactones were selectively formed starting from substrates with terminal alkyne units, while a mixture of the 5- and 6-membered ring lactone products was obtained from an internal diyne (Scheme 7).
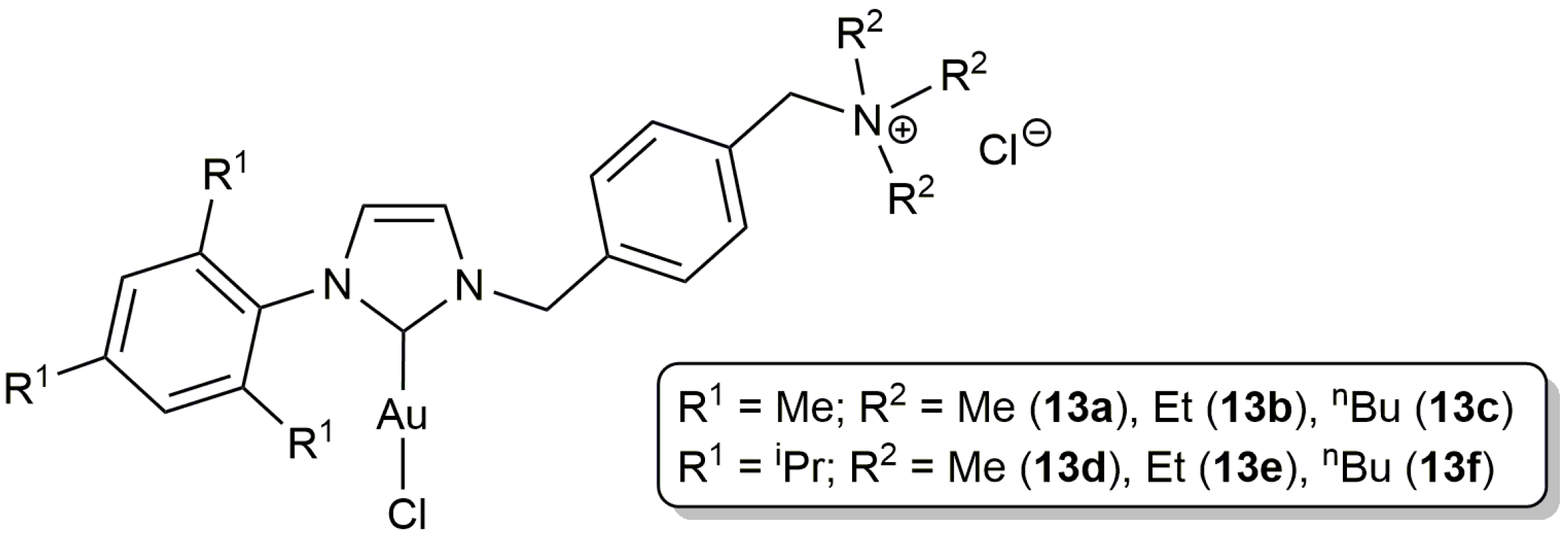
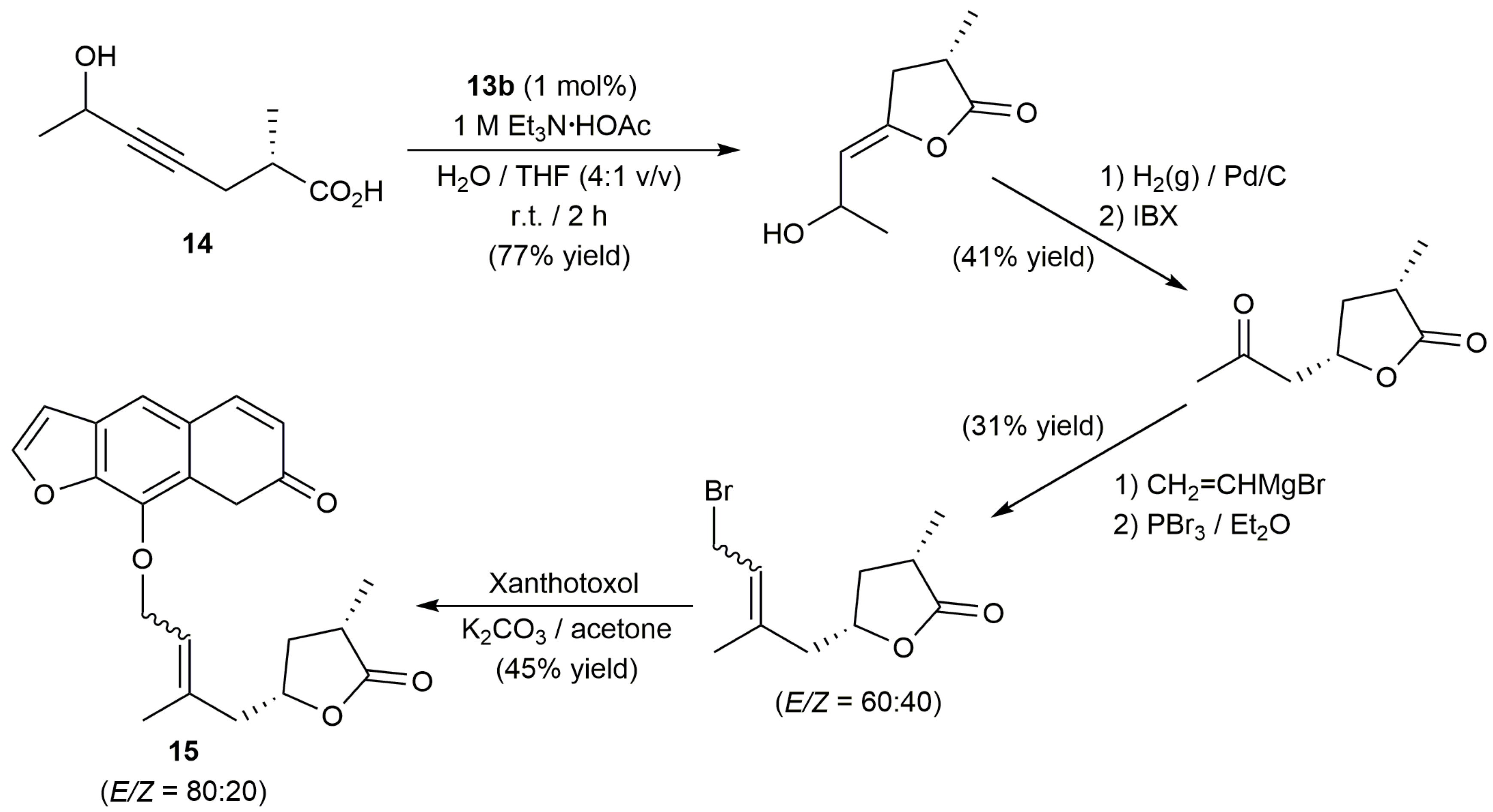
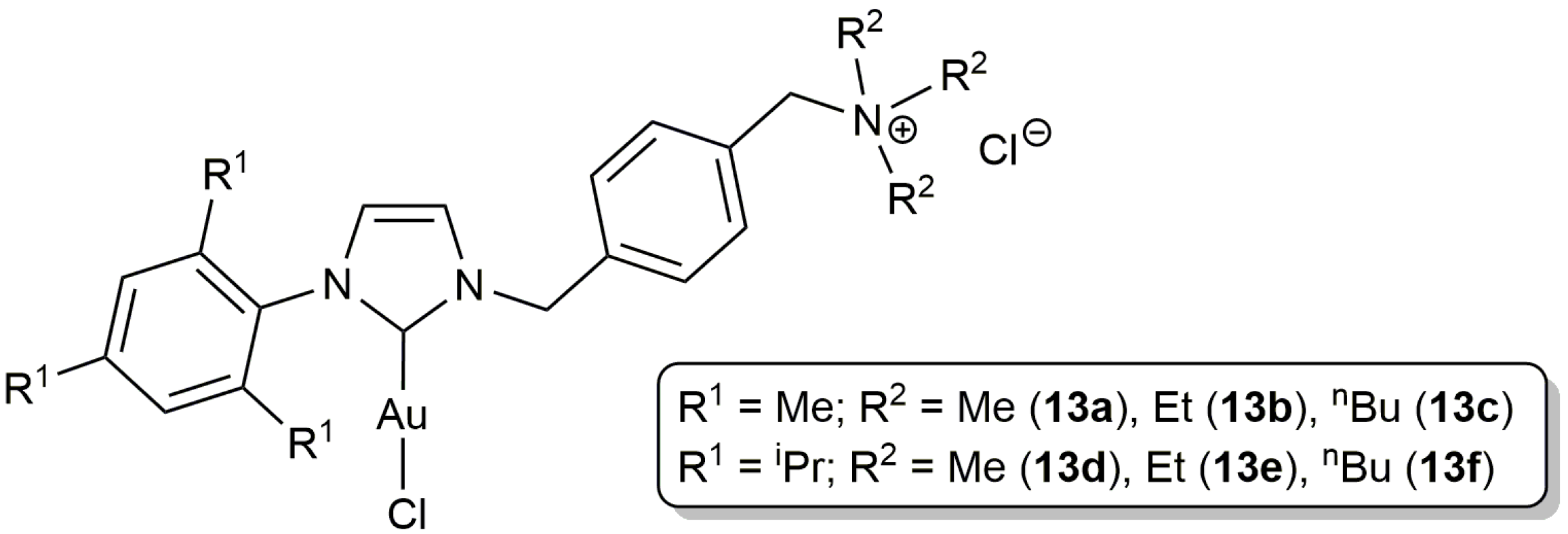
For its part, the group of Krause described the preparation of the ammonium salt-tagged Au(I)-NHC complexes 13a–f (Figure 4) and their application in the selective 5-exo-dig cyclization of γ-alkynoic acids bearing a terminal alkyne unit in pure water, or in aqueous triethylammonium buffer solution [47]. The catalytic reactions proceeded cleanly at r.t., in short time spans (0.5–6 h), employing 2.5 mol % of these complexes, with yields higher in general when the buffer solution was employed as the reaction medium (partial decomposition of the gold complexes was observed in pure water). Once again, no Ag(I) co-catalysts were needed and the catalysts could be reused after extraction of the lactone product from the aqueous solution with diethyl ether (up to 5 times).
Interestingly, starting from the internal alkynoic acid 14, a synthetic route to obtain the furanocoumarin-functionalized lactone 15, an epimer of the natural product clausemarine A, could be developed by Krause and co-workers [47]. As shown in Scheme 8, formation of the lactone ring was successfully achieved by cycloisomerization of 14 with 1 mol % of 13b in an aqueous triethylammonium buffer solution containing THF (tetrahydrofuran) as co-solvent.
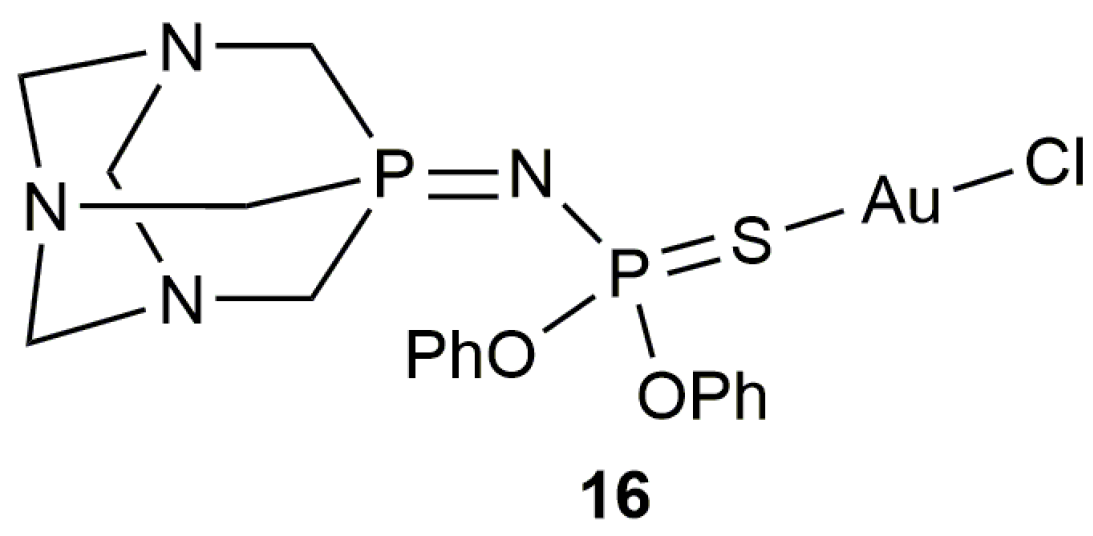
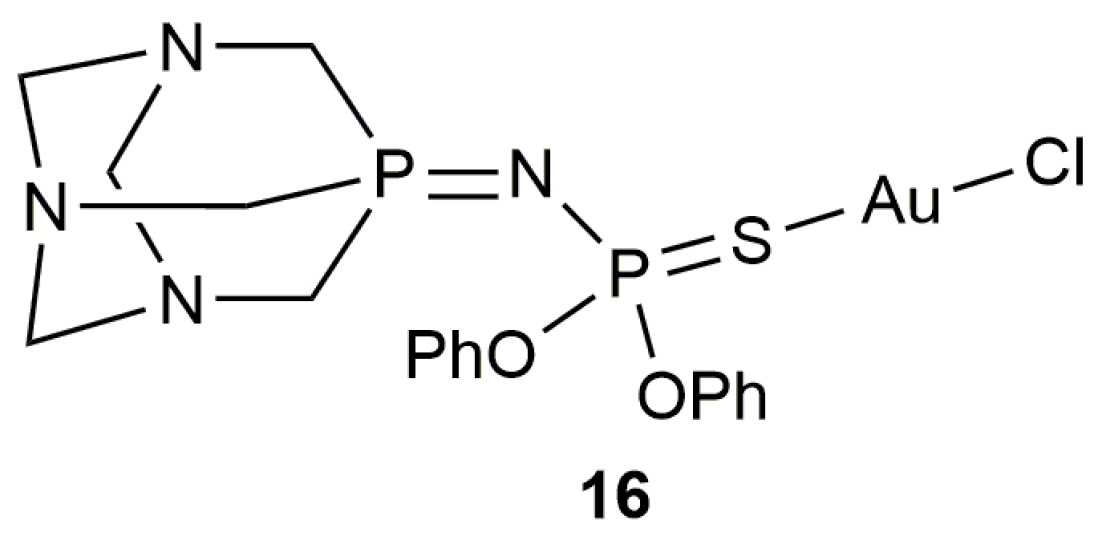
Besides the Au-NHC complexes commented above, the PTA-derived iminophosphorane-Au(I) derivative 16 (Figure 5) proved to be also an active and selective catalyst for the 5-exo-dig cyclization of 4-pentynoic acid in water (89% yield after 30 min at r.t. with 1 mol % of 16) [48]. However, this particular catalyst showed a higher reactivity in the eutectic mixture 1ChCl/2Urea (ChCl = choline chloride; 99% yield after 15 min under identical reaction conditions) and, consequently, its scope was explored only in this alternative and biorenewable reaction medium.
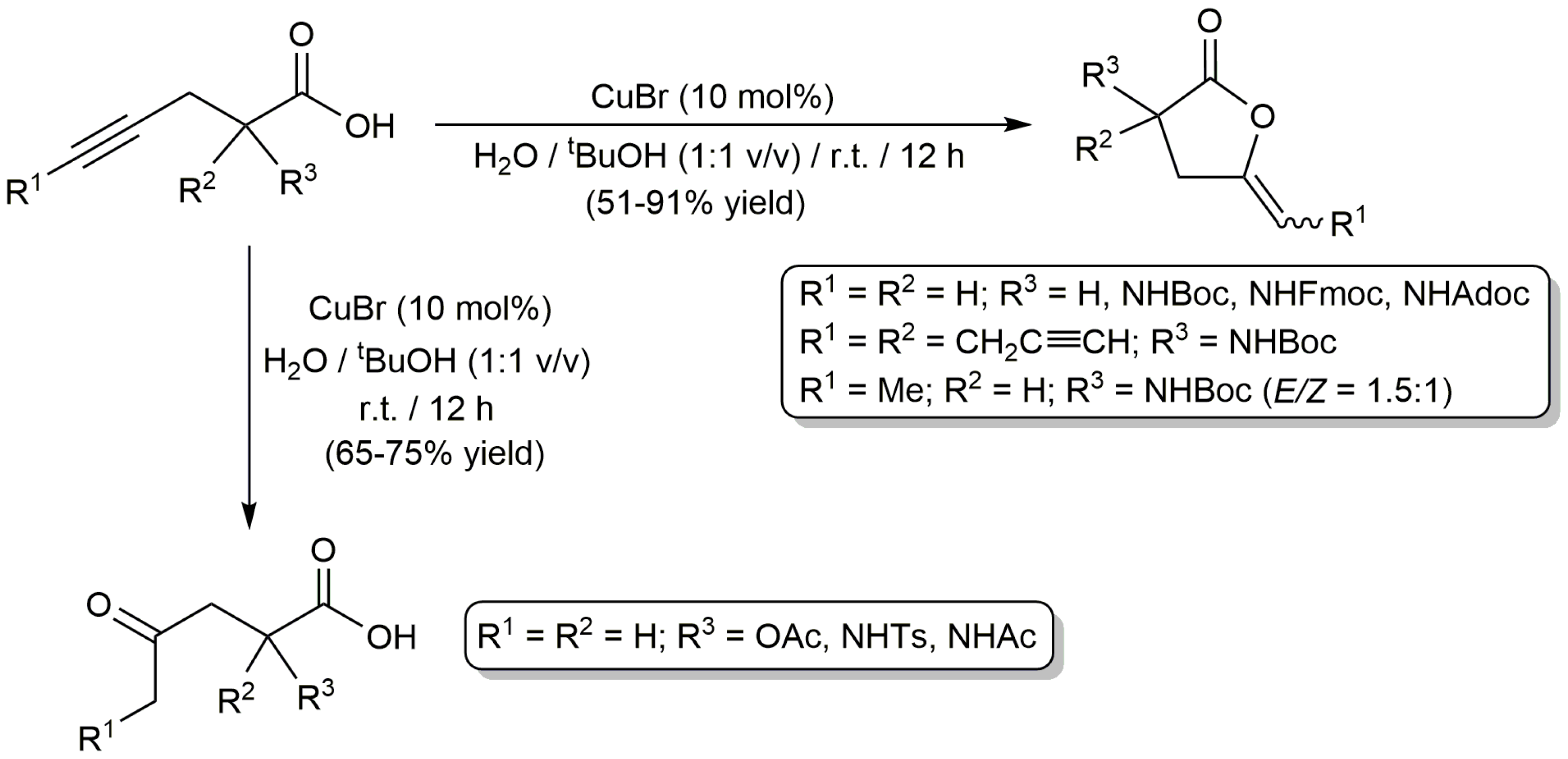
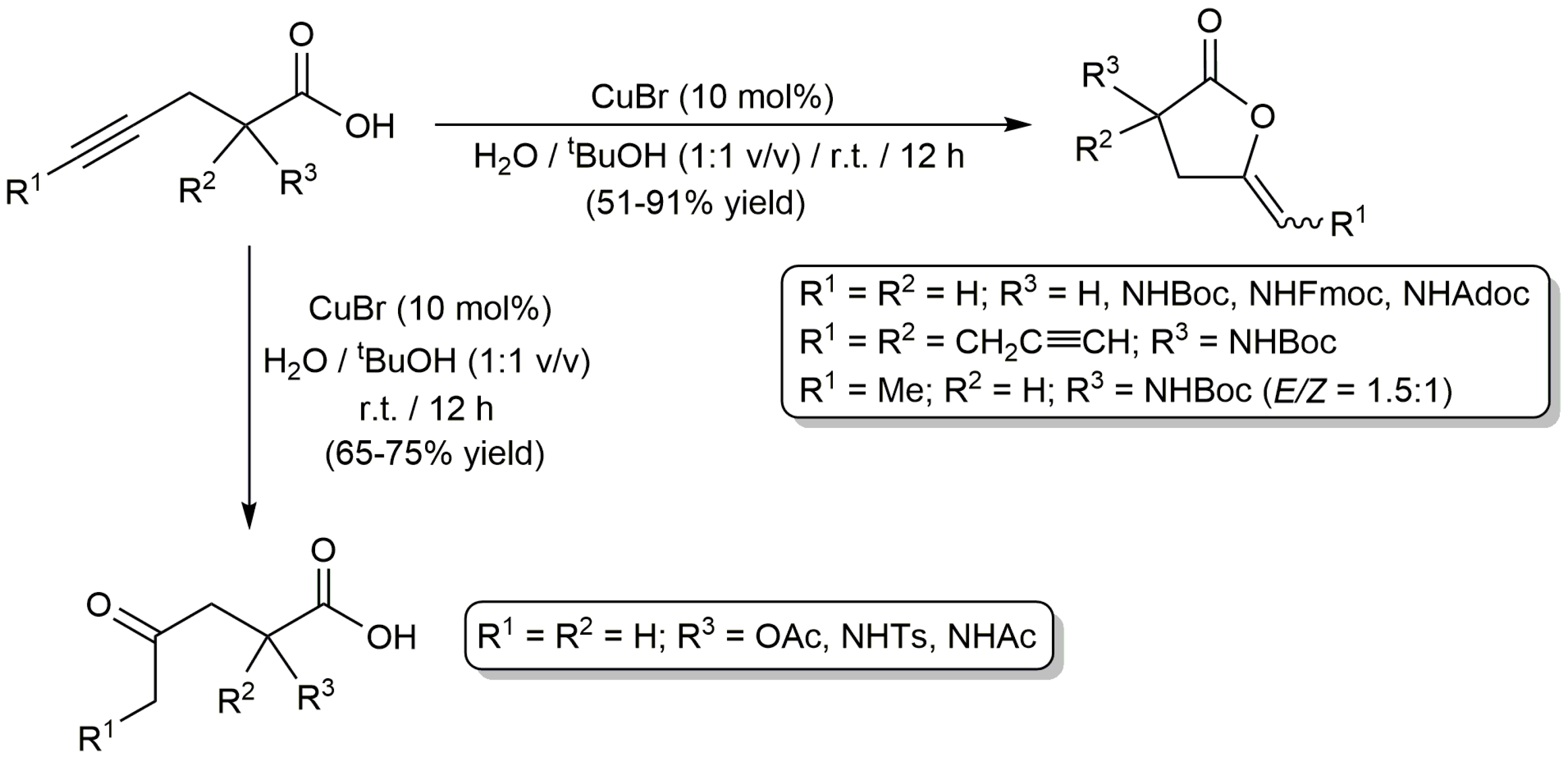
In addition to palladium, platinum and gold, cheaper copper catalysts have also been used in the cycloisomerization of alkynoic acids in aqueous media. Thus, while studying the CuBr-catalyzed cycloaddition of alkynes with azides in tBuOH/H2O mixtures, Mindt and Schibli observed the formation of enol-lactone by-products when employing γ-alkynoic acids as substrates, so they decided to explore separately the cyclization of these compounds [49]. Their results are shown in Scheme 9. Performing the reactions with 10 mol % of CuBr at r.t., different γ-alkynoic acids containing both terminal and internal C≡C units could be efficiently transformed into the corresponding γ-alkylidene butyrolactones. However, in some cases, γ-keto acids were selectively obtained due to the rapid hydrolysis of the lactone products under the aqueous conditions employed. On the other hand, the attempts made to extend the scope of this aqueous protocol to six- and seven-membered ring enol-lactones by cyclization of 5-hexynoic acids and 6-heptynoic acids, respectively, failed. It should be noted, however, that the former could be cyclizated by carrying out the reactions in acetonitrile instead of the tBuOH/H2O mixture.
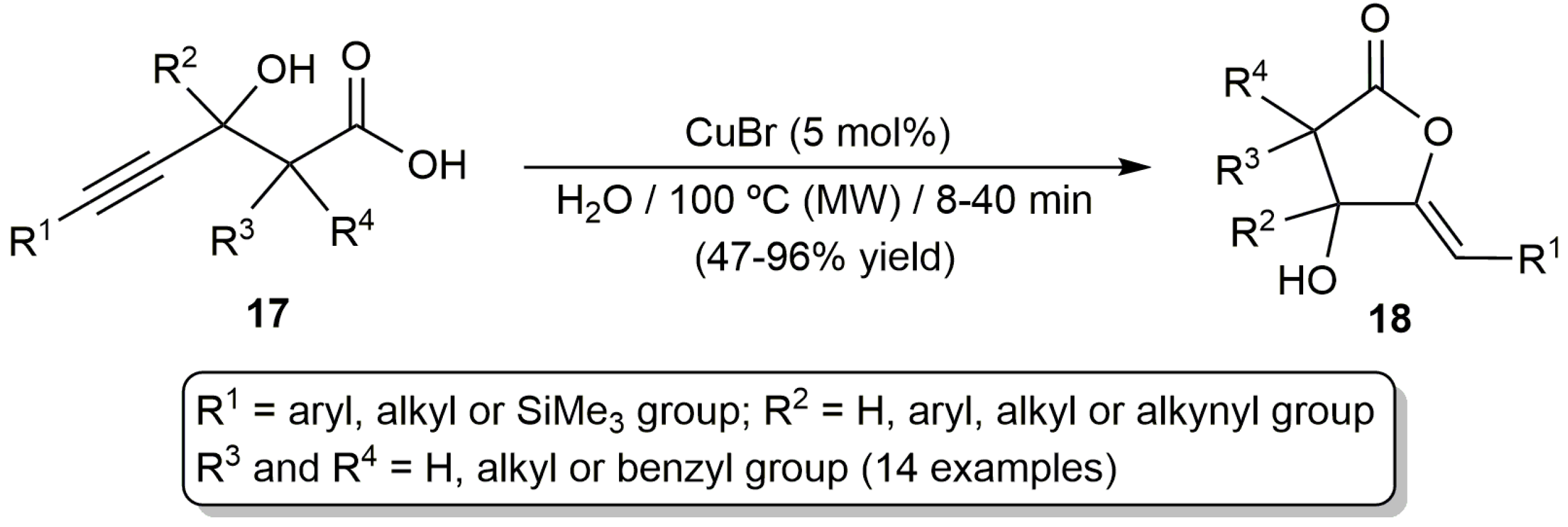
More recently, CuBr was also employed to promote the cyclization of a variety of β-hydroxy-γ-alkynoic acids 17 [50]. As shown in Scheme 10, the reactions proceeded cleanly in pure water under microwave (MW) irradiation at 100 °C, leading to the regio- and stereoselective formation of the five membered ring Z-enol-lactones 18 without observing hydrolysis products. In addition, the beneficial effect of water was evidenced by the authors, who obtained markedly lower yields when performing the same reactions in classical organic solvents such as THF or acetonitrile. The same must be said of the use of MW irradiation, since lower yields were also observed when conventional oil-bath heating or ultrasound irradiation was employed. The process was quite general, tolerating a broad range of substitution patterns on the substrate skeleton. However, it should be noted that when an alkynoic acid featuring a hydrogen atom and a phenyl group, as the R3 and R4 substituents, was employed in this reaction, the spontaneous dehydration of 18 to form a 5-alkylidene-furan-2-one product took place.
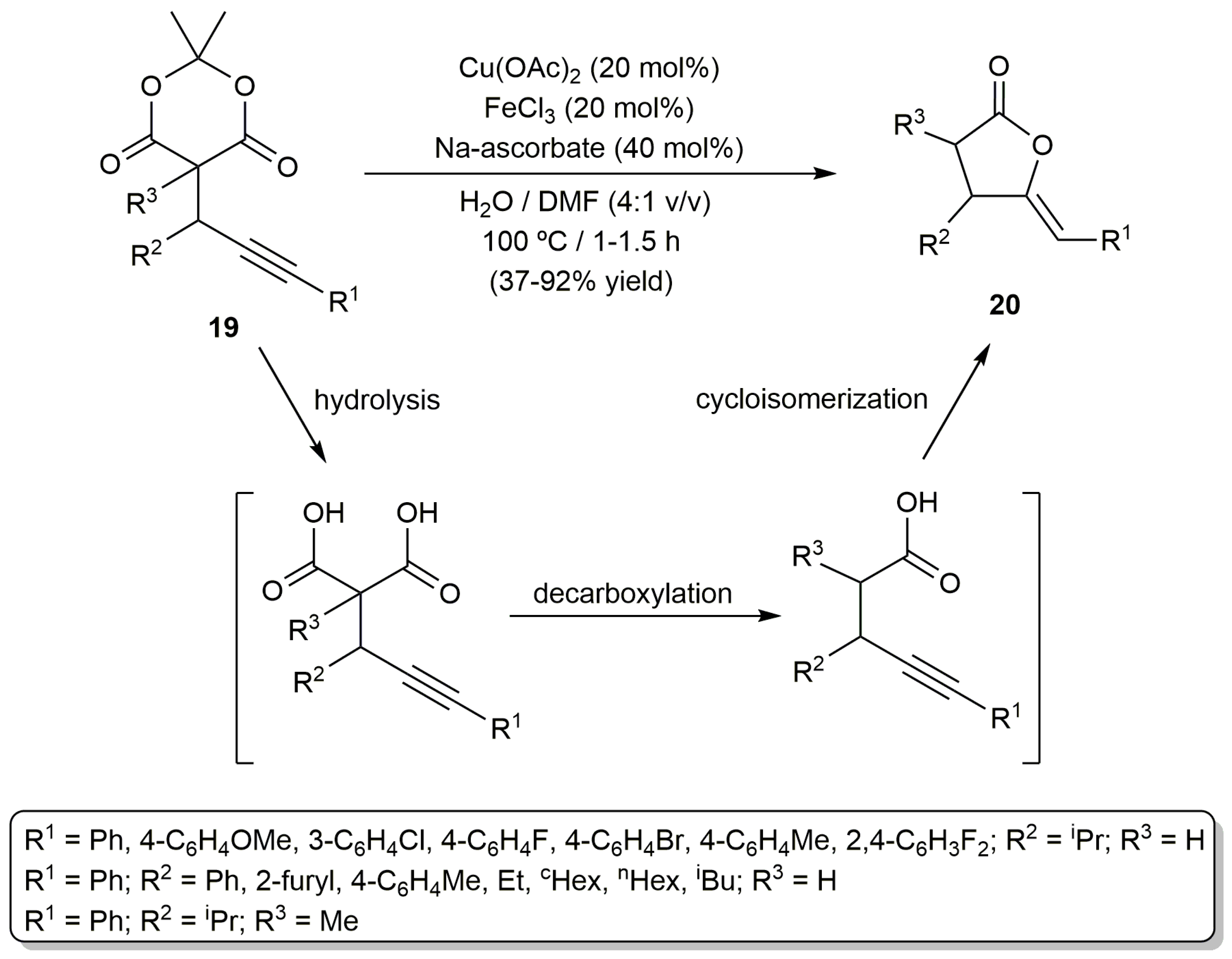
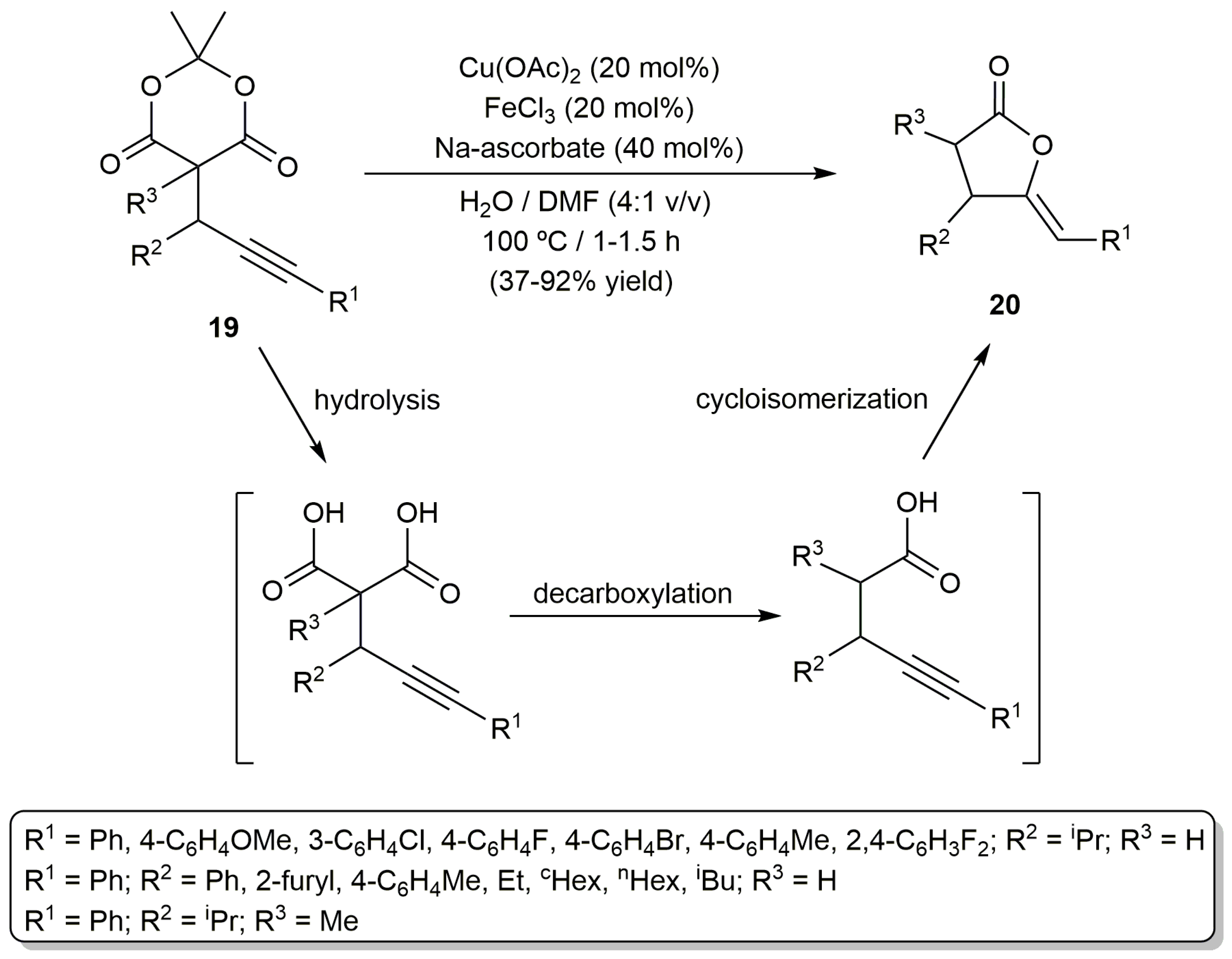
For its part, the group of Jiao described the cyclization of the propargylic Meldrum´s acids 19 into the (Z)-γ-alkylidene butyrolactones 20, in a basic aqueous environment, through the combined use of Cu(OAc)2 and FeCl3 (Scheme 11) [51]. A clear cooperative effect of the two metals was observed, the yields decreasing drastically when Cu(OAc)2 or FeCl3 was used as the sole catalyst. The process most probably involves the in situ generation of a γ-alkynoic acid intermediate through a hydrolysis/decarboxylation sequence. On the other hand, although the mechanism could not be unambiguously established, it was assumed that the Cu2+ ion is the one responsible for the activation of the alkyne unit during the final cycloisomerization step, with Fe3+ probably facilitating the intramolecular nucleophilic addition of the carboxylate on the C≡C bond by coordination to the carbonyl group.
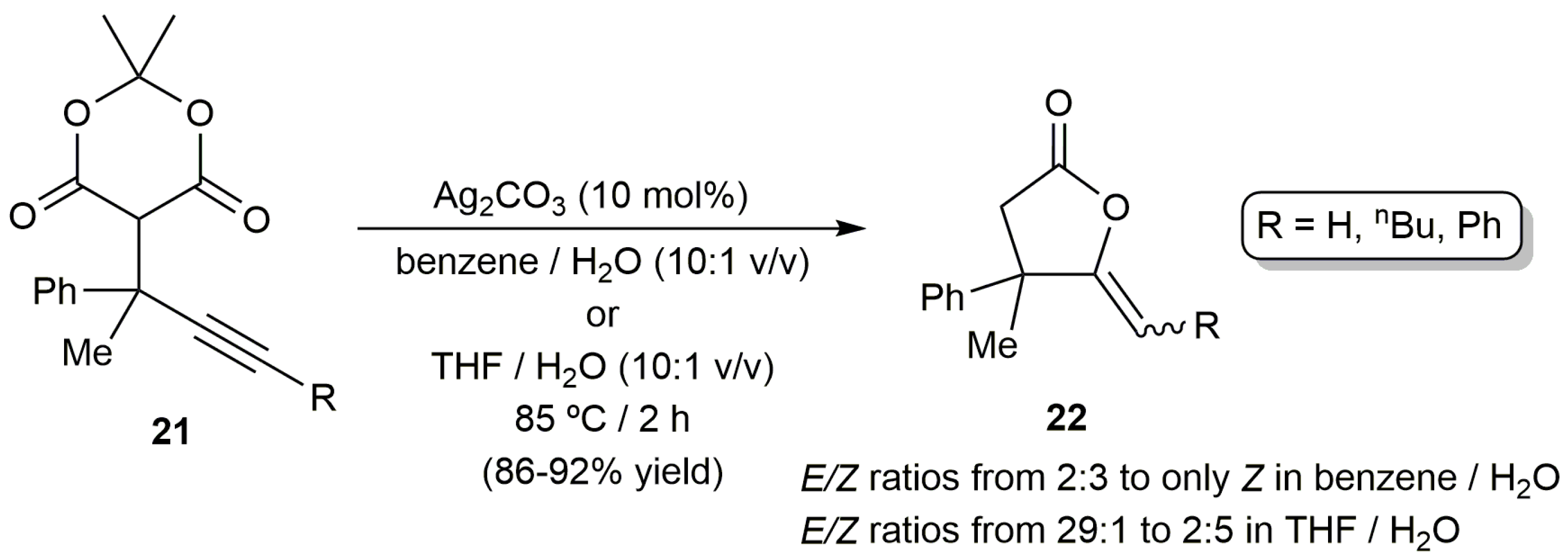
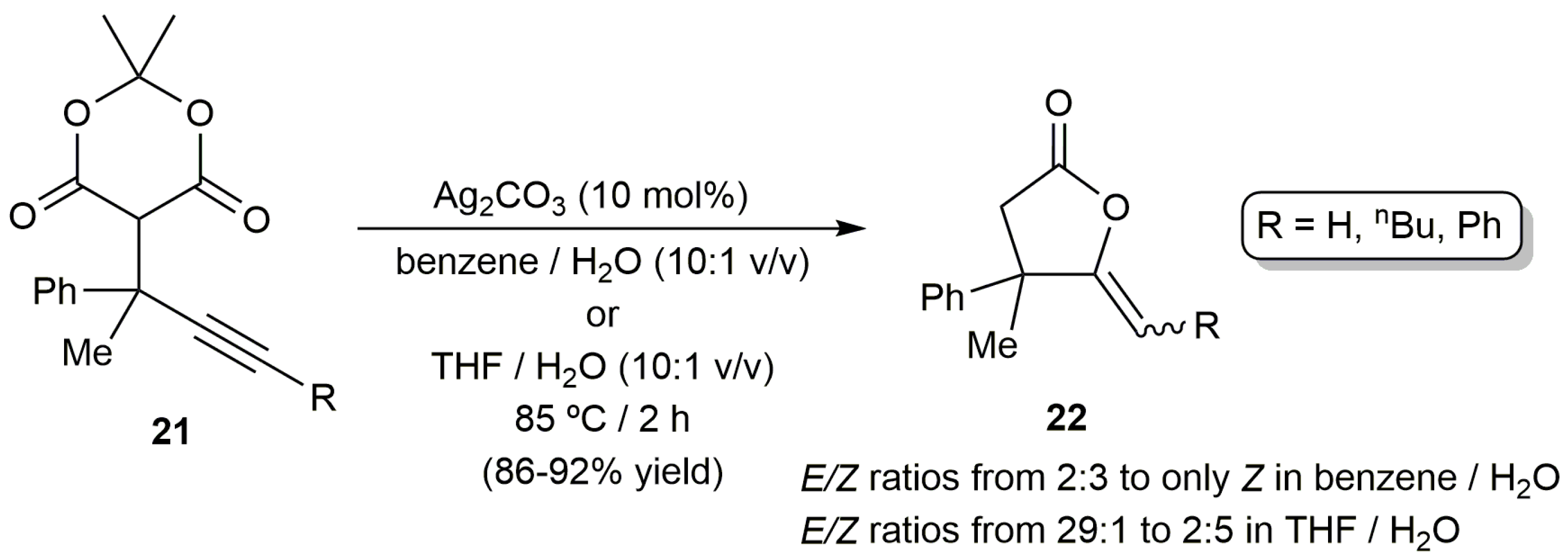
Further studies by the same group showed that the same catalytic reactions can be more conveniently carried out employing AgNO3 (5 mol %) instead of the Cu/Fe system [52]. The reactions, performed at 100 °C in a H2O/DMF (N,N-dimethylformamide) mixture under air, afforded regio- and stereoselectively the (Z)-γ-alkylidene butyrolactone products 20 in 45–87% isolated yields after ca. 1 h. Remarkably, no co-catalysts or bases were in this case needed. On the other hand, transformation of the related Meldrum´s acids 21 into lactones 22, containing an all-carbon quaternary center at the C-4 position, was also described by Ahmar and Fillion using catalytic amounts of Ag2CO3, and mixtures THF/H2O or toluene/H2O as the reaction media (Scheme 12) [53]. Again, the 5-exo-dig cyclization products were exclusively formed and the reactions proceeded cleanly in the absence of base. However, we must note that mixtures of E/Z isomers were in most cases obtained starting from those substrates containing an internal C≡C bond.
2.2. Tandem and Cascade Processes Involving the Cycloisomerization of an Alkynoic Acid
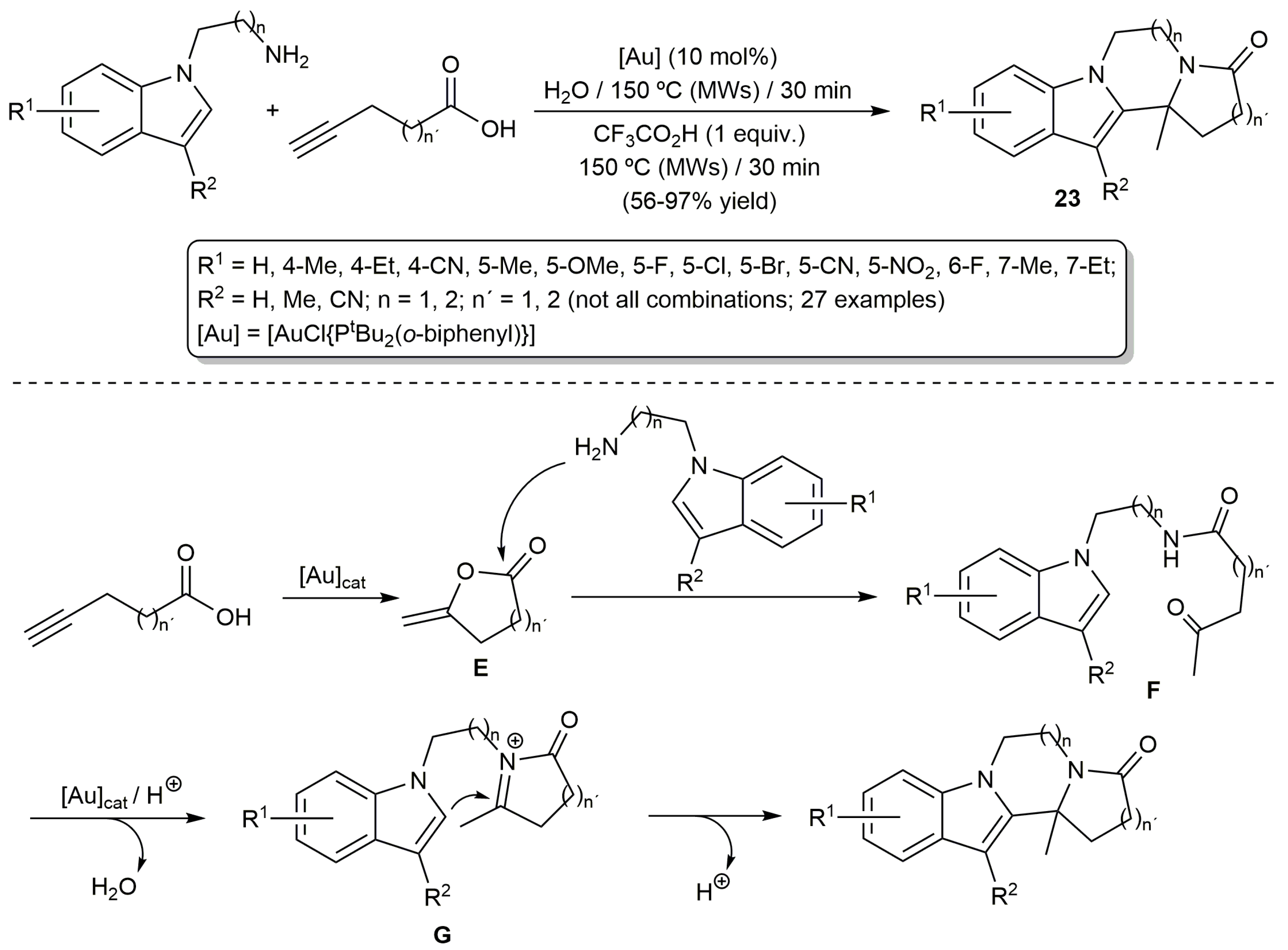
The aminolysis of lactones is a common transformation in organic synthesis which allows their direct conversion into linear amides [54,55]. The combination of this reaction with the cycloisomerization of alkynoic acids has been extensively studied in the last years giving access to a huge number of nitrogen-containing heterocyclic compounds, through different cascade processes, by appropriate selection of the functionalized amine partner [56,57,58,59]. In this context, Liu and co-workers developed an efficient and broad scope gold-catalyzed reaction for the synthesis of fused polycyclic indoles 23 in water, starting from 2-(1H-indol-1-yl)alkylamines and alkynoic acids (Scheme 13) [60].
The process involves the aminolysis of the in situ formed enol-lactones E to generate the linear keto-amides F, which subsequently evolve into G via a gold-catalyzed N-acyliminium ion formation/cyclization. Final intramolecular nucleophilic attack of the C-2 carbon of the indolic unit to the iminium carbon yields the products 23. To facilitate the cyclization step leading to G, the addition trifluoroactic acid was in some cases required. As shown in Scheme 13, the reactions proceeded in short times under microwave irradiation, tolerating the presence of different functional groups.
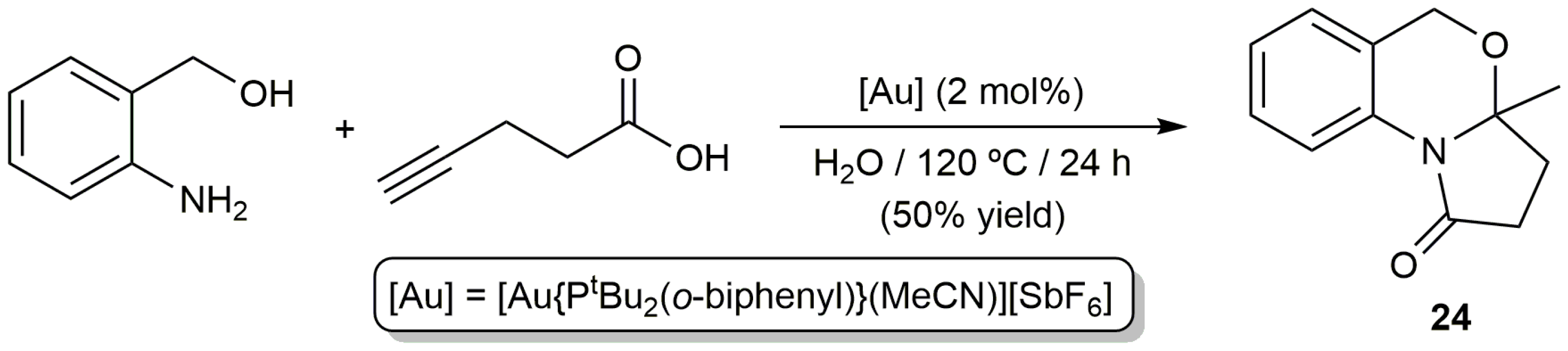
The same group also described the coupling of 4-pentynoic acid with o-aminophenylmethanol in water catalyzed by the cationic gold(I) complex [Au{PtBu2(o-biphenyl)}(MeCN)][SbF6] (Scheme 14) [61]. The reaction afforded the pyrrolo[2,1-b]benzo[d][1,3]oxazin-1-one 24 in 50% yield. However, we must note that this value was much lower to that obtained in THF under identical reaction conditions (91% yield), so it is not surprising that the generality of the process was studied in the latter solvent. As in the precedent case, the reaction is initiated by the cycloisomerization of the alkynoic acid, subsequent aminolysis of the enol lactone intermediate, and final cyclization of the resulting keto amide catalyzed by the gold complex.
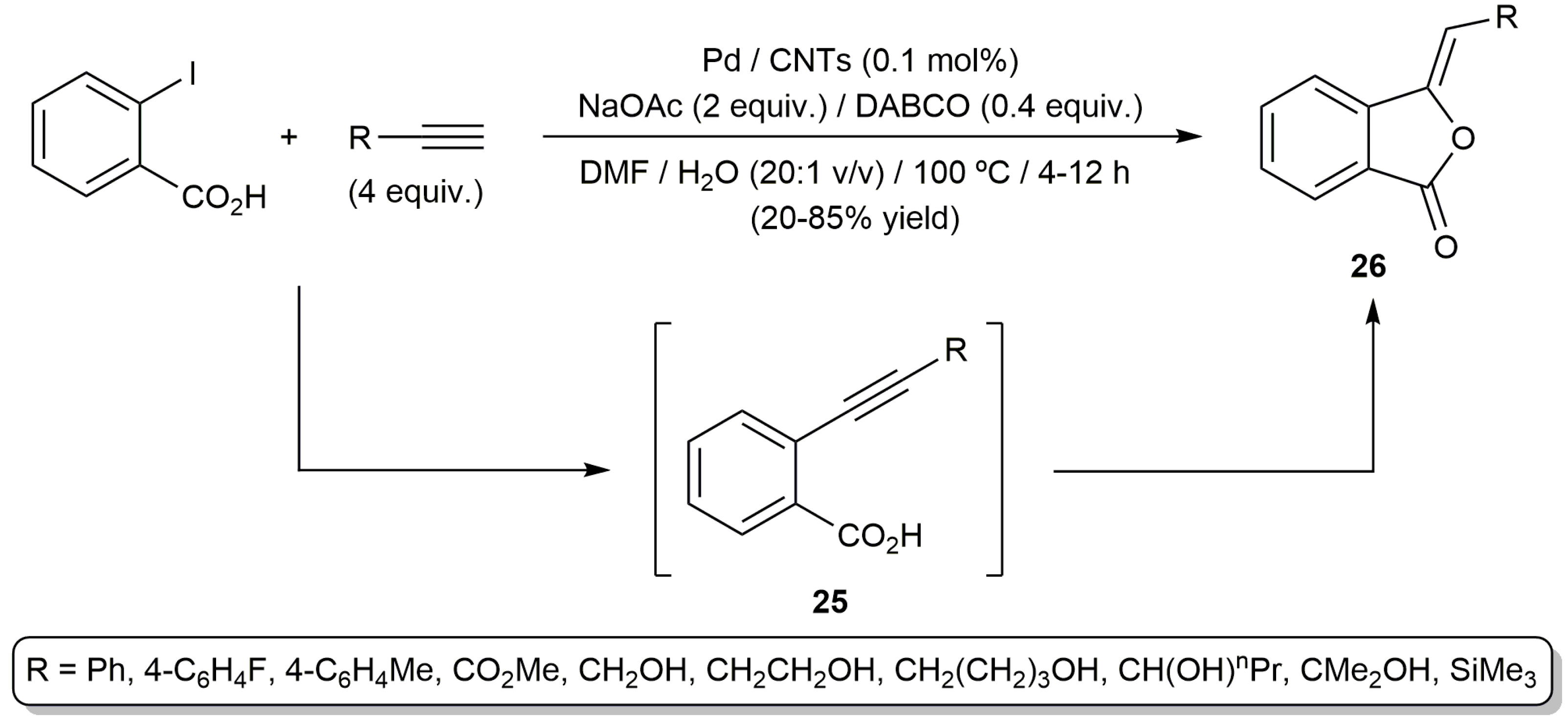
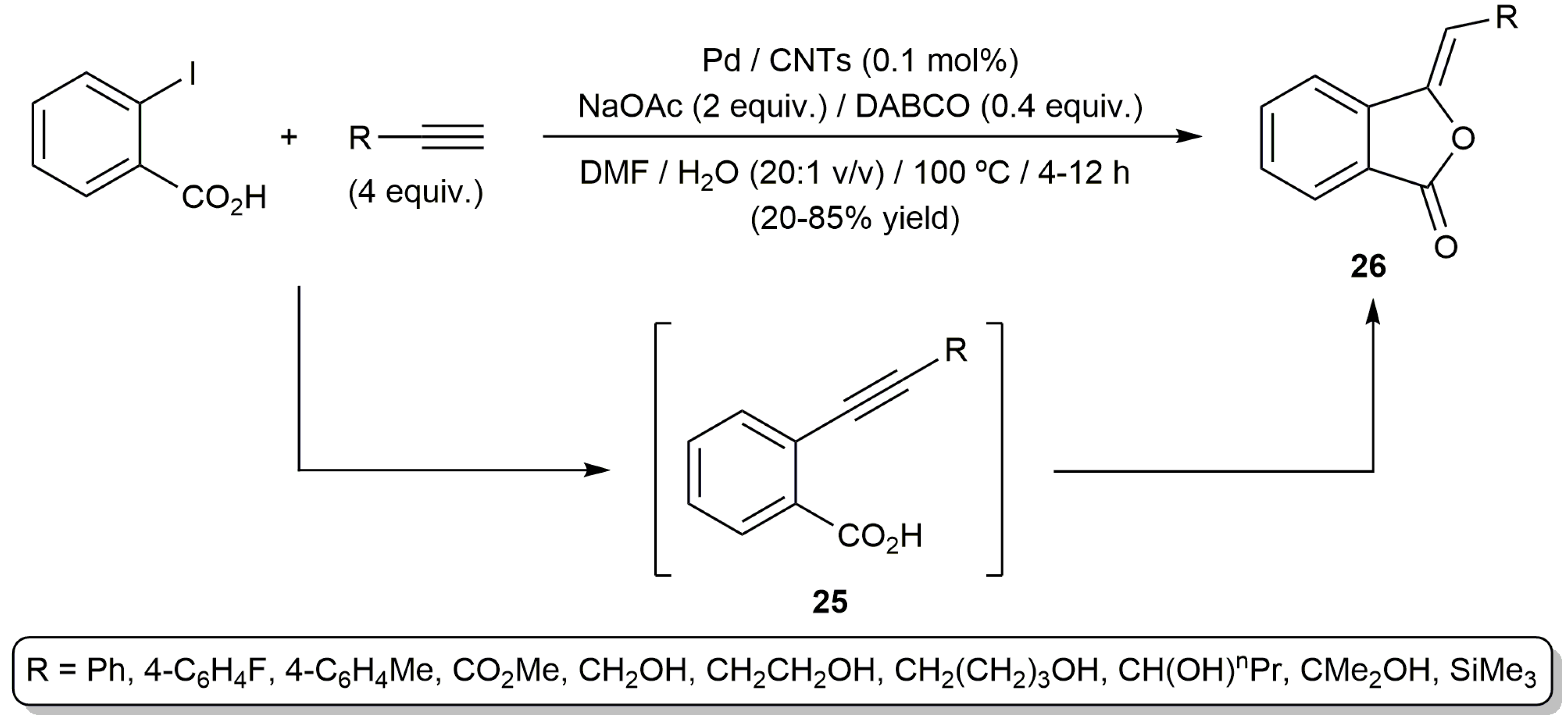
In another vein, the group of Jiang developed an aqueous protocol for the one-pot synthesis of phthalides 26 from terminal alkynes and o-iodobenzoic acid (Scheme 15) [62]. The process, which involves the initial Sonogashira coupling of the substrates and subsequent stereoselective 5-exo-dig cyclization of the resulting ethynyl-benzoic acid intermediates 25, was catalyzed by palladium immobilized on carbon nanotubes (CNTs) in a DMF:H2O mixture. Unlike previous examples of this tandem reaction, formation of isocoumarin by-products (6-endo-dig cyclization) was in this case not observed, and no additives such as phosphine ligands or CuI were needed.
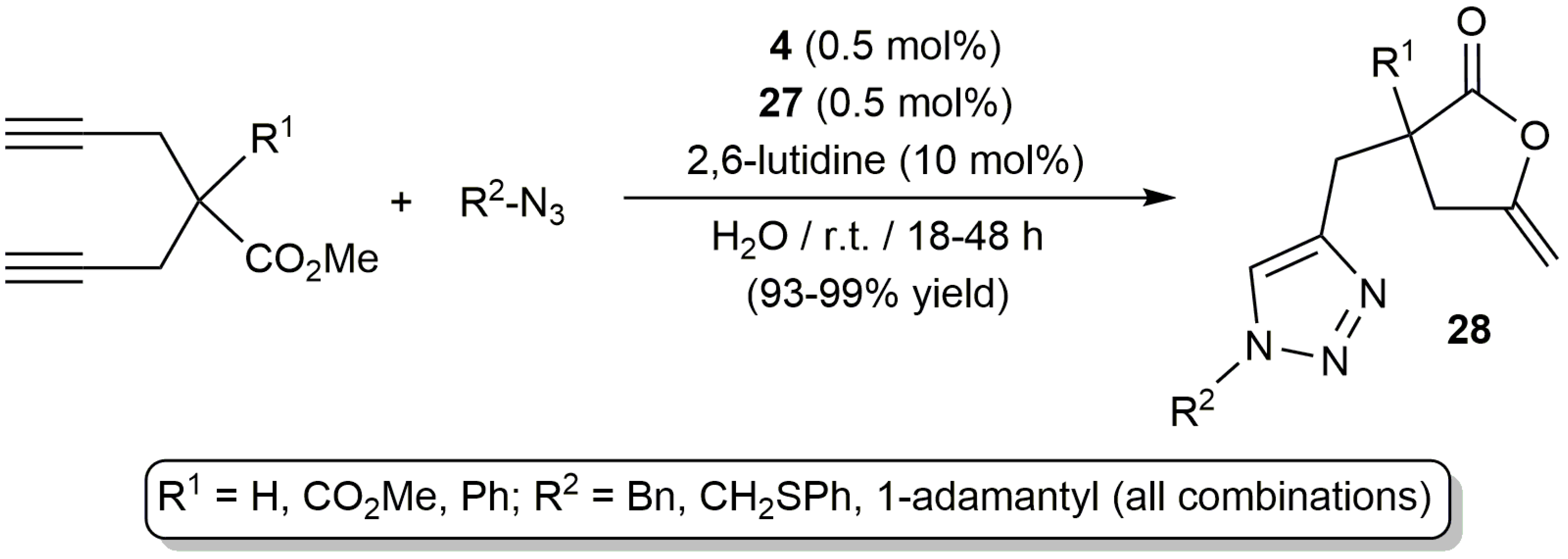
Taking advantage of the ability of the dinuclear iminophosphorane-palladium(II) complex 4 to promote the fast and selective transformation of bispropargylic carboxylic acids 5 into enol-lactones 6 (Scheme 4), García-Álvarez and co-workers could set up an unprecedented one-pot tandem process combining the aforementioned reaction with a copper-catalyzed 1,3-dipolar cycloaddition of azides with the terminal alkyne arm of the enol-lactone products (Scheme 16) [30]. To promote the 1,3-dipolar cycloaddition step the polymeric Cu(I) catalyst [Cu{µ2-N,S-(PTA)=NP(=S)(OEt)2}]x[SbF6]x (27) containing the same hydrophilic iminophosphorane ligand [63], in combination with the 2,6-lutidine base, was employed. The tandem process proceeded in pure water under mild conditions, affording the bicyclic triazol-enol-lactones 28 in excellent yields after a simple extraction with diethyl ether (no chromatographic purification was needed).
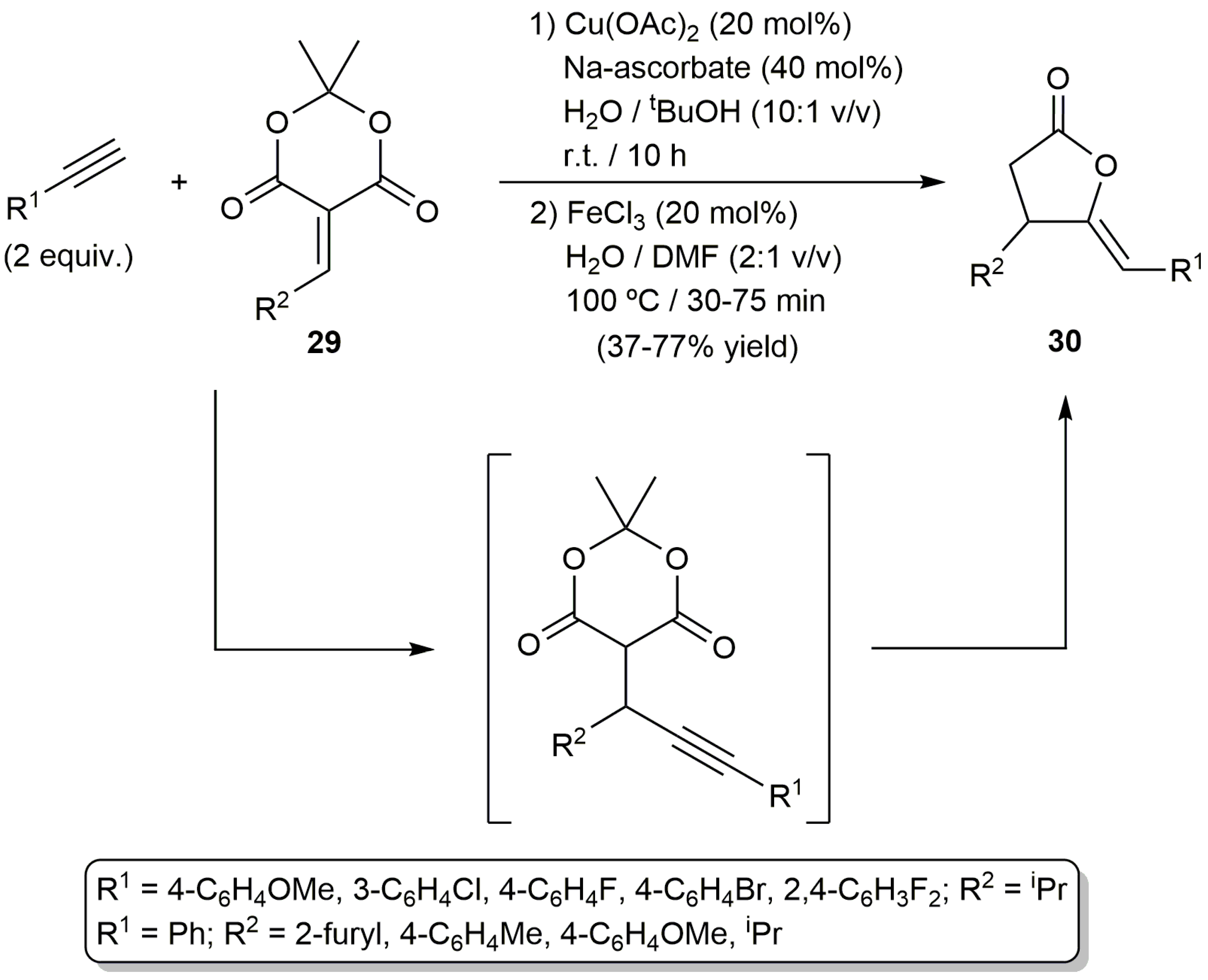
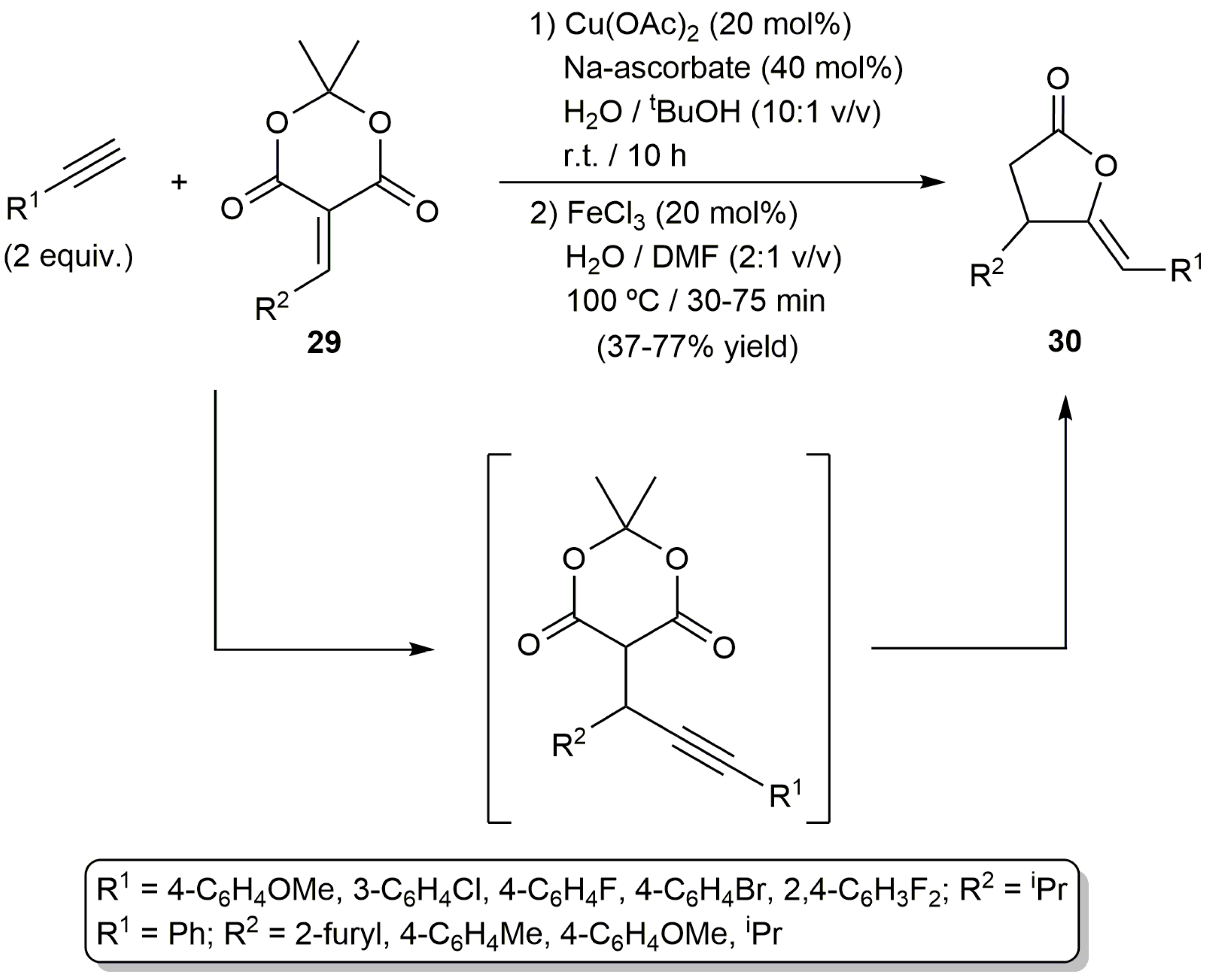
On the other hand, based on the capability of copper salts to promote the conjugate alkynylation of electron-deficient olefins, the (Z)-γ-alkylidene butyrolactones 30 could be synthesized in a one-pot manner starting from the corresponding terminal alkynes and the 5-alkylidene-Meldrun’s acids 29, via in situ formation of the corresponding propargylic Meldrun’s acids (Scheme 17). The process was promoted by the Cu(OAc)2/FeCl3 combination discussed above for the cyclization of Meldrun´s acids 19 (Scheme 11) [51].
Finally, the combination of metal catalysis and biocatalysis has emerged in recent years as a powerful tool for developing new synthetic methodologies merging the advantages of both disciplines in terms of reaction scope and selectivity [64,65,66]. In this context, García-Álvarez, González-Sabín and co-workers described very recently the one-pot conversion of 4-pentynoic acid into enantiopure γ-hydroxyvaleric acid in aqueous medium, through the combined use of KAuCl4 and ketoreductases (KREDs) (Scheme 18) [67]. The process involves the initial gold-catalyzed cycloisomerization of the substrate, concomitant hydrolysis of the lactone to form levulinic acid, and final bioreduction of the keto group of the latter. Remarkably, no isolation or purification steps were required, the reaction medium coming from the metal-catalyzed reaction being directly employed in the enzymatic step. Also of note is the fact that, just by selecting the adequate KRED, both enantiomers of the γ-hydroxyvaleric acid could be obtained with excellent ee values.
3. Intermolecular Processes
3.1. Catalytic Addition of Carboxylic Acids to Terminal and Internal Alkynes
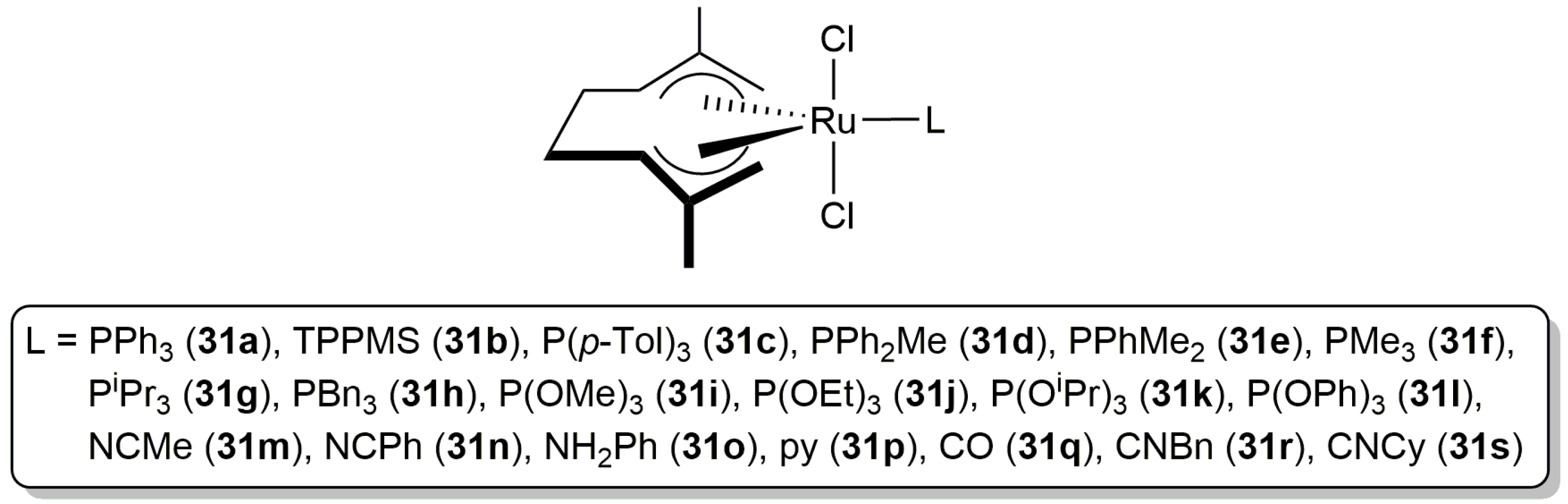
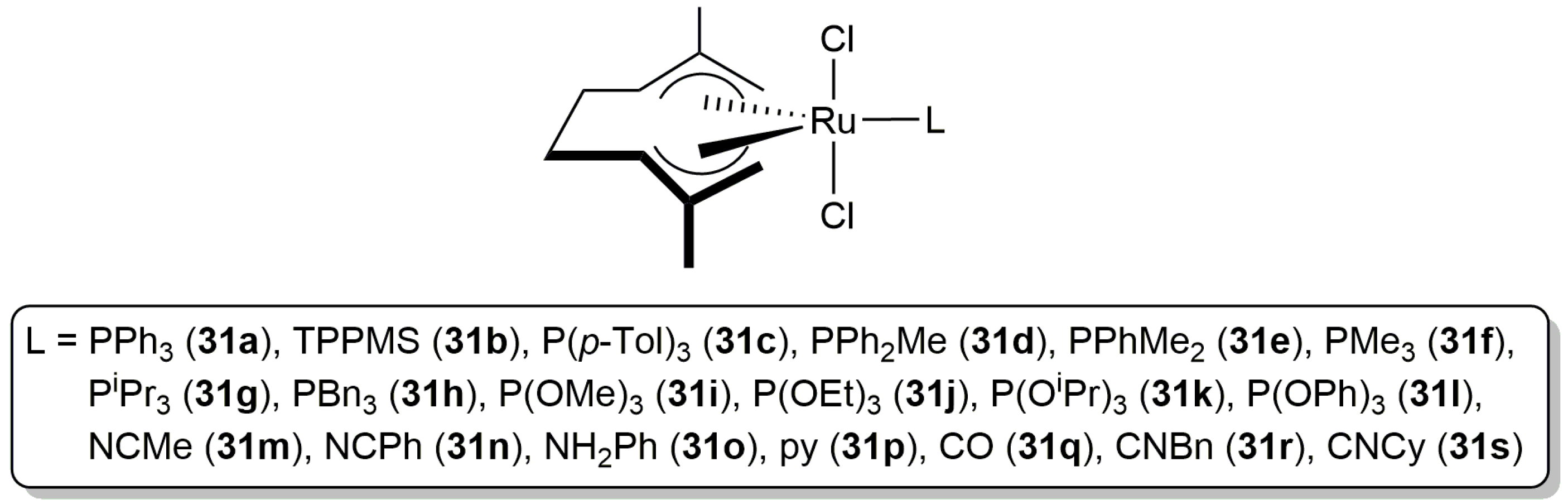
Although the intermolecular hydro-oxycarbonylation of alkynes has been widely studied in organic media [1,2,3,4,5,6], to date there are very few examples of this catalytic transformation described in water. In this regard, Cadierno, Gimeno and co-workers evaluated in 2011 the catalytic potential of a diverse family of ruthenium(IV) complexes, of general composition trans-[RuCl2(η3:η3-C10H16)(L)] (31a–s in Figure 6), for the hydro-oxycarbonylation of terminal alkynes in aqueous medium [68].
All these compounds were found to be active catalysts in the addition of benzoic acid to 1-hexyne in pure water, providing the corresponding enol esters nBuC(OBz)=CH2 (Markovnikov addition product) and (E/Z)-nBuCH=CH(OBz) (anti-Markovnikov addition products) in moderate to good yields after 3–24 h of heating at 60 °C (with a Ru loading of 2 mol %). A high selectivity towards the Markovnikov addition product was in general observed, except in the case of catalysts 31m–p containing a labile amine or nitrile ligand which generated preferentially (E/Z)-nBuCH=CH(OBz), albeit only in moderate yields. The best results in terms of activity and regioselectivity were obtained with [RuCl2(η3:η3-C10H16)(PPh3)] (31a), which was able to generate the enol ester nBuC(OBz)=CH2 in 96% yield (by gas chromatography) after only 3 h of heating. However, from the data obtained, no relationships between the steric and/or electronic nature of the auxiliary ligand L and the catalytic activity observed could be drawn. On the other hand, it must be also highlighted that, with the exception of [RuCl2(η3:η3-C10H16)(TPPMS)] (31b; TPPMS = 3-(diphenylphosphino)benzenesulfonate sodium salt) and [RuCl2(η3:η3-C10H16){P(OR)3}] (R = Me (31i), Et (31j), iPr (31k)), these Ru(IV) complexes are completely insoluble in water. Accordingly, in most of the reactions the catalyst remained dissolved in the organic phase, forming an emulsion with water under stirring. In other words, the reactions proceeded under the so-called “on-water” conditions [69,70]. In addition, when the same reactions were carried out under homogeneous conditions using organic solvents, the results were much worse in terms of yields (the regioselectivity remained unaffected), pointing out the marked positive effect of water in the process. Although to a lesser extent, the beneficial effect of water in the catalytic addition of carboxylic acids to terminal alkynes promoted by tethered arene-ruthenium(II) complexes was also observed by Demonceau and co-workers, who obtained higher yields when performing the reactions in moist vs. dry toluene [71].
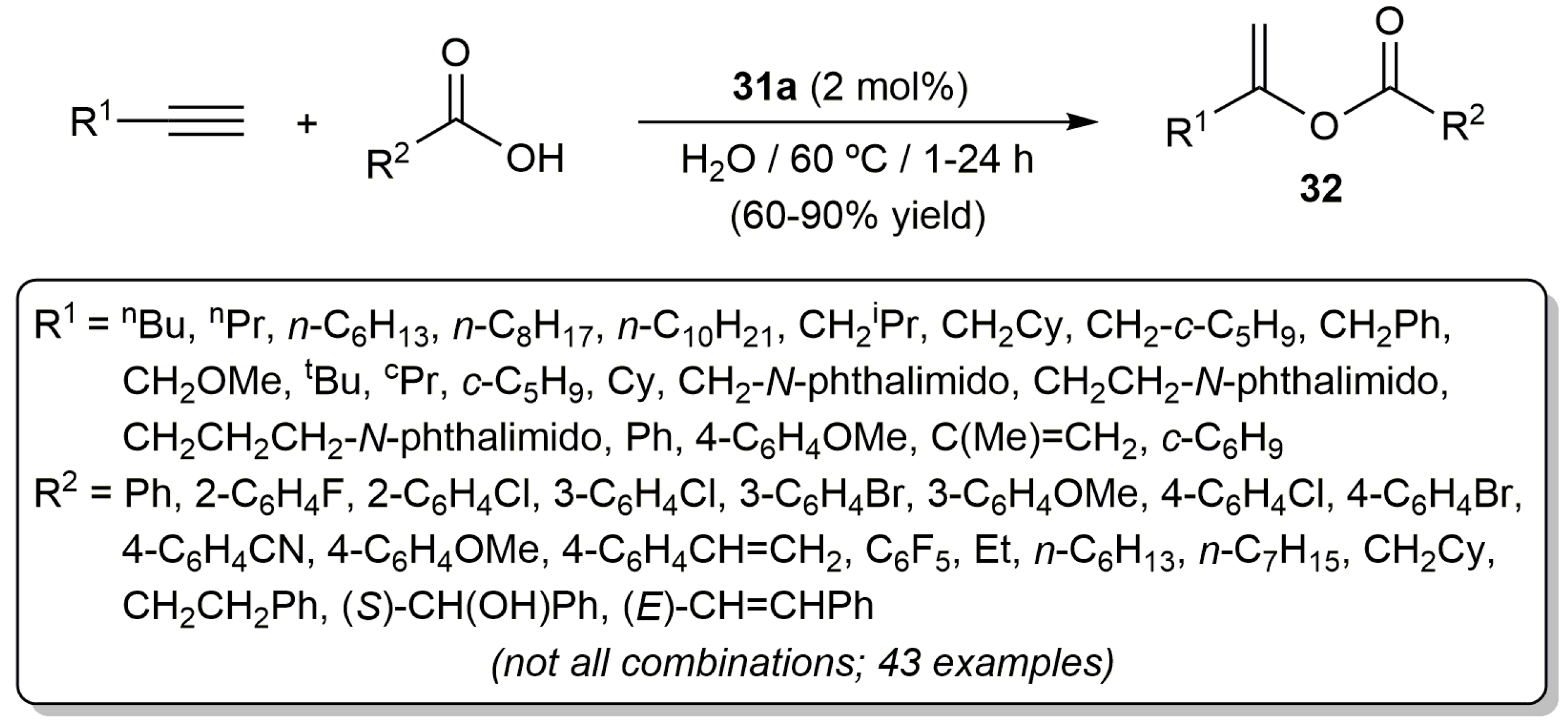
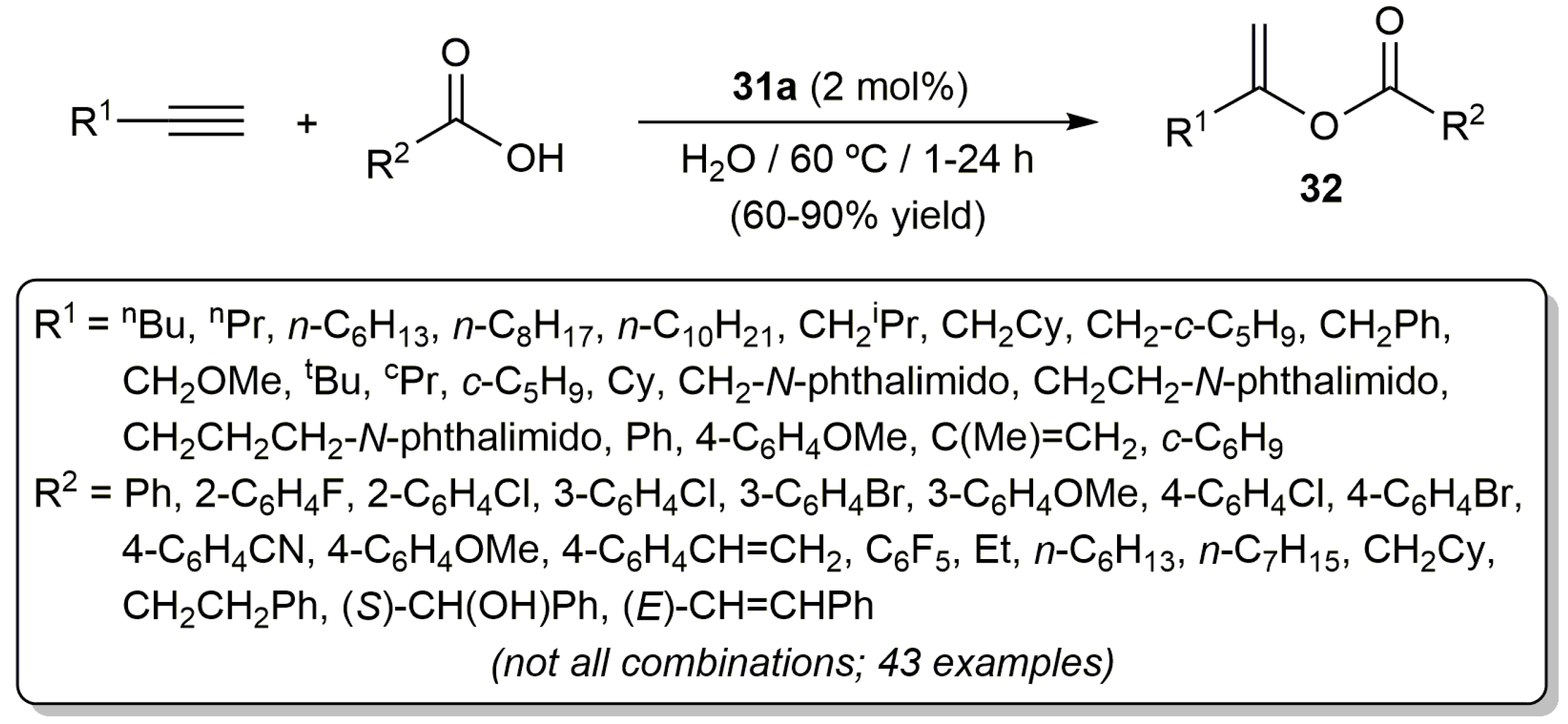
The most active catalyst of the series, i.e., [RuCl2(η3:η3-C10H16)(PPh3)] (31a), showed also a wide scope allowing the preparation of a large variety of enol esters 32 by selective Markovnikov addition of different carboxylic acids to both aliphatic and aromatic terminal alkynes, as well as to 1,3-enynes (Scheme 19) [68,72,73]. As a general trend, the reactions proceeded faster, and with higher yields, when aliphatic terminal alkynes were employed as substrates. Also notable are the high functional group compatibility showed by complex 31a and the absence of competing processes of hydration of the alkynes.
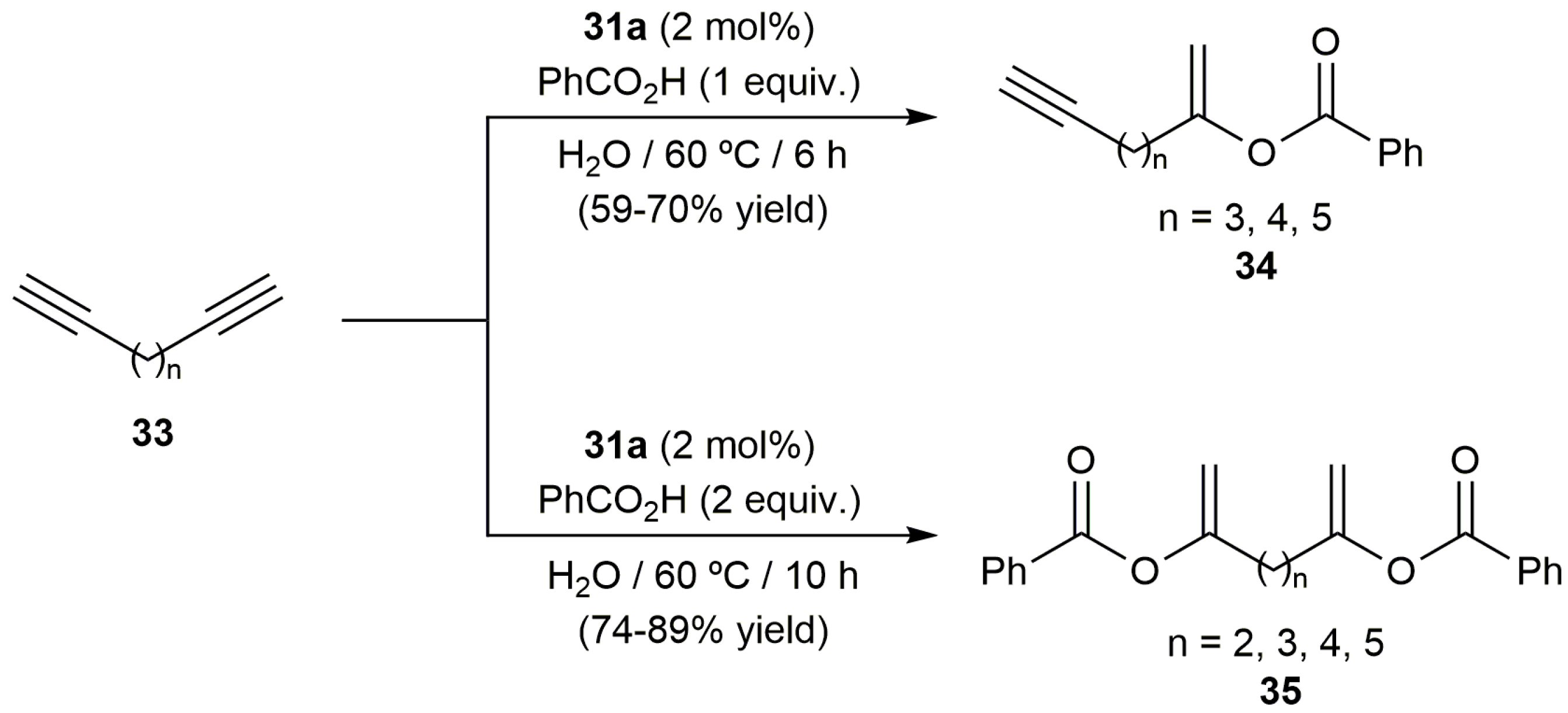
Further evidence of the versatility of [RuCl2(η3:η3-C10H16)(PPh3)] (31a) was gained in the reactions of the terminal diynes 33 with benzoic acid. Thus, as shown in Scheme 20, the corresponding enynes 34 or the diesters 35 could be selectively synthesized with 31a just by adjusting the diyne/benzoic acid ratio employed [68,72,73].
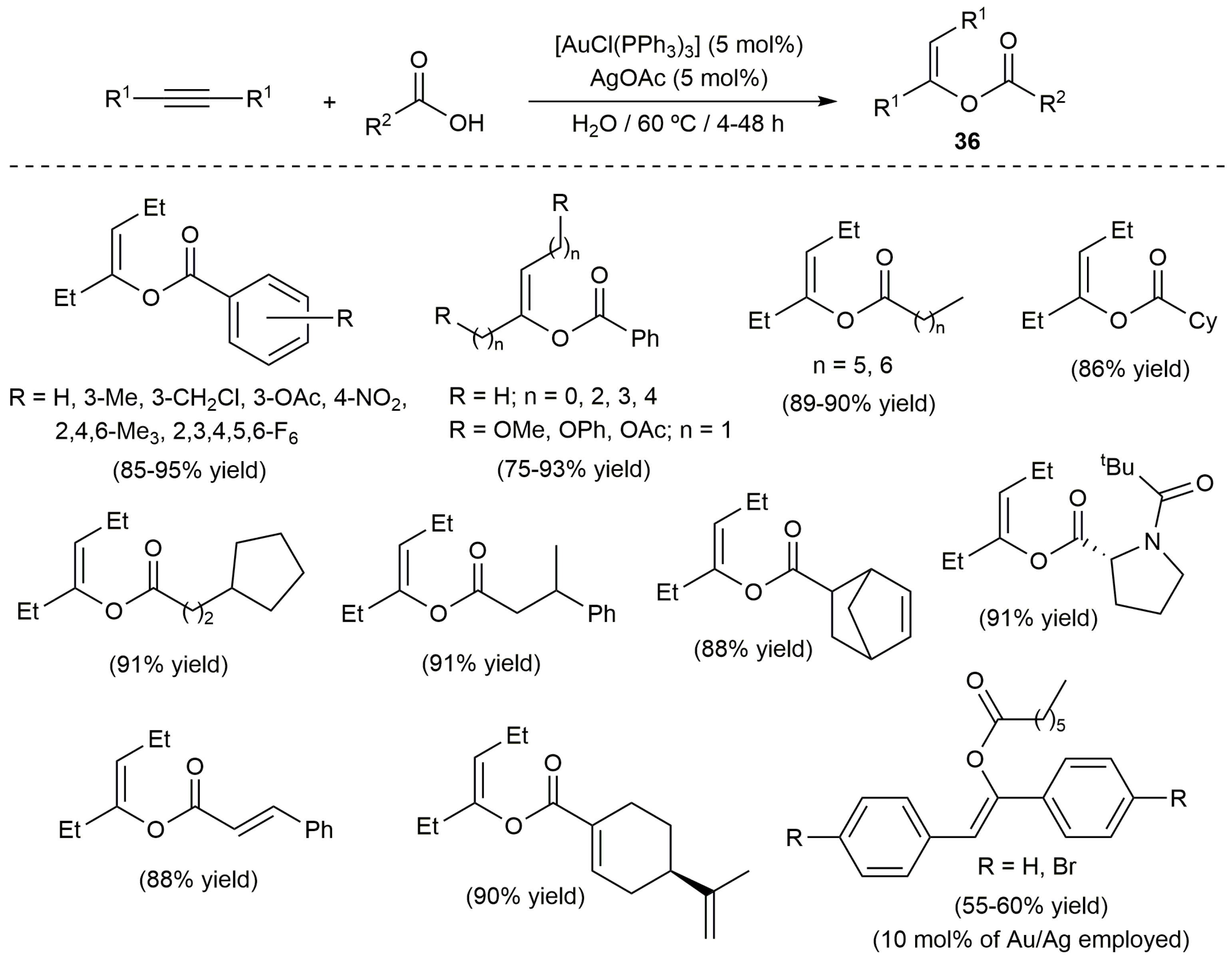
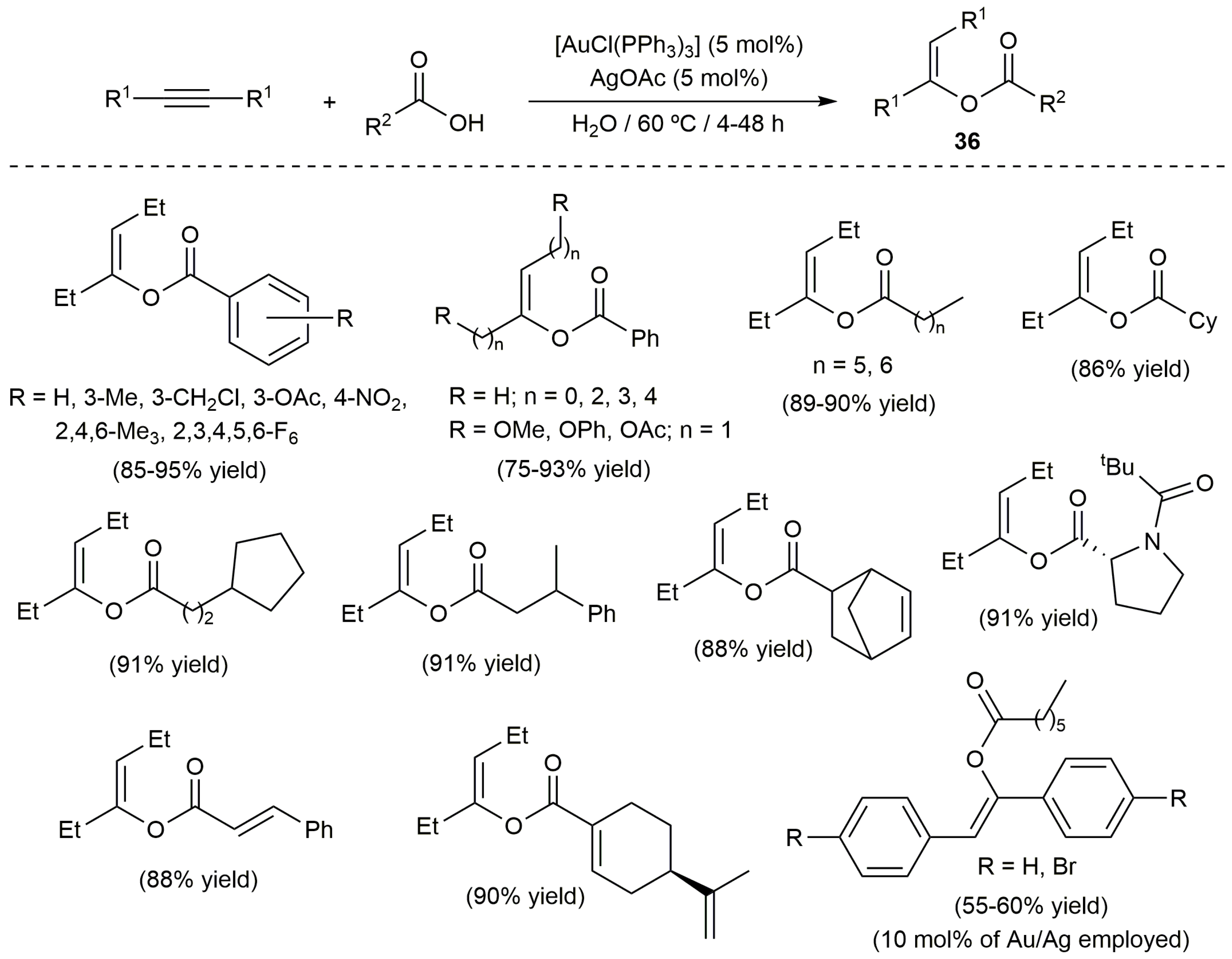
The only limitation encountered with the ruthenium catalyst 31a is that it is completely inactive with internal alkynes; substrates which, as commented in the introduction of this article, show a much lower reactivity in these intermolecular hydrocarboxylation reactions. For this particular class of alkynes, Cadierno, García-Garrido and co-workers developed very recently a protocol in water employing the catalytic system [AuCl(PPh3)]/AgOAc [74]. Thus, as depicted in Scheme 21, a broad range of trisubstituted enol esters 36 could be synthetized in good yields by addition of different aromatic, aliphatic, heterocyclic and α,β-unsaturated carboxylic acids to internal alkynes symmetrically substituted with aliphatic groups. Concerning the use of aromatic alkynes, they showed only a residual reactivity towards benzoic acid and, when an aliphatic carboxylic acid was employed, a higher temperature and catalyst loading were required to obtain the corresponding products in moderate yields. Interestingly, compounds 36 were obtained in a stereoselective manner (only Z isomers) as the result of the exclusive anti addition of the O-H bond of the carboxylic acid to the alkyne.
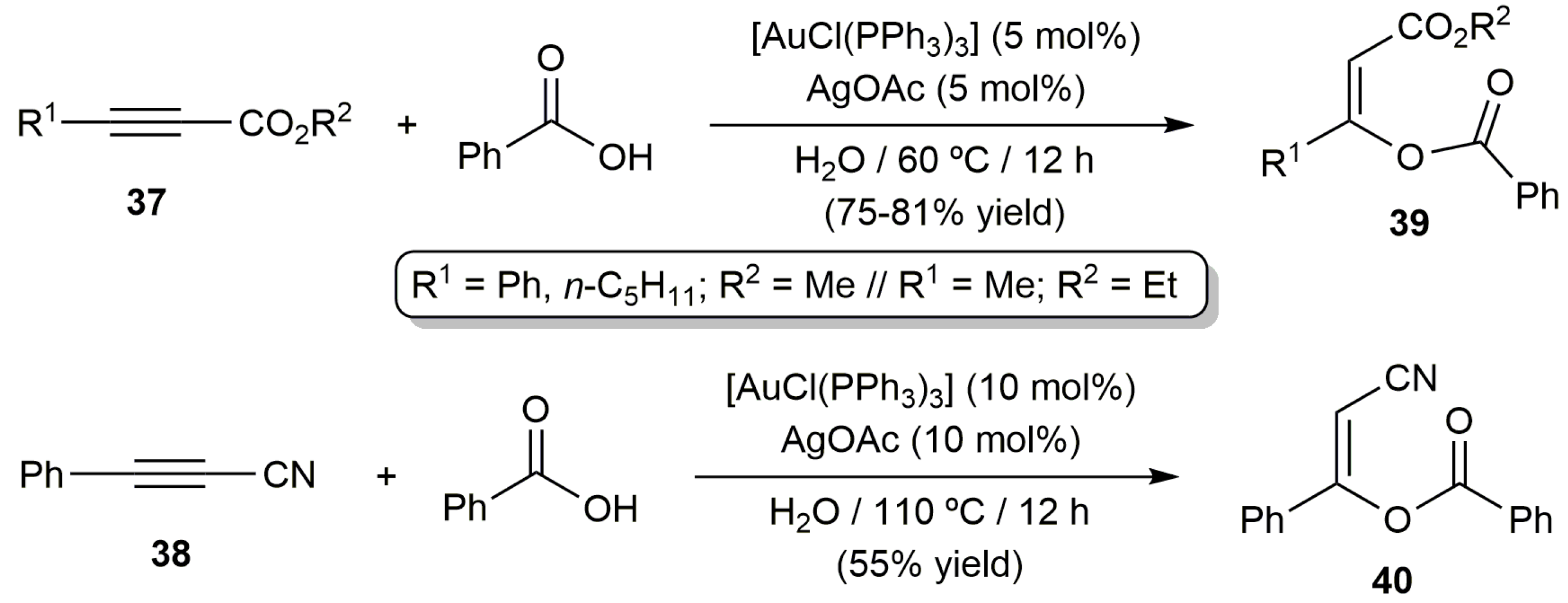
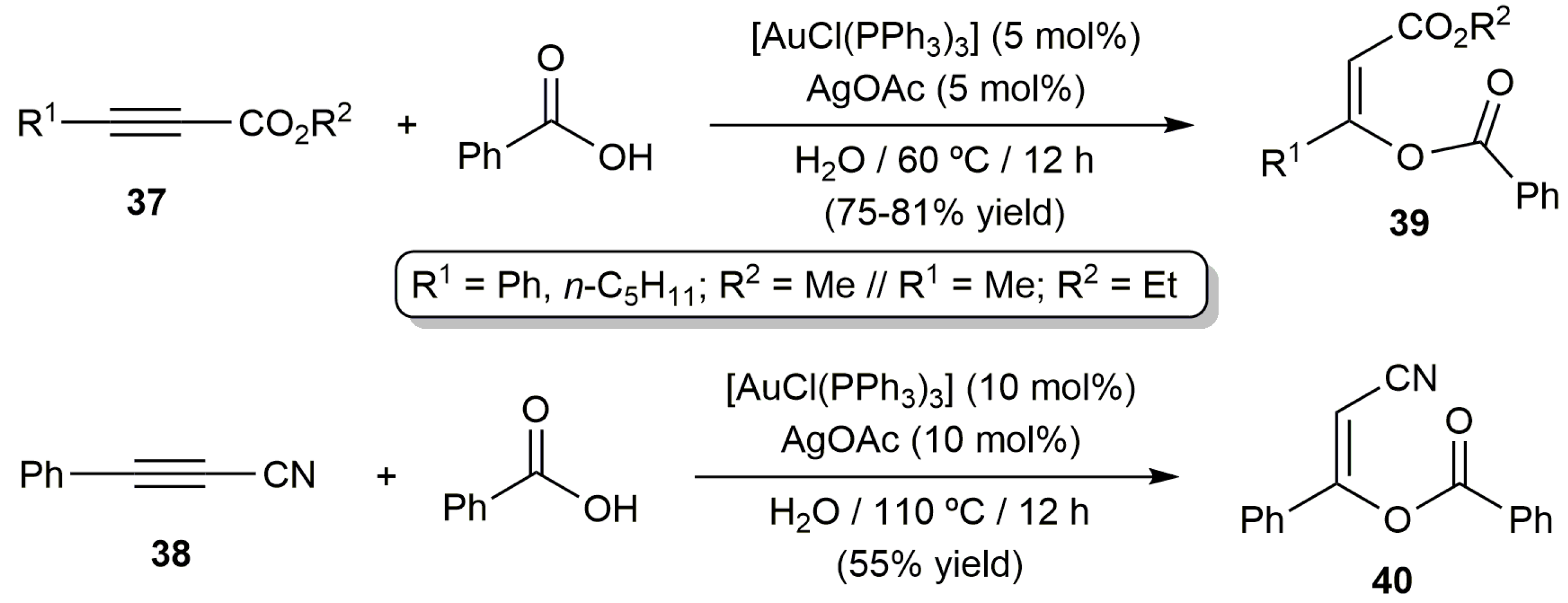
The reactivity of some unsymmetrically substituted internal alkynes towards benzoic acid in water was also explored using the catalytic system [AuCl(PPh3)]/AgOAc, the reations leading in most of the cases to mixtures of regioisomers derived from the attack of acid to both carbon atoms of the alkyne (complete Z-selectivity was again observed for both regioisomers). Only when the activated internal alkynes 37 and 38 were employed as substrates a complete regioselectivity to enol esters 39 and 40, respectively, was observed (Scheme 22) [74].
3.2. Catalytic Addition of Carboxylic Acids to Terminal Propargylic Alcohols
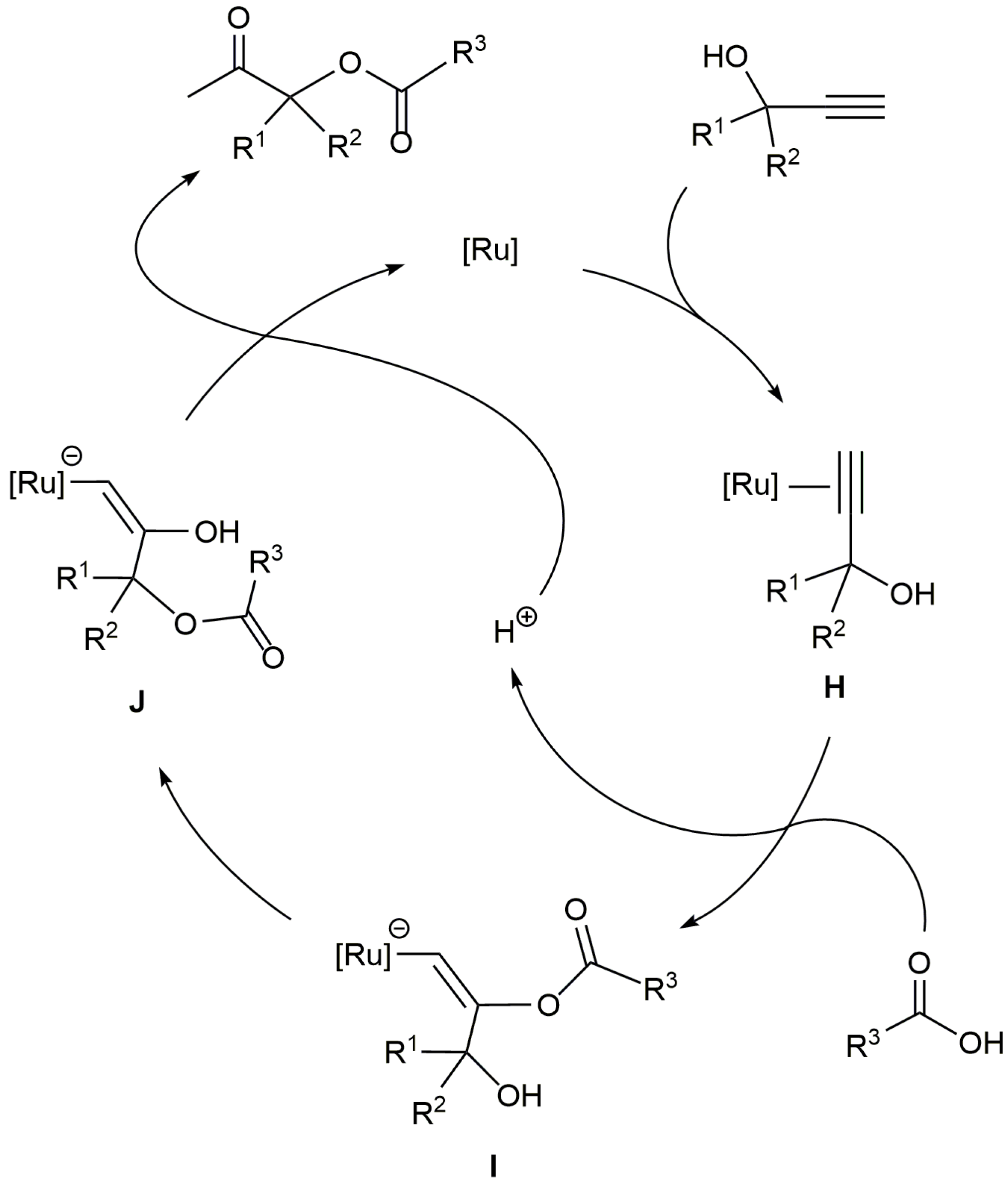
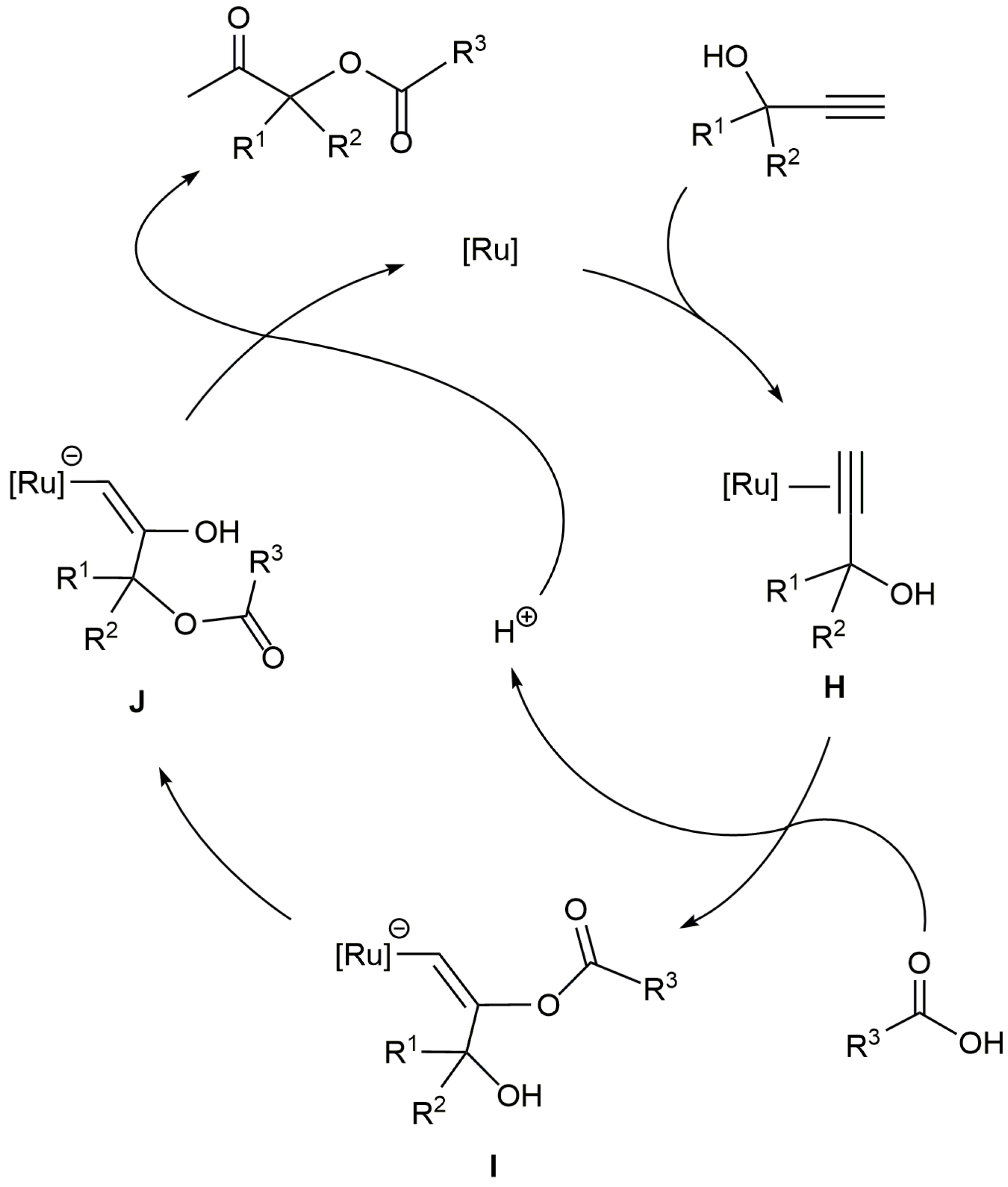
β-Oxo esters are useful intermediates in organic chemistry. For example, they have been employed in the synthesis of different pharmaceuticals [75,76,77], and can be easily transformed into the corresponding α-hydroxy ketones, which are structural units present in a large variety of biologically active molecules [78,79]. Among the different synthetic approaches to β-oxo esters [80], the ruthenium-catalyzed addition of carboxylic acids to terminal propargylic alcohols has emerged as one of the most straightforward and atom-economical routes (Scheme 23). The process involves the Markovnikov attack of the carboxylic acid to an initially formed π-alkyne-ruthenium complex H, which is followed by the intramolecular transesterification of the resulting intermediate I to form an alkenyl derivative J. The final protonolysis of J releases the β-oxo ester product and regenerates the catalytically active ruthenium species.
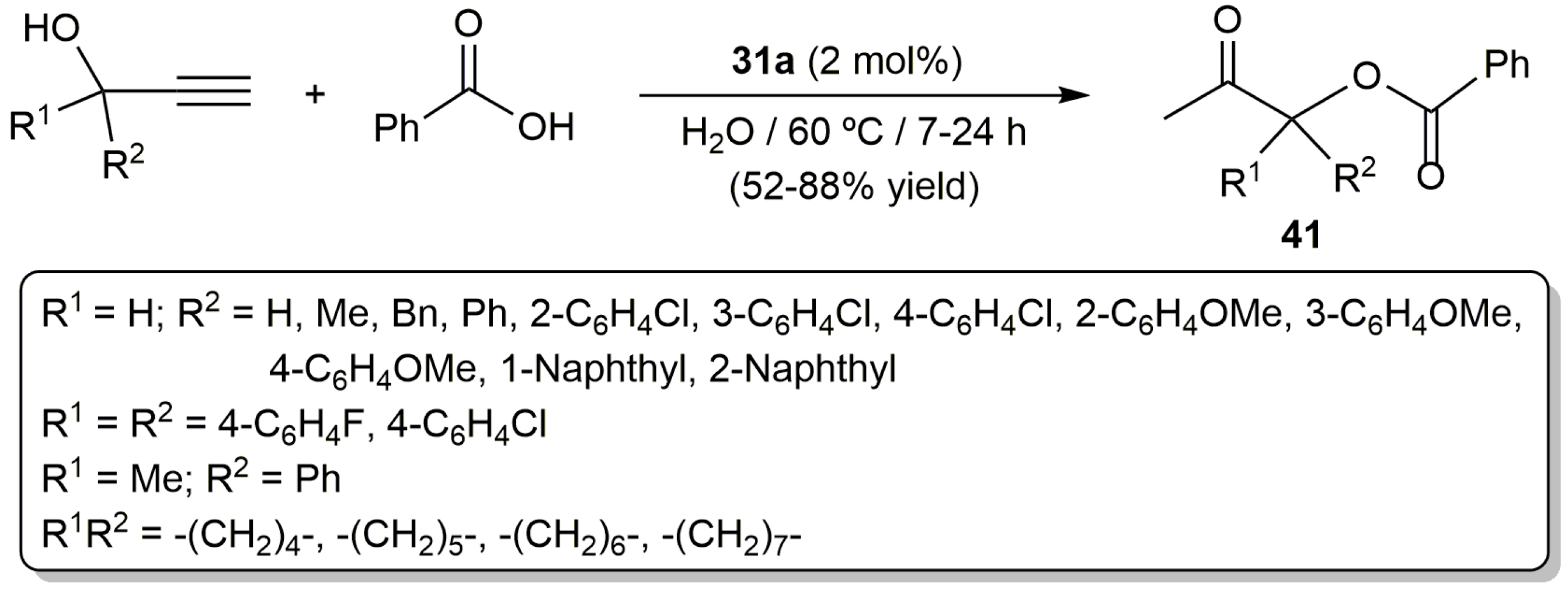
Since the pioneering work by Mitsudo and Watanabe in 1987 [81], a number of ruthenium catalysts for this transformation have been developed [80,82]. Among them, the bis(allyl)-ruthenium(IV) complex [RuCl2(η3:η3-C10H16)(PPh3)] (31a) (Figure 6) proved to be active in water [68]. Thus, as shown in Scheme 24, starting from different propargylic alcohols and benzoic acid, a family of β-oxo esters 41 could be synthetized in moderate to good yields employing identical experimental conditions to those applied in the preparation of the enol esters 32 (Scheme 19). Although the scope of the reaction concerning the carboxylic acid partner was not studied in much detail, the authors showed that it is not restricted to benzoic acid since the addition of 2-chlorobenzoic acid, pentafluorobenzoic acid, heptanoic acid and 3-cyclopentylpropionic acid to 1-phenyl-2-propyn-1-ol also afforded the corresponding β-oxo esters in 47–90% yield.
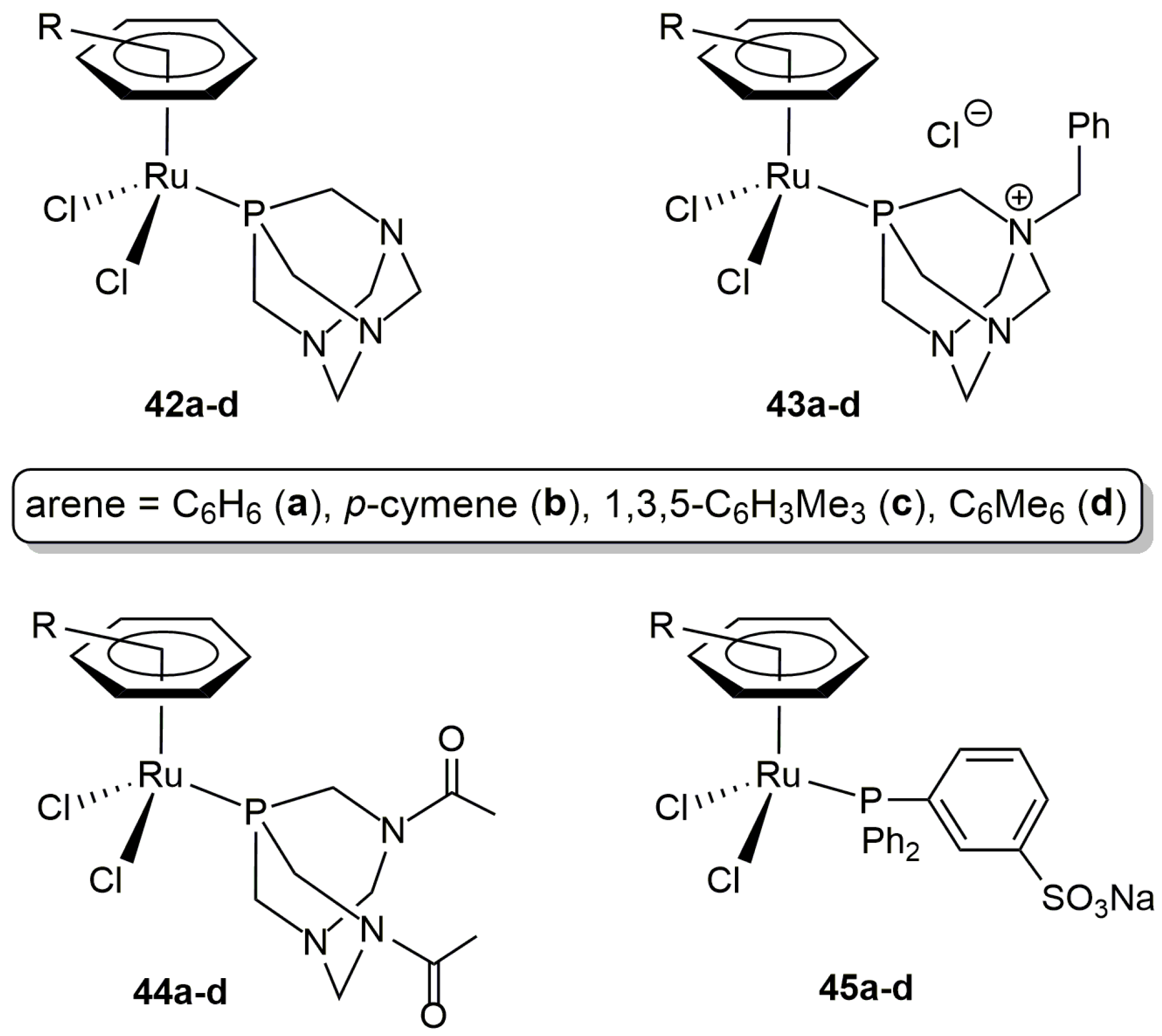
In an independent work, Cadierno, Gimeno and co-workers also explored the catalytic potential of a series of arene-ruthenium(II) complexes containing different water-soluble phosphine ligands (42–45a–d in Figure 7) [83]. All of them proved to the active in the addition of benzoic acid to 1-phenyl-2-propyn-1-ol in pure water, leading to the desired β-oxo ester, i.e., 1-phenyl-2-oxopropyl benzoate, as the major reaction product. Among them, best results in terms of activity and selectivity were obtained with the benzene derivative [RuCl2(η6-C6H6)(TPPMS)] (45a) (88% yield of 1-phenyl-2-oxopropyl benzoate after 3 h of heating at 100 °C using a ruthenium loading of 2 mol %). Concerning the scope of this complex, a variety of secondary propargylic alcohols, as well as prop-2-yn-1-ol, could be efficiently transformed. In contrast, tertiary propargylic alcohols resulted to be more challenging substrates and only those featuring low sterically-demanding substituents led to high conversions. On the other hand, 45a was operative with a large variety of aromatic carboxylic acids, bearing different functional group such as halide, alkoxy, ketone or sulfonamide. The use of heteroaromatic acids, with tetrahydrofuran, pyrrole, thiophene, indole or 2-oxo-2H-chromene fragments, as well as aliphatic acids was also tolerated.
4. Conclusions
Great attention is currently devoted to synthetic organic chemistry in water and research, particularly in the field of aqueous catalysis, is increasing exponentially since water is the most environmentally benign substitute for the volatile and toxic organic solvents commonly used in laboratories and industries. In this contribution, we have summarized the developments achieved in the field of metal-catalyzed additions of carboxylic acids to alkynes in aqueous media. Such processes now represent powerful tools for the construction of synthetically useful lactones, enol esters and β-oxo esters in an atom-economical manner. Throughout this review article, we have presented different catalytic systems based on Pd, Pt, Au, Cu and Ru, capable of promoting selectively these reactions in aqueous environments without observing the competing hydration of the alkyne substrates. The vast majority of the works discussed herein have been published during the last ten years, demonstrating clearly the current interest in this research field, which obviously remains open, with many opportunities for new discoveries.
Acknowledgments
The authors acknowledge the Spanish MINECO (projects CTQ2013-40591-P and CTQ2016-75986-P) and the Gobierno del Principado de Asturias (project GRUPIN14-006) for financial support. Javier Francos thanks The Ministry of Economy and Competitiveness (MINECO) and European Social Fund (ESF) for the award of a Juan de la Cierva contract.
Author Contributions
Both authors analyzed the data and jointly wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-metal-catalyzed addition of heteroatom-hydrogen bonds to alkynes. Chem. Rev. 2004, 104, 3079–3159. [Google Scholar] [CrossRef] [PubMed]
- Beller, M.; Seayad, J.; Tillack, A.; Jiao, H. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: Recent developments and trends. Angew. Chem. Int. Ed. 2004, 43, 3368–3398. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.T.; Kavthe, R.D.; Shinde, V.S. Transition metal-catalyzed addition of C-, N- and O-nucleophiles to unactivated C–C multiple bonds. Tetrahedron 2012, 68, 8079–8146. [Google Scholar] [CrossRef]
- Hintermann, L. Recent developments in metal-catalyzed additions of oxygen nucleophiles to alkenes and alkynes. Top. Organomet. Chem. 2010, 31, 123–155. [Google Scholar]
- Bruneau, C. Group 8 metals-catalyzed O–H bond addition to unsaturated molecules. Top. Organomet. Chem. 2013, 43, 203–230. [Google Scholar]
- Abbiati, G.; Beccalli, E.M.; Rossi, E. Groups 9 and 10 metals-catalyzed O-H bond addition to unsaturated molecules. Top. Organomet. Chem. 2013, 43, 231–290. [Google Scholar]
- Rao, Y.S. Recent advances in the chemistry of unsaturated lactones. Chem. Rev. 1976, 76, 625–694. [Google Scholar] [CrossRef]
- Laduwahetty, T. Saturated and unsaturated lactones. Contemp. Org. Synth. 1995, 2, 133–149. [Google Scholar] [CrossRef]
- Libiszewska, K. Lactones as biologically active compounds. Biotechnol. Food Sci. 2011, 75, 45–53. [Google Scholar]
- Janecki, T. (Ed.) Natural Lactones and Lactams: Synthesis, Occurrence and Biological Activity; Wiley-VCH: Weinheim, Germany, 2013; ISBN 9783527334148. [Google Scholar]
- Neaţu, F.; Toullec, P.Y.; Michelet, V.; Pârvulescu, V.I. Heterogeneous Au and Rh catalysts for the cycloisomerization reactions of γ-acetylenic carboxylic acids. Pure Appl. Chem. 2009, 81, 2387–2396. [Google Scholar] [CrossRef]
- Bruneau, C.; Neveux, M.; Kabouche, Z.; Ruppin, C.; Dixneuf, P.H. Ruthenium-catalyzed additions to alkynes: Synthesis of activated esters and their use in acylation reactions. Synlett 1991, 11, 755–763. [Google Scholar] [CrossRef]
- Kumar, M.; Bagchi, S.; Sharma, A. The first vinyl acetate mediated organocatalytic transesterification of phenols: A step towards sustainability. New J. Chem. 2015, 39, 8329–8336. [Google Scholar] [CrossRef]
- Liu, X.; Coutelier, O.; Harrison, S.; Tassaing, T.; Marty, J.-D.; Destarac, M. Enhanced solubility of polyvinyl esters in scCO2 by means of vinyl trifluorobutyrate monomer. ACS Macro Lett. 2015, 4, 89–93. [Google Scholar] [CrossRef]
- Foarta, F.; Landis, C.R. Condensation oligomers with sequence control but without coupling reagents and protecting groups via asymmetric hydroformylation and hydroacyloxylation. J. Org. Chem. 2016, 81, 11250–11255. [Google Scholar] [CrossRef] [PubMed]
- Jena, R.K.; Das, U.K.; Ghorai, A.; Bhattacharjee, M. Ruthenium-catalyzed addition of carboxylic acids to progargylic alcohols: An easy route to O-dienyl esters and their tandem atom-transfer radical polymerization. Eur. J. Org. Chem. 2016, 6015–6021. [Google Scholar] [CrossRef]
- Konrad, T.M.; Schmitz, P.; Leitner, W.; Franciò, G. Highly enantioselective Rh-catalysed hydrogenation of 1-alkyl vinyl esters using phosphine-phosphoramidite ligands. Chem. Eur. J. 2013, 19, 13299–13303. [Google Scholar] [CrossRef] [PubMed]
- González-Liste, P.J.; León, F.; Arribas, I.; Rubio, M.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Highly stereoselective synthesis and hydrogenation of (Z)-1-alkyl-2-arylvinyl acetates: A wide scope procedure for the preparation of chiral homobenzylic esters. ACS Catal. 2016, 6, 3056–3060. [Google Scholar] [CrossRef]
- León, F.; González-Liste, P.J.; García-Garrido, S.E.; Arribas, I.; Rubio, M.; Cadierno, V.; Pizzano, A. Broad scope synthesis of ester precursors of nonfunctionalized chiral alcohols based on the asymmetric hydrogenation of α,β-dialkyl-, α,β-diaryl-, and α-alkyl-β-aryl-vinyl esters. J. Org. Chem. 2017, 82, 5852–5867. [Google Scholar] [CrossRef] [PubMed]
- Takeno, M.; Kikuchi, S.; Morita, S.-I.; Nishiyama, Y.; Ishii, Y. A new coupling reaction of vinyl esters with aldehydes catalyzed by organosamarium compounds. J. Org. Chem. 1995, 60, 4974–4975. [Google Scholar] [CrossRef]
- Goossen, L.J.; Paetzold, J. Decarbonylative Heck olefination of enol esters: Salt-free and environmentally friendly access to vinyl arenes. Angew. Chem. Int. Ed. 2004, 43, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Geibel, I.; Dierks, A.; Schmidtmann, M.; Christoffers, J. Formation of δ-lactones by cerium-catalyzed, Baeyer-Villiger-type coupling of β-oxoesters, enol acetates, and dioxygen. J. Org. Chem. 2016, 81, 7790–7798. [Google Scholar] [CrossRef] [PubMed]
- González-Liste, P.J.; Francos, J.; García-Garrido, S.E.; Cadierno, V. The intermolecular hydro-oxycarbonylation of internal alkynes: Current state of the art. Arkivoc 2018, Part II. 17–39. [Google Scholar]
- Cornils, B.; Herrmann, W.A. (Eds.) Aqueous-Phase Organometallic Catalysis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004; ISBN 3527307125. [Google Scholar]
- Li, C.-J.; Chan, T.-H. Comprehensive Organic Reactions in Aqueous Media, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007; ISBN 9780471761297. [Google Scholar]
- Dixneuf, P.H.; Cadierno, V. (Eds.) Metal-Catalyzed Organic Reactions in Water; Wiley-VCH: Weinheim, Germany, 2013; ISBN 9783527331888. [Google Scholar]
- Benaglia, M. (Ed.) Recoverable and Recyclable Catalysts; John Wiley & Sons: Chichester, UK, 2009; ISBN 9780470681954. [Google Scholar]
- Chen, L.; Li, C.-J. Catalytic reactions of alkynes in water. Adv. Synth. Catal. 2006, 348, 1459–1484. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Ishii, Y.; Ishikawa, K.; Hidai, M. A novel catalyst with a cuboidal PdMo3S4 core for the cyclization of alkynoic acids to enol lactones. Angew. Chem. Int. Ed. 1996, 35, 2123–2124. [Google Scholar] [CrossRef]
- García-Álvarez, J.; Díez, J.; Vidal, C. Pd(II)-catalyzed cycloisomerisation of γ-alkynoic acids and one-pot tandem cycloisomerisation/CuAAC reactions in water. Green Chem. 2012, 14, 3190–3196. [Google Scholar] [CrossRef]
- Phillips, A.D.; Gonsalvi, L.; Romerosa, A.; Vizza, F.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phosphaadamantane (PTA): Transition metal complexes and related catalytic, medicinal and photoluminescent applications. Coord. Chem. Rev. 2004, 248, 955–993. [Google Scholar] [CrossRef]
- Bravo, J.; Bolaño, S.; Gonsalvi, L.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phosphaadamantane (PTA) and derivatives. Part II. The quest for tailored ligands, complexes and related applications. Coord. Chem. Rev. 2010, 254, 555–607. [Google Scholar] [CrossRef]
- Ogata, K.; Sasano, D.; Yokoi, T.; Isozaki, K.; Seike, H.; Takaya, H.; Nakamura, M. Pd-complex bound amino acid-based supramolecular gel catalyst for intramolecuar addition-cyclization of alkynoic acids in water. Chem. Lett. 2012, 41, 498–500. [Google Scholar] [CrossRef]
- Hamasaka, G.; Uozumi, Y. Cyclization of alkynoic acids in water in the presence of a vesicular self-assembled amphiphilic pincer palladium complex catalyst. Green Chem. 2014, 50, 14516–14518. [Google Scholar] [CrossRef] [PubMed]
- Michelet, V.; Toullec, P.Y.; Genêt, J.-P. Cycloisomerization of 1,n-enynes: Challenging metal-catalyzed rearrangements and mechanistic insights. Angew. Chem. Int. Ed. 2008, 47, 4268–4315. [Google Scholar] [CrossRef] [PubMed]
- Soriano, E.; Marco-Contelles, J. Mechanistic insights on the cycloisomerization of polyunsaturated precursors catalyzed by platinum and gold complexes. Acc. Chem. Res. 2009, 42, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Ke, D.; Espinosa, N.A.; Mallet-Ladeira, S.; Monot, J.; Martin-Vaca, B.; Bourissou, D. Efficient synthesis of unsaturated δ- and ε-lactones/lactams by catalytic cycloisomerization: When Pt outperforms Pd. Adv. Synth. Catal. 2016, 358, 2324–2331, and references therein. [Google Scholar] [CrossRef]
- Alemán, J.; del Solar, V.; Navarro-Ranninger, C. Anticancer platinum complexes as non-innocent compounds for catalysis in aqueous media. Chem. Commun. 2010, 46, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; del Solar, V.; Cubo, L.; Quiroga, A.G.; Navarro-Ranninger, C. New reactions of anticancer-platinum complexes and their intriguing behavior under various experimental conditions. Dalton Trans. 2010, 39, 10601–10607. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Toste, F.D. (Eds.) Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012; ISBN 9783527319527. [Google Scholar]
- Toste, F.D.; Michelet, V. (Eds.) Gold Catalysis: An Homogeneous Approach; Imperial College Press: London, UK, 2014; ISBN 9781848168527. [Google Scholar]
- Genin, E.; Toullec, P.Y.; Antoniotti, S.; Brancour, C.; Genêt, J.-P.; Michelet, V. Room temperature Au(I)-catalyzed exo-selective cycloisomerization of acetylenic acids: An entry to functionalized γ-lactones. J. Am. Chem. Soc. 2006, 128, 3112–3113. [Google Scholar] [CrossRef] [PubMed]
- Toullec, P.Y.; Genin, E.; Antoniotti, S.; Genêt, J.-P.; Michelet, V. Au2O3 as a stable and efficient catalyst for the selective cycloisomerization of γ-acetylenic carboxylic acids to γ-alkylidene-γ-butyrolactones. Synlett 2008, 707–711. [Google Scholar]
- Tomás-Mendivil, E.; Toullec, P.Y.; Díez, J.; Conejero, S.; Michelet, V.; Cadierno, V. Cycloisomerization versus hydration reactions in aqueous media: A Au(III)-NHC catalyst that makes the difference. Org. Lett. 2012, 14, 2520–2523. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Mendivil, E.; Toullec, P.Y.; Borge, J.; Conejero, S.; Michelet, V.; Cadierno, V. Water-soluble gold(I) and gold(III) complexes with sulfonated N-heterocyclic carbene ligands: Synthesis, characterization, and application in the catalytic cycloisomerization of γ-alkynoic acids into enol-lactones. ACS Catal. 2013, 3, 3086–3098. [Google Scholar] [CrossRef]
- Ferré, M.; Cattoën, X.; Wong Chi Man, M.; Pleixats, R. Sol-gel immobilized N-heterocyclic carbene gold complex as a recyclable catalyst for the rearrangement of allylic esters and the cycloisomerization of γ-alkynoic acids. ChemCatChem 2016, 8, 2824–2831. [Google Scholar] [CrossRef]
- Belger, K.; Krause, N. Smaller, faster, better: Modular synthesis of unsymmetrical ammonium salt-tagged NHC-gold(I) complexes and their application as recyclable catalysts in water. Org. Biomol. Chem. 2015, 13, 8556–8560. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Álvarez, M.J.; Vidal, C.; Díez, J.; García-Álvarez, J. Introducing deep eutectic solvents as biorenewable media for Au(I)-catalysed cycloisomerisation of γ-alkynoic acids: An unprecedented catalytic system. Chem. Commun. 2014, 50, 12927–12929. [Google Scholar] [CrossRef] [PubMed]
- Mindt, T.L.; Schibli, R. Cu(I)-catalyzed intramolecular cyclization of alkynoic acids in aqueous media: A “click side reaction”. J. Org. Chem. 2007, 72, 10247–10250. [Google Scholar] [CrossRef] [PubMed]
- López-Reyes, M.E.; Toscano, R.A.; López-Cortés, J.G.; Alvarez-Toledano, C. Fast and efficient synthesis of Z-enol-γ-lactones through a cycloisomerization reaction of β-hydroxy-γ-alkynoic acids catalyzed by copper(I) under microwave heating in water. Asian J. Org. Chem. 2015, 4, 545–551. [Google Scholar] [CrossRef]
- Li, S.; Jia, W.; Jiao, N. Copper/iron-cocatalyzed highly selective tandem reactions: Efficient approaches to Z-γ-alkylidene lactones. Adv. Synth. Catal. 2009, 351, 569–575. [Google Scholar] [CrossRef]
- Jia, W.; Li, S.; Yu, M.; Chen, W.; Jiao, N. AgNO3 catalyzed cyclization of propargyl-Meldrum´s acids in aqueous solvent: Highly selective synthesis of Z-γ-alkylidene lactones. Tetrahedron Lett. 2009, 50, 5406–5408. [Google Scholar] [CrossRef]
- Ahmar, S.; Fillion, E. Expedient synthesis of complex γ-butyrolactones from 5-(1-arylalkylidene) Meldrum´s acids via sequential conjugate alkynylation/Ag(I)-catalyzed lactonization. Org. Lett. 2014, 16, 5748–5751. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xu, D.D.; Repič, O.; Blacklock, T.J. A mild method for ring-opening aminolysis of lactones. Tetrahedron Lett. 2001, 42, 2439–2441. [Google Scholar] [CrossRef]
- Guo, W.; Gómez, J.E.; Martínez-Rodríguez, L.; Bandeira, N.A.G.; Bo, C.; Kleij, A.W. Metal-free synthesis of N-aryl amides using organocatalytic ring-opening aminolysis of lactones. ChemSusChem 2017, 10, 1969–1975. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Campbell, L.; Dixon, D.J. A Au(I)-catalyzed N-acyl iminium ion cyclization cascade. J. Am. Chem. Soc. 2007, 129, 12070–12071. [Google Scholar] [CrossRef] [PubMed]
- Feng, E.; Zhuo, Y.; Zhang, D.; Zhang, L.; Sun, H.; Jiang, H.; Liu, H. Gold(I)-catalyzed tandem transformation: A simple approach for the synthesis of pyrrolo/pyrido[2,1-a][1,3]benzoxazinones and pyrrolo/pyrido[2,1-a]quinazolinones. J. Org. Chem. 2010, 75, 3274–3282. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.T.; Lakshmi, P.G.V.V.; Sridhar, B.; Patra, S.; Bhadra, M.P.; Patra, C.R. New linearly and angularly fused quinazolinones: Synthesis through gold(I)-catalyzed cascade reactions and anticancer activities. Eur. J. Org. Chem. 2012, 1790–1799. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Yang, N.; Chen, Y.; Zhou, Y.; Ji, X.; Zhang, L.; Wang, J.; Xie, X.; Liu, H. Gold(I)-catalyzed cascade approach for the synthesis of tryptamine-based polycyclic privileged scaffolds as α1-adrenergenic receptor antagonists. J. Org. Chem. 2013, 76, 10802–10811. [Google Scholar] [CrossRef] [PubMed]
- Feng, E.; Zhou, Y.; Zhao, F.; Chen, X.; Zheng, L.; Jiang, H.; Liu, H. Gold-catalyzed tandem reaction in water: An efficient and convenient synthesis of fused polycyclic indoles. Green Chem. 2012, 14, 1888–1895. [Google Scholar] [CrossRef]
- Zhuo, Y.; Zhai, Y.; Ji, X.; Liu, G.; Feng, E.; Ye, D.; Zhao, L.; Jiang, H.; Liu, H. Gold(I)-catalyzed one-pot tandem coupling/cyclization: An efficient synthesis of pyrrolo-/pyrido[2,1-b]benzo[d][1,3]oxazin-1-ones. Adv. Synth. Catal. 2010, 352, 373–378. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, H.-F. Synthesis of phthalides via Pd/CNTs-catalyzed reaction of terminal alkynes and o-iodobenzoic acid under copper- and ligand-free conditions. Tetrahedron Lett. 2007, 48, 8449–8452. [Google Scholar] [CrossRef]
- García-Álvarez, J.; Díez, J.; Gimeno, J. A highly efficient copper(I) catalyst for the 1,3-dipolar addition of azides with terminal and 1-iodoalkynes in water: Regioselective synthesis of 1,4-disubstituted and 1,4,5-trisubstituted 1,2,3-triazoles. Green Chem. 2010, 12, 2127–2130. [Google Scholar] [CrossRef]
- Denard, C.A.; Hartwig, J.F.; Zhao, H. Multistep one-pot reactions combining biocatalysts and chemical catalysts for asymmetric synthesis. ACS Catal. 2013, 3, 2856–2864. [Google Scholar] [CrossRef]
- Gröger, H.; Hummel, W. Combining the ‘two worlds’ of chemocatalysis and biocatalysis towards multi-step one-pot processes in aqueous media. Curr. Opin. Chem. Biol. 2014, 19, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.; Balskus, E.P. Opportunities for merging chemical and biological synthesis. Curr. Opin. Biotechnol. 2014, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Álvarez, M.J.; Ríos-Lombardía, N.; Schumacher, S.; Pérez-Iglesias, D.; Morís, F.; Cadierno, V.; García-Álvarez, J.; González-Sabín, J. Combination of metal-catalyzed cycloisomerizations and biocatalysis in aqueous media: Asymmetric construction of chiral alcohols, lactones and γ-hydroxy-carbonyl compounds. ACS Catal. 2017, 7, 7753–7759. [Google Scholar] [CrossRef]
- Cadierno, V.; Francos, J.; Gimeno, J. Ruthenium(IV)-catalyzed Markovnikov addition of carboxylic acids to terminal alkynes in aqueous medium. Organometallics 2011, 30, 852–862. [Google Scholar] [CrossRef]
- Chanda, A.; Fokin, V.V. Organic synthesis ”on water”. Chem. Rev. 2009, 109, 725–748. [Google Scholar] [CrossRef] [PubMed]
- Butler, R.N.; Coyne, A.G. Water: Nature’s reaction enforcer—Comparative effects for organic synthesis “in-water” and “on-water”. Chem. Rev. 2010, 110, 6302–6337. [Google Scholar] [CrossRef] [PubMed]
- Nicks, F.; Aznar, R.; Sainz, D.; Muller, G.; Demonceau, A. Novel, highly efficient and selective ruthenium catalysts for the synthesis of vinyl esters from carboxylic acids and alkynes. Eur. J. Org. Chem. 2009, 5020–5027. [Google Scholar] [CrossRef]
- Kleman, P.; González-Liste, P.J.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Highly enantioselective hydrogenation of 1-alkylvinyl benzoates: A simple, nonenzymatic access to chiral 2-alkanols. Chem. Eur. J. 2013, 19, 16209–16212. [Google Scholar] [CrossRef] [PubMed]
- Kleman, P.; González-Liste, P.J.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Asymmetric hydrogenation of 1-alkyl and 1-aryl vinyl benzoates: A broad scope procedure for the highly enantioselective synthesis of 1-substituted ethyl benzoates. ACS Catal. 2014, 4, 4398–4408. [Google Scholar] [CrossRef]
- González-Liste, P.J.; García-Garrido, S.E.; Cadierno, V. Gold(I)-catalyzed addition of carboxylic acids to internal alkynes in aqueous medium. Org. Biomol. Chem. 2017, 15, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Scheid, G.; Kuit, W.; Ruijter, E.; Orru, R.V.A.; Henke, E.; Bornscheuer, U.; Wessjohann, L.A. A new route to protected acyloins and their enzymatic resolution with lipases. Eur. J. Org. Chem. 2004, 1063–1074. [Google Scholar] [CrossRef]
- Carpino, P.A.; Griffith, D.A.; Sakya, S.; Dow, R.L.; Black, S.C.; Hadcock, J.R.; Iredale, P.A.; Scott, D.O.; Fichtner, M.W.; Rose, C.R.; et al. New bicyclic cannabinoid receptor-1 (CB1-R) antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-L.; Wu, Z.-W.; Song, S.-Y.; Liao, X.-D.; Zhu, Y.; Huang, Y.-S. An improved synthesis of nomegestrol acetate. Org. Process Res. Dev. 2014, 18, 431–436. [Google Scholar] [CrossRef]
- Kaila, N.; Janz, K.; DeBernardo, S.; Bedard, P.W.; Camphausen, R.T.; Tam, S.; Tsao, D.H.H.; Keith, J.C.; Nickerson-Nutter, C.; Shilling, A.; Young-Sciame, R.; Wang, Q. Synthesis and biological evaluation of quinoline salicylic acids as P-Selectin antagonists. J. Med. Chem. 2007, 50, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.F.M.; Radics, G.; Windle, H.; Serra, H.O.; Simplício, A.L.; Kedziora, K.; Fallon, P.G.; Kelleher, D.P.; Gilmer, J.F. Design, synthesis, and pharmacological effects of a cyclization-activated steroid prodrug for colon targeting in inflammatory bowel disease. J. Med. Chem. 2009, 52, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, J.; Korb, M.; Rüffer, T.; Gäbler, C.; Lang, H. Atom economic ruthenium-catalyzed synthesis of bulky β-oxo esters. Adv. Synth. Catal. 2015, 357, 4069–4081, and references therein. [Google Scholar] [CrossRef]
- Mitsudo, T.; Hori, Y.; Yamakawa, Y.; Watanabe, Y. Ruthenium-catalyzed selective addition of carboxylic acids to alkynes. A new synthesis of enol esters. J. Org. Chem. 1987, 52, 2230–2239. [Google Scholar] [CrossRef]
- Bauer, E. Transition-metal-catalyzed functionalization of propargylic alcohols and their derivatives. Synthesis 2012, 44, 1131–1151. [Google Scholar] [CrossRef]
- Cadierno, V.; Francos, J.; Gimeno, J. Ruthenium-catalyzed synthesis of β-oxo esters in aqueous medium: Scope and limitations. Green Chem. 2010, 12, 135–143. [Google Scholar] [CrossRef]
Scheme 1.
The catalytic cycloisomerization of alkynoic acids.
Scheme 2.
The intermolecular addition of carboxylic acids to alkynes.

Scheme 3.
Cyclosiomerization of dipropargylmalonic acid 2 in water using a cluster catalyst.
Scheme 4.
Cycloisomerization of different γ-alkynoic acids in water catalyzed by a dinuclear Pd(II) complex.
Scheme 4.
Cycloisomerization of different γ-alkynoic acids in water catalyzed by a dinuclear Pd(II) complex.
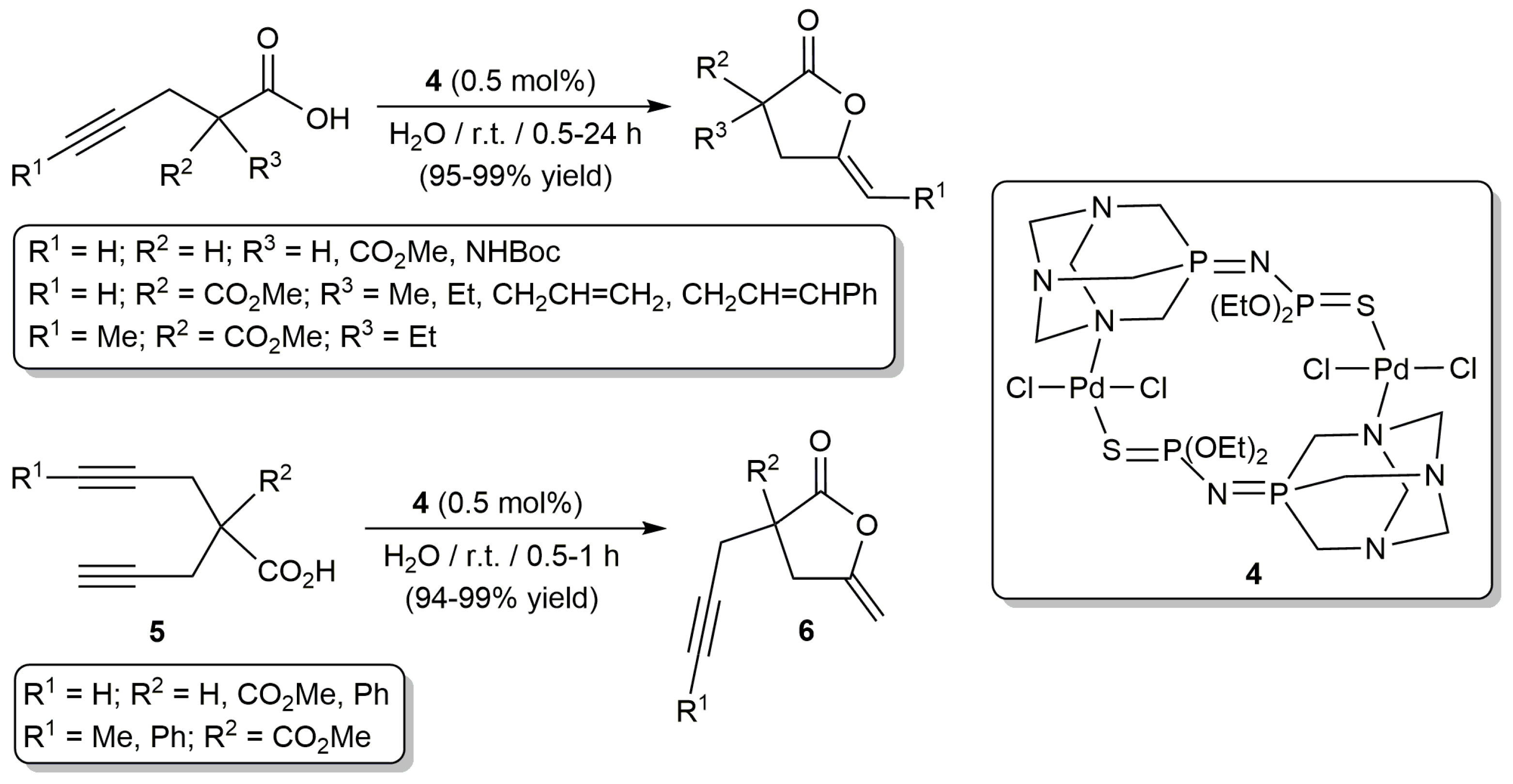
Figure 1.
Structure of the pincer palladium(II) complexes 7 and 8.
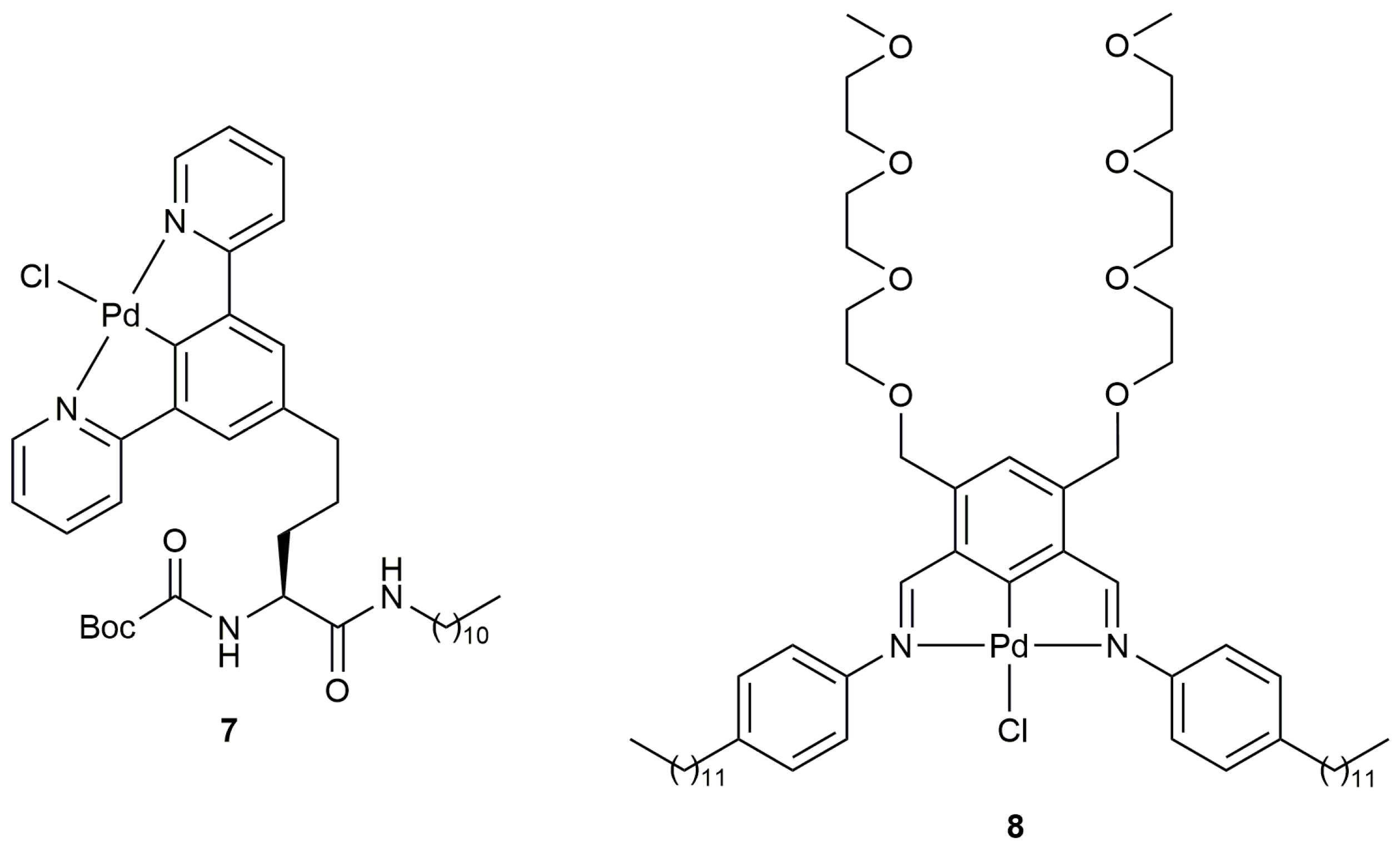
Figure 2.
Structure of the platinum complexes 9a–o.
Scheme 5.
Cyclization of different alkynoic acids in water using Pt(II) complexes.
Scheme 6.
Cyclosiomerization of different γ-alkynoic acids catalyzed by the Au(III)-N-heterocyclic carbenes (NHC) complex 10b.
Scheme 6.
Cyclosiomerization of different γ-alkynoic acids catalyzed by the Au(III)-N-heterocyclic carbenes (NHC) complex 10b.
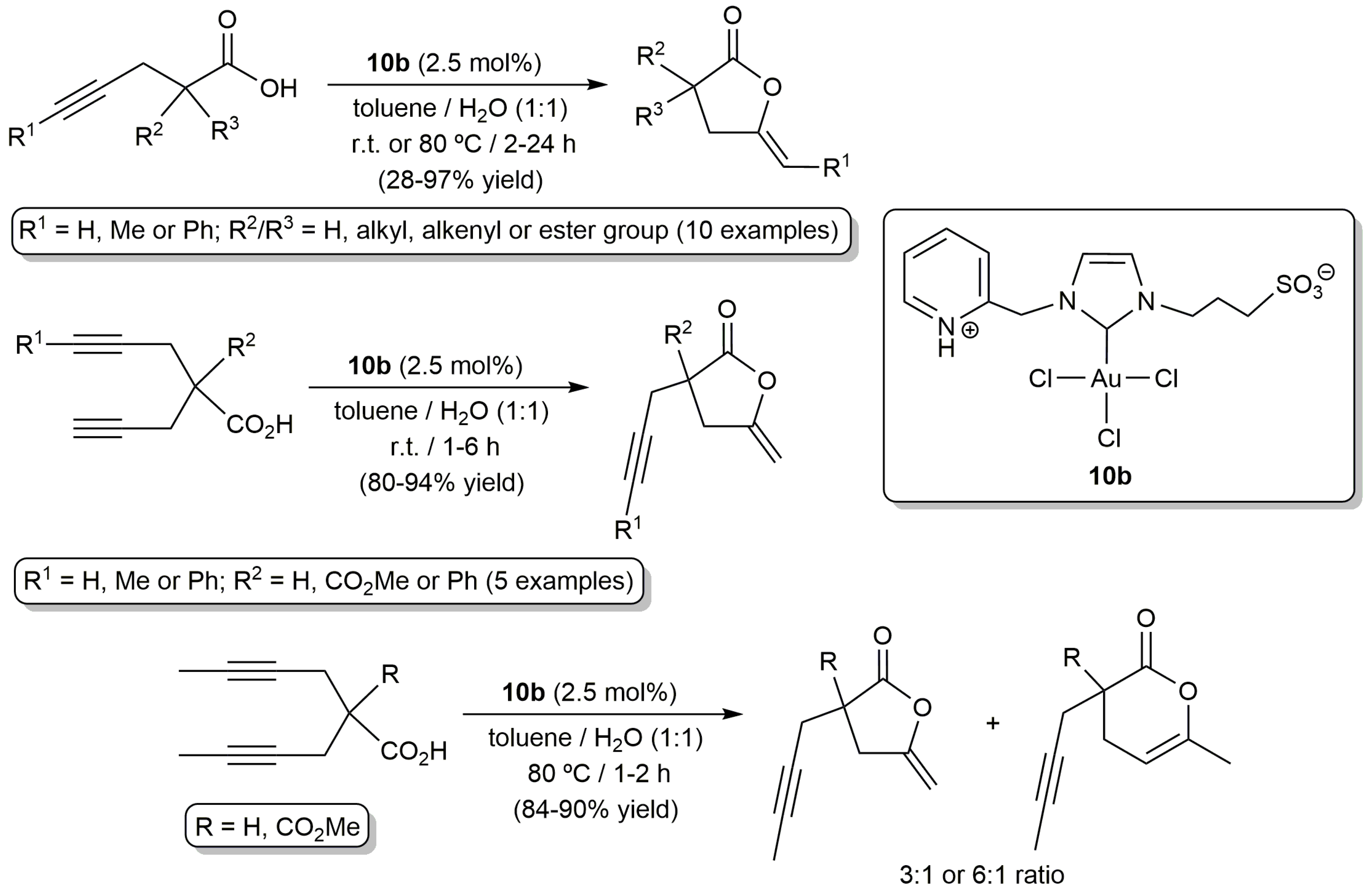
Figure 3.
Structure of the zwitterionic Au(III)- and Au(I)-NHC complexes 10,11a–c.
Scheme 7.
Cyclosiomerization of γ-alkynoic acids using a sol-gel immobilized Au(I)-NHC complex.

Figure 4.
Structure of the water-soluble gold(I)-NHC complexes 13a–f.
Scheme 8.
Synthesis of 2-epi-clausemarine A.
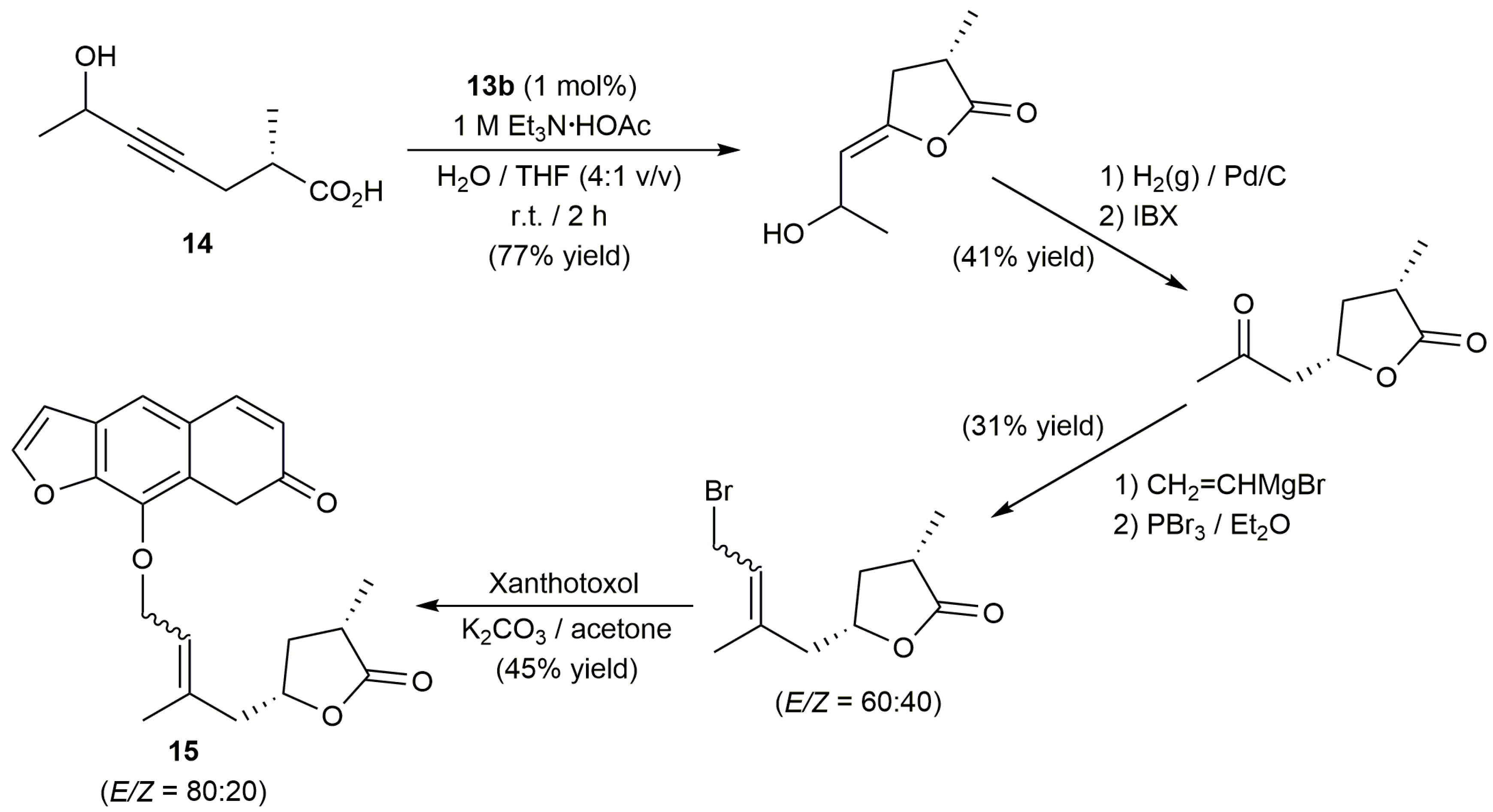
Figure 5.
Structure of the iminophosphorane-gold(I) complex 16.
Scheme 9.
CuBr-catalyzed transformations of different γ-alkynoic acids in aqueous medium.
Scheme 10.
Cu(I)-catalyzed cyclization of β-hydroxy-γ-alkynoic acids in water under microwave (MW) irradiation.
Scheme 10.
Cu(I)-catalyzed cyclization of β-hydroxy-γ-alkynoic acids in water under microwave (MW) irradiation.
Scheme 11.
Cu/Fe-cocatalyzed synthesis of lactones from propargylic Meldrum’s acids.
Scheme 12.
Silver-catalyzed cyclization of the propargylic Meldrum’s acids.
Scheme 13.
Gold-catalyzed synthesis of fused polycyclic indoles in water.
Scheme 14.
Gold-catalyzed synthesis of the pyrrolo[2,1-b]benzo[d][1,3]oxazin-1-one 24 in water.
Scheme 15.
Catalytic synthesis of phthalides from terminal alkynes and o-iodobenzoic acid.
Scheme 16.
Access to bicyclic triazol-enol-lactones from of bispropargylic carboxylic acids and azides.
Scheme 16.
Access to bicyclic triazol-enol-lactones from of bispropargylic carboxylic acids and azides.
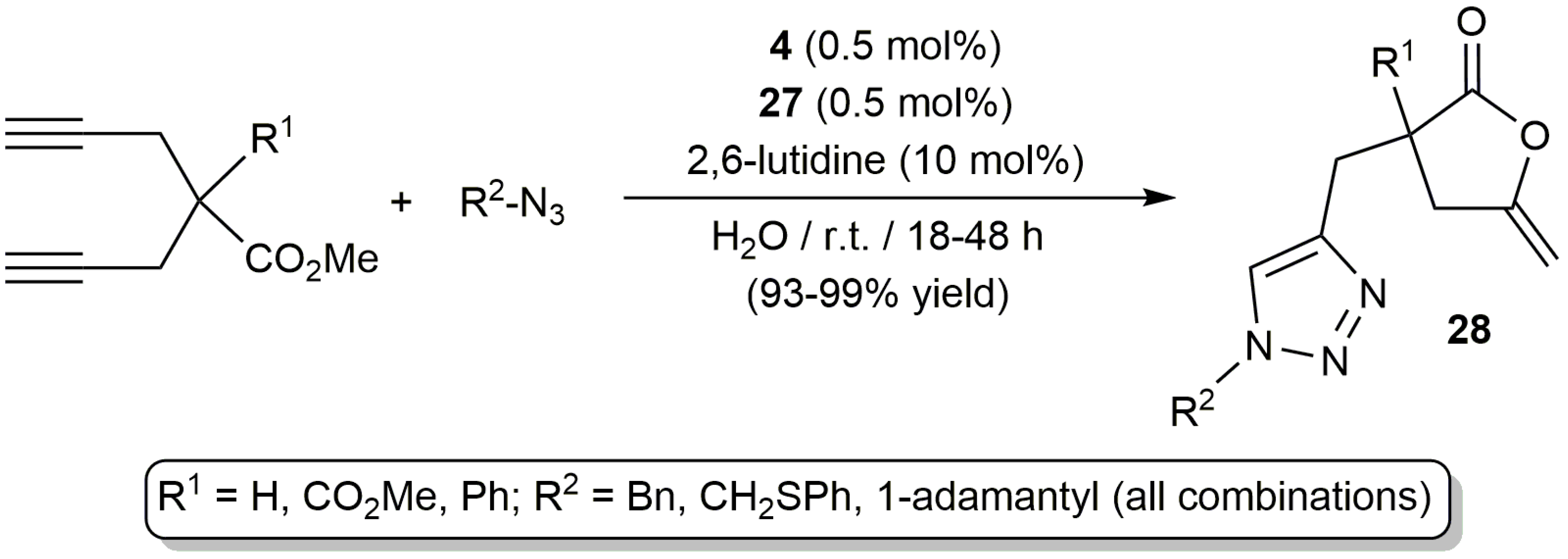
Scheme 17.
Cu/Fe-cocatalyzed synthesis of lactones from alkynes and 5-alkylidene-Meldrum’s acids.
Scheme 18.
Chemoenzymatic one-pot conversion of 4-pentynoic acid into enantiopure γ-hydroxyvaleric acid.
Scheme 18.
Chemoenzymatic one-pot conversion of 4-pentynoic acid into enantiopure γ-hydroxyvaleric acid.

Figure 6.
Structure of the bis(allyl)-ruthenium(IV) complexes 31a–s.
Scheme 19.
Ruthenium(IV)-catalyzed Markovnikov addition of carboxylic acids to terminal alkynes.
Scheme 20.
Ruthenium(IV)-catalyzed Markovnikov addition of benzoic acid to terminal diynes.
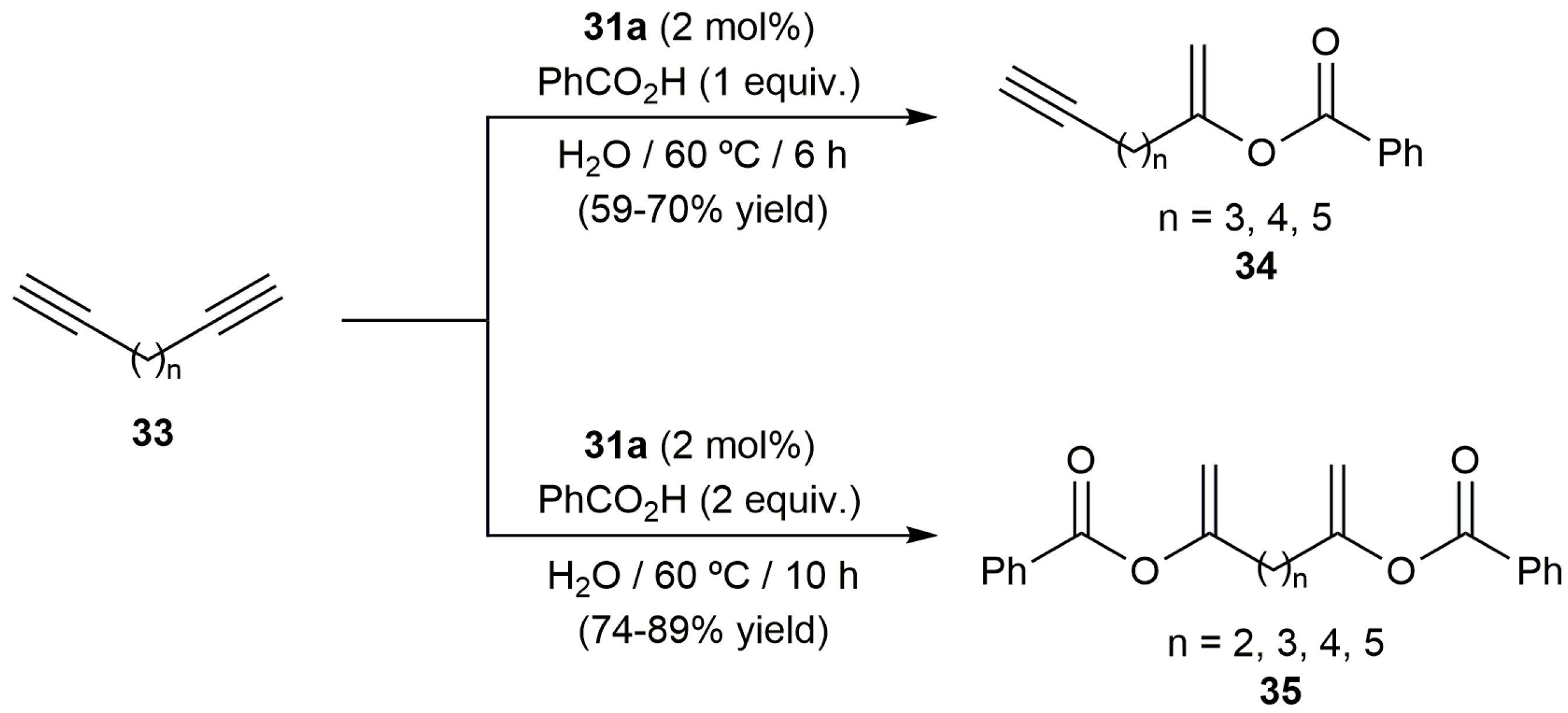
Scheme 21.
Gold-catalyzed addition of carboxylic acids to symmetrically substituted internal alkynes.
Scheme 21.
Gold-catalyzed addition of carboxylic acids to symmetrically substituted internal alkynes.
Scheme 22.
Regio- and stereoselective addition of benzoic acid to unsymmetrically substituted alkynes.
Scheme 22.
Regio- and stereoselective addition of benzoic acid to unsymmetrically substituted alkynes.
Scheme 23.
Mechanism of the Ru-catalyzed β-oxo esters formation reactions.
Scheme 24.
Synthesis of β-oxo esters in water catalyzed by the ruthenium(IV) complex 31a.
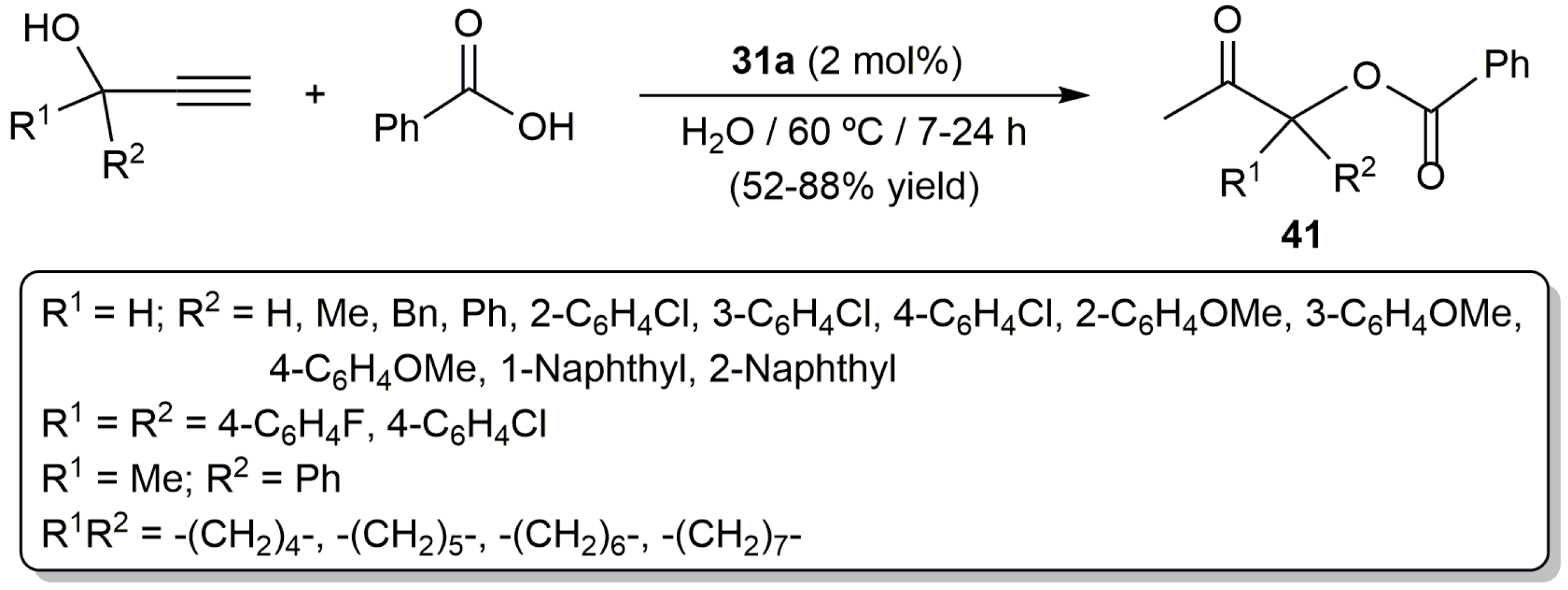
Figure 7.
Structure of the water-soluble arene-ruthenium(II) complexes 42–45a–d.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Francos, J.; Cadierno, V. Metal-Catalyzed Intra- and Intermolecular Addition of Carboxylic Acids to Alkynes in Aqueous Media: A Review. Catalysts 2017, 7, 328. https://doi.org/10.3390/catal7110328
AMA Style
Francos J, Cadierno V. Metal-Catalyzed Intra- and Intermolecular Addition of Carboxylic Acids to Alkynes in Aqueous Media: A Review. Catalysts. 2017; 7(11):328. https://doi.org/10.3390/catal7110328
Chicago/Turabian StyleFrancos, Javier, and Victorio Cadierno. 2017. "Metal-Catalyzed Intra- and Intermolecular Addition of Carboxylic Acids to Alkynes in Aqueous Media: A Review" Catalysts 7, no. 11: 328. https://doi.org/10.3390/catal7110328
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.