Catalytic Deoxygenation of Hexadecyl Palmitate as a Model Compound of Euglena Oil in H2 and N2 Atmospheres


National Institute of Advanced Industrial Science and Technology, AIST Tsukuba West, 16-1 Onogawa, Tsukuba, Ibaraki 305-8569, Japan
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(11), 333; https://doi.org/10.3390/catal7110333
Submission received: 15 October 2017
/
Revised: 3 November 2017
/
Accepted: 5 November 2017
/
Published: 9 November 2017
Abstract
:Hexadecyl palmitate (C15H31COOC16H33, used as a model compound for Euglena oil) was deoxygenated to hydrocarbons over various solid catalysts in autoclave reactors. In a H2 atmosphere, 1 wt.% of Pd/Mg(Al)O catalyst, derived from a hydrotalcite precursor, yielded a C15H31COOC16H33 conversion close to 100%, and a C10‒C16 (aviation fuel range) hydrocarbon yield of 90.2% for the deoxygenation of C15H31COOC16H33 at 300 °C for 2 h. In a N2 atmosphere, 1 wt.% of Pd/Mg(Al)O catalyst yielded a C10‒C16 hydrocarbon yield of 63.5%, which was much higher than those obtained with Mg(Al)O (15.1%), H-ZSM-5 (8.3%), and 1 wt.% Pd/C (26.2%) for the deoxygenation of C15H31COOC16H33 at 300 °C for 2 h. The Pd metal site and the solid base site in Mg(Al)O had a synergetic effect on the deoxygenation of C15H31COOC16H33 in N2 atmosphere over the Pd/Mg(Al)O catalyst. By prolonging the reaction time to 5 h for reaction at 300 °C in N2 atmosphere, the yield of C10‒C16 hydrocarbons increased to 80.4% with a C15H31COOC16H33 conversion of 99.1% over the 1 wt.% Pd/Mg(Al)O catalyst.
1. Introduction
The production of chemicals and transport fuels from renewable biomass is a sustainable and environment-friendly way to reduce the world’s dependence on crude oil. Starch and cellulose in wood can be converted to ethanol by yeast fermentation [1]. Lignin in wood and organic wastes can be converted to motor fuels by a biomass-to-liquid (BTL) process using a syngas platform [2,3]. Vegetable oils can be converted to fatty acid methyl esters (FAME, known as biodiesel) by transesterification with methanol [4]. Recently, the hydrotreatment of vegetable oils to produce hydrocarbon ‘drop-in’ fuels has attracted global research interest [5,6,7].
Algae oils have recently become a promising alternative feedstock for vegetable oils because the oil yields from algae are significantly higher than those from any other crop [8,9,10,11]. Euglena is a promising algae biomass because it is easily cultivated on a large scale. Most algae produce oils composed of triglycerides and fatty acids [8]. However, Euglena produces an oil wax that contains a mixture of saturated esters with long carbon chains (CmH2m+1COOCnH2n+1, m = 11–15, n = 12–16) [12,13]. While some studies have been reported on the deoxygenation of triglycerides and fatty acids (vegetable oils and algae oils), very few studies have reported on the deoxygenation of fatty acid esters to date [14].
Metal (NiMo, Pt, etc.)-supported catalysts possess high activities for the deoxygenation of triglycerides and fatty acids in H2 atmosphere [15,16,17]. However, the deoxygenation of triglycerides in N2 atmosphere (without H2) is an important subject, because H2 is expensive and may be difficult to obtain in areas of vegetable and algae cultivation. Solid acids and bases have been reported as catalysts for the deoxygenation of triglycerides in N2 atmosphere [18,19,20]. Solid acid catalysts possess relatively high activities; however, hydrocarbons with long carbon chains (heavy hydrocarbons) crack on the acid sites [18]. This is unfortunate, because, in general, heavy hydrocarbons have a high value as chemicals and transport fuels. In contrast, the activities of solid base catalysts are relatively low; moreover, they exhibit relatively high selectivity for heavy hydrocarbons [19,20]. Pd supported on active carbon (Pd/C) has been reported as an effective catalyst for the catalytic deoxygenation of fatty acids and their esters in He atmosphere [14,21].
In this study, we chose hexadecyl palmitate (C15H31COOC16H33) as a model compound of Euglena oil. We developed a highly active Pd/Mg(Al)O catalyst (derived from Pd-containing MgAl-type hydrotalcite) for the deoxygenation of C15H31COOC16H33 to hydrocarbons in H2 and N2 atmospheres.
2. Results and Discussion
2.1. Catalyst Characterization
Figure 1 shows a structural model of the Pd0.016Mg3Al(OH)16CO3∙xH2O hydrotalcite used in this study. Hydrotalcite is a kind of anion clay with the general formula [M2+1−mM3+m (OH)2]m+An−m/n·xH2O (M2+, M3+ = metal cations, An− = interlayer anion) [22]. The metal cations are distributed in brucite-like layers, and the interlayer anions are fixed between the brucite-like layers by electric charge interaction. The M2+ cation is usually Mg2+ and the M3+ cation is usually Al3+. The noble metal Pd2+ cation can be introduced into the M2+ position when Pd2+/(M2+ + M3+) < 5 mol % [23,24,25]. Basic MgAl-type hydrotalcite is the most common hydrotalcite, and has been used as an effective catalyst for some industrially important reactions [22,23,24,25,26,27,28,29].
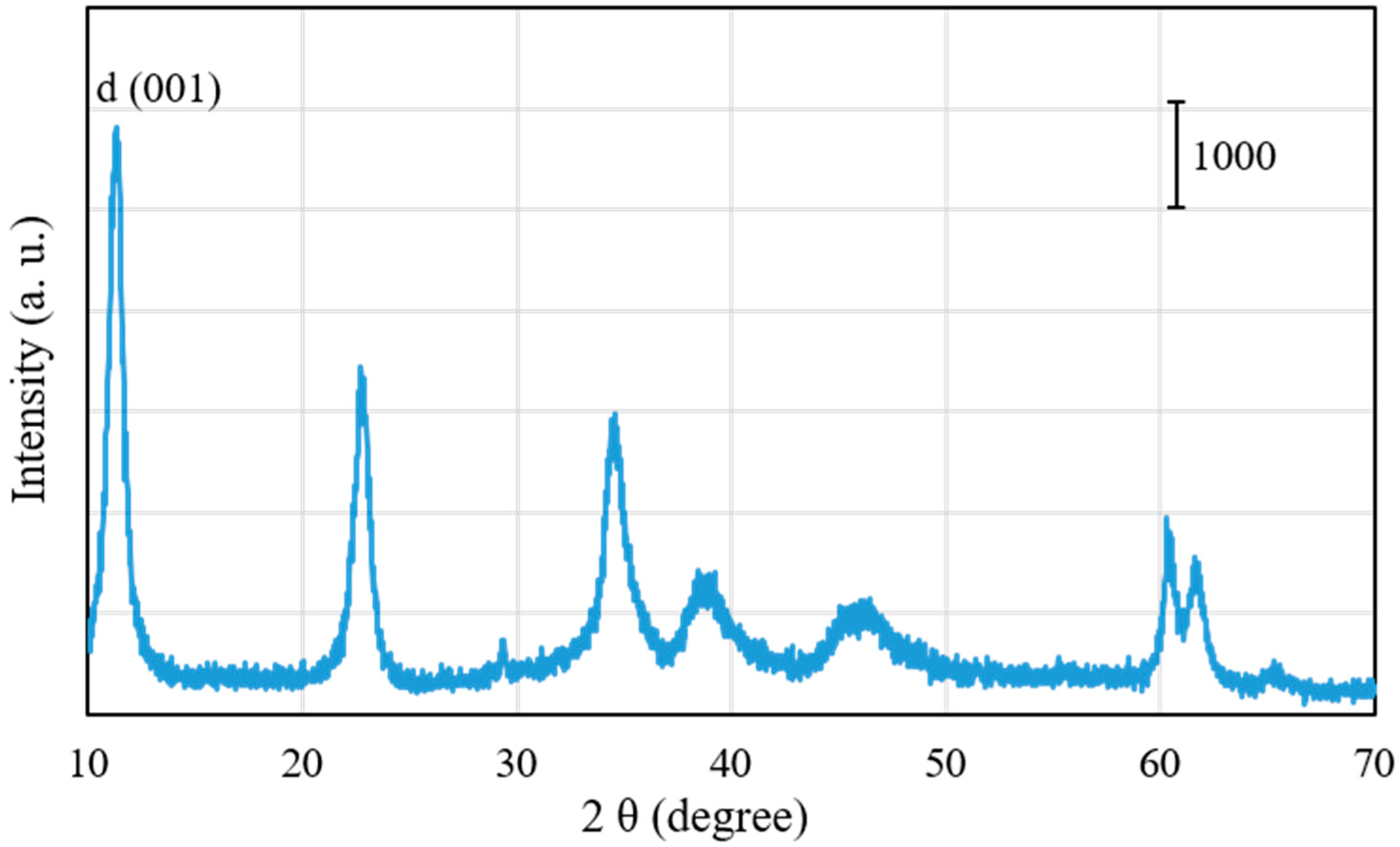
Figure 2 shows X-ray diffraction (XRD) patterns of Pd0.016Mg3Al(OH)16CO3∙xH2O after drying at 100 °C. By using a JICST database (from The Crystallographic Society of Japan) for identification, only the well-crystallized Mg3Al(OH)16CO3 phase was observed in the XRD pattern of Pd0.016Mg3Al(OH)16CO3∙xH2O. The peaks of Pd(OH)2 and PdO phases could not be observed in the XRD pattern, probably because they formed amorphous phases. It is also possible that the Pd2+ ions entered the Mg2+ position in Pd0.016Mg3Al(OH)16CO3∙xH2O at a low Pd content [25]. The d (001) (basal d) spacing at the lowest angle in the XRD pattern was 9.0 Å for Pd0.016Mg3Al(OH)16CO3∙xH2O. As shown in Figure 1, the thickness of the MgAl-type brucite-like layer was 4.7 Å [22]. The gallery height was 4.3 Å in Pd0.016Mg3Al(OH)16CO3∙xH2O after subtracting the thickness of the MgAl-type hydrotalcite layer (4.7 Å) from the basal d spacing (9.0 Å). This gallery height (4.3 Å) coincides with the size of the CO32− anion [22].
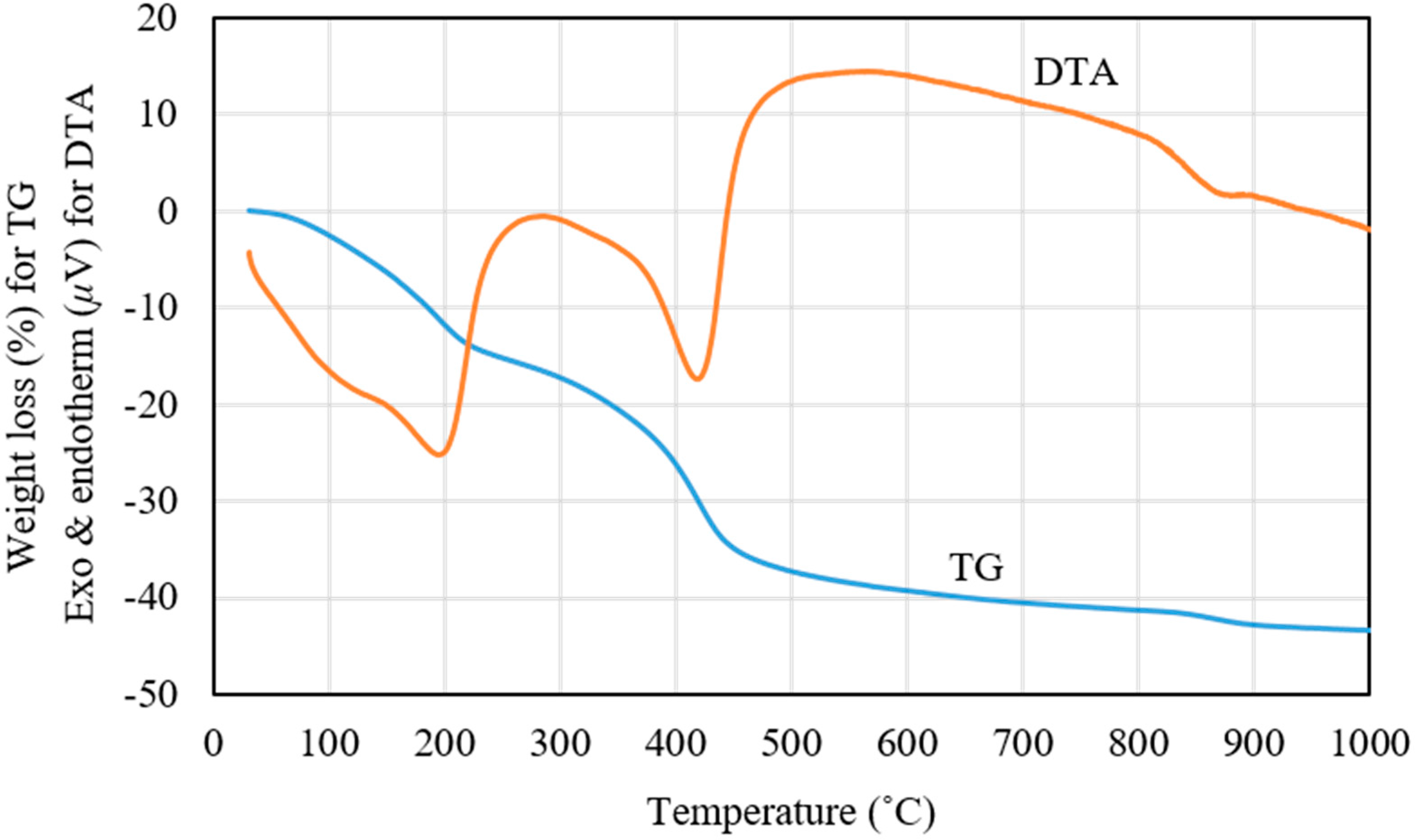
Figure 3 shows the thermogravimetric analysis (TGA) and differential thermal analysis (DTA) results for Pd0.016Mg3Al(OH)16CO3∙xH2O after drying at 100 °C. Three stages of sudden weight loss can be observed in the TGA curve. The first sudden weight loss, which appears at below 200 °C, was attributed to the loss of physically absorbed water and interlayer water. The second sudden weight loss, at 220–450 °C, was attributed to the removal of CO2 from the interlayer carbonate anions and hydroxyl groups from the brucite-like layers. The third sudden weight loss, at 850–890 °C, can be ascribed to the formation of the spinel-like phase of MgAl2O4 [20,24]. The results of DTA and TGA nearly coincide. The endotherm band at 130 °C in the DTA curve corresponds to the loss of physically absorbed water. After losing physically absorbed water, Pd0.016Mg3Al(OH)16CO3∙xH2O was converted to Pd0.016Mg3Al(OH)16CO3∙4H2O. The endotherm band at 190 °C in the DTA curve corresponds to the loss of interlayer water. After losing interlayer water, Pd0.016Mg3Al(OH)16CO3∙4H2O was converted to Pd0.016Mg3Al(OH)16CO3. The endotherm band at 420 °C in the DTA curve corresponds to the loss of interlayer carbonate anions and hydroxyl groups in the brucite-like layers. The layered structure of hydrotalcite was destroyed upon calcination at 420 °C, following which Pd0.016Mg3Al(OH)16CO3 was converted to a metal–oxide mixture Pd0.016Mg3AlOx, in which Pd2+ and Al3+ ions entered the MgO cubic lattices [22]. The small endotherm band at 860 °C in the DTA curve corresponds to the solid phase change of the metal–oxide mixture to a spinel-like phase of MgAl2O4 and crystalline MgO [22].
Figure 4 shows the dependence of the Brunauer-Emmett-Teller (BET) surface area of Pd0.016Mg3Al(OH)16CO3∙xH2O on calcination temperature from 100 °C to 1000 °C. Calcination was carried out at each temperature for 3 h in air. Pd0.016Mg3Al(OH)16CO3∙xH2O showed a BET surface area of 73 m2/g after calcination at 100 °C. The BET surface area increased with an increasing calcination temperature from 100 °C to 450 °C, and then decreased with increasing calcination temperature from 450 °C to 1000 °C. The layered structure of Pd0.016Mg3Al(OH)16CO3 hydrotalcite was destroyed, and a PdO-Mg(Al)O metal-oxide mixture was formed upon calcination at 450 °C. In the process of destruction, the removal of CO2 (from the interlayer carbonate anions) and hydroxyl groups (from the brucite-like layers) created pores in the PdO-Mg(Al)O metal-oxide mixture formed. These pores increased the BET surface area. Calcination at a temperature that was higher than 450 °C caused sintering of the metal-oxide mixture particles, which decreased the BET surface area. Moreover, an obvious decrease in the BET surface area was observed at 800–850 °C. The formation of spinel-like phases of MgAl2O4 from the PdO-Mg(Al)O metal-oxide mixture resulted in a decrease in the BET surface area upon calcination at 800–850 °C [25]. Because the highest BET surface area (143 m2/g) was obtained for calcination at 450 °C, we calcined Pd0.016Mg3Al(OH)16CO3∙xH2O at 450 °C for 3 h in air before use in this study.
The use of uniform multicomponent precursors results in well-dispersed metal particles on the surface of supports after calcination and reduction. This method, known as “solid phase crystallization” (SPC), is important in the preparation of highly active metal-supported catalysts [27,28,29,30,31,32,33,34,35]. In this study, Pd2+ could enter the Mg2+ position in Pd0.016Mg3Al(OH)16CO3∙xH2O hydrotalcite after drying at 100 °C in air. As a result, a well-distributed mixed oxide, PdO-Mg(Al)O, could be formed after calcining Pd0.016Mg3Al(OH)16CO3∙xH2O at 450 °C in air. Finally, a Pd/Mg(Al)O catalyst with highly dispersed Pd metal particles and strong interaction between the metal and support could be obtained after the pretreatment process of H2 flow reduction.
2.2. Catalytic Deoxygenation of C15H31COOC16H33 in H2 Atmosphere
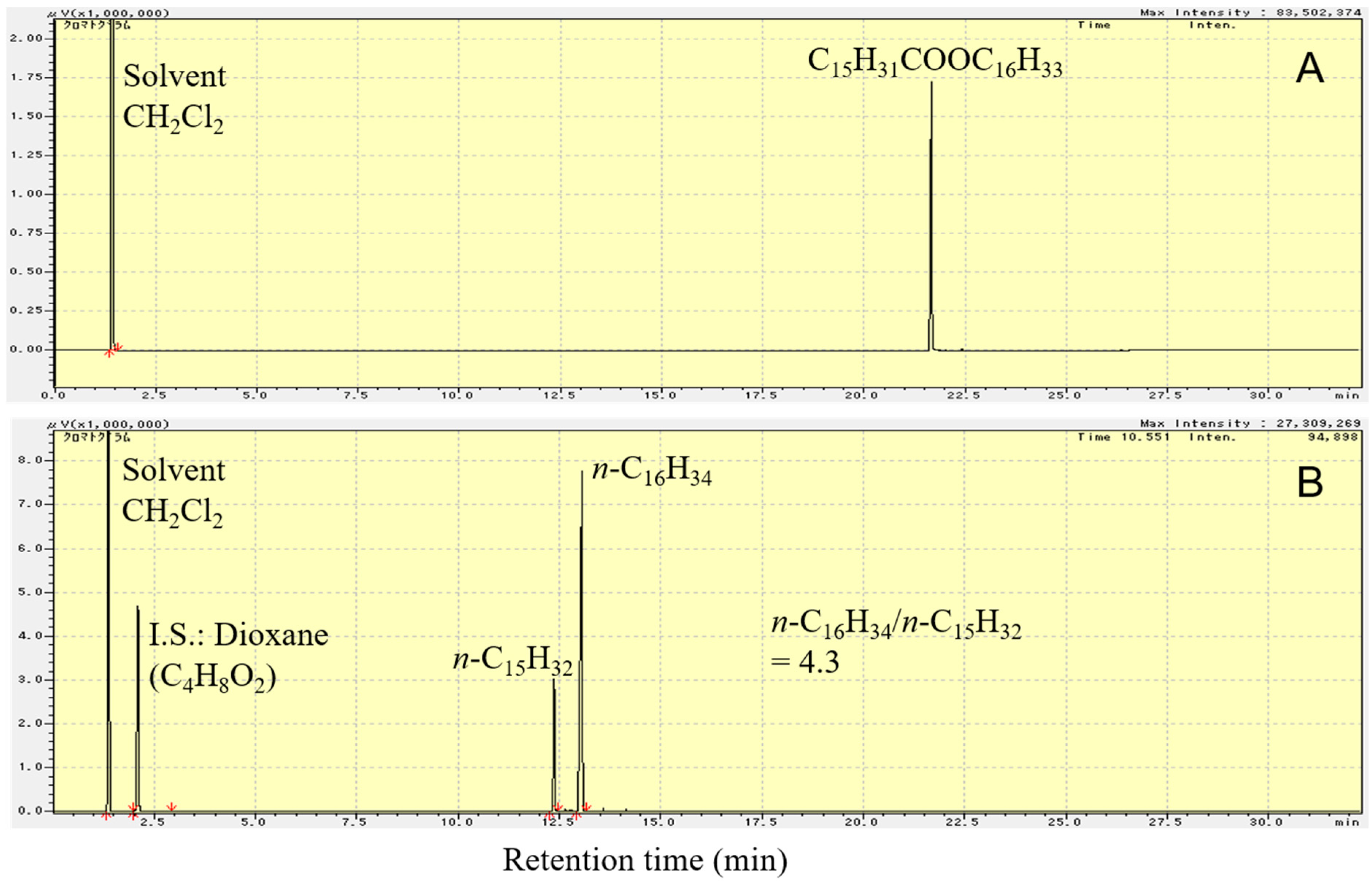
Figure 5 shows the flame ionization detector–gas chromatography (FID-GC) charts of C15H31COOC16H33 and the liquid product formed upon deoxygenation of C15H31COOC16H33 in H2 atmosphere, at 300 °C, for 2 h, over 1 wt.% Pd/Mg(Al)O catalyst. As shown in Figure 5B, the reactant (C15H31COOC16H33) peak is not observed in the GC chart after reaction at 300 °C for 2 h. The liquid products were almost saturated hydrocarbons n-C16H34 and n-C15H32. By calculating the amount of each component in the product using the GC chart, the molar ratio of n-C16H34 to n-C15H32 in the liquid product was found to be 4.3.
As shown in Reactions (1)–(3), deoxygenation of saturated fatty acids (such as stearic acid C17H35COOH) involves three parallel reactions: reduction, decarbonylation, and decarboxylation [7]. For a fatty acid having an even number of carbons, reduction produces a normal paraffin having an even number of carbons plus water; decarbonylation produces a normal paraffin having an odd number of carbons plus water and CO; and, decarboxylation produces a normal paraffin having an odd number of carbons plus CO2.
C17H35COOH + 3H2 = C18H38 + 2H2O Reduction
C17H35COOH + H2 = C17H36 + CO + H2O Decarbonylation
C17H35COOH = C17H36 + CO2 Decarboxylation
Based on Reactions (1)–(3), we believe that the deoxygenation of C15H31COOC16H33 in H2 atmosphere over 1 wt.% Pd/Mg(Al)O also involves three parallel reactions: reduction (Reaction (4)), decarbonylation (Reaction (5)), and decarboxylation (Reaction (6)).
C15H31COOC16H33 + 4H2 = 2C16H34 + 2H2O Reduction
C15H31COOC16H33 + 2H2 = C15H32 + C16H34 + CO + H2O Decarbonylation
C15H31COOC16H33 + H2 = C15H32 + C16H34 + CO2 Decarboxylation
As shown in Reactions (4)–(6), the reduction of one C15H31COOC16H33 molecule produces two C16H34 molecules. On the other hand, either decarbonylation or decarboxylation of one C15H31COOC16H33 molecule produces one C16H34 molecule and one C15H32 molecule. The decarbonylation of C15H31COOC16H33 produces CO, whereas the decarboxylation of C15H31COOC16H33 produces CO2. Both CO and CO2 were detected in the gas product from the reaction over 1 wt.% Pd/Mg(Al)O catalyst, indicating that both decarbonylation of C15H31COOC16H33 (Reaction (5)) and decarboxylation of C15H31COOC16H33 (Reaction (6)) occurred during the reaction. Moreover, among Reactions (4)–(6), the reduction of C15H31COOC16H33 (Reaction (4)) was the main reaction, because the quantity of n-C16H34 in the liquid product was much more than that of n-C15H32 (Figure 5B).
For deoxygenation of C15H31COOC16H33 in H2 atmosphere, the metal site alone could achieve a high activity, because deoxygenation was carried out via three routes: reduction, decarbonylation, and decarboxylation. We used an acid support for the metal in order to adjust the selectivity of the hydrocarbon products during deoxygenation in H2 atmosphere [6,7]. In this study, we used a base support (Mg(Al)O) for the metal (Pd) in order to suppress the hydrocracking of n-C15H32 and n-C16H34 products (to light hydrocarbons) during deoxygenation of C15H31COOC16H33 in H2 atmosphere.
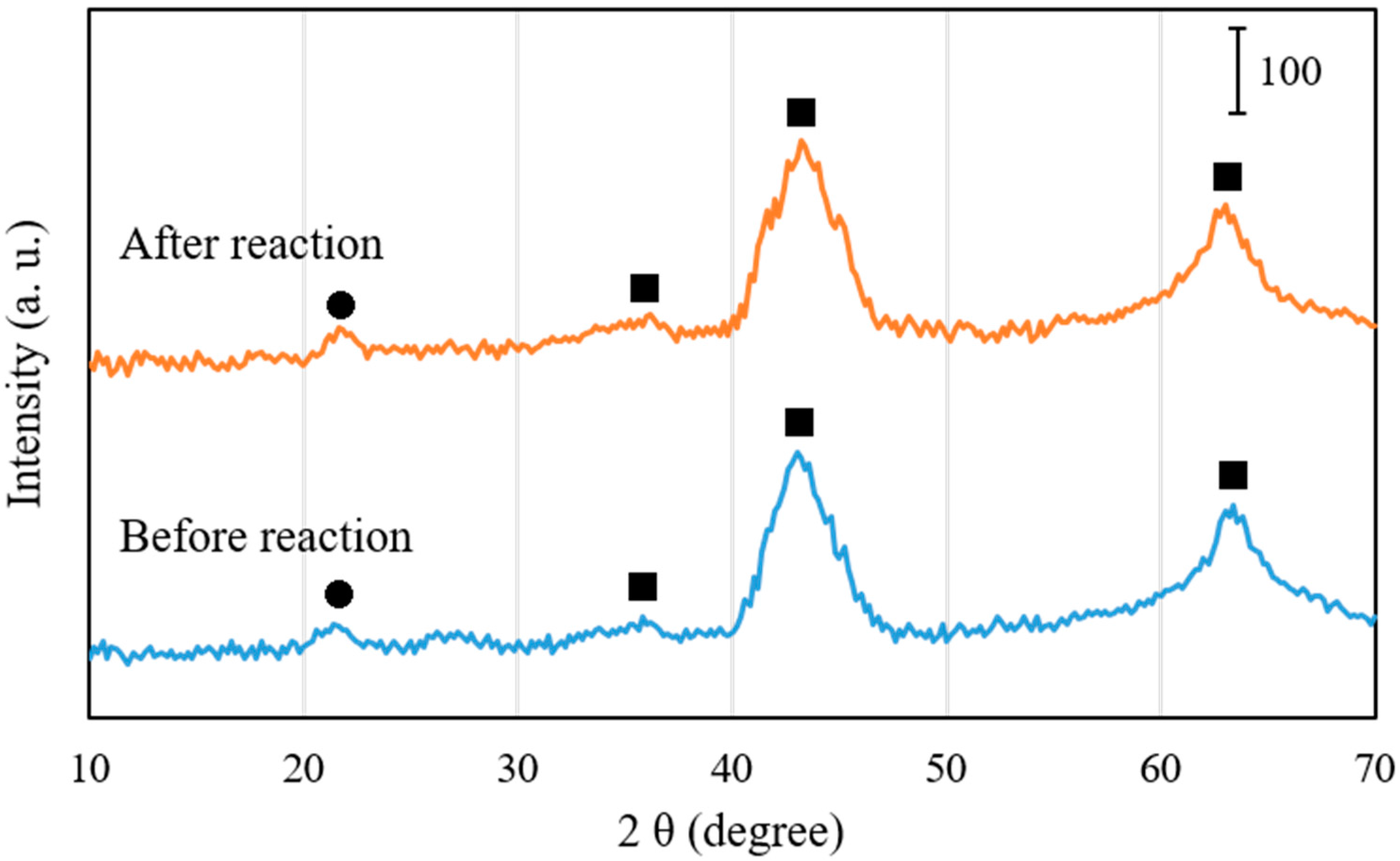
Figure 6 shows X-ray diffraction (XRD) patterns of 1 wt.% Pd/Mg(Al)O catalyst before reaction and after reaction (for deoxygenation of C15H31COOC16H33 in H2 atmosphere at 300 °C for 2 h). The sample before reaction was obtained by calcining Pd0.016Mg3Al(OH)16CO3∙xH2O in air at 450 °C for 3 h, and then reducing in H2 at 300 °C for 1 h. The XRD pattern of the sample before reaction proved the formation of mixed Mg-Al oxides phases, indicating that the decomposition of the layered structure occurred after calcination and reduction. The reflections in XRD pattern at about 43° and 63° corresponded to MgO-like phase (periclase), or rather MgO-Al2O3 solid solution Mg(Al)O [22]. The reflection of Al2O3 phase at 23° was very small, indicating that Al3+ cations were dispersed in the structure of MgO without the formation of spinel species. Because the Pd loading was low (1 wt.%) in the Pd/Mg(Al)O catalyst, the metal Pd crystals certainly formed in very small sizes, and thus the metal Pd phase could not be detected in the XRD pattern. The sample after reaction was obtained by filtrating out liquid products, and then drying in vacuum at room temperature for 10 h. From a comparison of the XRD pattern of Pd/Mg(Al)O catalyst before reaction, no obvious change could be observed in the XRD patterns of Pd/Mg(Al)O catalyst after reaction in H2 atmosphere at 300 °C for 2 h.
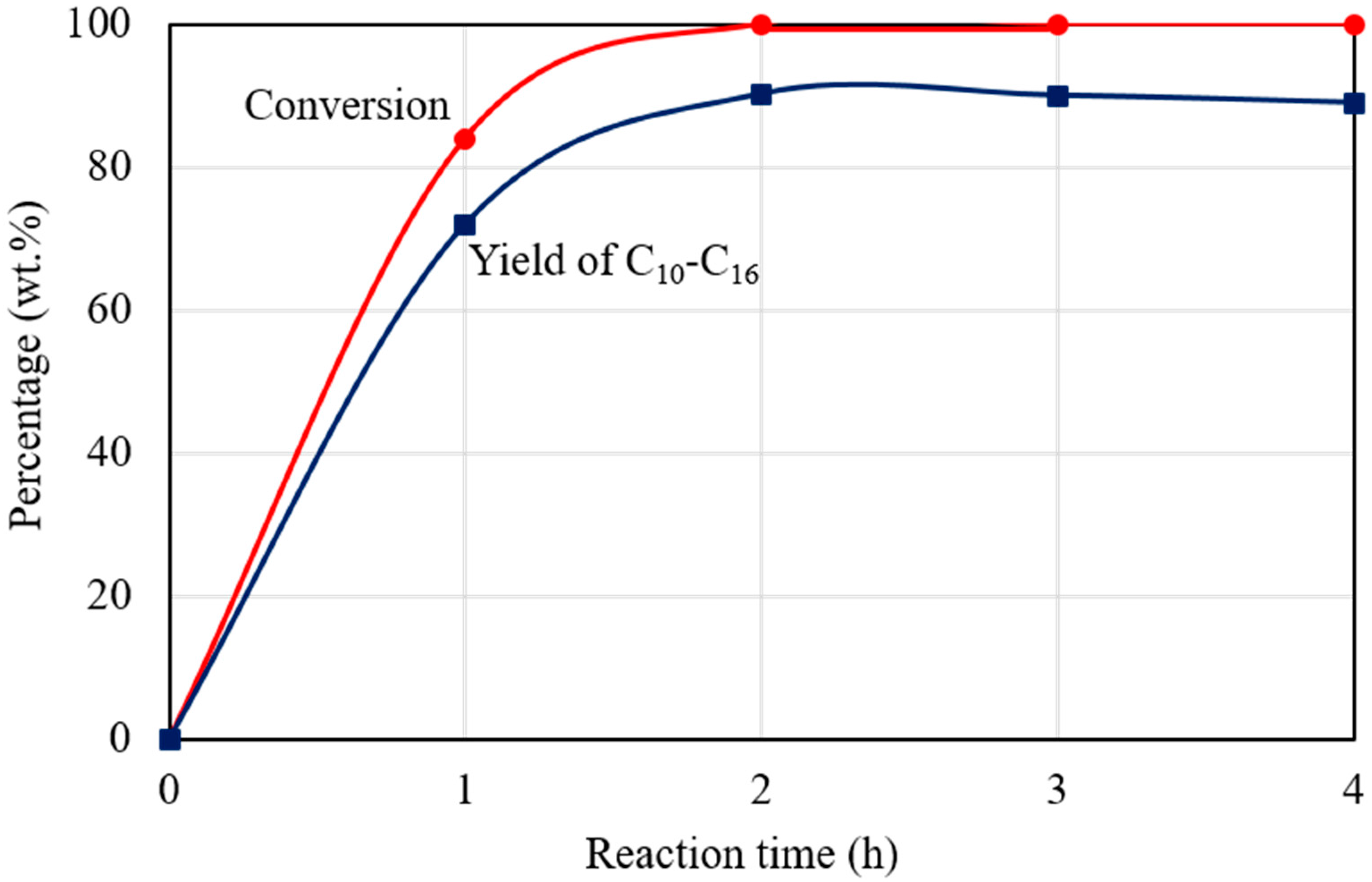
Figure 7 shows the dependence of C15H31COOC16H33 conversion and C10‒C16 hydrocarbon yield on the reaction time for the catalytic deoxygenation of C15H31COOC16H33 in H2 atmosphere, at 300 °C, over 1 wt.% Pd/Mg(Al)O catalyst. Four autoclave reactors were charged with the same reactant gases and the same amount of catalyst, and then operated at the same temperature (300 °C). The reaction in each autoclave reactor was finished in an hour. The products were analysed to determine the C15H31COOC16H33 conversion and the C10‒C16 hydrocarbon yield at that reaction time. The yield of C10‒C16 hydrocarbons is important as they can be used as aviation fuel [16]. As shown in Figure 7, C15H31COOC16H33 conversion increased with the reaction time, and approached 100% after reaction for 2 h in H2 atmosphere. CO and CO2, which formed from decarbonylation and decarboxylation of C15H31COOC16H33 (Reactions (5) and (6)) were the main by-products in the deoxygenation of C15H31COOC16H33 in H2 atmosphere [6,7]. The amounts of formed CO and CO2 almost kept constant after reaction for 2 h. A small amount of C1‒C4 gaseous hydrocarbons was also formed during the reaction in H2 atmosphere, and the amount of C1‒C4 gaseous hydrocarbons increased with prolonging the reaction time. As shown in Figure 7, the yield of C1‒C16 hydrocarbons increased to 90.2% after reaction for 2 h, and then decreased slightly with prolonging reaction time due to the formation of light hydrocarbons (<C9). On the whole, the Pd/Mg(Al)O catalyst provided a high catalytic performance for the catalytic deoxygenation of C15H31COOC16H33 to hydrocarbons in H2 atmosphere.
2.3. Catalytic Deoxygenation of C15H31COOC16H33 in N2 Atmosphere
Table 1 shows the C15H31COOC16H33 conversion and C10‒C16 hydrocarbons yield of the deoxygenation of C15H31COOC16H33 in N2 atmosphere at 300 °C for 2 h over various catalysts. The 1 wt.% Pd/Mg(Al)O catalyst resulted in a C15H31COOC16H33 conversion of 76.4% and a C10‒C16 yield of 63.5% for the reaction at 300 °C for 2 h in N2 atmosphere. Both, C15H31COOC16H33 conversion and the C10‒C16 yield obtained in N2 atmosphere, were much lower than those obtained in H2 atmosphere (Figure 6) over Pd/Mg(Al)O after reaction at 300 °C for 2 h. This indicates that H2 greatly improves the deoxygenation of C15H31COOC16H33, and that it is difficult to produce hydrocarbons from C15H31COOC16H33 in N2 atmosphere. The use of basic hydrotalcite, acidic H-ZSM-5, and Pd/C has been reported for the deoxygenation of triglycerides or fatty acids in N2 or Ar atmospheres [14,17,18]. As shown in Table 1, the basic catalyst Mg(Al)O without Pd gave low conversion (23.3%) and a low yield of C10‒C16 hydrocarbons (15.1%) for the reaction in N2 atmosphere. In contrast, the conversion was relatively high (55.6%), but the yield of C10‒C16 hydrocarbons was very low (8.3%) over the acidic catalyst H-ZSM-5. Heavy hydrocarbons formed in the reaction cracked to light hydrocarbons on the acidic sites of H-ZSM-5 during the reaction. The use of Pd/C resulted in a C15H31COOC16H33 conversion of 41.7% and a C10‒C16 hydrocarbons yield of 26.2% for the reaction at 300 °C for 2 h in N2 atmosphere. Therefore, Pd/Mg(Al)O shows a much higher catalytic performance than these catalysts that are reported in the literature, for the deoxygenation of C15H31COOC16H33 in N2 atmosphere. The development of multi-functional catalysts to improve catalyst performance is an important challenge in the catalyst field. The catalyst containing Pt metal and basic hydrotalcite has been reported as excellent for the aromatization of n-hexane [26]. We had developed some multi-functional catalysts containing metal and solid acid for some industrially important reactions [36,37,38,39,40,41,42]. Pd metal sites and solid base sites (in Mg(Al)O) achieved a synergetic effect for the deoxygenation of C15H31COOC16H33 in N2 atmosphere over the Pd/Mg(Al)O catalyst.
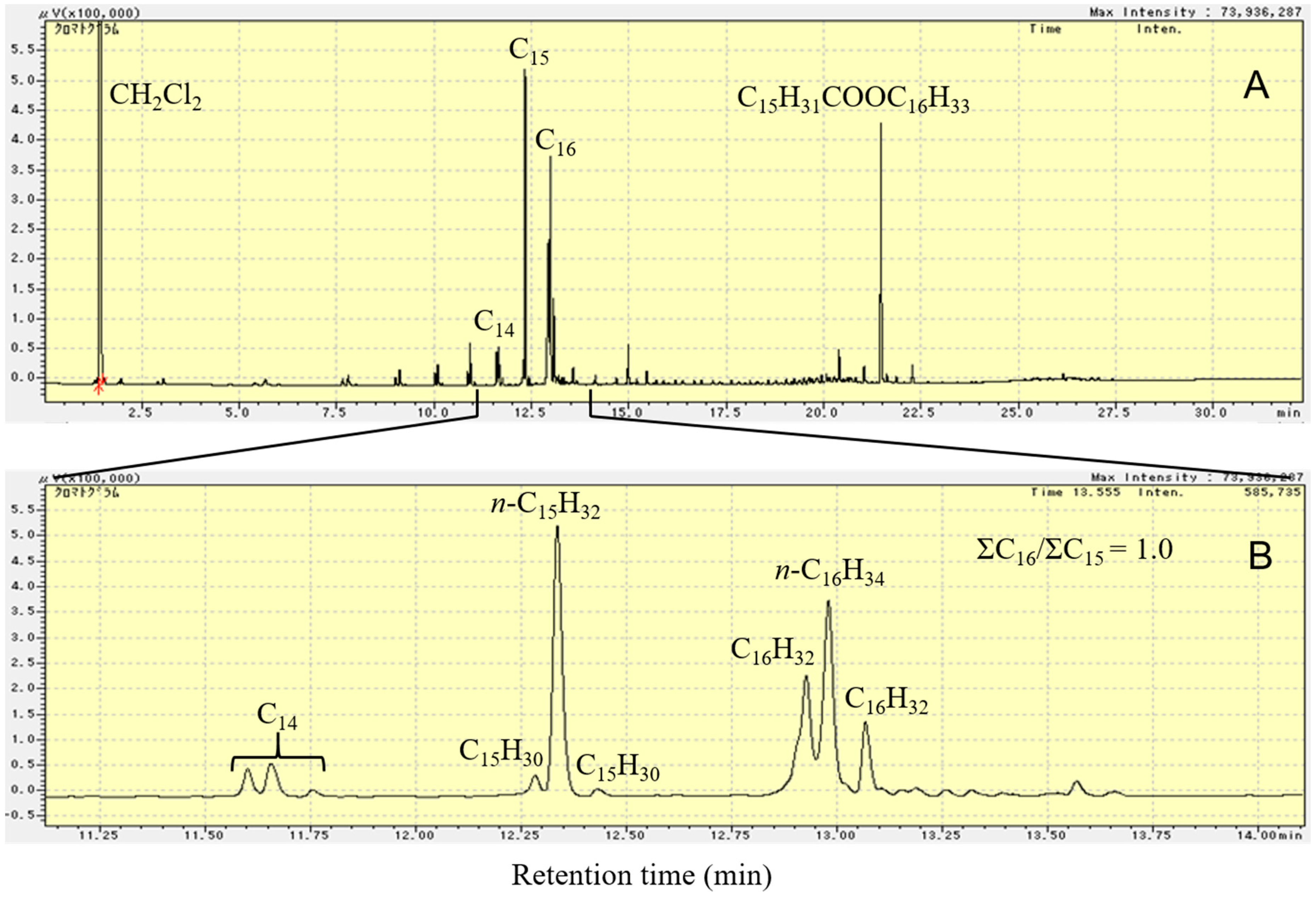
Figure 8 shows the FID-GC charts of the liquid product formed by deoxygenation of C15H31COOC16H33 in N2 atmosphere at 300 °C for 2 h over 1 wt.% Pd/Mg(Al)O catalyst. A comparison of Figure 5B and Figure 8A shows that the peak of the C15H31COOC16H33 reactant still appears in the GC chart after reaction in N2 atmosphere. For the deoxygenation of C15H31COOC16H33 over the Pd/Mg(Al)O catalyst, the reaction rate in N2 atmosphere was slow in comparison with that in the H2 atmosphere. The reaction in N2 atmosphere formed some compounds with a retention time ranging from 14 to 22 min in the GC chart (fatty acid, alcohol, and ketones). These compounds decreased the yield of hydrocarbons from the deoxygenation of C15H31COOC16H33. Moreover, peaks of C5‒C14 hydrocarbons appear in the GC chart of the 2 h reaction in N2 atmosphere (Figure 8A). Figure 8B shows the magnified area from 11.1 to 14.1 min in Figure 8A; this enables us to observe the composition of C15 and C16 hydrocarbons that are formed by the reaction in N2 atmosphere. As shown in Figure 5B, deoxygenation of C15H31COOC16H33 in H2 atmosphere only formed normal paraffin hydrocarbons. As shown in Figure 8B, the hydrocarbon products that are formed by the deoxygenation of C15H31COOC16H33 in N2 atmosphere contained n-paraffin and several kinds of olefins. A comparison of the GC-MS results with values in the NIST-11 database indicates that the peaks around n-paraffin were olefins with the same number of carbon chains. The initially formed olefins were converted into other olefins by double bond migration on the basic sites of Mg(Al)O. Solid base catalysts (such as MgO) have the catalytic ability for double bond migration of olefins [43]. By calculating the amount of each component in the product using the GC chart, the amount of total C16 hydrocarbons and the amount of total C15 hydrocarbons was found to be almost the same in the liquid product that was formed from the catalytic deoxygenation of C15H31COOC16H33 in N2 atmosphere, at 300 °C, for 2 h, over the Pd/Mg(Al)O catalyst.
Reactions (7) and (8) show the reactions for the deoxygenation of C15H31COOC16H33 in N2 (without H2) atmosphere. CO could not be detected in the gas products of the reaction in N2 atmosphere, thus, decarboxylation was deduced to be the only reaction in the deoxygenation of C15H31COOC16H33 in N2 atmosphere.
C15H31COOC16H33 = C15H32 + C16H32 + CO2 Decarboxylation
C15H31COOC16H33 = C15H30 + C16H34 + CO2 Decarboxylation
In N2 atmosphere, the number of H atoms in the C15H31COOC16H33 molecule was not enough to obtain the saturated hydrocarbons C16H34 and C15H32 in the decarboxylation of C15H31COOC16H33. One C15H31COOC16H33 molecule must form one paraffin and one olefin by decarboxylation without H2. The high reactivity of the olefin molecules formed resulted in the formation of light hydrocarbons. A few heavy hydrocarbons larger than C16 were also formed by the polymerization of light olefins. Because both Reactions (7) and (8) form the same amount of C15 and C16 hydrocarbons, the molar ratio of total C16 hydrocarbons to total C15 hydrocarbons was 1.0, as shown in Figure 8B. Moreover, the ratio of n-C15H32 in the total C15 hydrocarbons was much higher than the ratio of n-C16H34 in the total C16 hydrocarbons (Figure 8B). Thus, Reaction (7) was the main decarboxylation reaction (as compared to Reaction (8)) in the catalytic deoxygenation of C15H31COOC16H33 in N2 atmosphere.
For deoxygenation of C15H31COOC16H33 in N2 atmosphere, the metal site alone could not achieve enough activity, because deoxygenation was carried out via a single route, namely decarboxylation. In this study, we used a base support (Mg(Al)O) for the metal (Pd) in order to enhance the catalytic activity during deoxygenation of C15H31COOC16H33 in N2 atmosphere.
Figure 9 shows the dependence of C15H31COOC16H33 conversion and C10‒C16 hydrocarbons yield on reaction time for the catalytic deoxygenation of C15H31COOC16H33 in N2 atmosphere, at 300 °C, over the 1 wt.% Pd/Mg(Al)O catalyst. C15H31COOC16H33 conversion increased with prolonged reaction time and reached 100% after reaction for 6 h. The yield of C10‒C16 hydrocarbons increased with prolonged reaction until 5 h, and then decreased slightly due to the formation of light hydrocarbons. Although the catalytic activity in N2 atmosphere was lower than that in the H2 atmosphere, the Pd/Mg(Al)O catalyst yielded a C15H31COOC16H33 conversion of 99.1% and a C10‒C16 hydrocarbon yield of 80.4% by prolonging reaction time to 5 h for the reaction in N2 atmosphere at 300 °C.
Figure 10 shows the dependence of C15H31COOC16H33 conversion and C10–C16 hydrocarbon yield on reaction temperature for the catalytic deoxygenation of C15H31COOC16H33 in N2 atmosphere, for 2 h, over the 1 wt.% Pd/Mg(Al)O catalyst. C15H31COOC16H33 conversion increased with an increasing reaction temperature from 275 to 375 °C, and achieved a conversion of over 99% for the reaction at 375 °C for 2 h. The yield of C10‒C16 hydrocarbons increased with an increasing reaction temperature from 275 to 325 °C. At a reaction temperature higher than 325 °C, the yield of C10‒C16 hydrocarbons could not be improved by increasing the reaction temperature because the number of light hydrocarbons (<C9) in the product increased at high reaction temperatures.
3. Experimental Section
3.1. Reagents
Hexadecyl palmitate (C15H31COOC16H33) of purity higher than 95% was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan) Na-ZSM-5 zeolite (SiO2/Al2O3 = 50) was purchased from Wako Pure Chemical Industries, Ltd. (Tokyo, Japan) Activated carbon (C), with a particle size of 100 mesh, was purchased from Aldrich Chem. Co. Inc. (Milwaukee, WI, USA); the BET surface area of the C support was 593 m2/g by N2 adsorption measurement. Other chemical reagents were purchased from Wako Pure Chemical Industries Ltd. (Tokyo, Japan) with purities higher than 99%. Gas cylinders were purchased from Sumitomo Seika Chemicals Co., Ltd. (Tokyo, Japan) with purities higher than 99.995%.
3.2. Catalysts
Pd0.016Mg3Al(OH)16CO3∙xH2O hydrotalcite was prepared using a co-precipitation method at pH 10.0, by adding an aqueous solution containing Pd(NO3)2, Mg(NO3)2, and Al(NO3)3 (the molar ratio of Pd to Mg to Al was 0.016:3:1) to a solution containing a slight excess of Na2CO3 at 65 °C. A 1 M NaOH aqueous solution was added during the process to maintain the pH value at 10.0. After standing at 65 °C for 4 h, a precipitate was obtained by filtration. The resultant precipitate was dried in air at 100 °C for 24 h.
The Pd/Mg(Al)O catalyst was obtained by reducing PdO-Mg(Al)O mixed oxide under H2 flow (60 mL/min) at 300 °C for 1 h. The PdO-Mg(Al)O mixed oxide was obtained by calcining Pd0.016Mg3Al(OH)16CO3∙xH2O hydrotalcite in air at 450 °C for 3 h. By calculating the amount of Pd in the Pd0.016Mg3Al(OH)16CO3∙4H2O precursor, the designed Pd loading was determined to be about 1 wt.% in the Pd/Mg(Al)O catalyst. The actual amount of Pd loading (measured by elemental analysis) was 0.95 wt.% in the Pd/Mg(Al)O catalyst.
Mg(Al)O was obtained by calcination of Mg3Al(OH)16CO3∙xH2O hydrotalcite in air at 450 °C for 3 h. Mg3Al(OH)16CO3∙xH2O hydrotalcite was prepared by a co-precipitation method at pH 10.0, by adding an aqueous solution containing Mg(NO3)2 and Al(NO3)3 (molar ratio of Mg to Al was 3) to a solution containing a slight excess of Na2CO3 at 65 °C. A 1 M NaOH aqueous solution was added during the process to maintain the pH value at 10.0. The resultant precipitate was washed and dried in air at 100 °C for 24 h.
H-ZSM-5 was prepared using Na-ZSM-5 as follows. Na-ZSM-5 was treated with an aqueous solution of NH4NO3 (0.1 M) to form NH4-ZSM-5 by ion exchange. After filtering out the water, the NH4-ZSM-5 obtained was dried in air at 100 °C for 24 h, and then calcined in air at 550 °C for 3 h to form H-ZSM-5.
Pd/C was synthesized by using a wet impregnation method. The activated carbon support was impregnated with an aqueous solution of Pd(NO3)2 with stirring. After removal of the solvent by heating at 90 °C, the resultant solid product was dried in air at 100 °C for 24 h. Then, Pd/C was calcined under a N2 flow (60 mL/min) at 400 °C for 1 h to remove NO3− ions. In the Pd/C catalyst, the designed Pd loading was 1 wt.% and the actual amount of Pd loading (measured by element analysis) was 1.02 wt.%.
3.3. Instruments
Powder X-ray diffraction (XRD) patterns were measured using a MAC Science MXP-18 diffractometer (XrayScience Corp., Tokyo, Japan) with Cu Kα radiation operated at 40 kV and 50 mA. The solid phase was identified by referring to the JICST database (Version 6th, Japan Information Center of Science and Technology, Tokyo, Japan, 2012) from The Crystallographic Society of Japan. Thermogravimetric and differential thermal analyses (TG-DTA) were carried out using a Shimadzu TGA-50 instrument (Shimadzu Corp., Kyoto, Japan). The sample was heated under an atmosphere of N2 flow (60 mL/min) at a heating rate of 5 °C/min from room temperature to 1000 °C. N2 adsorption measurements were carried out at −196 °C using a Belsorp 28SA automatic adsorption instrument (MicrotracBEL Corp., Osaka, Japan). The surface areas of the samples were obtained from a Brunauer–Emmett–Teller (BET) plot. Elemental analysis of Pd was measured by an inductive coupled plasma analysis, using a Thermo Jarrell Ash IRIS/AP instrument (SpectraLab Scientific Inc., Markham, ON, Canada).
3.4. Reactions
Before reaction, Pd/Mg(Al)O and Pd/C were reduced in a H2 flow (60 mL/min) at 300 °C for 1 h. The reaction was carried out in a type of 100-mL stainless-steel autoclave reactor with a stirrer. In general, 0.2 g catalyst and 5 g hexadecyl palmitate (C15H31COOC16H33) were introduced into an autoclave reactor. Then, 0.9 MPa H2 (for the reaction in H2 atmosphere) or 0.9 MPa N2 (for the reaction in N2 atmosphere) was charged into the autoclave at room temperature (25 °C). The autoclave reactor was then heated and was kept at the reaction temperature (275–375 °C) for 1–6 h with vigorous stirring (600 rpm). After the reaction, the reactor was cooled down to room temperature before analysis.
3.5. Analyses
The gas products were collected into a plastic bag, from which the air had been taken out by using a pump. The total volume of the total gas was measured using a WS-1 integration flow meter (Shinagawa Corp., Tokyo, Japan). The composition of the gas products was analysed using gas chromatography (Shimadzu Corp., Kyoto, Japan). Inorganic gases (H2, N2, CO, and CO2) were analysed using a Shimadzu 14B type GC with a thermal conductivity detector (TCD) that was equipped with MS-5A and Porapak-Q columns. Gaseous hydrocarbons (C1‒C4) were analysed using a Shimadzu GC-2014 type FID-GC equipped with an RT-QPLOT (Agilent Technologies Japan, Ltd, Tokyo, Japan) capillary column. The factors of various gases (H2, N2, CO, CO2, C1‒C4 hydrocarbons) were obtained using a standard mixed gas (with known concentration for each component) from a cylinder.
The liquid products were taken out from the autoclave reactor. After filtering out the solid catalyst, a certain amount of dioxane (C4H8O2) was added to the liquid products as an internal standard. Dichloromethane (CH2Cl2) was used as a solvent to wash the reactor and the used catalyst, and then mixed with the liquid products. The liquid products were analysed by a Shimadzu GC-2014 type GC-FID equipped with a UA-DX30 capillary column (Frontier Laboratories Ltd., Koriyama, Fukushima, Japan). Gas chromatography–mass spectrometry (GC-MS) analysis was performed on a Shimadzu GCMS-QP2010 Ultra (Shimadzu Corp., Kyoto, Japan) to confirm the components of the liquid products. Each component in the liquid product was separated on a UA-DX30 capillary column, and was identified by GC-MS analysis, with reference to the NIST-11 database. The amount of each normal paraffin in the product was calculated from the results of GC-FID analysis, and the factor of each normal paraffin was obtained using a pure reagent with a certain amount of dioxane (C4H8O2) in the GC-FID analysis. Each olefin in the product was identified by GC-MS analysis, and the amount of each olefin was calculated from the results of GC-FID analysis by using the factor of a normal paraffin with the same number of carbon chains.
Using the results of the GC analyses, C15H31COOC16H33 conversion was calculated from the ratio of the decreased amount to the fed amount of C15H31COOC16H33; the yield of each carbon-containing product was calculated from the ratio of the formed amount of each product to the fed amount of C15H31COOC16H33. The carbon mass balance (before and after reaction) had an error less than ±5% for the reaction in H2 atmosphere, and had an error less than ±10% for the reaction in N2 atmosphere.
4. Conclusions
The Pd/Mg(Al)O catalyst derived from Pd0.016Mg3Al(OH)16CO3∙xH2O hydrotalcite precursor showed a high catalytic performance in both H2 and N2 atmospheres for the catalytic deoxygenation of C15H31COOC16H33. In H2 atmosphere, the reduction of C15H31COOC16H33 was the main reaction, and the main products were n-C16H34 and n-C15H32 from the deoxygenation of C15H31COOC16H33 over Pd/Mg(Al)O. In N2 atmosphere, the decarboxylation of C15H31COOC16H33 was the main reaction, and olefins and paraffins were formed as the products of deoxygenation of C15H31COOC16H33 over Pd/Mg(Al)O. The catalytic reaction in N2 atmosphere was slower than that in H2 atmosphere over Pd/Mg(Al)O. Pd/Mg(Al)O showed a much higher catalytic performance than that reported for other catalysts for the deoxygenation of C15H31COOC16H33 in N2 atmosphere. Prolonging the reaction time or increasing the reaction temperature improved the yield of C10‒C16 hydrocarbons until the C15H31COOC16H33 conversion approached 100% for the reaction in N2 atmosphere over the Pd/Mg(Al)O catalyst.
Author Contributions
The author Y.L. conceived and designed the experiments, Y.L. performed the experiments, Y.L, M.I., and K.M. analyzed the data, Y.L. drafted the paper. All authors have given approval for the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cesaro, A.; Belgiorno, V. Combined biogas and bioethanol production: Opportunities and challenge for industrial application. Energies 2015, 8, 8121–8144. [Google Scholar] [CrossRef]
- Hanaoka, T.; Liu, Y.; Matsunaga, K.; Miyazawa, T.; Hirata, S.; Sakanishi, K. Bench-scale production of liquid fuel from woody biomass via gasification. Fuel Process. Technol. 2010, 91, 859–865. [Google Scholar] [CrossRef]
- Liu, Y.; Hanaoka, T.; Miyazawa, T.; Murata, K.; Okabe, K.; Sakanishi, K. Fischer-Tropsch synthesis in slurry-phase reactors over Mn- and Zr-modified Co/SiO2 catalysts. Fuel Process. Technol. 2009, 90, 901–908. [Google Scholar] [CrossRef]
- Thanh, L.T.; Okitsu, K.; Boi, L.V.; Maeda, Y. Catalytic technologies for biodiesel fuel production and utilization of glycerol: A review. Catalysts 2012, 2, 191–222. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, L.; Cheng, S.; Julson, J. Review of heterogeneous catalysts for catalytically upgrading vegetable oils into hydrocarbon biofuels. Catalysts 2017, 7, 83. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyas, R.; Murata, K.; Minowa, T.; Sakanishi, K. Production of bio-hydrogenated diesel by hydrotreatment of high-acid-value waste cooking oil over ruthenium catalyst supported on Al-polyoxocation-pillared pontmorillonite. Catalysts 2012, 2, 171–190. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyas, R.; Murata, K.; Minowa, T.; Sakanishi, K. Hydrotreatment of vegetable oils to produce bio-hydrogenated diesel and liquefied petroleum gas fuel over catalysts containing sulfided Ni-Mo and solid Acids. Energy Fuels 2011, 25, 4675–4685. [Google Scholar] [CrossRef]
- Yoshida, M.; Tanabe, Y.; Yonezawa, N.; Watanabe, M.M. Energy innovation potential of oleaginous microalgae. Biofuels 2012, 3, 761–781. [Google Scholar] [CrossRef]
- Georgianna, D.R.; Mayfield, S.P. Exploiting diversity and synthetic biology for the production of algal biofuels. Nature 2012, 488, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Chisti, Y. Biodiesel from microalgae. Biotechnol. Adv. 2007, 25, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Milledge, J.J.; Smith, B.; Dyer, P.W.; Harvey, P. Macroalgae-derived biofuel: A review of method of energy extraction from seaweed biomass. Energies 2014, 7, 7194–7222. [Google Scholar] [CrossRef] [Green Version]
- Kanda, H.; Li, P.; Goto, M.; Makio, H. Energy-saving lipid extraction from wet Euglena gracilis by the low-boiling-point solvent dimethyl ether. Energies 2015, 8, 610–620. [Google Scholar] [CrossRef]
- Tani, Y.; Okumura, M.; Ii, S. Liquid wax ester production by euglena gracilis. Agric. Biol. Chem. 1987, 51, 225–230. [Google Scholar] [CrossRef]
- Maki-Arvela, P.; Kubickova, I.; Snare, M.; Eranen, K.; Murzin, D.Y. Catalytic deoxygenation of fatty acids and their derivatives. Energy Fuels 2007, 21, 30–41. [Google Scholar] [CrossRef]
- Sotelo-Boyas, R.; Liu, Y.; Minowa, T. Renewable diesel production from the hydrotreating of rapeseed oil with Pt/Zeolite and NiMo/Al2O3 catalysts. Ind. Eng. Chem. Res. 2011, 50, 2791–2799. [Google Scholar] [CrossRef]
- Murata, K.; Liu, Y.; Watanabe, M.M.; Inaba, M.; Takahara, I. Hydrocracking of algae oil into aviation fuel-range hydrocarbons using a Pt-Re catalyst. Energy Fuels 2014, 28, 6999–7006. [Google Scholar] [CrossRef]
- Sanchez-Cardenas, M.; Medina-Valtierra, J.; Kamaraj, S.-K.; Ramirez, R.R.M.; Sanchez-Olmos, L.A. Effect of size and distribution of Ni nanoparticles on γ-Al2O3 in oleic acid hydrodeoxygenation to produce n-alkanes. Catalysts 2016, 6, 156. [Google Scholar] [CrossRef]
- Charusiri, W.; Vitidsant, T. Catalytic cracking of used cooking oil to liquid fuels over HZSM-5. J. Energy 2003, 5, 58–68. [Google Scholar]
- Na, J.G.; Han, J.K.; Oh, Y.K.; Park, J.H.; Jung, T.S.; Han, S.S.; Yoon, H.C.; Chung, S.H.; Kim, J.N.; Ko, C.H. Decarboxylation of microalgal oil without hydrogen into hydrocarbon for the production of transportation fuel. Catal. Today 2012, 185, 313–317. [Google Scholar] [CrossRef]
- Tani, H.; Hasegawa, T.; Shimouchi, M.; Asami, K.; Fujimoto, K. Selective catalytic decarboxy-cracking of triglyceride to middle-distillate hydrocarbon. Catal. Today 2011, 164, 410–414. [Google Scholar] [CrossRef]
- Maki-Arvela, P.; Rozmyszowicz, B.; Lestari, S.; Simakova, O.; Eranen, K.; Salmi, T.; Murzin, D.Y. Catalytic deoxygenation of Tall oil fatty acid over palladium supported on mesoporous carbon. Energy Fuels 2011, 25, 2815–2825. [Google Scholar] [CrossRef]
- Cavani, F.; Trifiro, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Basile, F.; Basini, L.; Fornasari, G.; Gazzano, M.; Trifiro, F.; Vaccari, A. New hydrotalcite-type anionic clays containing noble metals. Chem. Commun. 1996, 2435–2436. [Google Scholar] [CrossRef]
- Narayanan, S.; Krishna, K. Structural influence of hydrotalcite on Pd dispersion and phenol hydrogenation. Chem. Commun. 1997, 1991–1992. [Google Scholar] [CrossRef]
- Basile, F.; Fornasari, G.; Gazzano, M.; Vaccari, A. Synthesis and thermal evolution of hydrotalcite-type compounds containing noble metals. Appl. Clay Sci. 2000, 16, 185–200. [Google Scholar] [CrossRef]
- Davis, R.J.; Derouane, E.G. A non-porous supported-platinum catalyst for aromatization of n-hexane. Nature 1991, 349, 313–315. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Hanaoka, T.; Inaba, M.; Sakanishi, K. Syntheses of new peroxo-polyoxometalates intercalated layered double hydroxides for propene epoxidation by molecular oxygen in methanol. J. Catal. 2007, 248, 277–287. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Direct epoxidation of proptlene by molecular oxygen over catalyst system containing palladium and peroxo-Heteropoly compound in methanol. Chem. Commun. 2004, 582–583. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Suzuki, K.; Hamakawa, S.; Hayakawa, T.; Murata, K.; Ishii, T.; Kumagai, M. Highly active methanol decomposition catalyst derived from Pd-hydrotalcite dispersed on mesoporous silica. Catal. Lett. 2000, 66, 205–213. [Google Scholar] [CrossRef]
- Liu, Y.; Suzuki, K.; Hamakawa, S.; Hayakawa, T.; Murata, K.; Ishii, T.; Kumagai, M. Catalytic methanol decomposition at low temperature over Pd catalyst derived from mesoporous silica carried Pd-hydrotalcite. Chem. Lett. 2000, 486–487. [Google Scholar] [CrossRef]
- Liu, Y.; Suzuki, K.; Hayakawa, T.; Tsunoda, T.; Suzuki, K.; Hamakawa, S.; Murata, K.; Shiozaki, R.; Ishii, T.; Kumagai, M. Steam reforming of methanol over Cu/CeO2 catalysts studied in comparison with Cu/ZnO and Cu/Zn(Al)O catalysts. Top. Catal. 2003, 22, 205–213. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Suzuki, K.; Hamakawa, S.; Tsunoda, T.; Ishii, T.; Kumagai, M. High active copper/ceria catalysts for the steam reforming of methanol. Appl. Catal. A Gen. 2002, 223, 137–145. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Ishii, T.; Kumagai, M.; Yasuda, H.; Suzuki, K.; Hamakawa, S.; Murata, K. Methanol decomposition to synthesis gas at low temperature over palladium support on ceria-zirconia solid solutions. Appl. Catal. A Gen. 2001, 210, 301–314. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Takahara, I.; Okabe, K. Mixed synthesis of mixed alcohols from syngas over Cs- and Ni-modified Cu/CeO2 catalysts. Fuel 2013, 104, 62–69. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Takahara, I.; Okabe, K. Synthesis of ethanol from syngas over Rh/Ce1−xZrxO2 catalysts. Catal. Today 2011, 164, 308–314. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Sakanishi, K. Hydroisomerization-cracking of gasoline distillate from Fischer–Tropsch synthesis over bifunctional catalysts containing Pt and heteropolyacids. Fuel 2011, 90, 3056–3065. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyás, R.; Murata, K.; Minowa, T.; Sakanishi, K. Hydrotreatment of Jatropha oil to produce green diesel over trifunctional Ni-Mo/SiO2-Al2O3 catalyst. Chem. Lett. 2009, 38, 552–553. [Google Scholar] [CrossRef]
- Liu, Y.; Koyano, G.; Misono, M. Hydroisomerization of n-hexane and n-heptane over platinum-promoted Cs2.5H0.5PW12O40 (Cs2.5) studied in comparison with several other solid acids. Top. Catal. 2000, 11, 239–246. [Google Scholar] [CrossRef]
- Liu, Y.; Na, K.; Misono, M. Skeletal isomerization of n-pentane over Pt-promoted cesium hydrogen salts of 12-tungstophosphoric acid. J. Mol. Catal. A Chem. 1999, 141, 145–153. [Google Scholar] [CrossRef]
- Liu, Y.; Misono, M. Hydroisomerization of n-butane over platinum-promoted cesium hydrogen salt of 12-tungstophosphoric acid. Materials 2009, 2, 2319–2336. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Okabe, K.; Inaba, M.; Takahara, I.; Hanaoka, T.; Sakanishi, K. Selective hydrocracking of Fischer-Tropsch waxes to high-quality diesel fuel over Pt-promoted polyoxocation-pillared montmorillonites. Top. Catal. 2009, 52, 597–608. [Google Scholar] [CrossRef]
- Liu, Y.; Koyano, G.; Na, K.; Misono, M. Isomerization of n-pentane and n-hexane over cesium hydrogen salt of 12-tungstophosphoric acid promoted by platinum. Appl. Catal. A Gen. 1998, 166, L263–L265. [Google Scholar] [CrossRef]
- Hattori, H. Heterogenous basic catalysis. Chem. Rev. 1995, 95, 537–558. [Google Scholar] [CrossRef]
Figure 1.
Structural model of the Pd0.016Mg3Al(OH)16CO3∙xH2O hydrotalcite used in this study.
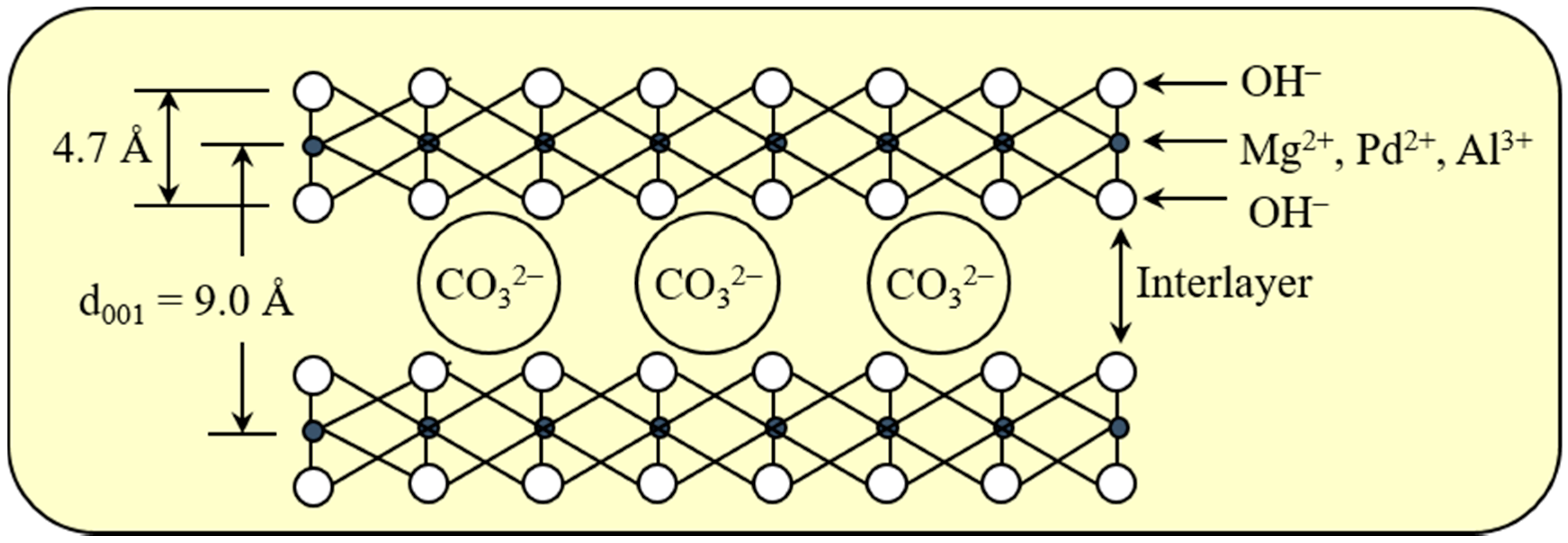
Figure 2.
X-ray diffraction (XRD) patterns of Pd0.016Mg3Al(OH)16CO3∙xH2O after drying at 100 °C.
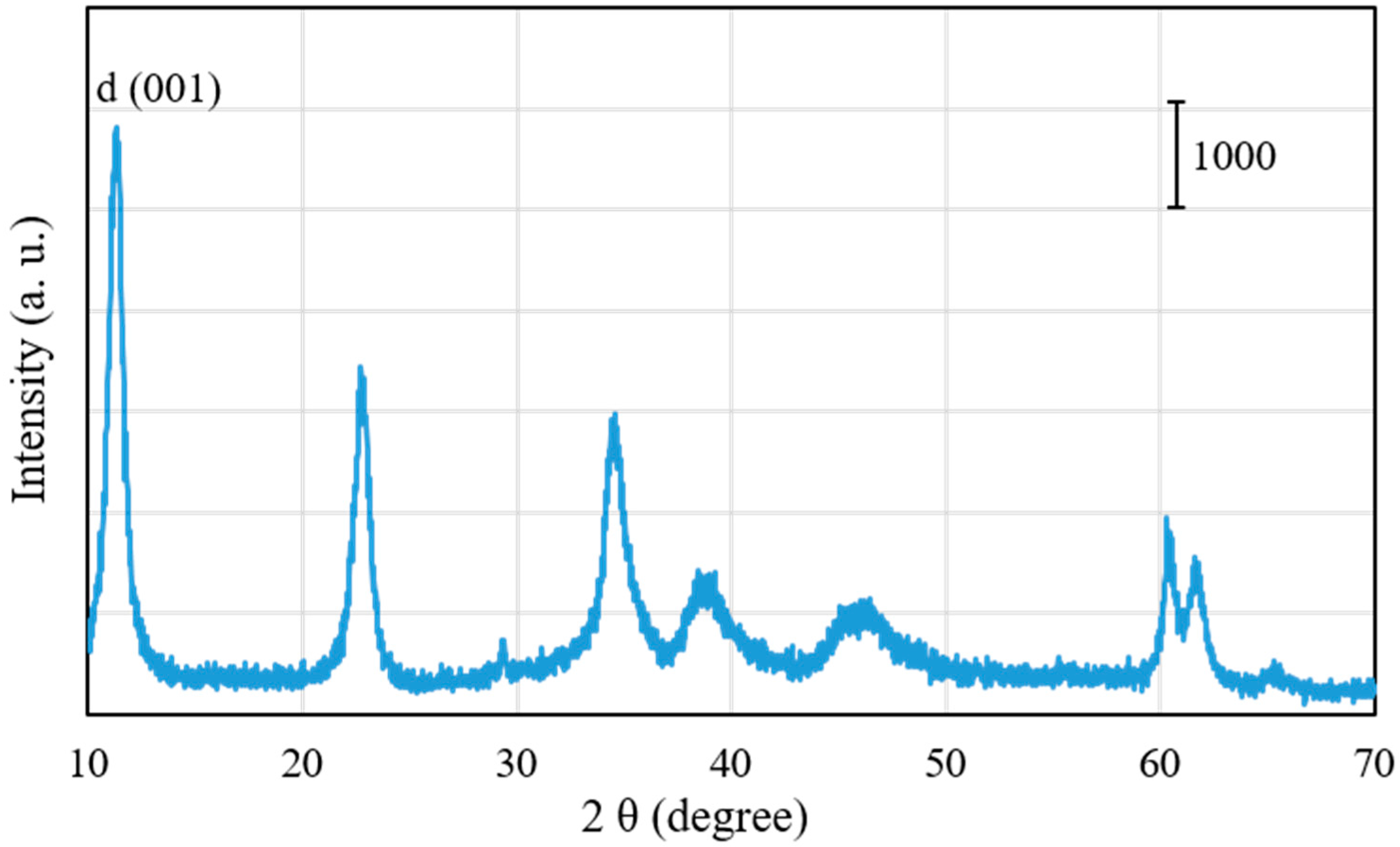
Figure 3.
The thermogravimetric analysis (TGA) and differential thermal analysis (DTA) results for Pd0.016Mg3Al(OH)16CO3∙xH2O after drying at 100 °C.
Figure 3.
The thermogravimetric analysis (TGA) and differential thermal analysis (DTA) results for Pd0.016Mg3Al(OH)16CO3∙xH2O after drying at 100 °C.

Figure 4.
The Brunauer-Emmett-Teller (BET) surface area of Pd0.016Mg3Al(OH)16CO3∙xH2O on calcination temperature from 100 °C to 1000 °C.
Figure 4.
The Brunauer-Emmett-Teller (BET) surface area of Pd0.016Mg3Al(OH)16CO3∙xH2O on calcination temperature from 100 °C to 1000 °C.
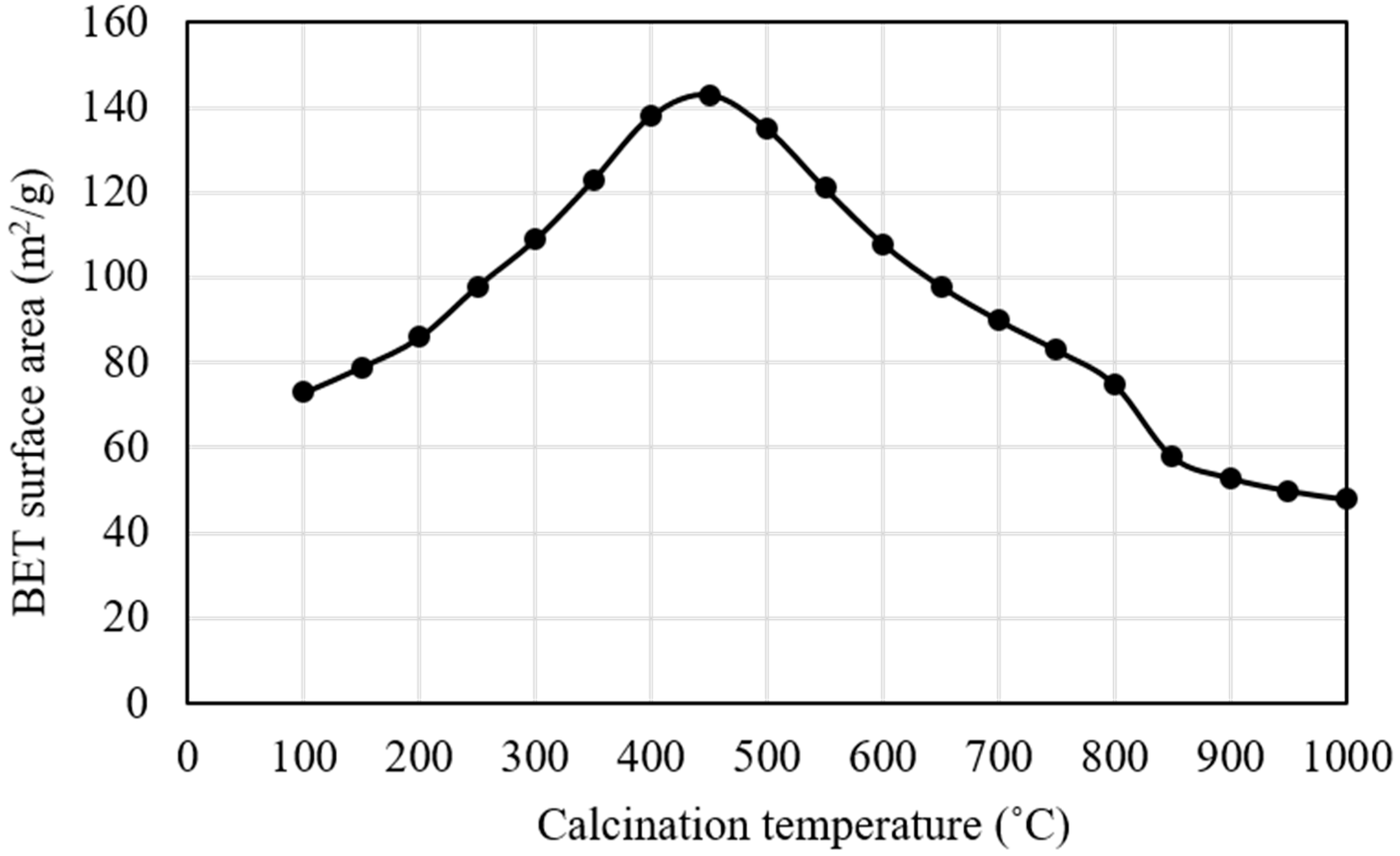
Figure 5.
The flame ionization detector–gas chromatography (FID-GC) charts of C15H31COOC16H33 (A) and the liquid product formed upon deoxygenation of C15H31COOC16H33 in H2 atmosphere, at 300 °C, for 2 h, over 1 wt.% Pd/Mg(Al)O catalyst (B). Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; and, H2 pressure at 25 °C: 0.9 MPa.
Figure 5.
The flame ionization detector–gas chromatography (FID-GC) charts of C15H31COOC16H33 (A) and the liquid product formed upon deoxygenation of C15H31COOC16H33 in H2 atmosphere, at 300 °C, for 2 h, over 1 wt.% Pd/Mg(Al)O catalyst (B). Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; and, H2 pressure at 25 °C: 0.9 MPa.
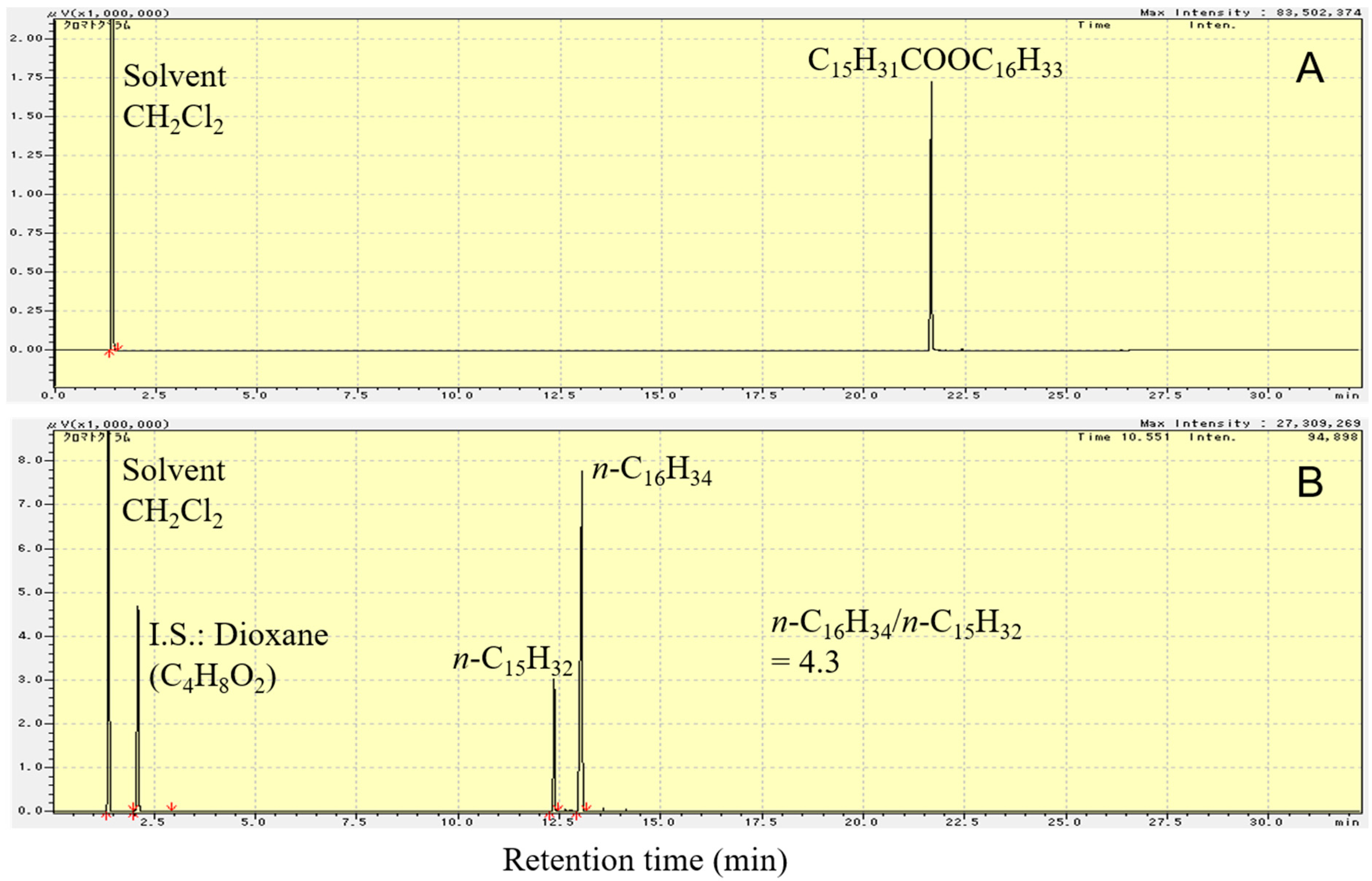
Figure 6.
X-ray diffraction (XRD) patterns of 1 wt.% Pd/Mg(Al)O catalyst before reaction and after reaction (for deoxygenation of C15H31COOC16H33 in H2 atmosphere at 300 °C for 2 h). ■, MgO; ●, Al2O3.
Figure 6.
X-ray diffraction (XRD) patterns of 1 wt.% Pd/Mg(Al)O catalyst before reaction and after reaction (for deoxygenation of C15H31COOC16H33 in H2 atmosphere at 300 °C for 2 h). ■, MgO; ●, Al2O3.

Figure 7.
The dependence of C15H31COOC16H33 conversion and C10‒C16 hydrocarbon yield on the reaction time for the catalytic deoxygenation of C15H31COOC16H33 in H2 atmosphere, at 300 °C, over 1 wt.% Pd/Mg(Al)O catalyst. Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; H2 pressure at 25 °C: 0.9 MPa.
Figure 7.
The dependence of C15H31COOC16H33 conversion and C10‒C16 hydrocarbon yield on the reaction time for the catalytic deoxygenation of C15H31COOC16H33 in H2 atmosphere, at 300 °C, over 1 wt.% Pd/Mg(Al)O catalyst. Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; H2 pressure at 25 °C: 0.9 MPa.
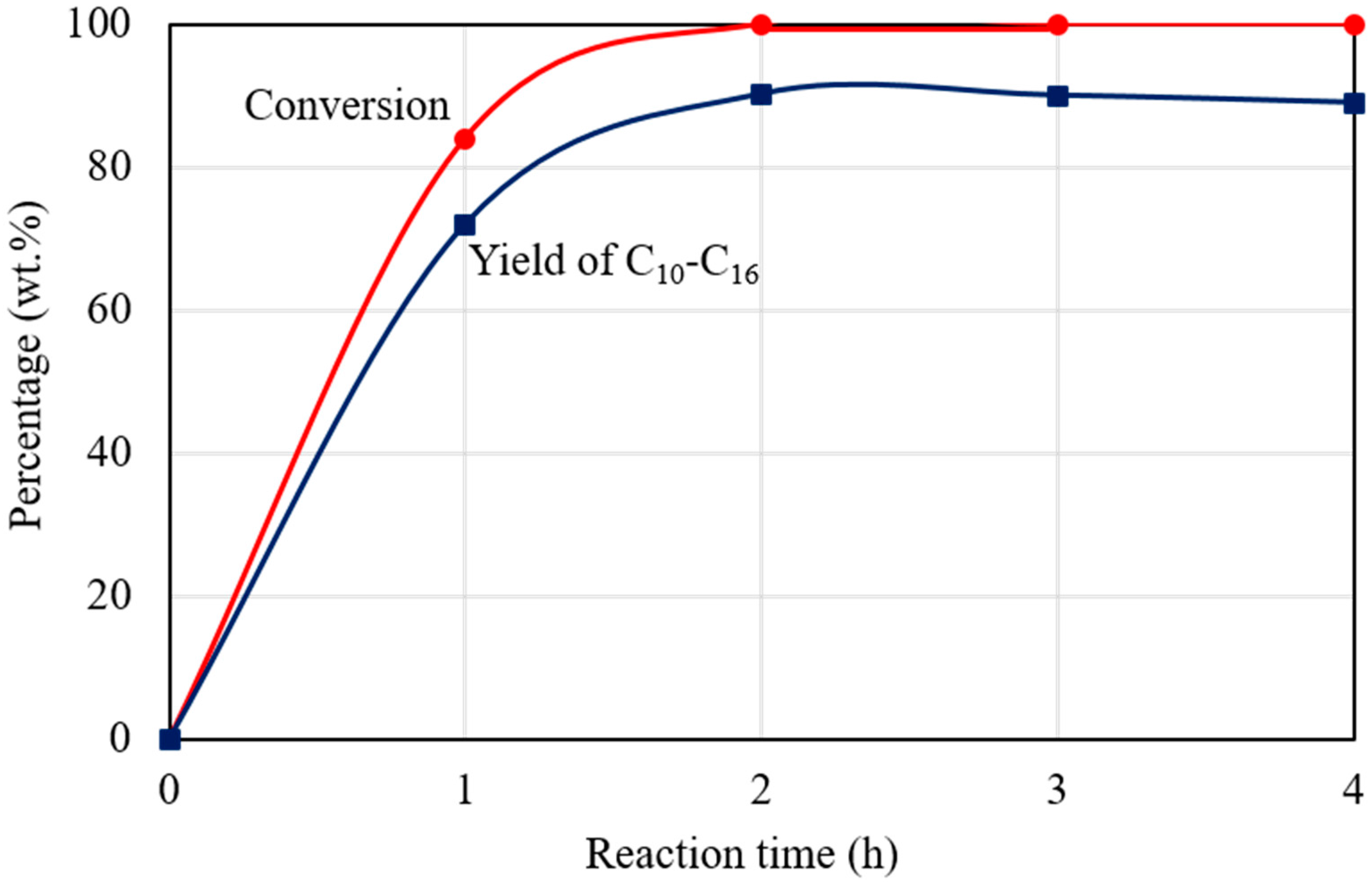
Figure 8.
The FID-GC charts of the liquid product formed by deoxygenation of C15H31COOC16H33 in N2 atmosphere at 300 °C for 2 h over 1 wt.% Pd/Mg(Al)O catalyst. (A): retention time from 0 to 32 min, (B): retention time from 11.1 to 14.1 min. Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; N2 pressure at 25 °C: 0.9 MPa.
Figure 8.
The FID-GC charts of the liquid product formed by deoxygenation of C15H31COOC16H33 in N2 atmosphere at 300 °C for 2 h over 1 wt.% Pd/Mg(Al)O catalyst. (A): retention time from 0 to 32 min, (B): retention time from 11.1 to 14.1 min. Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; N2 pressure at 25 °C: 0.9 MPa.
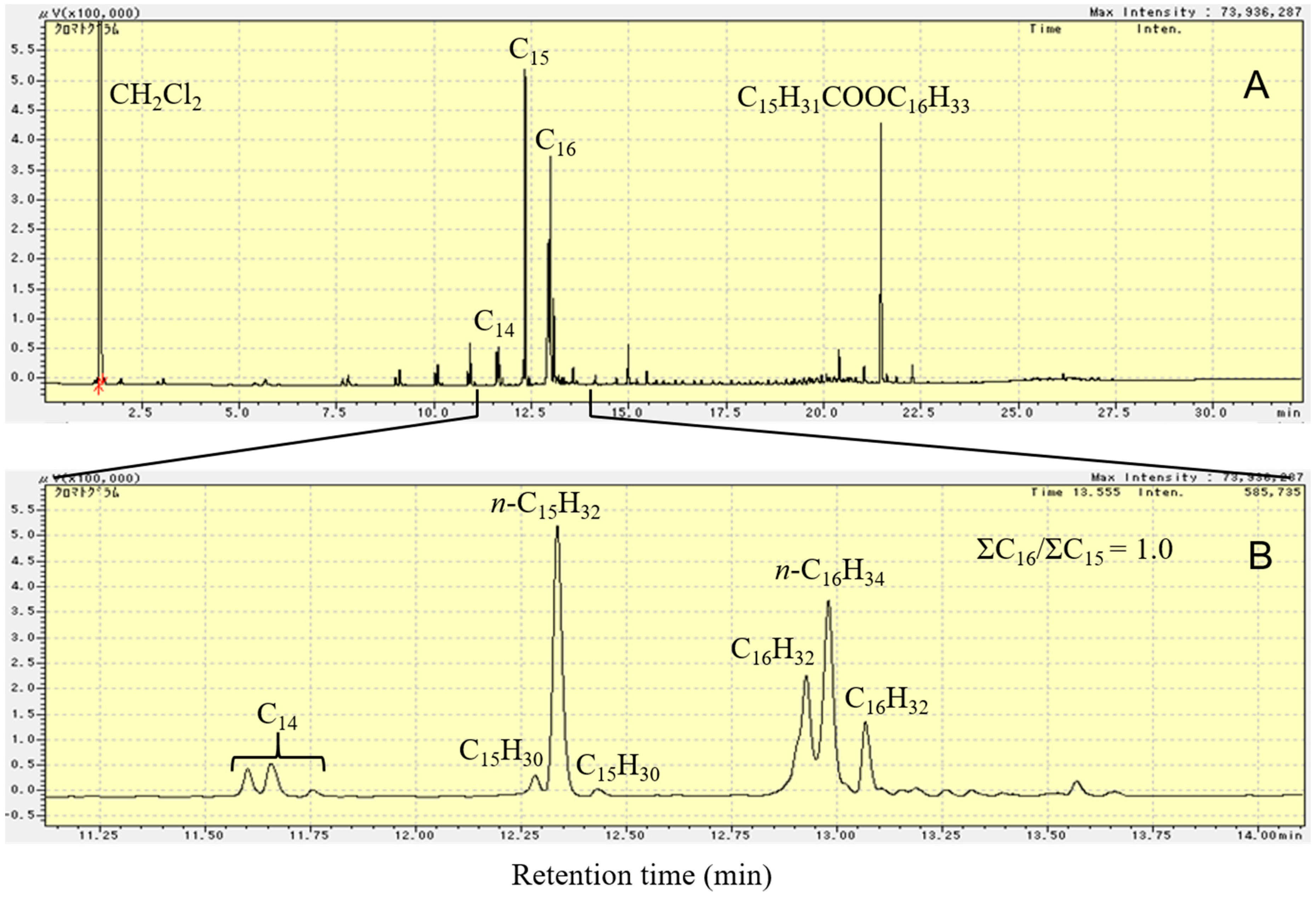
Figure 9.
The dependence of C15H31COOC16H33 conversion and C10–C16 hydrocarbons yield on reaction time for the catalytic deoxygenation of C15H31COOC16H33 in N2 atmosphere, at 300 °C, over the 1 wt.% Pd/Mg(Al)O catalyst. Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; N2 pressure at 25 °C: 0.9 MPa.
Figure 9.
The dependence of C15H31COOC16H33 conversion and C10–C16 hydrocarbons yield on reaction time for the catalytic deoxygenation of C15H31COOC16H33 in N2 atmosphere, at 300 °C, over the 1 wt.% Pd/Mg(Al)O catalyst. Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; N2 pressure at 25 °C: 0.9 MPa.
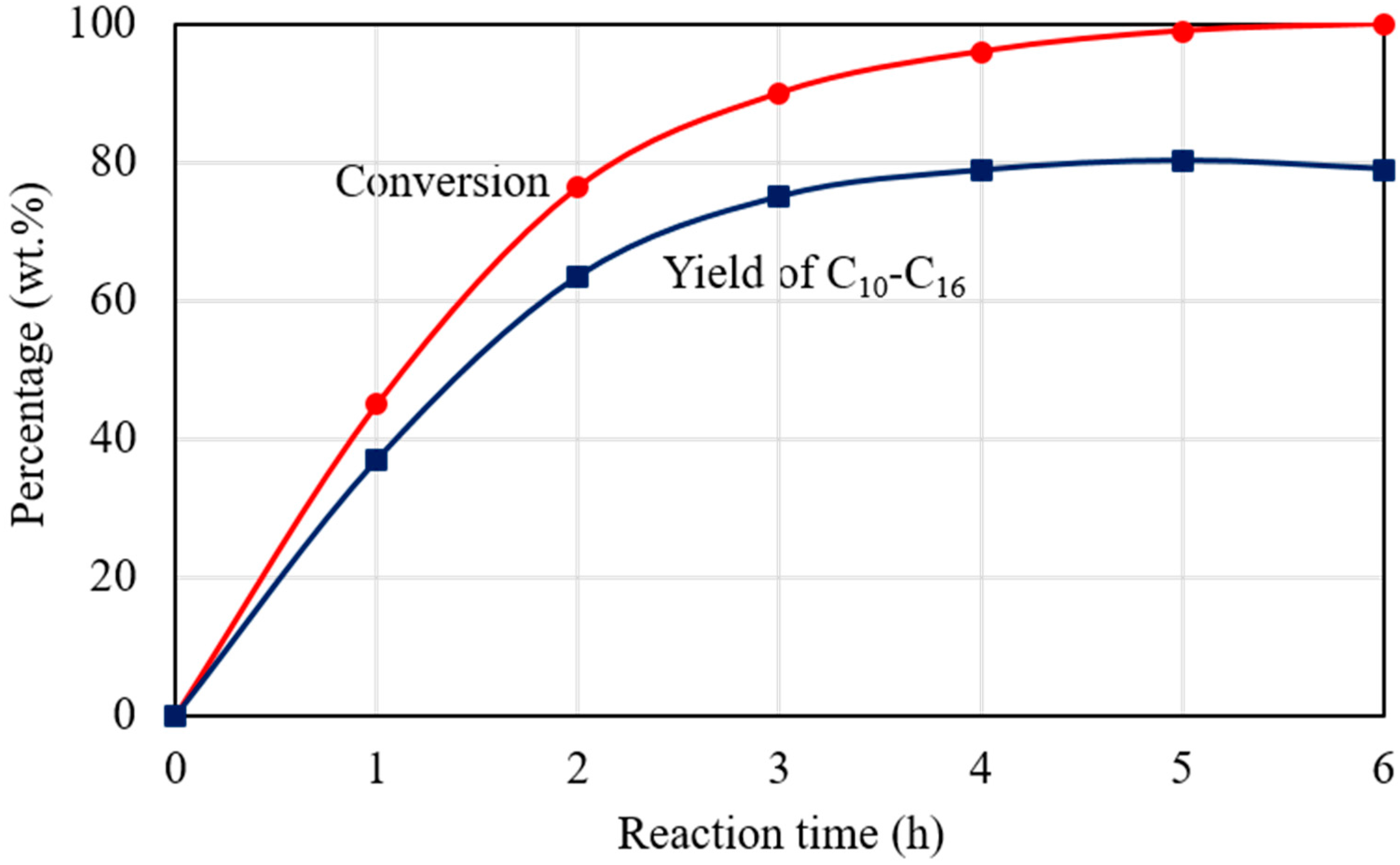
Figure 10.
The dependence of C15H31COOC16H33 conversion and C10‒C16 hydrocarbon yield on reaction temperature for the catalytic deoxygenation of C15H31COOC16H33 in N2 atmosphere, for 2 h, over the 1 wt.% Pd/Mg(Al)O catalyst. Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; N2 pressure at 25 °C: 0.9 MPa.
Figure 10.
The dependence of C15H31COOC16H33 conversion and C10‒C16 hydrocarbon yield on reaction temperature for the catalytic deoxygenation of C15H31COOC16H33 in N2 atmosphere, for 2 h, over the 1 wt.% Pd/Mg(Al)O catalyst. Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; N2 pressure at 25 °C: 0.9 MPa.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The C15H31COOC16H33 conversion and C10–C16 hydrocarbons yield of the deoxygenation of C15H31COOC16H33 in N2 atmosphere at 300 °C for 2 h over various catalysts a.
Table 1.
The C15H31COOC16H33 conversion and C10–C16 hydrocarbons yield of the deoxygenation of C15H31COOC16H33 in N2 atmosphere at 300 °C for 2 h over various catalysts a.
| Catalyst | Conversion (%) | Yield (%) | |||
|---|---|---|---|---|---|
| C1‒C4 | C5‒C9 | C10‒C16 | CO2 | ||
| Pd/Mg(Al)O b | 76.4 | 1.4 | 3.6 | 63.5 | 4.8 |
| Mg(Al)O | 23.3 | 0.5 | 1.7 | 15.1 | 1.6 |
| H-ZSM-5 | 55.6 | 5.8 | 26.3 | 8.3 | 3.1 |
| Pd/C b | 41.7 | 1.2 | 4.1 | 26.2 | 2.7 |
a Reaction conditions: C15H31COOC16H33: 5 g; catalyst: 0.2 g; N2 pressure at 25 °C: 0.9 MPa. b Pd loading: 1 wt.%.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, Y.; Inaba, M.; Matsuoka, K. Catalytic Deoxygenation of Hexadecyl Palmitate as a Model Compound of Euglena Oil in H2 and N2 Atmospheres. Catalysts 2017, 7, 333. https://doi.org/10.3390/catal7110333
AMA Style
Liu Y, Inaba M, Matsuoka K. Catalytic Deoxygenation of Hexadecyl Palmitate as a Model Compound of Euglena Oil in H2 and N2 Atmospheres. Catalysts. 2017; 7(11):333. https://doi.org/10.3390/catal7110333
Chicago/Turabian StyleLiu, Yanyong, Megumu Inaba, and Koichi Matsuoka. 2017. "Catalytic Deoxygenation of Hexadecyl Palmitate as a Model Compound of Euglena Oil in H2 and N2 Atmospheres" Catalysts 7, no. 11: 333. https://doi.org/10.3390/catal7110333
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.