Visible-Light Photocatalytic E to Z Isomerization of Activated Olefins and Its Application for the Syntheses of Coumarins

Department of Chemistry, Xi’an Jiaotong-Liverpool University, Suzhou 215123, China
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(11), 337; https://doi.org/10.3390/catal7110337
Submission received: 17 October 2017
/
Revised: 2 November 2017
/
Accepted: 7 November 2017
/
Published: 9 November 2017
Abstract
:Photocatalytic isomerization of thermodynamically stable E-alkene to less stable Z-alkene has been the subject of numerous studies, being successfully achieved mainly under UV irradiation. Recent development of visible light photoredox catalysis has witnessed it emerging as a powerful tool for the access of new structural complexity and many challenging targets. Herein, we report a visible light-promoted E to Z isomerization of cinnamates. When E-isomer of cinnamates was irradiated with blue light in the presence of an organo-photocatalyst, fac-Ir(ppy)3, Z-isomer was exclusively obtained in high yields and with good selectivity. The mild, convenient reaction condition has made this protocol an effective synthetic methodology, which was subsequently implemented in an efficient synthesis of coumarins.
1. Introduction
Photocatalysis under UV activation have been studied and developed for many decades and show capability of promoting the construction of a wide variety of nontraditional bonds in organic synthesis [1,2,3]. Recently, visible light photocatalysis emerged as an attractive strategy for activation of small molecules, when the chemical reaction is initiated mainly by the light-absorbing photocatalyst, providing mild conditions and high functional group tolerance [4,5,6,7,8].
Alkene has been widely used in organic synthesis as one of the most important building blocks and the essential intermediates in numerous organic transformations. The synthesis of alkene, especially alkene with specific geometry, had been the area of many efforts since the early era of organic chemistry, and is still the field of invention for modern organic synthesis. Over the decades, many methods have been established to obtain the thermodynamically more stable E-alkenes, however the reliable approaches to access the less stable Z-alkene are far less common [9,10,11,12]. For example, traditional Wittig reaction with unstable yield [13], alkyne hydrogenation, silicon-based stereospecific elimination and contemporary cross-coupling reaction [14], and cross-metathesis [15] are among a few methods which have so far enjoyed widespread applications, due to their simplicity, convenience, and relatively high geometrical control.
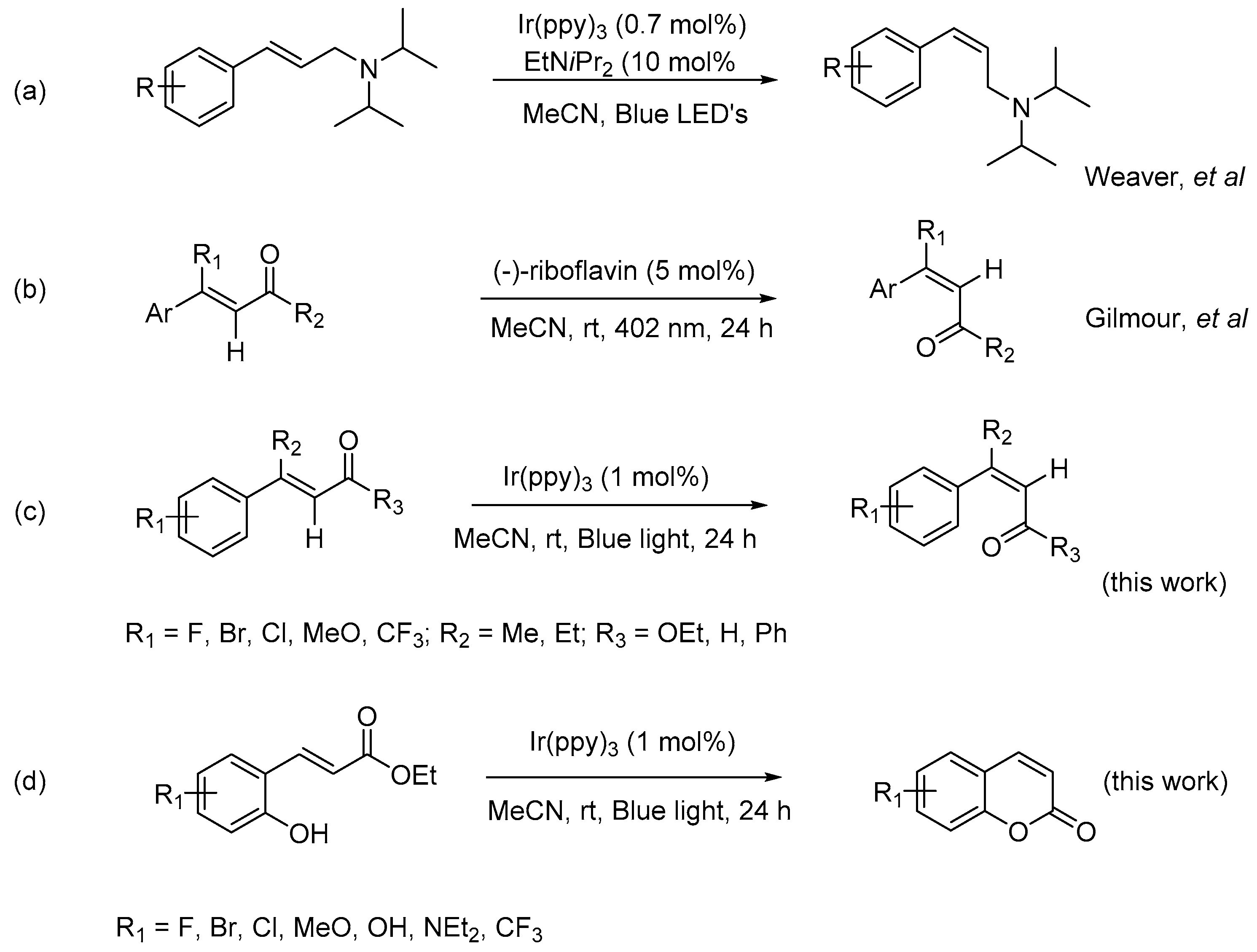
The direct photocatalytic E to Z isomerization of alkene, still considered as a fancy reaction in textbooks, has not yet won recognition as an orthodox method for the synthesis of Z-alkene and is a reaction of last resort. However, it has been well established that photochemical activation via a sensitizer, traditionally iodine [16], dyes; and currently photocatalyst [17], riboflavin [18], etc. can convert light into chemical energy and assist to circumvent the ‘uphill’ energy barrier of E to Z isomerization of alkene. The rotational barrier, which restricts the rotation about the double bond on the ground state, mitigates when the olefin is subjected to the photochemical excitation at an appropriate wavelength. Promotion of an electron by excitation from a π orbital to a π* orbital decreases the bond order and permits the rotation and subsequent relaxation to the ground state [19]. The isomerization of alkenes starts with triplet and singlet excited states, and a change in geometry leads to this twisted intermediate in form of zwitterionic or diradical. The sensitizers with a triplet state have higher energy than that of the excited state of the substrates, proving to be efficient for isomerization [20].
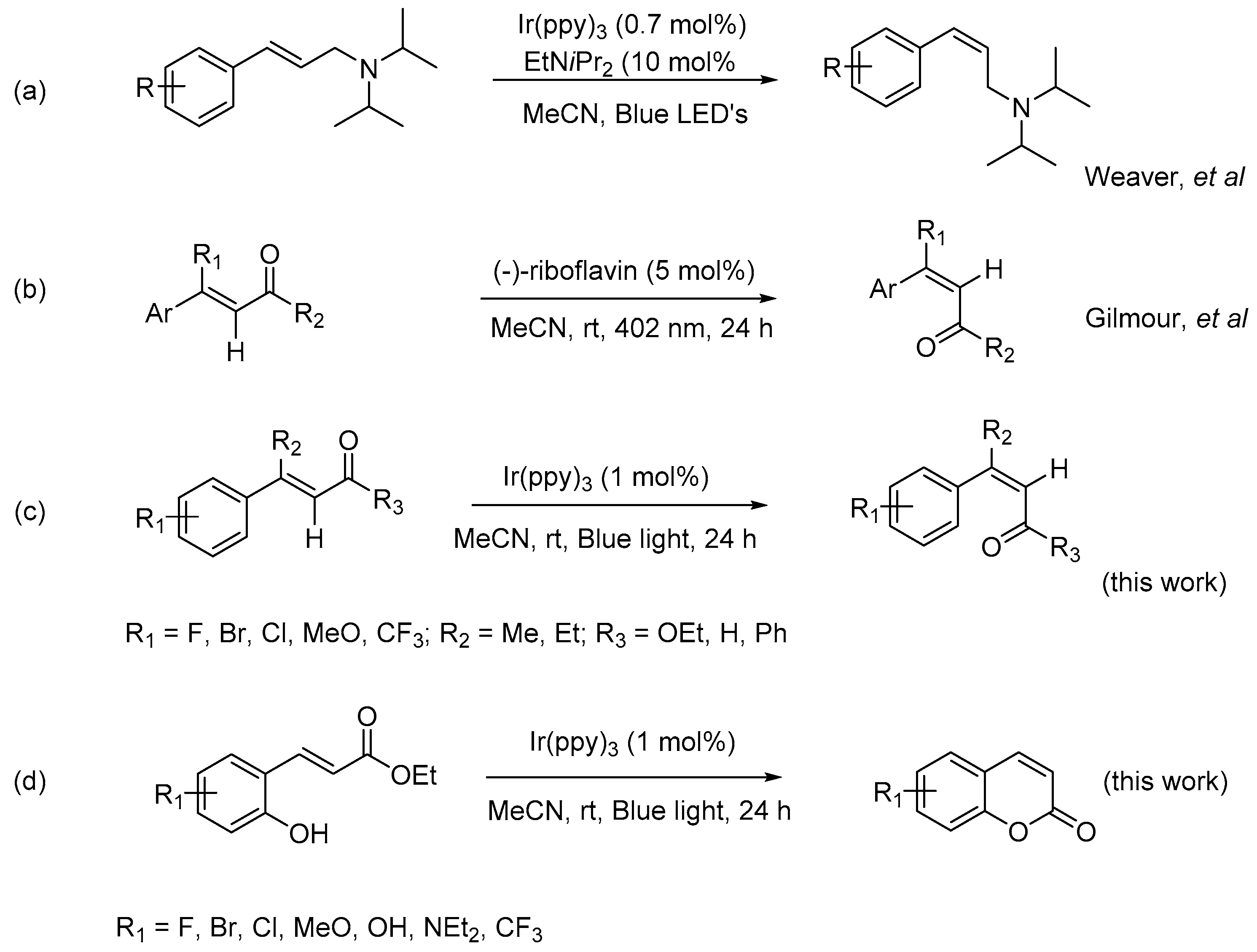
Numerous studies on theoretical perspectives and synthetic applications have been conducted. In 2014, Weaver discovered an isomerization of E-alkene to Z-isomer using blue LED (light-emitting diode) source, via a photochemical pumping mechanism, mediated by transition-metal photocatalyst [21]; however, the application of this method was limited to an allylamine system (Scheme 1a). In 2015, Gilmour reported a bio-inspired, catalytic E to Z isomerization of activated olefin, (Scheme 1b), translating Nature’s falvin-mediated photo-isomerization into the prototype of small molecule [18]. The transformation was successfully achieved via a selective UV-light irradiation at 402 nm. More recently, Wang and Zhai disclosed a blue-light-promoted carbon–carbon double bond isomerization and its application in the syntheses of quinolines in the absence of photocatalyst [22].
During our investigation of synthetic utilities of visible-light photoredox catalysts for carbon–carbon coupling reaction, it was noticed that the (E)-ethyl cinnamates could undergo an isomerization to form its Z-isomer in the presence of photoredox catalyst, this observation prompted us to explore the probability and potential of this transformation. Herein, we report the outcomes of this work, in which E to Z isomerization occurred when E-activated olefins were irradiated with blue light in the presence of organic photocatalyst, fac-Ir(ppy)3, to form its Z-isomer in high yields and good selectivity (Scheme 1c), and the synthetic applications of this transformation was perfectly demonstrated by a facile synthesis of coumarins (Scheme 1d). While the alkene isomerization has been achieved mostly with UV activation, this transformation could easily progress via the activation of visible-light with a photoredox catalyst.
2. Results and Discussion
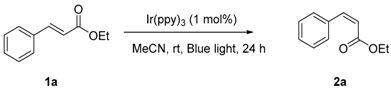
Starting from the first observation, we further investigated the photo-isomerization of (E)-ethyl cinnamates under various reaction conditions. It was verified that this transformation occurred in the presence of fac-Ir(ppy)3 (5 mol %) under visible-light irradiation with blue light, leading to the formation of its Z-isomer with a modest selectivity of Z/E 65:35 (Table 1, entry 1). The reaction was carried out at room temperature and finished in a quantitative yield, the products were purified after a simple filtration to remove the catalyst from the reaction mixture. Further experiments revealed that the photocatalyst was essential for the success of this reaction, no isomerization product could be detected in the absence of catalyst (entry 2). Encouraged by the initial results, we examined several catalysts, sensitizers for this photo-catalytic transformation and we found that transition-metal photoredox catalysts would generally catalyze this reaction, but produced diverse Z/E selectivity, iridium catalyst performed better than its ruthenium counterpart (entries 3 and 4), with fac-Ir(ppy)3 as the catalyst of choice to give the best selectivity. The varied catalytic activities could directly relate to the different emissive energies of the catalysts used in the investigation. fac-Ir(ppy)3 catalyst, which has a greater emissive energy (494 nm, 57.8 kcal mol−1) than the Ru(bpy)3Cl2 catalyst (615 nm, 46.5 kcal mol−1) [8,23], supposedly efficiently promotes the energy transfer to the substrate, its longer excited-state lifetime could also facilitate this process [24]. It is worth noting that (−)-riboflavin only showed marginal effect on the isomerization under visible light condition, compared with its significant performance under UV light (entry 5). Rose Bengal, the organic sensitizers, did not perform well either, the Z/E ratio could only reached 8:92 in 24 h under the reaction conditions (entry 6).
A screening of common solvents had identified acetonitrile as the best solvent for this reaction, providing the best efficiency and Z/E ratio of the products, the reaction carried out in other solvents either afforded worse selectivity (such as acetone) or were not productive at all (such as DMF (dimethylformamide), DCM (dichloromethane), and THF (tetrahydrofuran)) (entries 7–10). In contrast, Weaver et al. [21] described that the photochemical isomerization of cinnamyl derived amine worked well in all these solvents. Currently, it is not clear why the same trend was not observed in this case, as a reductive quenching mechanism was proposed in their studies. It is likely that these solvents stabilize the radical cation intermediate involved, while acetonitrile helps to elongate the lifetime of triplet excited states [25,26,27,28,29] or stabilize biradical intermediates in an energy transfer mechanism as suggested for our work. Acetone, a typical solvent which can be used as a sensitizer, was not effective enough by itself to achieve good transformation in this reaction. While the presence of photocatalyst was indispensable for the success of this reaction (entry 2), it is interesting to observe that the efficiency of E to Z isomerization was facilitated by the lower loading of photocatalyst for the reaction, which supposedly helps to reduce the self-quenching and avoid the general issue related to the lack of light penetration into solution, due to the high molar extinction coefficients of the photocatalyst (entries 11 and 12). The better results were obtained with 1 mol % of photocatalyst used in the reaction with the concentration of substrate at 0.1 M. The effect of external base on this isomerization was also studied with N,N-diisopropylethylamine (DIPEA, 10 mol %), (entry 13), there was no obvious influence noticed on the reaction efficiency. In their work, Weaver reported that the presence of air prevented the isomerization of allylamine from occurring [21], however, our experiments did not show that our reaction was air-sensitive, degassing the reaction mixture did not make the results any different (entry 14).
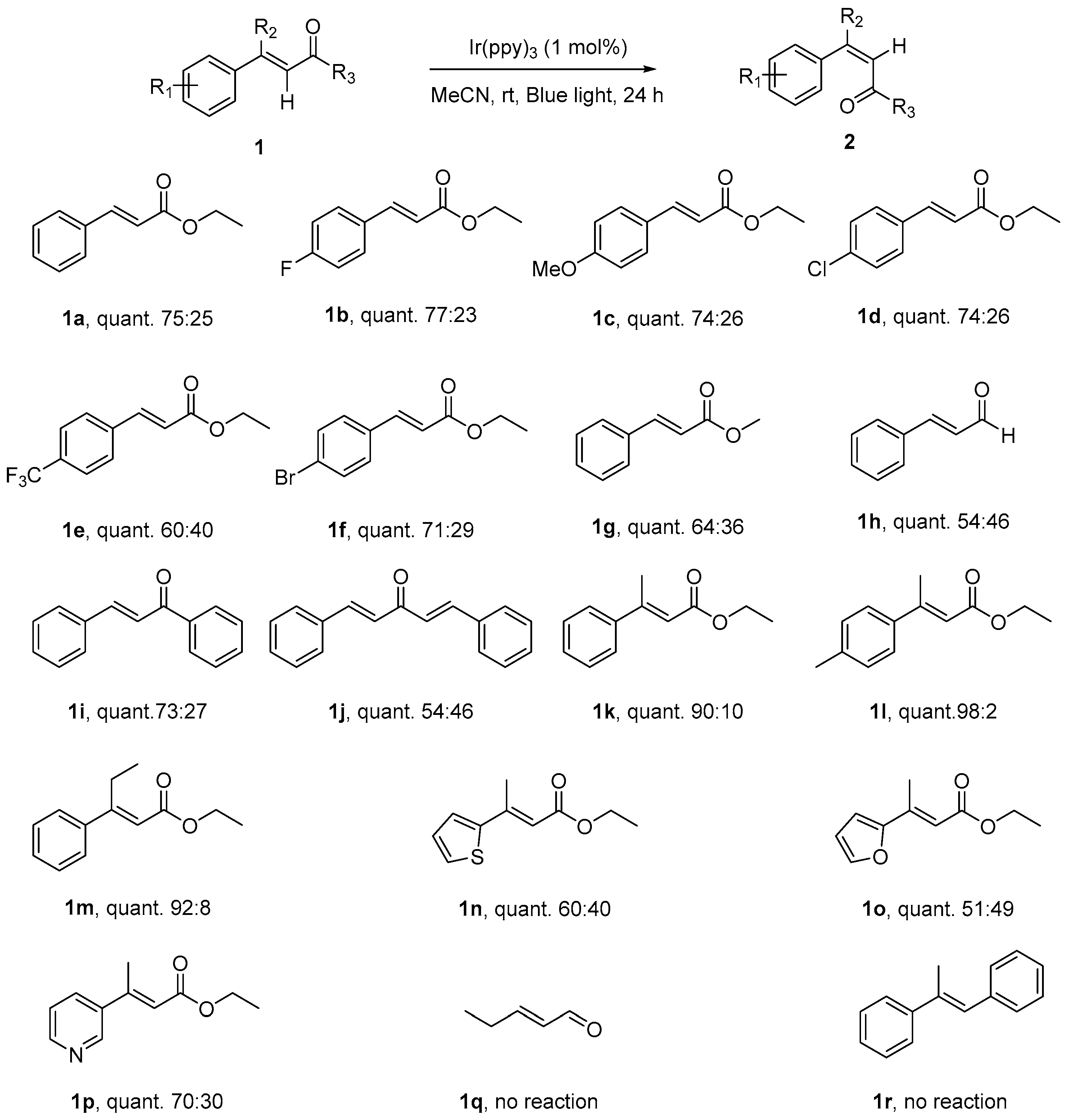
With the optimized reaction conditions in hand, we continued to examine the scope of E-olefins applicable in such transformation (Scheme 2). Various (E)-ethyl cinnamates—with electron-donating, electron-withdrawing, and neutral substituents at the para-position of benzene ring—could proceed effectively to give the isomerization product (Z)-isomers with modest of selectivity (1a–1g). The electronic properties of these substituents did not show apparent effects on the selectivity of this transformation, the Z/E selectivity of these reactions fell in the range from 60:40 to 75:25. Under the reaction condition, the substituents, such as F-, MeO-, Cl-, CF3-, and Br-, were well tolerated, there were no other byproducts could be detected along with the two isomers of the alkene, which made this reaction clean and practically very easy for workup. In addition, when (E)-cinnamyl aldehyde, (E)-α,β-unsaturated ketones were subjected to the reaction conditions, the corresponding Z-isomers were obtained with similar selectivity (1h–1j).
The selectivity of this isomerization could be greatly enhanced by small modifications of the structures, when a substituent, such as methyl- or ethyl- was introduced onto the β-position of double bond in (E)-ethyl cinnamates (1k–1m), a Z/E ratio as high as 98:2 could be attained. As reported by Gilmour in their seminal work [18], adjustment of the A 1,3-strain in the Z isomer by subtle alteration of the substitution on the double bond system could control efficiently the geometric outcome of this isomerization. Consistent with their findings, the isomerization of heterocycle substituted ethyl acrylates (2n, 2o, 2p), even with a β-substituent, could not reproduce the high selectivity shown by their aromatic counterparts, as the replacement of the six-membered aromatic ring with the less bulky five-membered heterocyclic ring reduces A 1,3-strain in the Z isomer, leading to an erosion of selectivity. Further investigation revealed that this visible light E to Z photoisomerization only occurred on the studied cinnamates system, the efforts on the other structural system, which lacks the essential conjugation when either the aromatic ring or the carbonyl functional group is missing (1q, 1r), were disappointingly futile, indicating the limitations of this isomerization.
The photoisomerization of alkene has been studied extensively using higher-energy UV light for the activation of the isolated double bond. The direct energy transfer generates the excited states of the substrate, which induces cleavage of the double bond to form zwitterions, biradicals, or radical pairs, providing a chance for the isomerization to proceed. On the other hand, the introduction of conjugated π system to the double bond can significantly decrease the energy gap between the ground state and excited state, making it possible for the isomerization to occur with low-energy light to be used as the source of activation, especially with the aid of photocatalysts, which have witnessed great development in the past decade.
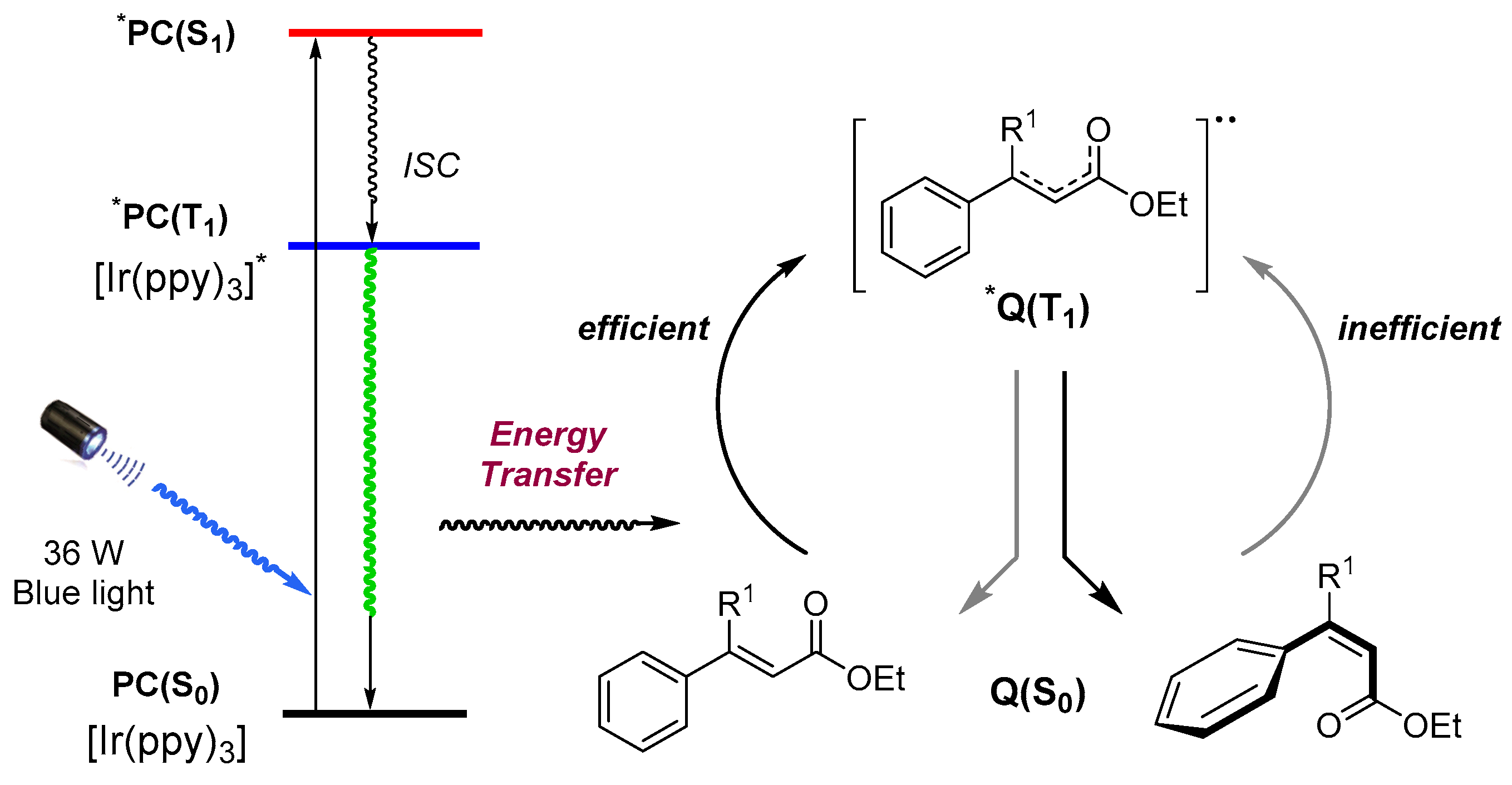
From the results of our work, it is clearly demonstrated that organo-photoredox catalyst—such as fac-Ir(ppy)3—can facilitate the energy transfer from visible light to the cinnamates system for the activation of the double bond. A plausible pathway was thus proposed, based on the literature precedents [18,30] and experimental data (Scheme 3). Absorbing a photon, the photocatalyst PC(S0) in its singlet ground state is excited to the first singlet excited state *PC(S1), which relaxes to the lowest energy triplet excited state *PC(T1) via successive fast intersystem crossing (ISC) (spin–orbital coupling) and internal conversion (vibrational relaxation). Since the transition of the triplet excited state to the singlet ground state is spin forbidden, the triplet excited state *PC(T1) is reasonably long lived. In photocatalysis, the photo-excited catalyst can be quenched by the substrates via outer-sphere single electron transfer (SET) or energy transfer (ET) processes, leading to productive transformation. It is difficult to completely exclude a single electron transfer (SET) pathway in our visible-light photocatalytic E-to-Z isomerization, However, this pathway was ruled out as the addition of a sacrificial reductant (e.g., trimethylamine, N,N-Diisopropylethylamine), the classical strategy to promote photoredox reaction [8,31], did not facilitate this transformation. Thus, we tentatively proposed that an energy transfer process occurs between the photo-excited triplet state *PC(T1) and the accessible low energy triplet state of cinnamates substrate in this isomerization. The triplet–triplet energy transfer regenerates the ground state of the photocatalyst and results in the substrate of an excited triplet state to engage in the photochemical isomerization reactions.
The selectivity of the isomerization is reliant on the different rates of the photoquenching of the excited photocatalyst by the two isomers. There is strong evidence [18,21] to suggest that E-isomer quenches the photoexcited state of the catalyst at a speed much faster that its counterpart of Z-isomer, thus resulting in the buildup of Z-isomer in this process, which, in turn, is related with physical structures of the two isomers, as better conjugation is possessed in the E-isomer and the substituents on the Z-double bond are forced out of plane by the steric hindrances to generate deconjugation in Z-isomer. Under the optimized reaction condition, the best Z/E ratio we could manage to achieve for the isomerization of (E)-ethyl cinnamates was 75:25 in 24 h. A time-course analysis of the reaction of (Z)-ethyl cinnamates, monitored by GC/MS, revealed that this photo-stationary state could be reached in 2 h. Interestingly, the selectivity of isomerization could be finely tuned by the introduction of more substituents on the double bond or the aromatic ring itself, which affects the degree of deconjugation in Z-isomer.
Encouraged by this successful isomerization of cinnamates, we reckoned that this facile isomerization of alkenes under mild reaction conditions created an attractive synthetic methodology to circumvent the encumbrance rendered by the double bond, thus useful for application in organic synthesis.
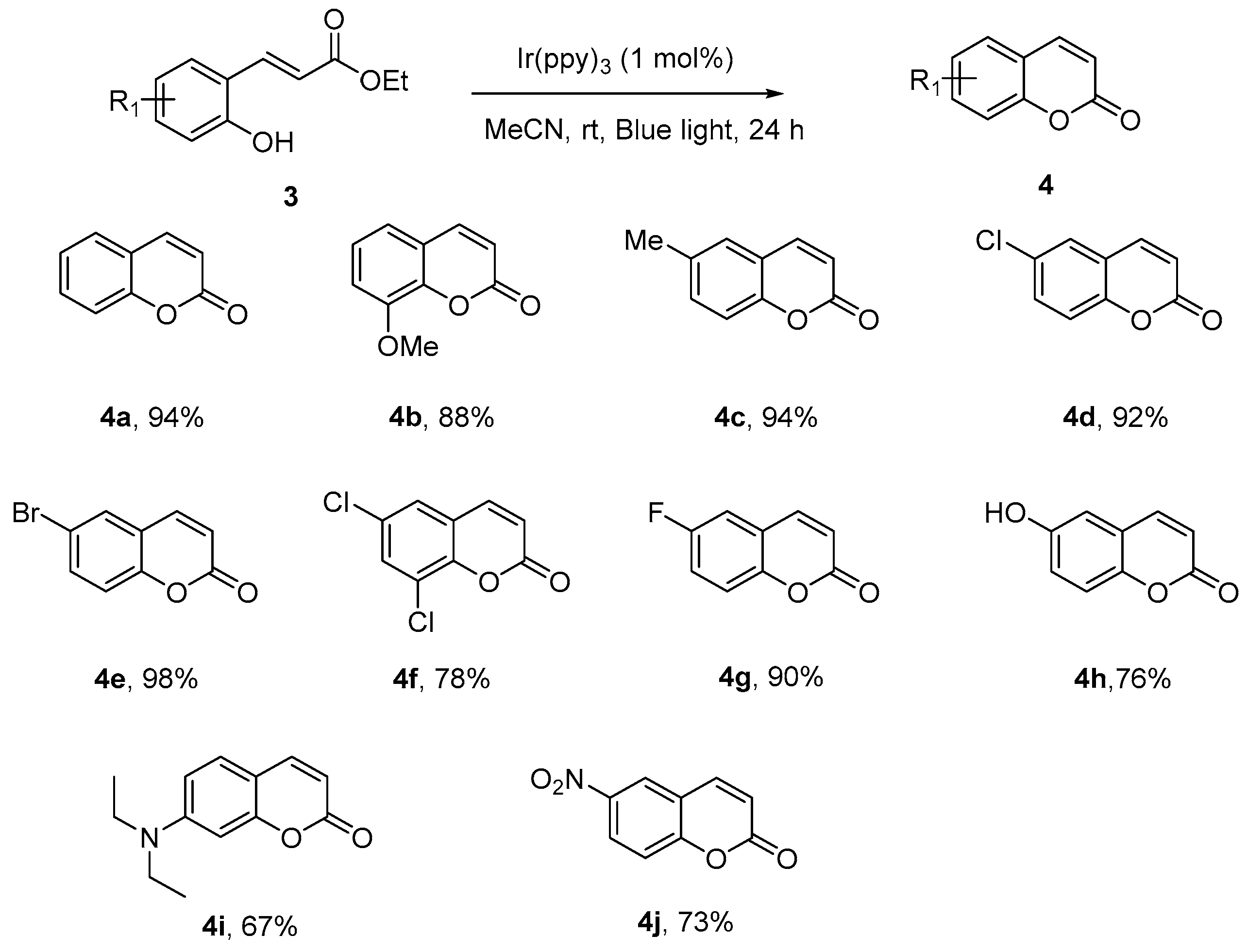
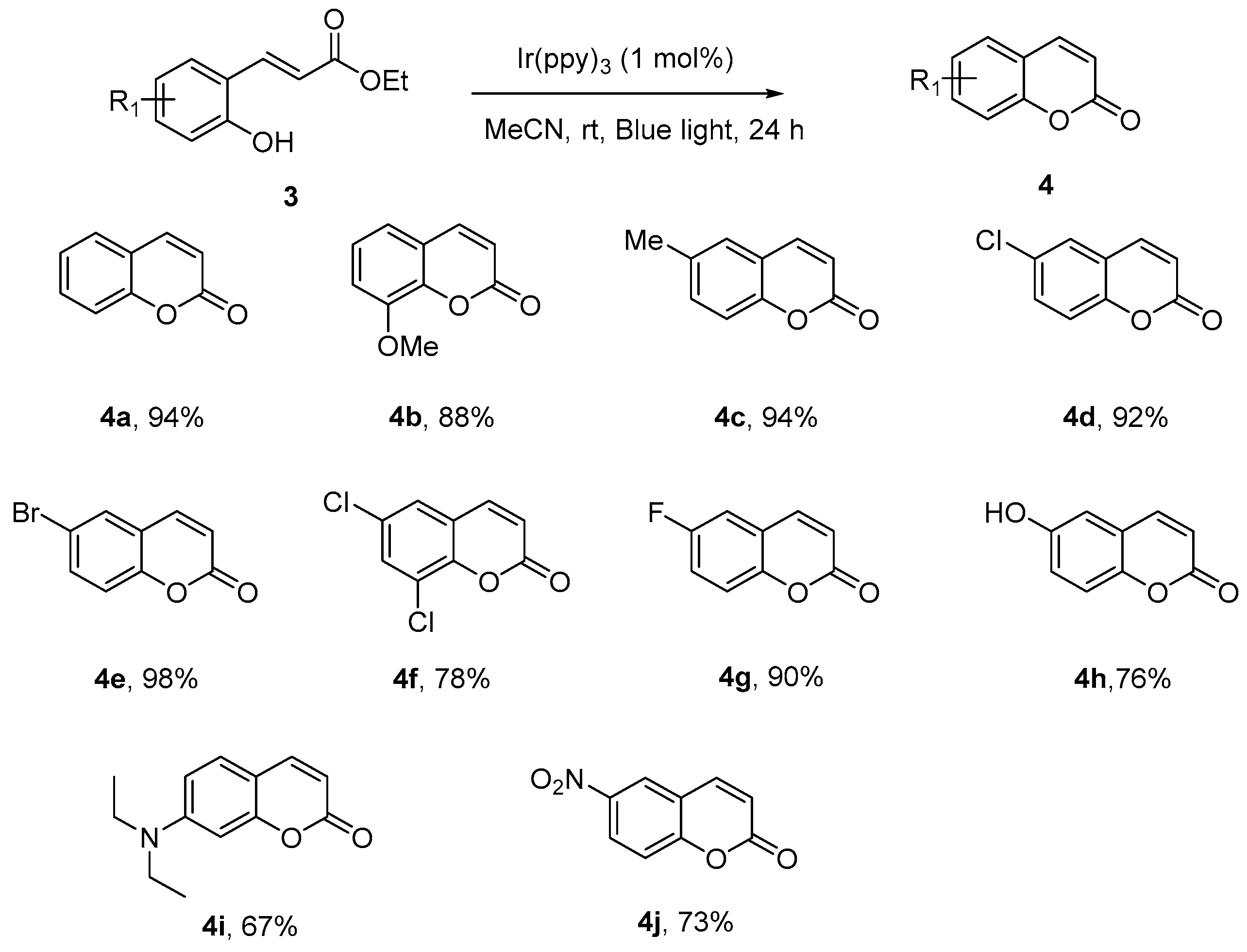
Coumarins, the important structural motifs in natural products and bioactive compounds, exhibiting broad biological activities, have been the targets for many organic syntheses [32,33,34]. Among many successful methods developed over the years, cyclization of ortho-hydroxycinnamates via double-bond isomerization was the one which was generally convenient from easily obtained starting material and afforded the coumarins in very good yields. A traditional method adopted to overcome the kinetic and energetic barrier against this isomerization includes high temperatures, boron tribromide, and nucleophile. The first synthesis via photochemical double-bond isomerization was achieved by Horaguchi [35], when a 400 W high-pressure mercury lamp was employed.
We decided to test our optimized reaction conditions on the synthesis of coumarin and were delighted to found that the transformations could completed in 24 h (Scheme 4). Generally ortho-(E)-hydroxycinnamates, which contain various substituents, underwent the visible light-promoted E-to-Z isomerization readily and the generated Z isomers could undergo lactonization smoothly to afford the coumarin compounds in very high yields (4a–4j). This effectively demonstrated the synthetic potential of this protocol for future applications in the target synthesis.
3. Experimental Details
3.1. General Procedure for the Isomerization of α,β-Unsaturated Carbonyl Compounds
The specified α,β-unsaturated carbonyl compound (0.2 mmol) and fac-Ir(ppy)3 (0.002 mmol, 1 mol %) were dissolved in 2 mL MeCN, the mixture was stirred under blue light (36 W, fluorescent bulb, peak wavelength 445 nm, FWHM (full width at half maximum) 50 nm) irradiation for 24 h at room temperature. The reaction was monitored by GC-MS (Agilent 5975C Triple Axis GC-MS, Santa Clara, CA, USA). After the trans-isomer reaches maximum conversion to cis-isomer, MeCN is removed under reduced pressure. The crude product was purified by flash chromatography using n-hexane and diethyl ether as eluent.
3.2. General Procedure for the Synthesis Ortho-Hydroxycinnamates (3)
To a solution of salicylaldehyde (10 mmol) in dry CH2Cl2 (40 mL) was added (carbethoxymethylene)triphenylphosphorane (15 mmol, 1.5 equiv.) and the reaction mixture was stirred at room temperature for 2 h. After the reaction completed, the solvent was concentrated under reduced pressure. The residue was purified by flash chromatography using n-hexane and ethyl acetate as eluent (see Supplementary Materials).
3.3. General Procedure for the Synthesis of Coumarin (4)
The specified ortho-hydroxycinnamates (0.2 mmol) and fac-Ir(ppy)3 (0.002 mmol, 1 mol %) were dissolved in 2 mL MeCN, the mixture was stirred under blue light (36 W, fluorescent bulb) irradiation for 24 h at room temperature. The reaction was monitored by GC-MS. When the reaction completed, MeCN was removed under reduced pressure. The crude product was purified by flash chromatography using n-hexane and ethyl acetate as eluent (see Supplementary Materials).
4. Conclusions
In summary, we have discovered that the isomerization of thermodynamically stable E-alkene to less-stable Z-alkene could be achieved under visible-light irradiation in the presence of organo- photocatalyst, fac-Ir(ppy)3. Z-isomer of cinnamates could be easily obtained in good yields and selectivity. Consistent with the previous report, the selectivity of isomerization was able to be tuned by the introduction of substituents on the double bond or the aromatic ring itself. The mild, convenient reaction conditions make this transformation an effective methodology for organic synthesis, which was properly demonstrated with a successful application in the synthesis of biological active coumarin compounds.
Supplementary Materials
The following are available online at www.mdpi.com/2073-4344/7/11/337/s1, Experimental procedure and spectral data for the precursors and final products.
Acknowledgments
The authors thank the Jiangsu Science and Technology Program (Basic Research Program) (Grant No. BK20131180) for the funding, and XJTLU RDF (Research Development Fund) for PhD scholarship to K.Z.
Author Contributions
Y.L. conceived and designed the experiments; K.Z. performed the experiments and analyzed the data; Y.L. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Snyder, J.J.; Tise, F.P.; Davis, R.D.; Kropp, P.J. Photochemistry of alkenes 7. E-reversible Z isomerization of alkenes sensitized with benzene and derivatives. J. Org. Chem. 1981, 46, 3609–3611. [Google Scholar]
- Dugave, C.; Demange, L. Cis-trans isomerization of organic molecules and biomolecules: Implications and applications. Chem. Rev. 2003, 103, 2475–2532. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Akita, M. Fine design of photoredox systems for catalytic fluoromethylation of carbon-carbon multiple bonds. Acc. Chem. Res. 2016, 49, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Zeitler, K. Photoredox catalysis with visible light. Angew. Chem. Int. Ed. Engl. 2009, 48, 9785–9789. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.U.; Strakova, K.; Slanina, T.; Konig, B. Eosin Y (EY) photoredox-catalyzed sulfonylation of alkenes: Scope and mechanism. Chem. Eur. J. 2016, 22, 8694–8699. [Google Scholar] [CrossRef] [PubMed]
- Ochola, J.R.; Wolf, M.O. The effect of photocatalyst excited state lifetime on the rate of photoredox catalysis. Org. Biomol. Chem. 2016, 14, 9088–9092. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Q.; Chen, J.R.; Xiao, W.J. Controllable remote C–H bond functionalization by visible-light photocatalysis. Angew. Chem. Int. Ed. Engl. 2017, 56, 1960–1962. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Twilton, J.; MacMillan, D.W. Photoredox catalysis in organic chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef] [PubMed]
- Goralski, C.T.; Singaram, B.; Rangaishenvi, M.V.; Brown, H.C. Stereospecific synthesis of pure [Z]-alkenes and [E]-alkenes from enamines via hydroboration. Abstr. Pap. Am. Chem. Soc. 1988, 195, 242. [Google Scholar]
- Hull, C.; Mortlock, S.V.; Thomas, E.J. Stereoselective synthesis of Z-alkenes from alpha-methylcrotylstannanes and aldehydes. Tetrahedron Lett. 1987, 28, 5343–5346. [Google Scholar] [CrossRef]
- Sano, S.; Takehisa, T.; Ogawa, S.; Yokoyama, K.; Nagao, Y. Stereoselective synthesis of tetrasubstituted (Z)-alkenes from aryl alkyl ketones utilizing the Horner-Wadsworth-Emmons reaction. Chem. Pharm. Bull. 2002, 50, 1300–1302. [Google Scholar] [CrossRef] [PubMed]
- Siau, W.Y.; Zhang, Y.; Zhao, Y. Stereoselective synthesis of Z-alkenes. Top. Curr. Chem. 2012, 327, 33–58. [Google Scholar] [PubMed]
- Pelter, A.; Buss, D.; Colclough, E. Stereoselective synthesis of E-alkene and Z-alkene by the boron-wittig reaction. J. Chem. Soc. Chem. Commun. 1987, 297–299. [Google Scholar] [CrossRef]
- Wang, K.K.; Chu, K.H. Preparation of (Z)-alkenes, ketones, and alkynes via organoboranes—Stereoselective synthesis of the sex-pheromones of the douglas-fir tussock moth, the gypsy-moth, and the wild silkmoth antheraea polyphemus. Abstr. Pap. Am. Chem. Soc. 1984, 188, 21. [Google Scholar]
- Wang, C.B.; Yu, M.; Kyle, A.F.; Jakubec, P.; Dixon, D.J.; Schrock, R.R.; Hoveyda, A.H. Efficient and selective formation of macrocyclic disubstituted z alkenes by ring-closing metathesis (RCM) reactions catalyzed by Mo- or W-based monoaryloxide pyrrolide (MAP) complexes: Applications to total syntheses of epilachnene, yuzu lactone, ambrettolide, epothilone c, and nakadomarin a. Chem. Eur. J. 2013, 19, 2726–2740. [Google Scholar] [PubMed]
- Hepperle, S.S.; Li, Q.B.; East, A.L.L. Mechanism of cis/trans equilibration of alkenes via iodine catalysis. J. Phys. Chem. A 2005, 109, 10975–10981. [Google Scholar] [CrossRef] [PubMed]
- Guignard, R.F.; Petit, L.; Zard, S.Z. A method for the net contra-thermodynamic isomerization of cyclic trisubstituted alkenes. Org. Lett. 2013, 15, 4178–4181. [Google Scholar] [CrossRef] [PubMed]
- Metternich, J.B.; Gilmour, R. A bio-inspired, catalytic E→Z isomerization of activated olefins. J. Am. Chem. Soc. 2015, 137, 11254–11257. [Google Scholar] [CrossRef] [PubMed]
- Metternich, J.B.; Gilmour, R. Photocatalytic E to Z isomerization of alkenes. Synlett 2016, 27, 2541–2552. [Google Scholar]
- Metternich, J.B.; Artiukhin, D.G.; Holland, M.C.; von Bremen-Kühne, M.; Neugebauer, J.; Gilmour, R. Photocatalytic E→Z isomerization of polarized alkenes inspired by the visual cycle: Mechanistic dichotomy and origin of selectivity. J. Org. Chem. 2017, 82, 9955–9977. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.D.; Singh, K.; Staig, S. Facile synthesis of Z-alkenes via uphill catalysis. J. Am. Chem. Soc. 2014, 136, 5275–5278. [Google Scholar]
- Chen, X.; Qiu, S.; Wang, S.; Wang, H.; Zhai, H. Blue-light-promoted carbon-carbon double bond isomerization and its application in the syntheses of quinolines. Org. Biomol. Chem. 2017, 15, 6349–6352. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.B.; Patel, N.R.; Primer, D.N.; Jouffroy, M.; Tellis, J.C.; Molander, G.A. Preparation of visible-light-activated metal complexes and their use in photoredox/nickel dual catalysis. Nat. Protoc. 2017, 12, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Oderinde, M.S.; Varela-Alvarez, A.; Aquila, B.; Robbins, D.W.; Johannes, J.W. Effects of molecular oxygen, solvent, and light on iridium-photoredox/nickel dual-catalyzed cross-coupling reactions. J. Org. Chem. 2015, 80, 7642–7651. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.R.; Umapathy, S. Solvent effects on the structure of the triplet excited state of xanthone: A time-resolved resonance raman study. J. Raman Spectrosc. 2016, 47, 1220–1230. [Google Scholar] [CrossRef]
- Narra, S.; Shigeto, S. Direct observation of the solvent effects on the low-lying nπ and ππ* excited triplet states of acetophenone derivatives in thermal equilibrium. J. Phys. Chem. B 2015, 119, 3808–3814. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.T.; Thomas, J.K. Laser and flash photolytic studies on effects of various solvents and solutes on excited singlet and triplet states of N,N,N’N’, tetramethyl paraphenylene diamine (TMPD). Radiat. Res. 1969, 39, 535. [Google Scholar]
- Ohashi, Y.; Kobayashi, T. Study on electronic excited-states of iridium(iii) complexes containing bipyridine and phenanthroline ligands—Solvent effect on triplet-triplet absorption-spectra. Bull. Chem. Soc. Jpn. 1979, 52, 2214–2217. [Google Scholar] [CrossRef]
- Schultz, D.M.; Yoon, T.P. Solar synthesis: Prospects in visible light photocatalysis. Science 2014, 343, 1239176. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Akita, M. Visible-light radical reaction designed by Ru- and Ir-based photoredox catalysis. Inorg. Chem. Front. 2014, 1, 562–576. [Google Scholar] [CrossRef]
- Boeck, F.; Blazejak, M.; Anneser, M.R.; Hintermann, L. Cyclization of ortho-hydroxycinnamates to coumarins under mild conditions: A nucleophilic organocatalysis approach. Beilstein J. Org. Chem. 2012, 8, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wang, Z.; Li, Y.; Wu, X.F. Iridium-catalyzed and ligand-controlled carbonylative synthesis of flavones from simple phenols and internal alkynes. Chem. Eur. J. 2017, 23, 3276–3279. [Google Scholar] [CrossRef] [PubMed]
- Metternich, J.B.; Gilmour, R. One photocatalyst, n activation modes strategy for cascade catalysis: Emulating coumarin biosynthesis with (–)-riboflavin. J. Am. Chem. Soc. 2016, 138, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Horaguchi, T.; Hosokawa, N.; Tanemura, K.; Suzuki, T. Photocyclization reactions. Part 8 [1]. Synthesis of 2-quinolone, quinoline and cournarin derivatives using trans-cis isomerization by photoreaction. J. Heterocycl. Chem. 2002, 39, 61–67. [Google Scholar] [CrossRef]
Scheme 1.
Photocatalytic E to Z isomerization of olefins.
Scheme 2.
Scope of photocatalytic isomerization of α,β-unsaturated carbonyl compounds. a Reaction conditions: (E)-α,β-unsaturated carbonyl compound (0.2 mmol) and fac-Ir(ppy)3 (0.002 mmol, 1 mol %) were dissolved in 2 mL MeCN, at room temperature under 24 h of blue light irradiation; b Isolated Yields; c Z/E ratios were determined by GC/MS.
Scheme 2.
Scope of photocatalytic isomerization of α,β-unsaturated carbonyl compounds. a Reaction conditions: (E)-α,β-unsaturated carbonyl compound (0.2 mmol) and fac-Ir(ppy)3 (0.002 mmol, 1 mol %) were dissolved in 2 mL MeCN, at room temperature under 24 h of blue light irradiation; b Isolated Yields; c Z/E ratios were determined by GC/MS.
Scheme 3.
Plausible pathway for the visible-light photocatalytic E to Z isomerization. PC: photocatalyst; Q: quencher (substrate); ISC: intersystem crossing; S0: singlet ground state; S1: first singlet excited state; and T1: first triplet excited state.
Scheme 3.
Plausible pathway for the visible-light photocatalytic E to Z isomerization. PC: photocatalyst; Q: quencher (substrate); ISC: intersystem crossing; S0: singlet ground state; S1: first singlet excited state; and T1: first triplet excited state.
Scheme 4.
Synthesis of coumarin compounds via photochemical isomerization. a Reaction conditions: Specified ortho-hydroxycinnamates (0.2 mmol) and fac-Ir(ppy)3 (0.002 mmol, 1 mol %) were dissolved in 2 mL MeCN, the mixture was stirred under blue light irradiation for 24 h at room temperature. b Isolated yields.
Scheme 4.
Synthesis of coumarin compounds via photochemical isomerization. a Reaction conditions: Specified ortho-hydroxycinnamates (0.2 mmol) and fac-Ir(ppy)3 (0.002 mmol, 1 mol %) were dissolved in 2 mL MeCN, the mixture was stirred under blue light irradiation for 24 h at room temperature. b Isolated yields.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected optimization of reaction conditions 1.
| Entry | Catalyst | Loading of Cat. (mol %) | Solvent | Other Conditions | Z/E Ratio 2 |
|---|---|---|---|---|---|
| 1 | fac-Ir(ppy)3 | 5 | MeCN | - | 65:35 |
| 2 | none | 5 | MeCN | - | 1:99 |
| 3 | Ru(bpy)3Cl2 | 5 | MeCN | - | 18:82 |
| 4 | Ir(ppy)2(dtbbpy)+ | 5 | MeCN | - | 31:69 |
| 5 | (−)-riboflavin | 5 | MeCN | - | 8:92 |
| 6 | Rose bengal | 5 | MeCN | - | 6:94 |
| 7 | fac-Ir(ppy)3 | 5 | acetone | - | 37:63 |
| 8 | fac-Ir(ppy)3 | 5 | DMF | - | 2:98 |
| 9 | fac-Ir(ppy)3 | 5 | DCM | - | 2:98 |
| 10 | fac-Ir(ppy)3 | 5 | THF | - | 2:98 |
| 11 | fac-Ir(ppy)3 | 3 | MeCN | - | 68:32 |
| 12 | fac-Ir(ppy)3 | 1 | MeCN | - | 75:25 |
| 13 | fac-Ir(ppy)3 | 1 | MeCN | DIPEA | 72:28 |
| 14 | fac-Ir(ppy)3 | 1 | MeCN | degassed | 73:27 |
1 Reaction conditions: Reactions were performed with 0.2 mmol of substrates in indicated solvent (0.1 M) at room temperature in the presence of catalyst under 24 h of blue light irradiation. 2 Z/E ratios were determined by GC/MS (gas chromatography–mass spectrometry).
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhan, K.; Li, Y. Visible-Light Photocatalytic E to Z Isomerization of Activated Olefins and Its Application for the Syntheses of Coumarins. Catalysts 2017, 7, 337. https://doi.org/10.3390/catal7110337
AMA Style
Zhan K, Li Y. Visible-Light Photocatalytic E to Z Isomerization of Activated Olefins and Its Application for the Syntheses of Coumarins. Catalysts. 2017; 7(11):337. https://doi.org/10.3390/catal7110337
Chicago/Turabian StyleZhan, Kun, and Yi Li. 2017. "Visible-Light Photocatalytic E to Z Isomerization of Activated Olefins and Its Application for the Syntheses of Coumarins" Catalysts 7, no. 11: 337. https://doi.org/10.3390/catal7110337
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.