Base Catalysis by Mono- and Polyoxometalates


1
Laboratory for Materials and Structures, Institute of Innovative Research, Tokyo Institute of Technology, Nagatsuta-cho 4259, Midori-ku, Yokohama 226-8503, Japan
2
Japan Science and Technology Agency (JST), Precursory Research for Embryonic Science and Technology (PRESTO), 4-1-8 Honcho, Kawaguchi 332-0012, Japan
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(11), 345; https://doi.org/10.3390/catal7110345
Submission received: 17 October 2017
/
Revised: 9 November 2017
/
Accepted: 13 November 2017
/
Published: 16 November 2017
(This article belongs to the Special Issue Recent Advances in Polyoxometalate-Catalyzed Reactions)
Abstract
:In sharp contrast with acid-, photo-, and oxidation-catalysis by polyoxometalates, base catalysis by polyoxometalates has scarcely been investigated. The use of polyoxometalates as base catalysts have very recently received much attention and has been extensively investigated. Numerous mono- and polyoxometalate base catalyst systems effective for the chemical fixation of CO2, cyanosilylation of carbonyl compounds, and C-C bond forming reactions have been developed. Mono- and polyoxometalate base catalysts are classified into four main groups with respect to their structures: (a) monomeric metalates; (b) isopolyoxometalates; (c) heteropolyoxometalates; and (d) transition-metal-substituted polyoxometalates. This review article focuses on the relationship among the molecular structures, the basic properties, and the unique base catalysis of polyoxometalates on the basis of groups (a)–(d). In addition, reaction mechanisms including the specific activation of substrates and/or reagents such as the abstraction of protons, nucleophilic action toward substrates, and bifunctional action in combination with metal catalysts are comprehensively summarized.
1. Introduction
Base-catalyzed reactions such as the isomerization of alkenes/alkynes, C-C bond forming reactions (aldol condensation, Michael addition, Henry reaction, etc.), the Tishchenko reaction, hydrogenation, (trans)esterification, and oxidation are important for the production of bulk and specialty chemicals [1,2,3,4,5,6]. Antiquated technologies with (super)stoichiometric amounts of inorganic and organic bases are still widely used for the manufacture of chemicals (especially fine-chemicals). However, these reagents are usually dissolved in the reaction media, so catalyst/product(s) separation (e.g., undesired formation of byproducts and product contamination during the neutralization process with acids) and reuse of the catalysts are very difficult. Therefore, the use and generation of toxic and hazardous substances can be reduced by replacement of the stoichiometric methodologies with easily recoverable and recyclable base catalysts. Inorganic solid base catalyst materials such as mixed oxides, zeolites, metal phosphates, and metal oxynitrides have been developed [4]. Among these, the basic properties of mixed oxides have been extensively investigated and various effective base catalysts have been reported. However, difficulty in the construction of electrically and structurally controlled uniform basic sites often leads to a problem where the fine-tuning of the catalyst structure and reactivity are limited. Therefore, the design and development of new high-performance inorganic base catalysts remains a strongly desired and challenging subject of research.
Polyoxometalates (POMs) are a large family of anionic metal-oxygen clusters that contain the early transition metals [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. POMs have stimulated many current research activities over a broad spectrum of science, such as in the fields of catalysis, materials, and medicine, because their chemical properties such as redox potential, acidity, and solubility in various media can be finely tuned by the selection of appropriate constituent elements and counter cations. POMs have the following advantages as catalysts: (i) the redox and acid-base properties can be fine-tuned by changing the chemical structures and compositions; (ii) POMs are less susceptible to oxidative and thermal degradation than organometallic complexes; and (iii) the catalytically active sites of POMs can be precisely controlled with an appropriate combination of transition metals and lacunary POMs as inorganic ligands. These superior properties allow the POMs-based catalysts to be designed at the atomic and molecular levels [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35].
Various POM catalysts have been developed for acid-catalyzed, photocatalytic, and oxidation reactions; however, base catalysis has scarcely been investigated [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. In this review article, the new aspects of POMs as base catalysts are comprehensively summarized [36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58]. POM base catalysts have the following advantages: (i) electrically and structurally controlled uniform basic sites can be constructed in contrast with solid bases; (ii) the thermal and oxidative stabilities of POMs are higher than those of organic bases (e.g., amines, phosphines, guanidines, amidines, phosphazenes, etc.); and (iii) the metal-oxo moiety (i.e., M=O and M-O-M) specifically activates nucleophilic substrates. In this review article, examples of POM base catalysts are highlighted with a focus on their molecular structures, basic properties, and catalytic applications to environmentally-friendly functional group transformations. Details on the syntheses, structures, properties, and applications of POMs have been summarized in many excellent books and review articles [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35].
2. Structure and Basic Property of Mono- and Polyoxometalates
In the field of solid base catalysts, the basic characteristics, which include the number, location, and strength of basic sites on solid catalysts, are not easily determined. Thus, the basic properties of solid surfaces are estimated by combining several characterization methods such as indicator/titration methods, spectroscopy of adsorbed probe molecules, and temperature programmed desorption (TPD) [4]. On the other hand, the basic properties of POMs can be discussed at the atomic and molecular levels due to their unique rigid and versatile structures. The basic sites of POMs are typically constituent oxygen atoms, and the basicity is correlated to the anion charges, sizes, molecular structures, and constituent elements. The surface POM oxygen atoms with the highest negative charges would presumably be active sites for base-catalyzed reactions. The negative charge densities of oxygen atoms cannot usually be measured experimentally; therefore, the most basic oxygen atoms have been assessed by X-ray crystallography analysis [59,60,61,62,63,64,65,66,67,68,69,70] and/or multinuclear nuclear magnetic resonance (NMR) [71,72,73,74,75,76,77,78,79] investigation of the protonated forms of POMs. On the other hand, the basic oxygen atoms are computationally assessed using molecular electrostatic potential maps, the relative energies of the various protonated forms, and anion charge [80,81,82,83,84].
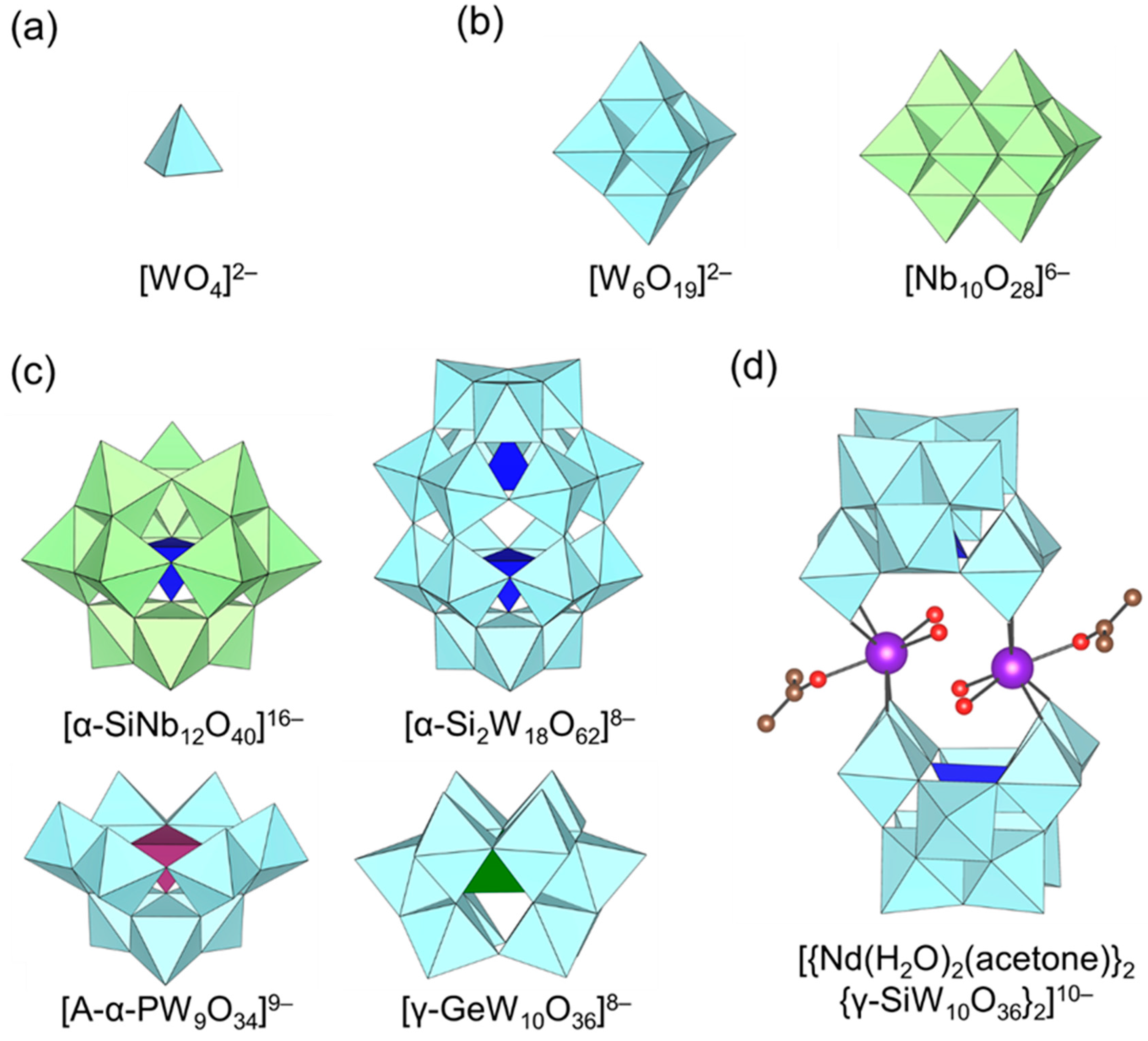
Several mono- and polyoxometalate catalyst systems effective for base-catalyzed reactions such as the chemical fixation of CO2, cyanosilylation of carbonyl compounds, and C-C bond forming reactions have recently been developed. POM base catalysts are classified into four main groups with respect to their structures: (a) monomeric metalates; (b) isopolyoxometalates; (c) heteropolyoxometalates; and (d) transition-metal-substituted POMs (Figure 1). This section focuses on the relationship between the molecular structures and basic properties (e.g., location of basic sites, amount, strength, etc.) estimated from experimental and theoretical data on the basis of groups (a)–(d).
2.1. Monomeric Metalates
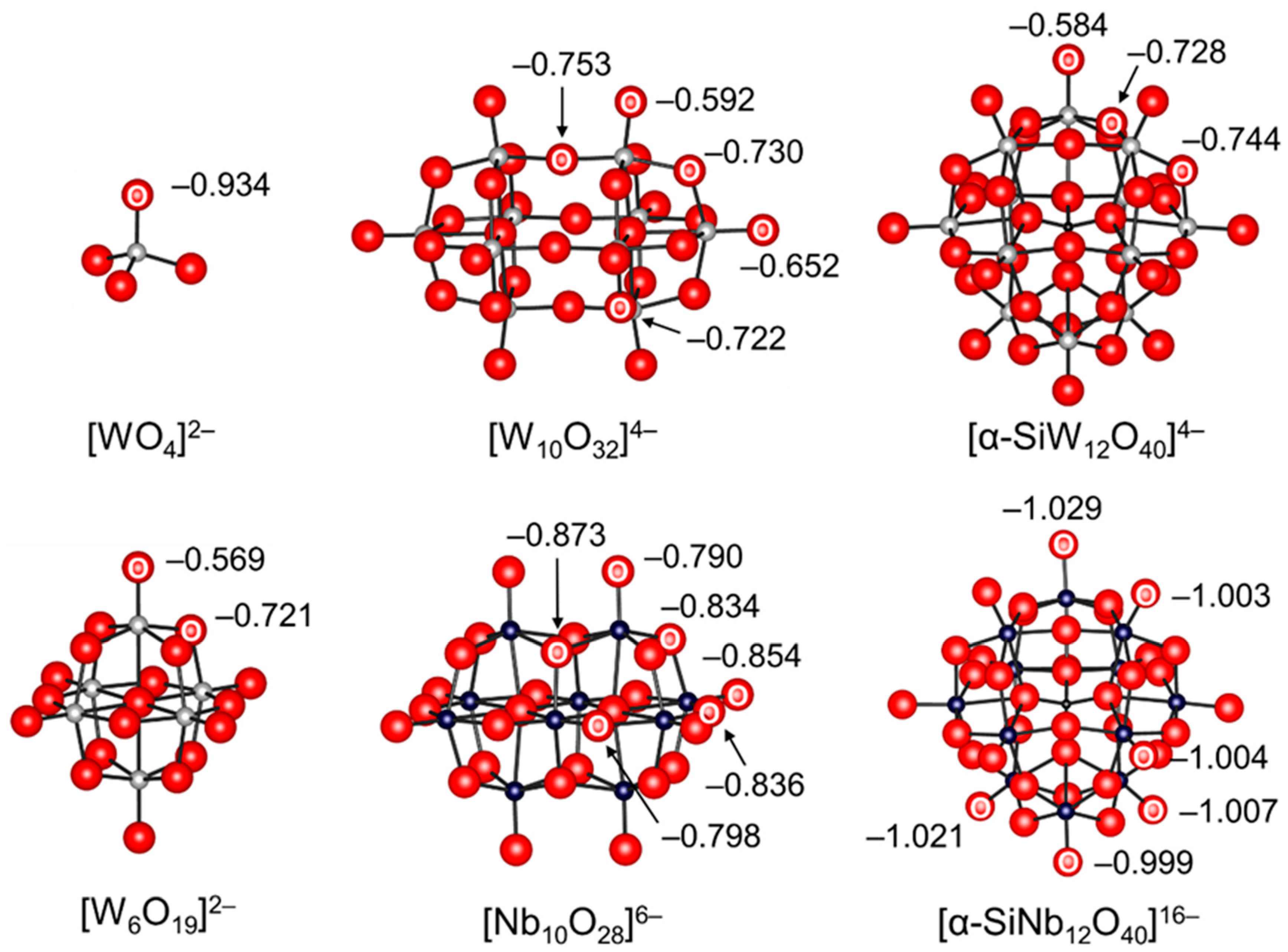
Monomeric metalates such as tetrahedral [WO4]2− and [MoO4]2− are not strictly a family of POMs but precursors for the synthesis of POMs [7,11,23,60]. While the sodium and potassium salts of monomeric metalates are typically soluble in water, the tetra-n-butyl ammonium (TBA) salts of monomeric metalates are soluble in common organic solvents. Thus, various types of POMs can be synthesized in both aqueous and organic media by control of the acidity, concentration, and temperature. Mizuno and co-workers were the first to focus on the high charge density of [WO4]2− in comparison with the other polytungstates and applied [WO4]2− to base-catalyzed reactions [38,42,43,44,45]. Various tungstate structures including [WO4]2− were optimized through density functional theory (DFT) calculations, and the basicities were estimated using the natural bond orbital (NBO) charges of the tungstate oxygen atoms. The lower NBO charge (−0.934) of an oxygen atom in [WO4]2− than those (−0.569 to −0.753) in the other POMs such as [W6O19]2−, [W10O32]4−, and [α-SiW12O40]4− indicates that [WO4]2− can likely act as a strong base catalyst (Figure 2). The pKa values of monomeric metalates and representative examples of strong inorganic and organic bases are shown in Table 1. Despite the weaker basicity of [WO4]2− (pKa of conjugate acid in water: 3.5), TBA2[WO4] exhibited much higher catalytic activity than the strong inorganic and organic bases for the chemical fixation of CO2 with various nucleophiles due to its bifunctional action towards CO2 and substrates [38,42,45]. The combination of TBA2[WO4] with various transition metal species such as Ag+ and Rh2+ was applicable to other base-catalyzed reactions that could not be accomplished with only [WO4]2− [43,44,48,54].
2.2. Isopolyoxometalates
Isopolyoxometalates with the general formula [MmOy]p− are formed by the reaction of protons and monomeric d0-early-transition-metal ions [MO4]n− (M = W, Mo, V, Nb, Ta, etc.) [7,11,23]. Equation (1) shows the typical reaction for the preparation of heptamolybdate:
7[MoO4]2− + 8H+ → [Mo7O24]6− + 4H2O.
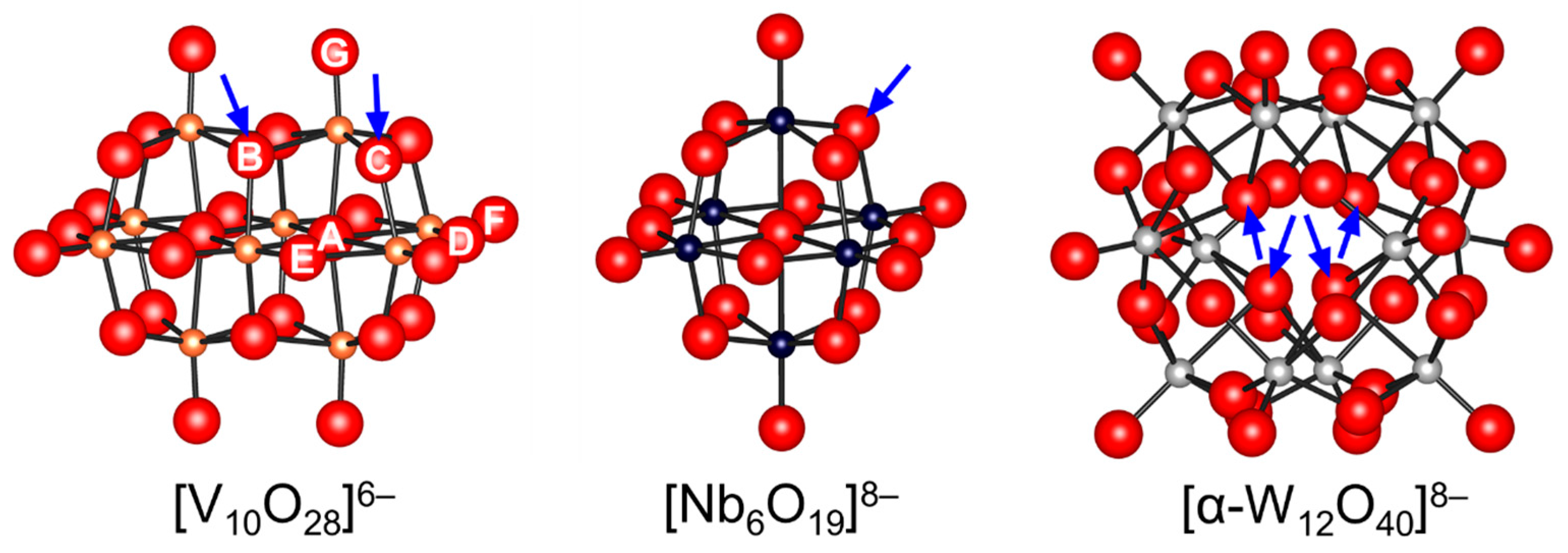
Isopolyoxometalates are often utilized as photocatalysts and building units for the synthesis of POM-based functional assemblies [12,13,14,17,35]. In addition, their basic properties have been extensively studied due to their simple compositions and structures. The bridging oxygens within the polyanion units themselves are generally more basic than the terminal oxygens and form monoprotonated μ-OH group(s) [11,23]. The protonation sites of [V10O28]6− have been widely investigated by Klemperer, Howarth, Yamase, and Ozeki by NMR and X-ray crystallography analyses [61,64,65,70,71,73,74]. There are seven types (OA–OG in Figure 3) of possible protonation sites, taking account of the D2h symmetry. The solution-state 17O NMR spectra of [V10O28]6− (enriched by the addition of H217O) show an up-field shift of the 17O NMR signals assignable to OB and OC upon acidification with HCl [71,74]. It has been reported that the protonation of oxygen atoms in POMs results in upfield shifts of 17O NMR signals. Thus, the OB and OC atoms are estimated to be the most basic oxygen atoms. While the protonation sites in the crystalline state are influenced by the nature of counter cations, the solvents incorporated as guest molecules in the crystals, and hydrogen bonds, protonation occurs at the OB and OC atoms in the solid state [64,65,70], which is in good agreement with the NMR results. Tsukuda and co-workers reported that the NBO charge of OB (−0.873) in [Nb10O28]6− was also the lowest among the constituent oxygen atoms (Figure 2) and that the tetraalkylammonium salts of [Nb10O28]6− were successfully applied to Knoevenagel condensation and CO2 fixation to epoxides [55,57]. Similarly, the protonation of surface bridging oxygen atoms in Lindqvist isopolyoxometalates such as [Nb6O19]8−, [Ta6O19]8−, and [Mo6O19]2− have been reported [79,80]. On the other hand, the metatungstate anion [α-W12O40]8− can act as an inorganic proton cryptate and capture up to three protons within the solvent-inaccessible interior [78].
2.3. Heteropolyoxometalates
Heteropolyoxometalates with the general formula [XxMmOy]q− (x < m) contains one or more heteroatoms X in addition to d0-early-transition-metal atoms M such as W, Mo, and V. The heteroatoms can be p-, d-, or f-block elements such as P, As, Si, Ge, and B [7,11,23]. A typical reaction for the preparation of phosphododecatungstate is given in Equation (2):
12[WO4]2− + [HPO4]2− + 23H+ → [PW12O40]3− + 12H2O.
Among a wide variety of heteropolyoxometalates, the Keggin structures are the most stable and more easily available. The Keggin anions (typically represented by the formula [Xn+M12O40](8−n)−) contain one central heteroatom surrounded by 12 addenda atoms in four M3O13 triads. Another typical variety of heteropolyoxometalate is the Wells-Dawson type POM. The Wells-Dawson structure (typically represented by the formula [X2n+M18O62](16−2n)−) consists of two trivacant A-type lacunary Keggin species, which are generated by the loss of the corner-shared group of MO6 octahedra, linked directly across the lacunae.
For heteropolyoxometalates (acid form) in the solid state, protons play an important role in the structure of the crystal, by linking the neighboring POM anions. Protons of crystalline H3PW12O40·6H2O are present as the hydrated species, H5O2+, each of which links four neighboring POMs by hydrogen bonding to the terminal W-O oxygen atoms [21]. For anhydrous H3PW12O40, the protons have been reported to be attached to the most basic bridging oxygen atoms based on infrared (IR) and 17O NMR spectroscopy results [21]. Himeno and co-workers reported that the potential difference between the first one- and two-electron redox waves of cyclic voltammograms served as a useful criterion for the basicity of Keggin anions [85,86]. The basicity of the Keggin anions decreased in the order of [XW12O40]5− (X = B, Al) > [XMo12O40]4− (X = Si, Ge) >> [XW12O40]4− (X = Si, Ge) > [XMo12O40]3− (X = P, As) > [XW12O40]3− (X = P, As) ≈ [SMo12O40]2−, which is in good agreement with that of the anion charges. Wang and co-workers reported that the NBO charges of the terminal oxygen atoms in [SiNb12O40]16− were much lower than those of other POMs (Figure 2), although the difference in the basic properties among oxygen atoms has not yet been clarified. The sodium salt of [SiNb12O40]16−, Na16[SiNb12O40], can act as an efficient heterogeneous catalyst for Knoevenagel condensation and CO2 cycloaddition [56].
The removal of one or more addenda atom from the fully-occupied POMs leads to the generation of lacunary POMs [7,11,23]. The most stable lacunary compounds are obtained with Si4+ as the heteroatom. While the silicododecatungstates are stable in acidic solution, the hydrolytic cleavages of W-O bonds occurs and well-defined lacunary POMs with 11, 10, and nine tungsten atoms are produced when the pH increases. The cis-dioxo moiety M(=O)2 at the lacunary site is sufficiently basic to be protonated, and some of them are diprotonated to form aqua ligands. Several structurally characterized POMs with aqua ligands at the vacant sites such as [γ-SiW10O34(H2O)2]4−, [{γ-SiW10O32(H2O)2}2(μ-O)2]4−, [{P2W15O54(H2O)2}2Zr]12−, [{P2W15O54(H2O)2}Zr{P2W17O61}]14−, α-[{[K(H2O)2](μ-H2O)Li(H2O)2]}2Si4W36O126(H2O)4]16−, and [PMo9O31(H2O)3]3− have been reported [59,66,67,68,69].
While lacunary POMs have been utilized as precursors for the preparation of various functionalized POMs, such as transition-metal-substituted POMs and inorganic-organic hybrid materials [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35], catalysis with lacunary POMs has not been extensively investigated. Mizuno and co-workers reported that the catalytic activity of a divacant silicodecatungstate, [γ-SiW10O36]8−, for the epoxidation of alkenes with H2O2 was strongly dependent on the protonation states [66]. A tetraprotonated silicodecatungstate with two aquo ligands at the vacant sites, [γ-SiW10O34(H2O)2]4−, efficiently catalyzed H2O2-based selective oxidation of alkenes. After the report, experimental and theoretical studies on the catalytic properties of lacunary POMs (especially [γ-SiW10O34(H2O)2]4−) were reported by several research groups [82,83,87,88,89,90]. The formula was suggested to be [γ-SiW10O34(H2O)2]4− based on X-ray crystallographic structure analysis; however, the structural assignment of the four protons on the lacunary sites remains controversial. DFT calculations on the structure of the tetraprotonated form of [γ-H4SiW10O36]4− were presented by Musaev and co-workers [83]. Based on DFT calculations at the B3LYP/Lanl2dz + d(Si) level, the structure of [γ-SiW10O32(OH)4]4− with four hydroxo ligands was calculated to be more stable by 8.0 kcal mol−1 than that of [γ-SiW10O34(H2O)2]4− with two aqua and two oxo(terminal) ligands (Figure 4). On the other hand, Bonchio and co-workers suggested that [γ-SiW10O34(H2O)2]4− was the active epoxidation catalyst [83]. Relativistic DFT calculations using the zeroth-order regular approximated (ZORA)-BP86/triple zeta with 1 polarization function (TZP) level theory including solvent effects with the Conductor-like screening model (COSMO) method indicated the energies of [γ-SiW10O34(H2O)2]4− and [γ-SiW10O32(OH)4]4− were so close as to be very sensitive to the method/basis set combination adopted. However, the optimized geometry of [γ-SiW10O34(H2O)2]4− was a better fit with the X-ray structure of [γ-SiW10O34(H2O)2]4− (Figure 4).
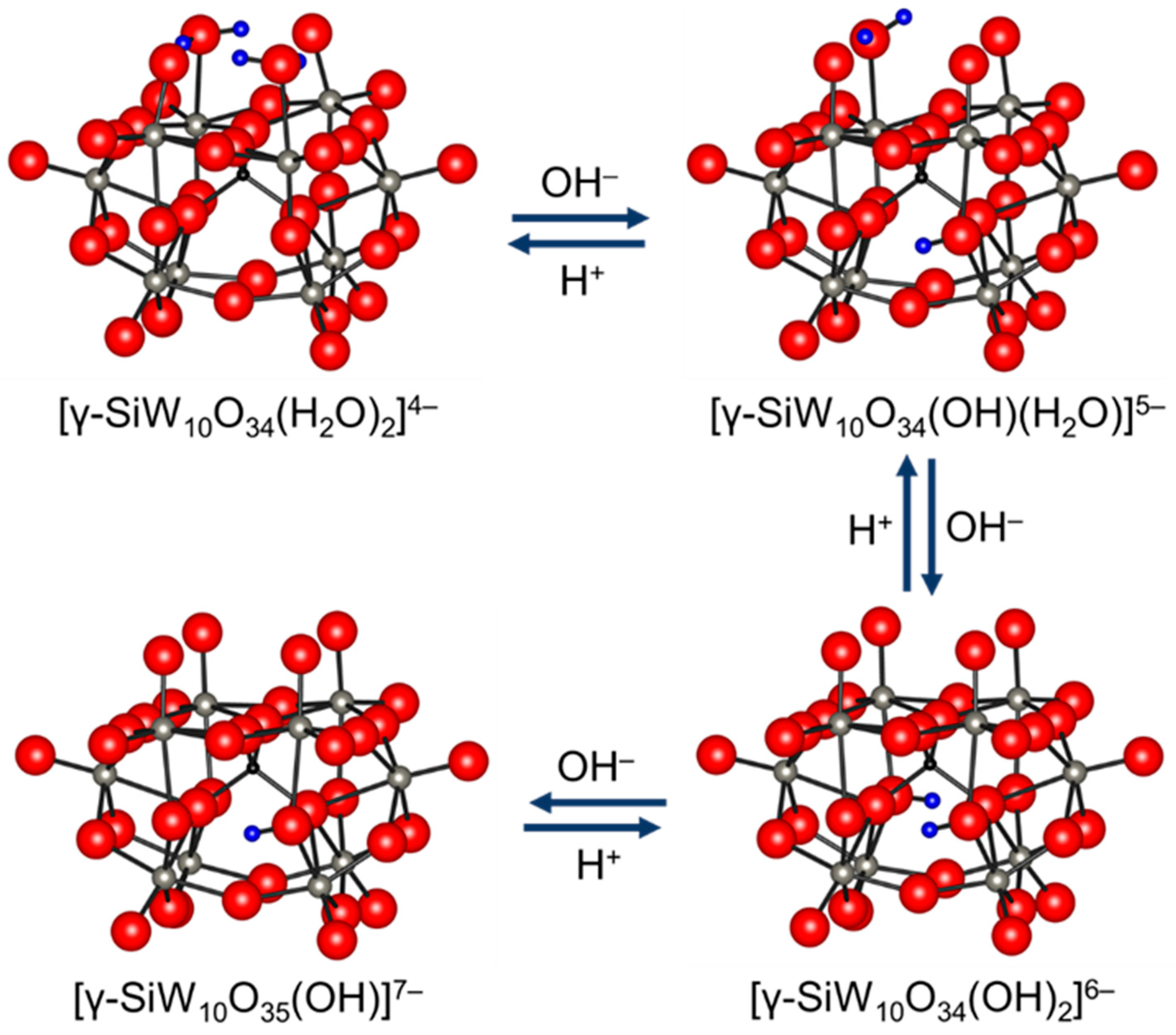
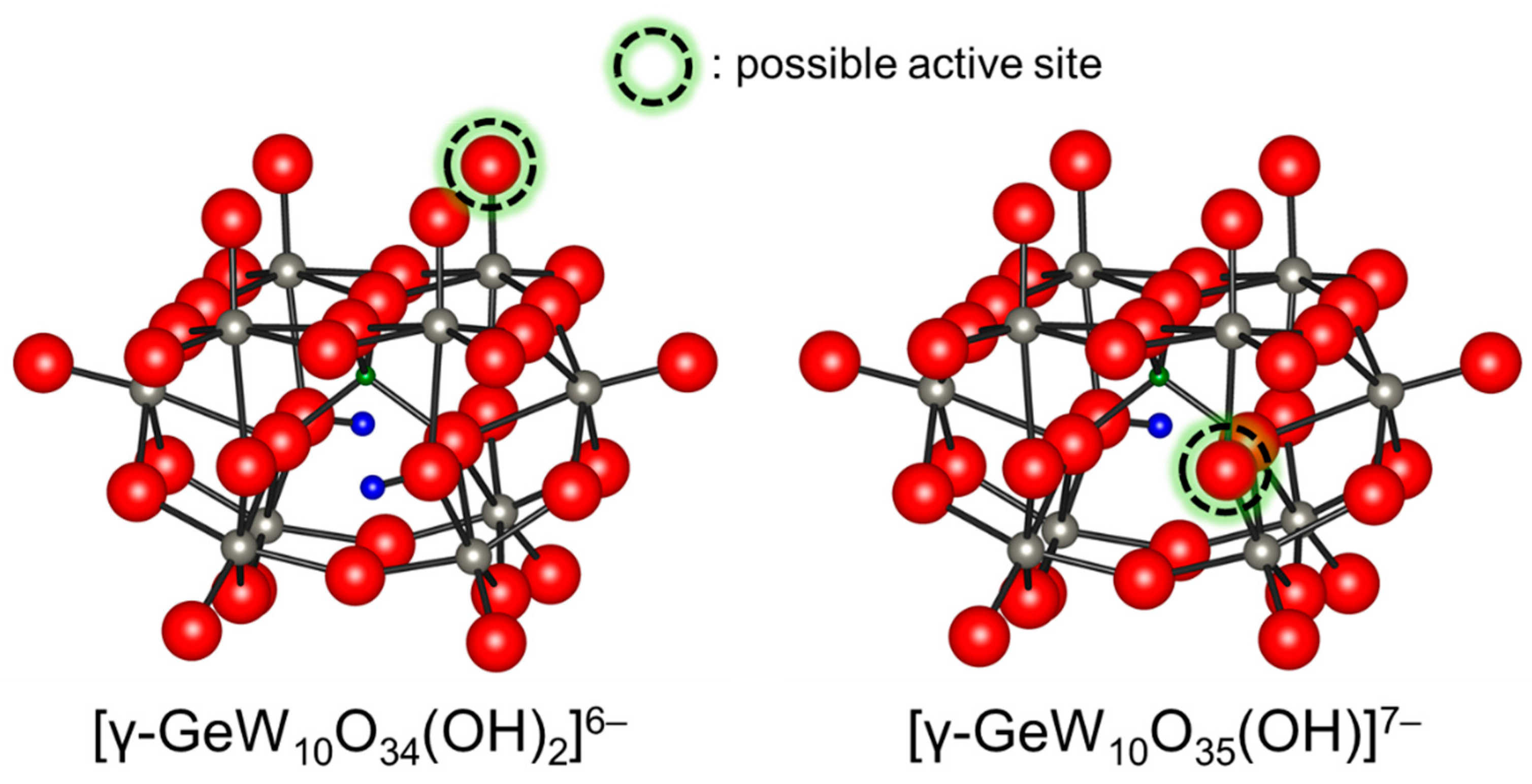
Potentiometric titration of [γ-SiW10O34(H2O)2]4− with TBAOH showed the formation of several deprotonated species; although, the reversibility and structures remain unclear [83]. Mizuno and co-workers reported the in situ formation of tri-, di-, and mono-protonated silicodecatungstates, [γ-SiW10O34(OH)(OH2)]5−, [γ-SiW10O34(OH)2]6−, and [γ-SiW10O35(OH)]7−, with C1, C2v, and C2 symmetries, respectively on the basis of the 1H, 29Si, and 183W NMR data (Figure 5) [91]. Single crystals of TBA6[γ-SiW10O34(OH)2] suitable for X-ray structure analysis were successfully obtained and the anion part was a monomeric γ-Keggin divacant silicodecatungstate with two protonated bridging oxygen atoms. Similar reversible deprotonation and protonation behaviors of germanodecatungstates were observed. Thus, the terminal lacunary oxygen atoms and the bridging oxygen atoms would be possible active sites for [γ-XW10O34(OH)2]6− (X = Si and Ge) and [γ-XW10O35(OH)]7− (X = Si and Ge), respectively (Figure 6). The di- and mono-protonated germanodecatungstates [γ-GeW10O34(OH)2]6− and [γ-GeW10O35(OH)]7− could act as homogeneous catalysts for Knoevenagel condensation and the chemoselective acylation of alcohols, respectively [39,46].
The α-Dawson-type silicotungstate, TBA8[α-Si2W18O62], which was synthesized by dimerization of a trivacant lacunary α-Keggin-type silicotungstate TBA4H6[α-SiW9O34] in an organic solvent, can reversibly capture protons inside the aperture by means of intramolecular hydrogen bonds [47]. This compound exhibited much higher catalytic performance for the Knoevenagel condensation of ethyl cyanoacetate with benzaldehyde than the starting material of trivacant TBA4H6[α-SiW9O34].
2.4. Transition-Metal-Substituted Polyoxometalates
Lacunary POM species can act as multidentate ligands with numerous metal cations, which leads to the formation of mono- and poly-nuclear transition-metal-substituted POMs according to the structure of the lacunary POMs and the type of transition-metal cations. The use of lacunary Keggin, Wells-Dawson, and other POMs and the introduction of metal cations (e.g., d- and f-block elements such as V, Fe, Cu, Ti, Zn, Al, Zr, Hf, Ag, Y, Pd, La, Ce, etc.) into the vacant sites has been used to synthesize several metal-substituted POMs with controlled multinuclear active sites in both aqueous and organic media [7,11,23]. These molecular catalysts exhibit significant activity and selectivity for a wide range of reactions [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. Some organotin-functionalized Dawson-type polyoxotungstates [P2W17O61(SnR)]7− have demonstrated strong nucleophilicity, while they have not yet acted as catalysts [92,93,94].
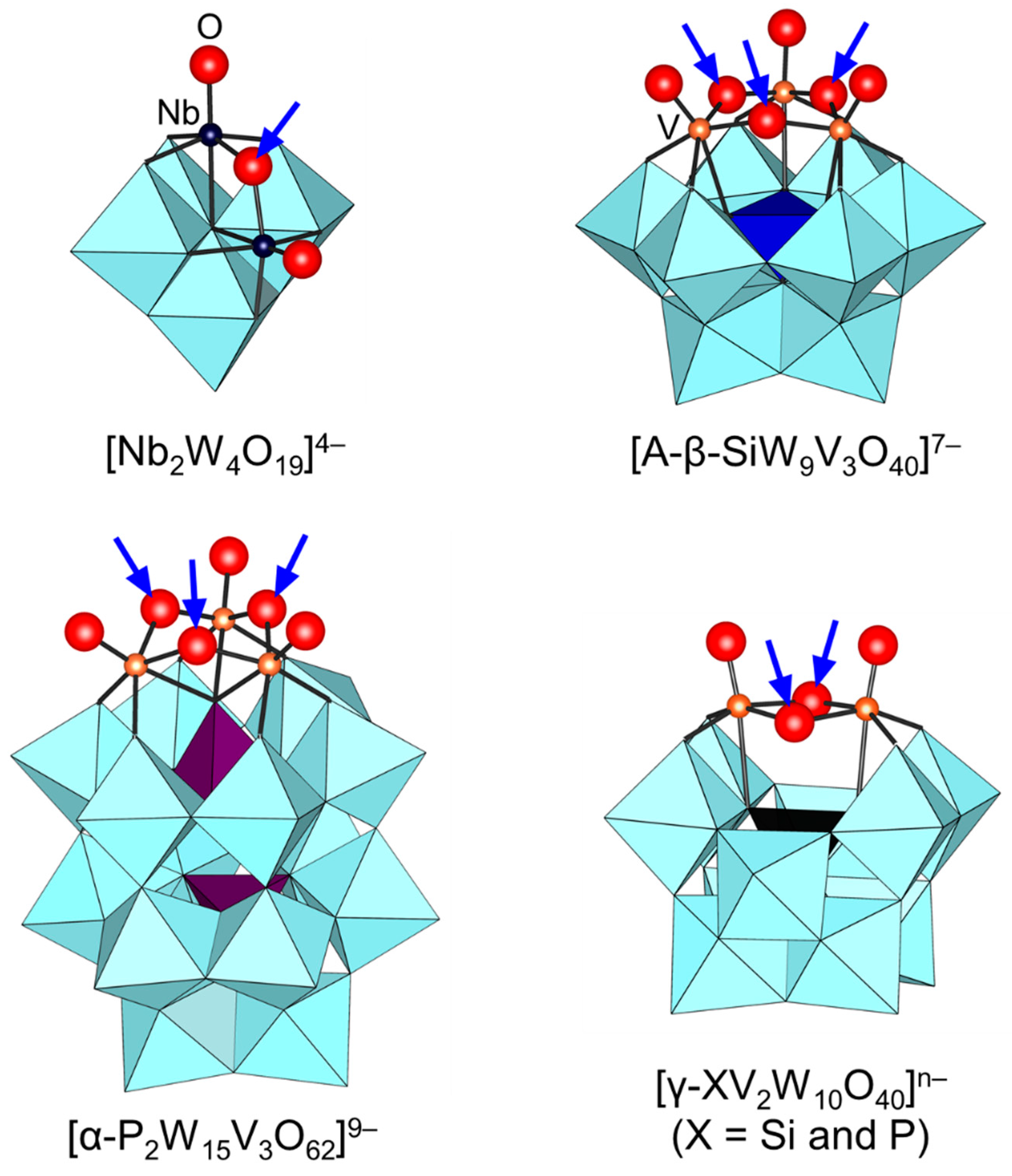
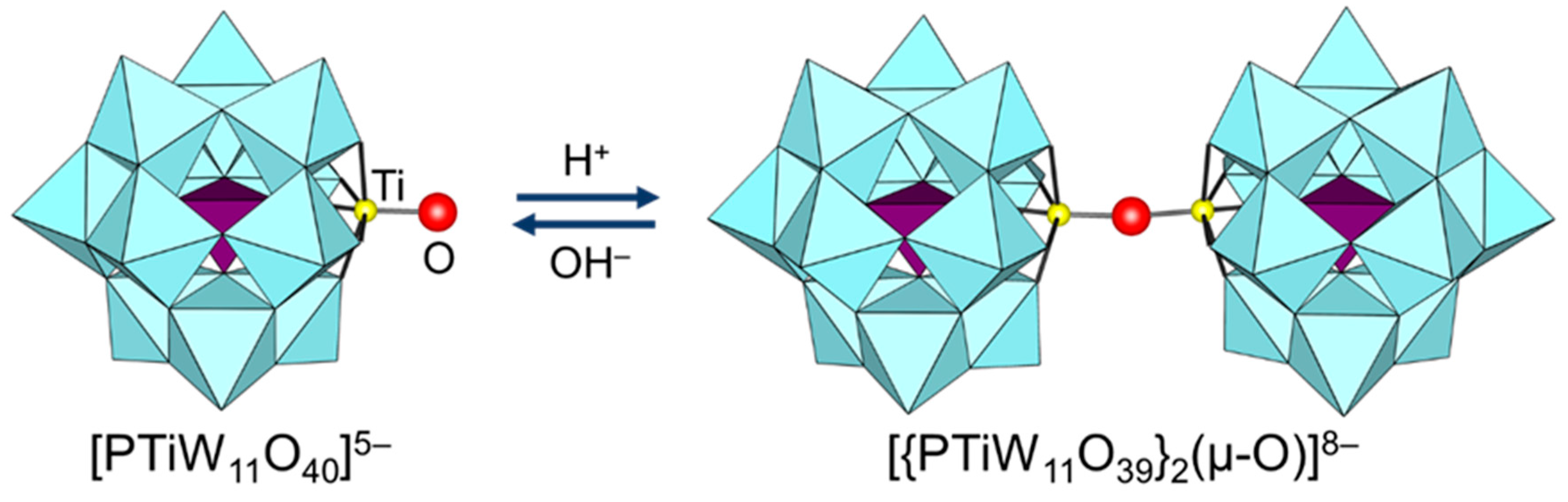
Since the protonation states of transition-metal-substituted POMs are dependent on their structures and compositions, various types of POMs have been reported. Thus, only several examples are described in this section. In the case of POMs containing group 5 metals such as [Nb2W4O19]4−, A-[β-SiW9V3O40]7−, [α-P2W15V3O62]9−, and [γ-XV2W10O40]n− (X = Si and P), protonation has been reported to occur at the oxygen atoms of Nb-O-Nb and V-O-V sites on the basis of NMR and X-ray crystallography results (Figure 7) [72,75,76,77,95,96,97,98,99,100,101]. On the other hand, the protonation of the Ti=O bond in monomeric [PTiW11O40]5− gives [PTi(OH)W11O39]4− followed by dimerization to form [{PTiW11O39}2(μ-O)]8− (Figure 8) [102]. Similar dimeric structures have been reported for POMs containing group 4 metals such as [{PZrW11O39}2(µ-OH)2]8− and [{α2-P2W17TiO61}2(μ-O)]14− [103,104]. These protonated sites play an important role in the activation of oxidants and the catalytic oxidation abilities are strongly dependent on the protonation states [95,96,97,98,99,100,102,103].
In contrast with small metal cations, full incorporation within lacunary sites to form “in-pocket” structures can be prevented for POMs containing rare-earth (RE) metals. RE cations with high coordination numbers (typically six–12) and flexible coordination geometries can also leave residual (open) coordination sites, which would act as effective Lewis acids for the activation of substrates. Several POMs containing REs have been reported to act not only as Lewis acid catalysts but also as hydrogen-bond acceptor for aldol, imino Diels−Alder reactions or the cyanosilylation of carbonyl compounds with trimethylsilyl cyanide (TMSCN) [37,41,105,106,107,108].
3. Base-Catalyzed Reactions by Mono- and Polyoxometalates
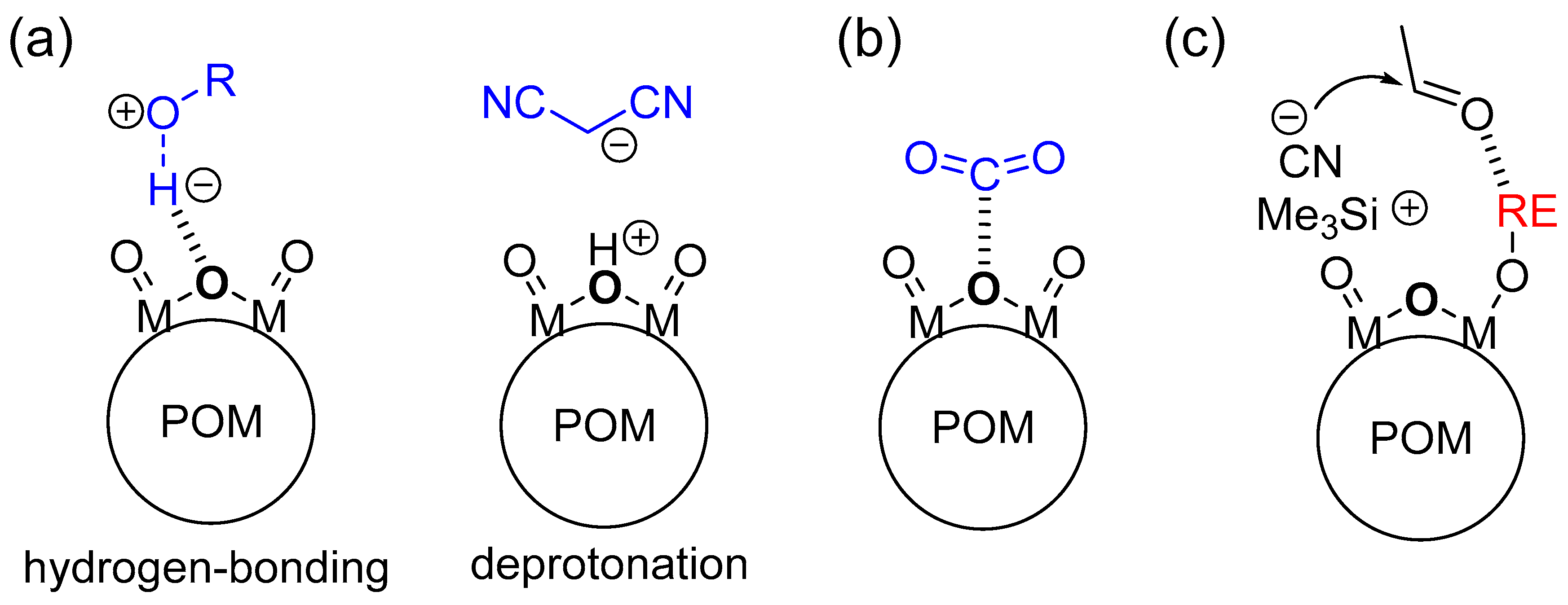
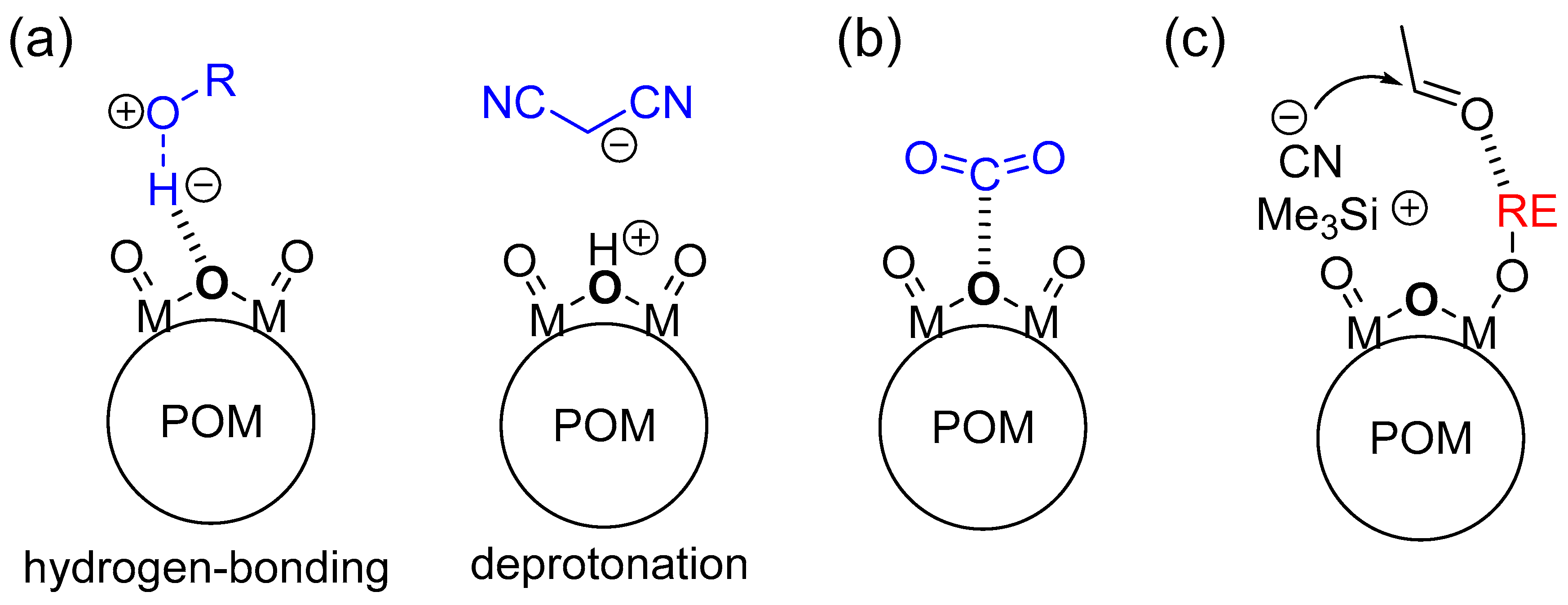
The basic properties of POM base catalysts are strongly dependent on the molecular structures, sizes, charges, and compositions. The structurally and electronically well-defined surface basic oxygen atoms exhibit the specific activation of substrates and/or reagents with basic POMs such as the abstraction of protons (Brønsted base), nucleophilic action toward substrates (Lewis base), and bifunctional action in combination with metal catalysts, which results in significant activity and selectivity (Figure 9). In this section, we focus on the unique base catalysis by POMs and its reaction mechanism.
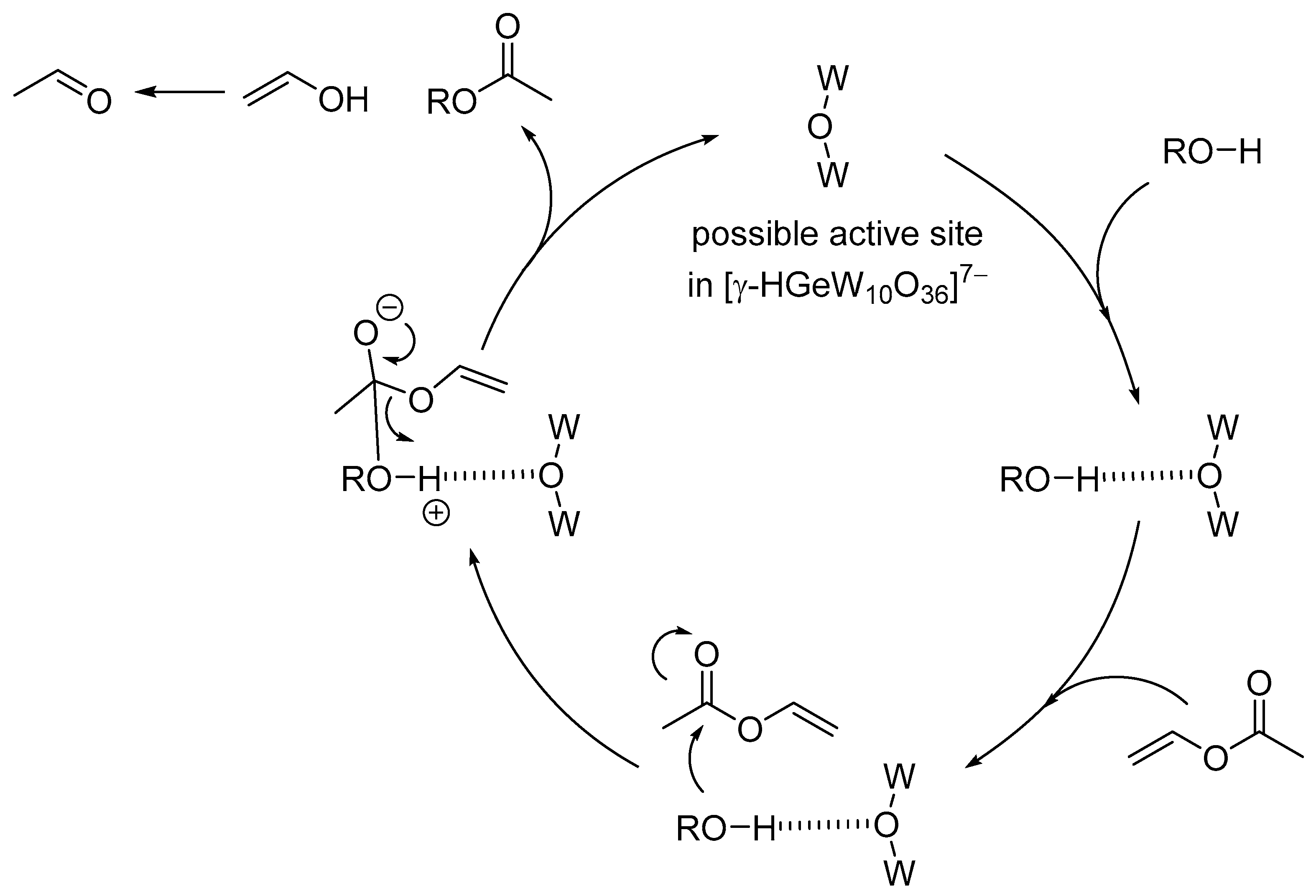
3.1. Chemical Fixation of CO2
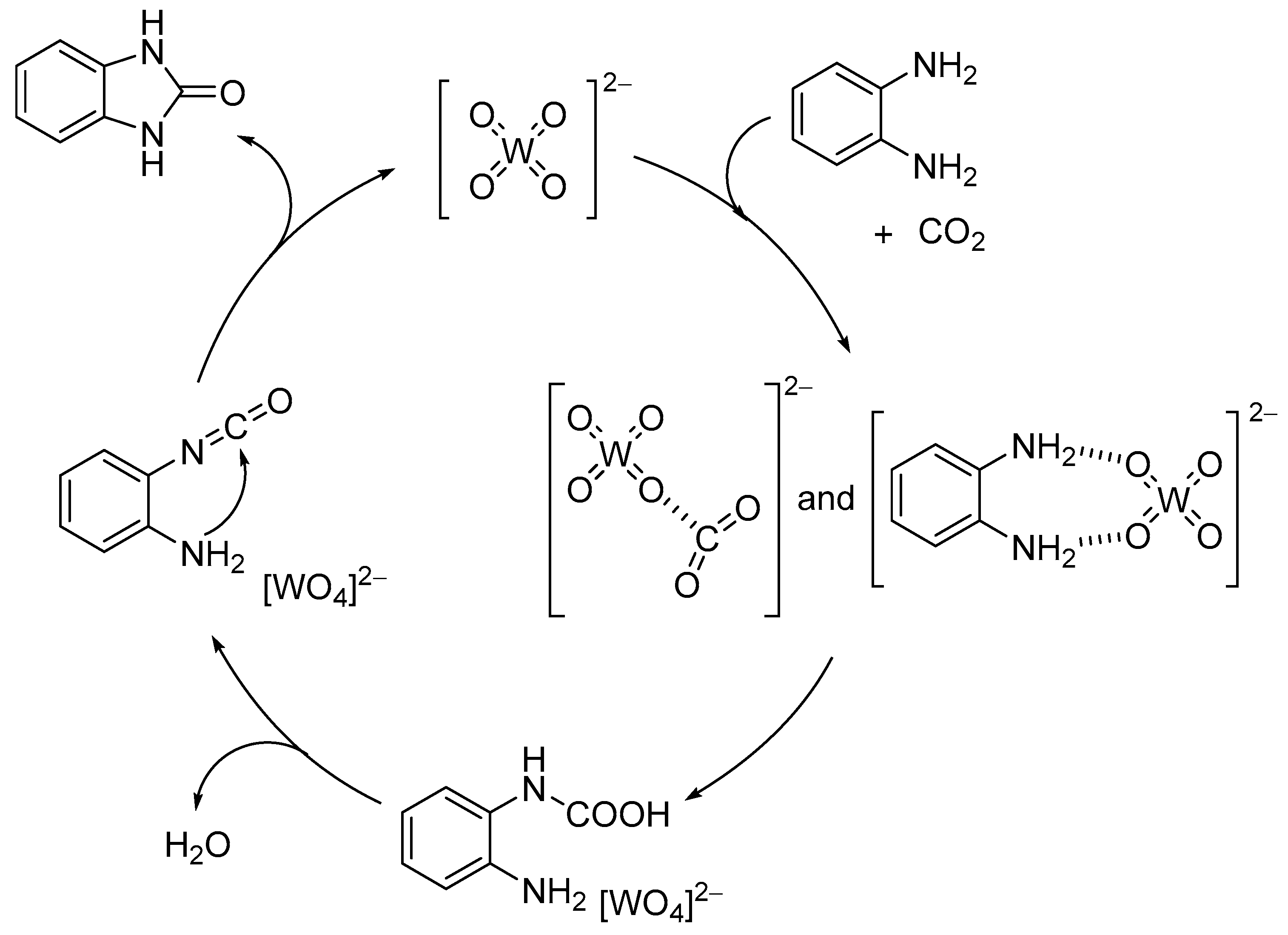
Chemical fixation of carbon dioxide (CO2) into useful and valuable chemicals is a key technology for a sustainable low-carbon society because CO2 is a renewable and environmentally-friendly C1 source in contrast with toxic CO and phosgene [109]. However, CO2 is much less reactive than CO and phosgene, and a large energy input (e.g., highly reactive reagents, high CO2 pressures, and stoichiometric amounts of strong acids or bases) is usually required to transform CO2 into various chemicals. Therefore, the low-energy catalytic fixation of CO2 is highly desirable, and several effective catalytic systems have been reported [110,111,112,113,114,115,116]. Mizuno and co-workers reported that a simple monomeric tungstate TBA2[WO4] could act as a highly efficient homogeneous catalyst for the chemical fixation of CO2 with amines, 2-aminobenzonitriles, and propargylic alcohols to urea derivatives, quinazoline-2,4(1H,3H)-diones, and cyclic carbonates, respectively (Scheme 1, Scheme 2 and Scheme 3) [38,42,45]. For the reaction of 1,2-phenylenediamine with CO2 at atmospheric pressure (0.1 MPa), the reaction rate of TBA2[WO4] was more than 48 times larger than those of other catalysts including other POMs and typical strong inorganic and organic bases. This study provides the first example of catalytic synthesis from aromatic diamines and CO2. The TBA2[WO4] catalyst shows a concerted action on both CO2 and the substrates, and the hydrogen bonding interaction between [WO4]2− and diamine and formation of Lewis base-CO2 adducts have been directly observed using 1H, 13C, and 183W NMR spectroscopy. The proposed reaction mechanism for CO2 fixation with TBA2[WO4] is shown in Figure 10. This bifunctionality facilitates the nucleophilic attack of the NH2 and OH groups on the carbon atom of CO2 to form the corresponding carbamic and carbonic acids followed by transformation to the final products. On the other hand, Cummins and co-workers reported that the reaction of [MoO4]2− with CO2 leads to the formation of the structurally characterized mono- and dicarbonate complexes, [MoO3(κ2-CO3)]2− and [MoO2(κ2-CO3)2]2− [49]. The monocarbonate species reacts with triethylsilane to produce formate together with silylated molybdate; however, catalytic CO2 fixation with [MoO4]2− has not been investigated yet.
After the report on the chemical fixation of CO2 with TBA2[WO4], various catalytic systems based on tungstates and niobates have been developed [48,53,54,55,56,57]. The Ag2[WO4] catalyst system in combination with the Ph3P ligand could efficiently catalyze CO2 fixation such as carboxylative cyclization of propargyl alcohols and one-pot synthesis of oxazolidinones and carbamates (Scheme 4 and Scheme 5) [48,54]. For example, the carboxylative cyclization of 2-methyl-3-butyn-2-ol proceeded under milder reaction conditions than those of TBA2[WO4] (i.e., under atmospheric pressure of CO2 at room temperature), and the Ag2[WO4]/Ph3P catalyst could be reused at least four times without significant loss of activity. A cooperative catalytic mechanism through the activation of the C≡C triple bonds and CO2 by silver and tungstate, respectively, are proposed. This Ag2[WO4] catalyst is also effective for the carboxylation of terminal alkynes with CO2 under ambient conditions even in the absence of Ph3P.
Hou and co-workers utilized a peroxoniobate salt of amidine, (DBUH)3[NbO3(O2)] (DBU = 1,8-diazabicyclo[5.4.0]-undec-7-ene) as a halogen-free catalyst for the synthesis of cyclic carbonates from epoxides and CO2 under solvent-free and halide-free conditions (Scheme 6) [53]. The structural transformation of peroxoniobate into stable niobates such as [NbO4]3− and [NbO3]− was observed during the catalytic recycles, and they did not play a crucial role in promoting the reaction as base catalysts but in immobilizing catalytically active organic cations (i.e., DBUH+). In contrast to monomeric niobates, polyoxoniobates such as TBA6[Nb10O28] and Na16[SiNb12O40] could efficiently catalyze CO2 cycloaddition to epoxides through a possible involvement of CO2 activation on the surface basic oxygen atoms (Scheme 7 and Scheme 8) [55,56,57]. Computational studies on the interaction of [Nb10O28]6− with CO2 indicated that CO2 activation occurs on the corner or edge O site(s) (i.e., OC–OG atoms in Figure 3), while the most basic sites in [M10O28]6− (M = V and Nb) are proposed to be OB and OC atoms as noted in Section 2.
3.2. Knoevenagel Condensation
Knoevenagel condensation of active methylene compounds with carbonyl compounds is one of the most important carbon-carbon bond forming reactions [3]. It is generally accepted that the reaction proceeds through the proton abstraction of active methylene compounds. Thus, Knoevenagel condensation is typically utilized as a test reaction to estimate the basic strength of the catalyst because there are various active methylene compounds with different pKa values. Table 2 summarizes the reactivities of POM base catalysts for the reaction of benzaldehyde with malononitrile (pKa in DMSO = 11.1), ethyl cyanoacetate (pKa = 13.1), and phenylacetonitrile (pKa = 21.9). Isopolyoxometalates (TBA2[W6O19] and TMA6[Nb10O28]), fully-occupied POMs (Na16[SiNb12O40] and TBA8[α-SiW18O62]), and lacunary POMs (TBA8−n[γ-HnGeW10O36] (n = 2 or 1), TBA4[γ-SiW10O34(H2O)2], and Na8H[PW9O34]) could efficiently catalyze the Knoevenagel condensation [36,39,40,46,50,55,56]. However, most catalytic systems exhibit narrow applicability to a limited number of active methylene compounds with low pKa values and there have been only a few reports on catalytic systems applicable to unreactive substrates. Among them, Na16[SiNb12O40] and TBA8−n[γ-HnGeW10O36] have exhibited higher catalytic activities [39,46,56], which indicates that highly negative charges of POMs play an important role in the Knoevenagel condensation. In particular, TBA6[γ-H2GeW10O36] could be applied to the Knoevenagel condensation of a wide range of substrates including unreactive phenylacetonitrile (as a donor) and ketones (as acceptors) (Scheme 9) [39]. The increase in the number of negative charges of germanodecatungstates from −6 to −7 significantly enhanced the catalytic activity for the condensation of phenylacetonitrile with benzaldehyde [46].
3.3. Cyanosilylation
Cyanosilylation of carbonyl compounds with TMSCN to cyanohydrin trimethylsilyl ethers is an important reaction because cyanohydrins are useful synthetic intermediates for the production of α-hydroxy acids, α-hydroxy aldehydes, and β-amino alcohols [37]. Several Lewis acid and base catalysts can activate carbonyl compounds and TMSCN, respectively, to promote cyanosilylation. Table 3 summarizes the reactivities of POM base catalysts for the cyanosilylation of ketones (adamantanone or acetophenone) and aldehyde (benzaldehyde) with TMSCN [36,37,39,41,50,51,52,53]. TBA4[γ-SiW10O34(H2O)2] can nucleophilically activate TMSCN on the basis of NMR and cold-spray ionization-mass spectrometry (CSI-MS) results [37]. Although the corresponding cyanohydrin trimethylsilyl ether was formed by the reaction with 2-adamantanone, the yield was still low. The combination of Lewis acid metal species with this lacunary POM could improve the reactivity, and Y3+ was effective for this reaction. The yttrium-containing POM, [{Y(H2O)2}2{γ-SiW10O36}2]10−, was successfully synthesized by the reaction of TBA4[γ-SiW10O34(H2O)2] with Y(acac)3, and this isolated species can effectively catalyze the cyanosilylation of various carbonyl compounds [37]. In this case, the nucleophilic surfaces of negatively charged POMs and the incorporated Y3+ species could act as Lewis bases and Lewis acids, respectively. Further examination of lanthanide species revealed that the Nd3+-containing POM, [{Nd(H2O)2}2{γ-SiW10O36}2]10−, was the most active because it possesses the highest C=O bond activation ability (i.e., the strongest Lewis acidity) [40]. The higher C=O activation ability of these RE-containing POMs with larger RE cations is likely explained by the weaker interaction between the RE cations and sandwiching [γ-SiW10O36]8− POMs. An and co-workers synthesized self-assemblies by the reaction of lanthanides with [Mo6V2O26]6− and [Co2Mo10H4O38]6− and then applied them to heterogeneous cyanosilylation [51,52]. These compounds could effectively catalyze the reaction; however, the reaction mechanism and applicability to inactive ketones are still unclear. Similarly, the lacunary POM of TBA6[γ-H2GeW10O36] also efficiently catalyzed the cyanosilylation of aldehydes and ketones with TMSCN, and the catalytic reactivity was comparable to those of RE-containing POMs [39], whereas the trivacant POM of Na8H[PW9O34] was not so active [50].
3.4. Other Reactions
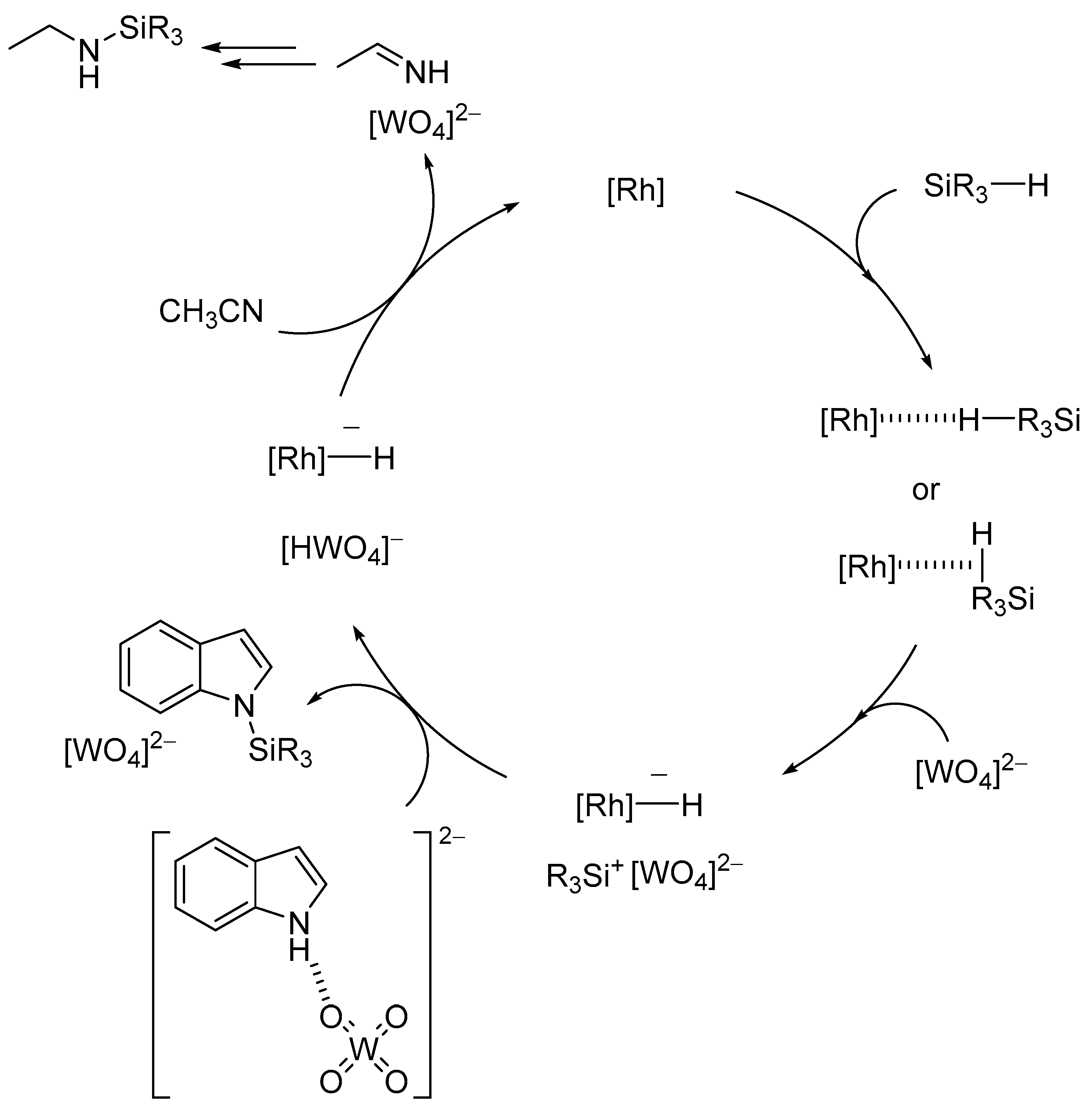
The specific activation of O-H and N-H bonds by basic surface oxygen atoms on POM base catalysts can be applied to various types of atom-efficient functional group transformations of nucleophiles such as alcohols, amines, and amides. Regioselective introduction of silyl and alkyl substituents at the N1 positions in indoles is generally difficult because the N1 nitrogen atoms are inert toward electrophilic reagents in comparison with the C3 carbon atoms. Therefore, relatively few examples are known for metal- or base-catalyzed N-silylation of indoles with hydrosilanes [117,118] and/or Michael-type addition of indoles to α,β-unsaturated compounds [119,120,121,122]. The combined catalytic system of Rh2(OAc)4 and TBA2[WO4] was efficient for site-selective N-silylation of indole derivatives with hydrosilanes into the corresponding N-silylated indoles which are important synthons that are widely utilized for the synthesis of indole-based natural products and drug candidates (Scheme 10) [43]. CSI-MS and NMR results revealed that hydrosilanes are activated to form silicon electrophiles and metal hydride species and that the specific hydrogen-bonding interaction between TBA2[WO4] and indole weaken the N-H bond of indole and facilitate the electrophilic attack of activated hydrosilanes on the nitrogen atom (Figure 11). This combined system could also efficiently catalyze the hydrosilylation of various substances including ketones, aldehydes, carbon dioxide, alkenes, nitriles, and furan derivatives into the corresponding hydrosilylation products [44].
The monoprotonated γ-Keggin germanodecatungstate TBA7[γ-HGeW10O36] with a −7 charge efficiently catalyzed the acylation of various alcohols with acid-sensitive functional groups such as acetal, epoxide, and disulfide groups, even under stoichiometric conditions (Scheme 11) [46]. The electronically and sterically well-defined basic site in this POM is effective for chemoselective acylation. For the intermolecular competitive acylation of a primary alcohol and secondary alcohol with vinyl acetate, only the primary alcohol was selectively acylated without formation of the esters from the secondary alcohol (Scheme 12). In this acylation system, a hydrogen-bonding interaction between the POM and benzyl alcohol was also evident from the 1H NMR results, and this interaction facilitates the nucleophilic attack of the hydroxy group in benzyl alcohol on the carbonyl carbon atom in vinyl acetate (Figure 12).
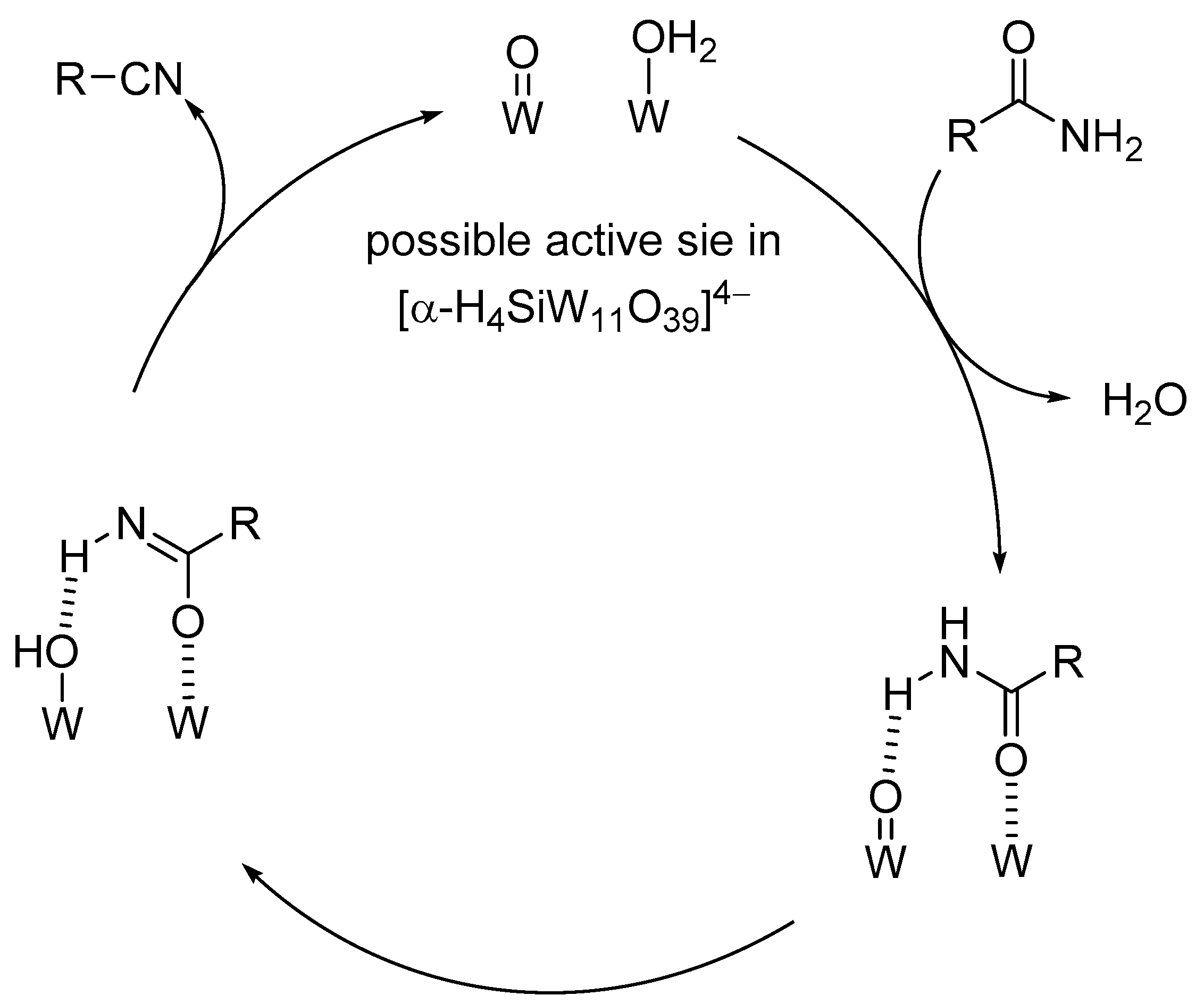
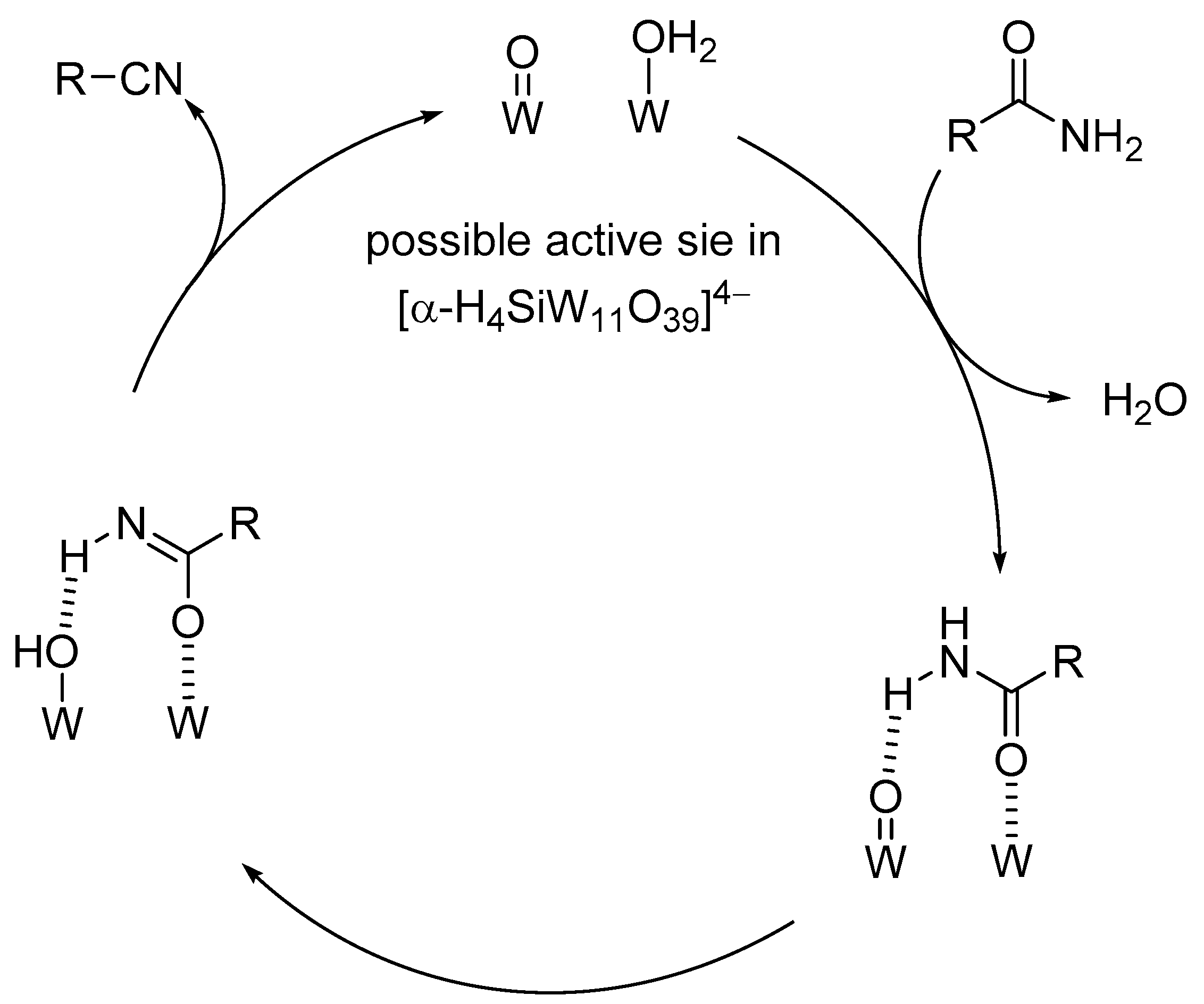
As discussed in Section 2, protonated lacunary POMs possess both labile aqua ligands (diprotonated oxo species, M-OH2) and oxo species (M-O-M and M=O species) at the vacant sites which can act as Lewis acid sites (by elimination of the aqua ligands) and basic sites, respectively. Among the protonated lacunary silicotungstates, TBA4[α-H4SiW11O39] could act as an efficient heterogeneous catalyst for the dehydration of primary amides to the corresponding nitriles in the presence of 3A molecular sieves (MS-3A) (Scheme 13) [123]. The proposed reaction mechanism is shown in Figure 13. The formation of the amide-POM adduct weakens the N-H bond and facilitates proton transfer from the coordinated amide to the oxo species to form the corresponding deprotonated species (e.g., imidate species) followed by further deprotonation to afford the corresponding nitrile.
4. Conclusions and Future Opportunities
Structurally and electronically controlled POMs exhibit unique base catalysis in contrast with typical strong inorganic and organic base catalysts due to their specific action toward substrates and/or reagents. Various types of POM base catalysts including monomeric metalates, isopoly- and heteropolyoxometalates, and RE-containing POMs can catalyze the atom-efficient conversion of substrates into value-added chemicals. Monomeric tungstate can interact with nucleophiles as well as CO2 to promote various types of chemical fixation of CO2, and the further combination of other transition metal catalysts can be effective for the development of new reactions. Isopoly- and heteropolyoxometalates with highly negative charges are intrinsically effective for C-C bond forming reactions due to their strong basicity. In particular, the activity and selectivity can be significantly improved by changing the anion charge of lacunary POMs. RE-containing POMs can also act as effective acid-base catalysts for cyanosilylation of carbonyl compounds with TMSCN.
In the cases of solid base catalysts, such as mixed metal oxide materials, the construction of electrically and structurally controlled uniform basic sites is generally difficult. Thus, these unique base catalyses result from the advantages of POMs where the structure can be controlled at the atomic and/or molecular levels. However, the reaction mechanisms, including the relationship between the substrate activation modes and basic properties of POMs, are still unclear. Further elucidation of the mechanistic aspects should lead to the design and development of highly active POM base catalysts. In addition, most POM base catalysts are homogeneous and the practical application of POMs to base catalysis will require methods for perfect catalyst recovery and recycling. Several heterogeneous catalysts such as alkali metal salts of basic POMs have been developed. However, the surface basic oxygen atoms (possible active site(s)) are strongly coordinated to alkali metal cations, which limits the catalytic performance. Therefore, future targets in this area will require novel catalyst design strategies to overcome these problems. Porous POM materials, POM nanoparticles with various shapes, and combined assemblies with active transition metal species are thus expected to be promising solid POM base catalysts that are workable under mild reaction conditions for practical base-catalyzed reactions.
Acknowledgments
This work was supported in part by the Precursory Research for Embryonic Science and Technology (PRESTO) program (No. JPMJPR15S3) of the Japan Science and Technology Agency (JST) and Kakenhi Grants-in-Aid (No. 24655187 and 15K13802) from the Japan Society for the Promotion of Science (JSPS).
Conflicts of Interest
The authors declare no conflict of interest
References
- Climent, M.J.; Corma, A.; Iborra, S.; Primo, J. Base catalysis for fine chemicals production: Claisen-schmidt condensation on zeolites and hydrotalcites for the production of chalcones and flavanones of pharmaceutical interest. J. Catal. 1995, 151, 60–66. [Google Scholar] [CrossRef]
- Tanabe, K. Industrial application of solid acid–base catalysts. Appl. Catal. A Gen. 1999, 181, 399–434. [Google Scholar] [CrossRef]
- Ono, Y.; Hattori, H. Solid Base Catalysis; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Ono, Y. Solid base catalysts for the synthesis of fine chemicals. J. Catal. 2003, 216, 406–415. [Google Scholar] [CrossRef]
- Hattori, H. Solid base catalysts: Fundamentals and their applications in organic reactions. Appl. Catal. A Gen. 2015, 504, 103–109. [Google Scholar] [CrossRef]
- Helwani, Z.; Othman, M.R.; Aziz, N.; Kim, J.; Fernando, W.J.N. Solid heterogeneous catalysts for transesterification of triglycerides with methanol: A review. Appl. Catal. A Gen. 2009, 363, 1–10. [Google Scholar] [CrossRef]
- Pope, M.T. Heteropoly and Isopoly Oxometalates; Springer: Berlin, Germany, 1983. [Google Scholar]
- Dickman, M.H.; Pope, M.T. Peroxo and superoxo complexes of chromium, molybdenum, and tungsten. Chem. Rev. 1994, 94, 569–584. [Google Scholar] [CrossRef]
- Hill, C.L. Thematic issue on “Polyoxometalates”. Chem. Rev. 1998, 98, 1–389. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Kögerler, P.; Dress, A.W.M. Giant metal-oxide-based spheres and their topology: From pentagonal building blocks to keplerates and unusual spin systems. Coord. Chem. Rev. 2001, 222, 193–218. [Google Scholar] [CrossRef]
- Pope, M.T. Polyoxoanions: Synthesis and structure. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier Pergamon: Amsterdam, The Netherlands, 2004; Volume 4, pp. 635–678. [Google Scholar]
- Uchida, S.; Mizuno, N. Design and syntheses of nano-structured ionic crystals with selective sorption properties. Coord. Chem. Rev. 2007, 251, 2537–2546. [Google Scholar] [CrossRef]
- Proust, A.; Matt, B.; Villanneau, R.; Guillemot, G.; Gouzerh, P.; Izzet, G. Functionalization and post-functionalization: A step towards polyoxometalate-based materials. Chem. Soc. Rev. 2012, 41, 7605–7622. [Google Scholar] [CrossRef] [PubMed]
- Long, D.-L.; Tsunashima, R.; Cronin, L. Polyoxometalates: Building blocks for functional nanoscale systems. Angew. Chem. Int. Ed. 2010, 49, 1736–1758. [Google Scholar] [CrossRef] [PubMed]
- Bassil, B.S.; Kortz, U. Divacant polyoxotungstates: Reactivity of the gamma-decatungstates [γ-XW10O36]8− (X = Si, Ge). Dalton Trans. 2011, 40, 9649–9661. [Google Scholar] [CrossRef] [PubMed]
- Dolbecq, A.; Mialane, P.; Sécheresse, F.; Keita, B.; Nadjo, L. Functionalized polyoxometalates with covalently linked bisphosphonate, n-donor or carboxylate ligands: From electrocatalytic to optical properties. Chem. Commun. 2012, 48, 8299–8316. [Google Scholar] [CrossRef] [PubMed]
- Miras, H.N.; Vilà-Nadal, L.; Cronin, L. Polyoxometalate based open-frameworks (POM-OFs). Chem. Soc. Rev. 2014, 43, 5679–5699. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-W.; Li, Y.-Z.; Chen, L.-J.; Yang, G.-Y. Research progress on polyoxometalate-based transition-metal-rare-earth heterometallic derived materials: Synthetic strategies, structural overview and functional applications. Chem. Commun. 2016, 52, 4418–4445. [Google Scholar] [CrossRef] [PubMed]
- Bijelic, A.; Rompel, A. Ten good reasons for the use of the tellurium-centered Anderson-Evans polyoxotungstate in protein crystallography. Acc. Chem. Res. 2017, 50, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.L.; Prosser-McCartha, C.M. Homogeneous catalysis by transition metal oxygen anion clusters. Coord. Chem. Rev. 1995, 143, 407–455. [Google Scholar] [CrossRef]
- Okuhara, T.; Mizuno, N.; Misono, M. Catalytic chemistry of heteropoly compounds. Adv. Catal. 1996, 41, 113–252. [Google Scholar]
- Kozhevnikov, I.V. Catalysts for Fine Chemical Synthesis, Volume 2, Catalysis by Polyoxometalates; John Wiley & Sons: Chichester, UK, 2002. [Google Scholar]
- Hill, C.L. Polyoxometalates: Reactivity. In Comprehensive Coordination Chemistry II, Vol. 4; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier Science: New York, NY, USA, 2004; Volume 4, pp. 679–759. [Google Scholar]
- Mizuno, N.; Yamaguchi, K.; Kamata, K. Epoxidation of olefins with hydrogen peroxide catalyzed by polyoxometalates. Coord. Chem. Rev. 2005, 249, 1944–1956. [Google Scholar] [CrossRef]
- Neumann, R.; Khenkin, A.M. Molecular oxygen and oxidation catalysis by phosphovanadomolybdates. Chem. Commun. 2006, 2529–2538. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Kamata, K. Catalytic oxidation of hydrocarbons with hydrogen peroxide by vanadium-based polyoxometalates. Coord. Chem. Rev. 2011, 255, 2358–2370. [Google Scholar] [CrossRef]
- Mizuno, N.; Kamata, K.; Yamaguchi, K. Liquid-Phase Selective Oxidation by Multimetallic Active Sites of Polyoxometalate-Based Molecular Catalysts in Topics in Organometallic Chemistry: Chemistry of Bifunctional Molecular Catalysis; Ikariya, T., Shibasaki, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 37, pp. 127–160. [Google Scholar]
- Lv, H.; Geletii, Y.V.; Zhao, C.; Vickers, J.W.; Zhu, G.; Luo, Z.; Song, J.; Lian, T.; Musaev, D.G.; Hill, C.L. Polyoxometalate water oxidation catalysts and the production of green fuel. Chem. Soc. Rev. 2012, 41, 7572–7589. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, J.; Putaj, P.; Caps, V.; Lefebvre, F.; Pelletier, J.; Basset, J.-M. Catalytic oxidation of light alkanes (C1–C4) by heteropoly compounds. Chem. Rev. 2014, 114, 981–1019. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K. Design of highly functionalized polyoxometalate-based catalysts. Bull. Chem. Soc. Jpn. 2015, 88, 1017–1028. [Google Scholar] [CrossRef]
- Zhou, Y.; Guo, Z.; Hou, W.; Wang, Q.; Wang, J. Polyoxometalate-based phase transfer catalysis for liquid-solid organic reactions: A review. Catal. Sci. Technol. 2015, 5, 4324–4335. [Google Scholar] [CrossRef]
- Narkhede, N.; Singh, S.; Patel, A. Recent progress on supported polyoxometalates for biodiesel synthesis via esterification and transesterification. Green Chem. 2015, 17, 89–107. [Google Scholar] [CrossRef]
- Sanchez, L.M.; Thomas, H.J.; Climent, M.J.; Romanelli, G.P.; Iborra, S. Heteropolycompounds as catalysts for biomass product transformations. Catal. Rev. Sci. Eng. 2016, 58, 497–586. [Google Scholar] [CrossRef]
- Ravelli, D.; Protti, S.; Fagnoni, M. Decatungstate anion for photocatalyzed “window ledge” reactions. Acc. Chem. Res. 2016, 49, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Yang, G.Y. Recent advances in polyoxometalate-catalyzed reactions. Chem. Rev. 2015, 115, 4893–4962. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Hikichi, S.; Mizuno, N. Acid–base catalyses by dimeric disilicoicosatungstates and divacant γ-Keggin-type silicodecatungstate parent: Reactivity of the polyoxometalate compounds controlled by step-by-step protonation of lacunary W=O sites. J. Organomet. Chem. 2007, 692, 455–459. [Google Scholar] [CrossRef]
- Kikukawa, Y.; Suzuki, K.; Sugawa, M.; Hirano, T.; Kamata, K.; Yamaguchi, K.; Mizuno, N. Cyanosilylation of carbonyl compounds with trimethylsilyl cyanide catalyzed by an yttrium-pillared silicotungstate dimer. Angew. Chem. Int. Ed. 2012, 51, 3686–3690. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kamata, K.; Mizuno, N. A bifunctional tungstate catalyst for chemical fixation of CO2 at atmospheric pressure. Angew. Chem. Int. Ed. 2012, 51, 6700–6703. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Kimura, T.; Kamata, K.; Yamaguchi, K.; Mizuno, N. A highly negatively charged γ-Keggin germanodecatungstate efficient for knoevenagel condensation. Chem. Commun. 2012, 48, 8422–8424. [Google Scholar] [CrossRef] [PubMed]
- Davoodnia, A. An efficient method for knoevenagel condensation catalyzed by tetrabutylammonium hexatungstate [TBA]2[W6O19] as novel and reusable heterogeneous catalyst. Synth. React. Inorg. Met.-Org. Chem. 2012, 42, 1022–1026. [Google Scholar] [CrossRef]
- Suzuki, K.; Sugawa, M.; Kikukawa, Y.; Kamata, K.; Yamaguchi, K.; Mizuno, N. Strategic design and refinement of Lewis acid-base catalysis by rare-earth-metal-containing polyoxometalates. Inorg. Chem. 2012, 51, 6953–6961. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Sunaba, H.; Kamata, K.; Mizuno, N. Efficient [WO4]2−-catalyzed chemical fixation of carbon dioxide with 2-aminobenzonitriles to quinazoline-2,4(1H,3H)-diones. Inorg. Chem. 2012, 51, 13001–13008. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, S.; Kamata, K.; Yamaguchi, K.; Mizuno, N. Rhodium acetate/base-catalyzed N-silylation of indole derivatives with hydrosilanes. Chem. Commun. 2012, 48, 9269–9271. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, S.; Sunaba, H.; Kamata, K.; Yamaguchi, K.; Mizuno, N. Hydrosilylation of various multiple bonds by a simple combined catalyst of a tungstate monomer and rhodium acetate. Chem. Lett. 2013, 42, 980–982. [Google Scholar] [CrossRef]
- Kamata, K.; Kimura, T.; Sunaba, H.; Mizuno, N. Scope of chemical fixation of carbon dioxide catalyzed by a bifunctional monomeric tungstate. Catal. Today 2014, 226, 160–166. [Google Scholar] [CrossRef]
- Sugahara, K.; Satake, N.; Kamata, K.; Nakajima, T.; Mizuno, N. A basic germanodecatungstate with a –7 charge: Efficient chemoselective acylation of primary alcohols. Angew. Chem. Int. Ed. 2014, 53, 13248–13252. [Google Scholar] [CrossRef] [PubMed]
- Minato, T.; Suzuki, K.; Kamata, K.; Mizuno, N. Synthesis of α-Dawson-type silicotungstate [α-Si2W18O62]8− and protonation and deprotonation inside the aperture through intramolecular hydrogen bonds. Chem. Eur. J. 2014, 20, 5946–5952. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.-W.; Yu, B.; Li, X.-D.; Ma, R.; Diao, Z.-F.; Li, R.-G.; Li, W.; He, L.-N. Efficient chemical fixation of CO2 promoted by a bifunctional Ag2WO4/Ph3P system. Green Chem. 2014, 16, 1633–1638. [Google Scholar] [CrossRef]
- Knopf, I.; Ono, T.; Temprado, M.; Tofan, D.; Cummins, C.C. Uptake of one and two molecules of CO2 by the molybdate dianion: A soluble, molecular oxide model system for carbon dioxide fixation. Chem. Sci. 2014, 5, 1772–1776. [Google Scholar] [CrossRef]
- Zhao, S.; Chen, Y.; Song, Y.-F. Tri-lacunary polyoxometalates of Na8H[PW9O34] as heterogeneous Lewis base catalysts for Knoevenagel condensation, cyanosilylation and the synthesis of benzoxazole derivatives. Appl. Catal. A Gen. 2014, 475, 140–146. [Google Scholar] [CrossRef]
- Fei, F.; An, H.; Meng, C.; Wang, L.; Wang, H. Lanthanide-supported molybdenum–vanadium oxide clusters: Syntheses, structures and catalytic properties. RSC Adv. 2015, 5, 18796–18805. [Google Scholar] [CrossRef]
- An, H.; Wang, L.; Hu, Y.; Fei, F. Temperature-induced racemic compounds and chiral conglomerates based on polyoxometalates and lanthanides: Syntheses, structures and catalytic properties. CrystEngComm 2015, 17, 1531–1540. [Google Scholar] [CrossRef]
- Chen, A.; Chen, C.; Xiu, Y.; Liu, X.; Chen, J.; Guo, L.; Zhang, R.; Hou, Z. Niobate salts of organic base catalyzed chemical fixation of carbon dioxide with epoxides to form cyclic carbonates. Green Chem. 2015, 17, 1842–1852. [Google Scholar] [CrossRef]
- Guo, C.-X.; Yu, B.; Xie, J.-N.; He, L.-N. Silver tungstate: A single-component bifunctional catalyst for carboxylation of terminal alkynes with CO2 in ambient conditions. Green Chem. 2015, 17, 474–479. [Google Scholar] [CrossRef]
- Hayashi, S.; Yamazoe, S.; Koyasu, K.; Tsukuda, T. Application of group V polyoxometalate as an efficient base catalyst: A case study of decaniobate clusters. RSC Adv. 2016, 6, 16239–16242. [Google Scholar] [CrossRef]
- Ge, W.; Wang, X.; Zhang, L.; Du, L.; Zhou, Y.; Wang, J. Fully-occupied Keggin type polyoxometalate as solid base for catalyzing CO2 cycloaddition and Knoevenagel condensation. Catal. Sci. Technol. 2016, 6, 460–467. [Google Scholar] [CrossRef]
- Hayashi, S.; Yamazoe, S.; Koyasu, K.; Tsukuda, T. Lewis base catalytic properties of [Nb10O28]6− for CO2 fixation to epoxide: Kinetic and theoretical studies. Chem. Asian J. 2017, 12, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; An, H.; Ding, B.; Li, Y. Evans-showell-type polyoxometalate constructing novel 3d inorganic architectures with alkaline earth metal linkers: Syntheses, structures and catalytic properties. Dalton Trans. 2017, 46, 8439–8450. [Google Scholar] [CrossRef] [PubMed]
- Hedman, B. Multicomponent polyanions. 18. A neutron diffraction study of Na3Mo9PO31(OH2)3.12–13H2O, a compound containing 9-molybdomonophosphate anions with molybdenum-coordinated water molecules. Acta Chem. Scand. A 1978, 32, 439–446. [Google Scholar] [CrossRef]
- Che, T.M.; Day, V.W.; Francesconi, L.C.; Fredrich, M.F.; Klemperer, W.G.; Shum, W. Synthesis and structure of the [(η5-C5H5)Ti(Mo5O18)]3− and [(η5-C5H5)Ti(W5O18)]3− anions. Inorg. Chem. 1985, 24, 4055–4062. [Google Scholar] [CrossRef]
- Day, V.W.; Klemperer, W.G.; Maltbie, D.J. Where are the protons in H3V10O283−? J. Am. Chem. Soc. 1987, 109, 2991–3002. [Google Scholar] [CrossRef]
- Day, V.W.; Klemperer, W.G.; Schwartz, C. Synthesis, characterization, and interconversion of the niobotungstic acid Nb2W4O19H3− and its anhydride and alkyl/silyl esters. J. Am. Chem. Soc. 1987, 109, 6030–6044. [Google Scholar] [CrossRef]
- Che, T.M.; Day, V.W.; Francesconi, L.C.; Klemperer, W.G.; Main, D.J.; Yagasaki, A.; Yaghi, O.M. Mono- and diprotonation of the [(η5-C5H5)Ti(W5O18)]3− and [(η5-C5Me5)Ti(W5O18)]3− anions. Inorg. Chem. 1992, 31, 2920–2928. [Google Scholar] [CrossRef]
- Ozeki, T.; Yamase, T.; Naruke, H.; Sasaki, Y. X-ray structural characterization of the protonation sites in the dihydrogenhexaniobate anion. Bull. Chem. Soc. Jpn. 1994, 67, 3249–3253. [Google Scholar] [CrossRef]
- Nakamura, S.; Ozeki, T. Hydrogen-bonded aggregates of protonated decavanadate anions in their tetraalkylammonium salts. J. Chem. Soc. Dalton Trans. 2001, 472–480. [Google Scholar] [CrossRef]
- Kamata, K.; Yonehara, K.; Sumida, Y.; Yamaguchi, K.; Hikichi, S.; Mizuno, N. Efficient epoxidation of olefins with ≥99% selectivity and use of hydrogen peroxide. Science 2003, 300, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Leclerc-Laronze, N.; Haouas, M.; Marrot, J.; Taulelle, F.; Hervé, G. Step-by-step assembly of trivacant tungstosilicates: Synthesis and characterization of tetrameric anions. Angew. Chem. Int. Ed. 2005, 45, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Yoshimura, M.; Uehara, K.; Hikichi, S.; Mizuno, N. Formation of S-shaped disilicoicosatungstate and efficient baeyer-villiger oxidation with hydrogen peroxide. Angew. Chem. Int. Ed. 2006, 45, 1956–1960. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Hill, C.L. Multiple reversible protonation of polyoxoanion surfaces: Direct observation of dynamic structural effects from proton transfer. Angew. Chem. Int. Ed. 2007, 46, 3877–3880. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Ozeki, T. Guest driven rearrangements of protonation and hydrogen bonding in decavanadate anions as their tetraalkylammonium salts. Dalton Trans. 2008, 6135–6140. [Google Scholar] [CrossRef] [PubMed]
- Klemperer, W.G.; Shum, W. Charge distribution in large polyoxoanions: Determination of protonation sites in vanadate (V10O286−) by 17O nuclear magnetic resonance. J. Am. Chem. Soc. 1977, 99, 3544–3545. [Google Scholar] [CrossRef]
- Klemperer, W.G.; Shum, W. Isomerism and charge distribution in mixed-metal polyoxoanion clusters: 17O nuclear magnetic resonance structure determinations of cis-V2W4O194− and cis-HV2W4O193−. J. Am. Chem. Soc. 1978, 100, 4891–4893. [Google Scholar] [CrossRef]
- Filowitz, M.; Ho, R.K.C.; Klemperer, W.G.; Shum, W. 17O nuclear magnetic resonance spectroscopy of polyoxometalates. 1. Sensitivity and resolution. Inorg. Chem. 1979, 18, 93–103. [Google Scholar] [CrossRef]
- Harrison, A.T.; Howarth, O.W. Oxygen exchange and protonation of polyanions: A multinuclear magnetic resonance study of tetradecavanadophosphate(9–) and decavanadate(6–). J. Chem. Soc. Dalton Trans. 1985, 1953–1957. [Google Scholar] [CrossRef]
- Finke, R.G.; Rapko, B.; Saxton, R.J.; Domaille, P.J. Trisubstituted heteropolytungstates as soluble metal oxide analogs. 3. Synthesis, characterization, 31P, 29Si, 51V, 31P, and 1-and 2-D 183W NMR, deprotonation, and proton mobility studies of organic solvent solute forms of HxSiW9V3O40x−7 and HxP2W15V3O62x−9. J. Am. Chem. Soc. 1986, 108, 2947–2960. [Google Scholar]
- Pohl, M.; Finke, R.G. Polyoxoanion-supported, atomically dispersed transition-metal precatalyst [(1,5-COD)Ir·P2W15Nb3O62]8−: Direct 17O NMR evidence for Ir-ONb2 bonding and for a C3V average symmetry, iridium-to-polyoxoanion support interaction. Organometallics 1993, 12, 1453–1457. [Google Scholar] [CrossRef]
- Weiner, H.; Aiken, J.D.; Finke, R.G. Polyoxometalate catalyst precursors. Improved synthesis, H+-titration procedure, and evidence for 31P NMR as a highly sensitive support-site indicator for the prototype polyoxoanion−organometallic-support system [(n-C4H9)4]9P2W15Nb3O62. Inorg. Chem. 1996, 35, 7905–7913. [Google Scholar] [CrossRef]
- Sprangers, C.R.; Marmon, J.K.; Duncan, D.C. Where are the protons in α-[HxW12O40](8−x)− (x = 2–4)? Inorg. Chem. 2006, 45, 9628–9630. [Google Scholar] [CrossRef] [PubMed]
- Balogh, E.; Anderson, T.M.; Rustad, J.R.; Nyman, M.; Casey, W.H. Rates of oxygen-isotope exchange between sites in the [HxTa6O19](8−x)−(aq) Lindqvist ion and aqueous solutions: Comparisons to [HxNb6O19](8−x)−(aq). Inorg. Chem. 2007, 46, 7032–7039. [Google Scholar] [CrossRef] [PubMed]
- Dolbecq, A.; Guirauden, A.; Fourmigué, M.; Boubekeur, K.; Batail, P.; Rohmer, M.-M.; Bénard, M.; Coulon, C.; Sallé, M.; Blanchard, P. Relative basicities of the oxygen atoms of the Linquist polyoxometalate [Mo6O19]2− and their recognition by hydroxyl groups in radical cation salts based on functionalized tetrathiafulvalene π donors. J. Chem. Soc. Dalton Trans. 1999, 1241–1248. [Google Scholar] [CrossRef]
- López, X.; Bo, C.; Poblet, J.M. Electronic properties of polyoxometalates: Electron and proton affinity of mixed-addenda Keggin and Wells−Dawson anions. J. Am. Chem. Soc. 2002, 124, 12574–12582. [Google Scholar] [CrossRef] [PubMed]
- Musaev, D.G.; Morokuma, K.; Geletii, Y.V.; Hill, C.L. Computational modeling of di-transition-metal-substituted γ-Keggin polyoxometalate anions. Structural refinement of the protonated divacant lacunary silicodecatungstate. Inorg. Chem. 2004, 43, 7702–7708. [Google Scholar] [CrossRef] [PubMed]
- Sartorel, A.; Carraro, M.; Bagno, A.; Scorrano, G.; Bonchio, M. Asymmetric tetraprotonation of γ-[(SiO4)W10O32]8− triggers a catalytic epoxidation reaction: Perspectives in the assignment of the active catalyst. Angew. Chem. Int. Ed. 2007, 46, 3255–3258. [Google Scholar] [CrossRef] [PubMed]
- Efremenko, I.; Neumann, R. Protonation of phosphovanadomolybdates H3+xPVxMo12−xO40: Computational insight into reactivity. J. Phys. Chem. A 2011, 115, 4811–4826. [Google Scholar] [CrossRef] [PubMed]
- Himeno, S.; Takamoto, M.; Santo, R.; Ichimura, A. Redox properties and basicity of Keggin-type polyoxometalate complexes. Bull. Chem. Soc. Jpn. 2005, 78, 95–100. [Google Scholar] [CrossRef]
- Nakajima, K.; Eda, K.; Himeno, S. Effect of the central oxoanion size on the voltammetric properties of Keggin-type [XW12O40]n− (n = 2–6) complexes. Inorg. Chem. 2010, 49, 5212–5215. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Sandei, L.; Sartorel, A.; Scorrano, G.; Bonchio, M. Hybrid polyoxotungstates as second-generation POM-based catalysts for microwave-assisted H2O2 activation. Org. Lett. 2006, 8, 3671–3674. [Google Scholar] [CrossRef] [PubMed]
- Sartorel, A.; Carraro, M.; Bagno, A.; Scorrano, G.; Bonchio, M. H2O2 activation by heteropolyacids with defect structures: The case of γ-[(XO4)W10O32]n− (X = Si, Ge, n = 8; X = P, n = 7). J. Phys. Org. Chem. 2008, 21, 596–602. [Google Scholar] [CrossRef]
- Phan, T.D.; Kinch, M.A.; Barker, J.E.; Ren, T. Highly efficient utilization of H2O2 for oxygenation of organic sulfides catalyzed by [γ-SiW10O34(H2O)2]4−. Tetrahedron Lett. 2005, 46, 397–400. [Google Scholar] [CrossRef]
- Berardi, S.; Bonchio, M.; Carraro, M.; Conte, V.; Sartorel, A.; Scorrano, G. Fast catalytic epoxidation with H2O2 and [γ-SiW10O36(PhPO)2]4− in ionic liquids under microwave irradiation. J. Org. Chem. 2007, 72, 8954–8957. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Kuzuya, S.; Hirano, T.; Kamata, K.; Mizuno, N. Reversible deprotonation and protonation behaviors of a tetra-protonated γ-Keggin silicodecatungstate. Inorg. Chem. 2012, 51, 7932–7939. [Google Scholar] [CrossRef] [PubMed]
- Boglio, C.; Micoine, K.; Derat, É.; Thouvenot, R.; Hasenknopf, B.; Thorimbert, S.; Lacôte, E.; Malacria, M. Regioselective activation of oxo ligands in functionalized Dawson polyoxotungstates. J. Am. Chem. Soc. 2008, 130, 4553–4561. [Google Scholar] [CrossRef] [PubMed]
- Thorimbert, S.; Hasenknopf, B.; Lacôte, E. Cross-linking organic and polyoxometalate chemistries. Isr. J. Chem. 2011, 51, 275–280. [Google Scholar] [CrossRef]
- Micoine, K.; Malacria, M.; Lacôte, E.; Thorimbert, S.; Hasenknopf, B. Regioselective double organic functionalization of polyoxotungstates through electrophilic addition of aromatic isocyanates to [P2W17O61(SnR)]7−. Eur. J. Inorg. Chem. 2013, 2013, 1737–1741. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Kamata, K.; Kotani, M.; Yamaguchi, K.; Mizuno, N. Polyoxovanadometalate-catalyzed selective epoxidation of alkenes with hydrogen peroxide. Angew. Chem. Int. Ed. 2005, 44, 5136–5141. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Mizuno, N. Mechanism of [γ-H2SiV2W10O40]4−-catalyzed epoxidation of alkenes with hydrogen peroxide. Inorg. Chem. 2007, 46, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Yonehara, K.; Nakagawa, Y.; Uehara, K.; Mizuno, N. Efficient stereo- and regioselective hydroxylation of alkanes catalysed by a bulky polyoxometalate. Nat. Chem. 2010, 2, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Yonehara, K.; Kamata, K.; Yamaguchi, K.; Mizuno, N. An efficient H2O2-based oxidative bromination of alkenes, alkynes, and aromatics by a divanadium-substituted phosphotungstate. Chem. Commun. 2011, 47, 1692–1694. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Sugahara, K.; Yonehara, K.; Ishimoto, R.; Mizuno, N. Efficient epoxidation of electron-deficient alkenes with hydrogen peroxide catalyzed by [γ-PW10O38V2(μ-OH)2]3−. Chem. Eur. J. 2011, 17, 7549–7559. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Yamaura, T.; Mizuno, N. Chemo- and regioselective direct hydroxylation of arenes with hydrogen peroxide catalyzed by a divanadium-substituted phosphotungstate. Angew. Chem. Int. Ed. 2012, 51, 7275–7278. [Google Scholar] [CrossRef] [PubMed]
- Uehara, K.; Miyachi, T.; Mizuno, N. Amphiprotic properties of a bis(μ-hydroxo)divanadium(IV)-substituted γ-Keggin-type silicodecatungstate containing two different kinds of hydroxyl moieties. Inorg. Chem. 2014, 53, 5341–5347. [Google Scholar] [CrossRef] [PubMed]
- Kholdeeva, O.A.; Maksimov, G.M.; Maksimovskaya, R.I.; Kovaleva, L.A.; Fedotov, M.A.; Grigoriev, V.A.; Hill, C.L. A dimeric titanium-containing polyoxometalate. Synthesis, characterization, and catalysis of H2O2-based thioether oxidation. Inorg. Chem. 2000, 39, 3828–3837. [Google Scholar] [CrossRef] [PubMed]
- Kholdeeva, O.A.; Maksimov, G.M.; Maksimovskaya, R.I.; Vanina, M.P.; Trubitsina, T.A.; Naumov, D.Y.; Kolesov, B.A.; Antonova, N.S.; Carbó, J.J.; Poblet, J.M. Zr(IV)-monosubstituted keggin-type dimeric polyoxometalates: Synthesis, characterization, catalysis of H2O2-based oxidations, and theoretical study. Inorg. Chem. 2006, 45, 7224–7234. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Murakami, H.; Sakai, Y.; Nomiya, K. Syntheses, molecular structures and pH-dependent monomer–dimer equilibria of dawson α2-monotitanium(IV)-substituted polyoxometalates. Dalton Trans. 2008, 4630–4638. [Google Scholar] [CrossRef]
- Xuan, W.-J.; Botuha, C.; Hasenknopf, B.; Thorimbert, S. Addition of carbon nucleophiles to hemiaminals promoted by a Lewis acidic polyoxotungstate. Org. Chem. Front. 2014, 1, 1091–1095. [Google Scholar] [CrossRef]
- Dupré, N.; Rémy, P.; Micoine, K.; Boglio, C.; Thorimbert, S.; Lacôte, E.; Hasenknopf, B.; Malacria, M. Chemoselective catalysis with organosoluble lewis acidic polyoxotungstates. Chem. Eur. J. 2010, 16, 7256–7264. [Google Scholar] [CrossRef] [PubMed]
- Boglio, C.; Lemière, G.; Hasenknopf, B.; Thorimbert, S.; Lacôte, E.; Malacria, M. Lanthanide complexes of the monovacant dawson polyoxotungstate [α1-P2W17O61]10− as selective and recoverable Lewis acid catalysts. Angew. Chem. Int. Ed. 2006, 45, 3324–3327. [Google Scholar] [CrossRef] [PubMed]
- El Moll, H.; Nohra, B.; Mialane, P.; Marrot, J.; Dupré, N.; Riflade, B.; Malacria, M.; Thorimbert, S.; Hasenknopf, B.; Lacôte, E.; et al. Lanthanide polyoxocationic complexes: Experimental and theoretical stability studies and Lewis acid catalysis. Chem. Eur. J. 2011, 17, 14129–14138. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M. Carbon Dioxide as Chemical Feedstock; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Shi, F.; Deng, Y.; SiMa, T.; Peng, J.; Gu, Y.; Qiao, B. Alternatives to phosgene and carbon monoxide: Synthesis of symmetric urea derivatives with carbon dioxide in ionic liquids. Angew. Chem. Int. Ed. 2003, 42, 3257–3260. [Google Scholar] [CrossRef] [PubMed]
- Ion, A.; Parvulescu, V.; Jacobs, P.; Vos, D.D. Synthesis of symmetrical or asymmetrical urea compounds from CO2 via base catalysis. Green Chem. 2007, 9, 158–161. [Google Scholar] [CrossRef]
- Jiang, T.; Ma, X.; Zhou, Y.; Liang, S.; Zhang, J.; Han, B. Solvent-free synthesis of substituted ureas from CO2 and amines with a functional ionic liquid as the catalyst. Green Chem. 2008, 10, 465–469. [Google Scholar] [CrossRef]
- Kayaki, Y.; Yamamoto, M.; Ikariya, T. N-heterocyclic carbenes as efficient organocatalysts for CO2 fixation reactions. Angew. Chem. Int. Ed. 2009, 48, 4194–4197. [Google Scholar] [CrossRef] [PubMed]
- Lalrempuia, R.; Iglesias, M.; Polo, V.; Sanz Miguel, P.J.; Fernández-Alvarez, F.J.; Pérez-Torrente, J.J.; Oro, L.A. Effective fixation of CO2 by iridium-catalyzed hydrosilylation. Angew. Chem. Int. Ed. 2012, 51, 12824–12827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyzavi, M.H.; Klet, R.C.; Tussupbayev, S.; Borycz, J.; Vermeulen, N.A.; Cramer, C.J.; Stoddart, J.F.; Hupp, J.T.; Farha, O.K. A hafnium-based metal-organic framework as an efficient and multifunctional catalyst for facile CO2 fixation and regioselective and enantioretentive epoxide activation. J. Am. Chem. Soc. 2014, 136, 15861–15864. [Google Scholar] [CrossRef] [PubMed]
- Li, P.Z.; Wang, X.J.; Liu, J.; Lim, J.S.; Zou, R.; Zhao, Y. A triazole-containing metal-organic framework as a highly effective and substrate size-dependent catalyst for CO2 conversion. J. Am. Chem. Soc. 2016, 138, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.W.; Boebel, T.A.; Hartwig, J.F. Iridium-catalyzed, silyl-directed borylation of nitrogen-containing heterocycles. J. Am. Chem. Soc. 2010, 132, 4068–4069. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimoto, T.; Iketani, Y.; Sekine, M. Zinc-catalyzed dehydrogenative N-silylation of indoles with hydrosilanes. Chem. Eur. J. 2012, 18, 9500–9504. [Google Scholar] [CrossRef] [PubMed]
- Dassonville, A.; Lardic, M.; Breteche, A.; Nourrisson, M.R.; Le Baut, G.; Grimaud, N.; Petit, J.Y.; Duflos, M. N-pyridinyl(methyl)-indole-1- or 3-propanamides and propenamides acting as topical and systemic inflammation inhibitors. J. Enzyme Inhib. Med. Chem. 2008, 23, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Ferorelli, S.; Abate, C.; Pedone, M.P.; Colabufo, N.A.; Contino, M.; Perrone, R.; Berardi, F. Synthesis and binding assays of novel 3,3-dimethylpiperidine derivatives with various lipophilicities as sigma(1) receptor ligands. Bioorg. Med. Chem. 2011, 19, 7612–7622. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.; Acher, F.C.; McCort-Tranchepain, I. Microwave-promoted michael addition of azaheterocycles to α,β-unsaturated esters and acid under solvent-free conditions. Synlett 2012, 23, 791–795. [Google Scholar] [CrossRef]
- Sevov, C.S.; Zhou, J.S.; Hartwig, J.F. Iridium-catalyzed, intermolecular hydroamination of unactivated alkenes with indoles. J. Am. Chem. Soc. 2014, 136, 3200–3207. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, S.; Kamata, K.; Yamaguchi, K.; Mizuno, N. A monovacant lacunary silicotungstate as an efficient heterogeneous catalyst for dehydration of primary amides to nitriles. ChemCatChem 2013, 5, 1725–1728. [Google Scholar] [CrossRef]
Figure 1.
Molecular structures of the representative polyoxometalate (POM) anions used for base-catalyzed reaction. (a) Monomeric metalate, (b) isopolyoxometalate, (c) heteropolyoxometalate and (d) transition-metal-substituted polyoxometalate.
Figure 1.
Molecular structures of the representative polyoxometalate (POM) anions used for base-catalyzed reaction. (a) Monomeric metalate, (b) isopolyoxometalate, (c) heteropolyoxometalate and (d) transition-metal-substituted polyoxometalate.
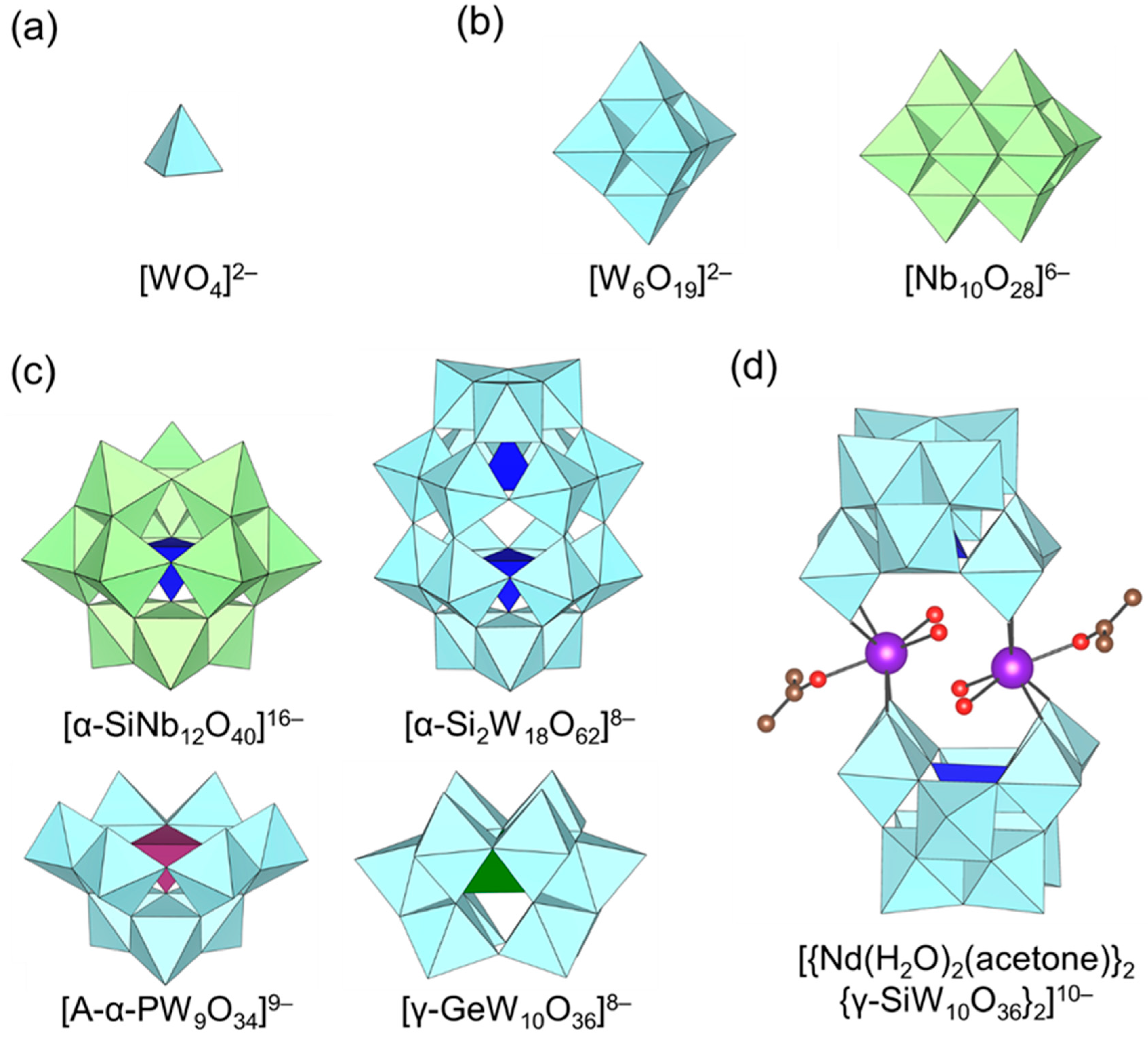
Figure 2.
Ball and stick representations of the anion parts of POM base catalysts and natural bond orbital (NBO) charges of oxygen atoms [38,55,56].
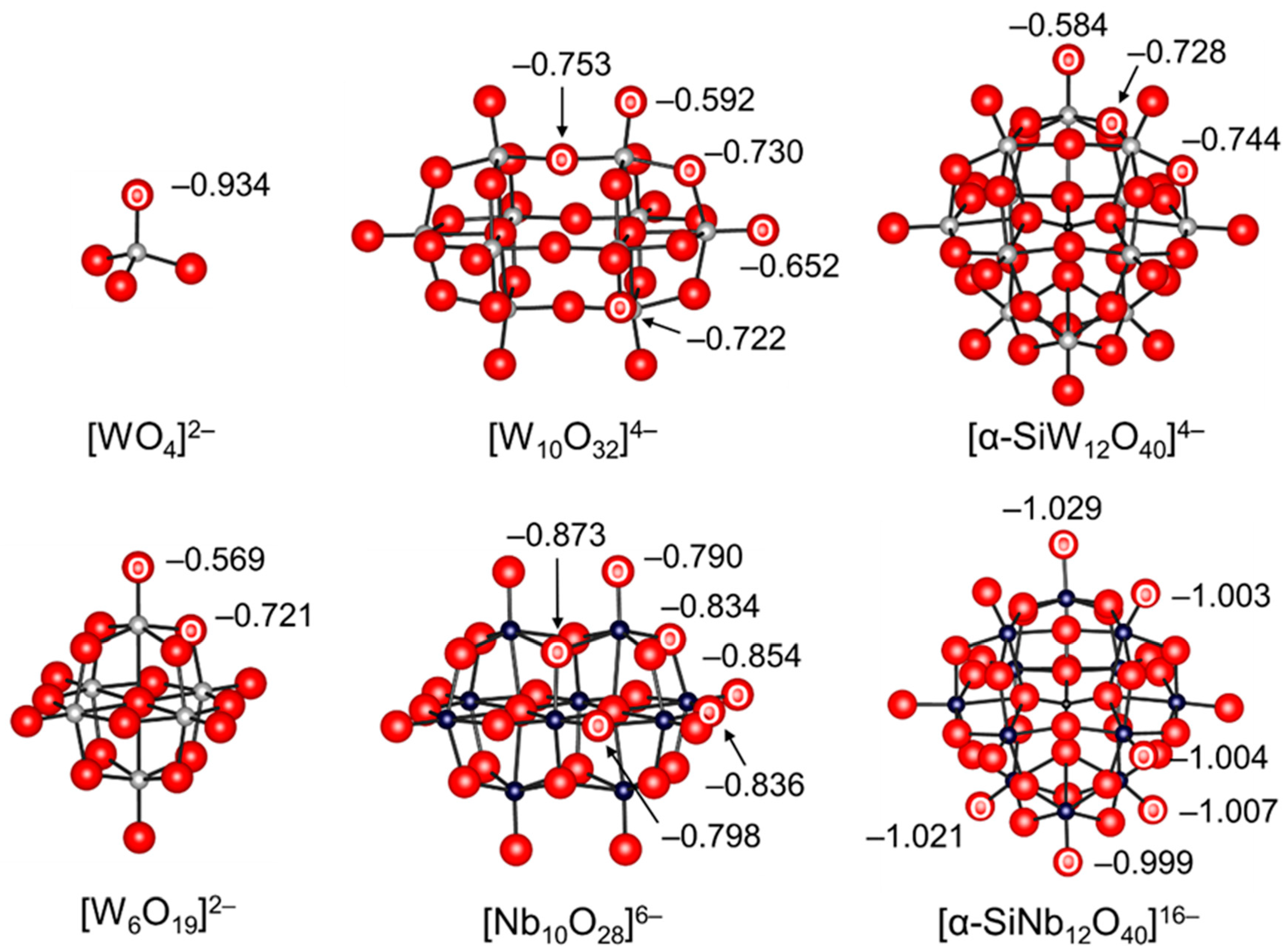
Figure 3.
Ball and stick representations of isopolyoxometalates. The arrows indicate the possible protonation sites proposed on the basis of the structure, NMR, and computational analysis [61,64,65,70,71,73,74,78,79,80].
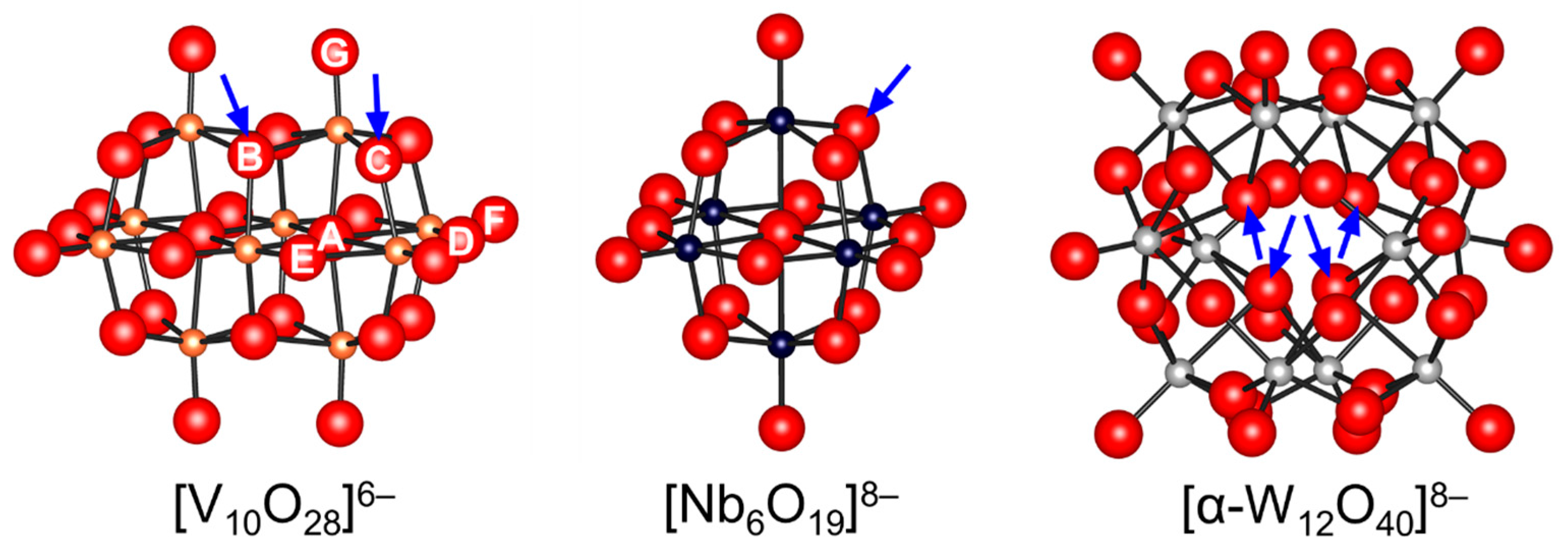
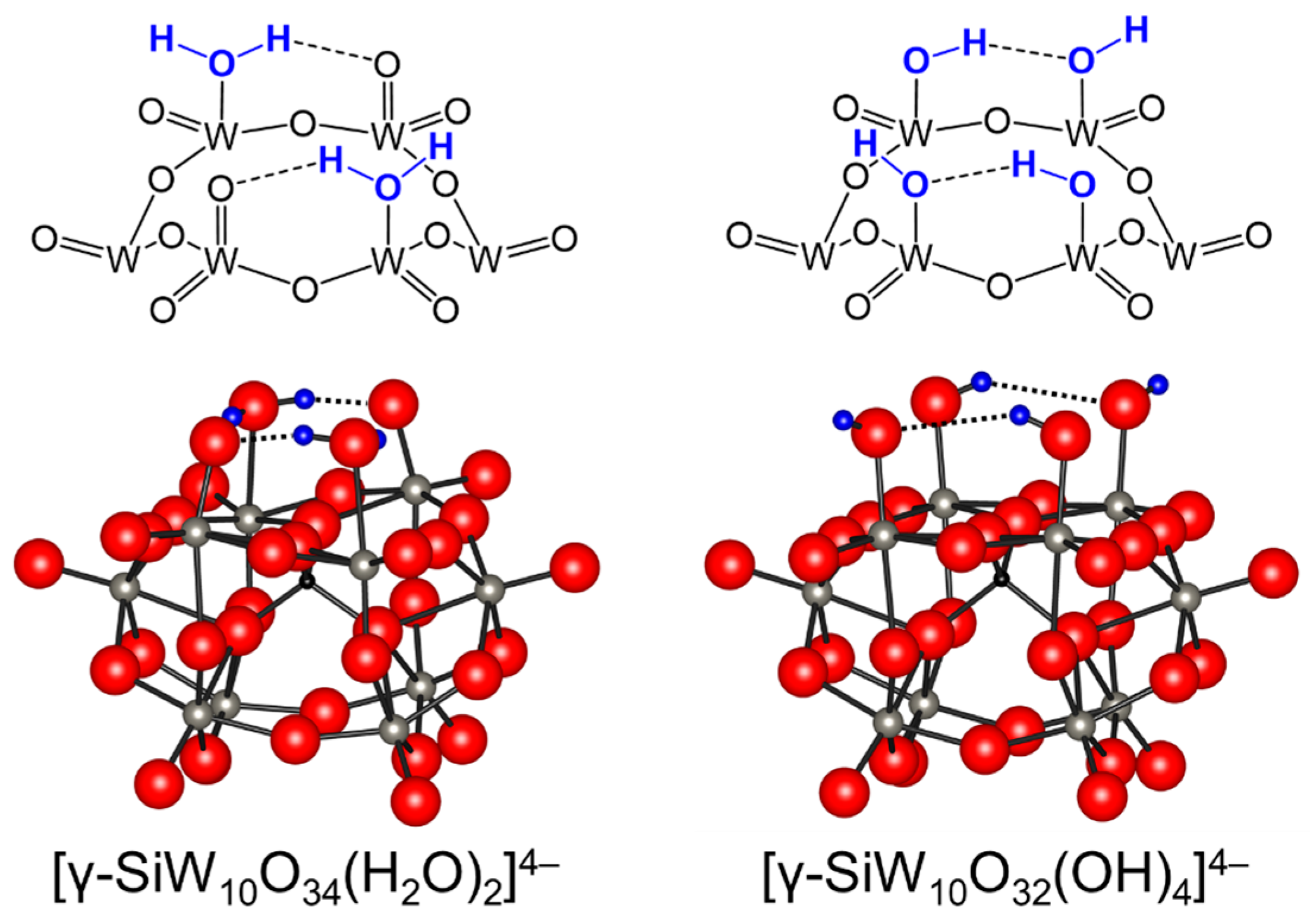
Figure 4.
Schematic representations of the lacunary sites on [γ-SiW10O34(H2O)2]4− and [γ-SiW10O32(OH)4]4− [82,83].
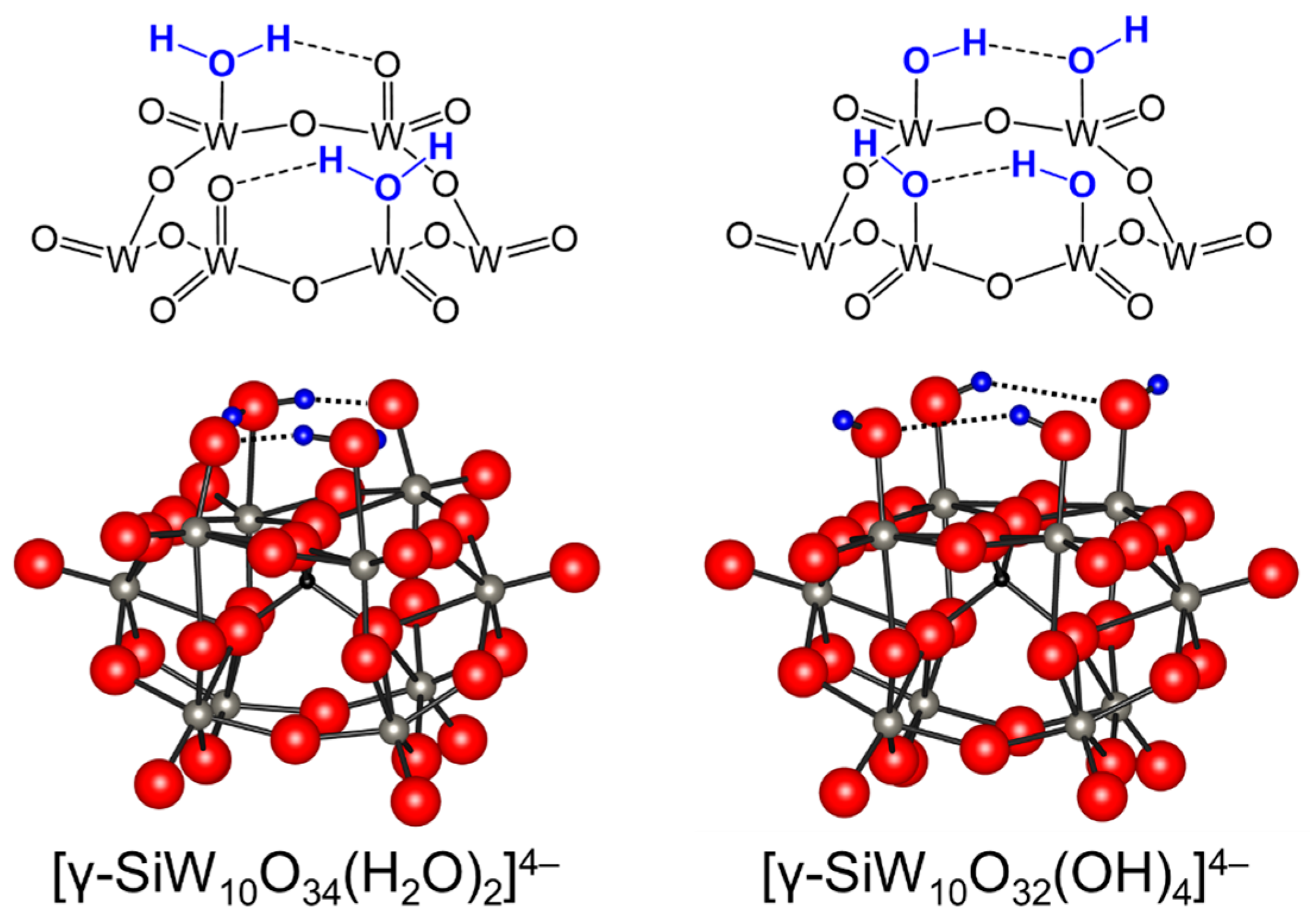
Figure 5.
Reversible changes among [γ-SiW10O34(OH)(OH2)]5−, [γ-SiW10O34(OH)2]6−, and [γ-SiW10O35(OH)]7− thorough protonation/deprotonation [91].
Figure 5.
Reversible changes among [γ-SiW10O34(OH)(OH2)]5−, [γ-SiW10O34(OH)2]6−, and [γ-SiW10O35(OH)]7− thorough protonation/deprotonation [91].

Figure 6.
Ball and stick representations of [γ-GeW10O34(OH)2]6− and [γ-GeW10O35(OH)]7− and possible active sites for base-catalyzed reactions [46].
Figure 6.
Ball and stick representations of [γ-GeW10O34(OH)2]6− and [γ-GeW10O35(OH)]7− and possible active sites for base-catalyzed reactions [46].
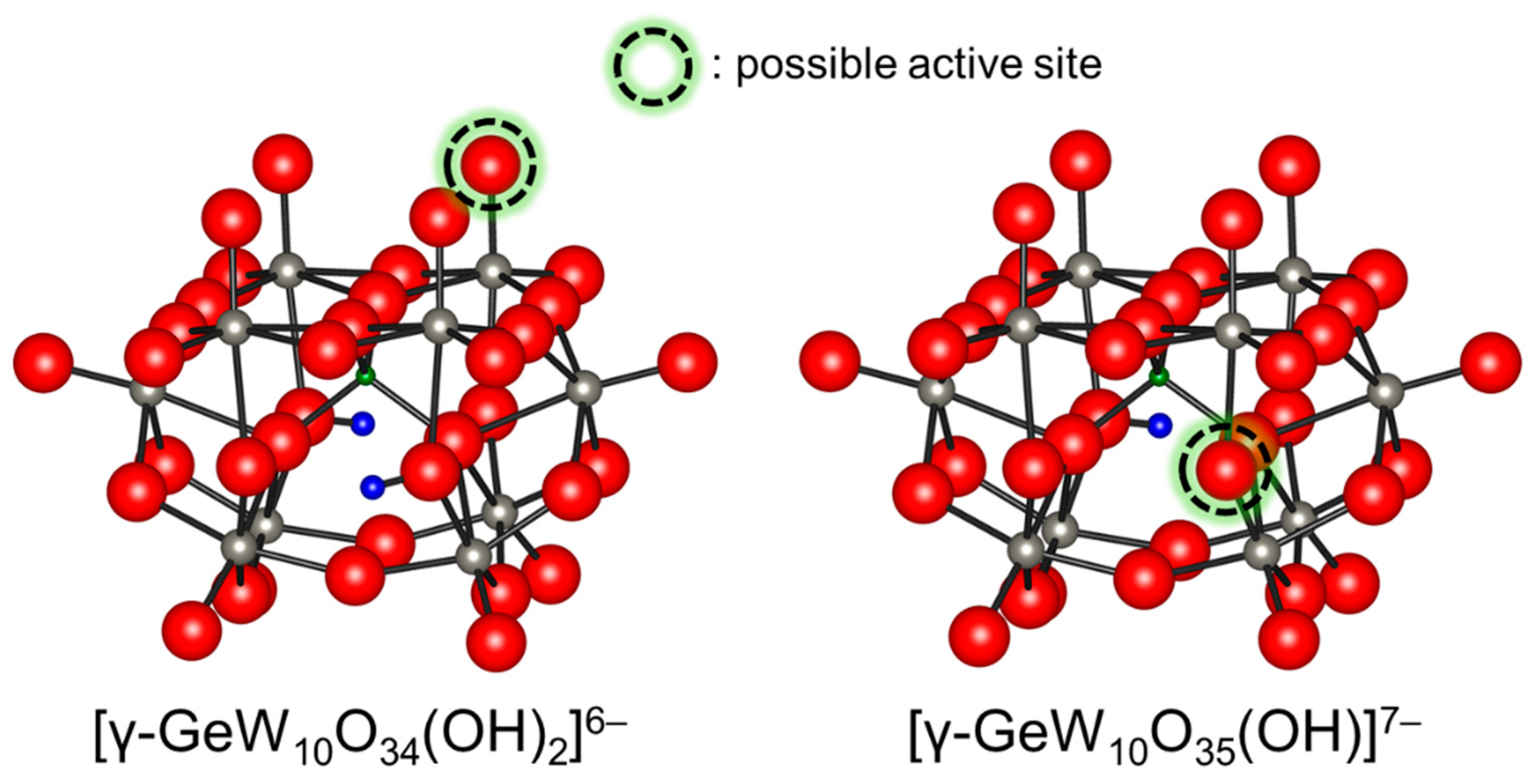
Figure 7.
Molecular structures of vanadium- and niobium-containing POMs. The arrows indicate the possible protonation sites proposed on the basis of the structure, NMR, and computational analysis [72,75,76,77,95,96,97,98,99,100,101].
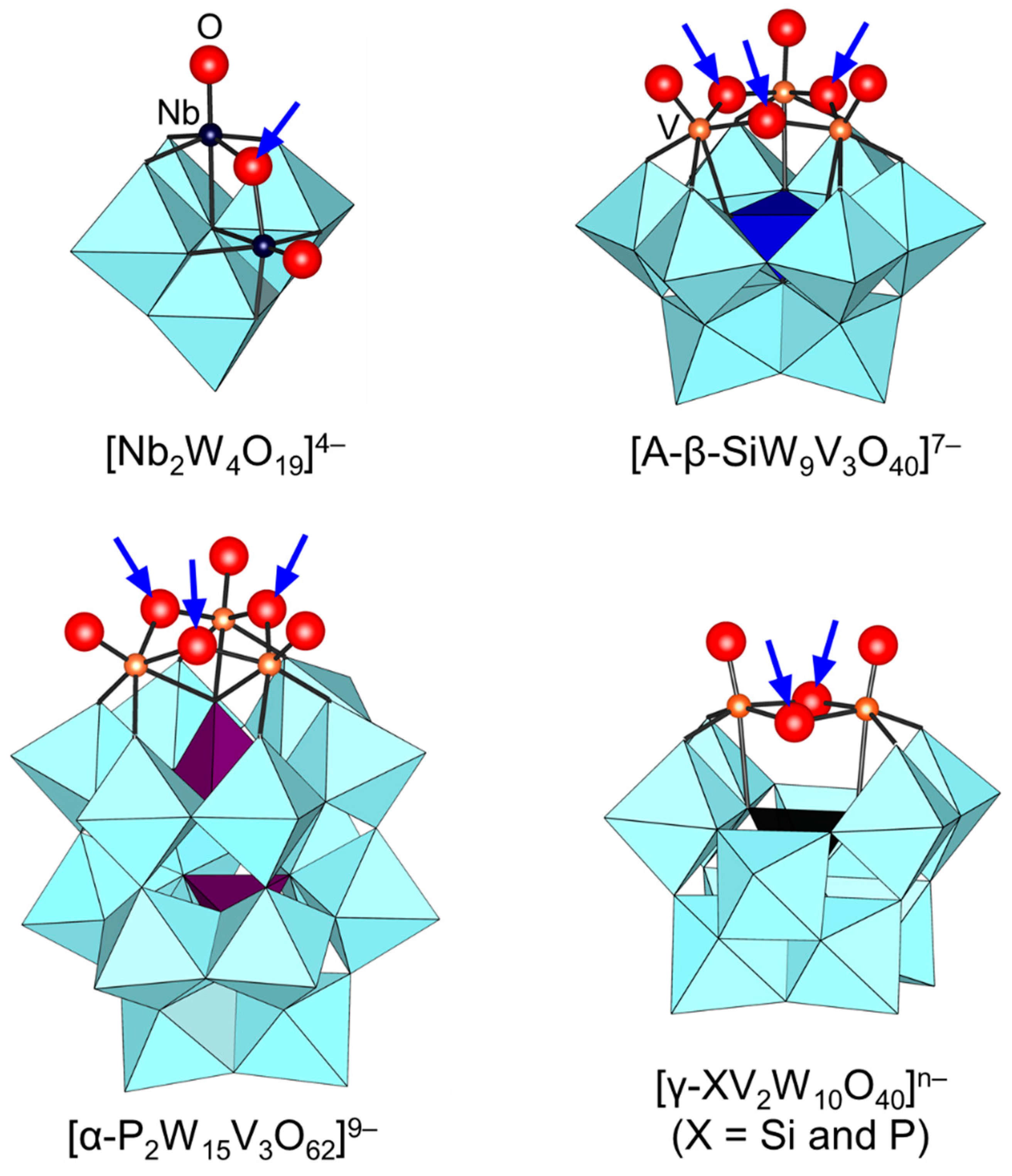
Figure 8.
Reversible change between [PTiW11O40]5− and [{PTiW11O39}2(μ-O)]8− [102].
Figure 8.
Reversible change between [PTiW11O40]5− and [{PTiW11O39}2(μ-O)]8− [102].
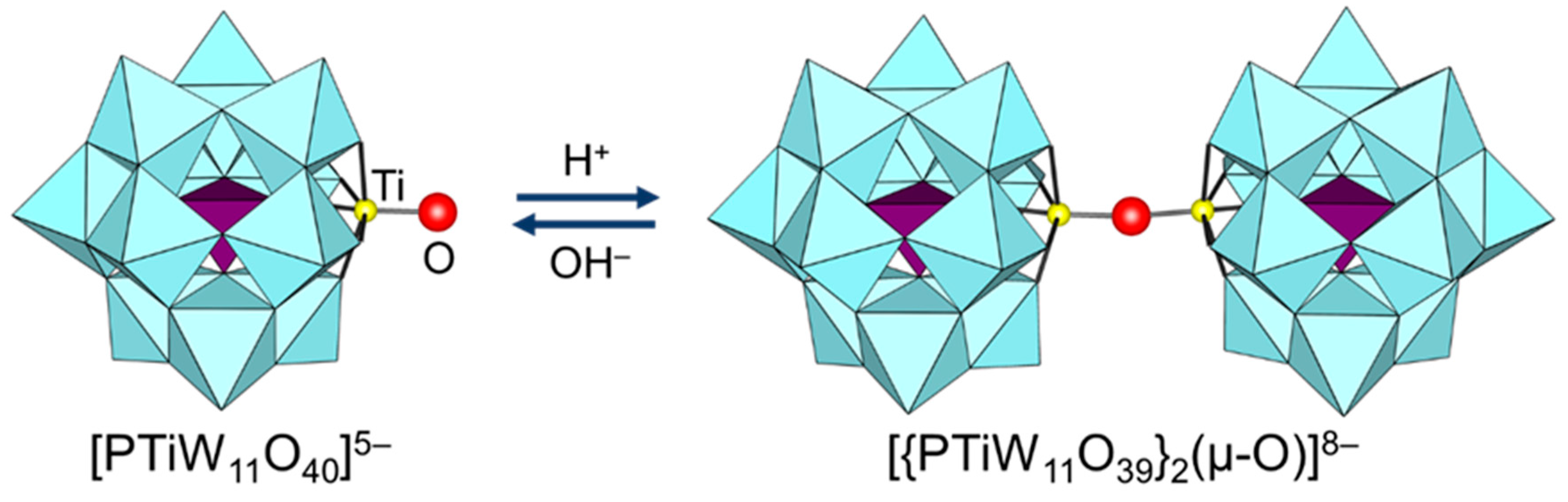
Figure 9.
Schematic representations for activation of substrates by POM base catalysts. (a) Abstraction of protons (Brønsted base); (b) nucleophilic action to substrates (Lewis base); and (c) bifunctional action in combination with metal catalysts.
Figure 9.
Schematic representations for activation of substrates by POM base catalysts. (a) Abstraction of protons (Brønsted base); (b) nucleophilic action to substrates (Lewis base); and (c) bifunctional action in combination with metal catalysts.
Scheme 1.
Chemical fixation of CO2 with 1,2-phenylenediamine catalyzed by TBA2[WO4].

Scheme 2.
Chemical fixation of CO2 with 2-methyl-3-butyn-2-ol catalyzed by TBA2[WO4].
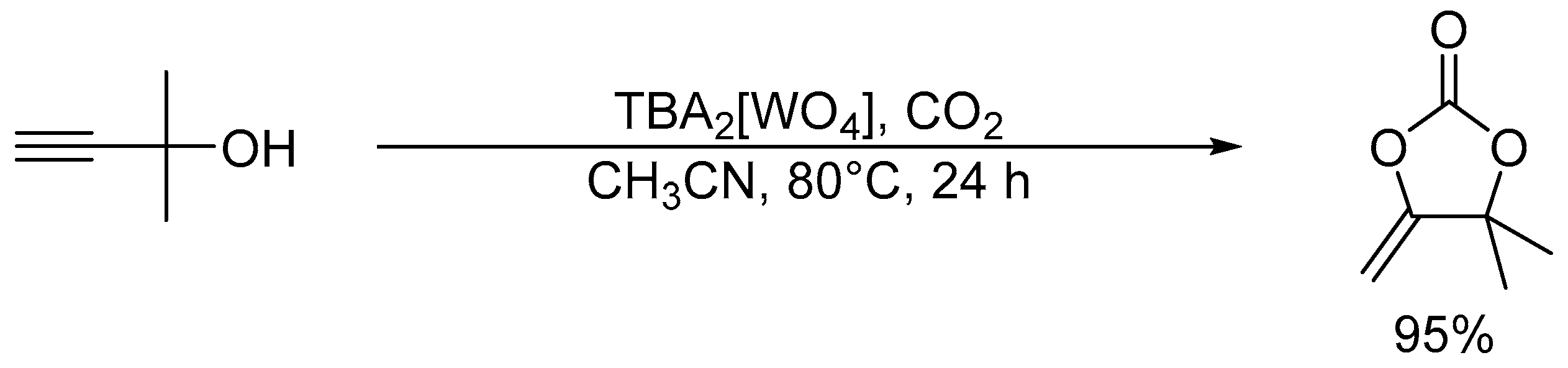
Scheme 3.
Chemical fixation of CO2 with 2-amino-5-chlorobenzonitrile catalyzed by TBA2[WO4].
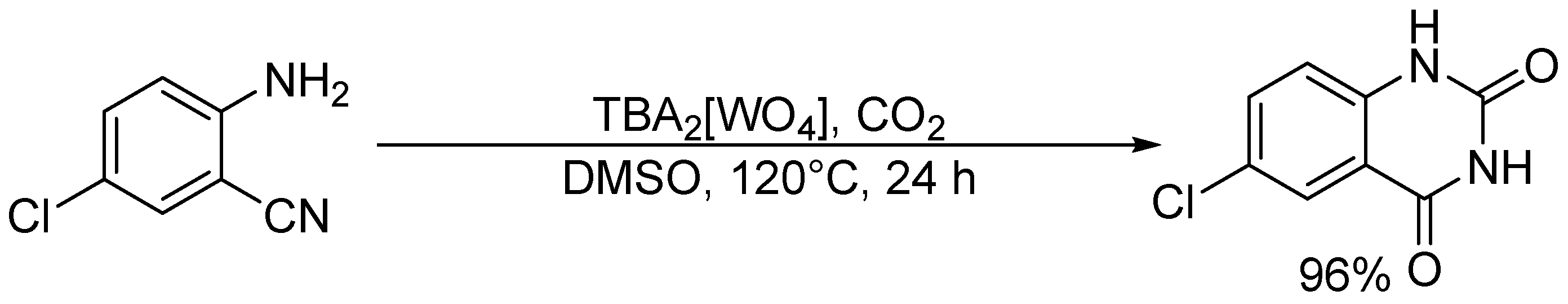
Figure 10.
Proposed mechanism for the chemical fixation of CO2 with aryldiamines catalyzed by TBA2[WO4] [38].
Figure 10.
Proposed mechanism for the chemical fixation of CO2 with aryldiamines catalyzed by TBA2[WO4] [38].
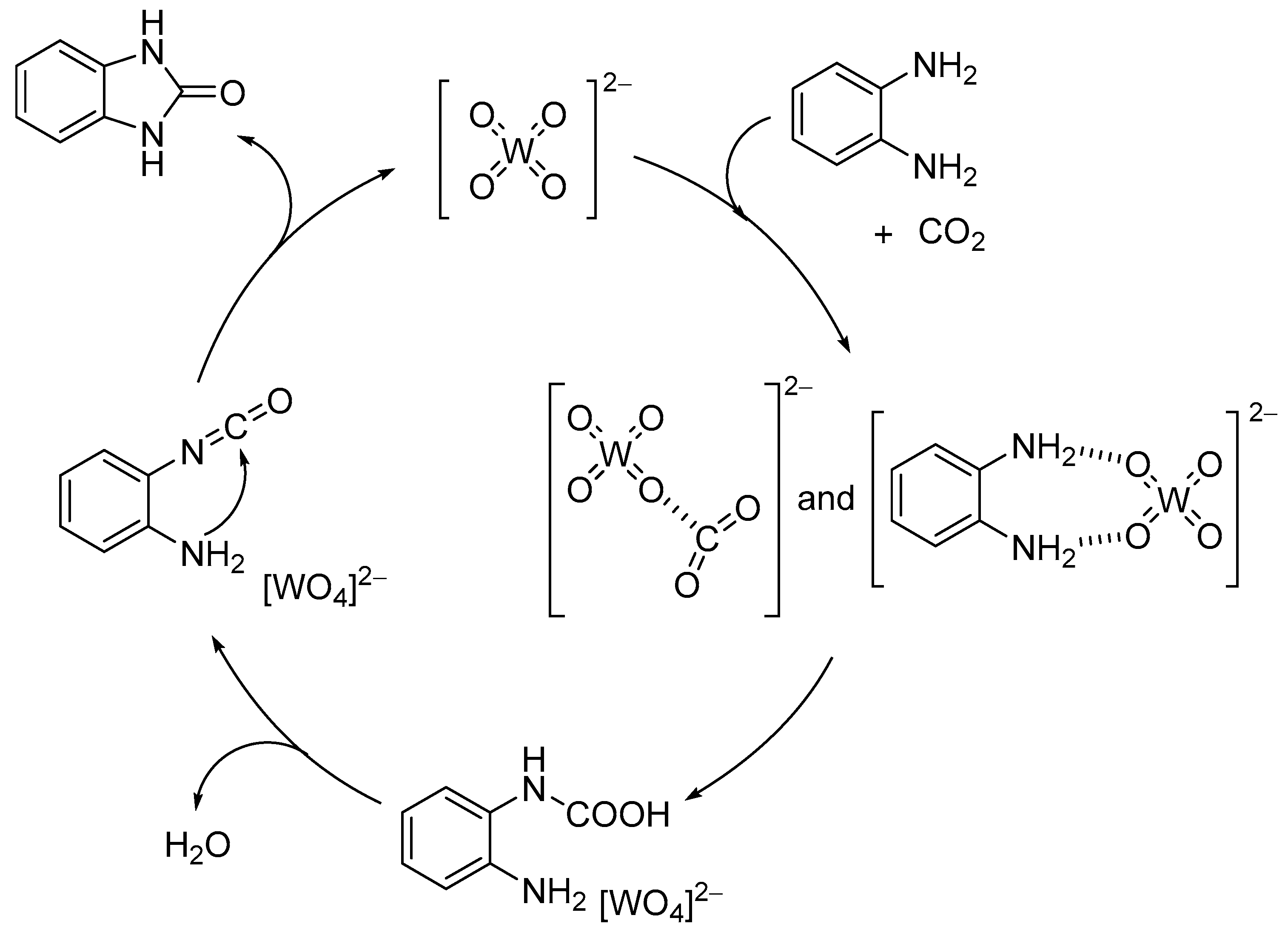
Scheme 4.
Chemical fixation of CO2 with 2-methyl-3-butyn-2-ol catalyzed by Ag2[WO4]/Ph3P.
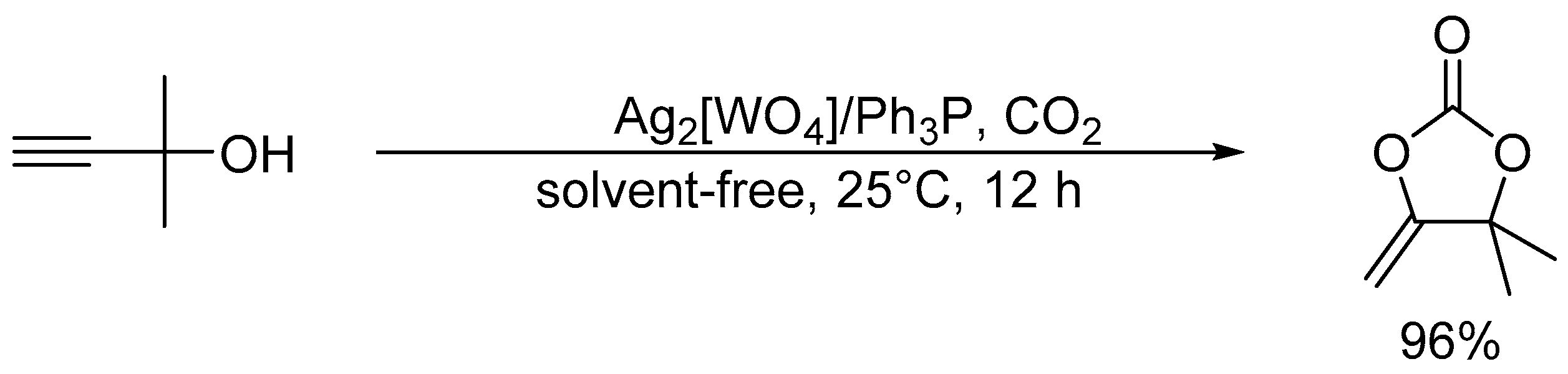
Scheme 5.
Carboxylation of phenylacetylene with n-butyl iodide catalyzed by Ag2[WO4]/Cs2CO3.
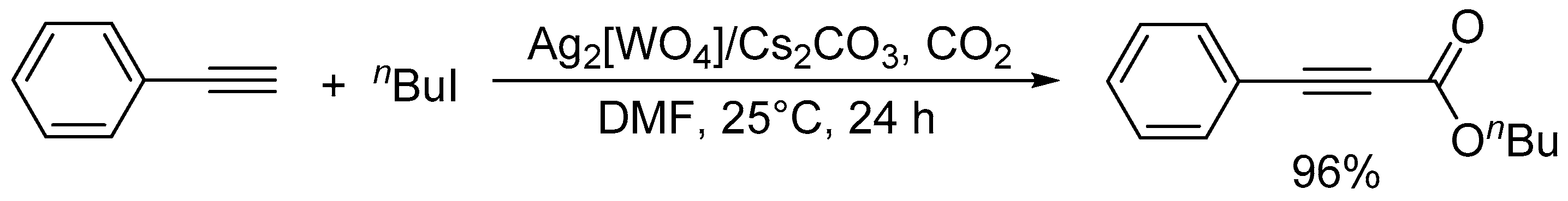
Scheme 6.
Cycloaddition of propylene oxide and CO2 catalyzed by (DBUH)3[NbO3(O2)].

Scheme 7.
Cycloaddition of epichlorohydrin and CO2 catalyzed by Na16[SiNb12O40].
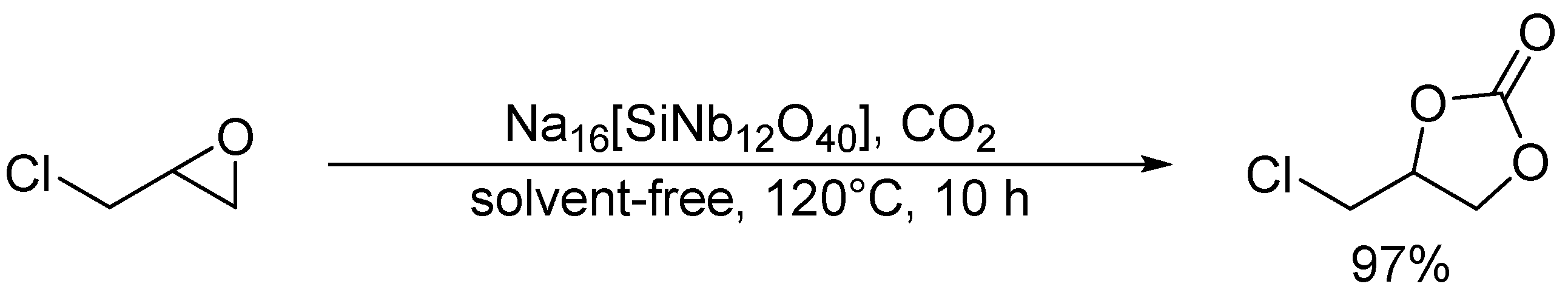
Scheme 8.
Cycloaddition of styrene oxide and CO2 catalyzed by TBA6[Nb10O28].
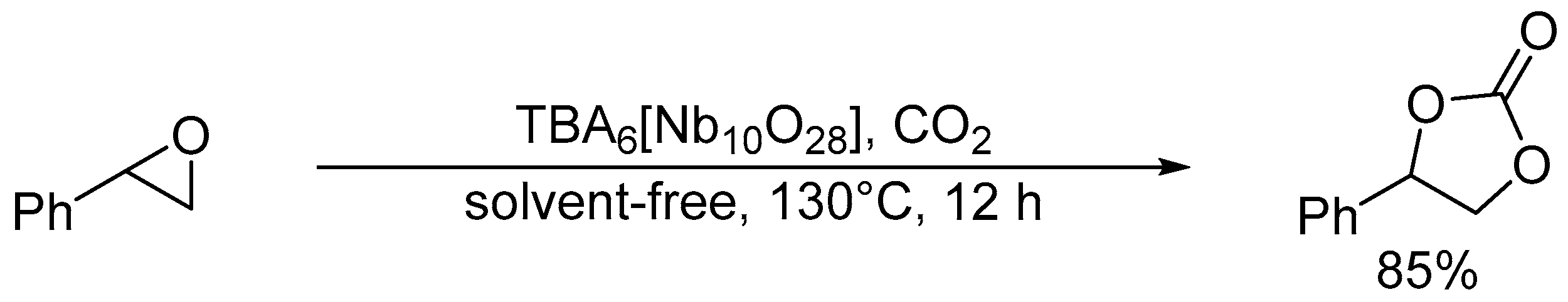
Scheme 9.
Knoevenagel condensation of ethyl cyanoacetate with cyclohexanone catalyzed by TBA6[γ-H2GeW10O36]
Scheme 9.
Knoevenagel condensation of ethyl cyanoacetate with cyclohexanone catalyzed by TBA6[γ-H2GeW10O36]
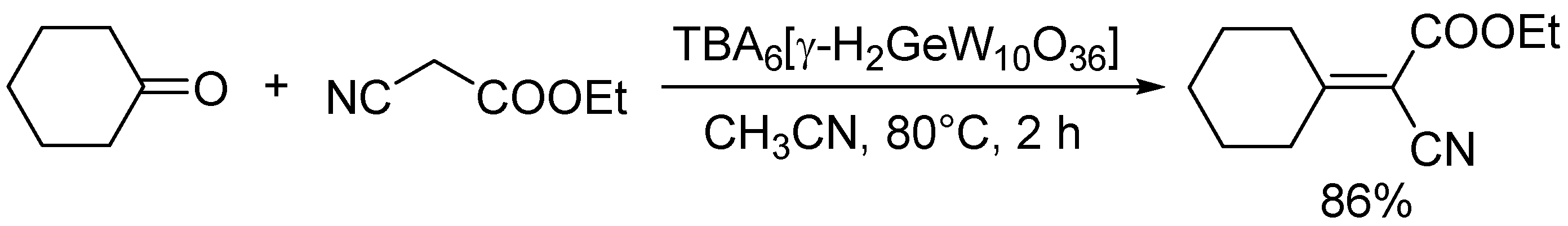
Scheme 10.
N-Silylation of indole with dimethylphenylsilane catalyzed by Rh2(OAc)4/TBA2[WO4].
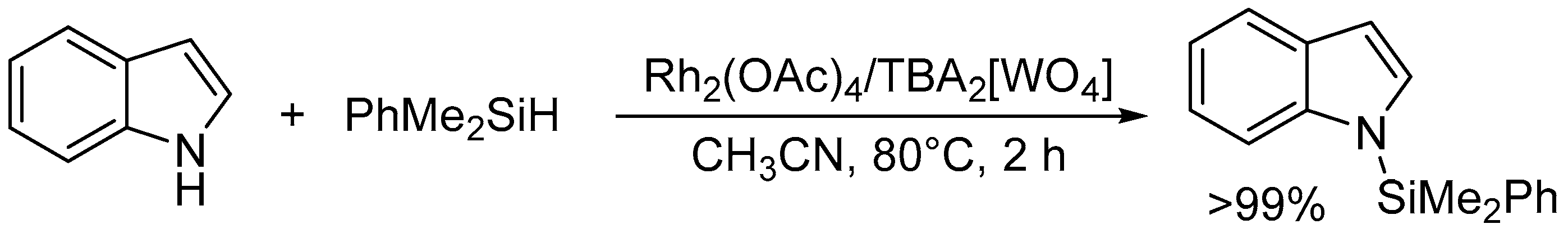
Figure 11.
Proposed mechanism for the site-selective N-silylation of indole derivatives with hydrosilanes by combined catalyst of Rh2(OAc)4 and [WO4]2− [43].
Figure 11.
Proposed mechanism for the site-selective N-silylation of indole derivatives with hydrosilanes by combined catalyst of Rh2(OAc)4 and [WO4]2− [43].
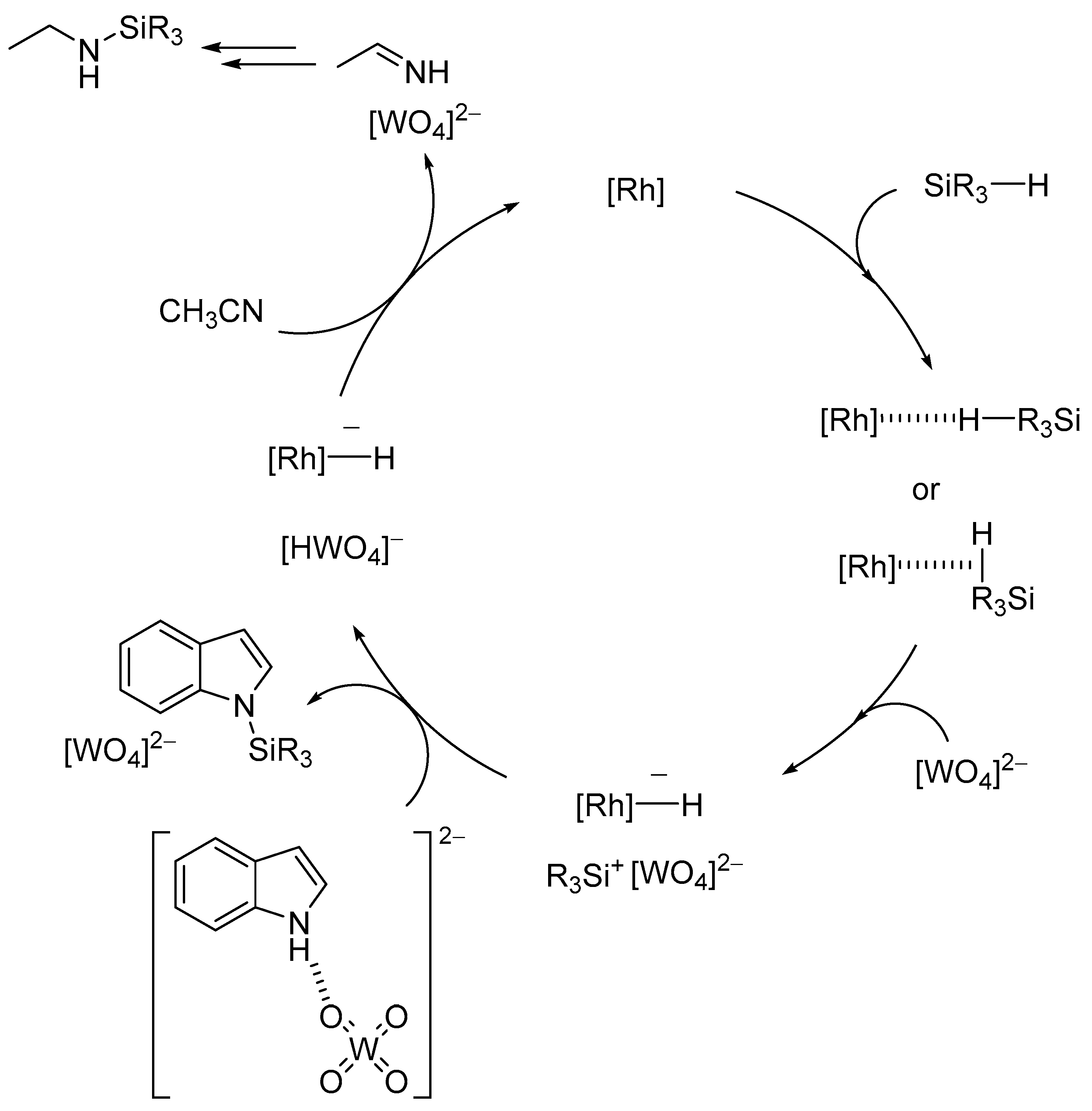
Scheme 11.
Acylation of 2,2-dimethyl-1,3-dioxolane-4-methanol with vinyl acetate catalyzed by TBA7[γ-HGeW10O36].
Scheme 11.
Acylation of 2,2-dimethyl-1,3-dioxolane-4-methanol with vinyl acetate catalyzed by TBA7[γ-HGeW10O36].
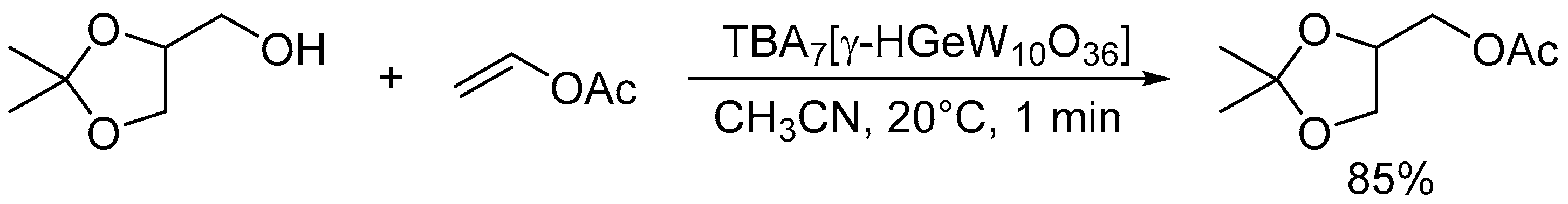
Scheme 12.
Intermolecular competitive acylation of primary alcohol (benzyl alcohol) and secondary alcohols (1-phenylethanol or 2-butanol) with vinyl acetate catalyzed by TBA7[γ-HGeW10O36].
Scheme 12.
Intermolecular competitive acylation of primary alcohol (benzyl alcohol) and secondary alcohols (1-phenylethanol or 2-butanol) with vinyl acetate catalyzed by TBA7[γ-HGeW10O36].
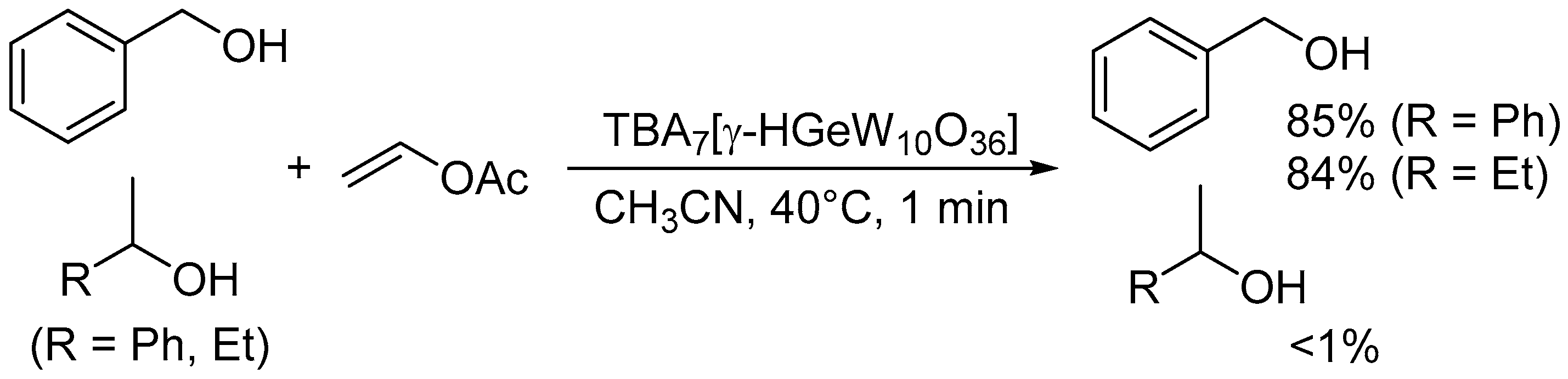
Figure 12.
Proposed mechanism for the chemoselective acylation of alcohols with vinyl acetate catalyzed by [γ-HGeW10O36]7− [46].
Figure 12.
Proposed mechanism for the chemoselective acylation of alcohols with vinyl acetate catalyzed by [γ-HGeW10O36]7− [46].

Scheme 13.
Dehydration of benzamide in the presence of MS-3A catalyzed by TBA4[α-H4SiW11O39].

Figure 13.
Proposed mechanism for the dehydration of amides to nitriles catalyzed by [α-H4SiW11O39]4− [123].
Figure 13.
Proposed mechanism for the dehydration of amides to nitriles catalyzed by [α-H4SiW11O39]4− [123].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
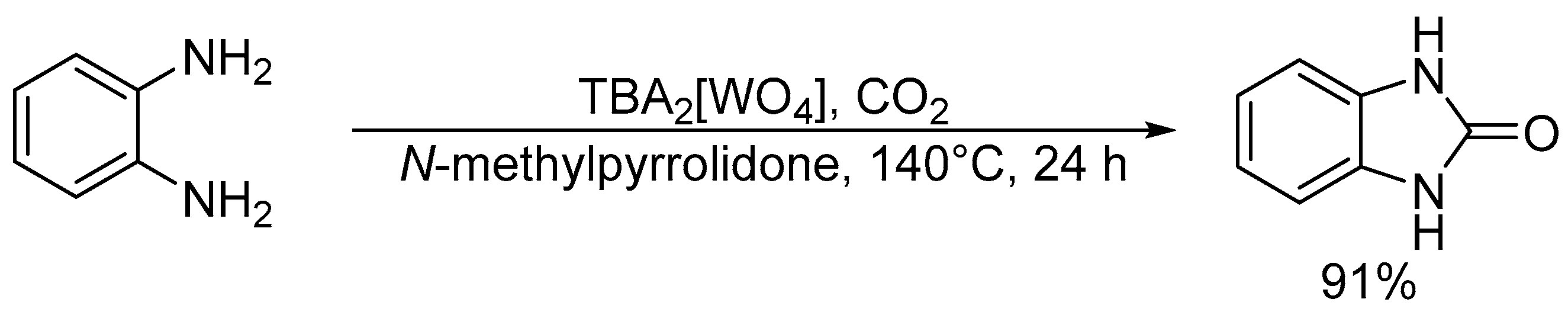{kind=link}
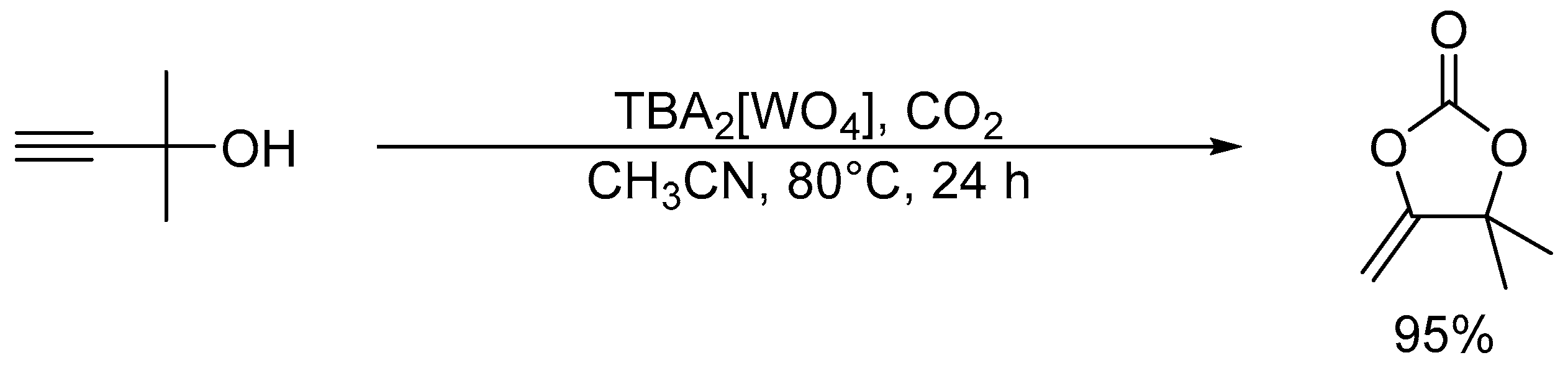{kind=link}
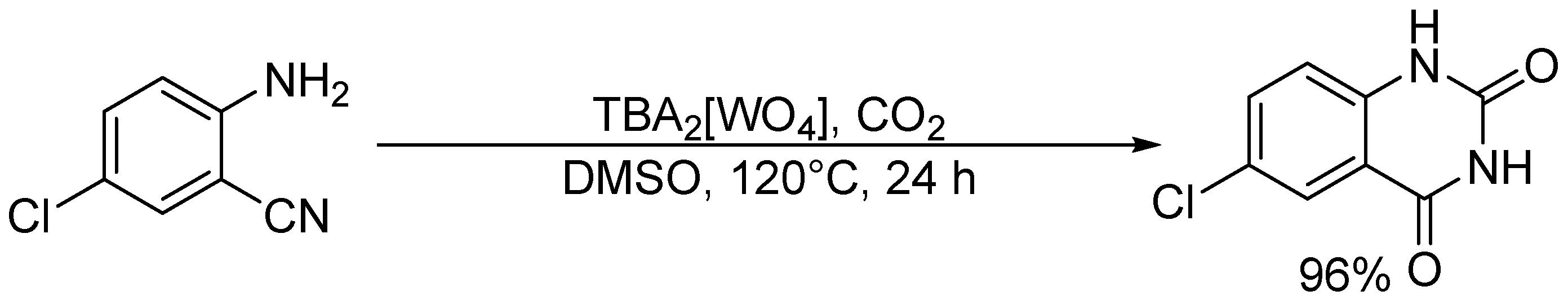{kind=link}
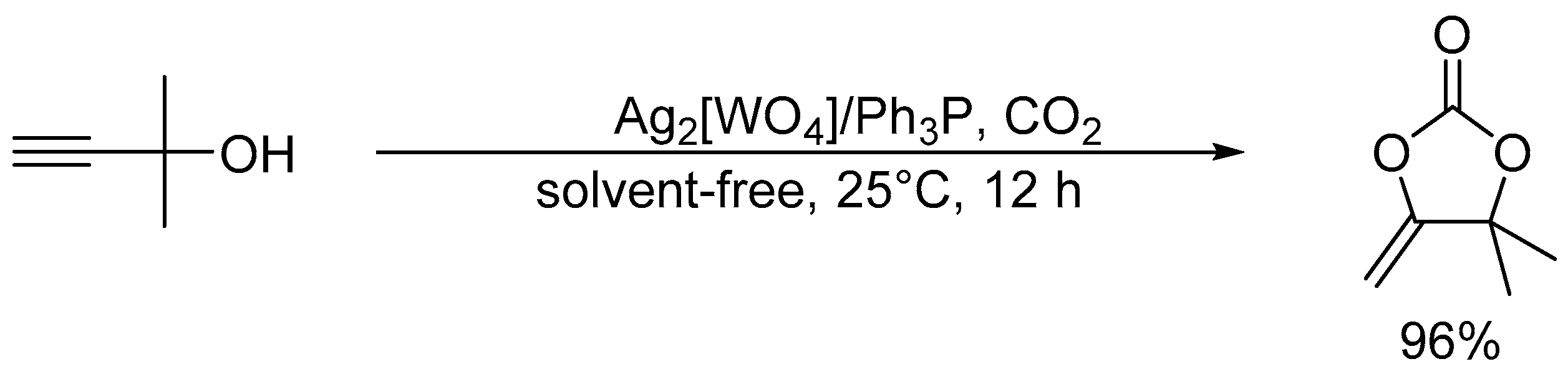{kind=link}
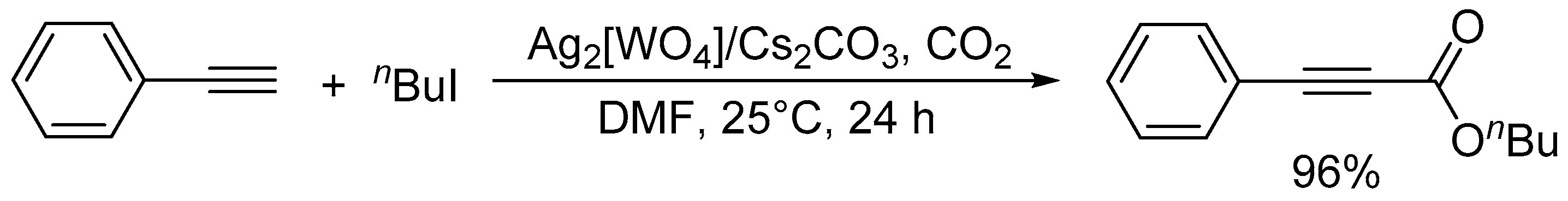{kind=link}
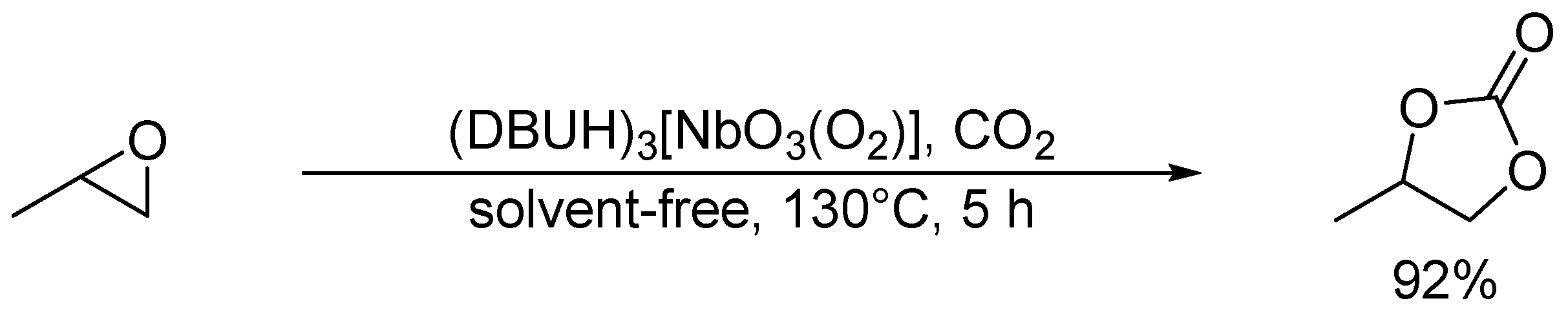{kind=link}
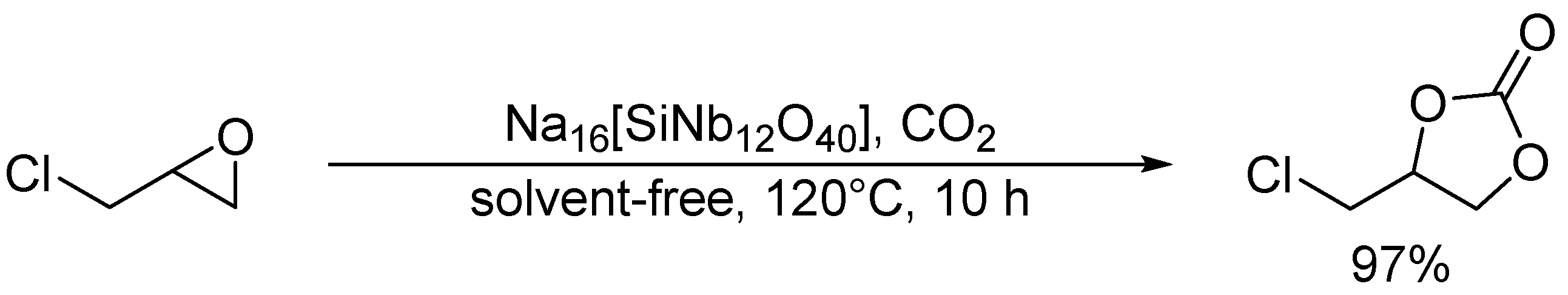{kind=link}
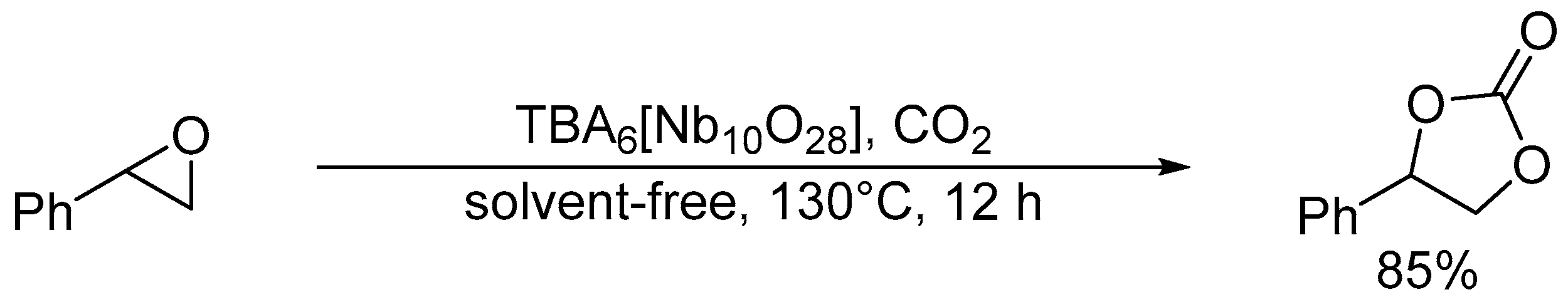{kind=link}
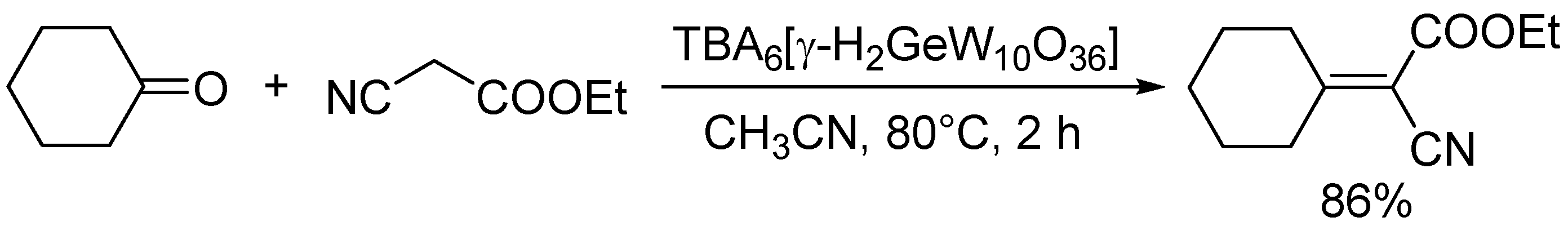{kind=link}
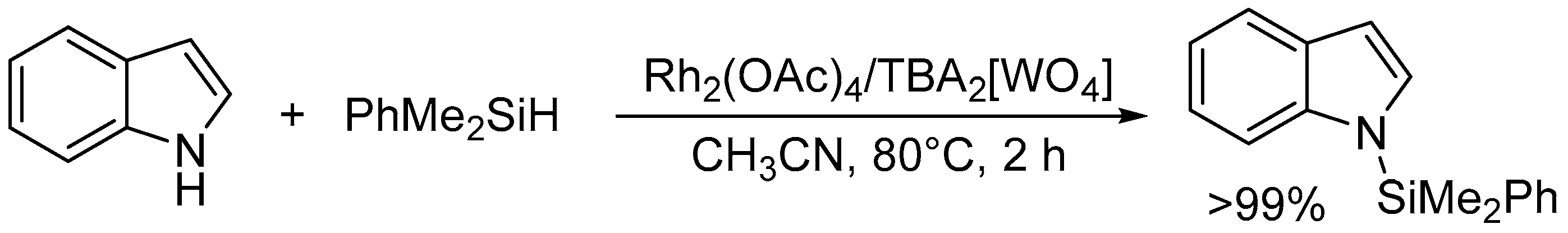{kind=link}
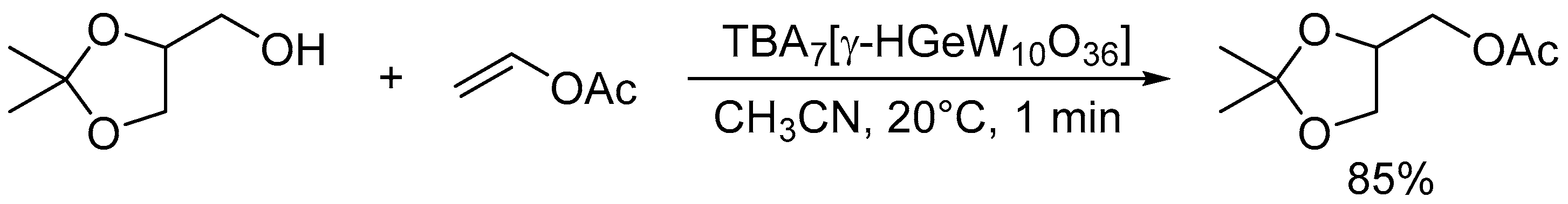{kind=link}
{kind=link}
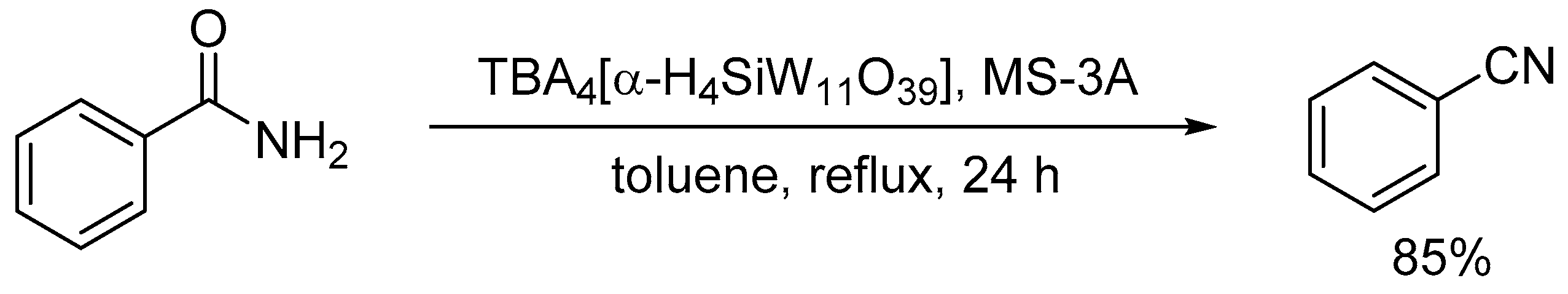{kind=link}
Table 1.
pKa values of the conjugate acids of various inorganic and organic bases.
| Compound | pKa | Refs. |
|---|---|---|
| [WO4]2− | 3.5 | [7] |
| [MoO4]2− | 3.9 | [7] |
| [VO4]3− | 13.2 | [7] |
| [CO3]2− | 10.3 | [38] |
| 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) | 12.0 | [38] |
| 1,1,3,3-Tetramethylguanidine (TMG) | 13.6 | [38] |
Table 2.
Knoevenagel condensation of benzaldehyde with active methylene compounds by POM base catalysts.
Table 2.
Knoevenagel condensation of benzaldehyde with active methylene compounds by POM base catalysts.

| Catalyst (mol %) | Donor (Equiv.) | Solvent | Temp. (°C) | Time | Yield (%) | TON [a] | TOF [b] (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| TBA6[γ-H2GeW10O36] (0.5) | Malononitrile (1.5) | CH3CN | 32 | 0.5 h | 99 | 198 | 396 | [39] |
| TBA6[γ-H2GeW10O36] (1) | Ethyl cyanoacetate (1.5) | CH3CN | 32 | 2 h | 98 | 98 | 49 | [39] |
| TBA6[γ-H2GeW10O36] (1) | Ethyl cyanoacetate (1.5) | – | 40 | 5 min | 85 | 85 | 1020 (>3000) [c] | [39] |
| TBA6[γ-H2GeW10O36] (5) | Phenylacetonitirle (1.5) | CH3CN | 80 | 2 h | 96 | 19 | 10 | [39] |
| TBA2[W6O19] (3.7) | Malononitrile (1) | EtOH | reflux | 7 min | 92 | 25 | 214 | [40] |
| TBA8[α-Si2W18O62] (0.5) | Ethyl cyanoacetate (1.5) | CH3CN | 32 | 3 h | 96 | 192 | 64 | [47] |
| TMA6[Nb10O28] (1) | Phenylacetonitirle (1) | CH3OH | 70 | 24 h | 35 | 35 | 1.5 | [55] |
| TBA7[γ-HGeW10O36] (2) | Phenylacetonitirle (1.5) | CH3CN | 80 | 5 min | 83 | 42 | 504 | [46] |
| Na16[SiNb12O40] (0.23) | Ethyl cyanoacetate (1) | – | 70 | 2 h | 67 | 288 | 144 | [56] |
| TBA4[γ-SiW10O34(H2O)2] (0.5) | Malononitrile (0.67) | CH3CN | 32 | 2.5 h | 90 | 180 | 72 | [36] |
| Na8H[PW9O34] (0.25) | Malononitrile (1.5) | CH3OH | 25 | 6 h | 92 | 368 | 61 | [50] |
| Na8H[PW9O34] (0.25) | Ethyl cyanoacetate (1.5) | CH3OH | 25 | 6 h | 80 | 320 | 53 | [50] |
[a] TON = Products (mol)/catalyst (mol). [b] TOF (h−1) = TON/time (h). [c] The TOF value in the parenthesis is determined by the initial rate.
Table 3.
Cyanosilylation of carbonyl compounds with trimethylsilyl cyanide (TMSCN) by POM base catalysts.
Table 3.
Cyanosilylation of carbonyl compounds with trimethylsilyl cyanide (TMSCN) by POM base catalysts.

| Catalyst (mol %) | Substrate | TMSCN (Equiv.) | Solvent | Temp. (°C) | Time | Yield (%) | TON [c] | TOF [d] (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| TBA4[γ-SiW10O34(H2O)2] (1) | 2-Adamantanone | 2 | DCE [a] | 32 | 72 h | 30 | 30 | 0.4 | [36] |
| TBA8H2[{Y(H2O)2}2(γ-SiW10O36)2] (1) | 2-Adamantanone | 1.5 | DCE [a] | 30 | 1.5 h | 91 | 91 | 61 | [37] |
| TBA8H2[{Y(H2O)2}2(γ-SiW10O36)2] (0.01) | Benzaldehyde | 1.5 | DCE [a] | 30 | 15 min | 94 | 9400 | 37,600 | [37] |
| TBA6[γ-H2GeW10O36] (0.1) | Acetophenone | 1.5 | CH3CN | 30 | 30 min | 99 | 990 | 1980 | [39] |
| TBA8H2[{Nd(H2O)2}2(γ-SiW10O36)2] (0.5) | 2-Adamantanone | 1.5 | DCE [a] | 30 | 7 min | 98 | 196 | 1680 | [41] |
| TBA8H2[{Nd(H2O)2}2(γ-SiW10O36)2] (0.1) | Benzaldehyde | 1.5 | DCE [a] | 30 | 2 min | 99 | 990 | 29,700 | [41] |
| Na8H[PW9O34] (0.25) | 2-Adamantanone | 1.5 | EtOAc [b] | 25 | 6 h | 92 | 368 | 61 | [50] |
| Na8H[PW9O34] (0.25) | Benzaldehyde | 1.5 | EtOAc [b] | 25 | 6 h | 99 | 396 | 66 | [50] |
| [Nd(H2O)5]2MoV2O26 (1) | Benzaldehyde | 3 | – | 25 | 7 h | 96 | 96 | 14 | [51] |
| [Sm(H2O)5][Sm(H2O)7][Co2Mo10H4O38] (2) | Benzaldehyde | 3 | – | 25 | 5 h | 98 | 49 | 10 | [52] |
| (C2N2H12)[Ba(H2O)3][Co2Mo10H4O38] (2) | Benzaldehyde | 3 | – | 25 | 6 h | 99 | 50 | 8 | [52] |
[a] DCE = 1,2-Dichloroethane. [b] EtOAc = Ethylacetate. [c] TON = Products (mol)/catalyst (mol). [d] TOF (h−1) = TON/time (h).
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kamata, K.; Sugahara, K. Base Catalysis by Mono- and Polyoxometalates. Catalysts 2017, 7, 345. https://doi.org/10.3390/catal7110345
AMA Style
Kamata K, Sugahara K. Base Catalysis by Mono- and Polyoxometalates. Catalysts. 2017; 7(11):345. https://doi.org/10.3390/catal7110345
Chicago/Turabian StyleKamata, Keigo, and Kosei Sugahara. 2017. "Base Catalysis by Mono- and Polyoxometalates" Catalysts 7, no. 11: 345. https://doi.org/10.3390/catal7110345
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.