Dehydrogenation of Formic Acid over a Homogeneous Ru-TPPTS Catalyst: Unwanted CO Production and Its Successful Removal by PROX

Abstract
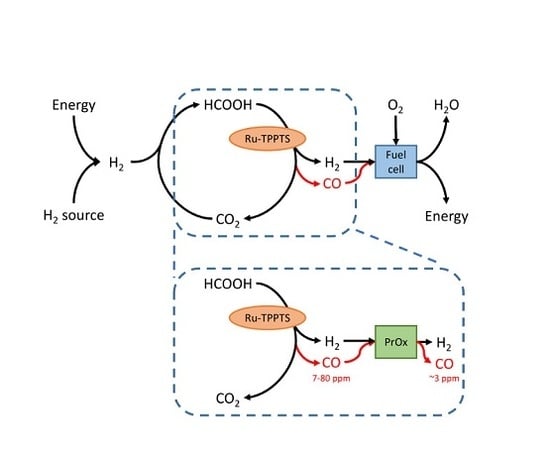
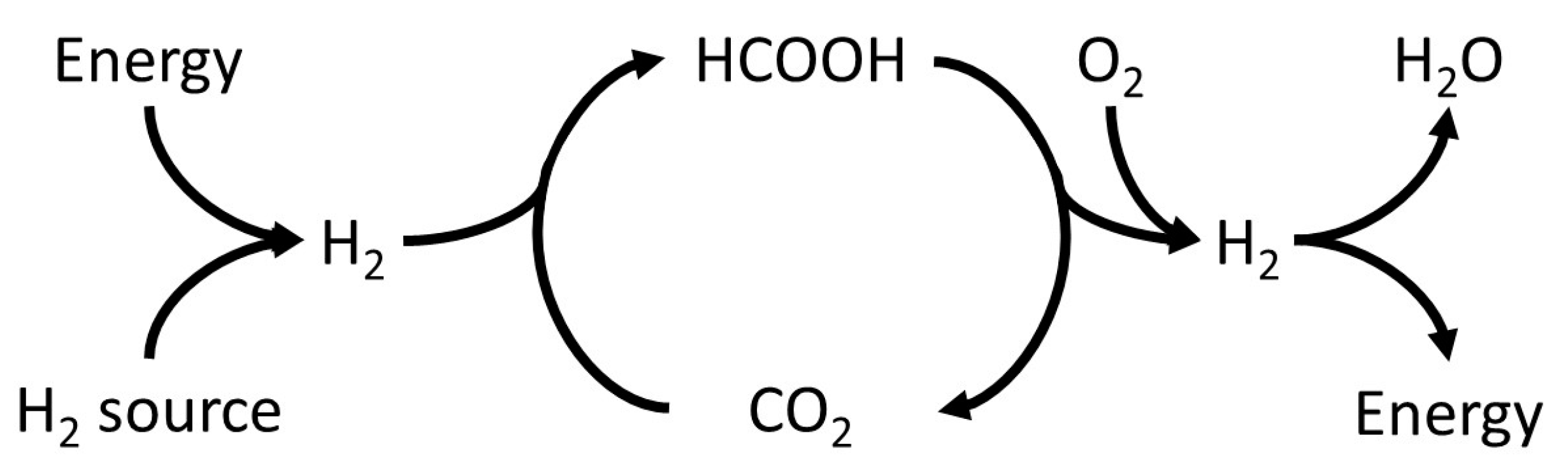
:
1. Introduction
2. Results and Discussion
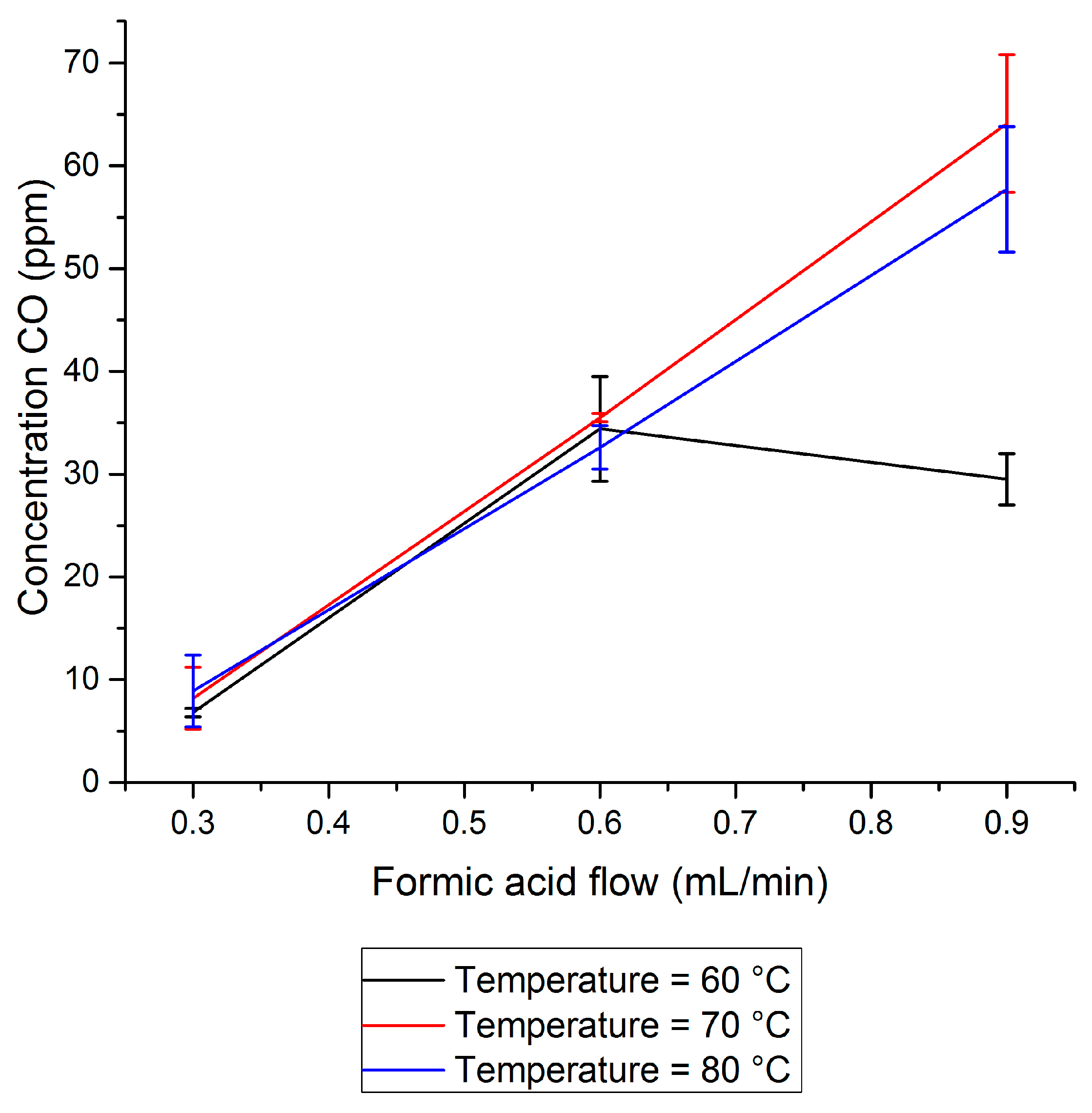
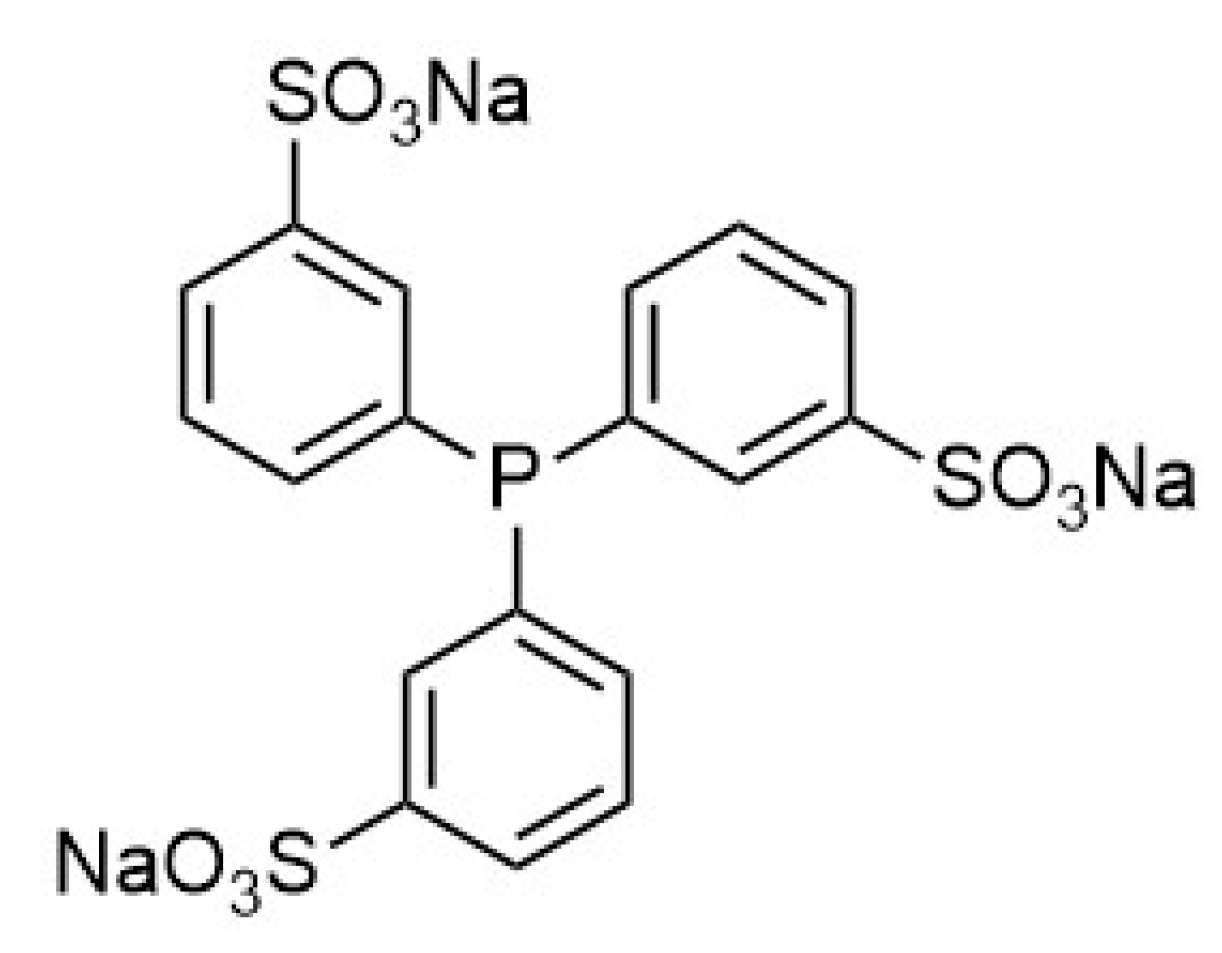
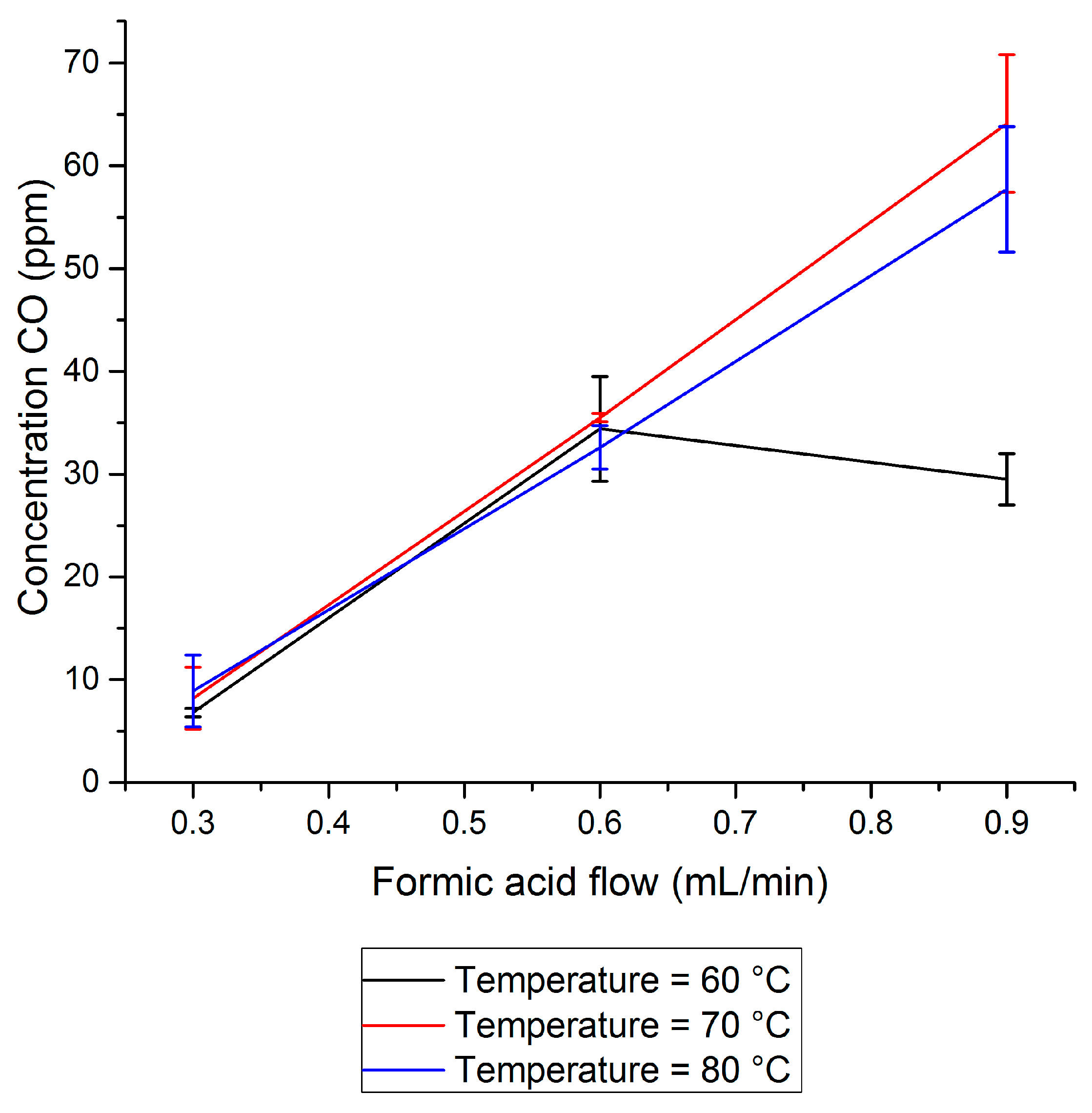
2.1. CO Side-Production in FA Dehydrogenation
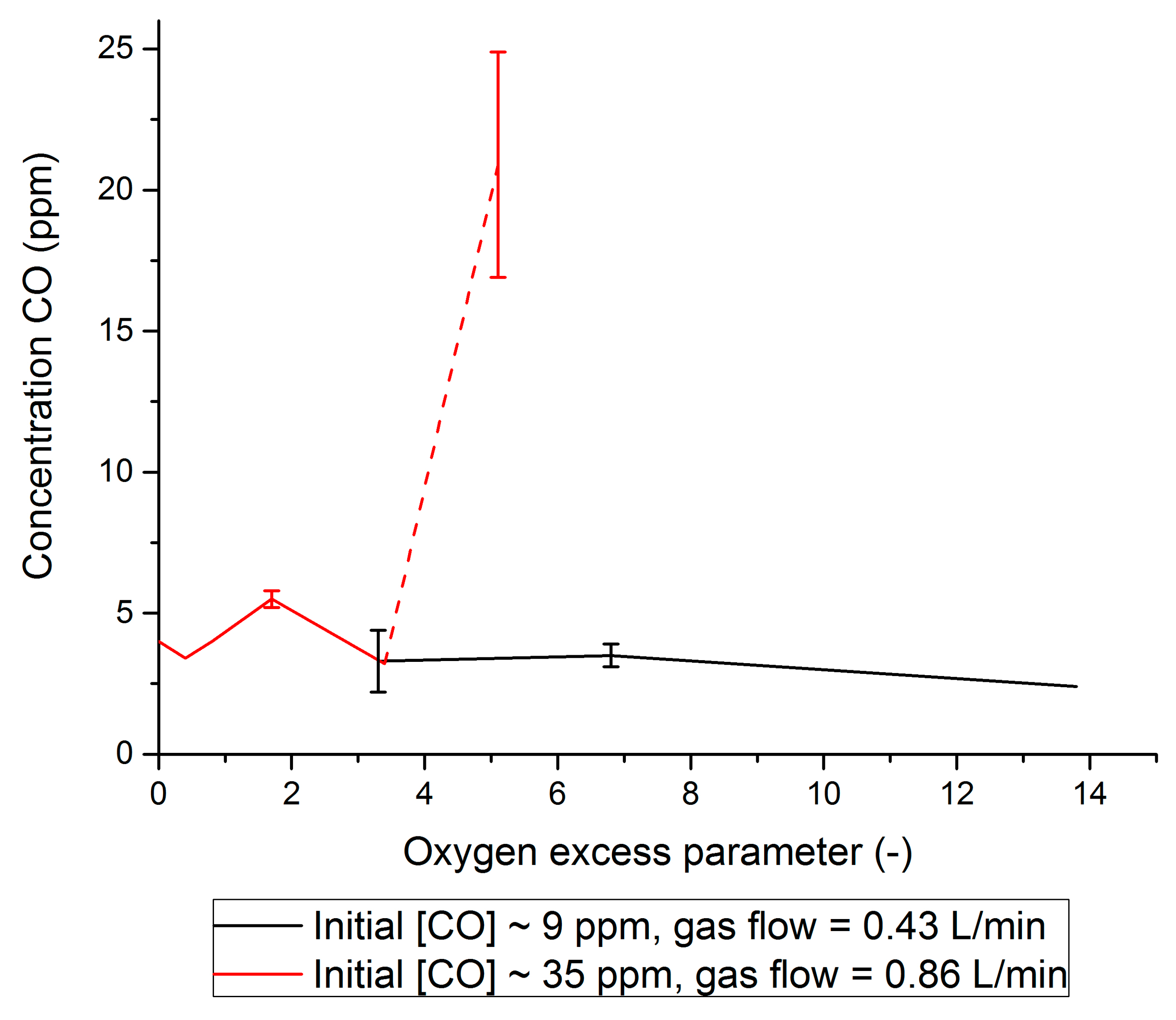
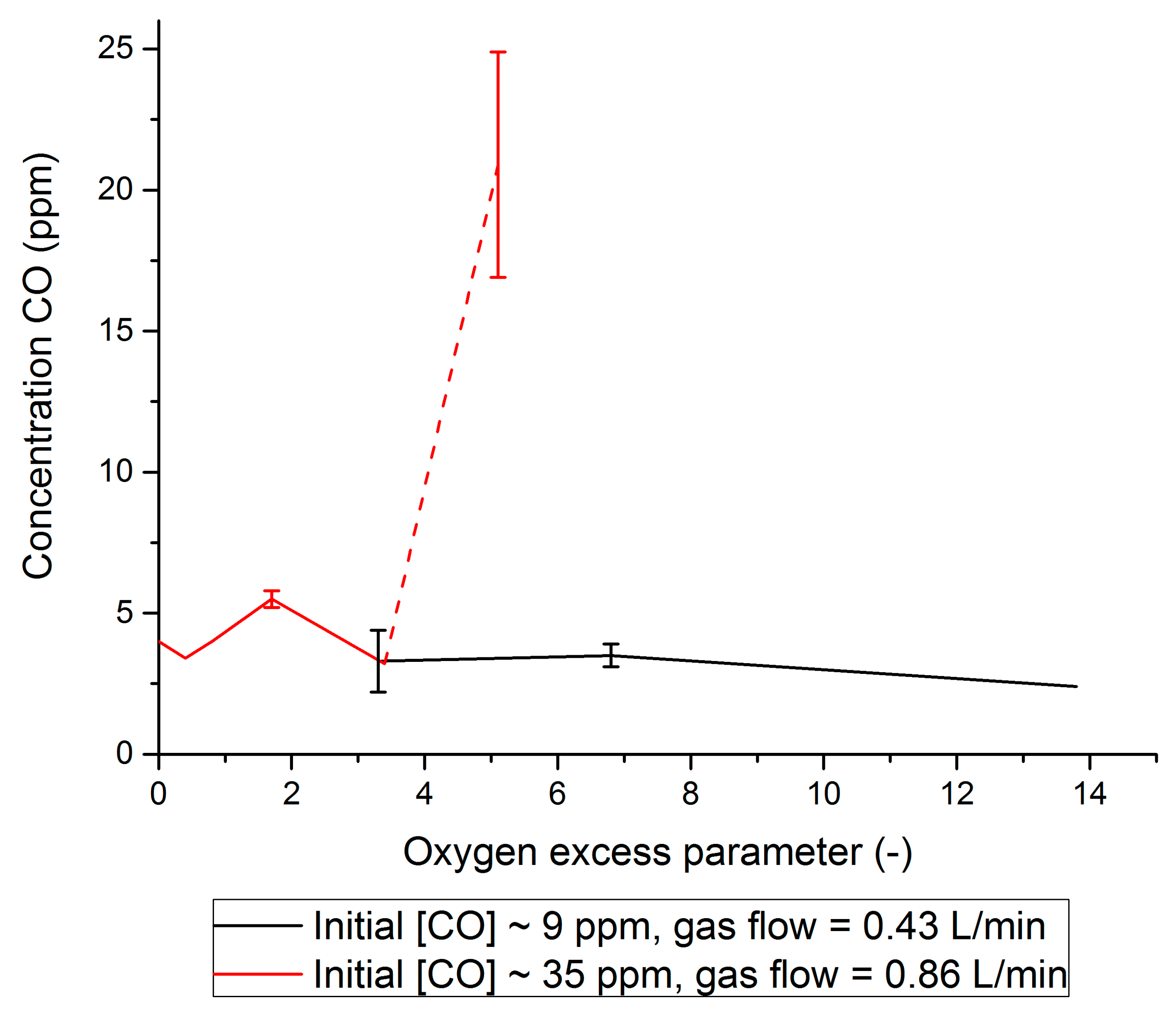
2.2. PROX
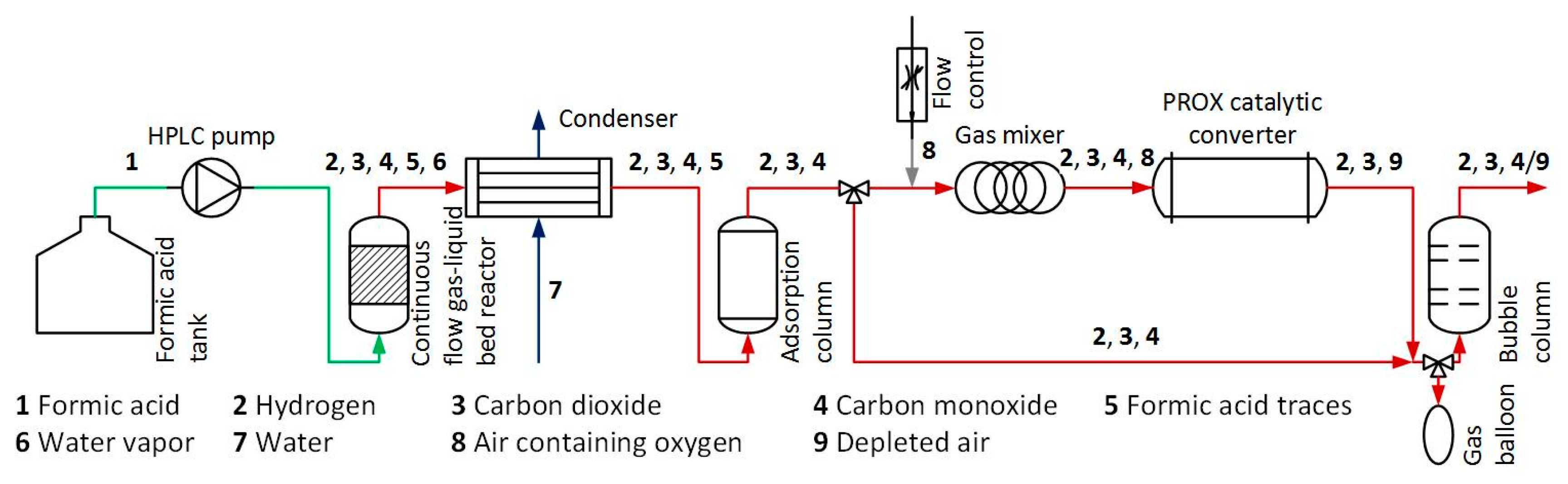
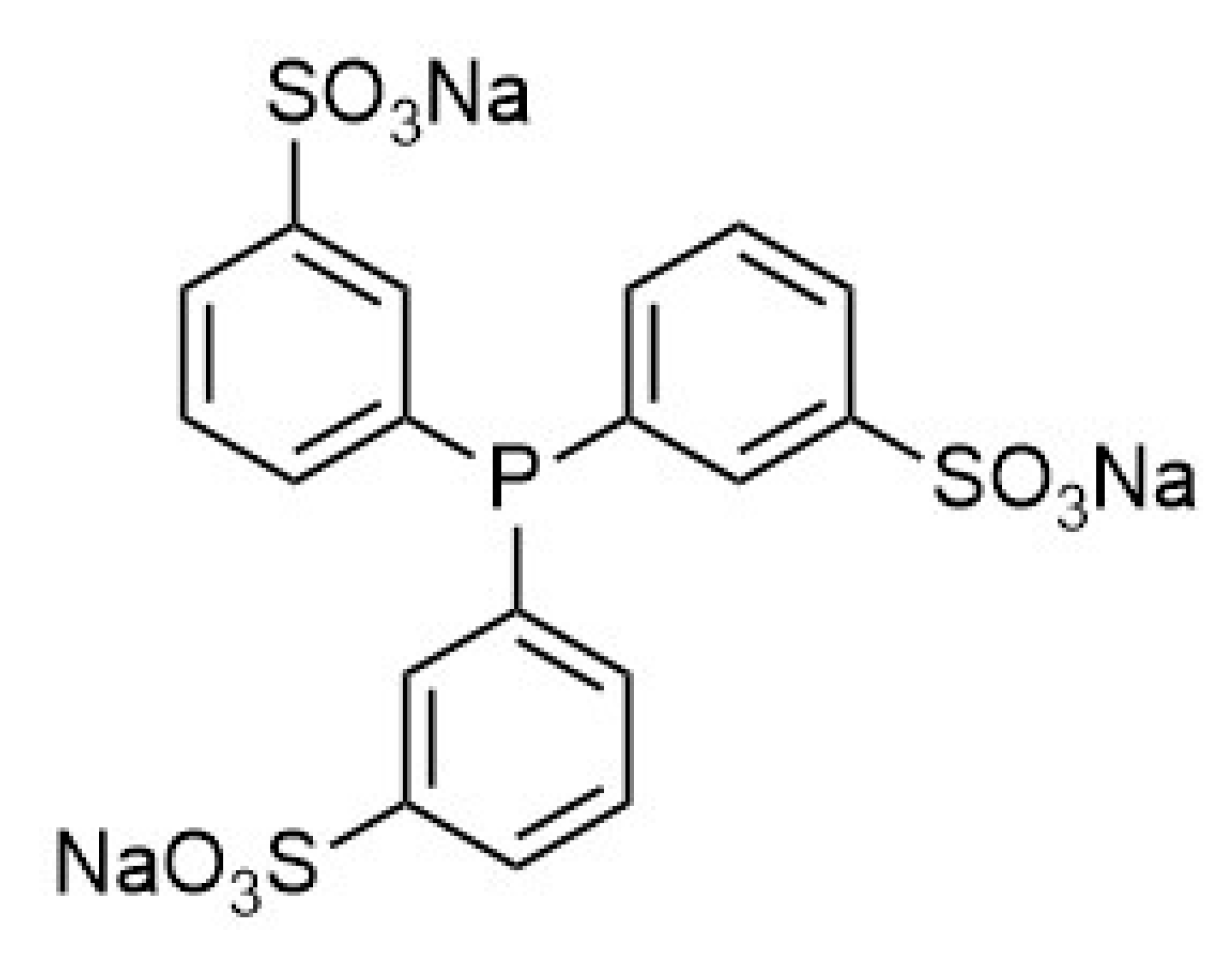
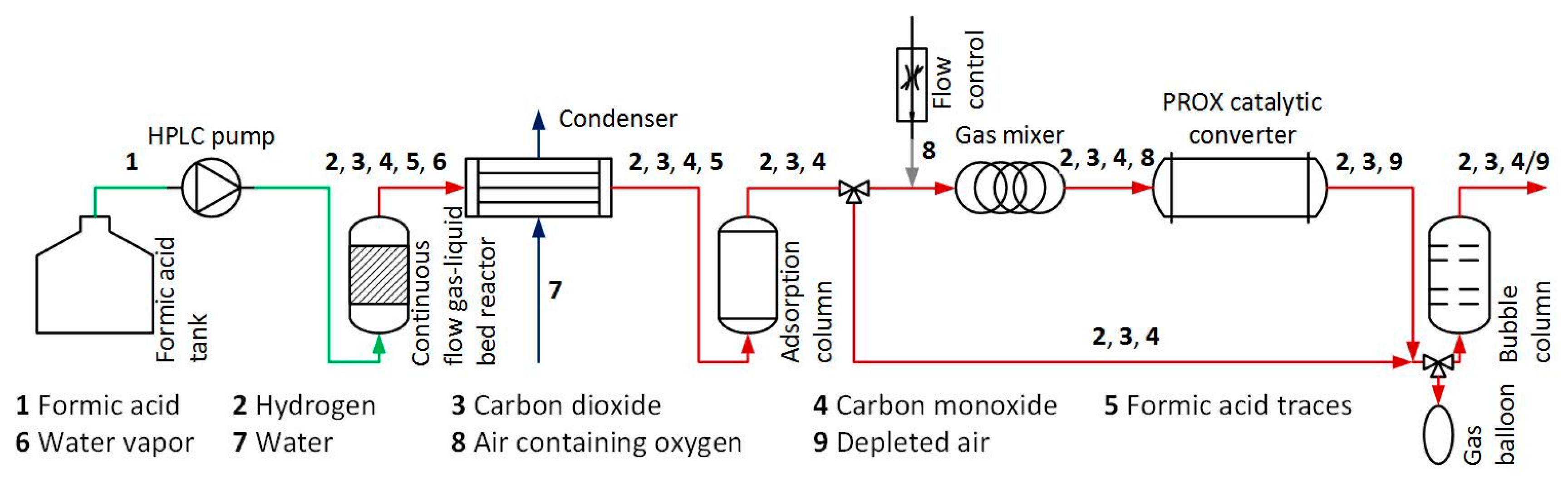
3. Materials and Methods
4. Conclusions
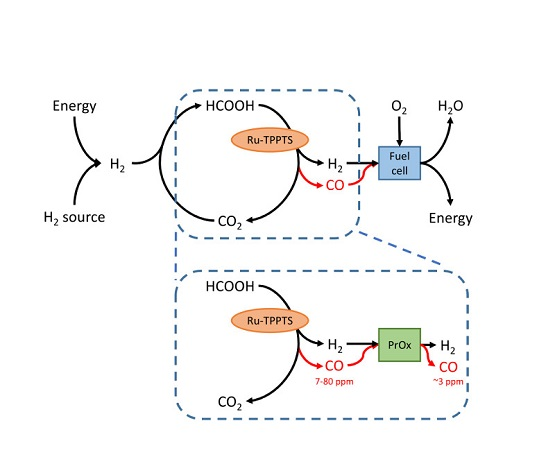
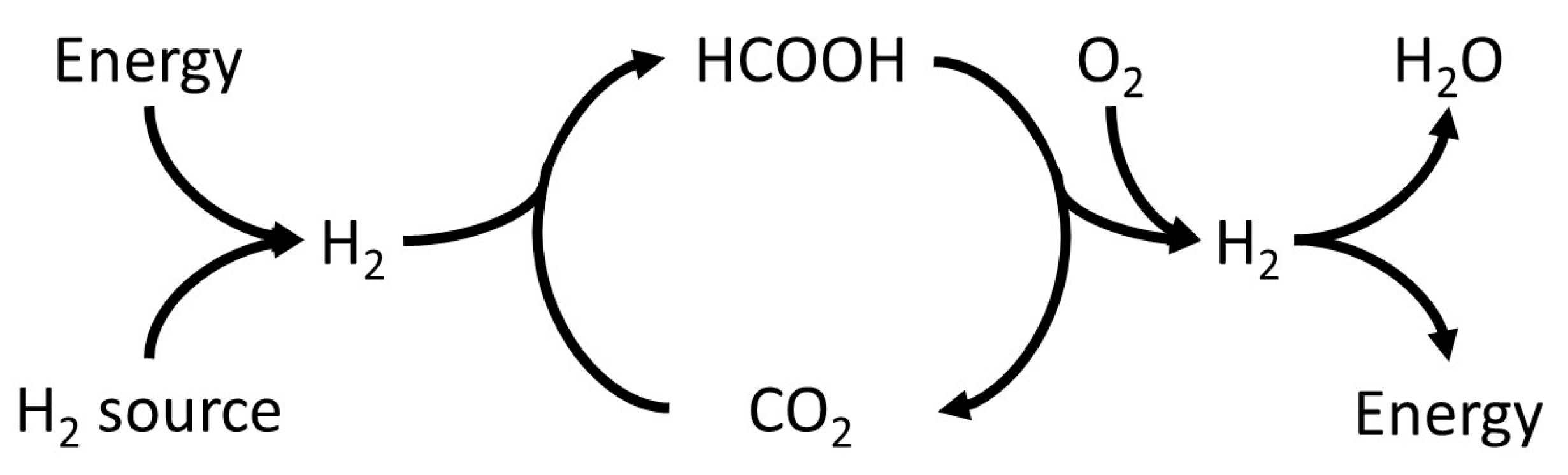
- It was found that unwanted CO concentrations in H2 + CO2 gas streams catalytically produced through FA dehydrogenation over a homogeneous Ru-mTPPTS catalyst in a continuous-flow gas–liquid bed reactor (60–80 °C) was too high (8–70 ppm) to be used directly in a fuel cell. The CO concentration was dependent on the FA feed flow rate and reaction design (mixing).
- It was shown that the Pt/Al2O3 catalyst can be easily implemented to clean H2 + CO2 gas streams from CO by PROX. It was shown that the O2 supply should be fine-tuned well to prevent both a water clogging of the PROX catalyst and a H2 loss due to its oxidation. Experimentally, CO concentration was confidently decreased by PROX at O2 concentrations close to stoichiometry (0.008–0.065 v % O2) to the level acceptable for fuel cell applications (<5 ppm). To our knowledge, this is the first example of successful CO removal from a FA reformate by PROX.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhu, Q.-L.; Xu, Q. Liquid Organic and Inorganic Chemical Hydrides for High-Capacity Hydrogen Storage. Energy Environ. Sci. 2015, 8, 478–512. [Google Scholar] [CrossRef]
- Dalebrook, A.F.; Gan, W.; Grasemann, M.; Moret, S.; Laurenczy, G. Hydrogen Storage: Beyond Conventional Methods. Chem. Commun. 2013, 49, 8735–8751. [Google Scholar] [CrossRef] [PubMed]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Moret, S.; Dyson, P.J.; Laurenczy, G. Direct Synthesis of Formic Acid from Carbon Dioxide by Hydrogenation in Acidic Media. Nat. Commun. 2014, 5, 4017. [Google Scholar] [CrossRef] [PubMed]
- Eppinger, J.; Huang, K.-W. Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2017, 2, 188–195. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Q. Metal-Nanoparticle-Catalyzed Hydrogen Generation from Formic Acid. Acc. Chem. Res. 2017, 50, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Fellay, C.; Dyson, P.J.; Laurenczy, G. A viable hydrogen-storage system based on selective formic acid decomposition with a ruthenium catalyst. Angew. Chem. Int. Ed. 2008, 47, 3966–3968. [Google Scholar] [CrossRef] [PubMed]
- Boddien, A.; Mellmann, D.; Gärtner, F.; Jackstell, R.; Junge, H.; Dyson, P.J.; Laurenczy, G.; Ludwig, R.; Beller, M. Efficient Dehydrogenation of Formic Acid Using an Iron Catalyst. Science 2011, 333, 1733–1736. [Google Scholar] [CrossRef] [PubMed]
- Himeda, Y.; Miyazawa, S.; Hirose, T. Interconversion between Formic Acid and H2/CO2 Using Rhodium and Ruthenium Catalysts for CO2 Fixation and H2 Storage. ChemSusChem 2011, 4, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Hull, J.F.; Himeda, Y.; Wang, W.-H.; Hashiguchi, B.; Periana, R.; Szalda, D.J.; Muckerman, J.T.; Fujita, E. Reversible Hydrogen Storage Using CO2 and a Proton-Switchable Iridium Catalyst in Aqueous Media under Mild Temperatures and Pressures. Nat. Chem. 2012, 4, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Dyson, P.J.; Laurenczy, G. Heterogeneous silica-supported ruthenium phosphine catalysts for selective formic acid decomposition. ChemCatChem 2013, 5, 3124–3130. [Google Scholar] [CrossRef]
- Sharaf, O.Z.; Orhan, M.F. An overview of fuel cell technology: Fundamentals and applications. Renew. Sustain. Energy Rev. 2014, 32, 810–853. [Google Scholar] [CrossRef]
- Baschuk, J.J.; Li, X. Modelling CO poisoning and O2 bleeding in a PEM fuel cell anode. Int. J. Energy Res. 2003, 27, 1095–1116. [Google Scholar] [CrossRef]
- Bröll, D.; Kaul, C.; Krämer, A.; Krammer, P.; Richter, T.; Jung, M.; Vogel, H.; Zehner, P. Chemistry in Supercritical Water. Angew. Chem. Int. Ed. Engl. 1999, 38, 2998–3014. [Google Scholar] [CrossRef]
- Wakai, C.; Yoshida, K.; Tsujino, Y.; Matubayasi, N.; Nakahara, M. Effect of Concentration, Acid, Temperature, and Metal on Competitive Reaction Pathways for Decarbonylation and Decarboxylation of Formic Acid in Hot Water. Chem. Lett. 2004, 33, 572–573. [Google Scholar] [CrossRef]
- Bjerre, A.B.; Sorensen, E. Thermal Decomposition of Dilute Aquaous Formic Acid Solutions. Ind. Eng. Chem. Res. 1992, 31, 1574–1577. [Google Scholar] [CrossRef]
- Akiya, N.; Savage, P.E. Role of water in formic acid decomposition. AIChE J. 1998, 44, 405–415. [Google Scholar] [CrossRef]
- Koper, M.T.M.; Shubina, T.E.; Van Santen, R.A. Periodic Density Functional Study of CO and OH Adsorption on Pt–Ru Alloy Surfaces: Implications for CO Tolerant Fuel Cell Catalysts. J. Phys. Chem. B 2002, 106, 686–692. [Google Scholar] [CrossRef]
- Yano, H.; Ono, C.; Shiroishi, H.; Okada, T. New CO tolerant electro-catalysts exceeding Pt–Ru for the anode of fuel cells. Chem. Commun. 2005, 1212–1214. [Google Scholar] [CrossRef] [PubMed]
- Sung, L.Y.; Hwang, B.J.; Hsueh, K.L.; Tsau, F.H. Effects of anode air bleeding on the performance of CO-poisoned proton-exchange membrane fuel cells. J. Power Sources 2010, 195, 1630–1639. [Google Scholar] [CrossRef]
- Sung, L.-Y.; Hwang, B.-J.; Hsueh, K.-L.; Su, W.-N.; Yang, C.-C. Comprehensive study of an air bleeding technique on the performance of a proton-exchange membrane fuel cell subjected to CO poisoning. J. Power Sources 2013, 242, 264–272. [Google Scholar] [CrossRef]
- Schmittinger, W.; Vahidi, A. A review of the main parameters influencing long-term performance and durability of PEM fuel cells. J. Power Sources 2008, 180, 1–14. [Google Scholar] [CrossRef]
- Mariño, F.; Descorme, C.; Duprez, D. Noble metal catalysts for the preferential oxidation of carbon monoxide in the presence of hydrogen (PROX). Appl. Catal. B Environ. 2004, 54, 59–66. [Google Scholar] [CrossRef]
- Bion, N.; Epron, F.; Moreno, M.; Mariño, F.; Duprez, D. Preferential oxidation of carbon monoxide in the presence of hydrogen (PROX) over noble metals and transition metal oxides: Advantages and drawbacks. Top. Catal. 2008, 51, 76–88. [Google Scholar] [CrossRef]
- Formanski, V.; Kalk, T.; Roes, J. Compact hydrogen production systems for solid polymer fuel cells. J. Power Sources 1998, 71, 199–207. [Google Scholar]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective methanation of CO over supported noble metal catalysts: Effects of the nature of the metallic phase on catalytic performance. Appl. Catal. A Gen. 2008, 344, 45–54. [Google Scholar] [CrossRef]
- Fellay, C.; Yan, N.; Dyson, P.J.; Laurenczy, G. Selective formic acid decomposition for high-pressure hydrogen generation: A mechanistic study. Chem. Eur. J. 2009, 15, 3752–3760. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FA Feed (mL/min) | Total Gas Flow (L/min) |
|---|---|
| 0.3 | 0.43 |
| 0.6 | 0.86 |
| 0.9 | 1.29 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henricks, V.; Yuranov, I.; Autissier, N.; Laurenczy, G. Dehydrogenation of Formic Acid over a Homogeneous Ru-TPPTS Catalyst: Unwanted CO Production and Its Successful Removal by PROX. Catalysts 2017, 7, 348. https://doi.org/10.3390/catal7110348
Henricks V, Yuranov I, Autissier N, Laurenczy G. Dehydrogenation of Formic Acid over a Homogeneous Ru-TPPTS Catalyst: Unwanted CO Production and Its Successful Removal by PROX. Catalysts. 2017; 7(11):348. https://doi.org/10.3390/catal7110348
Chicago/Turabian StyleHenricks, Vera, Igor Yuranov, Nordahl Autissier, and Gábor Laurenczy. 2017. "Dehydrogenation of Formic Acid over a Homogeneous Ru-TPPTS Catalyst: Unwanted CO Production and Its Successful Removal by PROX" Catalysts 7, no. 11: 348. https://doi.org/10.3390/catal7110348