Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts
by
 ,
,
Robert Wojcieszak
1,* ,
,
Camila P. Ferraz
1,2,
Jin Sha
1,
Sarah Houda
1,
Liane M. Rossi
2 and
Sébastien Paul
1
1
Université Lille, CNRS, Centrale Lille, ENSCL, Université Artois, UMR 8181—UCCS—Unité de Catalyse et Chimie du Solide, F-59000 Lille, France
2
Departamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, São Paulo 05508-000, Brazil
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(11), 352; https://doi.org/10.3390/catal7110352
Submission received: 19 September 2017
/
Revised: 10 November 2017
/
Accepted: 15 November 2017
/
Published: 21 November 2017
(This article belongs to the Special Issue Catalytic Sustainable Processes in Biorefineries)
Abstract
:The oxidation of bio-based molecules in general, and of carbohydrates and furanics in particular, is a highly attractive process. The catalytic conversion of renewable compounds is of high importance. Acids and other chemical intermediates issued from oxidation processes have many applications related, especially, to food and detergents, as well as to pharmaceutics, cosmetics, and the chemical industry. Until now, the oxidation of sugars, furfural, or 5-hydroxymethylfurfural has been mainly conducted through biochemical processes or with strong inorganic oxidants. The use of these processes very often presents many disadvantages, especially regarding products separation and selectivity control. Generally, the oxidation is performed in batch conditions using an appropriate catalyst and a basic aqueous solution (pH 7–9), while bubbling oxygen or air through the slurry. However, there is a renewed interest in working in base-free conditions to avoid the production of salts. Actually, this gives direct access to different acids or diacids without laborious product purification steps. This review focuses on processes applying gold-based catalysts, and on the catalytic properties of these systems in the base-free oxidation of important compounds: C5–C6 sugars, furfural, and 5-hydroxymethylfurfural. A better understanding of the chemical and physical properties of the catalysts and of the operating conditions applied in the oxidation reactions is essential. For this reason, in this review we put emphasis on these most impacting factors.
1. Introduction
The growing concern regarding the gradual depletion of fossil oil reservoirs and the awareness of climate change has stimulated recent research activities for the utilization of renewable biomass resources for the sustainable production of fuels and chemicals [1,2,3,4,5]. Low molecular weight sugars (the so called C5–C6 sugars) are among the most abundant natural compounds with broad applications. These important chemicals can be produced in bulk quantities with very high purity from biomass transformation, and they can also be then transformed into various platform molecules.
Recently, it has been reported that glucose, xylose, and other sugars can be obtained from plant dry matter biomass, which includes any renewable organic matter such as crops and trees, agricultural wastes, and other residues from different agro-industrial processes, the so-called lignocellulosic biomass. The use of the lignocellulosic waste streams has the advantage of not competing with the production of food, and, therefore, the production of high added-value products can be a promising option to reduce CO2 emission and to achieve a green and sustainable production of fuels and chemicals [1,2,3,4]. The lignocellulosic biomass, which is rich in carbohydrates, yields fermentable sugars that can be used for different chemical processes [6,7,8,9,10,11]. Different types of carbohydrates could be obtained depending on the type of the organic matter used. Glucose, for example, can be obtained from starch; polysaccharides with hexoses monomers can be extracted from cellulose. Hemicellulose, a hetero polysaccharide, exhibits a broader distribution of sugars, constituted by hexoses, pentoses, and sugar acids, that are branched and amorphous.
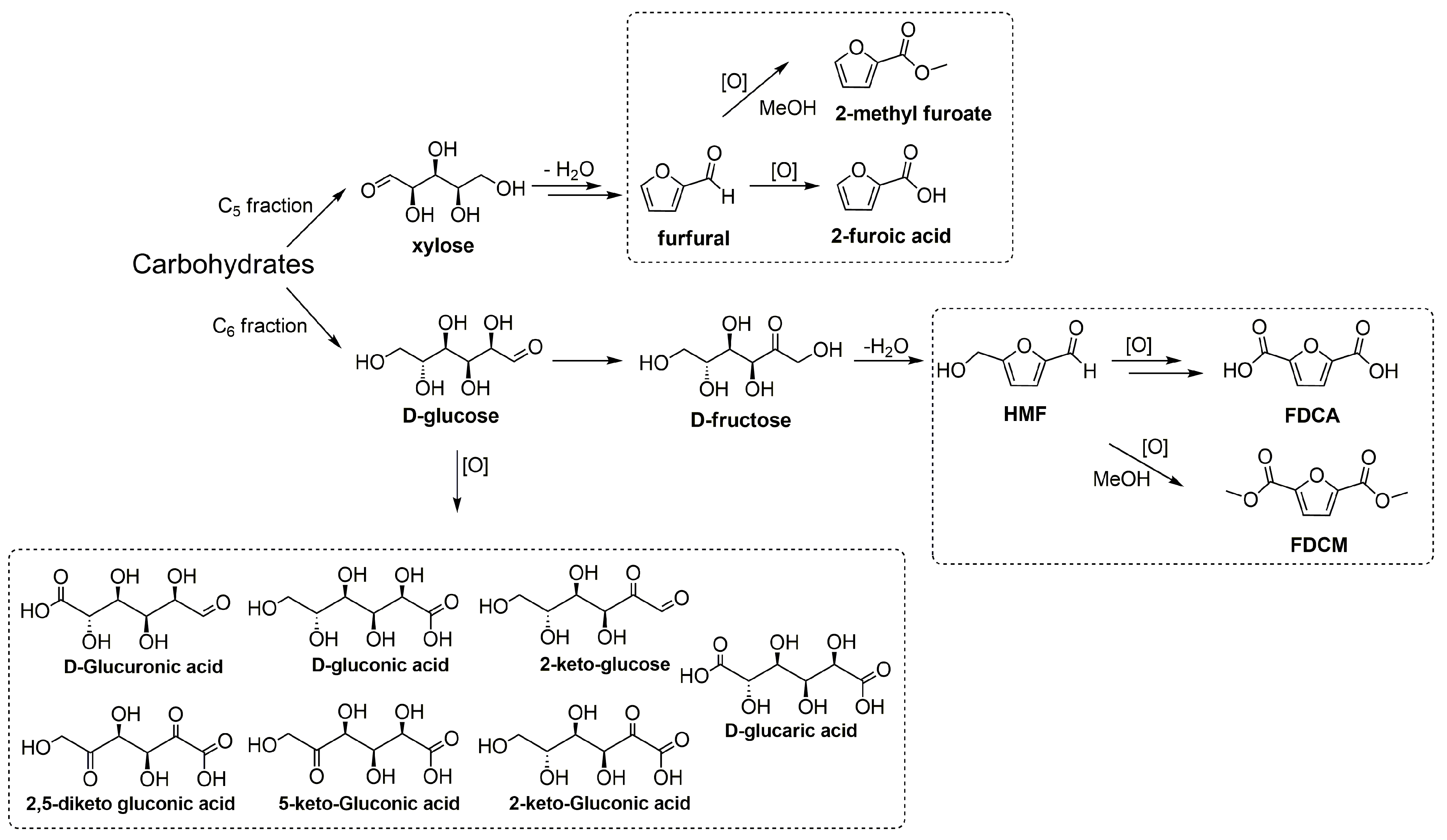
One of the most important transformations of sugars is the oxidation process. It yields different products for pharmaceutical and food industries through various biological and chemical processes. C5 and C6 sugars can also be transformed into furan derivatives that are important platform compounds, such as 5-hydroxymethylfurfural (HMF) and furfural [2,4,12] (Scheme 1). HMF is among the most important platform molecules in the development of biorefineries [1,2,3,4]. It is obtained from the dehydration of fructose, sucrose, and inulin, and, more recently, from glucose and cellulose through the isomerization of glucose to fructose [2,4,13]. The increasing interest in HMF arises from its great versatility, since it yields a wide variety of high value-added fuels and chemicals [2,4,13]. In particular, its oxidation products are of great interest for the polymer industry. Its derivatives can be used as six-carbon monomers that could replace other petrochemical-based monomers [2,14,15]. 2,5-Furandicarboxylic acid (FDCA) is a monomer for the synthesis of polyethylene furanoate (PEF) that is an alternative to the petroleum-based polyethylene terephthalate (PET). Besides being a green alternative, PEF is more easily degradable and does not bioaccumulate [14]. Furfural is another top 30 added-value substrates with a great potential to be developed in biorefineries [1,2,3,4,12]. It is produced from the hemicellulose portion of the biomass, mainly through the hydrolysis and dehydration of xylan, a polymer of xylose. Furoic acid (furan-2-carboxylic acid) has applications in the pharmaceutical, agrochemical, flavor, and fragrance industries. It is known as a preservative, acting as a bactericide and fungicide, and used to sterilize and pasteurize many foods. All these low molecular sugars and furan derivatives could be oxidized in different catalytic processes yielding very interesting chemical intermediates. Oxidation reactions can be performed using different methods, such as chemical and catalytic (homogeneous and heterogeneous) conversions [16,17,18,19,20]. Unfortunately, there are still a lot of drawbacks, mainly related to the difficulties in products’ separation and in the control of byproducts’ formation (selectivity issue).
In order to successfully carry out the process of carbohydrates oxidation, it is essential to select the best catalyst, to carefully control the operating conditions to keep a high reaction rate, and to prevent the catalyst from degradation or poisoning. Different solutions have been proposed to solve these problems, e.g., pH adjustment [2,21,22,23,24]. Indeed, most literature deals with the oxidation of sugars at a controlled, basic pH. At this pH, glucose is completely converted to gluconic acid. However, some studies have demonstrated that at a high pH, glucose is chemically unstable and its degradation occurs. Indeed, the full conversion of glucose was observed during the oxidation carried out with different bases [25]. Several authors claim high selectivity towards gluconic acid using gold catalysts under alkaline conditions [23,26,27]. However, different products could be also observed in the presence of a base and without a catalyst. Indeed, at 70 °C and after 4 h of reaction in the presence of K2CO3, a noncatalyzed (thermal) oxidative degradation of glucose was observed. Similar results already described in the literature [28,29,30] confirmed the formation of formic, acetic, and propionic acids, as well as of 24 hydrocarboxylic acids and 12 corresponding lactones. The aldehyde and alcohol functions of monosaccharides (such as glucose) can be oxidized giving high added-value compounds (see Scheme 1).
In addition, at alkaline pH, the isomerization of glucose to fructose occurs, and the two sugars then coexist in the media. Moreover, instead of leading to gluconate, the oxidation reaction can yield mixtures of low- and high-molecular-weight products such as carboxylates and aldehydes, thus resulting in a drop of selectivity. When a metallic catalyst is used for the oxidation of glucose at high pH, oxygen poisoning of the metal catalyst can also occur. Moreover, operating at high pH conditions leads to the necessity of neutralizing the solution to finally get the sugar acid, resulting in the coproduction of salts. Basic media are also a source of degradation of HMF and furfural under reaction conditions. Tests in the absence of a catalyst at high pH showed HMF conversion without any formation of detectable oxidation products. In addition, a color change from yellow to brown was noticed in the reaction mixture, as well as the formation of a black precipitate when HCl was added [31,32]. This precipitate was attributed to the formation of humic acids, although the formation of humins has also been reported. Humins are insoluble in water at all pHs, and humic acids are soluble only in a basic medium. A mixture of many different acids can form during the dehydration of sugars and from the condensation of sugars with furfural and HMF. As pointed out by some researchers, a basic medium also favors the formation of levulinic acid, formic acid, and some unknown byproducts. This behavior emphasizes how detrimental a high pH is for HMF stability in solution. According to the authors, the increase of NaOH concentration and temperature of the reaction accelerate the degradation process. In the presence of a catalyst, these degradation products formed in minor amounts or did not form [33]. Nevertheless, several papers report the need of a basic medium to perform the selective oxidation of HMF. According to them, the presence of a base allows higher reaction rates, higher feed concentrations (3%), better product solubility in the reaction media, and lower adsorption of the FDCA on the catalyst surface. However, the reaction performed under base-free conditions would simplify the downstream separation, decrease the number of added reagents, and produce less process waste. Moreover, it would minimize the occurrence of side reactions and eliminate the need for additional steps, such as the neutralization of FDCA salts and the removal of the resulting inorganic salts.
In summary, oxidation reactions of C5–C6 sugars and furan derivatives in basic media permit to obtain high rates; however, several problems related to the selectivity, the chemical stability of the reactants, and the stability or poisoning of the catalyst frequently occur. Consequently, it is highly important to develop base-free oxidation methods using heterogeneous catalysts and environmentally friendly conditions, which would allow the elimination of the neutralization step of the process. Recently, several new heterogeneous catalysts based on gold have been developed in this frame. These catalytic systems are believed to be able to maintain high activity and selectivity without the need of any base addition. They are discussed in the following sections of this review.
2. Base-Free Carbohydrate Oxidation
2.1. Supported Au-Based Catalysts
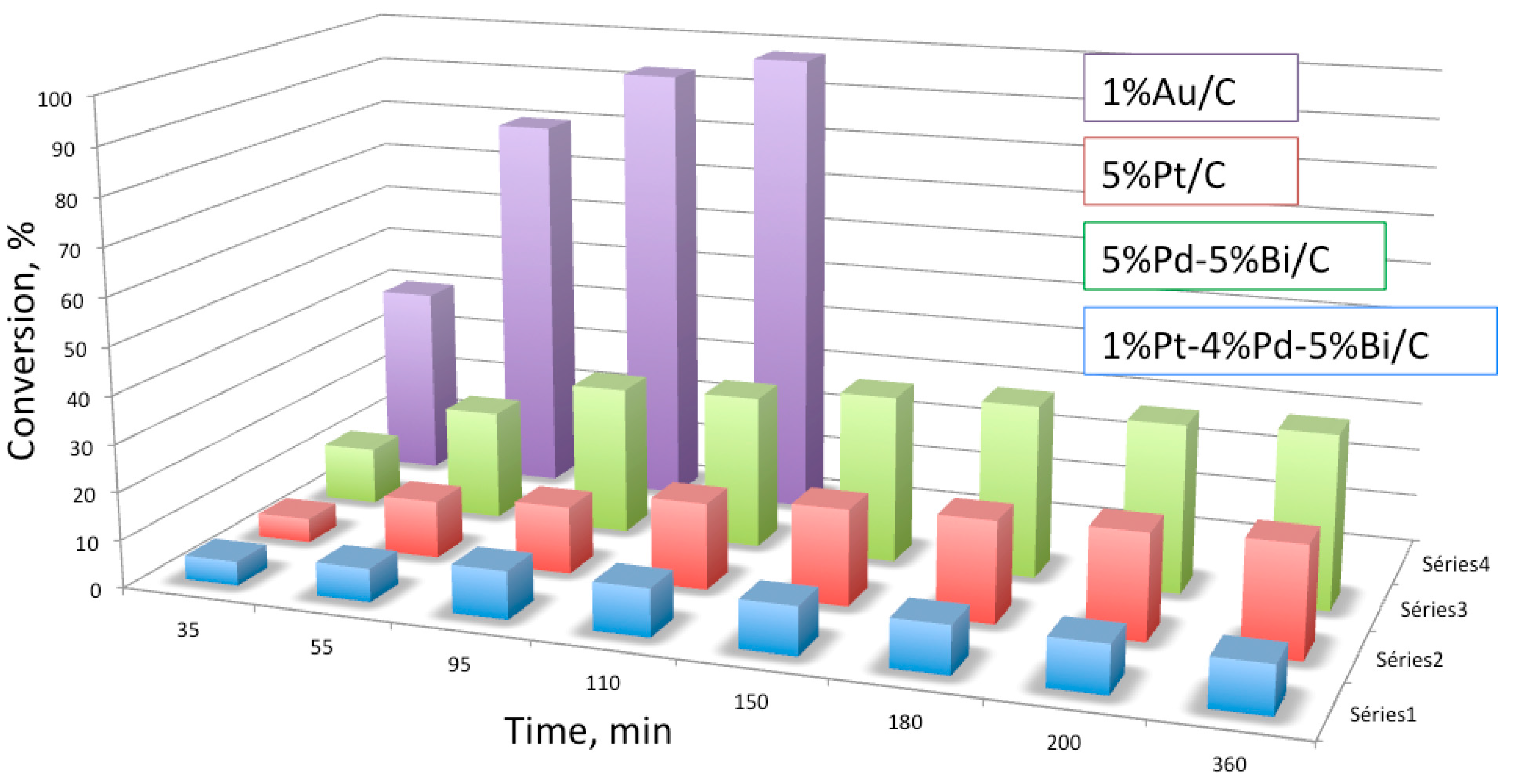
Gold-based catalysts have shown better performance than palladium or platinum-based ones for the oxidation of carbohydrates (Figure 1, [23]). One of the most interesting properties of Au is its ability to convert almost all types of aldoses (e.g., glucose, maltose, xylose) to the corresponding aldonic acids (e.g., gluconic acid). Comotti et al. [24,34] reported high activity and selectivity towards gluconic acid using unsupported Au nanoparticles (NPs). However, these systems showed relatively low stability [35], which was improved by immobilization on carbon [23,26].
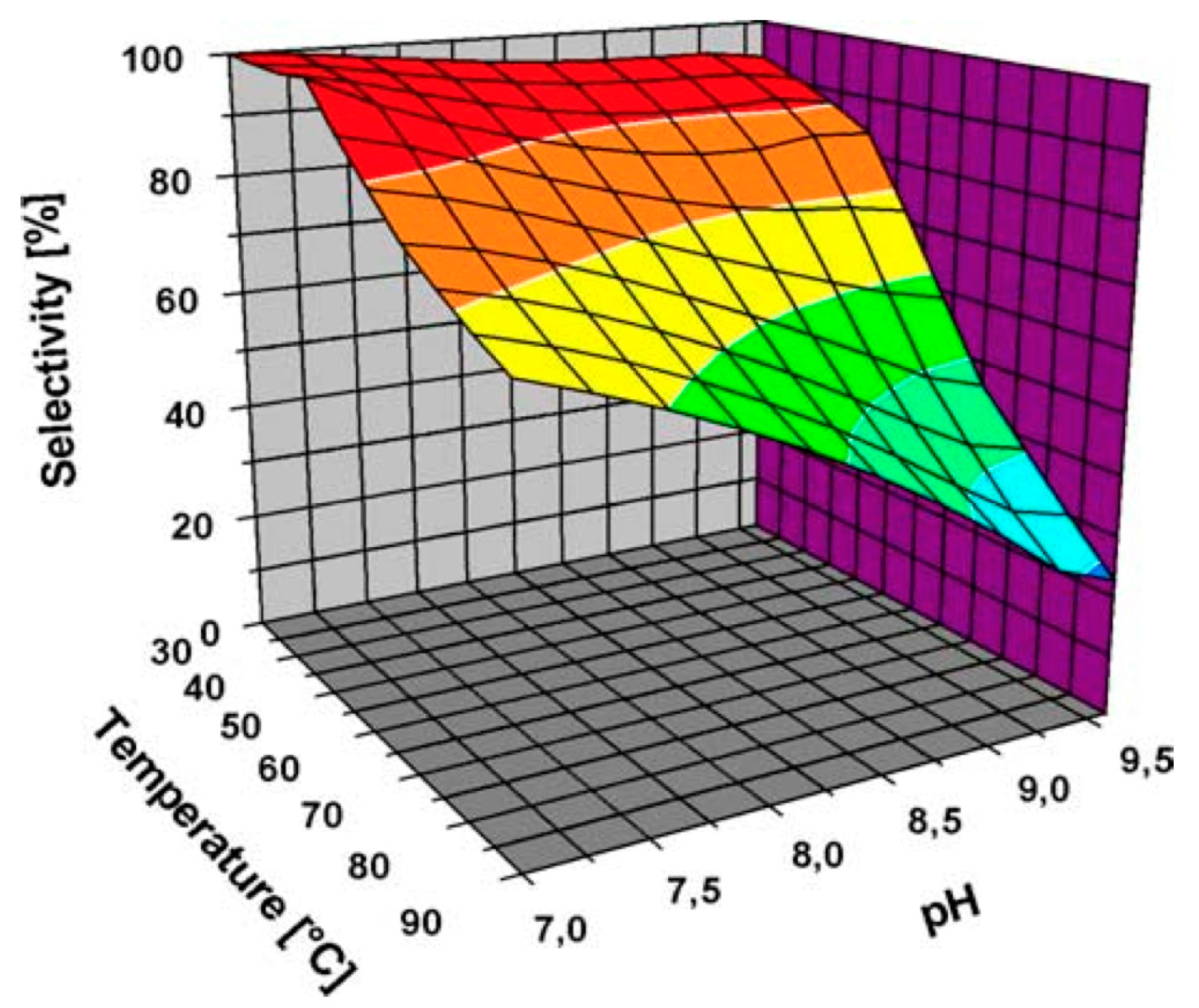
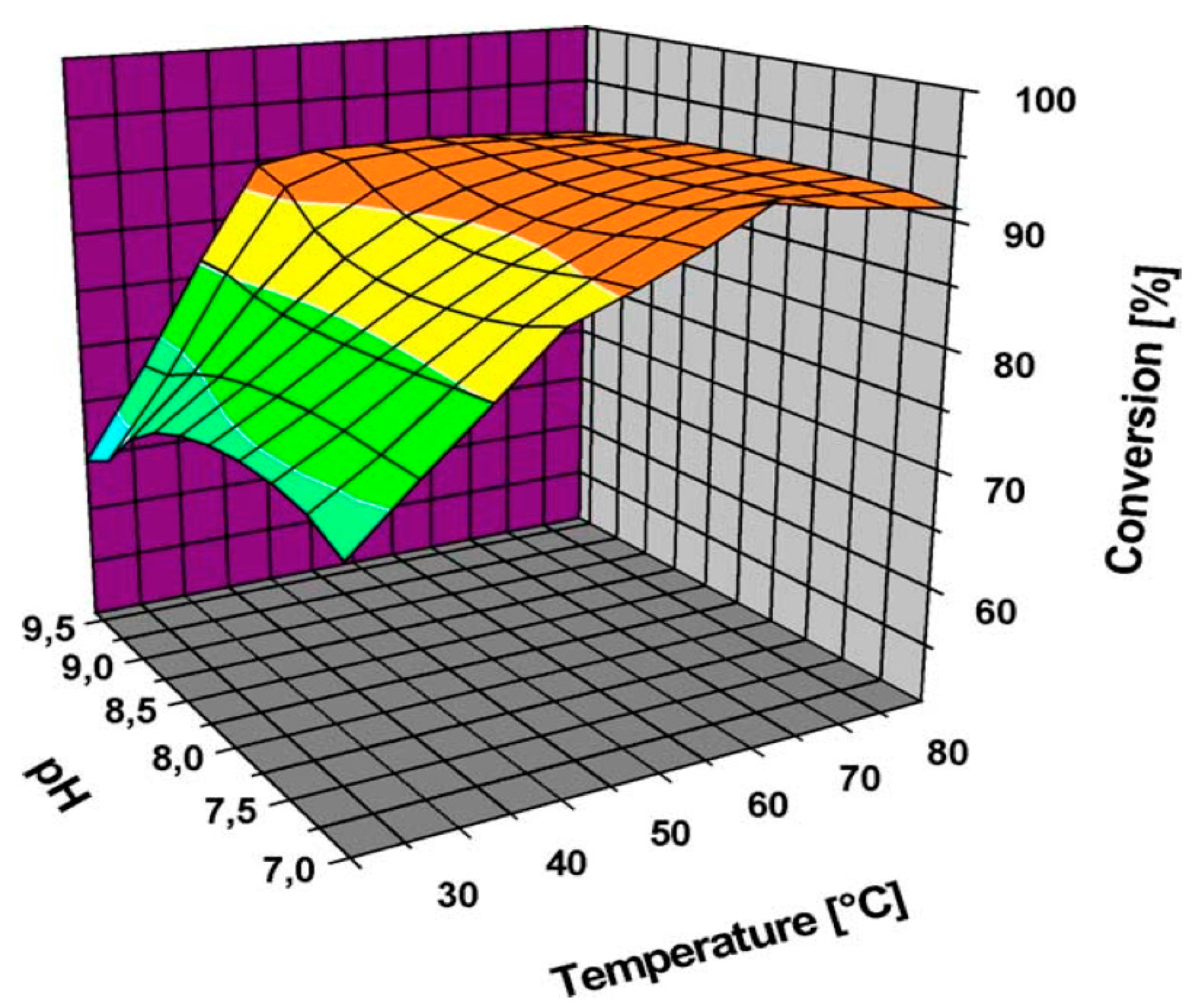
Önal et al. [36] also studied Au NPs supported on carbon (Figure 2 and Figure 3); however, the selectivity towards the desired acid was quite low at high conversions.
Taking into account this observation, the gold–support interaction was declared to play a crucial role in the formation of a stable catalytic system [37,38,39]. Various supports were studied using Au NPs of the same size. Different catalytic activities were observed indicating that a specific metal–support interaction between Au and the support was governing NPs’ activity [36]. On the contrary, Ishida et al. observed that the size of the Au NPs plays a more essential role than the nature of the support [39,40]. Excellent performance in terms of activity and recycling was observed for the Au/TiO2 system. Cao et al. [41] prepared 1 wt % Au/TiO2 catalysts using different methods. Once prepared, the catalysts were tested in base-free oxidation of glucose, giving good yields of gluconic acid (64%). The catalyst prepared by the sol immobilization method, using polyvinylalcohol (PVA) as a stabilizing ligand and NaBH4 as a reductant, showed the highest catalytic activity. The glucose reaction conditions were 0.3 MPa O2, 160 °C for 1 h. The effects of post-synthesis treatments, i.e., heating in air (at 250–550 °C for 3 h) or treatment with water, were studied. The effect of the quantity of the stabilizing ligand PVA added during the preparation was also studied. Post-synthesis treatments were also applied to remove the residual PVA. Lower activity of Au/TiO2 catalysts in the presence of PVA resulting from the formation of core–shell structures was reported by Villa et al. [42]. Indeed, as described in their work, the maximum catalytic activity was reached for the catalysts calcined at 250–350 °C. A higher temperature permits to remove more PVA from the catalyst surface. Moreover, the best PVA to Au ratio was 0.1, as observed by the authors. The group also reported results obtained with magnesium oxide modified with Au using the sol immobilization method. It showed an excellent selectivity towards gluconic acid [6]. Puyu Qi et al. [43] used an ordered mesoporous carbon (CMK-3)-supported Au catalyst in the aerobic oxidation of glucose with O2 under base-free conditions. Catalytic tests showed that the conversion remarkably increased, but the selectivity decreased when oxygen pressure and reaction temperature were increased. Glucose conversion to gluconic acid reached over 92% with 85% selectivity in 2 h, at 110 °C and 0.3 MPa oxygen pressure. Hydrogen peroxide was generated during the reaction, and the relationship between the hydrogen peroxide produced in situ and the formation of fructose as a byproduct was discussed. A low glucose/Au molar ratio minimized fructose formation. A 92% gluconic acid yield was obtained after 15 min of reaction when the molar ratio of glucose/Au was set to 100. The spent catalyst treated with an aqueous solution of NaOH at 90 °C could convert glucose up to 87%, which was close to the result obtained with the as-prepared catalyst, and excluded the effect of any alkaline residue. Wang et al. [44] studied Au catalysts supported on nano- or micro-sized metal oxides in the base-free oxidation of glucose to gluconic acid. The pH of the reaction solution was kept uncontrolled or, in certain cases, lowered by the addition of a mineral acid. The authors also studied the stability of these catalysts. They observed that the irreversible deactivation of the Au catalysts was due to the leaching and sintering of the gold nanoparticles. To improve the stability and counteract the hydrothermal sintering, they studied lower Au loadings. They observed that, during the oxidation reaction, the Au surface density significantly affected their agglomeration tendency. The best stability was observed for the sample composed of 0.02 wt % Au/μCeO2. The adsorption of different reactive species on the catalyst surface could also be responsible for the reversible deactivation of the catalyst. However, this adsorption could be removed by the calcination of the used catalyst (preferentially at 325 °C under static air) [44]. The base-free oxidation of glucose using microwave heating was studied by Rautiainen et al. [45]. High conversion and selectivity towards gluconic acid were obtained on Au-supported carbon catalysts. Moreover, the use of microwave heating permitted a remarkably short reaction time. Indeed, after only 10 min of reaction, a high yield of gluconic acid was obtained (76%) using H2O2 as an oxidant and Au/Al2O3 (0.09 mol % Au) as a catalyst. In addition, high turnover frequencies (TOFs = 10,000 h−1) due to the microwave-assisted oxidation were observed. In terms of stability, the catalyst activity remained constant after four runs, even though some increase in Au particle size was observed. Miedziak et al. [6] reported the selective synthesis of gluconic acid from glucose under mild conditions. They showed that this reaction could give good results even without any sacrificial base addition or pH control if a basic support was used. To conclude, the oxidation in a base-free medium is quite challenging, and there is still a need to enhance the efficiency of the catalysts used at low pH. Gold NPs-based catalysts permit to obtain high efficiencies in neutral or low pH conditions [44,45]. Table 1 summarizes the previous reports on base-free glucose oxidation on supported Au NPs catalysts.
2.2. Multimetallic Au-Based Catalysts
The combination of gold with one or more other metals in the oxidation of sugars under base-free conditions has been reported to enhance the activity and the selectivity towards the desired acid. The oxidation of glucose using Au–Pd catalyst at 60 °C gave better results than the monometallic gold (Table 1, entry 11). The advantage of the addition of a second metal was clearly shown when working in an acidic medium. Pt showed a better effect if compared to Pd, which means that Pt had a boosting effect when only gluconic acid is produced [47]. Furthermore, a high catalytic activity was also attributed to the presence of an Ag–Au core–shell structure, as charges could be easily transferred between the core and the shell leading to oxygen activation and to the attack of the desired functional group. The catalytic activity changed according to the composition of the system, and the best catalytic activity was obtained for 20% Ag and 80% Au. In addition to bimetallic nanoparticles, the use of trimetallic systems was also reported. One of the examples is the Au–Pt–Ag system prepared by reduction with NaBH4. Studies confirmed that the best catalytic activity and the highest TOF were observed with a mass ratio of 70/20/10 [48]. Recently, Hutchings’s group [28] studied several Au bimetallic catalysts for the oxidation of glucose and cellobiose. They found that the preparation method plays a crucial role in the catalytic activity of supported Au catalysts especially in the case of base-free oxidation processes. As can be seen from Table 2, the calcined catalysts exhibited higher glucose conversion than the dried ones. The authors concluded that the interaction between the substrate and the polyvinylpyrrolidone (PVP) used for the synthesis influences the catalytic activity.
The results obtained for bi- and trimetallic systems confirmed that the presence of multiple metals has a positive effect on the catalyst in comparison with a monometallic system. A simple change in the metal to metal ratio changes the catalytic results, which offers the advantage of fine-tuning the catalytic system. Moreover, it is possible to obtain a sufficient contact between the active sites and the reactants, even when using a protective agent (such as polymers) that can provide high stability [40].
2.3. Mechanism of the Base-Free Oxidation of Carbohydrates
The oxidation of an alcohol to an aldehyde over metal catalysts occurs in three main steps, as reported by Davis et al. [3]. The first step is the alcohol adsorption on the metal surface, which produces an adsorbed metal alkoxide and a metal hydride (or a water molecule, if metal hydroxide species are involved). After this adsorption, a carbonyl species and a metal hydride are produced via the β-hydride elimination. The final step is the oxidation of the metal hydride by O2 to regenerate the metal surface (or metal hydroxide species). As reported, this last step may also lead to the production of water (on metals that dissociatively adsorb oxygen) or hydrogen peroxide (on metals that do not dissociatively adsorb oxygen, such as gold). Both molecules would desorb to regenerate the active metal surface. In this way, a carboxylic acid is produced by the subsequent oxidation of the aldehyde through a geminal diol intermediate. Indeed, aldehydes may undergo reversible hydration to geminal diols. This hydrated diol can then also adsorb to a metal surface and undergo a β-hydride elimination to form a carboxylic acid. It was observed that, similar to an alkaline solution, the presence of surface-adsorbed hydroxides facilitates both the formation of an adsorbed alkoxide (or geminal diol) and the elimination step of a β-hydride towards the production of an aldehyde and a carboxylic acid. A similar mechanism corresponding to a modified oxidative dehydrogenation was also suggested as the most probable for glucose oxidation to gluconic acid using gold catalysts [50,51]. However, a deeper understanding was reported by Rossi et al. [52], who developed one of the best molecular models for carbohydrates oxidation in the presence of gold catalysts. Taking into account that this reaction was considered to be first-order (for oxygen), the authors suggested a fast adsorption of glucose on gold. In this way, the rate-determining step would be glucose oxidation by the oxygen dissolved in the liquid (aqueous) phase. Hydrogen peroxide could be detected, but it decomposed quickly under the reaction conditions. In a subsequent investigation, Rossi et al. [51,52] showed that these oxidations do not proceed by a radical pathway, but deliver gluconate and hydrogen peroxide by a two-electron mechanism. The hydrogen peroxide decomposes because of the basic medium before it reaches a concentration at which it would efficiently compete with oxygen as the oxidant.
As could be noted, various transformations are involved in the process. In the case of gold catalysts, these steps were shown to be highly dependent on the presence of the hydroxyl species. As, with no doubt, the formation of Au–alkoxide is crucial for further oxidation of glucose [53], in a base-free oxidation the presence of local basicity from the support, from the second metal, or from both is decisive [25]. It could be expected that the oxidation of glucose may proceed through the formation of gluconic acid (oxidation of the aldehyde at the carbon atom C1). The following step of this reaction is the oxidation of the primary hydroxyl to yield glucaric acid. Although the oxidation of glucose directly to dicarboxylic acids was already demonstrated to occur on a Pt/C catalyst using oxygen as an oxidant [53,54,55], the oxidation of free gluconic acid into glucaric acid was also demonstrated on Au/C catalysts [28]. It has been reported that the rate of the reaction between the chemisorbed oxygen and the adsorbed gluconic acid is much lower than that of the reaction between the chemisorbed oxygen and the adsorbed glucose [55]. However, it was also demonstrated that, under the same reaction conditions, gluconic acid, formed from glucose oxidation, is oxidized much faster than if it is used as a starting molecule. This could be explained by the low pH of the initial solution in the case of gluconic acid oxidation and by its adsorption on the catalysts surface, which could deactivate the active sites [25].
3. Base-Free 5-Hydroxymethylfurfural Oxidation
The aerobic oxidation of HMF to FDCA requires several tandem oxidation steps, thus highly active catalysts are essential to reach the final dicarboxylic acid product (Scheme 2).
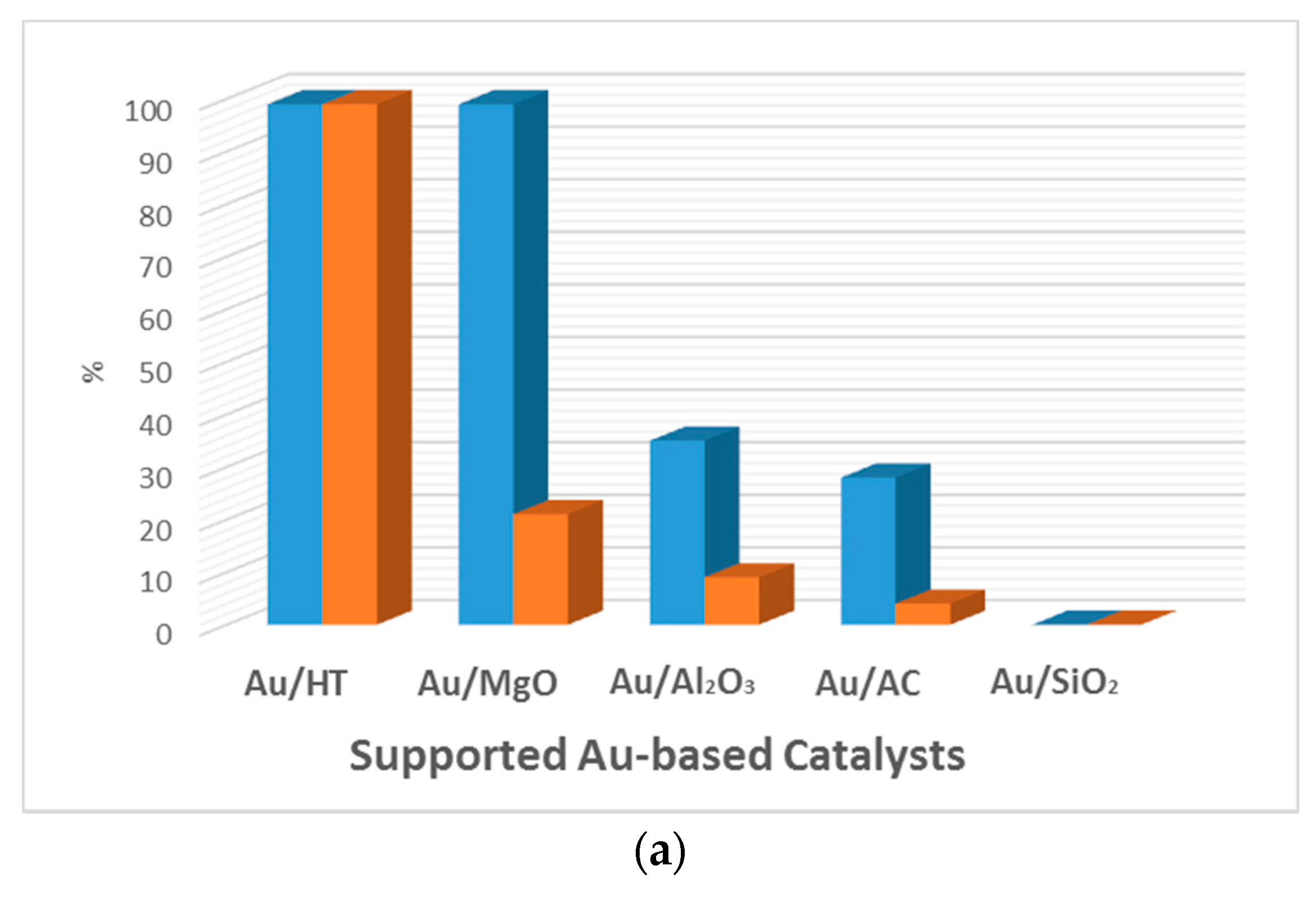
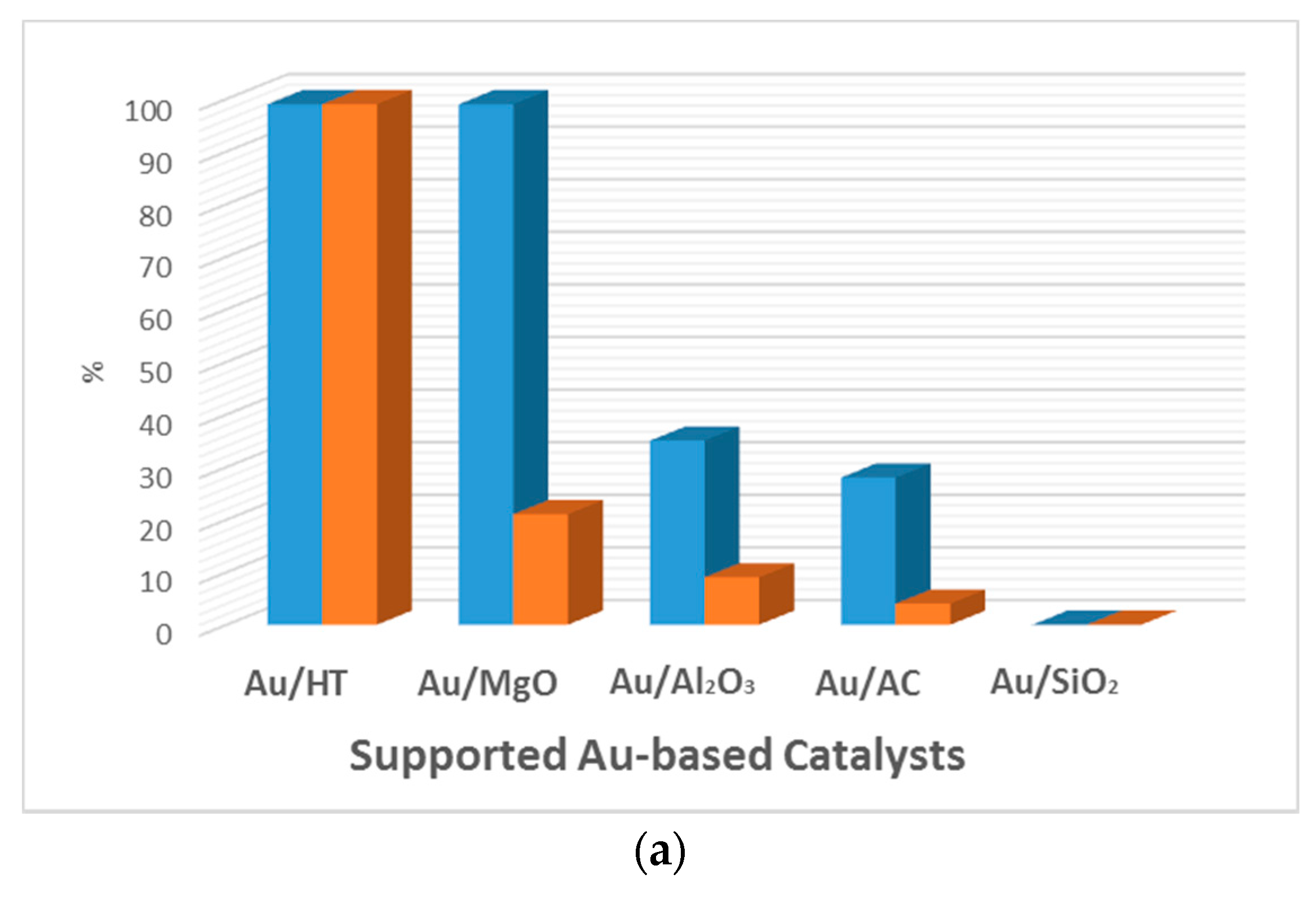
Au-based catalysts have been reported to perform this reaction in water and in the presence of an excess of base with high yields of FDCA [56,57,58,59,60,61]. Au-containing bimetallic catalysts were developed in order to improve the catalytic performance, but required the use of a homogenous base [33,58,62,63,64,65,66,67]. The basic medium provides higher reaction rates and, in the majority of cases, is essential to get a high catalytic activity. However, the basic medium also favors HMF degradation and requires a final neutralization step, which is not desirable from the environmental and economical points of view [3]. The development of active catalysts for the base-free aerobic oxidation of HMF is highly desired and challenging. Only few papers report Au-based catalysts for the oxidation of HMF to FDCA in the absence of a base. Erbitani et al. were the first to report a hydrotalcite-supported Au catalyst (Au/HT) for the aerobic base-free oxidation of HMF in water [68]. High activity was observed under mild conditions, in which FDCA was produced with 99% yield after 7 h under O2 flow (50 mL min−1) at 95 °C. The Au/HT was found to be more efficient than any of the other Au-supported catalysts tested in this work (Figure 4a). If catalysts based on neutral or acidic supports, such as Au/Al2O3 and Au/SiO2, displayed low or no yield of FDCA, those based on basic supports, such as Au/HT and Au/MgO, proved to be very active, achieving full HMF conversion. However, the selectivity for FDCA when using the MgO-based catalyst was lower (only 21%), although MgO is a strong basic support. The authors suggest that the difference might be due to the Au particle size. The Au/HT catalyst was recycled three times without a significant loss in activity (>99%, 92%, and 90%, respectively). However, further studies from Zope et al. with Au/TiO2 and hydrotalcite as a solid base for oxidation of HMF showed that leaching of Mg2+ from HT occurred [69]. According to the authors, leaching resulted from an acid–base reaction occurring once the formed acid lowered the pH of the reaction medium and consumed the basic solid support. The leaching test was not performed for the Au/HT catalyst previously described.
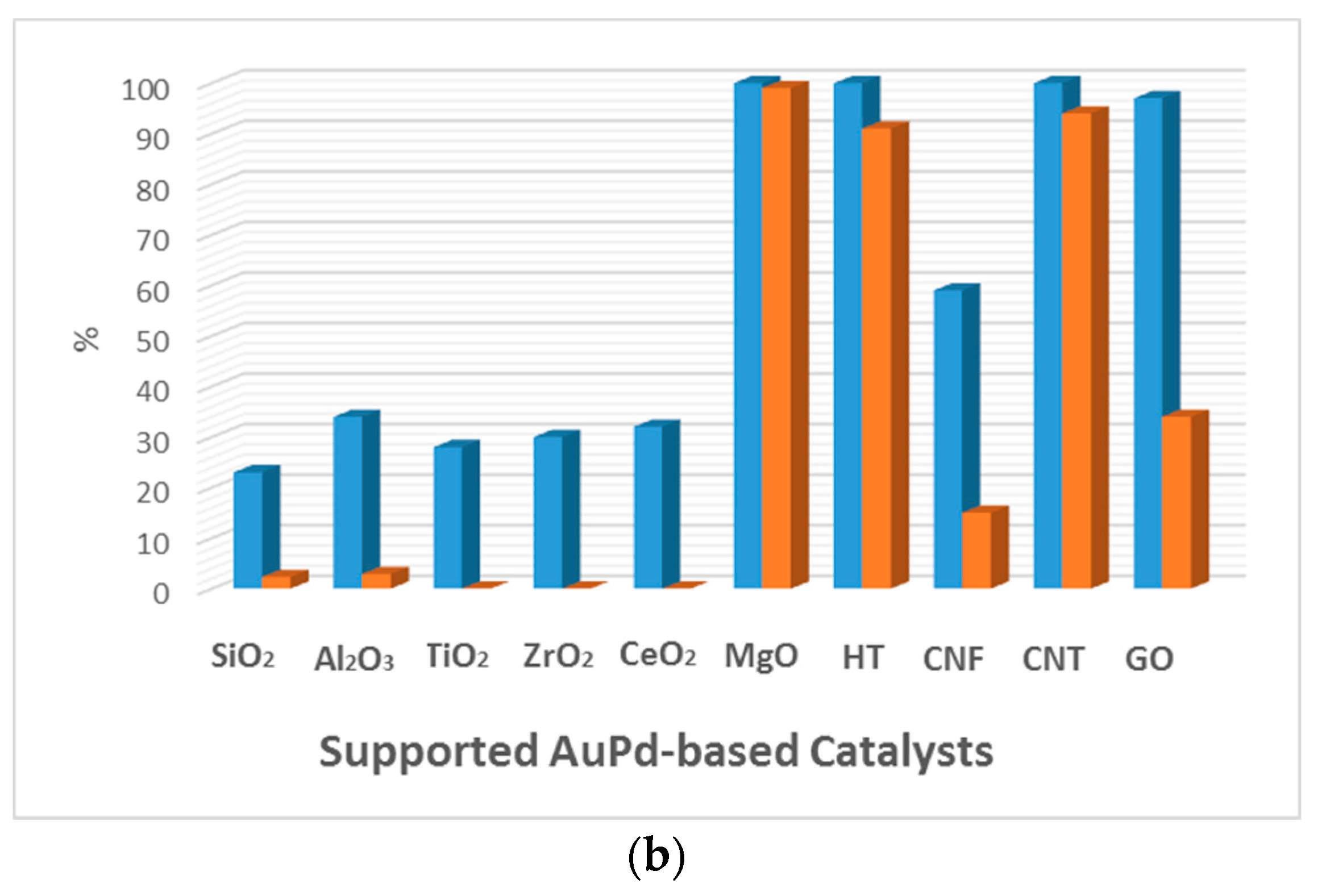
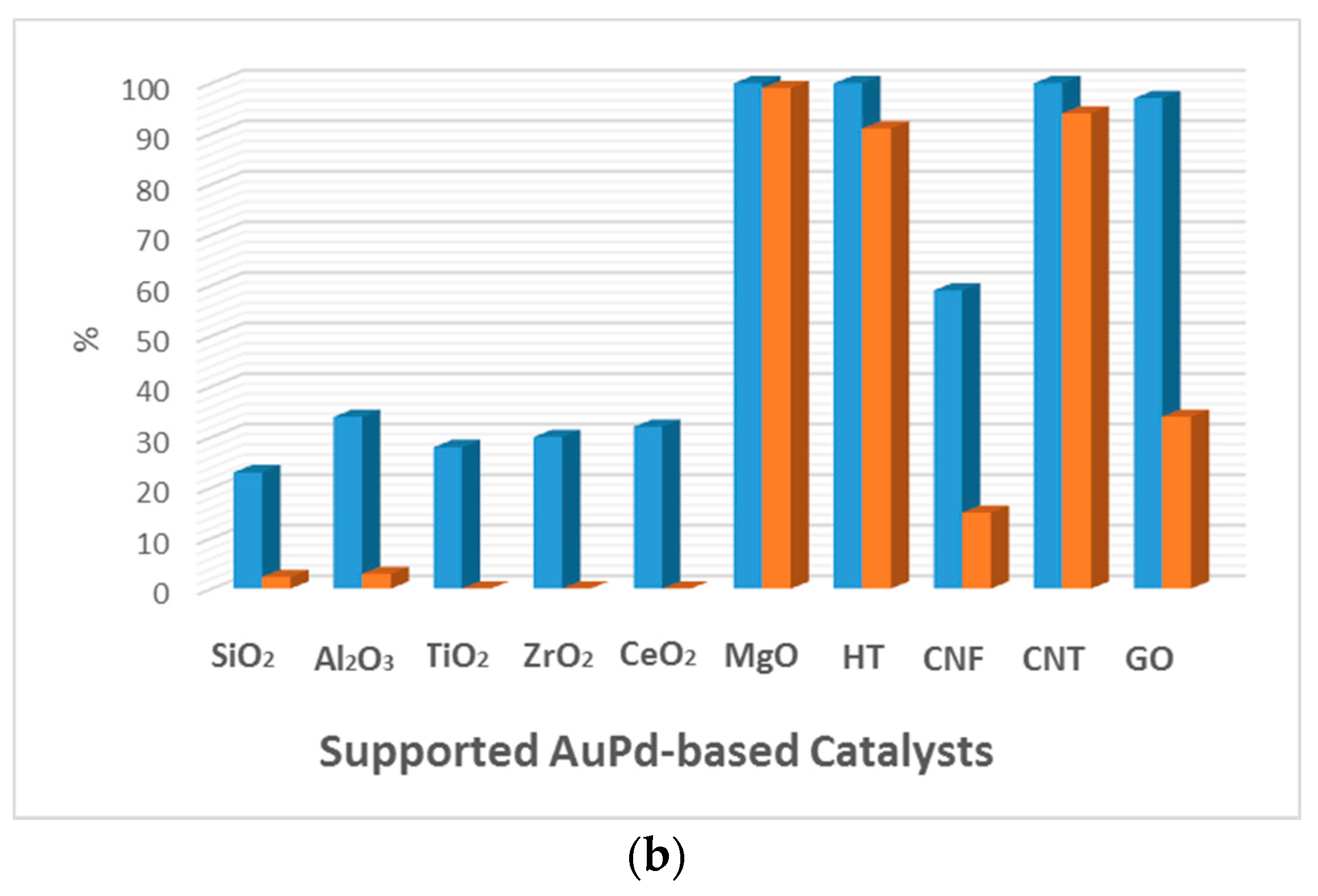
Wang et al. [70] reported an Au-containing bimetallic catalyst for base-free oxidation of HMF consisting of a carbon nanotube-supported Au–Pd catalyst (Au–Pd/CNT) that reached 100% conversion within 12 h, with 94% selectivity for FDCA. The authors also compared Au–Pd NPs supported on different supports (Figure 4b). The catalytic activity followed a similar trend to that reported by Erbitani and coworkers, with catalysts using basic supports being more active than those using neutral or acidic supports. Interestingly, Au-based catalysts with carbon-based supports like CNT and graphene oxide (GO) also presented high catalytic activity, with Au/CNT being the most active. The catalysts with basic supports were very active but deactivate during recycling tests. This was not the case for the Au/CNT catalyst that remained active for up to five cycles. According to the authors, the organic groups on the support surface that formed during the oxidative treatment of the support can affect, among other properties, the adsorption of the substrate, the intermediates, and the final products. The characterization of the CNT revealed the presence of carbonyl–quinone and phenol on the surface, which favored the adsorption of HMF but not of FDCA. This was consistent with the test of HMF conversion, which could be easily carried out without poisoning the catalytic surface. The authors also emphasized the synergistic effect of the Au–Pd catalyst, which presented much higher activity than the monometallic Au/CNT or Pd/CNT catalysts.
The direct use of FDCA in the industry is difficult because of its low solubility in most of the commonly used solvents. The furan-2,5-dimethylcarboxylate (FDMC) ester is actually more suitable for the subsequent polymerization reaction thanks to its better solubility [56]. For this reason, the development of catalytic systems capable of producing FDMC directly from HMF (Scheme 3) received attention recently. Au-based catalysts have been successfully applied for the oxidative esterification of alcohols [71,72,73,74] and were also explored for the transformation of HMF.
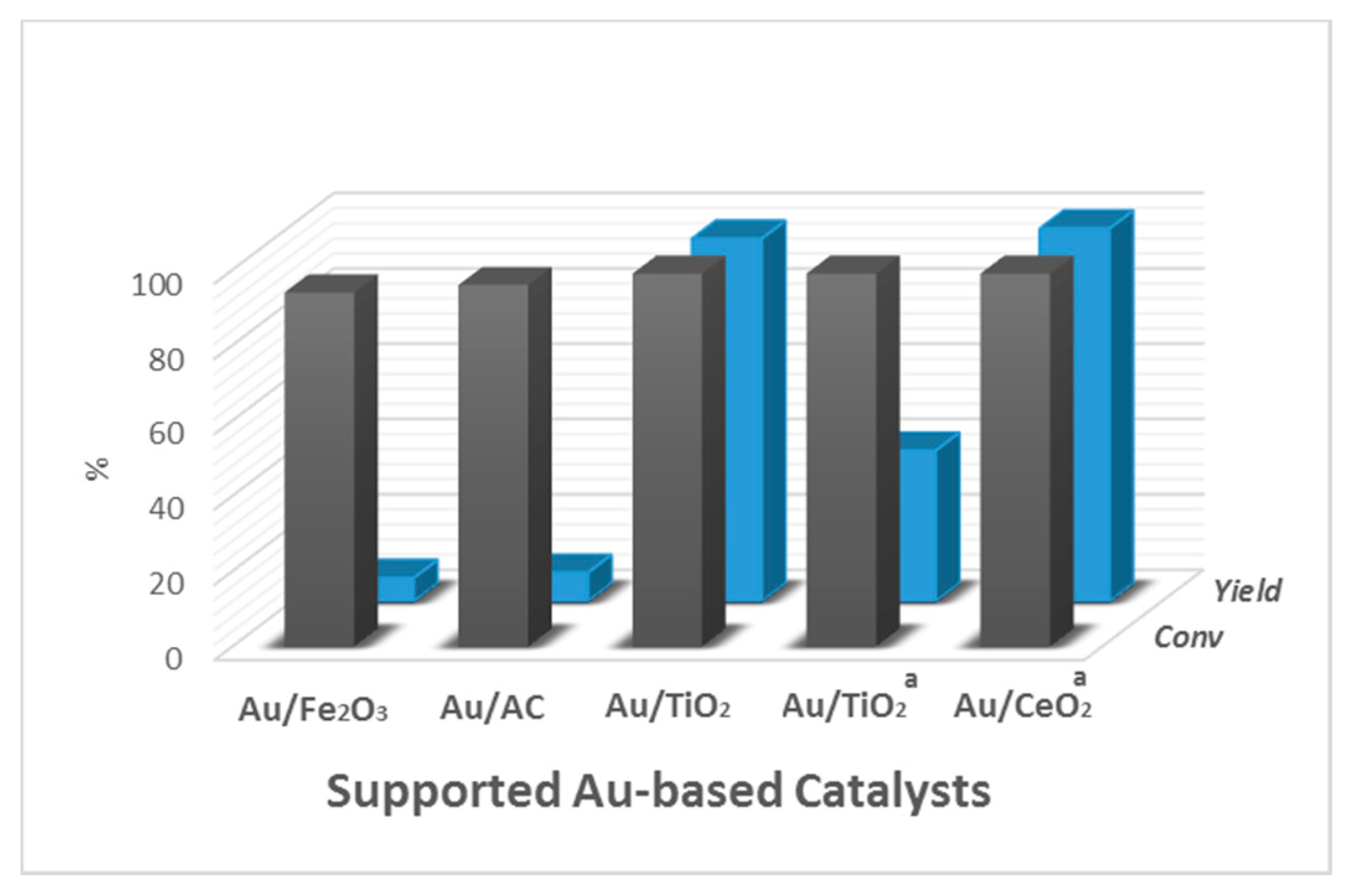
The first example of esterification of HMF to produce dimethyl-2-furoate required the use of a base [75]. Subsequently, Corma et al. reported an Au/CeO2 catalyst able to perform the reaction under base-free conditions [56]. This catalyst displayed full conversion of HMF in a 2 h reaction, but formed FDMC with a yield >99% within 5 h of reaction. The catalyst was compared with other Au-based catalysts using different supports (TiO2, activated carbon (AC), and Fe2O3), as shown in Figure 5. Au/AC and Au/Fe2O3 presented negligible yields of FDMC after 24 h, even if the latter presented the highest TOF for the conversion of HMF. Au/TiO2 was able to provide 96% yield of FDMC after 24 h, but only 40% after 5 h, meaning that the reaction rate was lower compared to Au/CeO2. Experiments with pure CeO2 showed up to 61% conversion after 24 h with the formation of large amounts of acetal, but no ester. In addition, the test performed in the absence of an oxidant (under N2 flow) resulted in 58% conversion with similar formation of acetal and 3.8% yield of the oxidation product (monoester alcohol). These results indicate some intriguing characteristic of this support, because the substrate is converted even in the absence of oxygen. The authors assumed that this nanoparticulated ceria acts as an oxygen supplier to the system by releasing and adsorbing oxygen through a redox process involving Ce4+/Ce3+ [76]. Previous studies detected the formation of peroxo and superoxo species in monoeletronic defects of nanoceria [69]. Thus, the improved activity in the oxidative esterification of HMF was attributed to the support containing appropriate Lewis acid sites that are capable of forming those intermediates during the reaction. The addition of water to the reaction medium showed a negative effect, as lower initial rates were observed when increasing the amount of water to 10% and 20%. With 20% of water, the reaction was not completed even after 30 h, reaching only 68% of FDMC. Although possible, the addition of water did not favor the formation of FDCA. The authors assumed that the presence of water may prevent the formation of metal–alcoholate adducts on the active sites, which are essential for the reaction to occur, and consequently, it disfavors the formation of hemiacetal. Therefore, this explains why it is so challenging to develop catalysts that work in a base-free aqueous phase.
Mechanism of HMF Oxidation
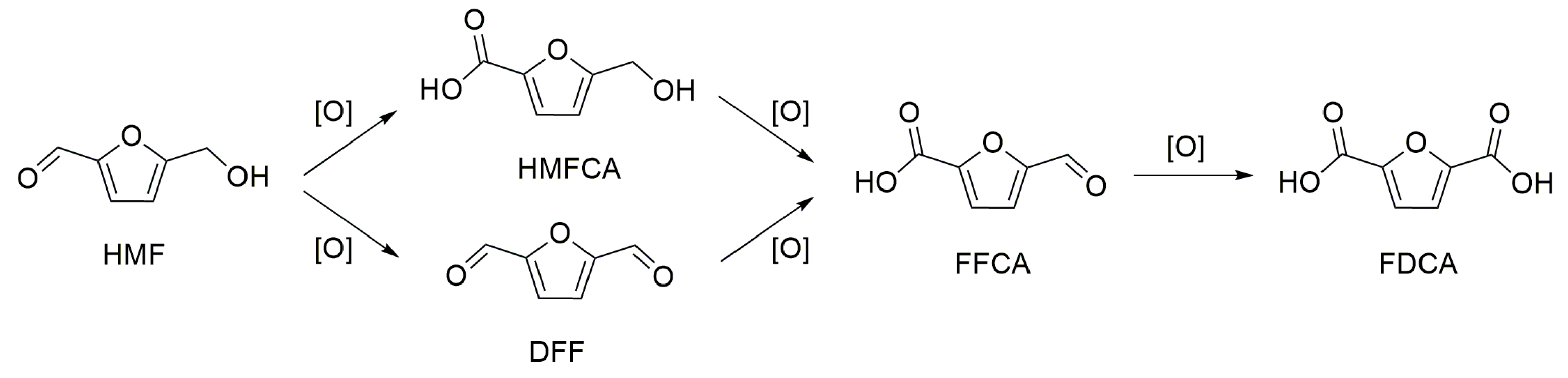
The mechanism of HMF oxidation can follow two routes as shown in Scheme 2 (1) the carbonyl group is first oxidized to a carboxylic group to form the monoacid 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), and then the hydroxyl group is oxidized to an aldehyde group, forming the 5-formyl-2-furancaboxylic acid (FFCA); (2) the alcohol group is first oxidized to a carbonyl group, forming 2,5-diformylfuran (DFF), and then the carbonyl group is oxidized to a carboxylic group, forming 5-formyl-2-furancaboxylic acid (FFCA); FDCA is formed through the oxidation of a carbonyl group in FFCA. The reaction pathways over different supported metal catalysts depend on the reaction medium and on the metal used. Apparently, pathway 1 (Scheme 2) is favored under basic conditions and in the presence of Au-based catalysts [77]. The oxidation of the alcohol group is the rate-determining step when using an Au catalyst, thus the formation of HMFCA is favored. Casanova et al. observed the formation of HMFCA when using Au/CeO2 and Au/TiO2 catalysts for the oxidation of HMF in a basic medium [56,78]. HMFCA was the only intermediate detected, which, according to the authors, might derive from the rapid conversion of FFCA to FDCA through a hemiacetal intermediate. The basic medium favors the formation of HMFCA, because the aldehyde group is believed to undergo a rapid, reversible hydration to a geminal diol through the nucleophilic addition of a hydroxide ion. The subsequent step, in which the geminal diol undergoes β-hydride elimination to form a carboxylic acid, is also facilitated by the hydroxide ions adsorbed on the metal surface [3]. Recently, some insights into the mechanisms were reported [3,79]. Zope and co-workers used an isotope-labeling technology in order to understand the formation of the intermediates. According to their study, the role of O2 is to remove electrons from the metal surface, oxidize metal–hydride bonds, and regenerate the hydroxide ions. According to their study, the four oxygen atoms incorporated in the FDCA molecule arise from water. In the absence of a base, however, a different mechanism had to be considered, and the characteristics of the support could drive the reaction to a different pathway. Erbitani and co-workers [68] studied the reaction mechanism for the synthesis of FDCA using the Au/HT catalyst. They showed that FDCA did not form through a free radical mechanism, but the reaction rather proceeded through the formation of HMFCA. HMF is readily oxidized to HMFCA even at room temperature through the formation of a hemiacetal intermediate. The authors claimed that the oxidation of HMF follows this pathway because of the basicity of HT. The rate-determining step is the transformation of HMFCA to FFCA, in which the alcohol group is oxidized via a β-hydride elimination, assisted by the HT basic sites. Once FFCA is formed, it is rapidly converted to FDCA through the formation of a second hemiacetal intermediate. This mechanism was supported by the observation of a temperature dependence of the products distribution. With the increase of temperature, the HMFCA yield decreased, while the FDCA yield increased. Therefore, the observations regarding the mechanism of HMF oxidation under base-free conditions using Au/HT were similar to those reported by Casanova et al. On the other hand, Wang et al. [56,69] observed DFF and FFCA as intermediates for the base-free oxidation of HMF using the Au–Pd/CNT catalyst. In this system, the oxidation of the alcohol group was faster than that of the aldehyde group. For the monometallic version of the catalyst Au/CNT, the aldehyde portion was oxidized forming HMFCA similarly to what observed for the other Au-based catalysts, whether in base-free (Au/HT) or basic medium (Au/CeO2 and Au/TiO2). However, HMFCA mainly underwent ring opening and degradation reactions to byproducts. The monometallic Pd/CNT catalyst followed the same reaction pathway as the bimetallic Au–Pd/CNT. The addition of Pd to the Au/CNT catalyst changed the reaction pathway towards the formation of DFF by facilitating the oxidation of the alcohol group. Moreover, the oxidation of FFCA to FDCA was favored by the incorporation of Pd in the catalyst, and this, according to the authors, is a difficult elementary step over Au-based catalysts under base-free conditions.
4. Base-Free Furfural Oxidation
For decades, studies have been directed towards the hydrogenation of furfural for the production of fuels, such as tetrahydrofuran and 2-methyltetrahydrofuran [12]. However, the oxidation of furfural can also lead to very interesting products, such as furoic acid, maleic acid, and succinic acid [80]. Considering that Au-based catalysts have shown great potential for the aerobic oxidation of many substrates in water, their use in the oxidation of furfural would be advantageous. To the best of our knowledge, Au-based catalysts for the production of furoic acid, maleic acid, or succinic acids under base-free conditions have not been reported yet. On the other hand, Au-based catalysts have been successfully applied for the oxidative esterification of furfural to produce alkylfuroates. Methyl-2-furoate is used as a flavor and fragrance as well as in other processes in fine chemical industry [81,82]. The first example of esterification of furfural to produce methyl-2-furoate required the use of a base [75]. However, the high performance of the Au/CeO2 catalyst under base-free conditions, reported by Casanova et al., inspired the development of Au-based catalysts for the base-free oxidative esterification of furfural [56]. Results obtained by different research groups for this reaction with Au-based catalysts with different supports are shown in Table 3. Studies from Signoretto’s group reveals that the catalysts’ performances in the absence of a base followed the order: Au/ZrO2 > Au/CeO2 >> Au/TiO2 [82]. According to the authors, ZrO2 is able to provide high Au dispersion and suitable acid–base properties. For this reason, Au/ZrO2 has been widely explored also for this reaction [82,83,84,85]. Apparently, the addition of dopants, such as sulfates [82,83,84], increased the catalytic activity. The authors assumed that the dopants in the support enhance the support’s surface area and acidity and retard crystallization [85]. The effect of calcination on the catalytic activity was also evaluated [82,83,85]. The studies indicate that high temperatures provided more active catalysts. However, calcination temperatures higher than 500 °C resulted in particles growth and in the increase of particle size distribution. Studies on Au/CeO2 were also performed by Ampelli et al. regarding the calcination of the support prior to the preparation of the catalyst, and the calcination of the catalyst as a post treatment step [86]. The results revealed that the thermal treatment was beneficial in both cases; therefore, the catalyst in entry 21 (pretreatment at 300 °C and post-treatment at 500 °C) was the most active.
Mechanism of Furfural Oxidation
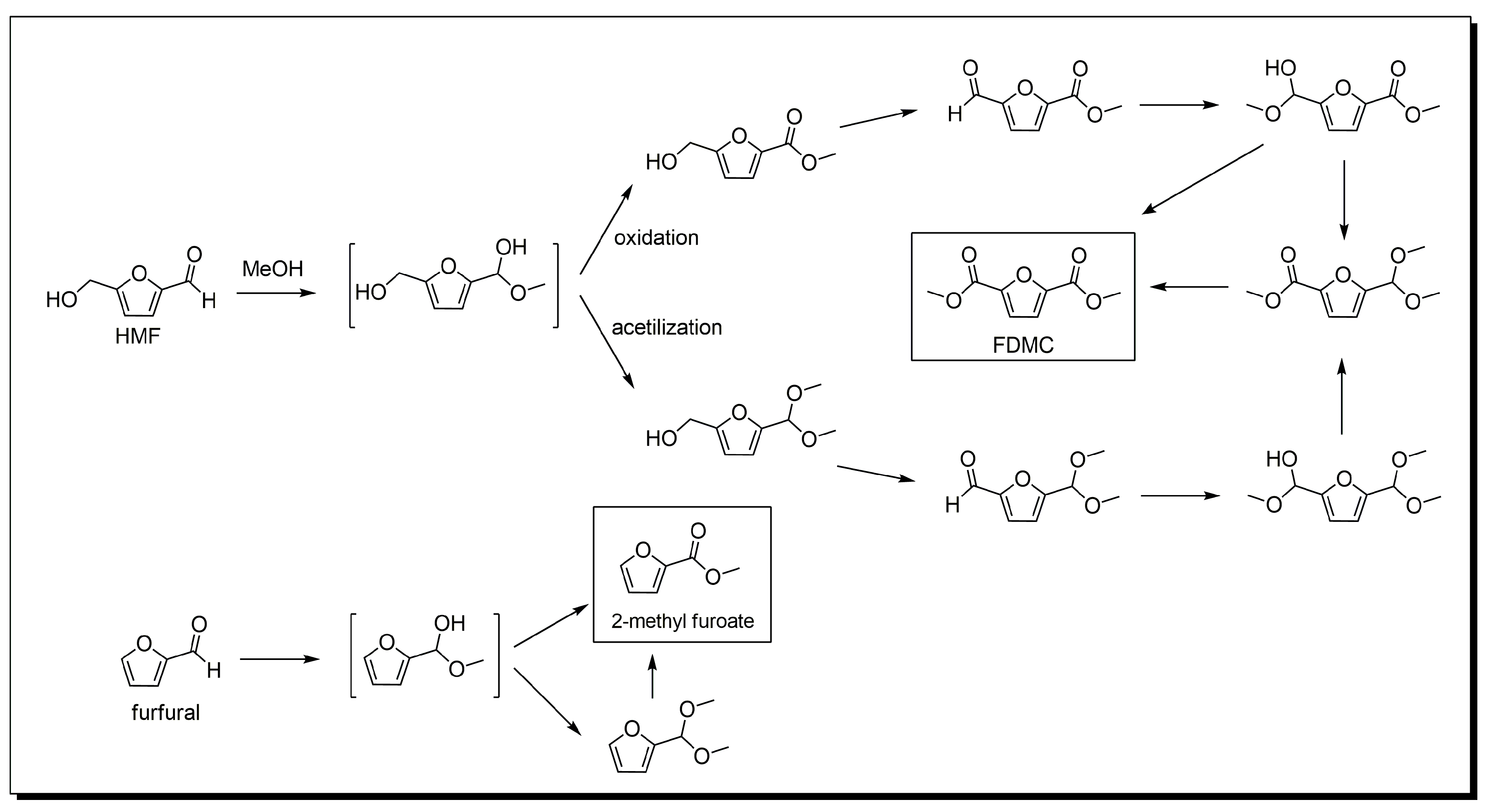
The understanding of the mechanism of the base-free oxidation of furfural is as highly desirable as the development of active Au-based catalytic systems. For the oxidative esterification of furfural, a mechanism was proposed taking into account the pathways suggested by Casanova et al. [56] for the oxidative esterification of HMF (Scheme 3). According to this study, furfural can first form a hemiacetal intermediate that could not be detected by gas chromatography (GC). This intermediate can then follow two pathways: (1) oxidation, or (2) acetylation. In pathway 1, the hemiacetal is directly oxidized to the ester 2-methylfuroate, whereas in pathway 2, the hemiacetal is first converted to 2-furaldehyde-dimethyl-acetal and then oxidized to the ester. In Casanova’s study, the Au/CeO2 catalyst favored the first pathway rather than the second pathway, although the latter is thermodynamically favored [56]. Menegazzo et al. investigated the reaction mechanism of the oxidative esterification of furfural using the Au/ZrO2 catalyst [84]. The catalyst allowed almost full conversion in 90 min, with 100% selectivity towards the ester. To clarify the mechanism, some tests were performed: (1) a blank test where no conversion was observed in the absence of a catalyst, indicating that the presence of a catalyst is crucial for the catalytic activity; (2) a reaction performed using furoic acid as a substrate, which did not produce the ester, indicating that the reaction pathway does not pass through the formation of the acid; (3) a support test where the full conversion of furfural was obtained with ZrO2, but acetal was the only product (this result highlights the importance of Lewis acidity of ZrO2 in providing the specific sites for the reaction); and (4) an acetal test where the acetal formed in the previous test was filtered and reacted with methanol in the presence of the Au/ZrO2 catalyst, in order to verify if the ester could form from the acetal. The results indicated that the acetal was not converted, confirming the stability of this product, already reported by Casanova et al. This means that for Au/ZrO2 the reaction occurs through pathway 1, i.e., by direct oxidation of the hemiacetal intermediate.
5. Factors Influencing the Activity in the Oxidation on Gold-Based Catalysts
5.1. Effect of the Size of the Gold Particles
Many studies have shown that the catalytic activity of gold is dependent on the crystal planes step density because the energy barrier of oxygen on these steps is lower, allowing the easy release of active oxygen and accelerating the oxidation reaction [87,88,89,90,91,92]. Therefore, the catalytic activity of Au NPs will be directly governed by their particle size, and this parameter should be effectively controlled. Different preparative methods for gold colloids are known, most of them based on the reduction of tetrachloroauric acid HAuCl4 [87]. Luo et al. [93] found that the size and shape of the nanoparticles are generally controlled by external factors such as time, precursor, reaction temperature, and surfactant concentration. It was stated that the lack of an external influence eventually leads to the uncontrolled growth of the nanoparticles. This could be responsible for the loss of the nanoscale properties of materials. Various authors found that glucose oxidation catalyzed by gold nanoparticles produces hydrogen peroxide (H2O2) in situ. In the presence of the gold precursor (HAuCl4), it provokes the particles’ growth, which is responsible for the decreased activity and surface passivation with gluconic acid (the product of the reaction), leading to the deactivation of the system [93]. Megías et al. [94] prepared and immobilized a series of gold colloids on commercial activated carbon. The influence and the stability of the colloid preparation were studied and related to the gold particle size in the final catalyst. The ratios between PVA and Au, and NaBH4 and Au were studied. Two limiting values were found for the achievement of a stable colloid and of a homogeneous particles size distribution: the PVA/Au ratio that should be superior to 0.85, and the NaBH4/Au ratio that should be superior to 3. The catalysts showed an important activity in the glucose to gluconic acid oxidation reaction, leading to a yield of gluconic acid close to 90% in base-free, mild conditions. The size–activity correlation and probable mechanism were also discussed. The structure and size sensitivity indicated the optimal size, showing maximum conversion for particles of around 9 nm, although after normalization to the exposed surface gold atoms (expressed in turnover frequency (TOF)) this optimal size of gold particles moved in the 15–20 nm range, pointing out a possible influence of the glucose adsorption step on the reaction mechanism. In addition, the catalysts showed a very good recyclability with a loss of activity of only 10% after the forth cycle [94]. Ishida et al. [39,40,95] also demonstrated that the size of Au nanoparticles supported on metal oxides during glucose oxidation strongly influences the overall TOFs. They concluded that the reaction occurs at the surface of the Au particles. The same trends were observed for the base-free catalytic oxidation reactions.
5.2. Effect of the Support
Au NPs can be immobilized on metal oxides, activated carbons, zeolites, and other supports by different methods of preparation, including impregnation, precipitation, sol immobilization, or microemulsion. Depending on the catalytic ability of the support itself, oxide supports are divided into active supports, such as Fe2O3, TiO2, Co3O4, etc., and inert supports, like MgO, Al2O3, and SiO2. For Au-based catalysts supported on active materials, the dominant reaction pathway involves the adsorption of a mobile, molecular oxygen species on the support and the dissociation at the interface, which supplies reactive oxygen. For Au supported on inert materials, where the oxygen supply most likely proceeds via a direct, dissociative adsorption on the Au particles, the size of the latter plays a crucial role. Moreover, the impact of the support on the catalytic activity also depends on the type of catalyzed reaction. Ishida et al. [39,40] reviewed the impact of supported gold catalysts on the catalytic oxidation of glucose. Contrary to the gas-phase CO oxidation, for glucose oxidation, the catalytic activity of Au NPs was influenced more significantly by the size than by the nature of the support. The TOF per surface Au atom confirmed these results (Table 4). The selectivity towards gluconic acid remained constant and high (above 98%). Therefore, after choosing a suitable support, more attention should be paid to the influence of the preparation method on the size of the Au NPs.
Although the size dependencies on the TOF values was not the same as that observed by Comotti et al. [47], the size of the Au particles plays a leading role. In the case of oxidation carried out in the liquid phase, the oxidation occurs at the Au NP surfaces and not at the perimeter interfaces between the Au particles and the supports, as suggested for the CO oxidation. The activation energy is not the same on different supports. It could be concluded that metal oxide supports affect the gold clusters electronically.
Megias-Sayago et al. [46] have reported the base-free oxidation of glucose using molecular oxygen over various Au-supported catalysts. The different supports screened were Al2O3 and CeO2 as simple oxides, and CeO2 (20 wt %)/Al2O3, CeO2 (25 wt %)/ZrO2, and CeO2 (50 wt %)/ZrO2 as mixed oxides. The effects of the support, reaction time, temperature, and stirring rate on the catalytic activity and product distribution were evaluated. Catalysts were prepared using the direct anionic exchange method (DAE) assisted by ammonia. X-ray diffraction (XRD) and transmission electron microscopy (TEM) were performed for the different catalysts. Catalytic tests for the different catalysts showed approximately the same conversion of glucose with a value of 75% in all cases, after 18 h of reaction at 120 °C. Both the selectivity and the activity were influenced by the support. The TOF calculated on the basis of Au dispersion for the different catalysts showed that the activity was in the following order: Au/Al > Au/Ce/Al > Au/Ce/Zr > Au/Ce50/Zr > Au/Ce. When comparing the selectivity towards gluconic acid, the results showed that alumina-supported samples presented the highest selectivity (95%), followed by Au/Ce/Al and Au/Ce. Conversely, zirconia containing samples showed the lowest selectivity towards gluconic acid, favoring some lactic acid formation. Moreover, lactic acid formation was favored at low temperatures and low stirring rates. Au/Al was tested for reusability, and gold metal leaching led to a drop in the conversion of glucose and a loss of catalytic activity.
5.3. Stability
Wang et al. [44] studied the stability of Au supported on metal oxides for the oxidation of glucose without base addition. Reactions were carried out at 65 °C and 0.23 MPa for 6 h with a glucose/Au molar ratio of 140. Approximately 30% and 60% decreases in conversion were observed for Au/n-CeO2 (nanosized CeO2) and Au/μ-CeO2, respectively. According to the report, the relatively more stable nature of the Au/n-CeO2 catalyst was due to the low surface density of the Au NPs and to the presence of more anchoring sites that could prevent the growth of the Au NPs. Generally, in the case of the base-free oxidation of glucose, deactivation of the catalysts is observed following the adsorption of acids on the catalysts' surface (Table 5). Some authors claimed that the reactivation of the catalysts could be done by calcination. However, the calcination step would lead to a severe loss of activity because the diameter of the Au NPs is obviously larger than that of the as-prepared catalyst. Miedziak et al. [6] also reported a drastic decrease in activity for a bimetallic Au–Pd NP catalyst, giving a conversion of less than 20% when used a second time. The conversion could be improved by only 5% through a calcination treatment.
Qi et al. [43] discussed two different regeneration methods, including NaOH treatment and calcination, for an Au/CMK-3 catalyst that had gradually lost its activity upon recycling during glucose oxidation. On the basis of their results, the used catalyst treated with 50 mL of 0.1 mol/L NaOH for 30 min at 90 °C showed the best performance, with 87% conversion. The characterization indicated that after the recycling experiments, abundant carboxylic acids were adsorbed on the surface of the catalyst, and this was the predominant factor that caused deactivation. Both NaOH treatment and calcination could effectively remove the adsorbates, and atmospheric calcination was more efficient in this frame. However, given the appearance of large particles during the calcination procedure, the NaOH treatment method seems more appropriate. Therefore, one can conclude that NaOH treatment is a very simple and useful way to regenerate used catalysts that suffered from adsorbate deactivation during glucose oxidation.
5.4. Effect of the Reaction Temperature
The conversion of glucose was found to be highly dependent on the temperature, and 100% conversion was reached at 150 °C. However, as shown in Table 6, the yield of acid decreased drastically. Indeed, the formation of different byproducts was observed (several peaks in the HPLC spectra). The highest yield of the corresponding sugar acid was achieved at the temperature of 70 °C. Above this temperature, several other overoxidation products appeared, as a result of thermal decomposition and polymerization. A similar trend of decreased selectivity as the temperature increased was reported on Au/CeO2 [25] and Au/CMK-3 catalysts [43] for base-free oxidation of glucose. At a relatively low temperature (namely, 70 °C), no byproduct was observed (Table 6). When the temperature was increased, some byproducts appeared.
The effect of temperature on cellobiose conversion was studied by Cao et al. [49,96,97]. It was found that the increase in the temperature from 130 to 140 °C increased the conversion from 32 to 52%, and the gluconic acid yield from 7 to 21%. However, no change in the conversion was observed after a further increase of the temperature to 150 °C. As expected, the increase to 160 °C decreased the conversion by 10% and significantly decreased the gluconic acid yield (Table 7). According to the authors, this decline in activity and selectivity was explained by the deposition of carbon species on the catalyst surface. This deposit could be favored at high temperatures and could be responsible for a lower number of active sites. Moreover, other side products, such as glycolic acid and glucaric acid, were observed at high temperatures [49].
6. Conclusions
To sum, the oxidation of low molecular weight sugars and furan derivatives using gold-based catalysts and different operating conditions have been reviewed. When working in base-free conditions, it is necessary to engineer advanced heterogeneous catalysts to obtain highly efficient processes without the use of hazardous and dangerous chemicals. Progress in this field was made starting from the simple monometallic Au-supported catalysts that were followed by the incorporation of other metals into the system. The local basicity derived from the support or from the second metal plays a crucial role. It is now well established that supported gold nanoparticles exhibit the best catalytic performances in oxidation processes. The results obtained for bi- and trimetallic systems confirmed that the presence of multiple metals improves the catalysts in comparison with monometallic systems. A simple change in the metal-to-metal ratio changes the catalytic results, which offers the advantage of fine-tuning the catalytic system, and allows to obtain highly efficient catalysts for base-free oxidation. Moreover, gold catalysts have the potential to be used for the industrial oxidation of different kinds of compounds (such as carbohydrates, alcohols, furanics, aldehydes). However, more knowledge in this field is needed. Even though it was possible to obtain high performances and high selectivity, the catalysts developed up to now are not able to yield high rates or to maintain their activity in an acidic or neutral media. Even though gold catalysts can oxidize C5–C6 sugars, HMF, or furfural without the need of base addition, catalyst deactivation issues must be solved. The deactivation of the catalysts may occur because of the leaching of the metal in acidic media or because of the saturation of the catalyst surface with adsorbed products or byproducts. The oxidation in neutral or acidic media gives the advantage of the direct formation of the desired acid in a one-step reaction. The use of unwanted and environmentally dangerous chemicals, such as NaOH, could be eliminated, which is positive from an economical point of view (increase of the equipment lifetime as a result of the elimination of strong corrosive media, elimination of the final gluconate acid transformation). Thus, it is important to focus on the preparation of catalysts that will have better activity and stability in base-free conditions. Despite the importance of this field of research, the literature is still limited to few examples.
Author Contributions
R.W., C.F., J.S., S.H., L.R., and S.P. participated in all aspects of this work (writing, checking, bibliographic research).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-Phase Catalytic Processing of Biomass-Derived Oxygenated Hydrocarbons to Fuels and Chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Iborra, S.; Velty, A. Chemical Routes for the Transformation of Biomass into Chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.E.; Ide, M.S.; Davis, R.J. Selective oxidation of alcohols and aldehydes over supported metal nanoparticles. Green Chem. 2013, 15, 17–45. [Google Scholar] [CrossRef]
- Dumeignil, F.; Capron, M.; Katryniok, B.; Wojcieszak, R.; Löfberg, A.; Girardon, J.-S.; Desset, S.; Araque-Marin, M.; Jalowiecki-Duhamel, L.; Paul, S. Biomass-derived Platform Molecules Upgrading through Catalytic Processes: Yielding Chemicals and Fuels. J. Jpn. Pet. Inst. 2015, 58, 257–273. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, J.; Huang, J.; Zhang, C.; Hong, F.; Zhou, Y.; Li, G.; Haruta, M. Efficient Aerobic Oxidation of Glucose to Gluconic Acid over Activated Carbon-Supported Gold Clusters. ChemSusChem 2017, 10, 1976–1980. [Google Scholar] [CrossRef] [PubMed]
- Miedziak, P.J.; Alshammari, H.; Kondrat, S.A.; Clarke, T.J.; Davies, T.E.; Morad, M.; Morgan, D.J.; Willock, D.J.; Knight, D.W.; Taylor, S.H.; et al. Base-free glucose oxidation using air with supported gold catalysts. Green Chem. 2014, 16, 3132–3141. [Google Scholar] [CrossRef]
- Eloutassi, N.; Louaste, B.; Boudine, L.; Remmal, A. Valorisation de la biomasse lignocellulosique pour la production de bioéthanol de deuxième génération. Rev. Energies Renouv. 2014, 17, 600–609. [Google Scholar]
- Donate, P.M. Green synthesis from biomass. Chem. Biol. Technol. Agric. 2014, 1, 4. [Google Scholar] [CrossRef]
- Dedsuksophon, W.; Faungnawakij, K.; Champreda, V.; Laosiripojana, N. Hydrolysis/dehydration/aldol-condensation/hydrogenation of lignocellulosic biomass and biomass-derived carbohydrates in the presence of Pd/WO3–ZrO2 in a single reactor. Bioresour. Technol. 2011, 102, 2040–2046. [Google Scholar] [CrossRef] [PubMed]
- Tathod, A.; Kane, T.; Sanil, E.S.; Dhepe, P.L. Solid base supported metal catalysts for the oxidation and hydrogenation of sugars. J. Mol. Catal. A Chem. 2014, 388–389, 90–99. [Google Scholar] [CrossRef]
- Guimarães, L.H.S. Carbohydrates from Biomass: Sources and Transformation by Microbial Enzymes. In Carbohydrates-Comprehensive Studies on Glycobiology and Glycotechnology; Intech: London, UK, 2012; Chapter 20; pp. 441–456. [Google Scholar]
- Li, X.; Jia, P.; Wang, T. Furfural: A Promising Platform Compound for Sustainable Production of C4 and C5 Chemicals. ACS Catal. 2016, 6, 7621–7640. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Dumeignil, F. Biorefineries an Introduction; Walter de Gruyter GmbH & Co. KG: Berlin, Germany, 2015. [Google Scholar]
- Zhang, Z.; Deng, K. Recent Advances in the Catalytic Synthesis of 2,5-Furandicarboxylic Acid and Its Derivatives. ACS Catal. 2015, 5, 6529–6544. [Google Scholar] [CrossRef]
- Zhu, Y.; Shen, M.; Xia, Y.; Lu, M. Au/MnO2 nanostructured catalysts and their catalytic performance for the oxidation of 5-(hydroxymethyl)furfural. Catal. Commun. 2015, 64, 37–43. [Google Scholar] [CrossRef]
- Yang, B.; Dai, Z.; Ding, S.-Y.; Wyman, C.E. Enzymatic hydrolysis of cellulosic biomass. Biofuels 2011, 2, 421–449. [Google Scholar] [CrossRef]
- De Wit, D.; Maat, L.; Kieboom, A.P.G. Carbohydrates as industrial raw materials. Ind. Crops Prod. 1993, 2, 1–12. [Google Scholar] [CrossRef]
- Rauter, A.P.; Vogel, P.; Queneau, Y. Carbohydrates in Sustainable Development II; Springer: Brelin, Germany, 2010; p. 204. [Google Scholar]
- Aquino Neto, S.; Milton, R.D.; Crepaldi, L.B.; Hickey, D.P.; de Andrade, A.R.; Minteer, S.D. Co-immobilization of gold nanoparticles with glucose oxidase to improve bioelectrocatalytic glucose oxidation. J. Power Sources 2015, 285, 493–498. [Google Scholar] [CrossRef]
- Sharma, K.K.; Wang, Y.; Roman-Leshkov, Y. Gold supported on nanocrystalline metal oxides as highly effective catalysts for the oxidation of glucose under base-free conditions. In Abstracts of Papers of the American Chemical Society; American Chemical Society: Washington, DC, USA, 2012. [Google Scholar]
- Trombotto, S.; Violet-Courtens, E.; Cottier, L.; Queneau, Y. Oxidation of Two Major Disaccharides: Sucrose and Isomaltulose. Top. Catal. 2004, 27, 31–37. [Google Scholar] [CrossRef]
- Pasta, M.; Ruffo, R.; Falletta, E.; Mari, C.M.; Pina, C.D. Alkaline glucose oxidation on nanostructured gold electrodes. Gold Bull. 2010, 43, 57–64. [Google Scholar] [CrossRef]
- Biella, S.; Prati, L.; Rossi, M. Selective Oxidation of d-Glucose on Gold Catalyst. J. Catal. 2002, 206, 242–247. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of Biomass into Chemicals over Metal Catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Wojcieszak, R.; Cuccovia, I.M.; Silva, M.A.; Rossi, L.M. Selective oxidation of glucose to glucuronic acid by cesium-promoted gold nanoparticle catalyst. J. Mol. Catal. A Chem. 2016, 422, 35–42. [Google Scholar] [CrossRef]
- Biella, S.; Castiglioni, G.L.; Fumagalli, C.; Prati, L.; Rossi, M. Application of gold catalysts to selective liquid phase oxidation. Catal. Today 2002, 72, 43–49. [Google Scholar] [CrossRef]
- Delidovich, I.V.; Taran, O.P.; Matvienko, L.G.; Simonov, A.N.; Simakova, I.L.; Bobrovskaya, A.N.; Parmon, V.N. Selective Oxidation of Glucose over Carbon-supported Pd and Pt Catalysts. Catal. Lett. 2010, 140, 14–21. [Google Scholar] [CrossRef]
- Solmi, S.; Morreale, C.; Ospitali, F.; Agnoli, S.; Cavani, F. Oxidation of d-Glucose to Glucaric Acid Using Au/C Catalysts. ChemCatChem 2017, 9, 2797–2806. [Google Scholar] [CrossRef]
- Novotný, O.; Cejpek, K.; Velíšek, J. Formation of Carboxylic Acids during Degradation of Monosaccharides. Czech. J. Food Sci. 2008, 26, 117–131. [Google Scholar]
- Moulik, S.P.; Basu, D.; Bhattacharya, P.K. Effects of various conditions on the alkaline degradation of d-fructose and d-glucose. Carbohydr. Res. 1978, 63, 165–172. [Google Scholar] [CrossRef]
- Vuyyuru, K.R.; Strasser, P. Oxidation of biomass derived 5-hydroxymethylfurfural using heterogeneous and electrochemical catalysis. Catal. Today 2012, 195, 144–154. [Google Scholar] [CrossRef]
- Piccolo, A.; Conte, P.; Cozzolino, A. Effects of mineral and monocarboxylic acids on the molecular association of dissolved humic substances. Eur. J. Soil Sci. 1999, 50, 687–694. [Google Scholar] [CrossRef]
- Pasini, T.; Piccinini, M.; Blosi, M.; Bonelli, R.; Albonetti, S.; Dimitratos, N.; Lopez-Sanchez, J.A.; Sankar, M.; He, Q.; Kiely, C.J.; et al. Selective oxidation of 5-hydroxymethyl-2-furfural using supported gold-copper nanoparticles. Green Chem. 2011, 13, 2091–2099. [Google Scholar] [CrossRef]
- Comotti, M.; Della Pina, C.; Matarrese, R.; Rossi, M. The Catalytic Activity of “Naked” Gold Particles. Angew. Chem. Int. Ed. 2004, 43, 5812–5815. [Google Scholar] [CrossRef] [PubMed]
- Baatz, C.; Thielecke, N.; Prüße, U. Influence of the preparation conditions on the properties of gold catalysts for the oxidation of glucose. Appl. Catal. B Environ. 2007, 70, 653–660. [Google Scholar] [CrossRef]
- Önal, Y.; Schimpf, S.; Claus, P. Structure sensitivity and kinetics of d-glucose oxidation to d-gluconic acid over carbon-supported gold catalysts. J. Catal. 2004, 223, 122–133. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Gold-Catalyzed Organic Reactions. Chem. Rev. 2007, 107, 3180–3211. [Google Scholar] [CrossRef] [PubMed]
- Comotti, M.; Pina, C.D.; Matarrese, R.; Rossi, M.; Siani, A. Oxidation of alcohols and sugars using Au/C catalysts: Part 2. Sugars. Appl. Catal. A Gen. 2005, 291, 204–209. [Google Scholar] [CrossRef]
- Okatsu, H.; Kinoshita, N.; Akita, T.; Ishida, T.; Haruta, M. Deposition of gold nanoparticles on carbons for aerobic glucose oxidation. Appl. Catal. A Gen. 2009, 369, 8–14. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, N.; Okatsu, H.; Akita, T.; Takei, T.; Haruta, M. Influence of the Support and the Size of Gold Clusters on Catalytic Activity for Glucose Oxidation. Angew. Chem. Int. Ed. 2008, 120, 9405–9408. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, X.; Iqbal, S.; Miedziak, P.J.; Edwards, J.K.; Armstrong, R.D.; Morgan, D.J.; Wang, J.; Hutchings, G.J. Base-free oxidation of glucose to gluconic acid using supported gold catalysts. Catal. Sci. Technol. 2016, 6, 107–117. [Google Scholar] [CrossRef]
- Villa, A.; Wang, D.; Veith, G.M.; Vindigni, F.; Prati, L. Sol immobilization technique: A delicate balance between activity, selectivity and stability of gold catalysts. Catal. Sci. Technol. 2013, 3, 3036–3041. [Google Scholar] [CrossRef]
- Qi, P.; Chen, S.; Chen, J.; Zheng, J.; Zheng, X.; Yuan, Y. Catalysis and Reactivation of Ordered Mesoporous Carbon-Supported Gold Nanoparticles for the Base-Free Oxidation of Glucose to Gluconic Acid. ACS Catal. 2015, 5, 2659–2670. [Google Scholar] [CrossRef]
- Wang, Y.; Van de Vyver, S.; Sharma, K.K.; Roman-Leshkov, Y. Insights into the stability of gold nanoparticles supported on metal oxides for the base-free oxidation of glucose to gluconic acid. Green Chem. 2014, 16, 719–726. [Google Scholar] [CrossRef]
- Rautiainen, S.; Lehtinen, P.; Vehkamäki, M.; Niemelä, K.; Kemell, M.; Heikkilä, M.; Repo, T. Microwave-assisted base-free oxidation of glucose on gold nanoparticle catalysts. Catal. Commun. 2016, 74, 115–118. [Google Scholar] [CrossRef]
- Megías-Sayago, C.; Ivanova, S.; López-Cartes, C.; Centeno, M.A.; Odriozola, J.A. Gold catalysts screening in base-free aerobic oxidation of glucose to gluconic acid. Catal. Today 2017, 279, 148–154. [Google Scholar] [CrossRef]
- Comotti, M.; Pina, C.D.; Rossi, M. Mono- and bimetallic catalysts for glucose oxidation. J. Mol. Catal. A Chem. 2006, 251, 89–92. [Google Scholar] [CrossRef]
- Zhang, H.; Toshima, N. Preparation of novel Au/Pt/Ag trimetallic nanoparticles and their high catalytic activity for aerobic glucose oxidation. Appl. Catal. A Gen. 2011, 400, 9–13. [Google Scholar] [CrossRef]
- Cao, Y.; Iqbal, S.; Miedziak, P.J.; Jones, D.R.; Morgan, D.J.; Liu, X.; Wang, J.; Hutchings, G.J. An investigation into bimetallic catalysts for base free oxidation of cellobiose and glucose. J. Chem. Technol. Biotechnol. 2017, 92, 2246–2253. [Google Scholar] [CrossRef]
- Prüße, U.; Heidinger, S.; Baatz, C. Catalytic conversion of renewables: Kinetic and mechanistic aspects of the gold-catalyzed liquid-phase glucose oxidation. Agric. For. Res. 2011, 16, 261–272. [Google Scholar]
- Comotti, M.; DellaPina, C.; Falletta, E.; Rossi, M. Aerobic Oxidation of Glucose with Gold Catalyst: Hydrogen Peroxide as Intermediate and Reagent. Adv. Synth. Catal. 2006, 348, 313–316. [Google Scholar] [CrossRef]
- Beltrame, P.; Comotti, M.; Della Pina, C.; Rossi, M. Aerobic oxidation of glucose: II. Catalysis by colloidal gold. Appl. Catal. A Gen. 2006, 297, 1–7. [Google Scholar] [CrossRef]
- Larew, L.A.; Johnson, D.C. Concentration dependence of the mechanism of glucose oxidation at gold electrodes in alkaline media. J. Electroanal. Chem. Interfacial Electrochem. 1989, 262, 167–182. [Google Scholar] [CrossRef]
- Dirkn, J.M.H.; van der Baan, H.S.; van den Broen, J.M.A.J.J. The preparation of d-glucaric acid by the oxidation of d-gluconic acid catalysed by platinum on carbon. Carbohydr. Res. 1977, 59, 63–72. [Google Scholar] [CrossRef]
- Dirkx, J.M.H.; van der Baan, H.S. The oxidation of gluconic acid with platinum on carbon as catalyst. J. Catal. 1981, 67, 14–20. [Google Scholar] [CrossRef]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into Chemicals: Aerobic Oxidation of 5-Hydroxymethyl-2-furfural into 2,5-Furandicarboxylic Acid with Gold Nanoparticle Catalysts. ChemSusChem 2009, 2, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Albonetti, S.; Lolli, A.; Morandi, V.; Migliori, A.; Lucarelli, C.; Cavani, F. Conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over Au-based catalysts: Optimization of active phase and metal–support interaction. Appl. Catal. B Environ. 2015, 163, 520–530. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, Y.; Pan, X.; Wu, T.; Zhang, B.; Li, J.; Yi, T.; Zhang, Z.; Yang, X. Superior catalytic performance of Ce1-xBixO2-[small delta] solid solution and Au/Ce1-xBixO2-[small delta] for 5-hydroxymethylfurfural conversion in alkaline aqueous solution. Catal. Sci. Technol. 2015, 5, 1314–1322. [Google Scholar] [CrossRef]
- Cai, J.; Ma, H.; Zhang, J.; Song, Q.; Du, Z.; Huang, Y.; Xu, J. Gold Nanoclusters Confined in a Supercage of Y Zeolite for Aerobic Oxidation of HMF under Mild Conditions. Chem. Eur. J. 2013, 19, 14215–14223. [Google Scholar] [CrossRef] [PubMed]
- Gorbanev, Y.Y.; Klitgaard, S.K.; Woodley, J.M.; Christensen, C.H.; Riisager, A. Gold-Catalyzed Aerobic Oxidation of 5-Hydroxymethylfurfural in Water at Ambient Temperature. ChemSusChem 2009, 2, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.E.; Houk, L.R.; Tamargo, E.C.; Datye, A.K.; Davis, R.J. Oxidation of 5-hydroxymethylfurfural over supported Pt, Pd and Au catalysts. Catal. Today 2011, 160, 55–60. [Google Scholar] [CrossRef]
- Ardemani, L.; Cibin, G.; Dent, A.J.; Isaacs, M.A.; Kyriakou, G.; Lee, A.F.; Parlett, C.M.A.; Parry, S.A.; Wilson, K. Solid base catalysed 5-HMF oxidation to 2,5-FDCA over Au/hydrotalcites: Fact or fiction? Chem. Sci. 2015, 6, 4940–4945. [Google Scholar] [CrossRef]
- Albonetti, S.; Pasini, T.; Lolli, A.; Blosi, M.; Piccinini, M.; Dimitratos, N.; Lopez-Sanchez, J.A.; Morgan, D.J.; Carley, A.F.; Hutchings, G.J.; et al. Selective oxidation of 5-hydroxymethyl-2-furfural over TiO2-supported gold–copper catalysts prepared from preformed nanoparticles: Effect of Au/Cu ratio. Catal. Today 2012, 195, 120–126. [Google Scholar] [CrossRef]
- Villa, A.; Schiavoni, M.; Campisi, S.; Veith, G.M.; Prati, L. Pd-modified Au on Carbon as an Effective and Durable Catalyst for the Direct Oxidation of HMF to 2,5-Furandicarboxylic Acid. ChemSusChem 2013, 6, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Gao, T.; Fang, W.; Cao, Q. Base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid in water by hydrotalcite-activated carbon composite supported gold catalyst. Mol. Catal. 2017, 439, 171–179. [Google Scholar] [CrossRef]
- Gao, Z.; Xie, R.; Fan, G.; Yang, L.; Li, F. Highly Efficient and Stable Bimetallic AuPd over La-Doped Ca–Mg–Al Layered Double Hydroxide for Base-Free Aerobic Oxidation of 5-Hydroxymethylfurfural in Water. ACS Sustain. Chem. Eng. 2017, 5, 5852–5861. [Google Scholar] [CrossRef]
- Gui, Z.; Cao, W.; Saravanamurugan, S.; Riisager, A.; Chen, L.; Qi, Z. Efficient Aerobic Oxidation of 5-Hydroxymethylfurfural in Aqueous Media with Au–Pd Supported on Zinc Hydroxycarbonate. ChemCatChem 2016, 8, 3636–3643. [Google Scholar] [CrossRef]
- Gupta, N.K.; Nishimura, S.; Takagaki, A.; Ebitani, K. Hydrotalcite-supported gold-nanoparticle-catalyzed highly efficient base-free aqueous oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid under atmospheric oxygen pressure. Green Chem. 2011, 13, 824–827. [Google Scholar] [CrossRef]
- Zope, B.N.; Davis, S.E.; Davis, R.J. Influence of Reaction Conditions on Diacid Formation during Au-Catalyzed Oxidation of Glycerol and Hydroxymethylfurfural. Top. Catal. 2012, 55, 24–32. [Google Scholar] [CrossRef]
- Wan, X.; Zhou, C.; Chen, J.; Deng, W.; Zhang, Q.; Yang, Y.; Wang, Y. Base-Free Aerobic Oxidation of 5-Hydroxymethyl-furfural to 2,5-Furandicarboxylic Acid in Water Catalyzed by Functionalized Carbon Nanotube-Supported Au–Pd Alloy Nanoparticles. ACS Catal. 2014, 4, 2175–2185. [Google Scholar] [CrossRef]
- Nielsen, I.S.; Taarning, E.; Egeblad, K.; Madsen, R.; Christensen, C.H. Direct aerobic oxidation of primary alcohols to methyl esters catalyzed by a heterogeneous gold catalyst. Catal. Lett. 2007, 116, 35–40. [Google Scholar] [CrossRef]
- Gallas-Hulin, A.; Kotni, R.K.; Nielsen, M.; Kegnæs, S. Catalytic Oxidation of Allylic Alcohols to Methyl Esters. Top. Catal. 2017. [Google Scholar] [CrossRef]
- Whiting, G.T.; Kondrat, S.A.; Hammond, C.; Dimitratos, N.; He, Q.; Morgan, D.J.; Dummer, N.F.; Bartley, J.K.; Kiely, C.J.; Taylor, S.H.; et al. Methyl Formate Formation from Methanol Oxidation Using Supported Gold–Palladium Nanoparticles. ACS Catal. 2015, 5, 637–644. [Google Scholar] [CrossRef]
- Oliveira, R.L.; Kiyohara, P.K.; Rossi, L.M. Clean preparation of methyl esters in one-step oxidative esterification of primary alcohols catalyzed by supported gold nanoparticles. Green Chem. 2009, 11, 1366–1370. [Google Scholar] [CrossRef]
- Taarning, E.; Nielsen, I.S.; Egeblad, K.; Madsen, R.; Christensen, C.H. Chemicals from Renewables: Aerobic Oxidation of Furfural and Hydroxymethylfurfural over Gold Catalysts. ChemSusChem 2008, 1, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.; Carrettin, S.; Fierro-Gonzalez, J.C.; Hao, Y.; Gates, B.C.; Corma, A. CO Oxidation Catalyzed by Supported Gold: Cooperation between Gold and Nanocrystalline Rare-Earth Supports Forms Reactive Surface Superoxide and Peroxide Species. Angew. Chem. Int. Ed. 2005, 44, 4778–4781. [Google Scholar] [CrossRef] [PubMed]
- Lilga, M.A.; Hallen, R.T.; Gray, M. Production of Oxidized Derivatives of 5-Hydroxymethylfurfural (HMF). Top. Catal. 2010, 53, 1264–1269. [Google Scholar] [CrossRef]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into chemicals: One pot-base free oxidative esterification of 5-hydroxymethyl-2-furfural into 2,5-dimethylfuroate with gold on nanoparticulated ceria. J. Catal. 2009, 265, 109–116. [Google Scholar] [CrossRef]
- Zope, B.N.; Hibbitts, D.D.; Neurock, M.; Davis, R.J. Reactivity of the Gold/Water Interface during Selective Oxidation Catalysis. Science 2010, 330, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Wojcieszak, R.; Santarelli, F.; Paul, S.; Dumeignil, F.; Cavani, F.; Gonçalves, R.V. Recent developments in maleic acid synthesis from bio-based chemicals. Sustain. Chem. Process. 2015, 3, 9. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Pinna, F.; Manzoli, M.; Aina, V.; Cerrato, G.; Boccuzzi, F. Oxidative esterification of renewable furfural on gold-based catalysts: Which is the best support? J. Catal. 2014, 309, 241–247. [Google Scholar] [CrossRef]
- Ampelli, C.; Barbera, K.; Centi, G.; Genovese, C.; Papanikolaou, G.; Perathoner, S.; Schouten, K.J.; van der Waal, J.K. On the nature of the active sites in the selective oxidative esterification of furfural on Au/ZrO2 catalysts. Catal. Today 2016, 278, 56–65. [Google Scholar] [CrossRef]
- Signoretto, M.; Menegazzo, F.; Contessotto, L.; Pinna, F.; Manzoli, M.; Boccuzzi, F. Au/ZrO2: An efficient and reusable catalyst for the oxidative esterification of renewable furfural. Appl. Catal. B Environ. 2013, 129, 287–293. [Google Scholar] [CrossRef]
- Menegazzo, F.; Fantinel, T.; Signoretto, M.; Pinna, F.; Manzoli, M. On the process for furfural and HMF oxidative esterification over Au/ZrO2. J. Catal. 2014, 319, 61–70. [Google Scholar] [CrossRef]
- Pinna, F.; Olivo, A.; Trevisan, V.; Menegazzo, F.; Signoretto, M.; Manzoli, M.; Boccuzzi, F. The effects of gold nanosize for the exploitation of furfural by selective oxidation. Catal. Today 2013, 203, 196–201. [Google Scholar] [CrossRef]
- Manzoli, M.; Menegazzo, F.; Signoretto, M.; Cruciani, G.; Pinna, F. Effects of synthetic parameters on the catalytic performance of Au/CeO2 for furfural oxidative esterification. J. Catal. 2015, 330, 465–473. [Google Scholar] [CrossRef]
- Daniel, M.-C.; Astruc, D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jiang, X.; Dong, S.; Wang, E. One-Step Synthesis and Size Control of Dendrimer-Protected Gold Nanoparticles: A Heat-Treatment-Based Strategy. Macromol. Rapid Commun. 2003, 24, 1024–1028. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, T.; Zhang, L.; Wang, Q.; Zhang, R.; Ding, B. Morphological transition of gold nanostructures induced by continuous ultraviolet irradiation. Nanotechnology 2006, 17, 5639–5643. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Mahima, S.; Kakade, B.A.; Pasricha, R.; Mandale, A.B.; Vijayamohanan, K. Solvent-Assisted One-Pot Synthesis and Self-Assembly of 4-Aminothiophenol-Capped Gold Nanoparticles. J. Phys. Chem. B 2004, 108, 13280–13286. [Google Scholar] [CrossRef]
- Roy, P.; Dhara, K.; Manassero, M.; Banerjee, P. A new organic compound for the synthesis of gold nanoparticles. Polyhedron 2008, 27, 3085–3090. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, G.S.; Lee, S.Y. Effects of PVP and KCl concentrations on the synthesis of gold nanoparticles using a solution plasma processing. Mater. Lett. 2008, 62, 4354–4356. [Google Scholar] [CrossRef]
- Luo, W.; Zhu, C.; Su, S.; Li, D.; He, Y.; Huang, Q.; Fan, C. Self-Catalyzed, Self-Limiting Growth of Glucose Oxidase-Mimicking Gold Nanoparticles. ACS Nano 2010, 4, 7451–7458. [Google Scholar] [CrossRef] [PubMed]
- Megías-Sayago, C.; Santos, J.L.; Ammari, F.; Chenouf, M.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. Influence of gold particle size in Au/C catalysts for base-free oxidation of glucose. Catal. Today. [CrossRef]
- Ishida, T.; Haruta, M. Gold Catalysts: Towards Sustainable Chemistry. Angew. Chem. Int. Ed. 2007, 46, 7154–7156. [Google Scholar] [CrossRef] [PubMed]
- Morawa Eblagon, K.; Pereira, M.F.R.; Figueiredo, J.L. One-pot oxidation of cellobiose to gluconic acid. Unprecedented high selectivity on bifunctional gold catalysts over mesoporous carbon by integrated texture and surface chemistry optimization. Appl. Catal. B Environ. 2016, 184, 381–396. [Google Scholar] [CrossRef]
- An, D.; Ye, A.; Deng, W.; Zhang, Q.; Wang, Y. Selective Conversion of Cellobiose and Cellulose into Gluconic Acid in Water in the Presence of Oxygen, Catalyzed by Polyoxometalate-Supported Gold Nanoparticles. Chem. Eur. J. 2012, 18, 2938–2947. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Examples of carbohydrates transformation to highly valued compounds.
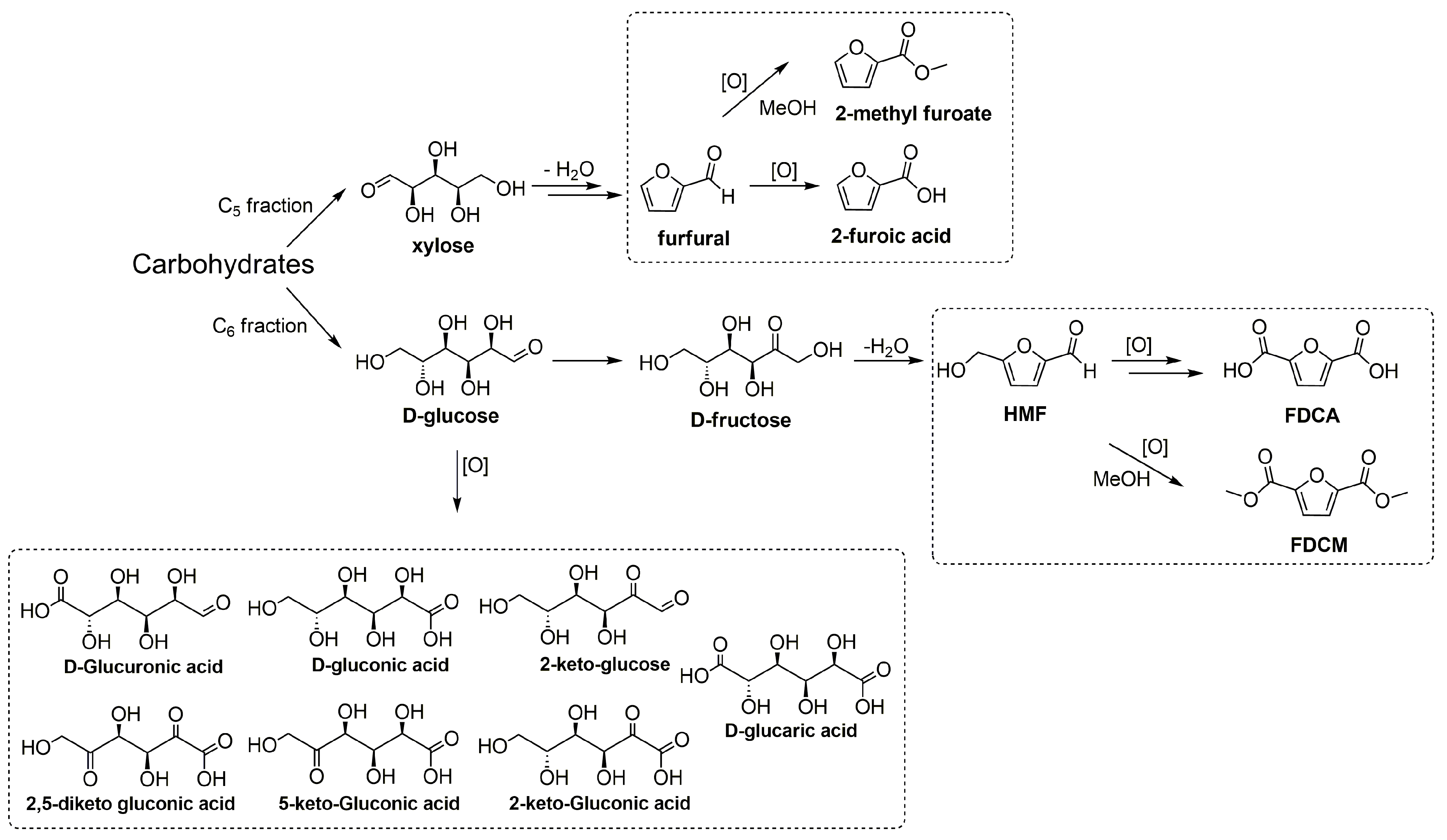
Figure 1.
Comparison of the catalytic activity of Au and different bi- and trimetallic catalysts for glucose oxidation at pH = 7 (Adapted from [23], Copyright 2002, Elsevier Science).
Figure 1.
Comparison of the catalytic activity of Au and different bi- and trimetallic catalysts for glucose oxidation at pH = 7 (Adapted from [23], Copyright 2002, Elsevier Science).

Figure 2.
Activity of Au nanoparticles (NPs) supported on carbon in glucose oxidation at different pHs and temperatures (Reprinted with permission from [36], Copyright 2004, Elsevier Inc.).
Figure 2.
Activity of Au nanoparticles (NPs) supported on carbon in glucose oxidation at different pHs and temperatures (Reprinted with permission from [36], Copyright 2004, Elsevier Inc.).
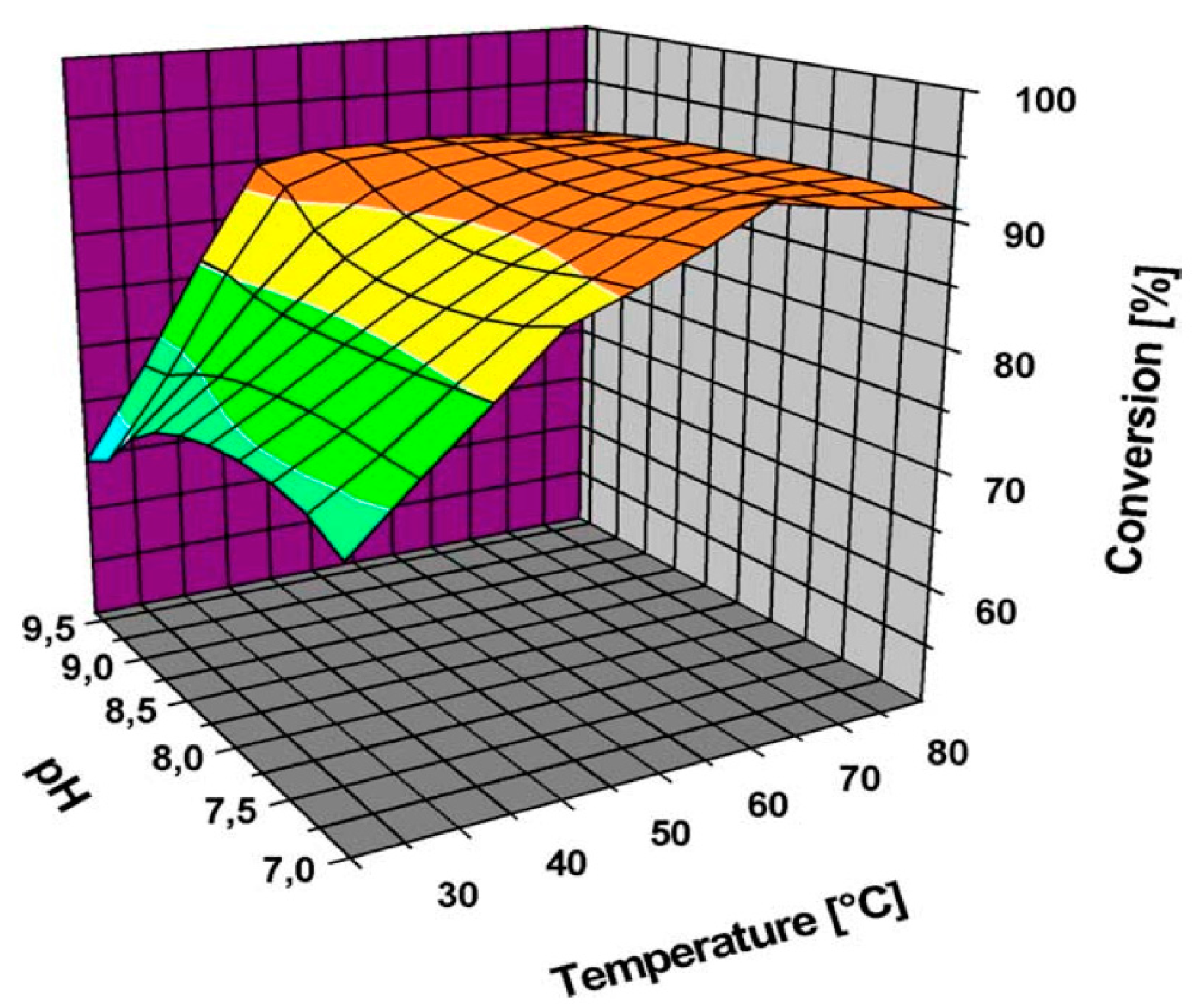
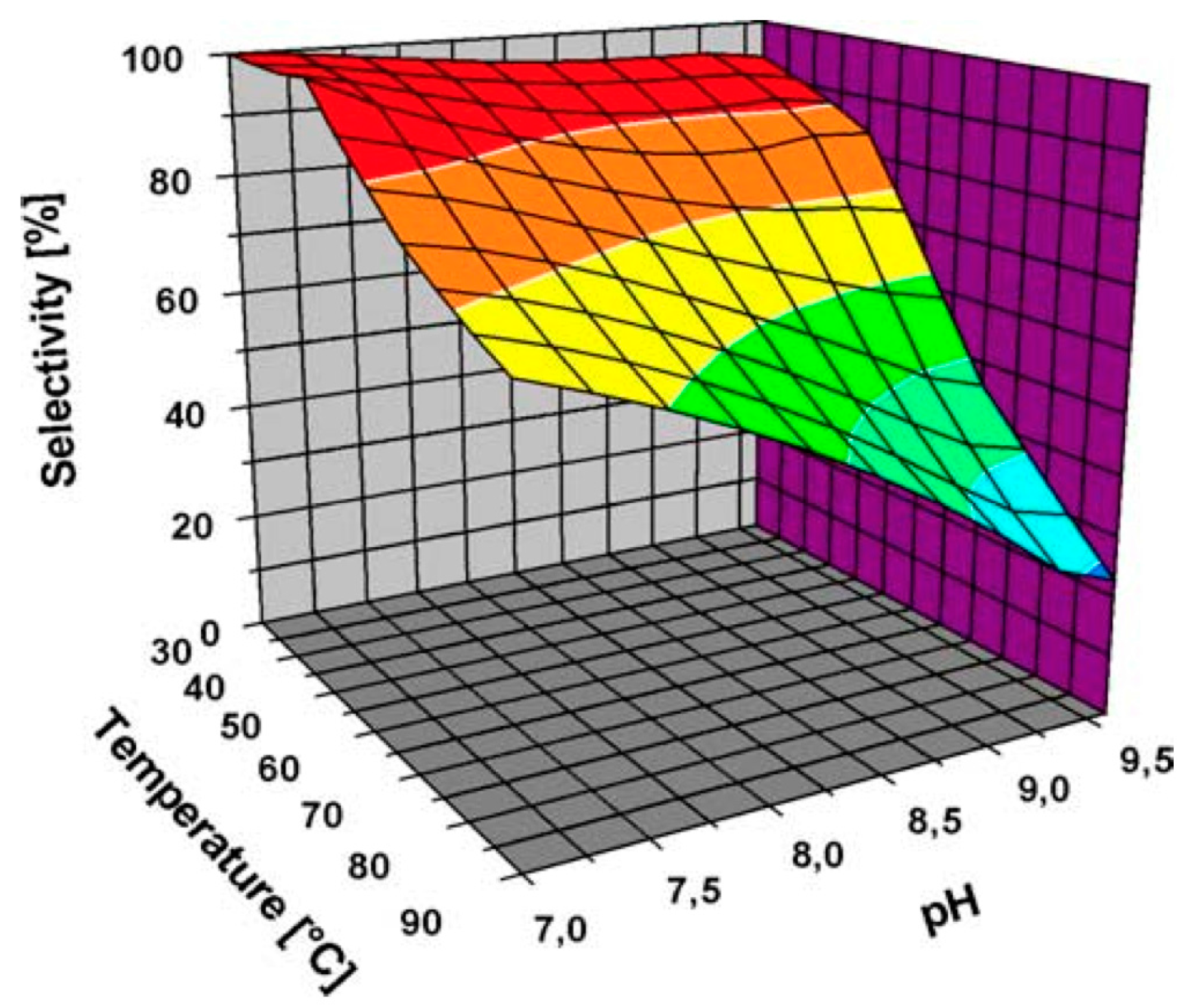
Figure 3.
Selectivity of Au NPs supported on carbon towards gluconic acid at different pH (Reprinted with permission from [36], Copyright 2004, Elsevier Inc.).
Figure 3.
Selectivity of Au NPs supported on carbon towards gluconic acid at different pH (Reprinted with permission from [36], Copyright 2004, Elsevier Inc.).
Scheme 2.
Major products from the oxidation of 5-hydroxymethylfurfural (HMF).
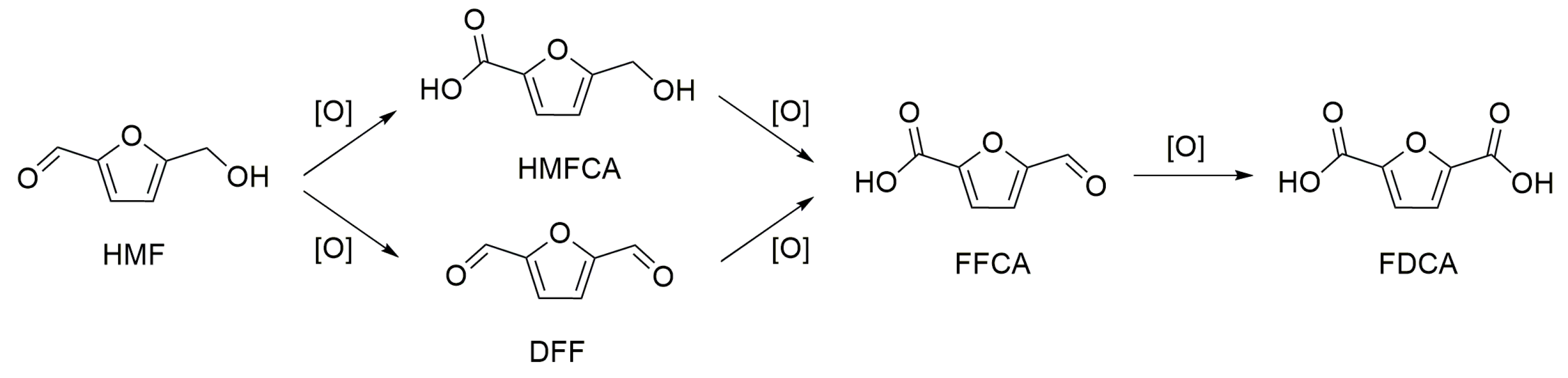
Figure 4.
Comparison of the catalytic activity of Au and AuPd catalysts for the aerobic base-free oxidation of HMF. Conversion (blue) and selectivity towards FDCA (orange). (a) HMF (1 mmol), H2O (6 mL), HMF/metal = 40 (mol/mol), under O2 flow (50 mL min−1), 368 K, 7 h. 1.92 wt % Au/HT. 2 wt % Au was used with different supports. (Adapted from [68], Copyright 2011, RSC Publishing); (b) HMF, 0.50 mmol; HMF/(Au + Pd) (molar ratio), 100/1; H2O, 20 mL; O2, 0.5 MPa; temperature, 373 K; reaction time, 12 h. (Adapted from [70], Copyright 2014, American Chemical Society). CNF: carbon nanofiber; CNT: carbon nanotube; GO: graphene oxide; HT: hydrotalcite; AC: activated carbon.
Figure 4.
Comparison of the catalytic activity of Au and AuPd catalysts for the aerobic base-free oxidation of HMF. Conversion (blue) and selectivity towards FDCA (orange). (a) HMF (1 mmol), H2O (6 mL), HMF/metal = 40 (mol/mol), under O2 flow (50 mL min−1), 368 K, 7 h. 1.92 wt % Au/HT. 2 wt % Au was used with different supports. (Adapted from [68], Copyright 2011, RSC Publishing); (b) HMF, 0.50 mmol; HMF/(Au + Pd) (molar ratio), 100/1; H2O, 20 mL; O2, 0.5 MPa; temperature, 373 K; reaction time, 12 h. (Adapted from [70], Copyright 2014, American Chemical Society). CNF: carbon nanofiber; CNT: carbon nanotube; GO: graphene oxide; HT: hydrotalcite; AC: activated carbon.
Scheme 3.
Proposed mechanism for the oxidative esterification of HMF and furfural. (Adapted from [56] Copyright 2004, Willey and [58], Copyright 2015, RSC Publishing).

Figure 5.
Comparison of the catalytic activity of Au-based catalysts for the aerobic base-free oxidative esterification of HMF. (a) HMF 0.2 mL (2 mmol), methanol (20 mL), HMF/metal = 300 (mol/mol), O2 (1.0 MPa), 403 K, reaction time 24 h. a. reaction time 5 h. (Adapted from [56], Copyright 2004, Willey).
Figure 5.
Comparison of the catalytic activity of Au-based catalysts for the aerobic base-free oxidative esterification of HMF. (a) HMF 0.2 mL (2 mmol), methanol (20 mL), HMF/metal = 300 (mol/mol), O2 (1.0 MPa), 403 K, reaction time 24 h. a. reaction time 5 h. (Adapted from [56], Copyright 2004, Willey).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Catalytic performances observed in base-free glucose oxidation with supported Au NPs.
| Entry | Catalyst | Particle Size (nm) | Preparation Method | Glucose/Au | T a (°C) | Time (h) | Oxidant | Conversion (%) | Gluconic Acid Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1% Au/C | 4.4 | SIM e | 1000 | 100 | 6 | 0.3 MPa O2 | 100 | 100 | [23] |
| 2 | 0.5% Au/nCeO2 f | 1.8 | DP e | 140 | 65 | 2 | 0.23 MPa O2 | 74 | 95 | [44] |
| 3 | 1.4% Au/nCeO2 | 2.5 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 75 | 99 | |
| 4 | 1.6% Au/nZrO2 | 1.7 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 89 | 99 | |
| 5 | 1.3% Au/nTiO2 | 3.0 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 91 | 99 | |
| 6 | 0.6% Au/μCeO2 f | 2.0 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 76 | 96 | |
| 7 | 1.1% Au/μCeO2 | 2.9 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 77 | 98 | |
| 8 | 0.8% Au/μZrO2 | 2.1 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 65 | 98 | |
| 9 | 1.2% Au/μTiO2 | 2.0 | DP | 140 | 65 | 2 | 0.23 MPa O2 | 89 | 98 | |
| 10 | 0.5% Au/MgO | SIM | 1600 | 60 | 24 | air b | 57 | 100 | [6] | |
| 11 | 0.5% Au–Pd/MgO | 6.5 | SIM | 1600 | 60 | 24 | air b | 62 | 100 | |
| 12 | 1% Au/CMK-3 c | 3.0 | See Ref. | 1000 | 110 | 2 | 0.3 MPa O2 | 92 | 88 | [43] |
| 13 | 1% Au/SBA-15 | 3.6 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 67 | 92 | |
| 14 | 1% Au/CNTs | 2.9 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 62 | 83 | |
| 15 | 1% Au/graphene | 3.2 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 56 | 74 | |
| 16 | 1% Au/graphite | 3.7 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 55 | 84 | |
| 17 | 1% Au/AC g | 3.5 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 21 | 91 | |
| 18 | 1% Au/ZrO2 | 2.5 | See Ref. | 100 | 110 | 2 | 0.3 MPa O2 | 13 | 92 | |
| 19 d | 2.3% Au/MgAl2O4 | 3.8 | DP | 870 | 120 | 1/6 | 2.2 eq H2O2 | 54 | 93 | [45] |
| 20 d | 1.8% Au/Al2O3 | 2.4 | DP | 1110 | 120 | 1/6 | 2.2 eq H2O2 | 83 | 87 | |
| 21 | 2% Au/Al h | 5.1 ± 1.5 | DAE e | 100 | 120 | 18 | 0.1MPa O2 | 76 | 95 | [46] |
| 22 | 2% Au/Ce/Al h | 3.4 ± 0.8 | DAE | 100 | 120 | 18 | 0.1 MPa O2 | 78 | 96 | |
| 23 | 2% Au/Ce h | 3.9 ± 1.5 | DAE | 100 | 120 | 18 | 0.1 MPa O2 | 73 | 90 | |
| 24 | 2% Au/Ce25/Zr h | 3.4 ± 0.8 | DAE | 100 | 120 | 18 | 0.1 MPa O2 | 75 | 86 | |
| 25 | 2% Au/Ce50/Zr h | 3.4 ± 0.8 | DAE | 100 | 120 | 18 | 0.1 MPa O2 | 77 | 87 | |
| 26 | 1% Au/TiO2 | 2.1 | SIM | 438 | 160 | 1 | 0.3 MPa O2 | 88 | 95 | [41] |
a Conventional heating; b Atmospheric air; c Au supported on mesoporous carbon; d microwave heating; e SIM: sol immobilization, DP: deposition–precipitation DAE: direct anionic exchange; f n: nanosized, μ: microsized; g AC: activated carbon; h Al: Al2O3, Ce/Al: CeO2(20 wt %)/Al2O3, Ce: CeO2, Ce25/Zr CeO2(25 wt %)/ZrO2, Ce50/Zr: CeO2(50 wt %)/ZrO2.
Table 2.
Oxidation of glucose over bimetallic Au catalysts (Adapted from [49], Copyright 2017, Willey).
Table 2.
Oxidation of glucose over bimetallic Au catalysts (Adapted from [49], Copyright 2017, Willey).
| Catalyst | PostSynthesis Treatment | Glucose Conversion (%) | Yield (%) |
|---|---|---|---|
| 0.5% Au–0.5% Pd/TiO2 | no | 65 | 60 |
| 0.5% Au–0.5% Pd/TiO2 | air | 70 | 63 |
| 0.5% Au–0.5% Pd/TiO2 | reflux a | 48 | 40 |
| 0.5% Au–0.5% Pt/TiO2 | no | 80 | 70 |
| 0.5% Au–0.5% Pt/TiO2 | air | 68 | 62 |
| 0.5% Au–0.5% Pt/TiO2 | reflux | 71 | 65 |
Reaction conditions: glucose 0.20 g, water 20.0 g, catalyst 0.05 g, reaction temperature 160 °C, reaction time 1 h, O2 0.3 MPa. a reflux: the dried catalyst was refluxed with hot water.
Table 3.
Base-free oxidative esterification of furfural with supported Au NPs.
| Entry | Catalyst | Particle Size (nm) | T Calc. (°C) | T a (°C) | Time (h) | Oxidant (MPa) | Conversion (%) | MF Select. (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.2% Au/TiO2 | 4.2 | 300 | 120 | 1.5 | 0.6 | 20 | 92 | [81] |
| 2 | 2.2% Au/CeO2 | 2.0 | 650 | 120 | 1.5 | 0.6 | 66 | 92 | |
| 3 | 1.0% Au/ZrO2 | 2.3 | 650 | 120 | 1.5 | 0.6 | 82 | 66 | |
| 4 | 0.4% Au/ZrO2 | 2–3 | 200 | 120 | 6 | 0.6 | 63 | 10 | [82] |
| 5 | 0.4% Au/ZrO2 | 2–3 | 400 | 120 | 6 | 0.6 | 99 | 95 | |
| 6 | 1.0% Au/TiO2 § | 2–3 | 250 | 120 | 6 | 0.6 | 73 | 31 | |
| 7 | 1.5% Au/ZrO2 | 1.8 | 150 | 120 | 3 | 0.6 | 99 | 94 | [83] |
| 8 | 1.5% Au/ZrO2 | 1.9 | 300 | 120 | 3 | 0.6 | 98 | 94 | |
| 9 | 1.5% Au/ZrO2 | 2.7 | 500 | 120 | 3 | 0.6 | 100 | 98 | |
| 10 | 1.5% Au/ZrO2 | 4.9 | 600 | 120 | 3 | 0.6 | 57 | 76 | |
| 11 | 1.2% Au/ZrO2 | - | 400 | 130 | 1.5 | 0.3 | 90 | 99 | [84] |
| 12 | 1.5% Au/TiO2 WGC | - | - | 130 | 1.5 | 0.3 | 63 | 62 | |
| 13 | 1.5% Au/ZrO2 | 1.7 | 180 | 120 | 1.5 | 0.6 | 100 | 100 | [85] |
| 14 | 1.5% Au/ZrO2 | 5.0 | 600 | 120 | 3.0 | 0.6 | 40 | 70 | |
| 15 | 1.5% Au/TiO2 WGC | 3.8 | 400 | 120 | 3.0 | 0.6 | 90 | 65 | |
| 16 | 2.1% Au/CeO2 (90 °C) | 3.1 | 300 | 120 | 1.5 | 0.6 | 29 | 100 | [86] |
| 17 | 1.3% Au/CeO2 (110 °C) | 7.7 | 300 | 120 | 1.5 | 0.6 | 29 | 100 | |
| 18 | 1.4% Au/CeO2 (300 °C) | 7.7 | 300 | 120 | 1.5 | 0.6 | 54 | 100 | |
| 19 | 2.1% Au/CeO2 (90 °C) | 3.2 | 500 | 120 | 1.5 | 0.6 | 6 | 100 | |
| 20 | 1.3% Au/CeO2 (110 °C) | 7.9 | 500 | 120 | 1.5 | 0.6 | 28 | 100 | |
| 21 | 1.5% Au/CeO2 (300 °C) | 8.4 | 500 | 120 | 1.5 | 0.6 | 74 | 100 |
a Reaction conditions: 100 mg of the catalyst, 300 μL of furfural (3.62 mmol) and 150 mL of methanol. Furfural/Au ratio = 300–1800 (2.2–0.4% Au, respectively), § AUROlite. WGC: World Gold Council. All catalysts were prepared by the deposition–precipitation (DP) method.
Table 4.
Turnover frequencies for glucose oxidation (glucose reacted (mol) per number of surface Au atoms (mol) per second) versus the diameter of the Au particles.
Table 4.
Turnover frequencies for glucose oxidation (glucose reacted (mol) per number of surface Au atoms (mol) per second) versus the diameter of the Au particles.
| Support | Al2O3 a | Al2O3 b | ZrO2 a | ZrO2 b | TiO2 b | CeO2 b | CeO2 a | NPC a | AC a |
|---|---|---|---|---|---|---|---|---|---|
| Au particles size/nm | 2.6 | 4.4 | 3.3 | 3.6 | 2.9 | 4 | 4.1 | 2.4 | 5.4 |
| TOF/molglucosemolsurfaceAu−1s−1 | 41 | 7 | 45 | 28.5 | 24 | 19 | 22 | 3 | 10 |
a,b indicate samples prepared by SG and DP, respectively. Reaction conditions: 5 wt % glucose solution (175 mL), Au catalyst (30 mg), O2 (120 mL/min), 50 °C, pH 9.0. Adapted from [40], Copyright 2008, Willey.
Table 5.
Recyclability of Au-based catalysts in base-free oxidation of glucose.
| Catalyst | First Run | Second Run | Third Run | Fourth Run | Ref. |
|---|---|---|---|---|---|
| AuPd/MgO | 62.0 | 24.4 | 18.7 | - | [6] |
| Au/CMK-3 | 92.0 | - | - | 70.0 | [4] |
| Au/nCeO2 | 88.0 | 69.0 | 62.0 | 56.0 | [44] |
| Au/CeO2 | 88.0 | 53.0 | 40.1 | 31.1 | [44] |
Table 6.
Oxidation of glucose by Au/CeO2 catalyst as adapted from [25], Copyright 2016, Elsevier.
Table 6.
Oxidation of glucose by Au/CeO2 catalyst as adapted from [25], Copyright 2016, Elsevier.
| Temperature (°C) | Time (min) | Conversion (%) | Acid Yield (%) |
|---|---|---|---|
| 70 | 240 | 12 | 12 |
| 110 | 240 | 65 | 53 |
| 130 | 240 | 89 | 48 |
| 150 | 240 | 100 | 23 |
Table 7.
Effect of the reaction temperature on cellobiose oxidation as adapted from [49], Copyright 2017, Willey.
Table 7.
Effect of the reaction temperature on cellobiose oxidation as adapted from [49], Copyright 2017, Willey.
| Temperature (°C) | Time (min) | Conversion (%) | Acid Yield (%) |
|---|---|---|---|
| 130 | 60 | 32 | 7 |
| 140 | 60 | 52 | 21 |
| 150 | 60 | 52 | 28 |
| 160 | 60 | 40 | 15 |
Reaction conditions: Cellobiose 0.20 g, water 20.0 g, catalyst 0.05 g, reaction time 1 h, O2 0.3 MPa.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wojcieszak, R.; Ferraz, C.P.; Sha, J.; Houda, S.; Rossi, L.M.; Paul, S. Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts. Catalysts 2017, 7, 352. https://doi.org/10.3390/catal7110352
AMA Style
Wojcieszak R, Ferraz CP, Sha J, Houda S, Rossi LM, Paul S. Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts. Catalysts. 2017; 7(11):352. https://doi.org/10.3390/catal7110352
Chicago/Turabian StyleWojcieszak, Robert, Camila P. Ferraz, Jin Sha, Sarah Houda, Liane M. Rossi, and Sébastien Paul. 2017. "Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts" Catalysts 7, no. 11: 352. https://doi.org/10.3390/catal7110352
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.