Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems

1
College of Chemistry, Liaoning University, Shenyang 110036, China
2
Materials Science & Engineeing, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
3
School of Chemistry and Chemical Engineering, Anhui University, Hefei 230039, China
4
Discipline of Chemistry, University of Newcastle, Callaghan, Newcastle, NSW 2308, Australia
5
School of Chemical Engineering, University of Adelaide, Adelaide, SA 5005, Australia
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Catalysts 2017, 7(12), 373; https://doi.org/10.3390/catal7120373
Submission received: 10 October 2017
/
Revised: 13 November 2017
/
Accepted: 27 November 2017
/
Published: 1 December 2017
(This article belongs to the Special Issue Nanostructured Materials for Applications in Heterogeneous Catalysis)
Abstract
:Global climate change and increasing demands for clean energy have brought intensive interest in the search for proper electrocatalysts in order to reduce carbon dioxide (CO2) to higher value carbon products such as hydrocarbons. Recently, transition-metal-centered molecules or organic frameworks have been reported to show outstanding electrocatalytic activity in the liquid phase. Their d-orbital electrons are believed to be one of the key factors to capture and convert CO2 molecules to value-added low-carbon fuels. In this review, recent advances in electrocatalytic CO2 reduction have been summarized based on the targeted products, ranging from homogeneous reactions to heterogeneous ones. Their advantages and fallbacks have been pointed out and the existing challenges, especially with respect to the practical and industrial application are addressed.
1. Introduction
Carbon dioxide (CO2) is a necessary substance for the growth of all living organisms on the earth and a vital raw material in many industrial processes [1]. Unfortunately, with the rising global population and the increasing usage of fossil fuels as a result of the dramatic development of industry, this has led to an unprecedented CO2 generation, making global warming a pressing issue. On this topic, a vast majority of governments all over the world have expressed their concerns by introducing a carbon tax and increasing their investment in the issue of climate change. Therefore, discovering a new energy for reducing CO2 emission and developing viable protocols for capturing and converting CO2 into useful materials has become critical and urgent with respect to sustainable development.
In order to fulfill the high energy demands of modern society, the conversion and utilization of CO2 [2] seem to be a more attractive and promising solution than merely capturing and geologically sequestrating CO2 [3,4]. Since the 1970s, many plausible strategies have proved that CO2 can be converted by chemical methods [5], by photocatalytic and electrocatalytic reduction [6], and by other methodologies [7]. However, after decades of investigation, there are still obstacles that hinder the practical application of transforming CO2 into value-added materials and fuels. Among them, the most urgent barriers confronted are the high cost of capturing CO2 and the high energy requirements for CO2 conversion. These challenges make the catalysts (both photo- and electro-) a feasible and promising cutting-edge topic in terms of energy sustainability and environmental protection.
In recent years, electrocatalytic CO2 reduction approaches have been investigated by many groups in the clean energy community [8]. The high thermodynamic stability of CO2 induces challenges for its reduction; therefore more stable molecules, such as CO, formic acid, formaldehyde, methanol, methane etc., are generally produced through proton-coupled multi-electron steps (Equations (1)–(6), Table 1) [6]. In contrast, single electron reduction of CO2 needs relatively high potential energy due to the large amount of transformation energy between the linear molecule and the bent radical anion (Equation (7), Table 1). In addition, the kinetics of CO2 electroreduction involves a complicated reaction mechanism, typically generating a mixture of the aforementioned products. Even in the presence of electrocatalysts, the reaction rates are still very low. In fact, it is the insufficient catalytic activity, selectivity, and stability of currently used electrocatalysts that restrain the practical usage and technological commercialization of the electrocatalytic CO2 conversion systems. Comparing with the gas phase transformation process, which simplifies product separation and is not bothered by CO2 solubility issues, liquid phase electrocatalytic reduction of CO2 displays advantages of higher productivity and less-chaotic outputs [9].
Transition-metal elements possessing partially filled d orbital electrons are believed to have the capability to facilitate bonding with CO2 in the formation of active intermediates. In 1975, Aresta and Nobile first discovered a crystal structure of CO2 bound to a transition metal complex [10]. In this finding, an η2-bidentate binding mode with significant bending in the CO2 structure, involving the carbon atom and one oxygen atom, was reported, which proved the advantages of transition-metal elements as electrocatalysts to mediate the reduction of CO2. Thus, these elements and their related compounds are most commonly explored as electrocatalysts.
Although many review articles have been published relating to CO2 reduction reactions [8,11,12,13], they mainly focus on electrocatalyst effects. In this review, selected recent advances in developing and applying the electrocatalysts with transition metal centers is presented and discussed. Meanwhile, the demonstration of electrocatalytic CO2 reduction is begun with homogeneous reactions discussing the influence of different ligands, followed by heterogeneous reactions with product categories.
2. Useful Notations
Basically, there are four fundamental parameters to compare and evaluate the performance of electrocatalysts for CO2 reduction. The definitions of them are shown as follows:
(1) Catalytic selectivity (CS).
CS refers to the parts occupied by the intended products from all the reaction products, and this parameter describes the effectiveness of the reaction. From the definition we can tell that a higher CS represents a better focusing on the formation of a given product.
(2) Turnover number (TON) and Turnover frequency (TOF).
TON is the total number of the substrate molecules that a catalyst converts into product molecules, and TOF is the TON in unit time. The parameters are used as a yard stick to measure the efficiency of the catalysts.
(3) Faradaic efficiency (FE).
FE is defined as the percentage of electrons employed to generate a desired product. The FE can be simply calculated by the quotient of moles of electrons consumed in reaction product formation and the total moles of electrons transferred from anode to cathode. This parameter is not only related to the product selectivity, but also refers to the energetic efficiency.
where, α is the number of electrons transferred (e.g., α = 8 for reduction of CO2 to CH4), n is the number of moles of a desired product, F is Faraday’s constant (96,485 C/mol), while Q is the total charge passed.
(4) The Tafel slope.
The Tafel plot is constructed by the logarithm of the current density changing with overpotential, and the Tafel slope is the gradient of the plot. It is noteworthy that a lower Tafel slope value indicates a better performance of electrocatalysts. As for CO2 electroreduction, the Tafel slope value can imply the reaction mechanism.
3. Homogeneous CO2 Reduction
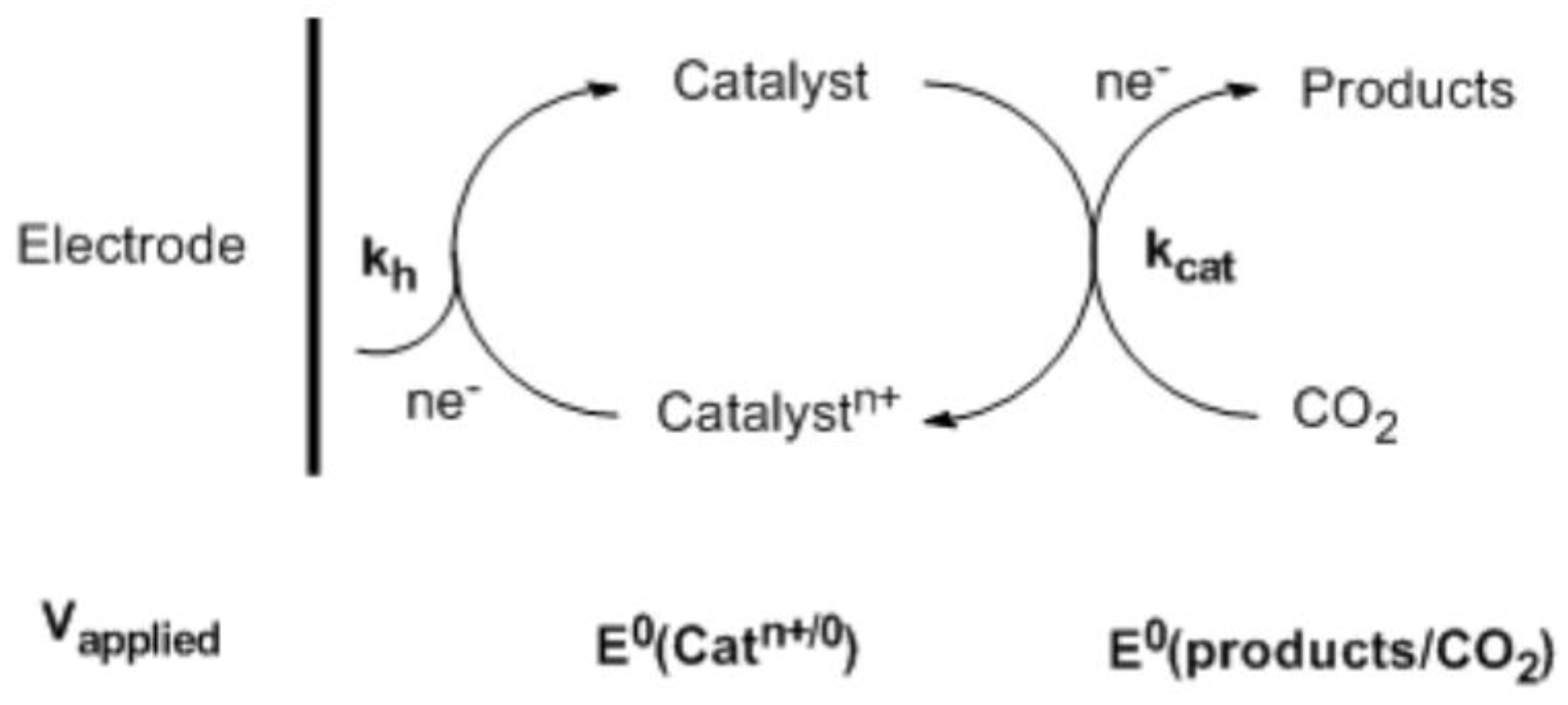
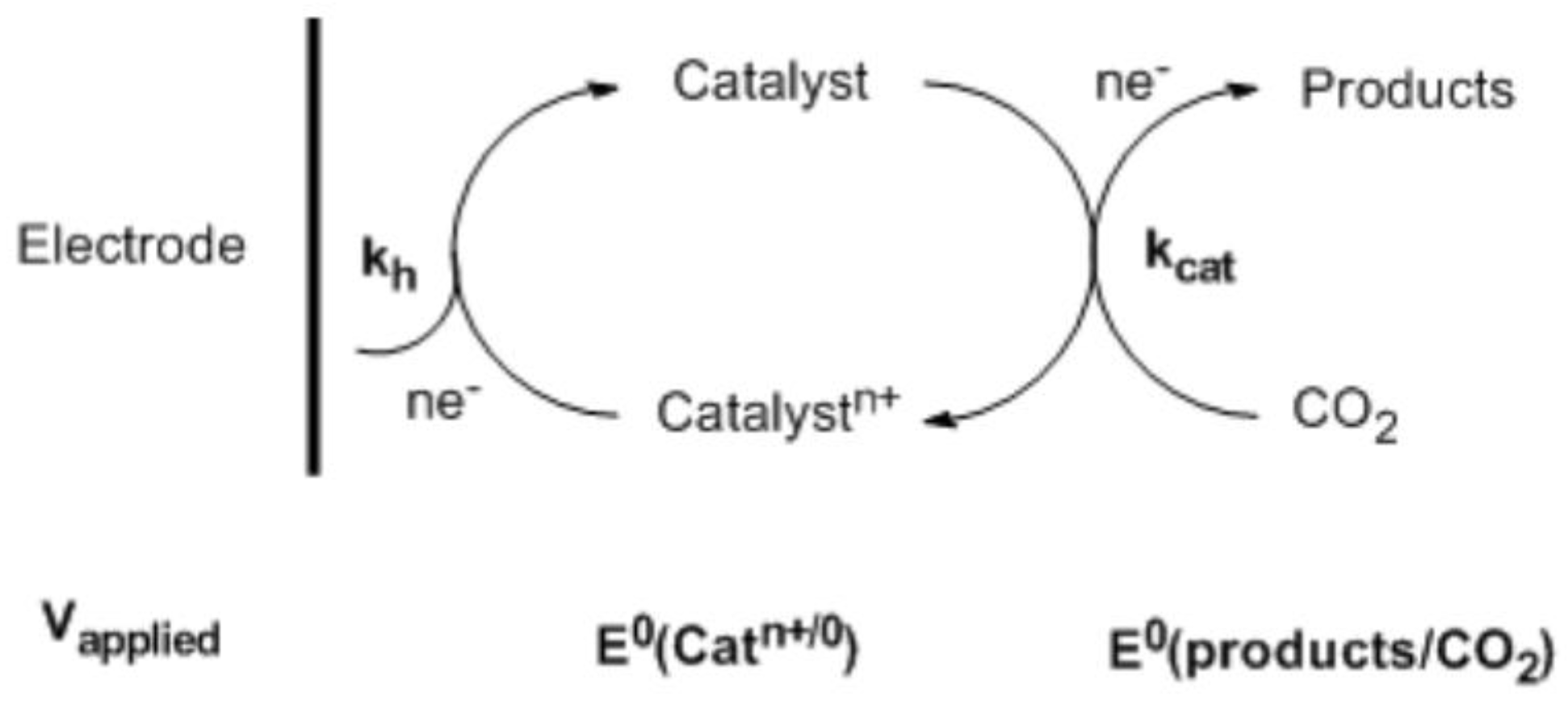
In the homogeneous electroreduction of CO2, the soluble electrocatalysts act as the electron transfer agents to facilitate and accelerate the reaction [14,15]. Ideally, the reaction can be driven near the thermodynamic potential, E° (products/substrates). However, in most cases, the difference between the applied electrode potential, Vapplied, and E° (products/substrates) cannot be eliminated. As such, a significant overvoltage is required in the process of direct electroreduction of CO2. In this situation, in order to increase the conversion efficiency, catalysts that possess formal potential, E° (Catn+/0), well matched to E° (products/substrates) with proper rate constant, kcat, need to be developed. Additionally, at the electrode, the heterogeneous rate constant, kh, must be high for Vapplied near E° (Catn+/0) to further boost the conversion efficiency. A general layout for a homogenous electrocatalytic reduction of CO2 is shown in Scheme 1.
The past decades have witnessed the appearance of many effective homogeneous CO2 reduction electrocatalysts [16]. In the following subsections, recently published seminal works employing these catalysts are summarized and classified into two major categories depending on the ligand type: (1) metal catalysts with macrocyclic ligands [17]; and (2) metal catalysts with polydentade ligands [18]. It should be noted that this section focuses mainly on introducing recent literature about transition metal centered homogeneous catalysts for CO2 electroreduction and thus may omit old literature and other types of CO2 activation reactions.
3.1. Metal Complexes with Macrocyclic Ligands
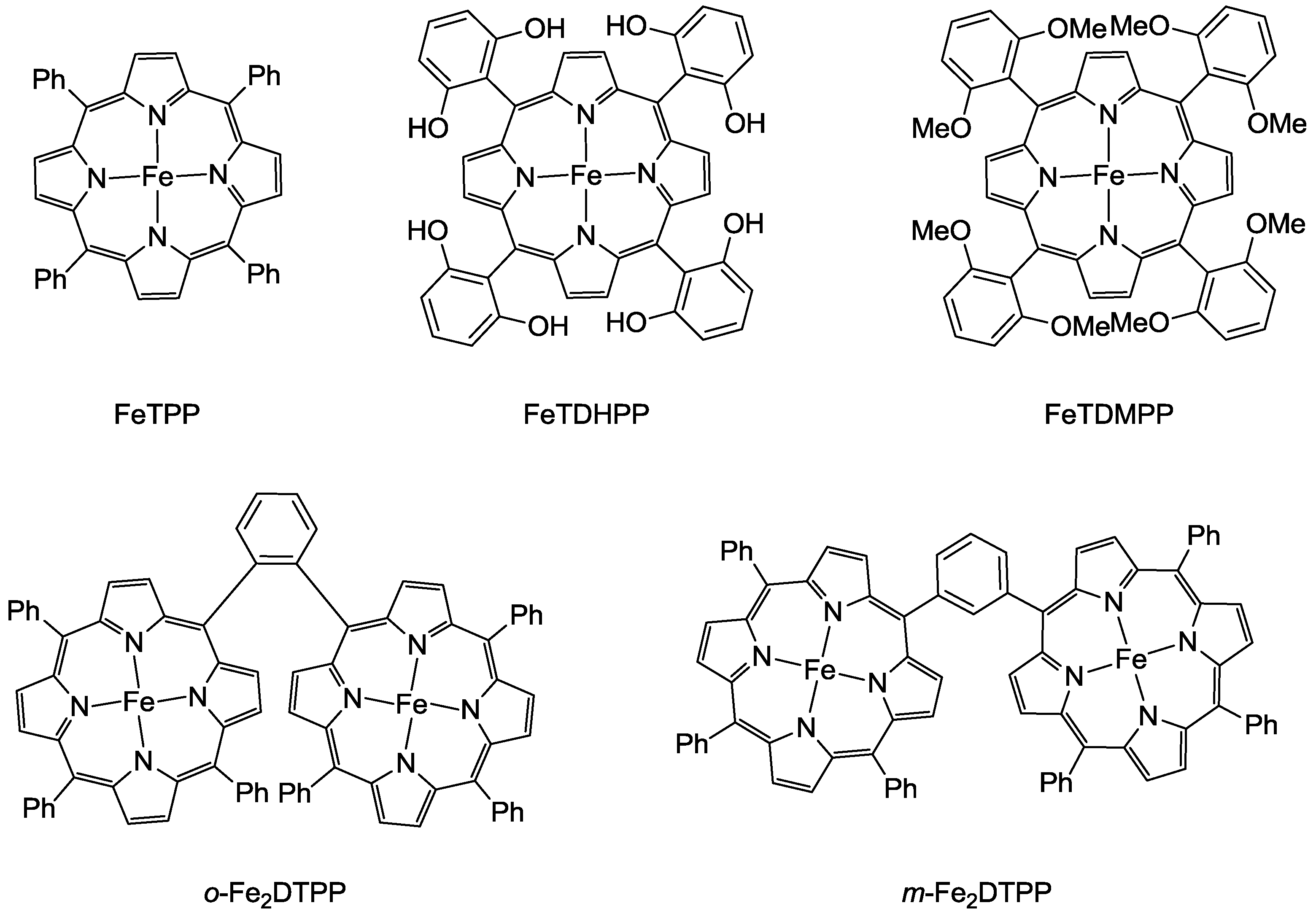
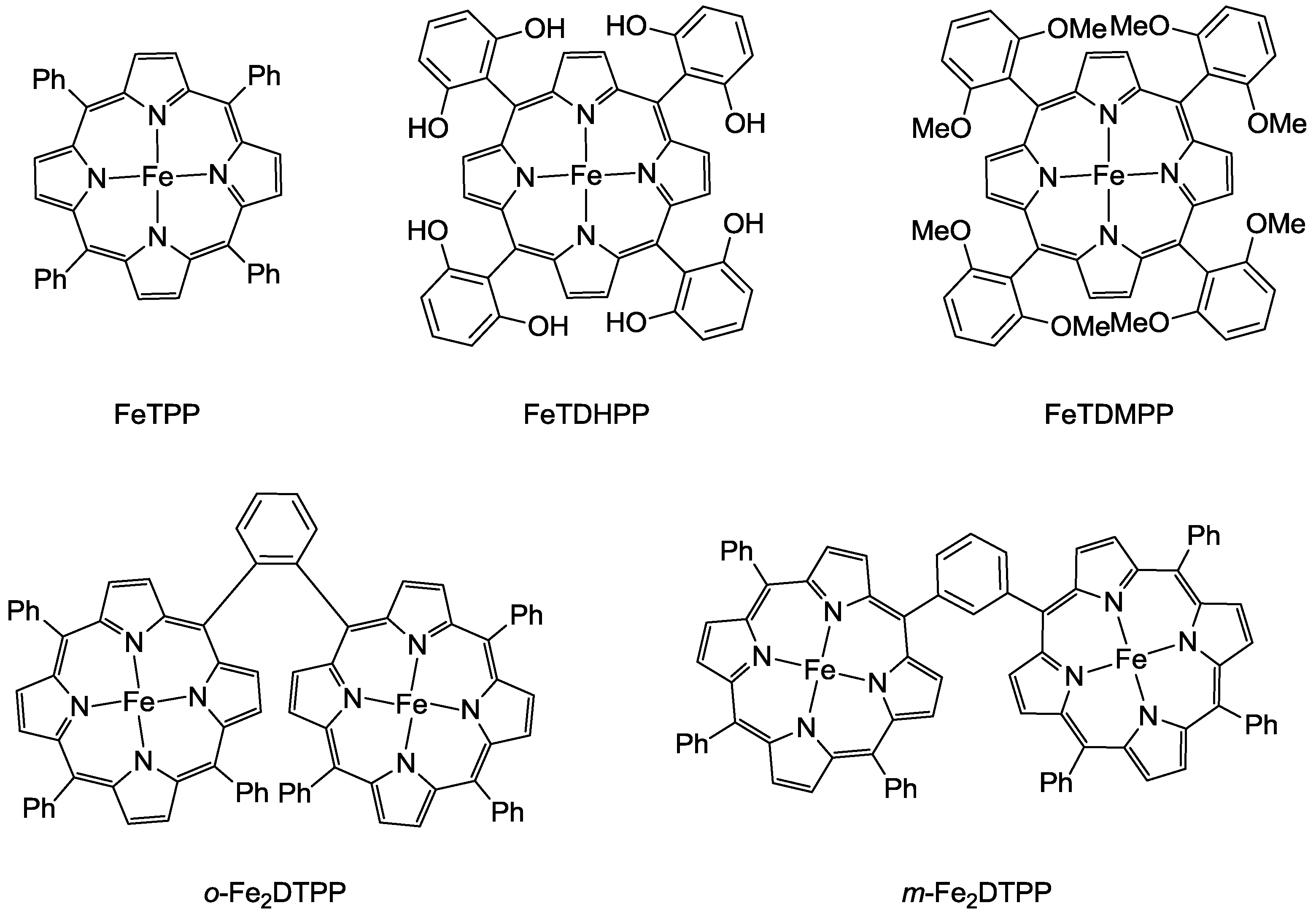
In 2012, Saveant and co-workers reported a superior molecular system that can efficiently reduce CO2 to produce CO by Fe complexes [19]. Introducing phenolic groups in all ortho and ortho’ positions of the phenyl groups for the modification of tetraphenylporphyrin ligand facilitated the catalysis efficiency of the reaction by the electrogenerated iron(0) complex (Scheme 2). At a low overpotential (0.47 V), the catalyst manifested a CO FE above 90% through 50 million turnovers over 4 h electrolysis with no degradation observed. Due to the phenolic hydroxyl substituents, the enhanced activity appeared to be due to the high local concentration of protons. Meanwhile, inspired by the Ni-Fe containing metalloenzyme, a cofacial iron(0) tetraphenyl porphyrin dimer, o-Fe2DTPP (Scheme 2), was also reported to be highly effective in the electroreduction of CO2 [20]. The electrogenerated Fe0(por) species exhibited excellent CO selectivity from the aspects of a high FE of 95% and a TOF of 4300 s−1 at a moderate overpotential of 0.66 V.
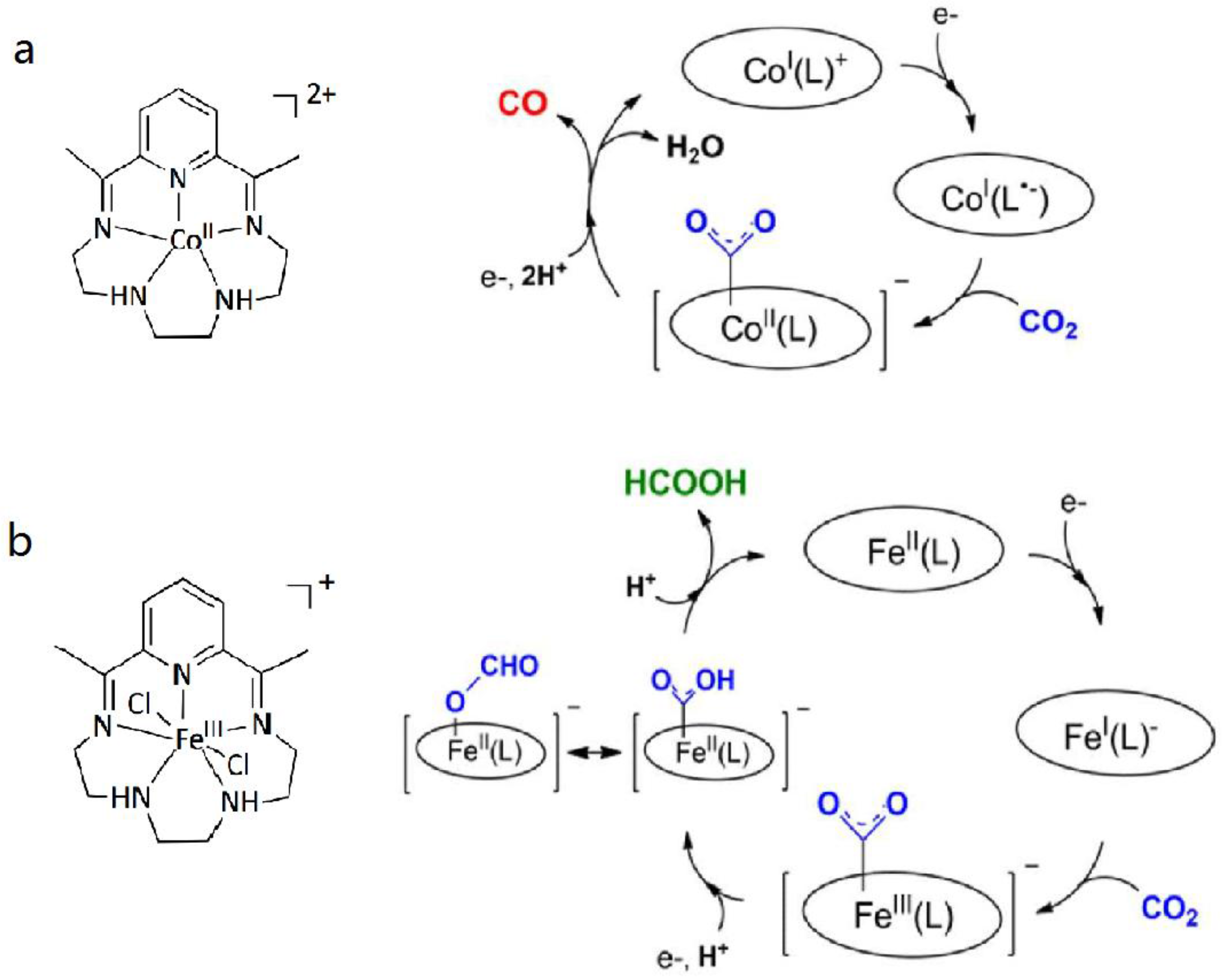
Another pentadendate N5 ligand (2,13-dimethyl-3,6,9,12,18-penta-azabicyclo[12.3.1]octadeca-1 (18),2,12,14,16-pentaene) was used to synthesize two effective electrocatalysts [21]. The synthesized CoII complex selectively catalyzed CO2 to generate CO with a high efficiency, while the FeIII centered electrocatalyst exhibited a high selectivity for the HCOOH product at low overpotential (Scheme 3). The phenomenon that changing the metal from Co to Fe allows the switching of catalysis product over two electrons reduction of CO2 is due to different protonation and reduction pathways. Within the Co-centered complex mediated process, a C-O bond cleavage could be expected which furnishes an OH− and a CO molecule as the final product (Scheme 3a). However, Fe complexes prefer to form a η1-OCOH coordinated intermediates through the catalytic pathway and eventually release a formate molecule (Scheme 3b). Besides, both catalysts showed sustainable catalytic activity over a long period of time and as well as turnover numbers.
3.2. Metal Complexes with Polydentade Ligands
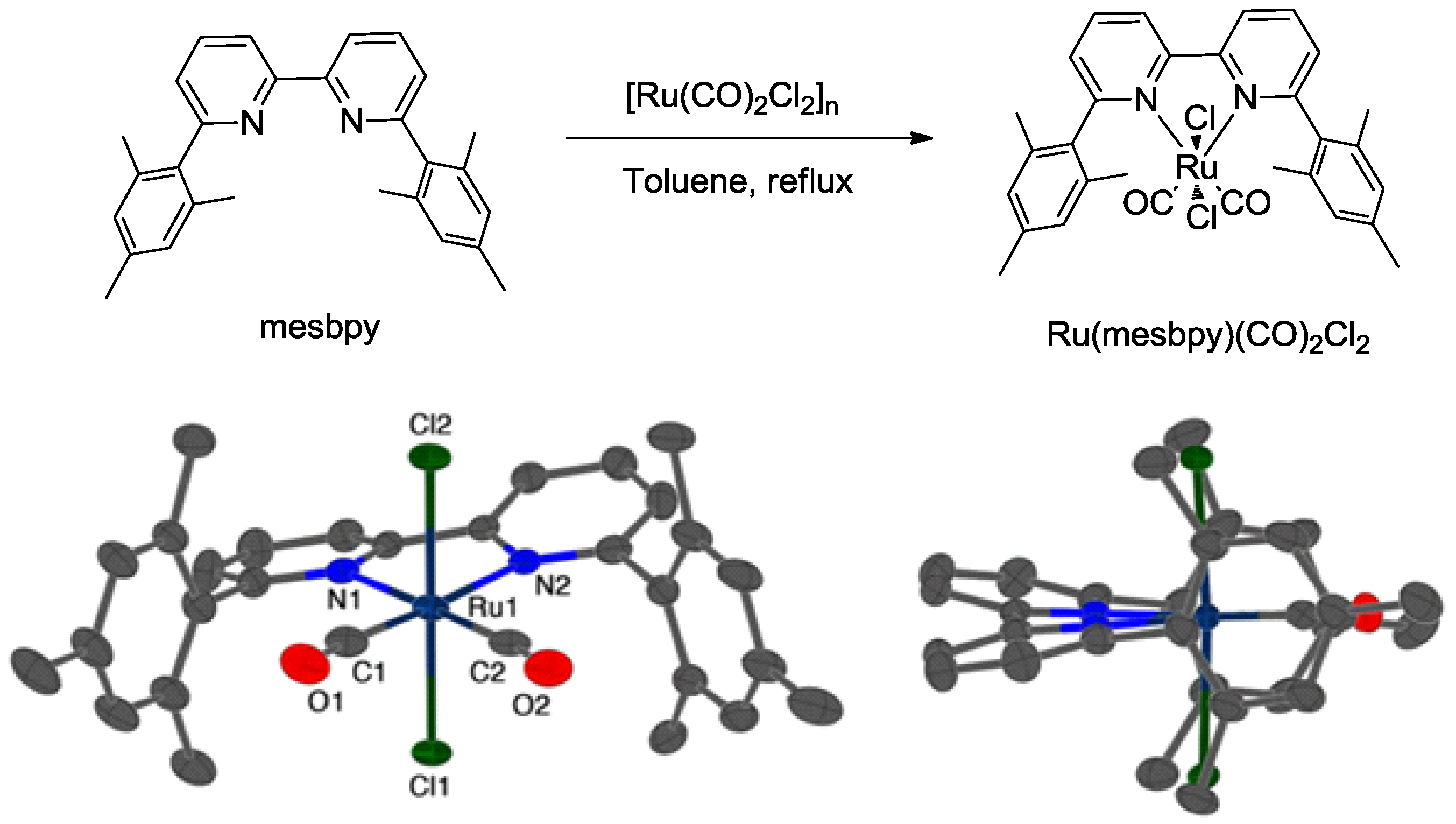
Selectively generating CO product through a Ru-based catalyst was achieved by the use of a bulky bipyridine ligand, 6,6′-dimesityl-2,2′-bipyridine (mesbpy) (Scheme 4) [22]. The Ru complex exhibited a TOF of 1300 s−1 and 95% FE for CO in the presence of Brønsted acids. Based on the mechanistic electrochemical and spectro-electrochemical studies, it was found that the cooperative redox response of the bipyridine ligand and Ru metal center at negative potentials, as well as the inhibition of Ru-Ru bond formation through steric interactions played critical roles in realizing such a remarkable performance.
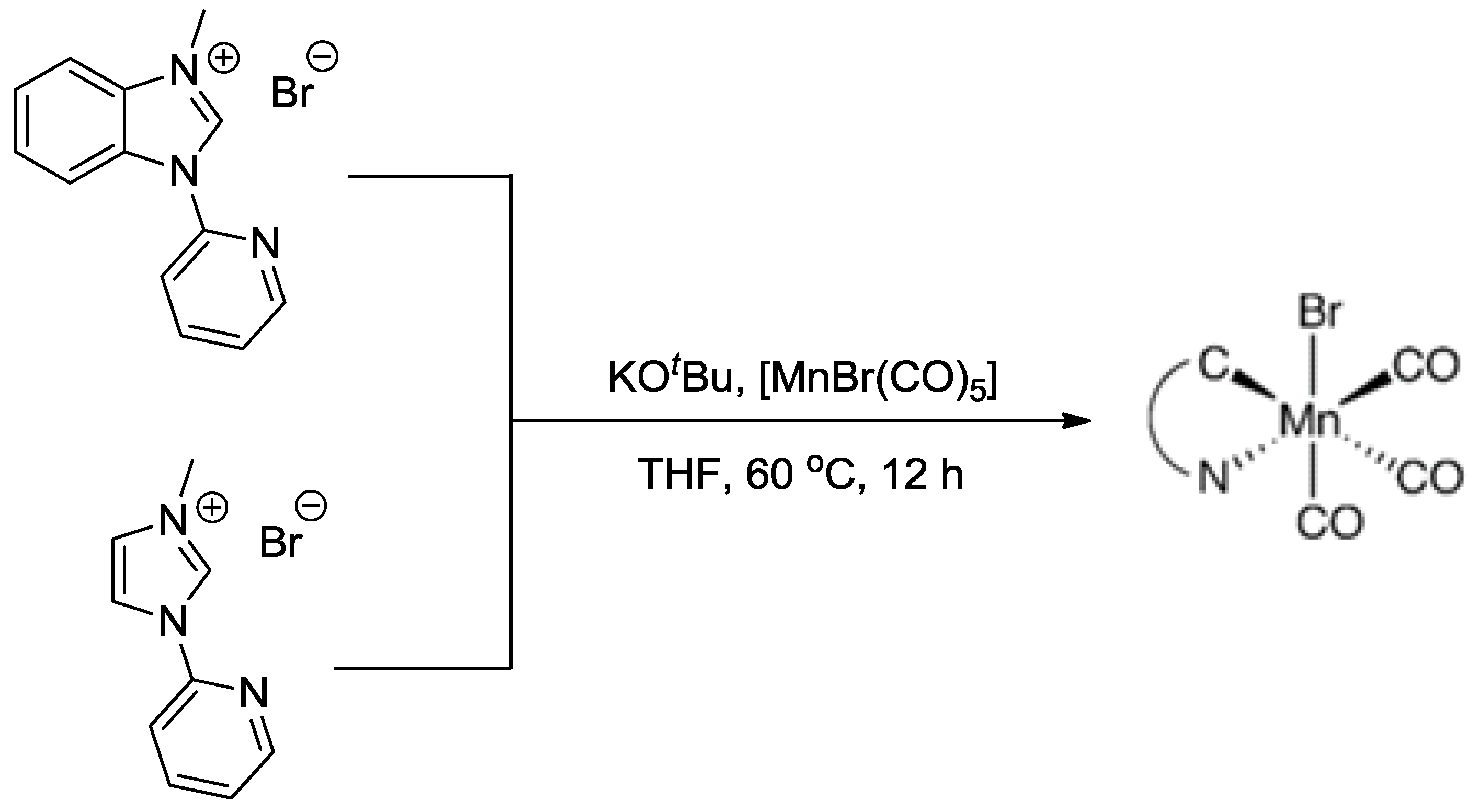
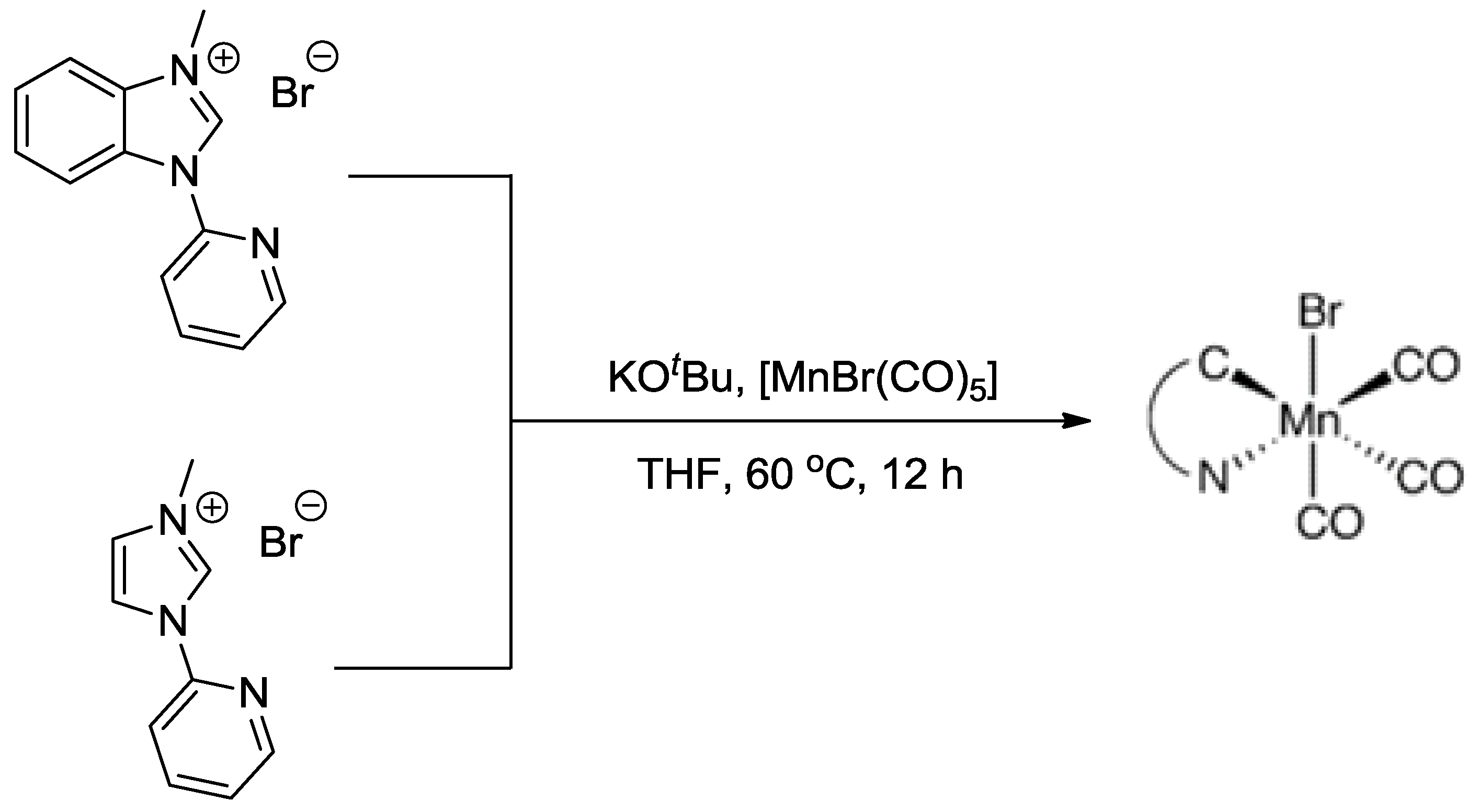
In addition, the manganese tricarbonyl bromide complexes incorporating bidentate ligands, were proved to be very effective and robust as a new group of catalysts for CO2 reduction [23,24]. In 2014, Agarwal et al. reported the electrocatalytic reduction of CO2 using two Mn complexes with N-heterocyclic carbene (NHC) ligands (Scheme 5) [25]. In a wet CH3CN (5% water) solution, the complexes were able to selectively transform CO2 to CO at the potentials of −1.35 V vs. standard calomel electrode (SCE) and −1.46 V vs. SCE, respectively. As compared to [MnBr(bpy)(CO)3], both species exhibited enhancement of catalytic current densities at the voltages. TOF values for converting CO2 to CO with the assistance of two Mn complexes were 0.08 s−1 and 0.07 s−1, respectively. It is noteworthy that throughout the electrolysis, H2 product was not observed, suggesting the excellent selectivity of the developed electrocatalysts, and an average Faradaic efficiency of 34.6% was obtained at the 4 h mark.
4. Heterogeneous Electrocatalysis of CO2 Reduction
Although homogeneous catalysts presented excellent activity and selectivity for electrocatalytic CO2 reduction, the non-recyclable character and high cost still impeded their application in practical industrial utilization. However, in the heterogeneous electroreduction of CO2, the selected electrode itself also performs as the electrocatalyst, which artfully overcomes the afore-mentioned difficulties.
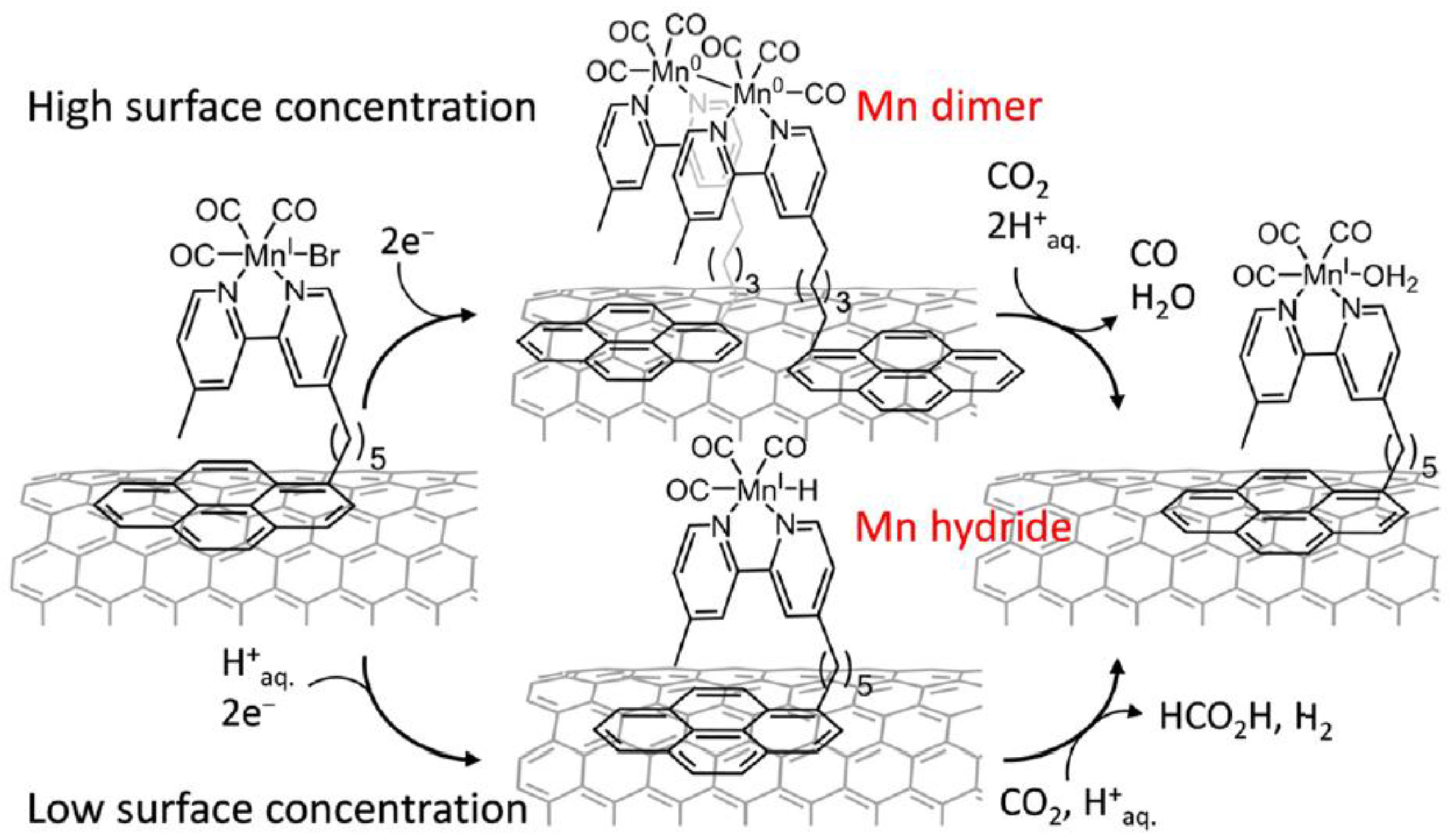
A simple alternative for the transition from homogeneous to heterogeneous system in the arena of electrocatalytic CO2 reduction is to functionalize or modify the electrode with catalytic effective organic metal complexes [26]. Recently, Reisner’s group reported a novel approach by immobilizing Mn(bipyridine) catalysts on a carbon nanotube electrode (Scheme 6) [27]. Anchored by a pyrene unit, the once robust homogeneous CO2 reduction catalyst dramatically transformed to a quasi-heterogeneous system. In such a hybrid system, the CO2 was effectively reduced with a catalytic onset overpotential of 0.36 V and with more than 1000 turnovers at 0.55 V. Selectively, CO as the main product was obtained at high catalyst loading, whereas formate was the dominant product when lower catalytic loading was adopted.
In addition, taking the advantage of a facile synthesis protocol without any further purification, with outstanding efficiency and great potential for large-scale applications, the heterogeneous electrocatalysts yield more promising materials than the homogeneous ones to be applied in real industrial processes. The carbon dioxide molecule is adsorbed onto the conductive materials and subsequent reduction occurs on the surface of the electrode. During the past decades, various transition-metal-centered heterogeneous electrocatalysts have been widely explored for mediating CO2 reduction reactions [28]. Cu, Au, Sn, Hg, Ag, Zn, Pd, etc. have proved to be robust and effective as both metal electrodes and electrocatalysts. Meanwhile, several transition metal oxides and chalcogenides such as TiO2, FeOx, Cu2O, ZnS, MoS2, and VS4, have been reported as versatile electrocatalysts for CO2 reductions as well [29]. Typically, heterogeneous electrocatalysis reduction of CO2 involves three major steps: (i) adsorption of CO2 molecules onto the surface of the electrocatalysts; (ii) electron transfer and/or proton migration; (iii) configuration rearrangement and desorption of products. However, not all the aforementioned electrodes are able to bind the key intermediates generating from the first step tightly. Different electrocatalysts have their own protocols to provide diversity of low-carbon products. Therefore, the following sections summarize recent works categorized by different main products.
4.1. Selective Production of Carbon Monoxide
CO is one of the important molecules applied in industrial and domestic usage. The syngas consisting of CO and H2 can be transferred to liquid hydrocarbons through the famous Fischer-Tropsch synthesis. For the formation of CO, the reaction initiates from the reductive adsorption of CO2 on the catalysts’ surface to construct a COOH* intermediate. After further reduction by another electron coupled to proton transfer, the COOH* intermediate is desorbed from the electrode, providing CO and H2O as the final products. From the reaction pathway, in order to selectively generate CO product, the catalysts should fulfill the following requirements: a strong binding with COOH* and a weak coordinating with the CO molecule. Recently, nanostructured catalysts with well-controlled size, composition and surface characteristics based on different active sites have been proven robust and effective for reducing CO2 to CO.
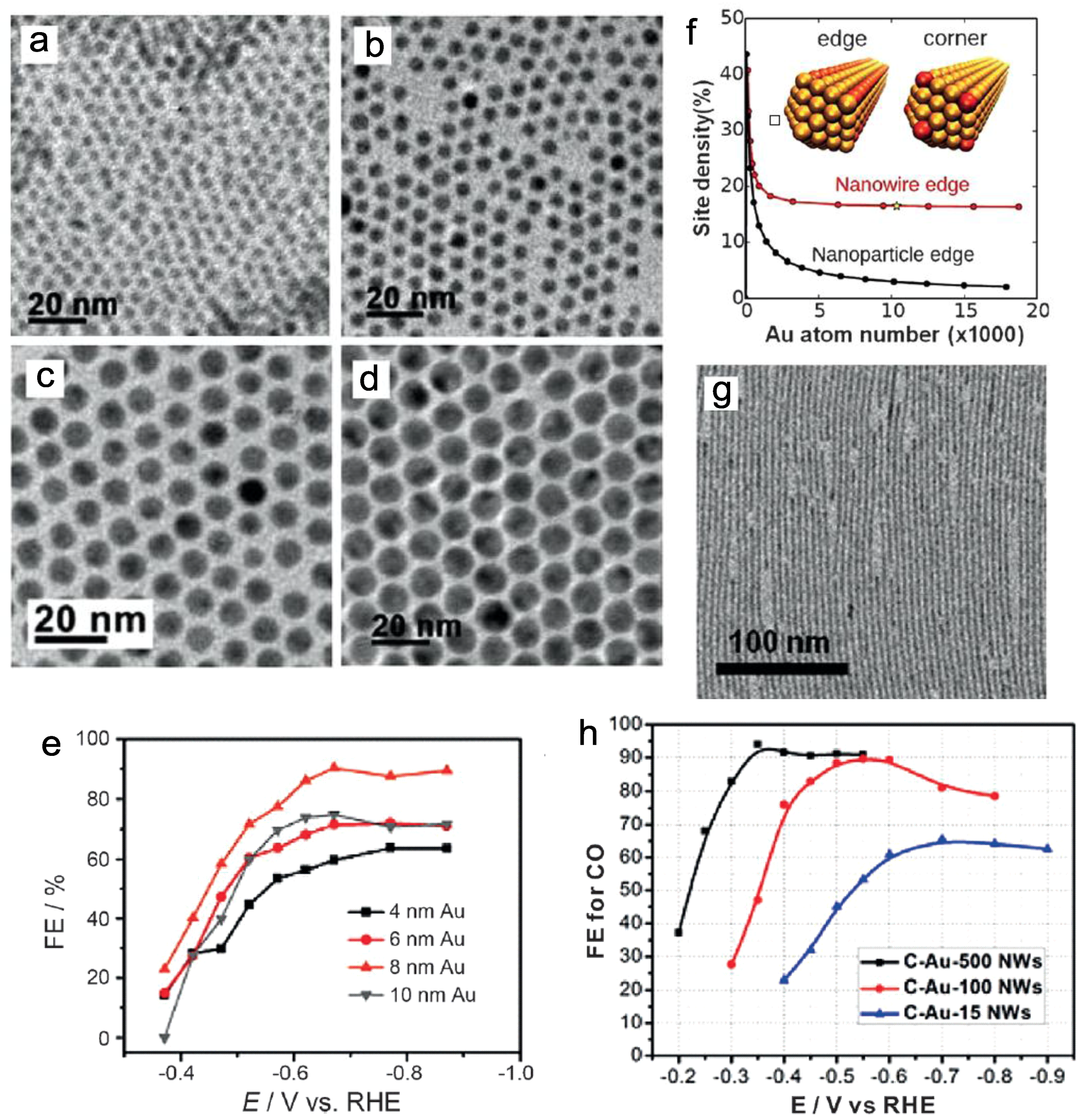
In 2013, Sun and co-workers studied the performance of monodispersed Au nanoparticles (NPs) of various sizes [30]. The edge sites of the NPs are considered to accelerate CO formation, and the 8 nm Au NPs with an optimal ratio of edge sites thus offered the maximum FE up to 90% at −0.67 V vs. reversible hydrogen electrode (RHE) (Figure 1a–e). Meanwhile, similar work was also reported by Strasser’s group with the sizes of Au NPs ranging from 1 nm to 8 nm [31]. As mentioned, the FE of CO declined with decreasing NP size. Both works suggested that 8 nm is the optimal size of Au NPs in CO2 reduction. In the follow up studies, Sun and co-workers designed Au nanowires with high aspect-ratios and conducted electroreduction of CO2 to CO at an onset potential of −0.2 V vs. RHE (Figure 1f,g) [32]. Under the catalysis of 500 nm length Au nanowires, the reduction FE reached 94% at −0.35 V, with a mass activity 1.84 A g−1 (Figure 1h). Furthermore, Nam and co-workers proposed concave Au rhombic dodecahedrons as the electrodes with a high density of atomic steps on the surface, obtaining enhanced efficiency over that of Au film [33]. After long-term electrocatalytic testing, the morphologies of such concave rhombic dodecahedrons were still maintained. In addition, Kanan et al. constructed Au NPs with a high density of grain boundaries, which was proven to be a powerful approach to improve the catalytic activity of metal NPs [34].
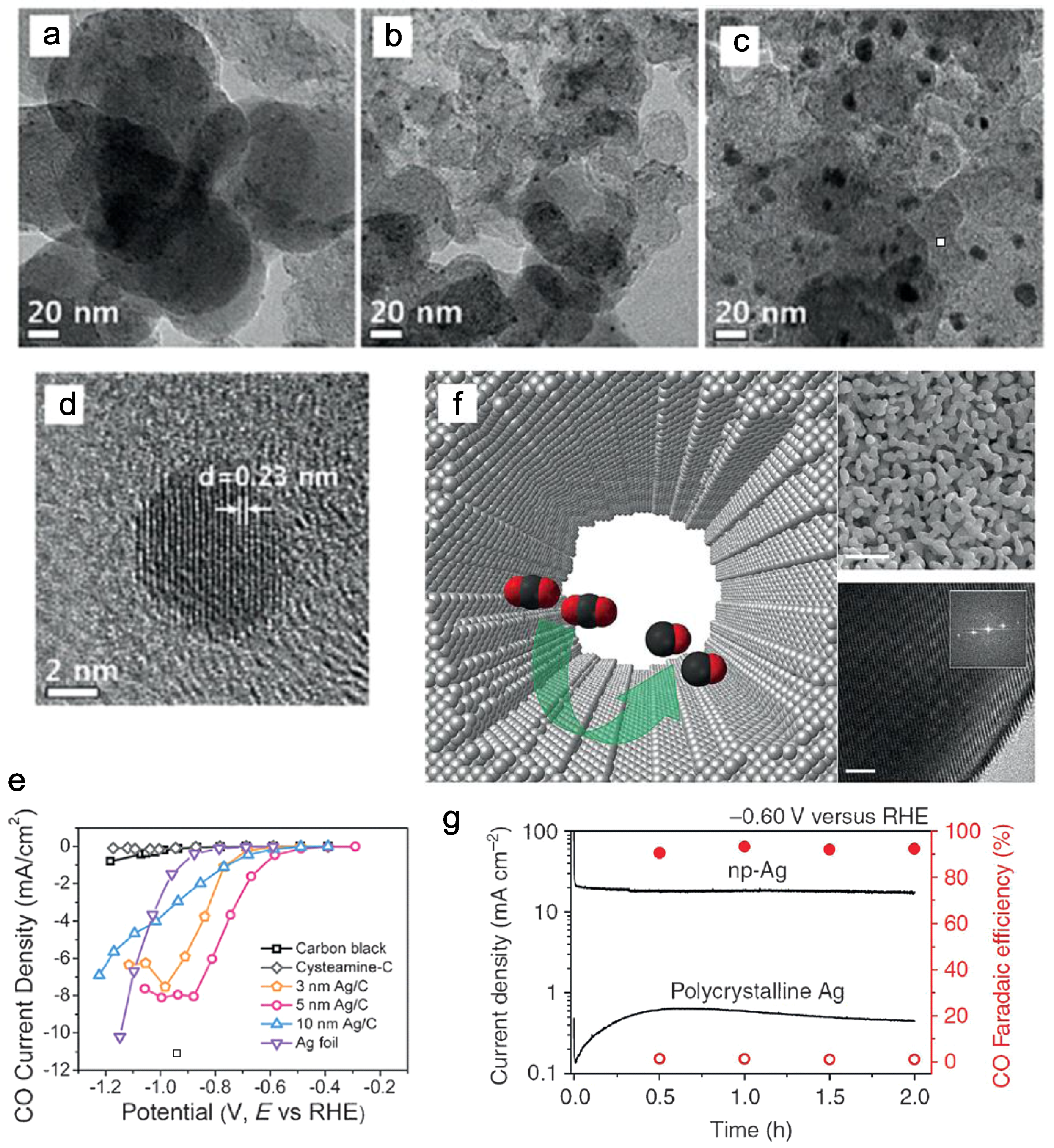
Similarly, relevant studies demonstrated that the optimal size of Ag NPs for CO2 reduction was 5 nm. Hwang and co-workers discovered the optimal choice by comparing three different sizes of easy-synthesized carbon-based Ag NPs (Figure 2a–d) [35]. Comparing to polycrystalline Ag foil, the 5 nm Ag/C catalysts provide four times enhanced CO FE at −0.75 V vs. RHE with the smallest overpotential about 0.63 V (Figure 2e). In Salehi-Khojin’s study, the 5 nm Ag NPs boosted the conversion of CO2 to CO at a 10 times higher conversion rate than the bulk silver electrode in an ionic liquid 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIM-BF4) solution [36]. Moreover, after two-step de-alloying in an aqueous HCl solution, a two-dimensional nanoporous Ag electrocatalyst could be prepared from an Ag-Al precursor (Figure 2f) [37]. The readily made catalyst delivered a nearly 150 times larger surface area and exhibited at least 20 times higher activity in comparison with that of the polycrystalline silver in the electroreduction of CO2 (Figure 2g). Such a remarkable performance was associated with the amazing stability of COOH* intermediates on the highly curved surface, thus overcoming the kinetic barrier to produce CO at a relatively small overpotential.
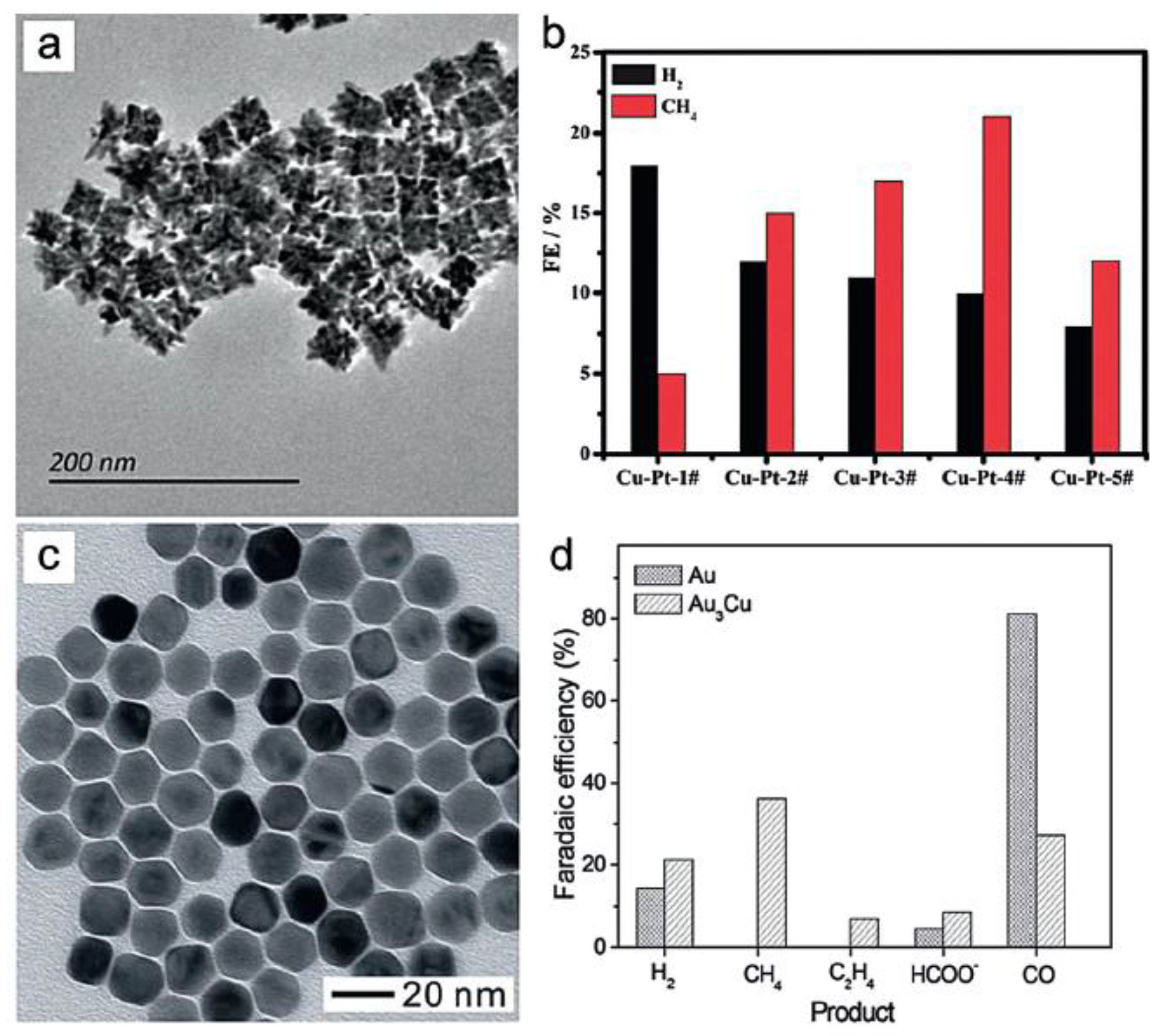
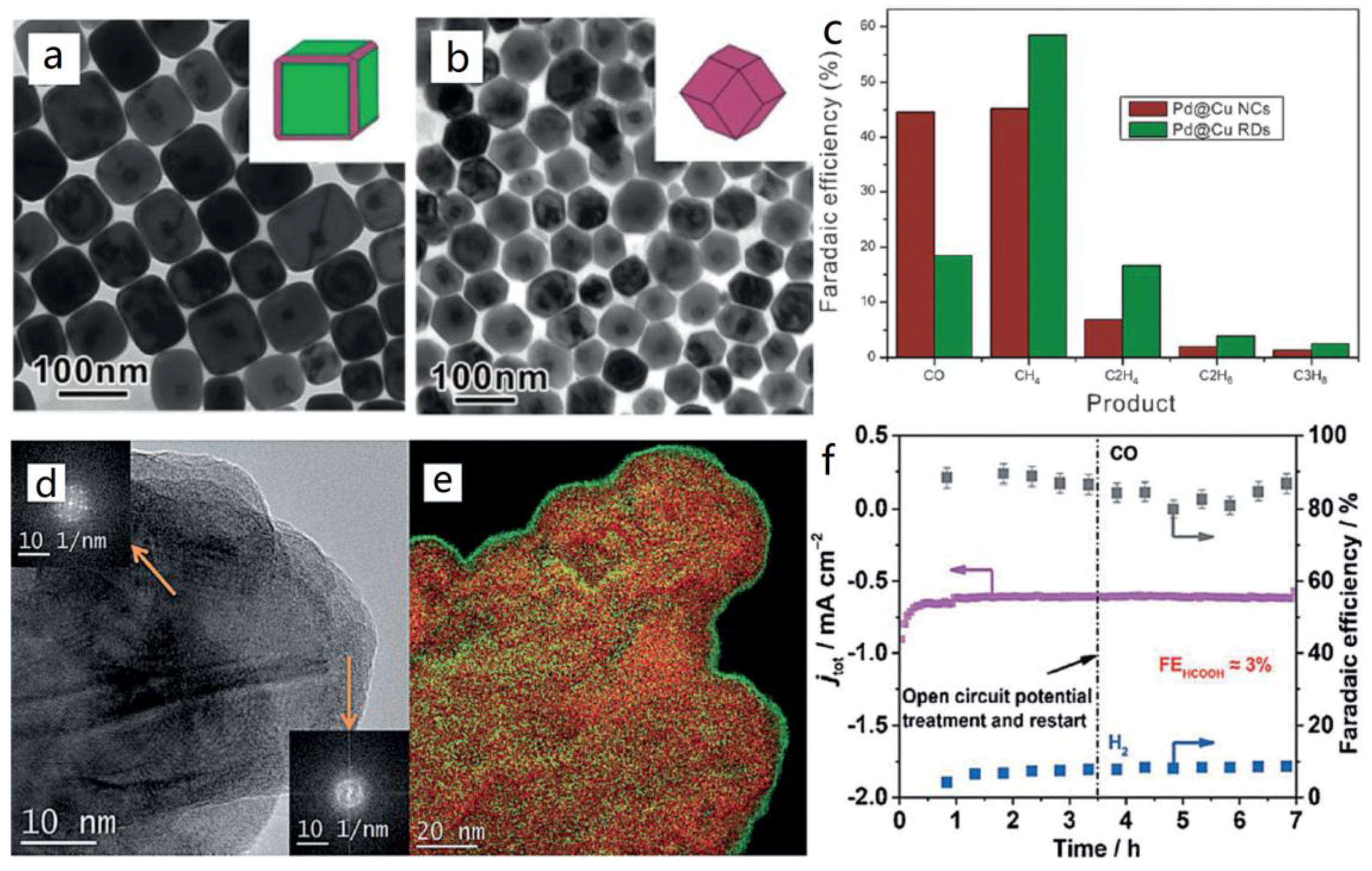
Apart from Au and Ag NPs, small Pd particles also presented good selectivity for CO formation in the CO2 reduction process, with the FE approaching a maximum value of 91.2% at −0.89 V vs. RHE [38]. In addition, the catalytic activities of the Cu NPs increased with the decreasing sizes of the particles, however, not as profound as with the noble metal NPs [39]. Intriguingly, effectiveness can be promoted by changing the morphology. The Yin group prepared high-energy Cu surfaces for the electroreduction of CO2 by an etching process [40]. The rhombic dodecahedra structure with abundant high-energy (110) facets originated from Cu nanocubes with exposed (100) facets (Figure 3a,b). It is noteworthy that the selectivity of specific binding agents is the key factor in determining the etched morphologies of Cu NPs. The formation of Cu rhombic dodecahedra was achieved in N,N-dimethylformamide solvent. Meanwhile, due to its strong capping of (100) facets of Cu, hexapod-shaped NPs with cubic arms formed under the promotion of the etching process on the corners and edges by oleylamine. The current density of the ready-made Cu electrode was nearly three times higher than that of the Cu nanocubes, at −1.4 V vs. RHE. Noticeably, the selectivity toward CH4, C2H4, C2H6, and C3H8 was also higher on Cu(110) facets than on the original Cu(100) facets. These results indicated the enhanced catalytic activity and C1, C2 selectivity of the Cu rhombic dodecahedra catalyst (Figure 3c).
According to recent studies, multi-component catalysts exhibited alternative properties to single-component catalysts. Takanabe and co-workers explored the CO2 electroreduction performance of a Cu-In electrode prepared by electrochemical reduction of the oxide-derived Cu in a two-electrode system (Figure 3d,e). The as-prepared electrode selectivity reduced CO2 to CO product, while suppressing the formation of H2 [41]. After extensive trials, the CO was produced as almost the sole product of CO2 reduction, reaching an FE up to 90% at −0.5 V vs. RHE (Figure 3f). The Tafel slope over 0.12 V dec−1 indicated that the rate-limiting step in the mechanism was the initial single-electron transfer. Density functional theory (DFT) calculation showed the replacement of one Cu by In atom on the four-fold site which disfavored H adsorption by 0.12 eV, while the CO adsorption energy was constant. Also, the In atom enhanced the stability of COOH* by roughly 0.1 eV, thereby reducing the trend to release formate as final product.
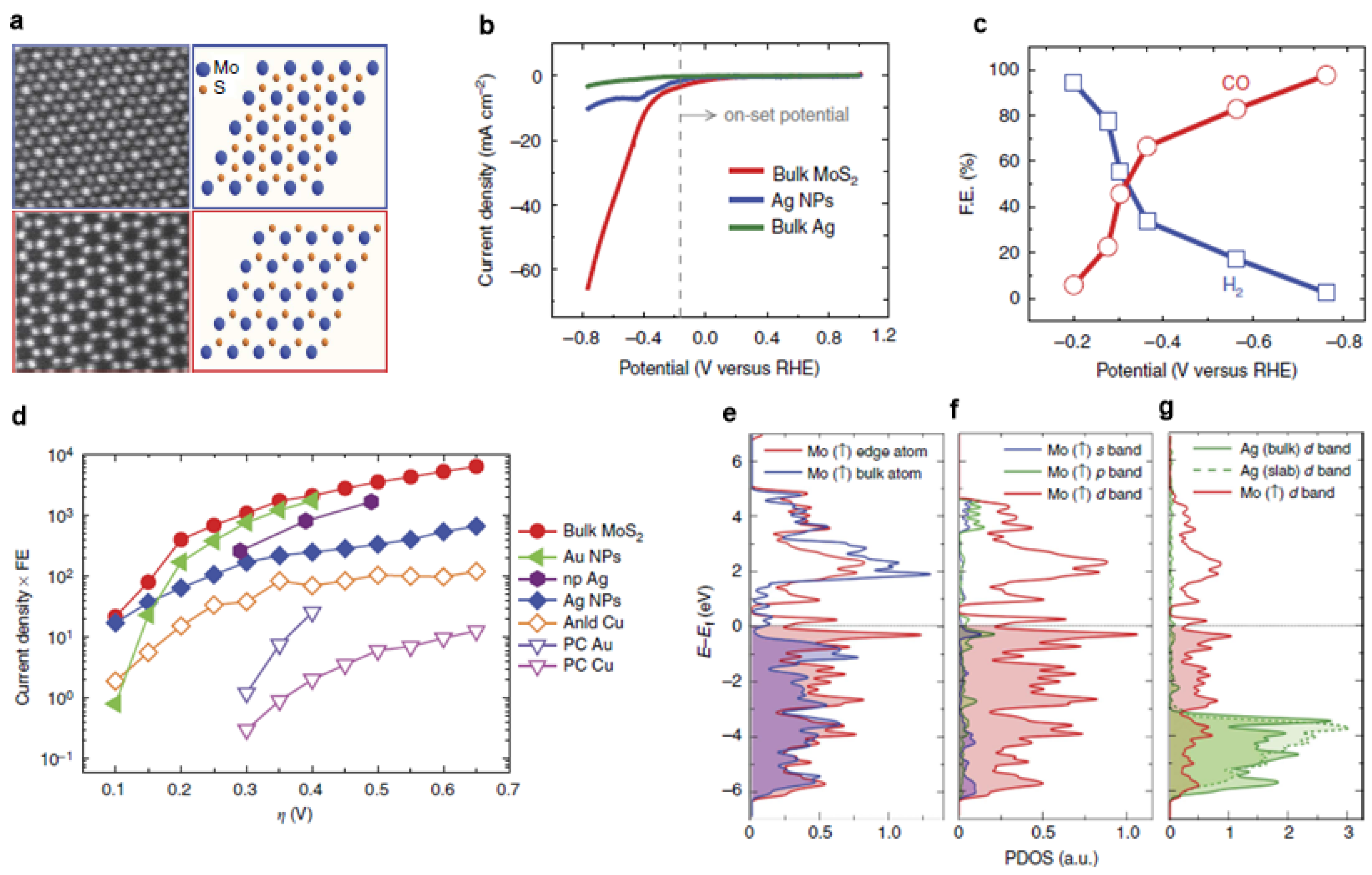
As a costless and promising alternative to replace noble-metal catalysts in hydrogen evolution reaction (HER), MoS2 also exhibited superior CO2 reduction performance (Figure 4a) [42]. Comparing with the noble metals, such a cost-effective substitute selectively reduced CO2 to CO with a high current density and low overpotential (0.054 V) in an ionic liquid (Figure 4b–d). Due to the metallic character and a high d-electron density, the Mo-terminated edges are the main characteristic related to its catalytic performance (Figure 4e–g). A thorough review on this material family was conducted by Jaramillo et al., so this review does not go into any details [43].
4.2. Selective Production of Formate
As one of the most commonly used raw materials in modern industrial processes, the selective production of formate through CO2 electroreduction is one of the facilitated ways to satisfy the supply requirement. Recent attention has been paid to further increase the selectivity and decrease the overpotential through the electrolytic process.
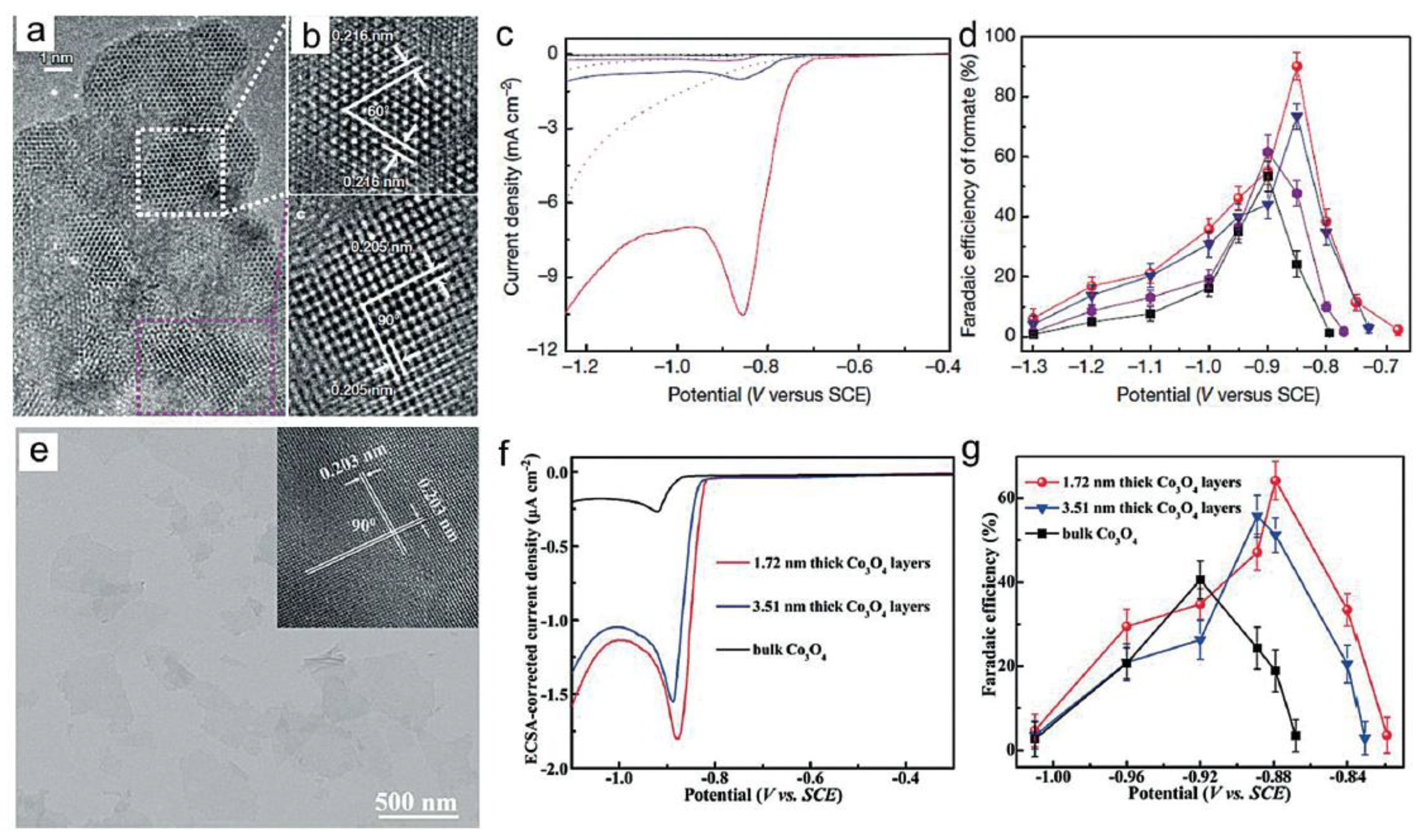
In 2016, ultra-thin Co related nanosheets of 4-atoms-thick were prepared through a ligand-confined growth strategy by Xie et al. (Figure 5a,b) [44]. Compared with bulky samples, the surface Co atoms of the atomically thin layers produced formate during CO2 electroreduction with higher intrinsic activity and selectivity at lower overpotential, 0.24 V (Figure 5c). The partially oxidized nanostructured cobalt electrocatalyst further attained stable current densities of about 100 A/m2 over 40 h, with almost 90% FE for formate at −0.85 V vs. SCE. Gas chromatography quantified the remaining 10% of the charge causing H2 evolution (Figure 5d). Based on mechanistic studies, it was speculated that the declined onset potential from 0.73 V to 0.68 V upon partial oxidation was related to the facilitated rate-determining chemical reaction by the cobalt oxides. Another fast-heating strategy was applied to generate 1.72 nm thick Co3O4 layers, which were ideal models of transition-metal-oxide-based atomic layers for CO2 reduction (Figure 5e) [45]. Abundant active sites and high electrical conductivity of the as-prepared nanosheets exhibited promotion for the electroreduction of CO2. At −0.88 V vs. SCE, the ultrathin Co3O4 nanosheet electrocatalyst had a current density of 6.8 A/m2 with a formate FE of over 60% in 20 h (Figure 5f,g).
Palmore and co-workers investigated the reduction of CO2 on Cu foams with a hierarchical porosity [46]. Both the distribution of products and their FE presented a significant difference from those provided by smooth electropolished Cu electrodes. The highest FE of formate was up to 29% through increasing the thickness of the copper nanofoams which could suppress the HER process. The main reasons for obtaining such results were the high surface roughness, hierarchical porosity, and confinement of the reactive species. In addition to non-precious metals, Pd has the ability to convert CO2 to formate with no overpotential. Unfortunately, within such an activity process, the low reaction rates and deactivation pathway have limited its application. Kanan et al. discovered that Pd NPs dispersed on a carbon support exhibited a high mass activity at an overpotential of 0.20 V [47]. In an aqueous solution containing CO2− and HCO3−, the FE of formate could reach up to 100%. It was assumed that an electrochemically generated Pd hydride surface could be the inducement for the reduction of CO2. The electrode could be contaminated by the formed CO products, and brief exposure to air would remove CO to restore activity. Meanwhile, Jaramillo and co-workers presented a facile tuning AuPd alloy thin film, as being effective in CO2 electroreduction [48]. The alloys were found to generate 2e− products (i.e., H2, CO, and formate) during the conversion process. Different from the pure Au or Pd metals, the alloy thin films were more active and selective for formate production. These results indicated that the synergistic effect of Au and Pd in AuPd alloys fructified new performances rather than simple addition of the individual components.
4.3. Selective Production of Methanol
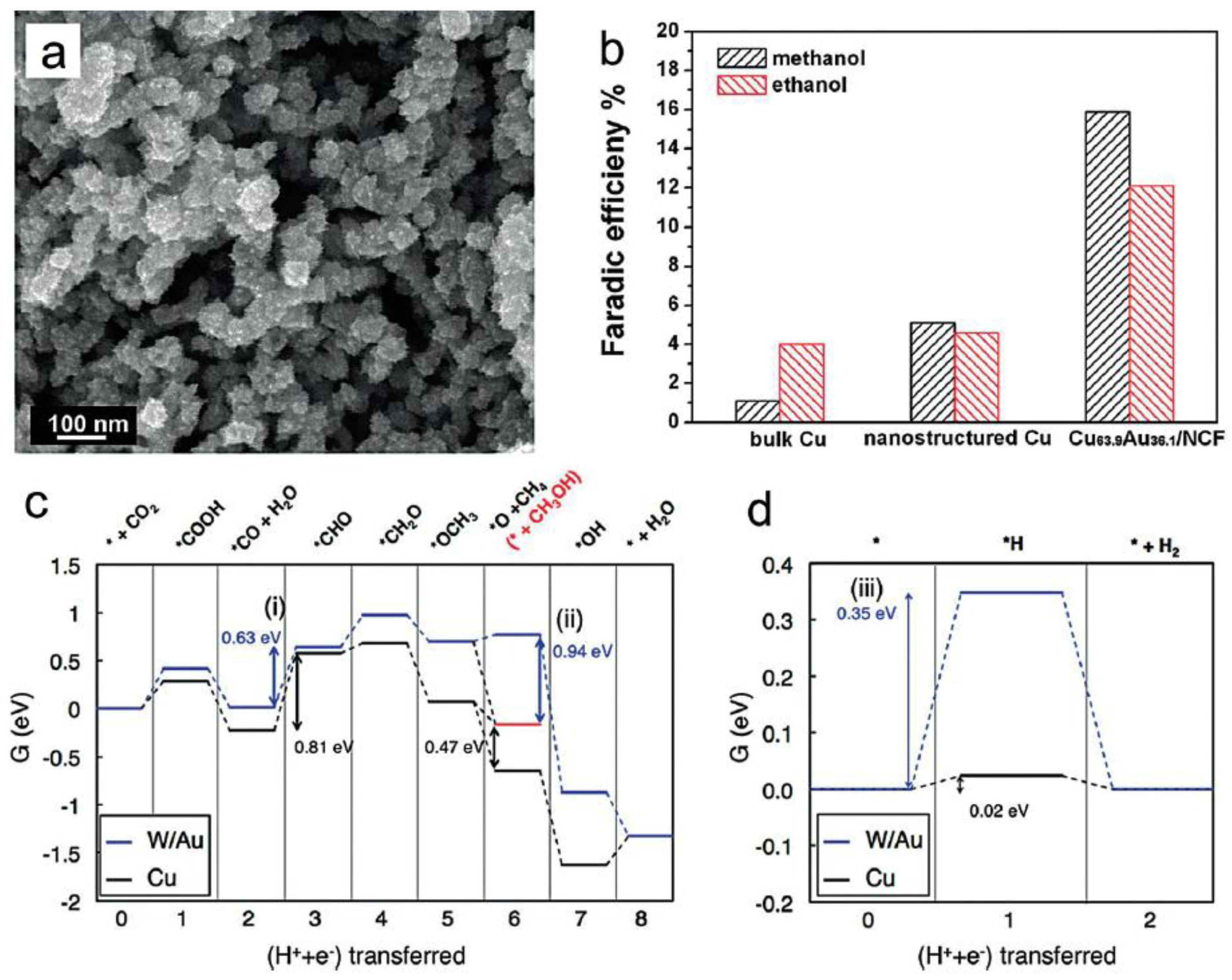
Methanol, as a liquid fuel, is one of the most important products generated directly from electrocatalysis reduction of the CO2 process. Based on the mechanism, the protonation of a key intermediate CH3O leads to the final formation of the methanol molecule. During this process, more protons could combine with carbon atoms, providing methane as a competitive product. Recently, nanostructured CuAu alloys with no organic additives were synthesized by electrochemical deposition with a nanoporous Cu film (NCF) template (Figure 6a) [49]. The alloy material exhibited superior catalytic activity and selectivity for the transformation of CO2 into alcohols. The FE of methanol was 15.9% on the Cu63.9Au36.1/NCF electrode, which is almost 18 times higher than that of pure Cu (Figure 6b). In addition, the FE for formate in such an apparatus decreased with the increase of atomic percentage of Au in CuAu alloys. Meanwhile, Jung and co-workers further investigated the details of electroreduction of CO2 to methanol, attempting to discover new effective catalysts through DFT calculations [50]. A few criteria, such as CO binding energy, OH binding energy, and H binding energy, were combined and collectively used as activity and selectivity determining descriptors [51]. Accordingly, the W/Au alloy was found to be a promising counterpart with increased efficiency and promoted selectivity toward methanol production in comparison to conventional Cu catalyst (Figure 6c,d).
4.4. Selective Production of Methane
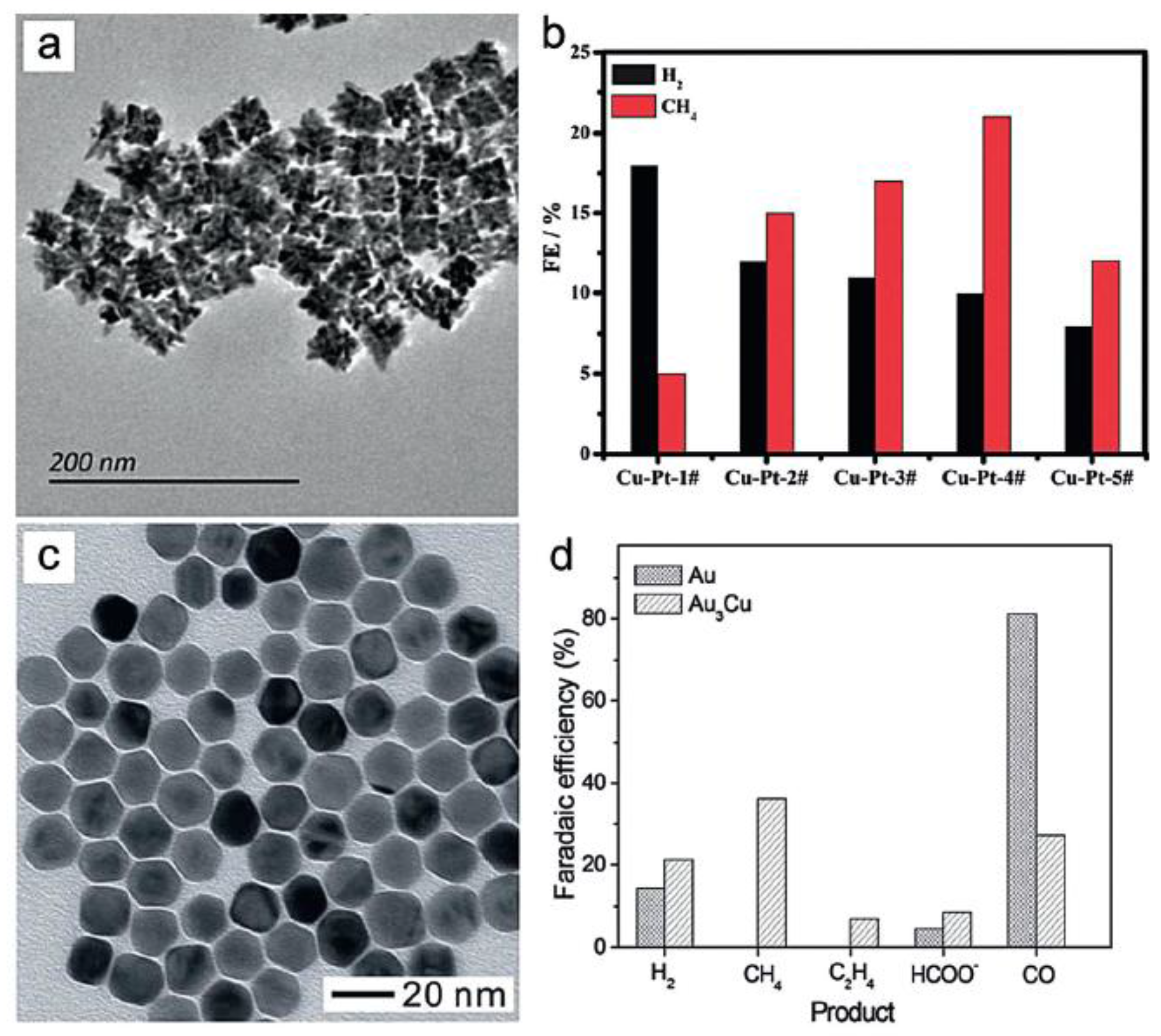
As the most significant component of natural gas, methane has become an alternative fuel in both industrial and domestic facilities. Therefore, methanizing CO2 through electrochemical methods is a promising process to convert greenhouse gases to valuable fuels. However, as the reduction of CO2 to CH4 involves an eight-electron process, the formation of a wide range of products, such as ethylene, hydrogen, carbon monoxide, and formate, can hardly be avoided. To date, the majority of studies on the CO2 electroreduction to CH4 adopted Cu-based materials as the catalyst. Alivisatos and co-workers proposed that the well-dispersed Cu NPs supported on glassy carbon exhibited up to 4-fold greater methanation current densities as compared to relatively pure copper foil electrodes [52]. The FE for CH4 on such Cu NPs reached 80% at −1.25 V, which is the highest value till now. Also, the Tafel plot showed the reaction pathway was CO2→CO2−→CO2-CO2−→CO→CH4, and the rate-limiting step was the formation of CO2-CO2−. Meanwhile, guided by the dramatic composition effect, various Cu catalysts were also investigated. Sun and Yan synthesized bimetallic CuPt NPs with diverse Cu/Pt ratios (Figure 7a) [53]. The highest FE for CH4 was 21% at −1.8 V obtained on the sample of Cu3Pt1 NPs (Figure 7b). DFT calculations suggested that the protonation of adsorbed CO* to form adsorbed HCO* was the key step in the reaction process. Jin et al. prepared new Au3Cu alloy NPs via a phase-stabilized synthesis, the size of which could be facilely tuned by controlling the amount of Au precursors (Figure 7c) [54]. Compared to Au NPs, the Au3Cu surface could absorb more CO molecules, which promoted the formation of CH4, thus leading to the distinct selectivity of CO2 reduction products (Figure 7d). In addition, Mul and co-workers investigated the influence of KHCO3 concentration and CO2 pressure on the production selectivity of Cu electrodes [55]. The results revealed that the selectivity of CH4 increased from 1% to 20% with the increase of electrolyte concentrations. Importantly, the low CO2 pressures could be better in selectively producing CH4 through the CO2 reduction process.
4.5. Selective Production of C2 Products
In the arena of electrochemical CO2 reduction, Cu catalysts not only showed effective catalytic performance in generating C1 products, the robust catalytic activity also presented potential for formation of C2 products. According to recent studies, the selectivity of C2H4 can be effectively improved by surface treatments of Cu materials. For instance, a roughened copper electrode with copper NPs coated surfaces was found to be very effective in electrochemical CO2 conversion towards ethylene production [56]. In addition, Baltrusaitis and co-workers investigated the effectiveness of cuprous oxide films of various thickness and [100], [110], and [111] orientation in the electrocatalytic reduction of CO2 [57]. The experimental results demonstrated that the parent Cu2O film thickness rather than the different orientation was the main factor that affected the selectivity of the process, which provided an FE up to 35% for C2H4 at −1.1 V. With thickness of Cu2O film increasing, the selectivity for ethane soared, while that for ethylene declined. Meanwhile, a novel 3D chrysanthemum-like structured Cu nanoflower catalyst was prepared by Yu and co-workers, which expressed electrocatalytic CO2 reduction selectively to C2H4 product with an FE value of about 10% at −0.9 V in aqueous solution [58].
Recently, Lewis and co-workers prepared Ni-Ga bimetallic films employing a drop-casting strategy with three different phases for reducing CO2 to the highly reduced C2 products including ethylene and ethane and C1-product methane [59]. In aqueous bicarbonate electrolytes at neutral pH, the onset potential was found to be −0.48 V vs. RHE, which was 0.25 V more positive than that of polycrystalline copper. The Ni5Ga3 phase provided the highest yield of 0.25 mol h−1 for acetate production. An isotope labeling experiment was also conducted with 13CO2 to confirm that the origin of the generated C2 products was from the reduction of CO2.
5. Conclusions and Outlook
In recent years, a number of novel transition-metal-centered electrocatalysts have been developed for the electrocatalytic reduction of CO2. Instead of focusing on optimization of the reaction conditions, most of the latest studies intended to purposefully design the structures and constitutions of the catalysts, as well as investigate the actual reaction mechanisms. This review summarizes the recent progress of CO2 electroreduction that is dependent on its catalytic phase states. Although numerous works have shown powerful strategies in converting CO2 to value-added C1 or C2 products, there are still several challenges and constraints remaining, such as moderate catalytic activity, insufficient product selectivity, and inadequate catalyst stability. It seems that the maturity of CO2 electroreduction technology is still far away from fulfilling the requirements for industrial and commercial applications. To overcome these deficiencies, enormous efforts should be made on the following aspects: (1) Innovation of electrocatalysts to enhance activity and stability; (2) Further fundamental understanding through experimental and theoretical modeling; (3) Optimization of electrolytes, reaction apparatus, and system designs for practical applications.
In summary, in order to overcome excessive and environmentally harmful CO2 emissions, and to remit energy shortage issues, it is of critical importance to develop electroreduction of CO2 to generate value-added low-carbon fuels. With continued and extensive efforts focusing on overcoming the confronted difficulties on insufficient catalyst activity, product selectivity, and catalyst stability, we believe that e CO2 electroreduction technology will become practical in the near future.
Acknowledgments
This work was supported by the Australian Research Council (ARC) Discovery Early Career Researcher Award (DE150101306) and the Linkage Project (LP160100927).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Spinner, N.S.; Vega, J.A.; Mustain, W.E. Recent Progress in the Electrochemical Conversion and Utilization of CO2. Catal. Sci. Technol. 2012, 2, 19–28. [Google Scholar] [CrossRef]
- Robert, M. Running the Clock: CO2 Catalysis in the Age of Anthropocene. ACS Energy Lett. 2016, 1, 281–282. [Google Scholar] [CrossRef]
- Schrag, D.P. Preparing to Capture Carbon. Science 2007, 315, 812–813. [Google Scholar] [CrossRef] [PubMed]
- Whipple, D.T.; Kenis, P.J.A. Prospects of CO2 Utilization via Direct Heterogeneous Electochemical Reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. [Google Scholar] [CrossRef]
- Omae, I. Recent Developments in Carbon Dioxide Utilization for the Production of Organic Chemicals. Coord. Chem. Rev. 2012, 256, 1384–1405. [Google Scholar] [CrossRef]
- Schneider, J.; Jia, H.F.; Muckerman, J.T.; Fujita, E. Thermodynamics and Kinetics of CO2, CO, and H+ Binding to the Metal Centre of CO2 Reduction Catalysts. Chem. Soc. Rev. 2012, 41, 2036–2051. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M. Carbon Dioxide Recovery and Utilization; Springer Netherlands: Berlin, Germany, 2003. [Google Scholar]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A Review of Catalysts for the Electroreduction of Carbon Dioxide to Produce Low-Carbon Fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Genovese, C.; Ampelli, C.; Marepally, B.C.; Papanikolaou, G.; Perathoner, S.; Centi, G. Electrocatalytic Reduction of CO2 for the Production of Fuels: A Comparison between Liquid and Gas Phase Conditions. Chem. Eng. Trans. 2015, 43, 2281–2286. [Google Scholar]
- Aresta, M.; Nobile, C.F.; Albano, V.G.; Forni, E.; Manassero, M. New Nickel-Carbon Dioxide Complex: Synthesis, Properties, and Crystallographic Characterization of (Carbon dioxide)-bis(tricyclehexyl phosphine)nickel. J. Chem. Soc. Chem. Commun. 1975, 636–637. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Saveant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Grice, K.A. Carbon Dioxide Reduction with Homogeneous Early Transition Metal Complexes: Opportunities and Challenges for Developing CO2 Catalysis. Coord. Chem. Rev. 2017, 336, 78–95. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and Homogeneous Approaches to Conversion of CO2 to Liquid Fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7, 70–88. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Saveant, J.-M. Current Issues in Molecular Catalysis Illustrated by Iron Porphyrins as Catalysts of the CO2-to-CO Electrochemical Conversion. Acc. Chem. Res. 2015, 48, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Elgrishi, N.; Chambers, M.B.; Wang, X.; Fontecave, M. Molecular Polypyridine-Based Metal Complexes as Catalysts for the Rreduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Saveant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.A.; Zahran, Z.N.; Naruta, Y. Efficient Electrocatalytic CO2 Reduction with a Molecular Cofacial Iron Porphyrin Dimer. Chem. Commun. 2015, 51, 16900–16903. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, Z.; Wei, X.G.; Gallenkamp, C.; Bonin, J.; Anxolabenhere-Mallart, E.; Lau, K.C.; Lau, T.C.; Robert, M. Molecular Catalysis of the Electrochemical and Photochemical Reduction of CO2 with Earth-Abundant Metal Complexes. Selective Production of CO vs. HCOOH by Switching of the Metal Center. J. Am. Chem. Soc. 2015, 137, 10918–10921. [Google Scholar] [CrossRef] [PubMed]
- Machan, C.W.; Sampson, M.D.; Kubiak, C.P. A Molecular Ruthenium Electrocatalyst for the Reduction of Carbon Dioxide to CO and Formate. J. Am. Chem. Soc. 2015, 137, 8564–8571. [Google Scholar] [CrossRef] [PubMed]
- Spall, S.J.P.; Keane, T.; Tory, J.; Cocker, D.C.; Adams, H.; Fowler, H.; Meijer, H.M.; Hartl, F.; Weinstein, J.A. Manganese Tricarbonyl Complexes with Asymmetric 2-Iminopyridine Ligands: Toward Decoupling Steric and Electronic Factors in Electrocatalytic CO2 Reduction. Inorg. Chem. 2016, 55, 12568–12582. [Google Scholar] [CrossRef] [PubMed]
- Bourrez, M.; Orio, M.; Molton, F.; Vezin, H.; Duboc, C.; Deronzier, A.; Chardon-Noblat, S. Pulsed-EPR Evidence of a Manganese(II) Hydroxycarbonyl Intermediate in the Electrocatalytic Reduction of Carbon Dioxide by a Manganese Bipyridyl Derivative. Angew. Chem. Int. Ed. 2014, 53, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.; Shaw, T.W.; Stanton, C.J., 3rd; Majetich, G.F.; Bocarsly, A.B.; Schaefer, H.F., 3rd. NHC-Containing Manganese(I) Electrocatalysts for Two-Electron Reduction of CO2. Angew. Chem. Int. Ed. 2014, 53, 5152–5155. [Google Scholar]
- Sun, C.; Gobetto, R.; Nervi, C. Recent Advances in Catalytic CO2 Reduction by Organometal Complexes Anchored on Modified Electrodes. New J. Chem. 2016, 40, 5656–5661. [Google Scholar] [CrossRef]
- Reuilard, B.; Ly, K.H.; Rosser, T.E.; Kuehnel, M.F.; Zebger, I.; Reisner, E. Tuning Product Selectivity for Aqueous CO2 Reduction with a Mn(bipyridine)-pyrene Catalyst Immobilized on a Carbon Nanotube Electrode. J. Am. Chem. Soc. 2017, 139, 14425–14435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.D.; Liu, J.L.; Qiao, S.Z. Recent Advances in Inorganic Heterogeneous Electrocatalysts for Reduction of Carbon Dioxide. Adv. Mater. 2017, 28, 3423–3452. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Kim, K.; Liu, C.; Addepalli, A.V.; Abbasi, P.; Yasaei, P.; Phillips, P.; Behranginia, A.; Cerrato, J.M.; Haasch, R.; et al. Nanostructured Transition Metal Dichalcogenide Electrocatalysts for CO2 Reduction in Ionic Liquid. Science 2016, 353, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Michalsky, R.; Metin, O.; Lv, H.; Guo, S.; Wright, C.J.; Sun, X.; Peterson, A.A.; Sun, S. Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.-J.; Greeley, J.; Strasser, P.; Cuenya, B.R. Exceptional Size-Dependnet Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, Y.-J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A.A.; Sun, S. Active and Selective Conversion of CO2 to CO on Ultrathin Au Nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-E.; Yang, K.D.; Yoon, S.M.; Ahn, H.-Y.; Lee, Y.Y.; Chang, H.; Jeong, D.H.; Lee, Y.-S.; Kim, M.Y.; Nam, K.T. Concave Rhombic Dodecahedral Au Nanocatalyst with Multiple High-Index Facets for CO2 Reduction. ACS Nano 2015, 9, 8384–8393. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jiang, K.; Fan, S.; Kanan, M.W. Grain-Boundary-Dependent CO2 Eletroreduction Activity. J. Am. Chem. Soc. 2015, 137, 4606–4609. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Jeon, H.S.; Eom, T.; Jee, M.S.; Kim, H.; Friend, C.M.; Min, B.K.; Hwang, Y.J. Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles. J. Am. Chem. Soc. 2015, 137, 13844–13850. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Khojin, A.; Jhong, H.-R.M.; Rosen, B.A.; Zhu, W.; Ma, S.; Kenis, P.J.A.; Masel, R.I. Nanoparticles Silver Catalysts that Show Enhanced Activity for Carbon Dioxide Electrolysis. J. Phys. Chem. C 2013, 117, 1627–1632. [Google Scholar] [CrossRef]
- Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G.S.; Kimmel, Y.C.; Chen, J.G.; Jiao, F. A Selective and Efficient Electrocatalyst for Carbon Dioxide Reduction. Nat. Commun. 2014, 5, 3242–3247. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhou, H.; Wang, J.; Miao, S.; Yang, F.; Wang, G.; Wang, J.; Bao, X. Size-Dependent Electrocatalytic Reduction of CO2 over Pd Nanoparticles. J. Am. Chem. Soc. 2015, 137, 4288–4291. [Google Scholar] [CrossRef] [PubMed]
- Reske, R.; Mistry, H.; Behafarid, F.; Cuenya, B.R.; Strasser, P. Particle Size Effects in the Catalytic Electroreduction of CO2 on Cu Nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, G.; Zhang, Z.; Jin, M.; Yin, Y. Selectivity on Etching: Creation of High-Energy Facets on Copper Nanocrystals for CO2 Electrochemical Reduction. ACS Nano 2016, 10, 4559–4564. [Google Scholar] [CrossRef] [PubMed]
- Rasul, S.; Anjum, D.H.; Jedidi, A.; Minenkov, Y.; Cavallo, L.; Takanabe, K. A Highly Selective Copper–Indium Bimetallic Electrocatalyst for the Electrochemical Reduction of Aqueous CO2 to CO. Angew. Chem. Int. Ed. 2015, 54, 2146–2150. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Kumar, B.; Behranginia, A.; Rosen, B.A.; Baskin, A.; Repnin, N.; Pisasale, D.; Phillips, P.; Zhu, W.; Haasch, R.; et al. Robust Carbon Dioxide Reduction on Molybdenum Disulphide Edges. Nat. Commun. 2014, 5, 4470–4477. [Google Scholar] [CrossRef] [PubMed]
- Benck, J.D.; Hellstern, T.R.; Kibsgaard, J.; Chakthranont, P.; Jaramillo, T.F. Catalyzing the Hydrogen Evolution Reaction (HER) with Molybdenum Sulfide Nanomaterials. ACS Catal. 2014, 4, 3957–3971. [Google Scholar] [CrossRef]
- Gao, S.; Lin, Y.; Jiao, X.; Sun, Y.; Luo, Q.; Zhang, W.; Li, D.; Yang, J.; Xie, Y. Partially Oxidized Atomic Cobalt Layers for Carbon Dioxide Electroreduction to Liquid Fuel. Nature 2016, 529, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Jiao, X.; Sun, Z.; Zhang, W.; Sun, Y.; Wang, C.; Hu, Q.; Zu, X.; Yang, F.; Yang, S.; et al. Ultrathin Co3O4 Layers Realizing Optimized CO2 Eletroreduction to Formate. Angew. Chem. Int. Ed. 2016, 55, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Liu, D.; Palmore, G.T.R. Electrochemical Reduction of CO2 at Copper Nanofoams. ACS Catal. 2014, 4, 3091–3095. [Google Scholar] [CrossRef]
- Min, X.; Kanan, M.W. Pd-Catalyzed Electrohydrogenation of Carbon Dioxide to Formate: High Mass Activity at Low Overpotential and Identification of the Deactivation Pathway. J. Am. Chem. Soc. 2015, 137, 4701–4708. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Abram, D.N.; Hansen, H.A.; Hatsukade, T.; Jackson, A.; Johnson, N.C.; Hellstern, T.R.; Kuhl, K.P.; Cave, E.R.; Feastera, J.T.; et al. Synthesis of Thin Film AuPd Alloys and Their Investigation for Electrocatalytic CO2 Reduction. J. Mater. Chem. A 2015, 3, 20185–20194. [Google Scholar] [CrossRef]
- Jia, F.; Yu, X.; Zhang, L. Enhanced Selectivity for the Electrochemical Reduction of CO2 to Alcohols in Aqueous Solution with Nanostructured Cu–Au Alloy as Catalyst. J. Power Sources 2014, 252, 85–89. [Google Scholar] [CrossRef]
- Back, S.; Kim, H.; Jung, Y. On the Selective Heterogeneous CO2 Electroreduction to Methanol. ACS Catal. 2015, 5, 965–971. [Google Scholar] [CrossRef]
- Hansen, H.A.; Montoya, J.H.; Zhang, Y.-J.; Shi, C.; Peterson, A.A.; Nørskov, J.K. Electroreduction of Methanediol on Copper. Catal. Lett. 2013, 143, 631–635. [Google Scholar] [CrossRef]
- Manthiram, K.; Beberwyck, B.J.; Alivisatos, A.P. Enhanced Electrochemical Methanation of Carbon Dioxide with a Dispersible Nanoscale Copper Catalyst. J. Am. Chem. Soc. 2014, 136, 13319–13325. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, Y.; Deng, C.; Li, X.; Xue, Y.; Yan, Y.-M.; Sun, K. Composition Dependent Activity of Cu–Pt Nanocrystals for Electrochemical Reduction of CO2. Chem. Commun. 2015, 51, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Yang, L.; Yin, Y.; Jin, M. Thermodynamic Controlled Synthesis of Intermetallic Au3Cu Alloy Nanocrystals from Cu Microparticles. J. Mater. Chem. A 2014, 2, 902–906. [Google Scholar] [CrossRef]
- Kas, R.; Kortlever, R.; Yilmaz, H.; Koper, M.T.M.; Mul, G. Manipulating the Hydrocarbon Selectivity of Copper Nanoparticles in CO2 Electroreduction by Process Conditions. ChemElectroChem 2015, 2, 354–358. [Google Scholar] [CrossRef]
- Tang, W.; Peterson, A.A.; Varela, A.S.; Jovanov, Z.P.; Bech, L.; Durand, W.J.; Dahl, S.; Norskov, J.K.; Chorkendorff, I. The Importance of Surface Morphology in Controlling the Selectivity of Polycrystalline Copper for CO2 Eletroreduction. Phys. Chem. Chem. Phys. 2012, 14, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Kas, R.; Kortlever, R.; Milbrat, A.; Koper, M.T.M.; Mul, G.; Baltrusaitis, J. Electrochemical CO2 Reduction on Cu2O-Derived Copper Nanoparticles: Controlling the Catalytic Selectivity of Hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.-F.; Huang, Y.-X.; Li, W.-W.; Song, X.-N.; Xiong, L.; Yu, H.-Q. Efficient Electrochemical CO2 Reduction on a Unique Chrysanthemum-Like Cu Nanoflower Electrode and Direct Observation of Carbon Deposite. Electrochim. Acta 2014, 139, 137–144. [Google Scholar] [CrossRef]
- Torelli, D.A.; Francis, S.A.; Crompton, J.C.; Javie, A.; Thompson, J.R.; Brunschwig, B.S.; Soriaga, M.P.; Lewis, N.S. Nickel-Gallium-Catalyzed Electrochemical Reduction of CO2 to Highly Reduced Products at Low Overpotentials. ACS Catal. 2016, 6, 2100–2104. [Google Scholar] [CrossRef]
Scheme 1.
Homogeneous electrocatalytic reduction of CO2. Reproduced with permission from Ref. [15]. Copyright 2009, Royal Society of Chemistry.
Scheme 1.
Homogeneous electrocatalytic reduction of CO2. Reproduced with permission from Ref. [15]. Copyright 2009, Royal Society of Chemistry.
Scheme 2.
Structure illustration of Iron(0) complex electrocatalysts.
Scheme 3.
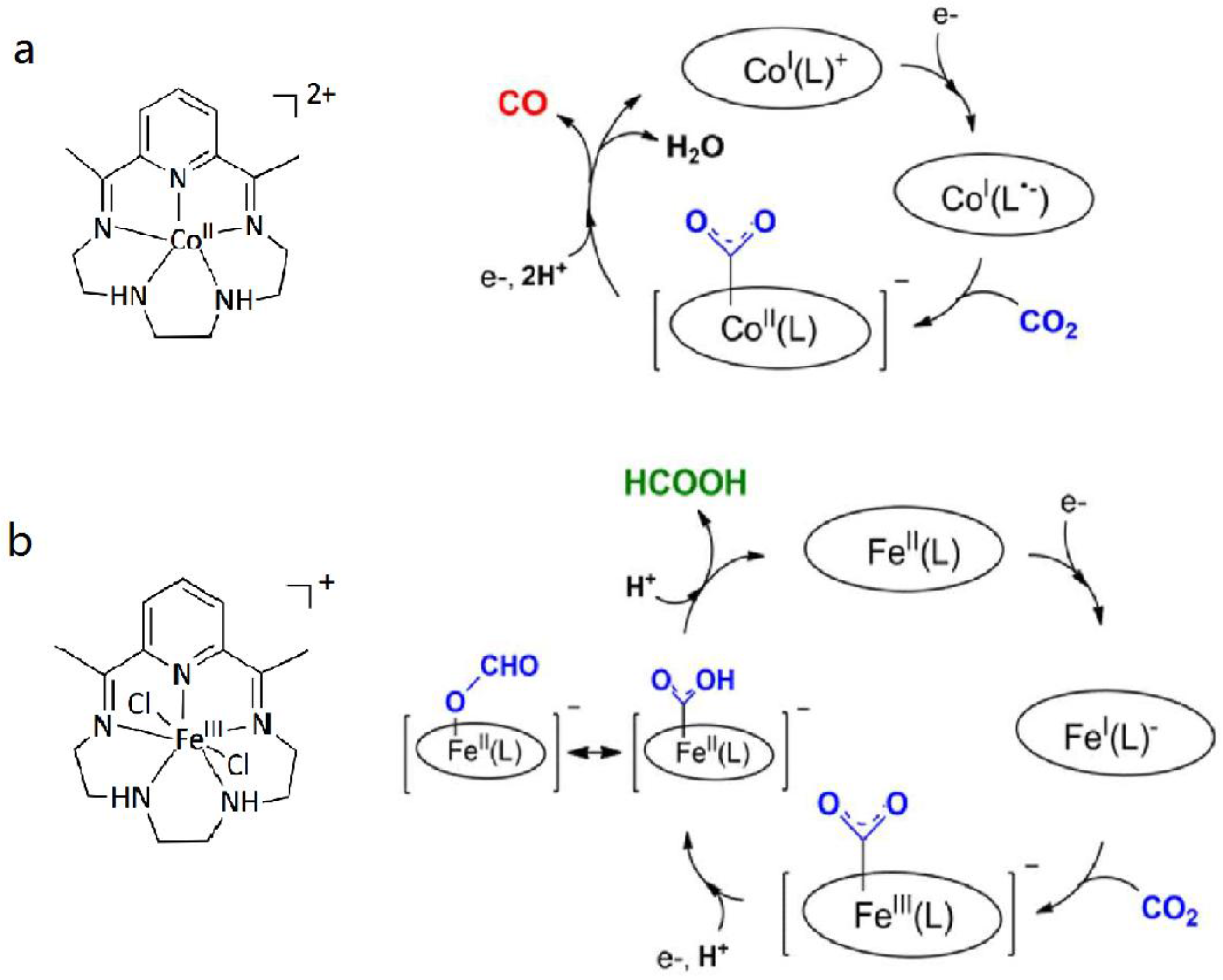
(a) Structure of CoII complexes and plausible mechanism for the reduction of CO2; (b) Structure of FeIII complexes and plausible mechanism for the reduction of CO2. Adapted with permission from Ref. [21]. Copyright 2015, American Chemical Society.
Scheme 3.
(a) Structure of CoII complexes and plausible mechanism for the reduction of CO2; (b) Structure of FeIII complexes and plausible mechanism for the reduction of CO2. Adapted with permission from Ref. [21]. Copyright 2015, American Chemical Society.
Scheme 4.
Synthesis of complex Ru(mesbpy)(CO)2Cl2 from ligand mesbpy and Ru salt [Ru(CO)2Cl2]n and the corresponding molecular structure of complex Ru(mesbpy)(CO)2Cl2. Reproduced with permission from Ref. [22]. Copyright 2015, American Chemical Society.
Scheme 4.
Synthesis of complex Ru(mesbpy)(CO)2Cl2 from ligand mesbpy and Ru salt [Ru(CO)2Cl2]n and the corresponding molecular structure of complex Ru(mesbpy)(CO)2Cl2. Reproduced with permission from Ref. [22]. Copyright 2015, American Chemical Society.

Scheme 5.
Synthesis of Mn complexes from NHC ligand and Mn salt. Reproduced with permission from Ref. [25], Copyright 2014, Wiley-VCH Verlag GmbH & Co. KGaA.
Scheme 5.
Synthesis of Mn complexes from NHC ligand and Mn salt. Reproduced with permission from Ref. [25], Copyright 2014, Wiley-VCH Verlag GmbH & Co. KGaA.
Scheme 6.
Schematic representation of [MnBr(bpypyr)(CO)3](Mnpyr) immobilized on a CNT sidewall, concentration-dependent dimerization or Mn-H formation, and intermediate-dependent reduction of CO2 to CO or HCOOH. Adapted with permission from Ref. [27]. Copyright 2017, American Chemical Society.
Scheme 6.
Schematic representation of [MnBr(bpypyr)(CO)3](Mnpyr) immobilized on a CNT sidewall, concentration-dependent dimerization or Mn-H formation, and intermediate-dependent reduction of CO2 to CO or HCOOH. Adapted with permission from Ref. [27]. Copyright 2017, American Chemical Society.
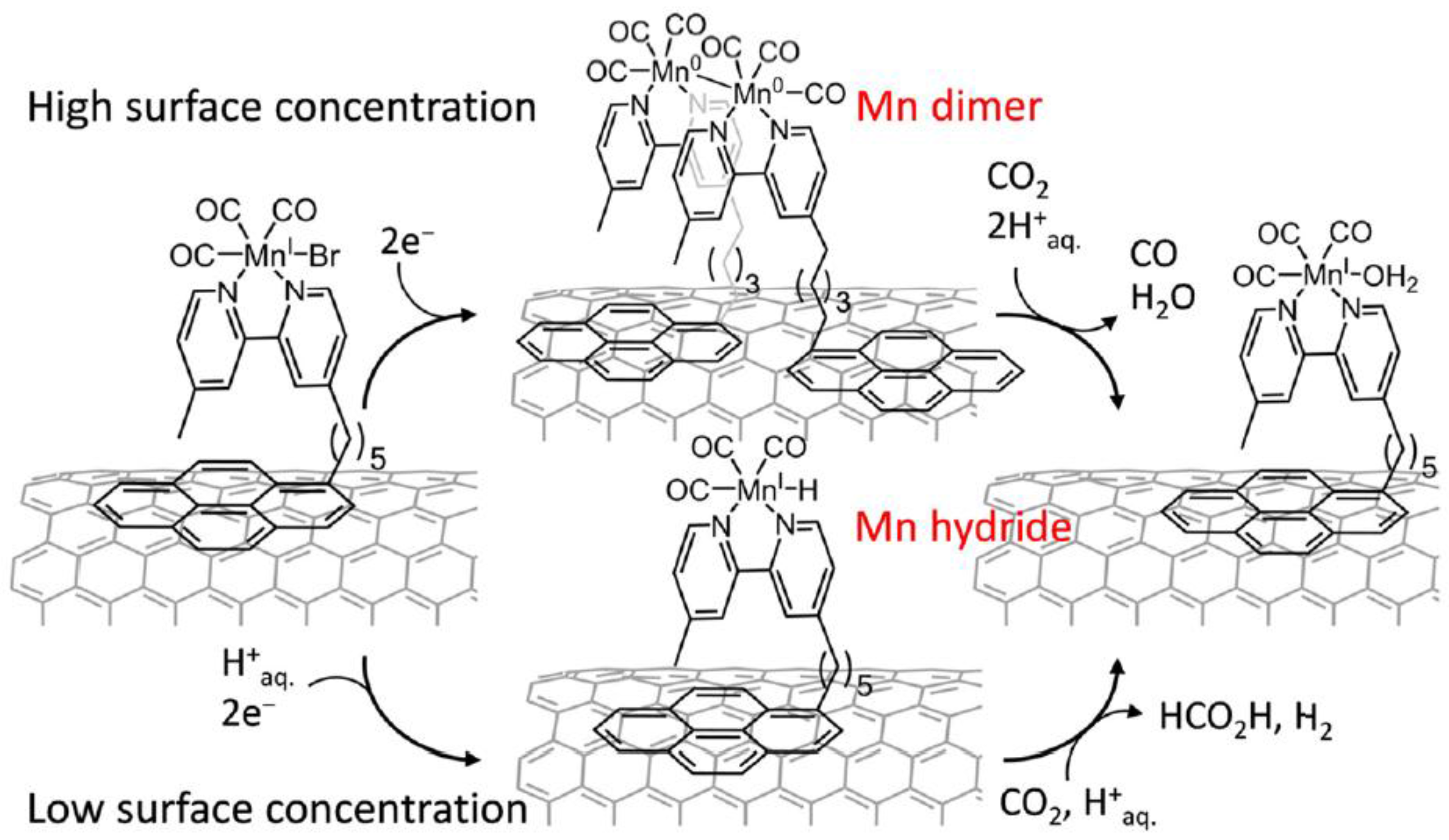
Figure 1.
(a–e) Transmission electron microscope (TEM) images and potential-dependent Faradaic efficiencies of the 4, 6, 8, and 10 nm Au nanoparticles (NPs). Adapted with permission from Ref. [30]. Copyright 2013 American Chemical Society; (f) Edge site weight percentage for the Au nanowires and NPs; (g,h) TEM image and potential-dependent Faradaic efficiencies of the Au nanowires. Adapted with permission from Ref. [32]. Copyright 2014, American Chemical Society.
Figure 1.
(a–e) Transmission electron microscope (TEM) images and potential-dependent Faradaic efficiencies of the 4, 6, 8, and 10 nm Au nanoparticles (NPs). Adapted with permission from Ref. [30]. Copyright 2013 American Chemical Society; (f) Edge site weight percentage for the Au nanowires and NPs; (g,h) TEM image and potential-dependent Faradaic efficiencies of the Au nanowires. Adapted with permission from Ref. [32]. Copyright 2014, American Chemical Society.
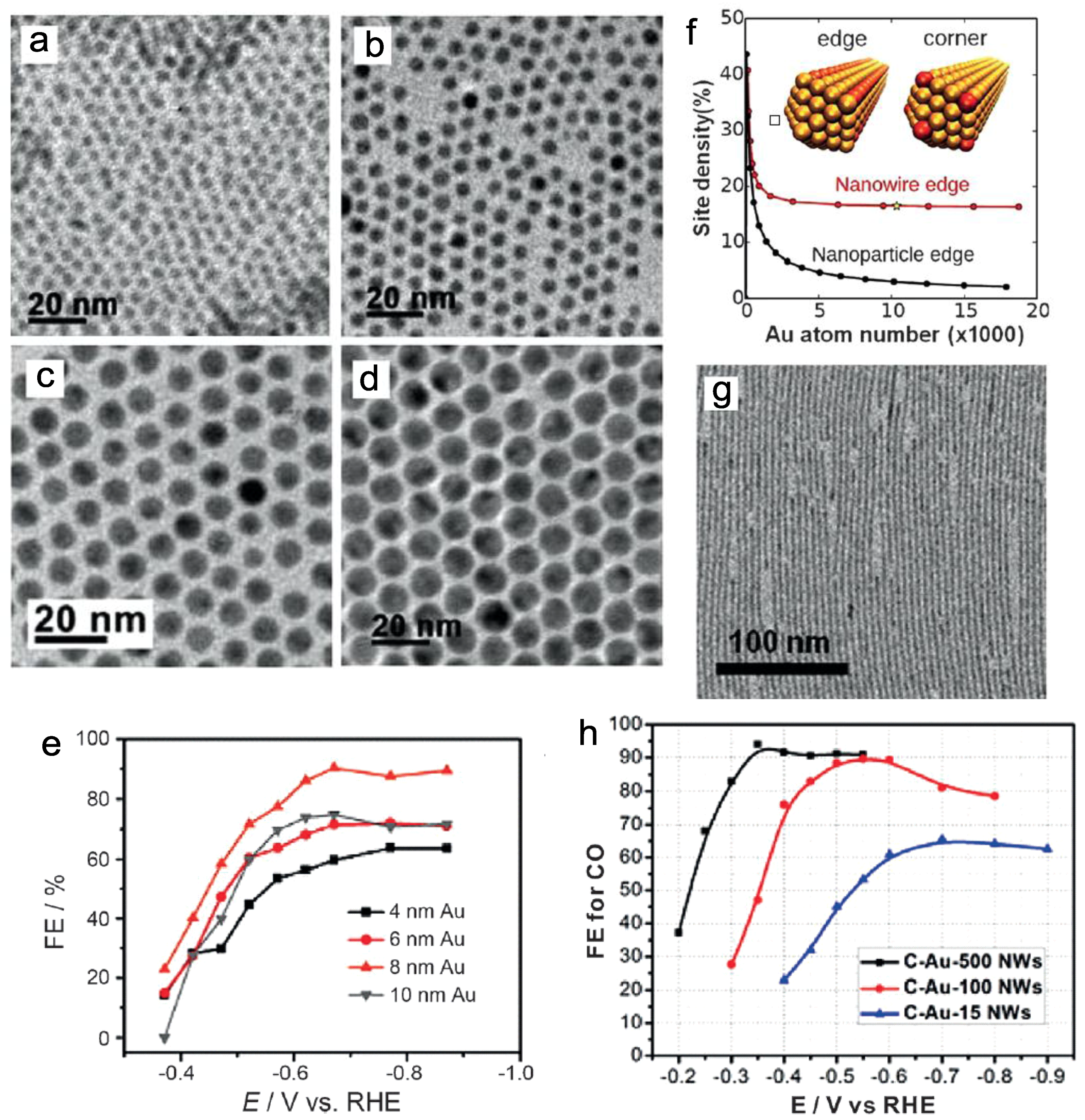
Figure 2.
(a–c) TEM images of the 3, 5, and 10 nm Ag NPs load on carbon supports; (d) High-resolution transmission electron microscope (HR-TEM) image of the 5 nm Ag/C catalyst; (e) CO partial current densities of three typical Ag/C catalysts with different NP sizes. Adapted with permission from Ref. [35]. Copyright 2015, American Chemical Society; (f) The schematic representation, scanning electron microscope (SEM), and HR-TEM images of a nanoporous silver electrocatalyst with highly curved internal surface (scale bar 500 nm & 2 nm); (g) CO2 reduction activity of nanoporous silver. Adapted with permission from Ref. [37]. Copyright 2014, Nature Publishing Group.
Figure 2.
(a–c) TEM images of the 3, 5, and 10 nm Ag NPs load on carbon supports; (d) High-resolution transmission electron microscope (HR-TEM) image of the 5 nm Ag/C catalyst; (e) CO partial current densities of three typical Ag/C catalysts with different NP sizes. Adapted with permission from Ref. [35]. Copyright 2015, American Chemical Society; (f) The schematic representation, scanning electron microscope (SEM), and HR-TEM images of a nanoporous silver electrocatalyst with highly curved internal surface (scale bar 500 nm & 2 nm); (g) CO2 reduction activity of nanoporous silver. Adapted with permission from Ref. [37]. Copyright 2014, Nature Publishing Group.

Figure 3.
(a) TEM imagies of Cu nanocubes; (b) TEM images of Cu rhombic dodecahedrons; (c) The FE of Cu nanocubes and Cu rhombic dodecahedra toward CO2 electroreduction. Adapted with permission from Ref. [40]. Copyright 2016, American Chemical Society; (d,e) HR-TEM images and energy dispersive spectrometer element mapping of Cu-In catalysts; (f) the long-term stability test for the Cu-In catalyst. Adapted with permission from Ref. [41]. Copyright 2015, Wiley-VCH Verlag GmbH & Co. KGaA.
Figure 3.
(a) TEM imagies of Cu nanocubes; (b) TEM images of Cu rhombic dodecahedrons; (c) The FE of Cu nanocubes and Cu rhombic dodecahedra toward CO2 electroreduction. Adapted with permission from Ref. [40]. Copyright 2016, American Chemical Society; (d,e) HR-TEM images and energy dispersive spectrometer element mapping of Cu-In catalysts; (f) the long-term stability test for the Cu-In catalyst. Adapted with permission from Ref. [41]. Copyright 2015, Wiley-VCH Verlag GmbH & Co. KGaA.

Figure 4.
(a) High-angle annular dark-field (HAADF) images (scale bar, 5 nm) of the MoS2 flakes; (b) Cyclic voltammogram curves for MoS2 catalysts; (c) CO and H2 Faradaic efficiency at different applied potentials; (d) Overview of different catalysts’ performance at different overpotential; (e) The Mo atom at the edge and Mo atom with in the lattice; (f) s, p, and d orbital of the Mo-edge atom; (g) Projected density of states of d band of the Mo-edge atom, Ag atom from bulk and from a Ag-slab. Adapted with permission from Ref. [42]. Copyright 2014, Nature Publishing Group.
Figure 4.
(a) High-angle annular dark-field (HAADF) images (scale bar, 5 nm) of the MoS2 flakes; (b) Cyclic voltammogram curves for MoS2 catalysts; (c) CO and H2 Faradaic efficiency at different applied potentials; (d) Overview of different catalysts’ performance at different overpotential; (e) The Mo atom at the edge and Mo atom with in the lattice; (f) s, p, and d orbital of the Mo-edge atom; (g) Projected density of states of d band of the Mo-edge atom, Ag atom from bulk and from a Ag-slab. Adapted with permission from Ref. [42]. Copyright 2014, Nature Publishing Group.
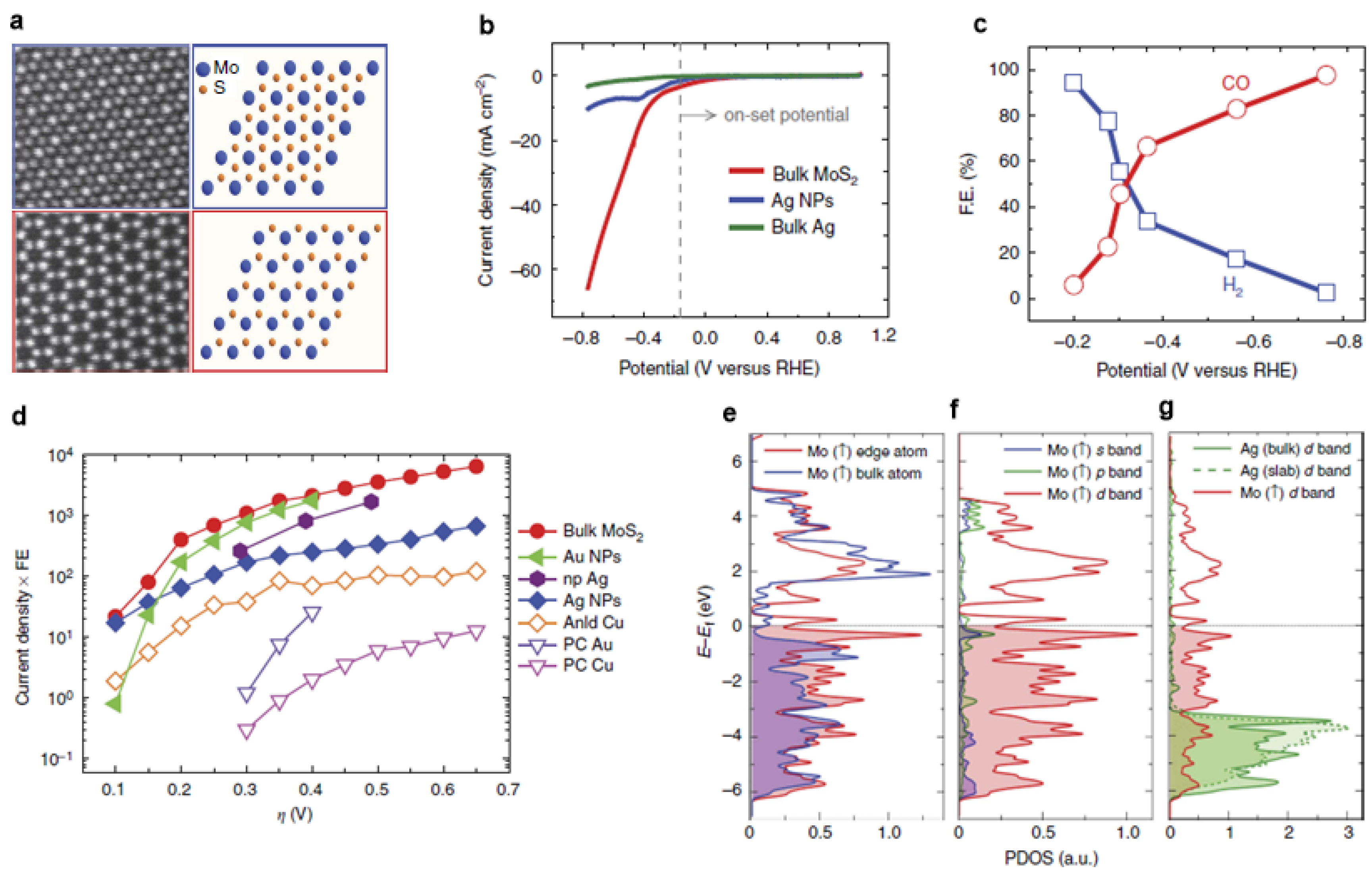
Figure 5.
(a,b) TEM images of the partially oxidized Co 4-atom-thick layers; (c,d) Linear sweep voltammograms and the Faradaic efficiency for formate on partially oxidized Co 4-atom-thick layers (red), Co 4-atom-thick layers (blue), partially oxidized bulk Co (violet), and bulk Co (black). Adapted with permission from Ref. [44]. Copyright 2016, Nature Publishing Group. (e) Typical TEM image for Co3O4 atomic layers with an average thickness of 1.72 nm; (f,g) Linear sweep voltammograms and Faradaic efficiency for formate on Co3O4 with different thicknesses. Adapted with permission from Ref. [45]. Copyright 2016, Wiley-VCH Verlag GmbH & Co. KGaA.
Figure 5.
(a,b) TEM images of the partially oxidized Co 4-atom-thick layers; (c,d) Linear sweep voltammograms and the Faradaic efficiency for formate on partially oxidized Co 4-atom-thick layers (red), Co 4-atom-thick layers (blue), partially oxidized bulk Co (violet), and bulk Co (black). Adapted with permission from Ref. [44]. Copyright 2016, Nature Publishing Group. (e) Typical TEM image for Co3O4 atomic layers with an average thickness of 1.72 nm; (f,g) Linear sweep voltammograms and Faradaic efficiency for formate on Co3O4 with different thicknesses. Adapted with permission from Ref. [45]. Copyright 2016, Wiley-VCH Verlag GmbH & Co. KGaA.

Figure 6.
(a) SEM image of CuAu sample deposited on nanoporous Cu film; (b) Faradaic efficiency of methanol and ethanol on different electrodes. Adapted with permission from Ref. [49]. Copyright 2015, Elsevier; (c) Free energy diagram for the CO2 electroreduction to CH4 or CH3OH (shown in red) and (d) Free energy diagram for the H2 evolution reactions at zero electrode potential for Cu (black) and W/Au (blue). Adapted with permission from Ref. [50]. Copyright 2014, American Chemical Society.
Figure 6.
(a) SEM image of CuAu sample deposited on nanoporous Cu film; (b) Faradaic efficiency of methanol and ethanol on different electrodes. Adapted with permission from Ref. [49]. Copyright 2015, Elsevier; (c) Free energy diagram for the CO2 electroreduction to CH4 or CH3OH (shown in red) and (d) Free energy diagram for the H2 evolution reactions at zero electrode potential for Cu (black) and W/Au (blue). Adapted with permission from Ref. [50]. Copyright 2014, American Chemical Society.
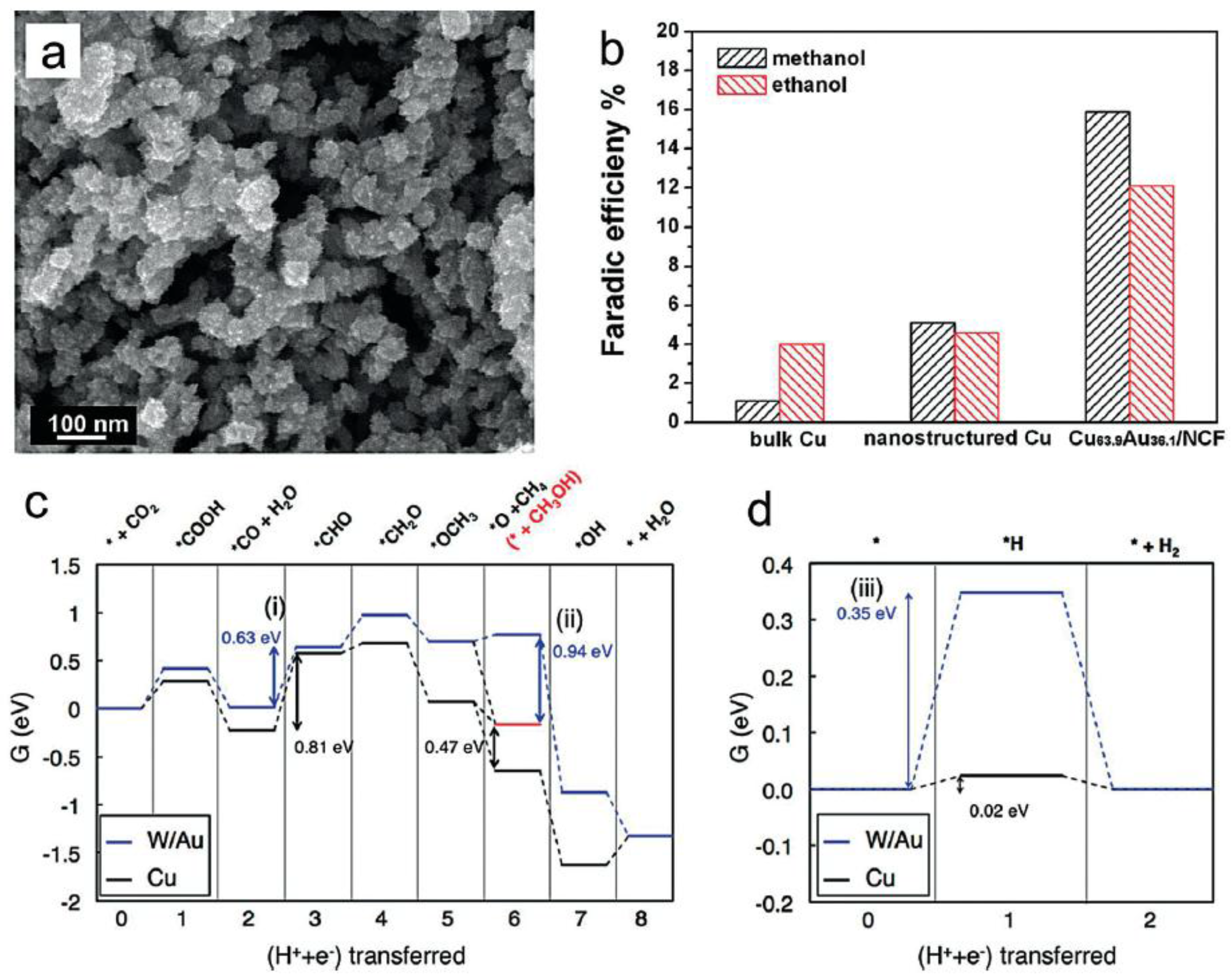
Figure 7.
(a) TEM images of the Cu-Pt alloy NPs; (b) The Faradaic efficiency of H2 and CH4 using different Cu-Pt NPs at −1.6 V. Adapted with permission from Ref. [53]. Copyright 2014, Royal Society of Chemistry; (c) TEM image of the Au3Cu truncated nanocubes; and (d) the Faradaic efficiency of Au3Cu and Au NPs for CO2 electroreduction. Adapted with permission from Ref. [54]. Copyright 2013, Royal Society of Chemistry.
Figure 7.
(a) TEM images of the Cu-Pt alloy NPs; (b) The Faradaic efficiency of H2 and CH4 using different Cu-Pt NPs at −1.6 V. Adapted with permission from Ref. [53]. Copyright 2014, Royal Society of Chemistry; (c) TEM image of the Au3Cu truncated nanocubes; and (d) the Faradaic efficiency of Au3Cu and Au NPs for CO2 electroreduction. Adapted with permission from Ref. [54]. Copyright 2013, Royal Society of Chemistry.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected standard potentials of CO2 reduction in aqueous solutions 1.
| Equation Number | Half-Electrochemical Thermodynamic Reactions | E° (V) |
|---|---|---|
| Equation (1) | CO2 + 2H+ + 2e− = CO(g) + H2O | −0.53 |
| Equation (2) | CO2 + 2H+ + 2e− = HCO2H | −0.61 |
| Equation (3) | CO2 + 4H+ + 4e− = HCHO + H2O | −0.48 |
| Equation (4) | CO2 + 6H+ + 6e− = CH3OH + H2O | −0.38 |
| Equation (5) | CO2 + 8H+ + 8e− = CH4 + 2H2O | −0.24 |
| Equation (6) | 2CO2 + 12H+ + 12e− = C2H4 + 4H2O | −0.34 |
| Equation (7) | CO2 + e− = CO2•− | −1.90 |
1 pH = 7 in aqueous solution vs. standard hydrogen electrode (SHE), 25 °C, 1 atmosphere gas pressure, and 1 M for other solutes.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Feng, D.-M.; Zhu, Y.-P.; Chen, P.; Ma, T.-Y. Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems. Catalysts 2017, 7, 373. https://doi.org/10.3390/catal7120373
AMA Style
Feng D-M, Zhu Y-P, Chen P, Ma T-Y. Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems. Catalysts. 2017; 7(12):373. https://doi.org/10.3390/catal7120373
Chicago/Turabian StyleFeng, Da-Ming, Yun-Pei Zhu, Ping Chen, and Tian-Yi Ma. 2017. "Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems" Catalysts 7, no. 12: 373. https://doi.org/10.3390/catal7120373
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.