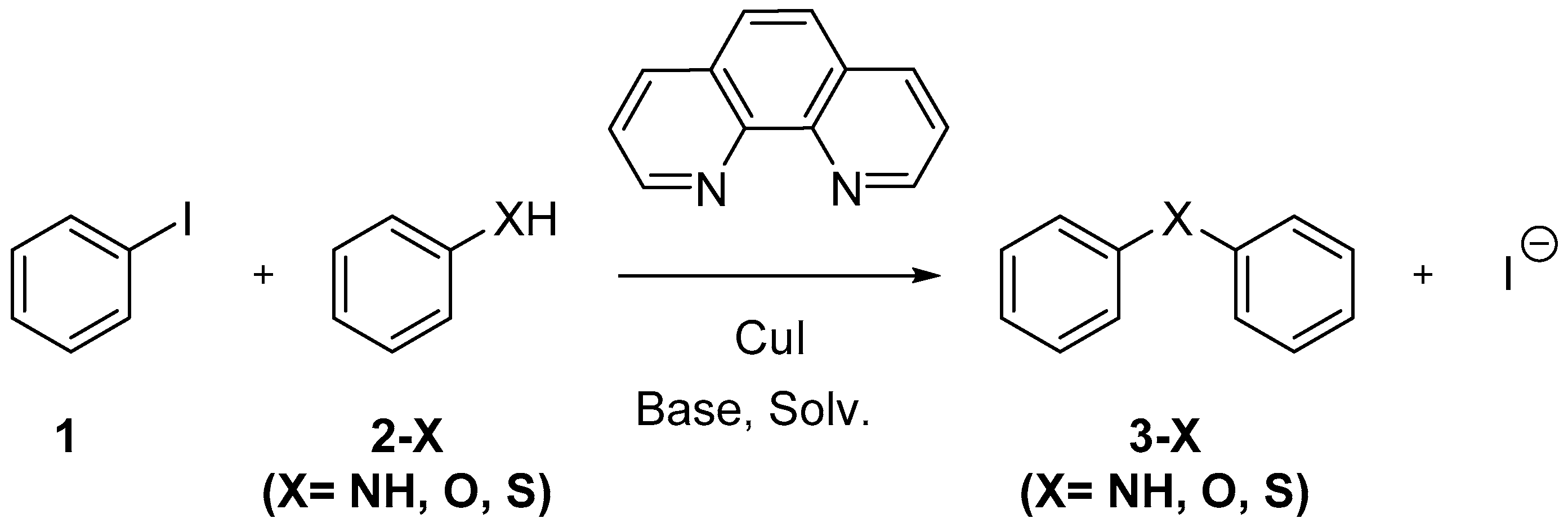
Understanding the Heteroatom Effect on the Ullmann Copper-Catalyzed Cross-Coupling of X-Arylation (X = NH, O, S) Mechanism

Abstract
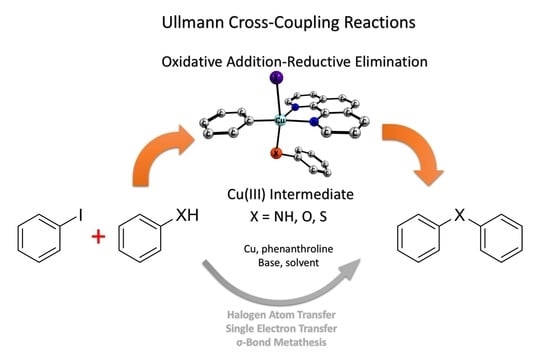
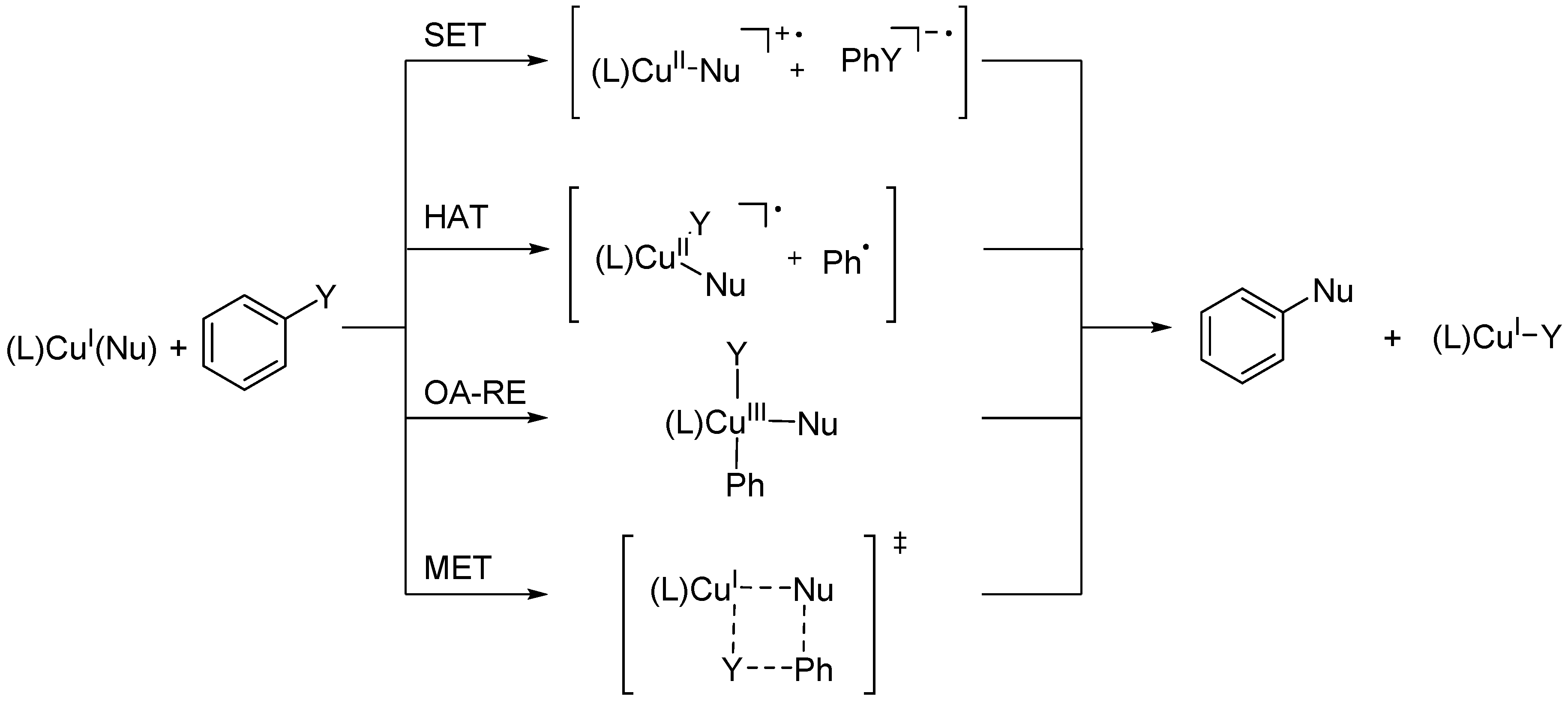
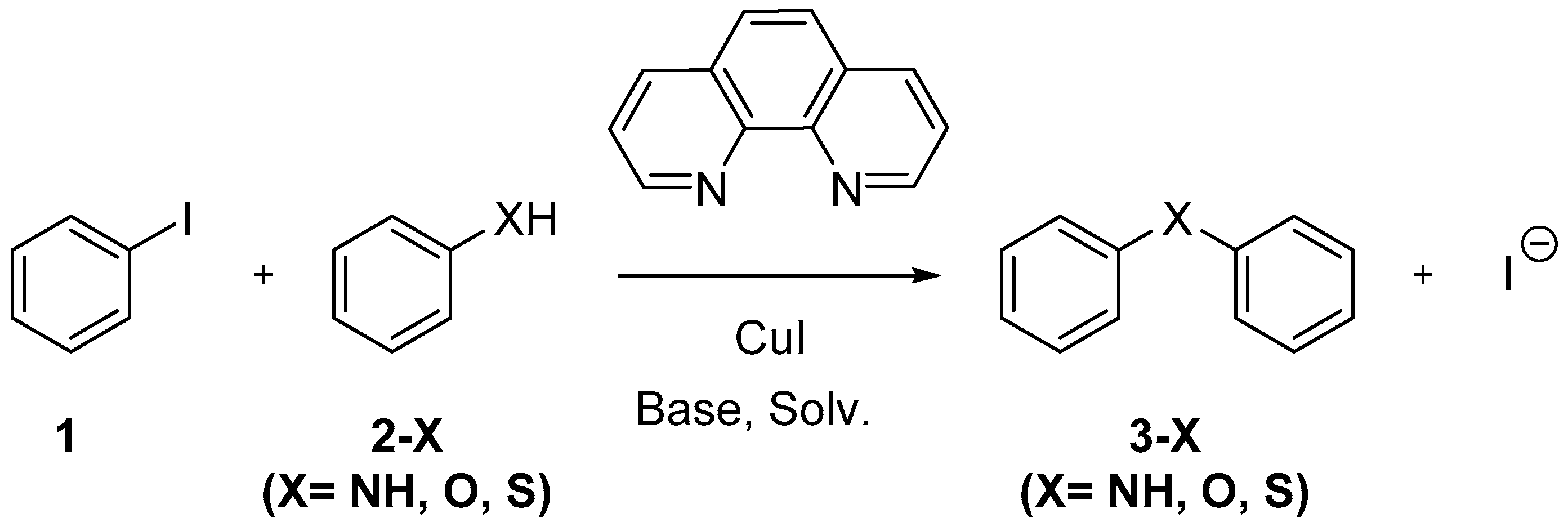
:
1. Introduction
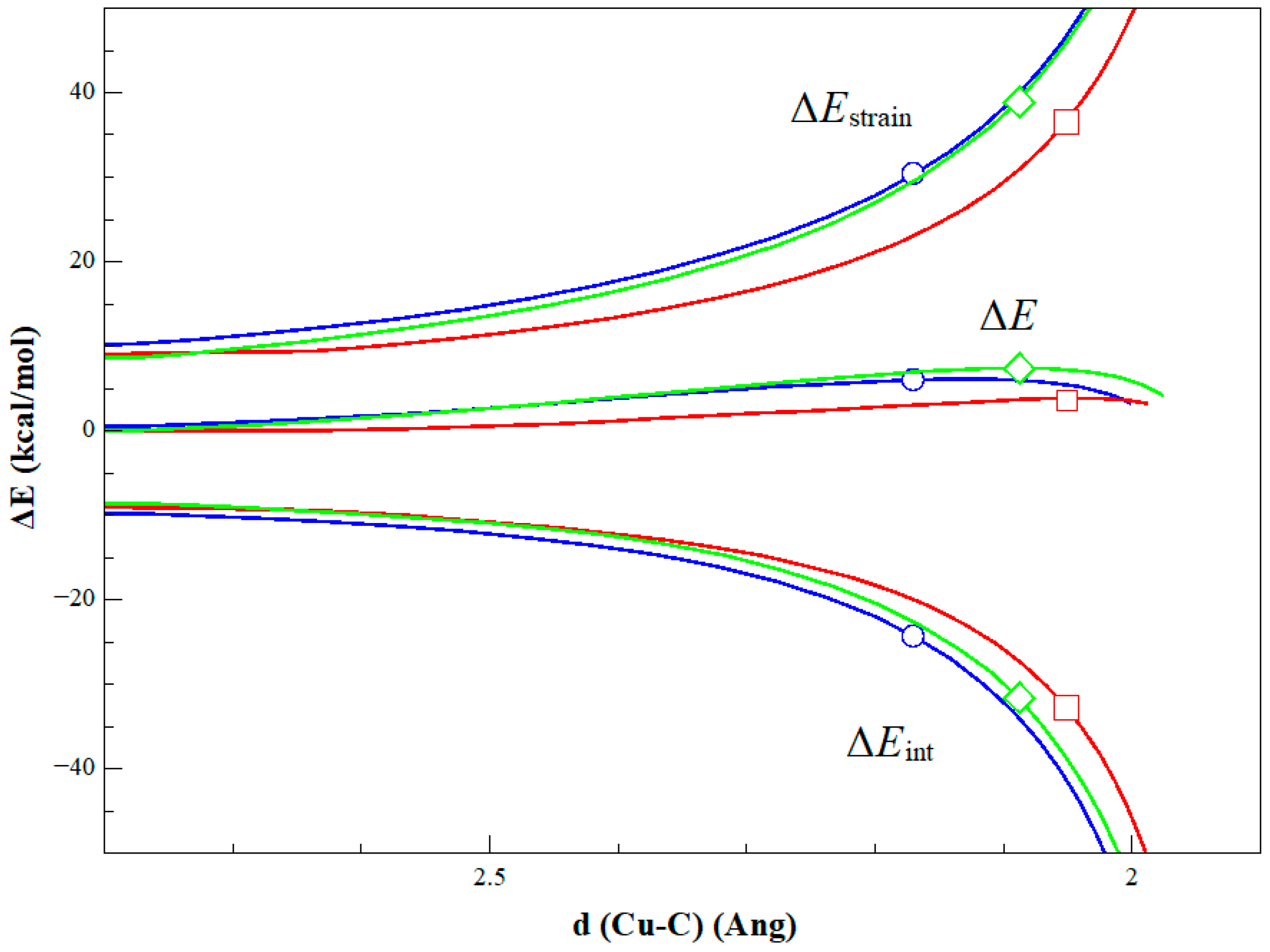
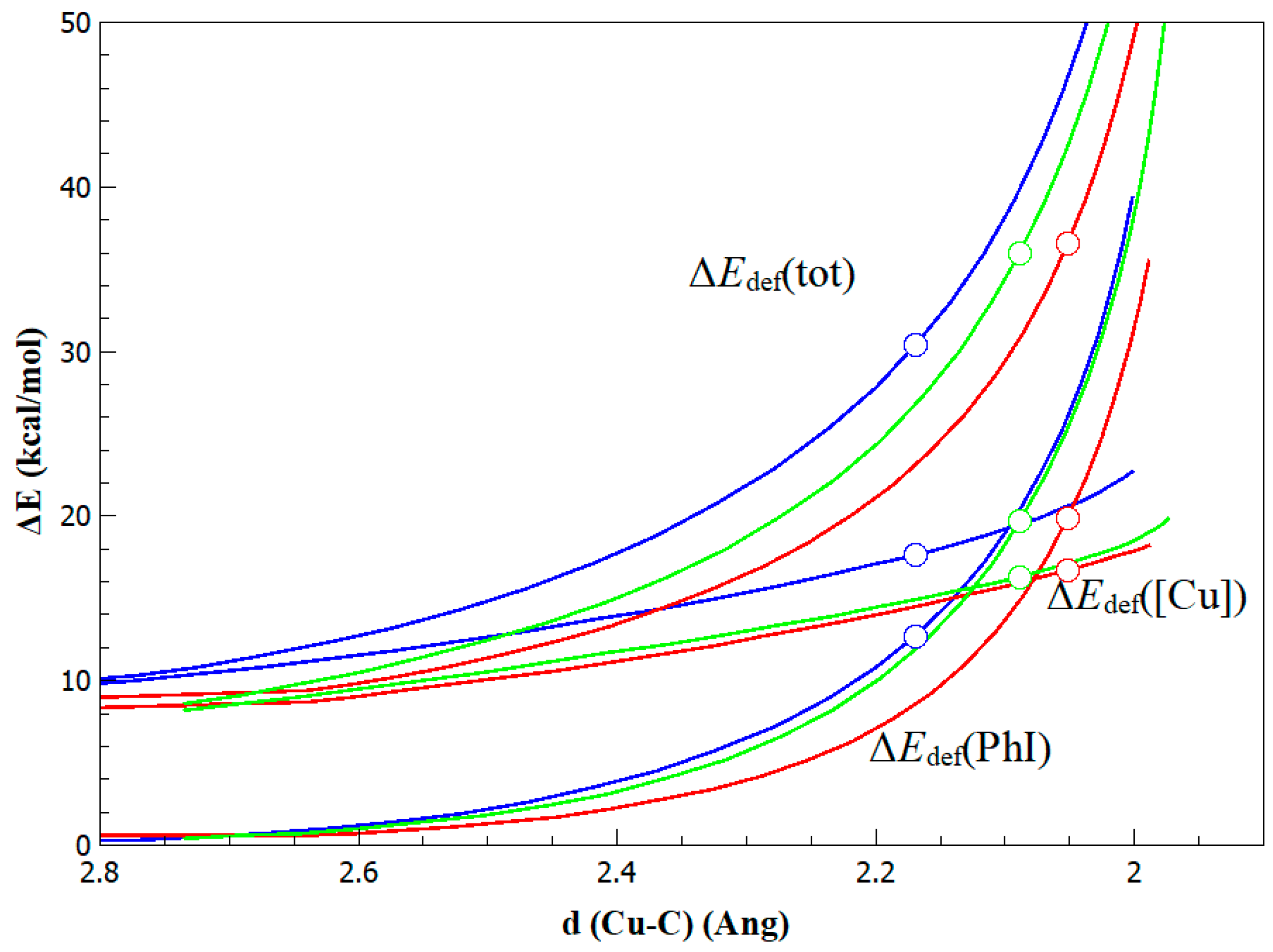
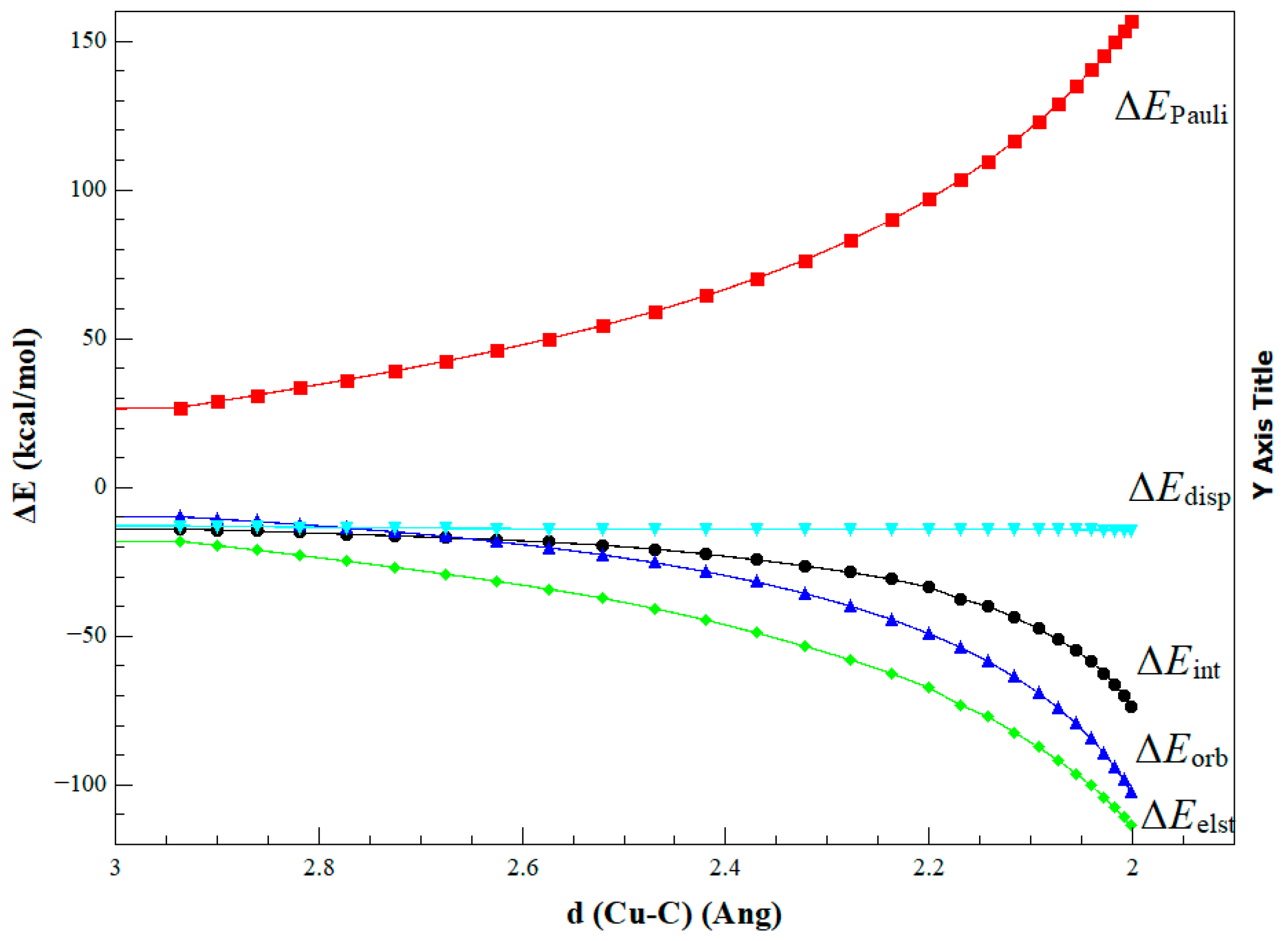
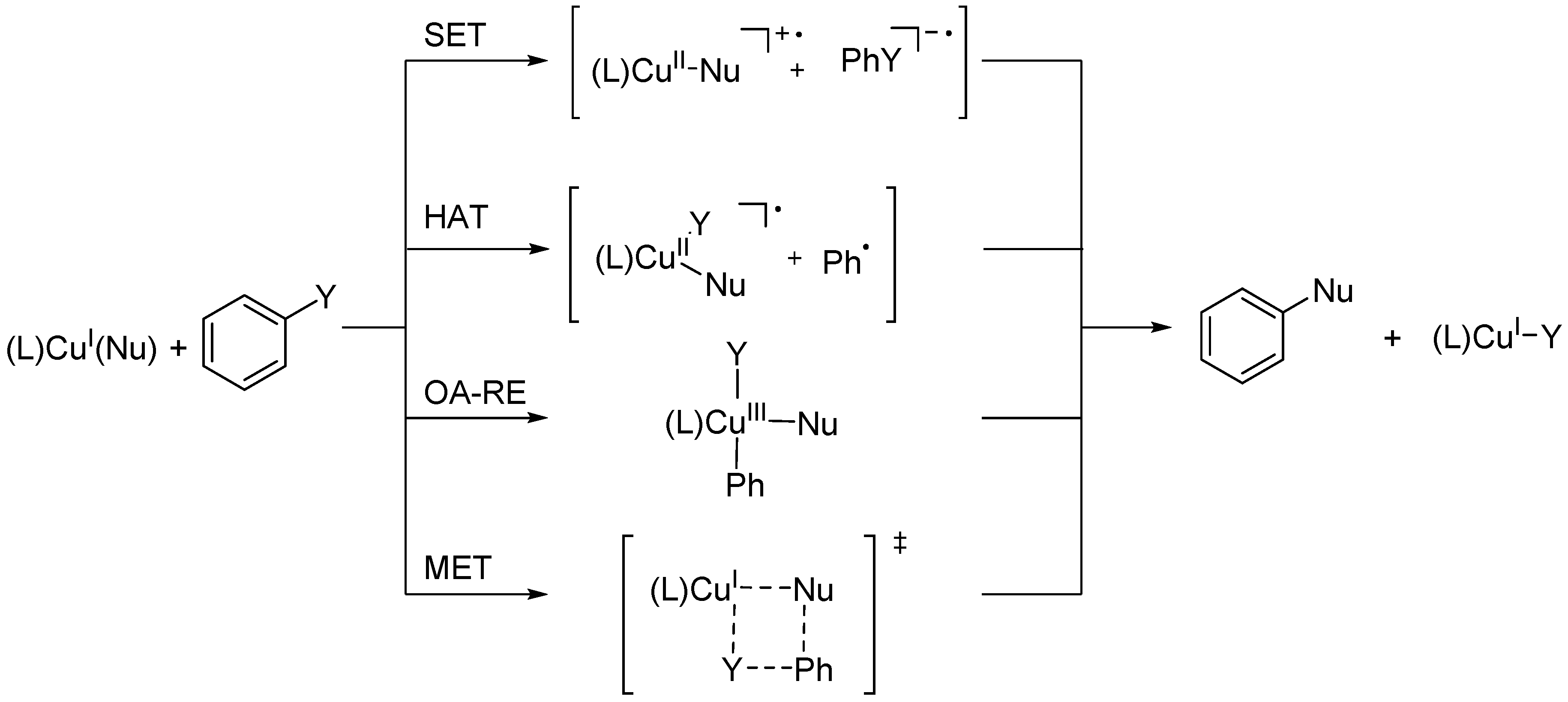
2. Results and Discussion
3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Beletskaya, I.P.; Cheprakov, A.V. Copper in cross-coupling reactions—The post-Ullmann chemistry. Coord. Chem. Rev. 2004, 248, 2337–2364. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Ananikov, V.P. Transition-metal-catalyzed C–S, C–Se, and C–Te bond formation via cross-coupling and atom-economic addition reactions. Chem. Rev. 2011, 111, 1596–1636. [Google Scholar] [CrossRef] [PubMed]
- Evano, G.; Theunissen, C.; Pradal, A. Impact of Copper-catalyzed cross-coupling reactions in natural product synthesis: The emergence of new retrosynthetic paradigms. Nat. Prod. Rep. 2013, 30, 1467–1489. [Google Scholar] [CrossRef] [PubMed]
- Sambiagio, C.; Marsden, S.P.; Blacker, A.J.; McGowan, P.C. Copper catalysed Ullmann type chemistry: From mechanistic aspects to modern development. Chem. Soc. Rev. 2014, 43, 3525–3550. [Google Scholar] [CrossRef] [PubMed]
- Monnier, F.; Taillefer, M. Catalytic C–C, C–N, and C–O Ullmann-type coupling reactions. Angew. Chem. Int. Ed. 2009, 48, 6954–6971. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, S.; Pawar, G.G.; Kumar, S.V.; Jiang, Y.; Ma, D. Selected copper-based reactions for C−N, C−O, C−S, and C-C bond formation. Angew. Chem. Int. Ed. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, F.; Bielecki, J. Ueber synthesen in der biphenylreihe. Ber. Dtsch. Chem. Ges. 1901, 34, 2174–2185. [Google Scholar] [CrossRef]
- Ullmann, F. Ueber eine neue bildungsweise von diphenylaminderivaten. Ber. Dtsch. Chem. Ges. 1903, 36, 2382–2384. [Google Scholar] [CrossRef]
- Ullmann, F.; Sponagel, P. Ueber die phenylirung von phenolen. Ber. Dtsch. Chem. Ges. 1905, 38, 2211–2212. [Google Scholar] [CrossRef]
- Goldberg, I. Ueber phenylirungen bei gegenwart von kupfer als katalysator. Ber. Dtsch. Chem. Ges. 1906, 39, 1691–1692. [Google Scholar] [CrossRef]
- Lindley, J. Tetrahedron report number 163: Copper assisted nucleophilic substitution of aryl halogen. Tetrahedron 1984, 40, 1433–1456. [Google Scholar] [CrossRef]
- Monnier, F.; Taillefer, M. Catalytic C–C, C–N, and C–O Ullmann-type coupling reactions: Copper makes a difference. Angew. Chem. Int. Ed. 2008, 47, 3096–3099. [Google Scholar] [CrossRef] [PubMed]
- Surry, D.S.; Buchwald, S.L. Diamine ligands in Copper-catalyzed reactions. Chem. Sci. 2010, 1, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Goodbrand, H.B.; Hu, N.X. Ligand-accelerated catalysis of the Ullmann condensation: Application to hole conducting triarylamines. J. Org. Chem. 1999, 64, 670–674. [Google Scholar] [CrossRef]
- Gujadhur, R.; Venkataraman, D.; Kintigh, J.T. Formation of aryl-nitrogen bonds using a soluble Copper(I) catalyst. Tetrahedron Lett. 2001, 42, 4791–4793. [Google Scholar] [CrossRef]
- Gujadhur, R.K.; Bates, C.G.; Venkataraman, D. Formation of aryl-nitrogen, aryl-oxygen, and aryl-carbon bonds using well-defined Copper(I)-based catalysts. Org. Lett. 2001, 3, 4315–4317. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Shen, R.C.; Su, S.; Porco, J.A. Copper-mediated synthesis of N-acyl vinylogous carbamic acids and derivatives: Synthesis of the antibiotic CJ-15,801. Org. Lett. 2004, 6, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wolter, M.; Klapars, A.; Buchwald, S.L. Synthesis of N-aryl hydrazides by copper-catalyzed coupling of hydrazides with aryl iodides. Org. Lett. 2001, 3, 3803–3805. [Google Scholar] [CrossRef] [PubMed]
- Klapars, A.; Huang, X.H.; Buchwald, S.L. A general and efficient Copper catalyst for the amidation of aryl halides. J. Am. Chem. Soc. 2002, 124, 7421–7428. [Google Scholar] [CrossRef] [PubMed]
- Kwong, F.Y.; Buchwald, S.L. Mild and efficient Copper-catalyzed amination of aryl bromides with primary alkylamines. Org. Lett. 2003, 5, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Kwong, F.Y.; Klapars, A.; Buchwald, S.L. Copper-catalyzed coupling of alkylamines and aryl iodides: An efficient system even in an air atmosphere. Org. Lett. 2002, 4, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Cai, Q. Copper/amino acid catalyzed cross-couplings of aryl and vinyl halides with nucleophiles. Acc. Chem. Res. 2008, 41, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, Q.; Ma, D.W. Amino acid promoted cui-catalyzed C-N bond formation between aryl halides and amines or N-containing heterocycles. J. Org. Chem. 2005, 70, 5164–5173. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, Y.F.; Zou, W.; Liu, L.; Guo, Q.X. Amino acid-mediated Goldberg reactions between amides and aryl iodides. Tetrahedron Lett. 2004, 45, 2311–2315. [Google Scholar] [CrossRef]
- Shafir, A.; Buchwald, S.L. Highly selective room-temperature Copper-catalyzed C–N coupling reactions. J. Am. Chem. Soc. 2006, 128, 8742–8743. [Google Scholar] [CrossRef] [PubMed]
- Shafir, A.; Lichtor, P.A.; Buchwald, S.L. N- versus O-arylation of aminoalcohols: Orthogonal selectivity in Copper-based catalysts. J. Am. Chem. Soc. 2007, 129, 3490–3491. [Google Scholar] [CrossRef] [PubMed]
- Cristau, H.J.; Cellier, P.P.; Spindler, J.F.; Taillefer, M. Highly efficient and mild Copper-catalyzed N- and C-arylations with aryl bromides and iodides. Chem. Eur. J. 2004, 10, 5607–5622. [Google Scholar] [CrossRef] [PubMed]
- Fagan, P.J.; Hauptman, E.; Shapiro, R.; Casalnuovo, A. Using intelligent/random library screening to design focused libraries for the optimization of homogeneous catalysts: Ullmann ether formation. J. Am. Chem. Soc. 2000, 122, 5043–5051. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Chen, H.-H. 1,1,1-tris(hydroxymethyl)ethane as a new, efficient, and versatile tripod ligand for Copper-catalyzed cross-coupling reactions of aryl iodides with amides, thiols, and phenols. Org. Lett. 2006, 8, 5609–5612. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.C.; Porco, J.A. Synthesis of enamides related to the salicylate antitumor macrolides using copper-mediated vinylic substitution. Org. Lett. 2000, 2, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Ley, S.V.; Thomas, A.W. Modern synthetic methods for copper-mediated C(aryl)–O, C(aryl)–N, and C(aryl)-S bond formation. Angew. Chem. Int. Ed. 2003, 42, 5400–5449. [Google Scholar] [CrossRef] [PubMed]
- Zweig, J.E.; Kim, D.E.; Newhouse, T.R. Methods utilizing first-row transition metals in natural product total synthesis. Chem. Rev. 2017, 117, 11680–11752. [Google Scholar] [CrossRef] [PubMed]
- Evano, G.; Blanchard, N.; Toumi, M. Copper-mediated coupling reactions and their applications in natural products and designed biomolecules synthesis. Chem. Rev. 2008, 108, 3054–3131. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T. The chemical versatility of natural-product assembly lines. Acc. Chem. Res. 2008, 41, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.R.; Frechet, J.M.J. Organic semiconducting oligomers for use in thin film transistors. Chem. Rev. 2007, 107, 1066–1096. [Google Scholar] [CrossRef] [PubMed]
- Daprano, G.; Leclerc, M.; Zotti, G.; Schiavon, G. Synthesis and characterization of polyaniline derivatives—Poly(2-alkoxyanilines) and poly(2,5-dialkoxyanilines). Chem. Mater. 1995, 7, 33–42. [Google Scholar] [CrossRef]
- Casitas, A.; King, A.E.; Parella, T.; Costas, M.; Stahl, S.S.; Ribas, X. Direct observation of Cu-I/Cu-III redox steps relevant to Ullmann-type coupling reactions. Chem. Sci. 2010, 1, 326–330. [Google Scholar] [CrossRef]
- Casitas, A.; Ribas, X. Insights into the mechanism of modern Ullmann–Goldberg coupling reactions. In Copper-Mediated Cross-Coupling Reactions; John Wiley & Sons, Inc.: New York, NY, USA, 2013; pp. 253–279. [Google Scholar]
- Oxidation potentials for Cu(I)/Cu(0), E° = 0.52 V; Cu(II)/Cu(I), E° = 0.162 V, vs. SCE.
- Giri, R.; Hartwig, J.F. Cu(I)-amido complexes in the Ullmann reaction: Reactions of Cu(I)-amido complexes with iodoarenes with and without autocatalysis by Cu(I). J. Am. Chem. Soc. 2010, 132, 15860–15863. [Google Scholar] [CrossRef] [PubMed]
- Bowman, W.R.; Heaney, H.; Smith, P.H.G. Copper(1) catalyzed aromatic nucleophilic-substitution—A mechanistic and synthetic comparison with the SRN1 reaction. Tetrahedron Lett. 1984, 25, 5821–5824. [Google Scholar] [CrossRef]
- Chen, C.H.; Weng, Z.Q.; Hartwig, J.F. Synthesis of Copper(I) thiolate complexes in the thioetherification of aryl halides. Organometallics 2012, 31, 8031–8036. [Google Scholar] [CrossRef] [PubMed]
- Furuta, H.; Maeda, H.; Osuka, A. Doubly n-confused porphyrin: A new complexing agent capable of stabilizing higher oxidation states. J. Am. Chem. Soc. 2000, 122, 803–807. [Google Scholar] [CrossRef]
- Ribas, X.; Jackson, D.A.; Donnadieu, B.; Mahia, J.; Parella, T.; Xifra, R.; Hedman, B.; Hodgson, K.O.; Llobet, A.; Stack, T.D.P. Aryl c-h activation by Cu(II) to form an organometallic aryl-Cu(III) species: A novel twist on copper disproportionation. Angew. Chem. Int. Ed. 2002, 41, 2991–2994. [Google Scholar] [CrossRef]
- Xifra, R.; Ribas, X.; Llobet, A.; Poater, A.; Duran, M.; Sola, M.; Stack, T.D.P.; Benet-Buchholz, J.; Donnadieu, B.; Mahia, J.; et al. Fine-tuning the electronic properties of highly stable organometallic Cu-III complexes containing monoanionic macrocyclic ligands. Chem. Eur. J. 2005, 11, 5146–5156. [Google Scholar] [CrossRef] [PubMed]
- Casitas, A.; Canta, M.; Sola, M.; Costas, M.; Ribas, X. Nucleophilic aryl fluorination and aryl halide exchange mediated by a Cu-I/Cu-III catalytic cycle. J. Am. Chem. Soc. 2011, 133, 19386–19392. [Google Scholar] [CrossRef] [PubMed]
- Huffman, L.M.; Stahl, S.S. Carbon-nitrogen bond formation involving well-defined aryl-Copper(III) complexes. J. Am. Chem. Soc. 2008, 130, 9196–9197. [Google Scholar] [CrossRef] [PubMed]
- Huffman, L.M.; Casitas, A.; Font, M.; Canta, M.; Costas, M.; Ribas, X.; Stahl, S.S. Observation and mechanistic study of facile C-O bond formation between a well-defined aryl-Copper(III) complex and oxygen nucleophiles. Chem. Eur. J. 2011, 17, 10643–10650. [Google Scholar] [CrossRef] [PubMed]
- Bacon, R.G.R.; Hill, H.A.O. Metal ions + complexes in organic reactions.I. Substitution reactions between aryl halides + cuprous salts in organic solvents. J. Chem. Soc. 1964, 1097–1107. [Google Scholar] [CrossRef]
- Bacon, R.G.R.; Karim, A. Metal-ions and complexes in organic reactions 15. Copper-catalyzed substitutions of aryl halides by phthalimide ion. J. Chem. Soc. Perkin Trans. 1973, 1, 272–278. [Google Scholar] [CrossRef]
- Jones, G.O.; Liu, P.; Houk, K.N.; Buchwald, S.L. Computational explorations of mechanisms and ligand-directed selectivities of Copper-catalyzed Ullmann-type reactions. J. Am. Chem. Soc. 2010, 132, 6205–6213. [Google Scholar] [CrossRef] [PubMed]
- Strieter, E.R.; Blackmond, D.G.; Buchwald, S.L. The role of chelating diamine ligands in the Goldberg reaction: A kinetic study on the Copper-catalyzed amidation of aryl iodides. J. Am. Chem. Soc. 2005, 127, 4120–4121. [Google Scholar] [CrossRef] [PubMed]
- Strieter, E.R.; Bhayana, B.; Buchwald, S.L. Mechanistic studies on the Copper-catalyzed N-arylation of amides. J. Am. Chem. Soc. 2009, 131, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Wolter, M.; Nordmann, G.; Job, G.E.; Buchwald, S.L. Copper-catalyzed coupling of aryl iodides with aliphatic alcohols. Org. Lett. 2002, 4, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, G.; Buchwald, S.L. A domino copper-catalyzed C–O coupling-claisen rearrangement process. J. Am. Chem. Soc. 2003, 125, 4978–4979. [Google Scholar] [CrossRef] [PubMed]
- Tye, J.W.; Weng, Z.; Johns, A.M.; Incarvito, C.D.; Hartwig, J.F. Copper complexes of anionic nitrogen ligands in the amidation and imidation of aryl halides. J. Am. Chem. Soc. 2008, 130, 9971–9983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-L.; Liu, L.; Fu, Y.; Guo, Q.-X. Theoretical study on Copper(I)-catalyzed cross-coupling between aryl halides and amides. Organometallics 2007, 26, 4546–4554. [Google Scholar] [CrossRef]
- Zhang, S.L.; Ding, Y.Q. Theoretical study on mechanism of Copper(I)-catalyzed cross-coupling between aryl halides and alkylamines. Organometallics 2011, 30, 633–641. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, Z.; Ding, Y. Proposal for halogen atom transfer mechanism for Ullmann O-arylation of phenols with aryl halides. Dalton Trans. 2012, 41, 13832–13840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-L.; Fan, H.-J. Theoretical study on copper-catalyzed S-arylation of thiophenols with aryl halides: Evidence supporting the LCu(I)-SPh active catalyst and halogen atom transfer mechanism. Organometallics 2013, 32, 4944–4951. [Google Scholar] [CrossRef]
- Zhang, S.L.; Bie, W.F.; Huang, L. Theoretical insights into mechanisms for Copper(I)-catalyzed C–P coupling of diarylphosphines with aryl halides: A combined solvent and ancillary ligand effect on the identity of active catalyst and reaction mechanism. Organometallics 2014, 33, 5263–5271. [Google Scholar] [CrossRef]
- Soria-Castro, S.M.; Penenory, A.B. Efficient Cu-catalyzed base-free C-S coupling under conventional and microwave heating. A simple access to S-heterocycles and sulfides. Beilstein J. Org. Chem. 2013, 9, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Soria-Castro, S.M.; Andrada, D.M.; Caminos, D.A.; Argüello, J.E.; Robert, M.; Peñéñory, A.B. Mechanistic insight into the Cu-catalyzed C–S cross-coupling of thioacetate with aryl halides: A joint experimental–computational study. J. Org. Chem. 2017, 82, 11464–11473. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-Z.; Jiang, Y.-Y.; Fu, Y.; Liu, L. Alternative mechanistic explanation for ligand-dependent selectivities in Copper-catalyzed N- and O-arylation reactions. J. Am. Chem. Soc. 2010, 132, 18078–18091. [Google Scholar] [CrossRef] [PubMed]
- Rout, L.; Saha, P.; Jammi, S.; Punniyamurthy, T. Efficient Copper(I)-catalyzed C–S cross coupling of thiols with aryl halides in water. Eur. J. Org. Chem. 2007, 640–643. [Google Scholar] [CrossRef]
- Fiaschi, P.; Floriani, C.; Pasquali, M.; Chiesivilla, A.; Guastini, C. Copper(I) phenoxide complexes—Synthesis and ligand-induced transformations of the Copper(I) phenoxo functionality. Inorg. Chem. 1986, 25, 462–469. [Google Scholar] [CrossRef]
- Tye, J.W.; Weng, Z.Q.; Giri, R.; Hartwig, J.F. Copper(I) phenoxide complexes in the etherification of aryl halides. Angew. Chem. Int. Ed. 2010, 49, 2185–2189. [Google Scholar] [CrossRef] [PubMed]
- Stange, A.F.; Sixt, T.; Kaim, W. Highly dissymmetric chelate coordination of 3,4,7,8-tetramethyl-1,10-phenanthroline to Cu-I(SR). Chem. Comm. 1998, 469–470. [Google Scholar] [CrossRef]
- We considered only the outer-sphere set mechanism. The inner-sphere set mechanism was not possible to compute since we could not find any stable complex between [(phen)CuI(XPh)] and PhI.
- Marcus, R.A. Theory of oxidation-reduction reactions involving electron transfer. 3. Applications to data on the rates of organic redox reactions. J. Chem. Phys. 1957, 26, 872–877. [Google Scholar] [CrossRef]
- Marcus, R.A. On the theory of electrochemical and chemical electron transfer processes. Can. J. Chem. 1959, 37, 155–163. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical + electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Marcus, R.A. On theory of electron-transfer reactions. 6. Unified treatment for homogeneous and electrode reactions. J. Chem. Phys. 1965, 43, 679. [Google Scholar] [CrossRef]
- Marcus, R.A. The 2nd Robinson, R.A. Memorial lecture—Electron, proton and related transfers. Faraday Discuss. 1982, 74, 7–15. [Google Scholar] [CrossRef]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Marcus, R.A. On the theory of oxidation-reduction reactions involving electron transfer. 1. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Marcus, R.A. Electrostatic free energy and other properties of states having nonequilibrium polarization. 1. J. Chem. Phys. 1956, 24, 979–989. [Google Scholar] [CrossRef]
- Saveant, J.M. A simple-model for the kinetics of dissociative electron-transfer in polar-solvents—Application to the homogeneous and heterogeneous reduction of alkyl-halides. J. Am. Chem. Soc. 1987, 109, 6788–6795. [Google Scholar] [CrossRef]
- Saveant, J.M. Dissociative electron-transfer—New tests of the theory in the electrochemical and homogeneous reduction of alkyl-halides. J. Am. Chem. Soc. 1992, 114, 10595–10602. [Google Scholar] [CrossRef]
- Saveant, J.M. Electron-transfer, bond breaking, and bond formation. Acc. Chem. Res. 1993, 26, 455–461. [Google Scholar] [CrossRef]
- Huang, W.K.; Chen, W.T.; Hsu, I.J.; Han, C.C.; Shyu, S.G. Cross C–S coupling reaction catalyzed by Copper(I) N-heterocyclic carbene complexes. RSC Adv. 2017, 7, 4912–4920. [Google Scholar] [CrossRef]
- Armstrong, A.; Boto, R.A.; Dingwall, P.; Contreras-Garcia, J.; Harvey, M.J.; Mason, N.J.; Rzepa, H.S. The Houk-list transition states for organocatalytic mechanisms revisited. Chem. Sci. 2014, 5, 2057–2071. [Google Scholar] [CrossRef]
- Kruse, H.; Goerigk, L.; Grimme, S. Why the standard B3LYP/6–31G* model chemistry should not be used in DFT calculations of molecular thermochemistry: Understanding and correcting the problem. J. Org. Chem. 2012, 77, 10824–10834. [Google Scholar] [CrossRef] [PubMed]
- Wolters, L.P.; van Zeist, W.-J.; Bickelhaupt, F.M. New concepts for designing d(10)-M(L)(N) catalysts: D regime, s regime and intrinsic bite-angle flexibility. Chem. Eur. J. 2014, 20, 11370–11381. [Google Scholar] [CrossRef] [PubMed]
- Hering, F.; Nitsch, J.; Paul, U.; Steffen, A.; Bickelhaupt, F.M.; Radius, U. Bite-angle bending as a key for understanding group-10 metal reactivity of d(10)-M(NHC)(2) complexes with sterically modest NHC ligands. Chem. Sci. 2015, 6, 1426–1432. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Turbomole GmbH. Turbomole Version 6.0 2015; a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007; TURBOMOLE GmbH: Karlsruhe, Germany, 2007; Available online: http://www.turbomole.com (accessed on 12 December 2017).
- Peng, C.Y.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- McIver, J.W.; Komornic, A. Structure of transition-states in organic reactions—General theory and an application to cyclobutene-butadiene isomerization using a semiempirical molecular-orbital method. J. Am. Chem. Soc. 1972, 94, 2625–2633. [Google Scholar] [CrossRef]
- Atkins, P.W.; De Paula, J. Physical Chemistry, 8th ed.; Oxford University Press: Oxford, NY, USA, 2006. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. Reaction-path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.M.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.M.; Perdew, J.P. Erratum: “Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes [J. Chem. Phys. 119, 12129 (2003)]”. J. Chem. Phys. 2004, 121, 11507. [Google Scholar] [CrossRef]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401–146404. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The pbe0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Erratum: Generalized gradient approximation made simple [phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum—A direct utilization of abinitio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of Pople-Santry-Segal CNDO method to cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural-population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. Gennbo 5.9; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2009. [Google Scholar]
- Wolters, L.P.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 2015, 5, 324–343. [Google Scholar] [CrossRef] [PubMed]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing reaction rates with the distortion/interaction-activation strain model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar] [CrossRef] [PubMed]
- Morokuma, K. Moluecular orbital studies of hydrogen bonds 3. C=O H–O hydrogen bond in H2CO and H2O and H2CO 2H2O. J. Chem. Phys. 1971, 55, 1236–1244. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. Theoretical-study of the ethylene-metal bond in complexes between Cu+, Ag+, Au+, Pt0, or Pt2+ and ethylene, based on the Hartree-Fock-slater transition-state method. Inorg. Chem. 1979, 18, 1558–1565. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. CO, CS, N2, PF3, and CNCH3 as sigma-donors and pi-acceptors—Theoretical-study by the Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Nibbering, N.M.M.; Van Wezenbeek, E.M.; Baerends, E.J. Central bond in the three CN.cntdot.dimers NC-CN, CN-CN and CN-NC: electron pair bonding and Pauli repulsion effects. J. Phys. Chem. 1992, 96, 4864–4873. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-Sham density functional theory: Predicting and understanding chemistry. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000; Volume 15, pp. 1–86. [Google Scholar]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Fernández, I.; Bickelhaupt, F.M. Deeper insight into the diels-alder reaction through the activation strain model. Chem. Asian J. 2016, 11, 3297–3304. [Google Scholar] [CrossRef] [PubMed]
- Fernández, I.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory: Understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef] [PubMed]
- Krijn, J.; Baerends, E.J. Fit Functions in the HFS-Method; Internal Report (in Dutch). Free University: Amsterdam, The Netherlands, 1981. [Google Scholar]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
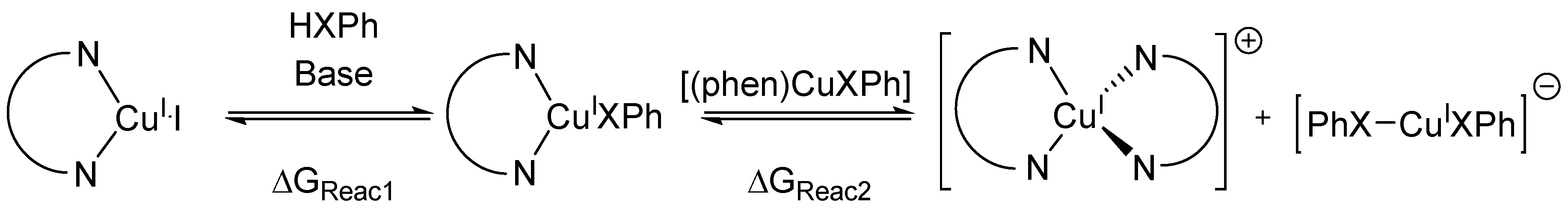
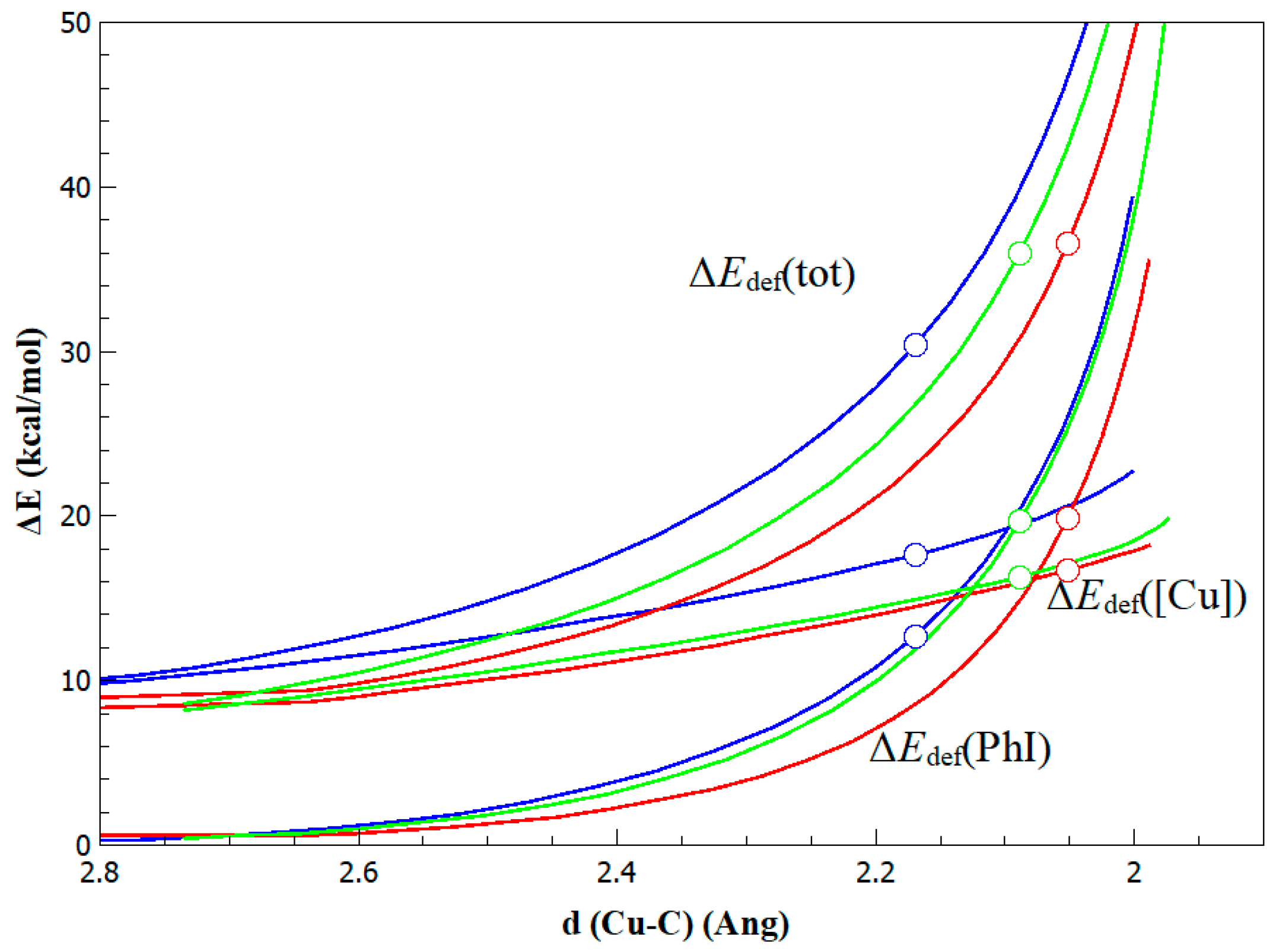


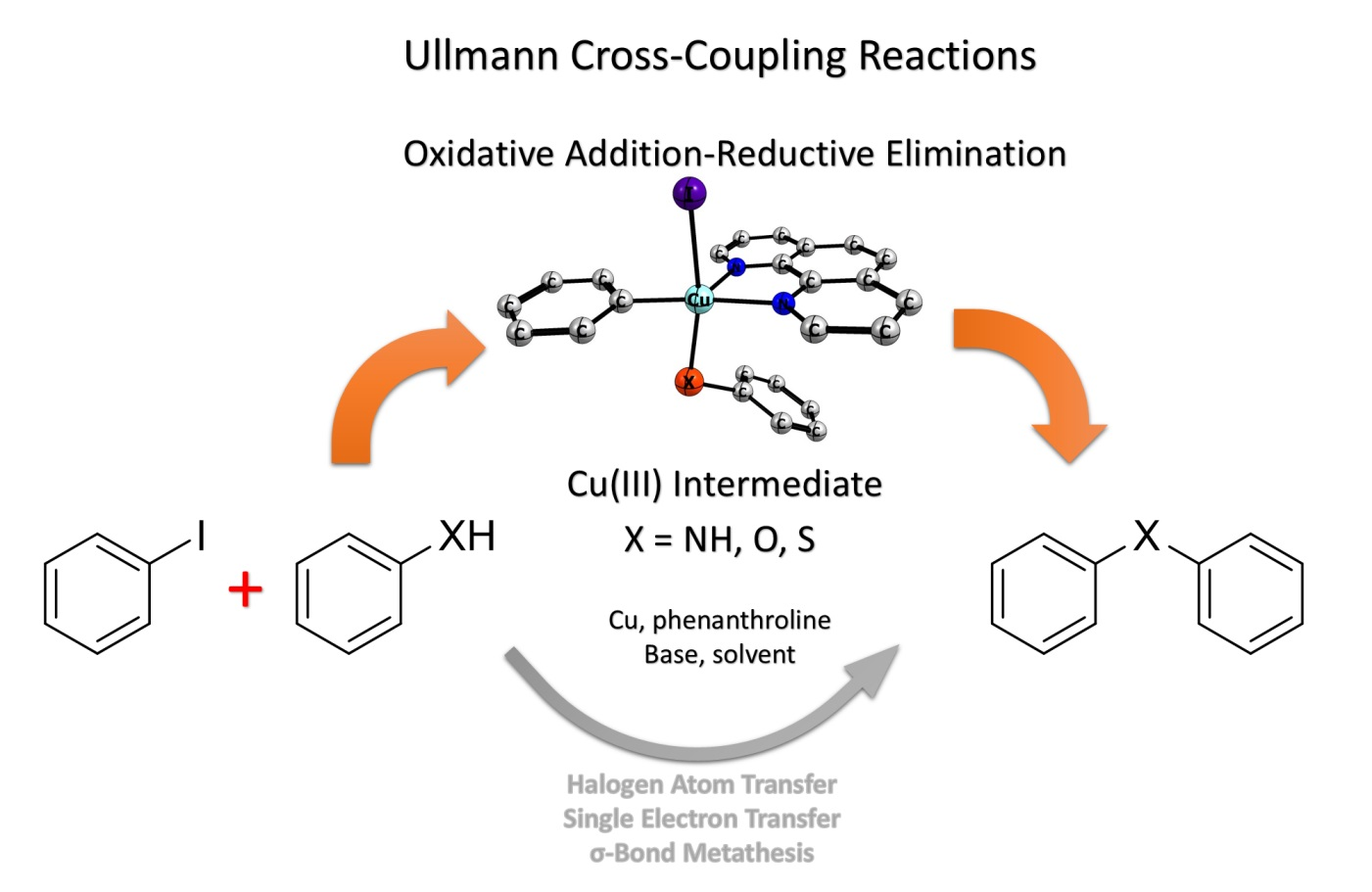{kind=link}
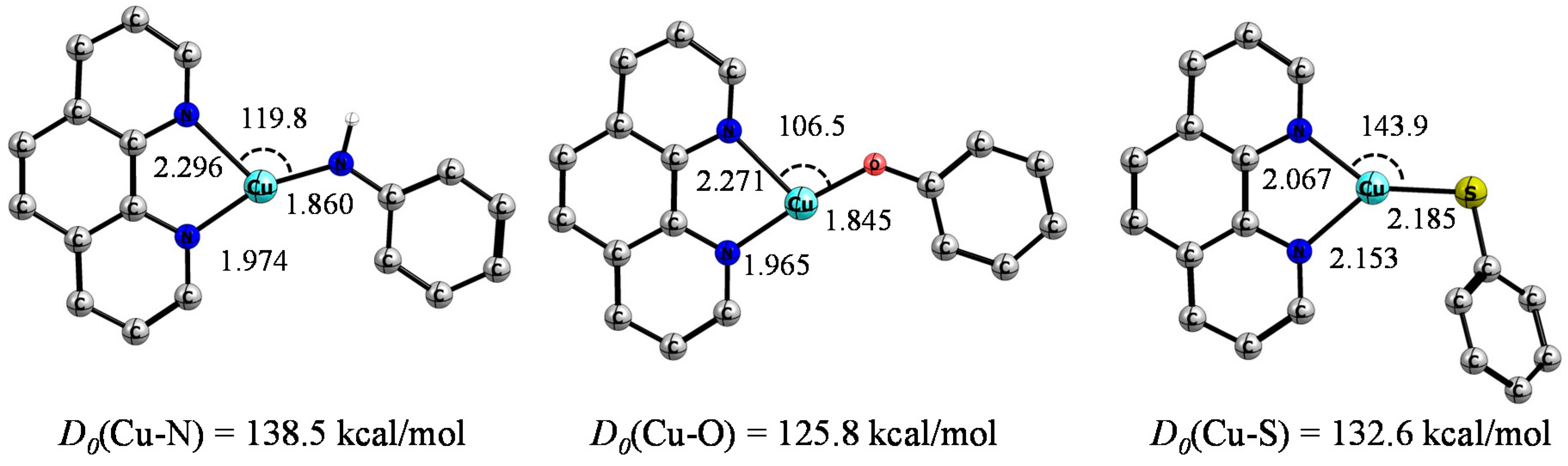{kind=link}
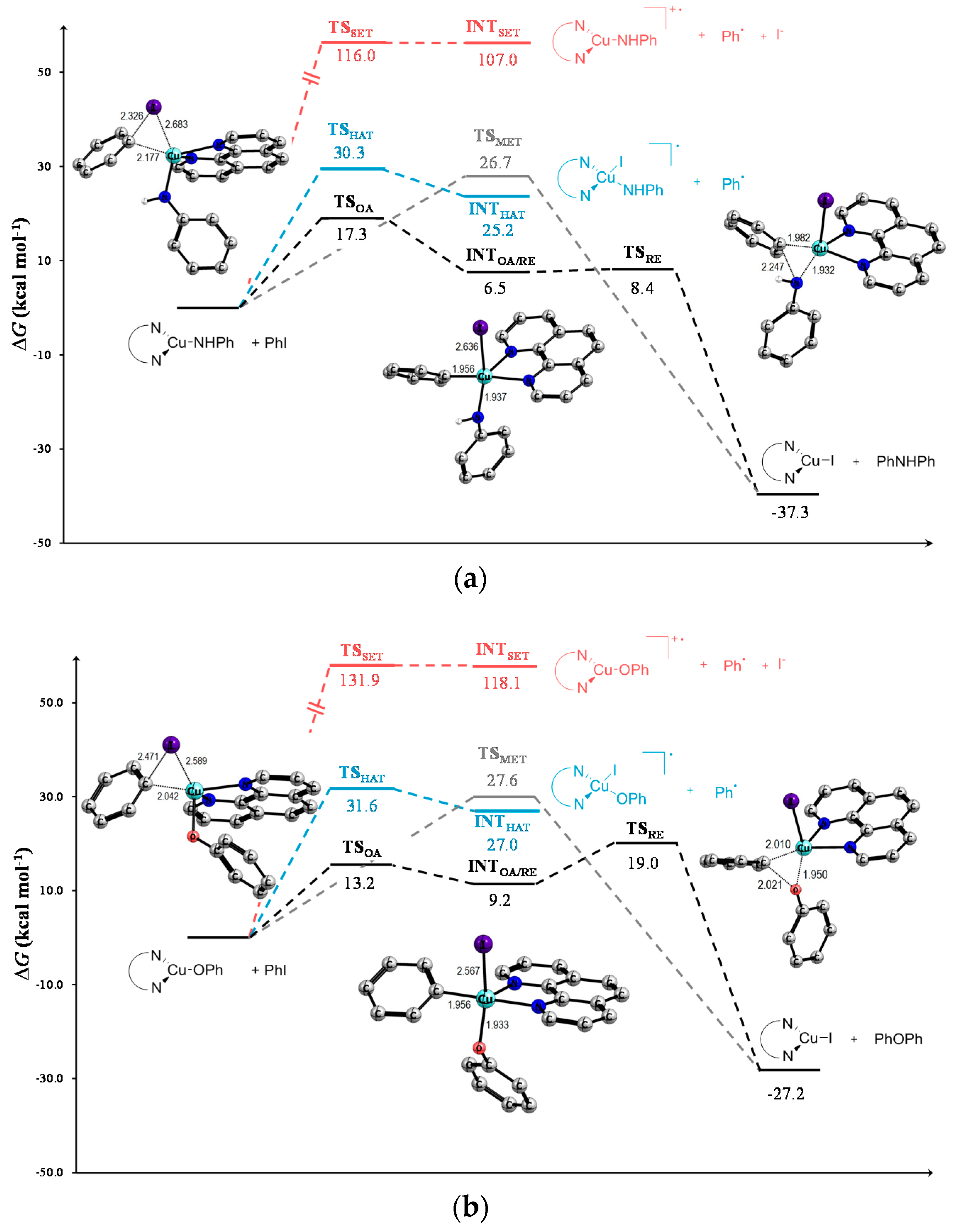{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
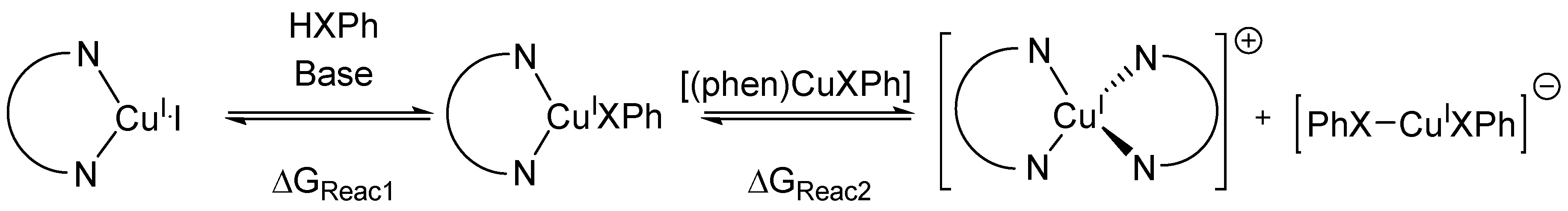{kind=link}
| Reaction | PhNH2 | PhOH | PhSH | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ΔGgas | ΔGtol | ΔGacn | ΔGgas | ΔGtol | ΔGacn | ΔGgas | ΔGtol | ΔGacn | |
| 1 (Cs2CO3) | 15.2 | 8.2 | 2.3 | 8.5 | 0.6 | −5.8 | −4.2 | −12.4 | −19.4 |
| 1 (KOtBu) | 0.2 | −3.2 | −7.7 | −6.5 | −10.8 | −15.8 | −19.2 | −23.8 | −29.4 |
| 2 | 44.7 | 13.7 | −3.4 | 40.4 | 10.6 | −6.0 | 46.9 | 16.4 | −0.7 |
| Entry | SP | PhNH2 | PhOH | PhSH | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ΔGgas | ΔGtol | ΔGacn | ΔGgas | ΔGtol | ΔGacn | ΔGgas | ΔGtol | ΔGacn | ||
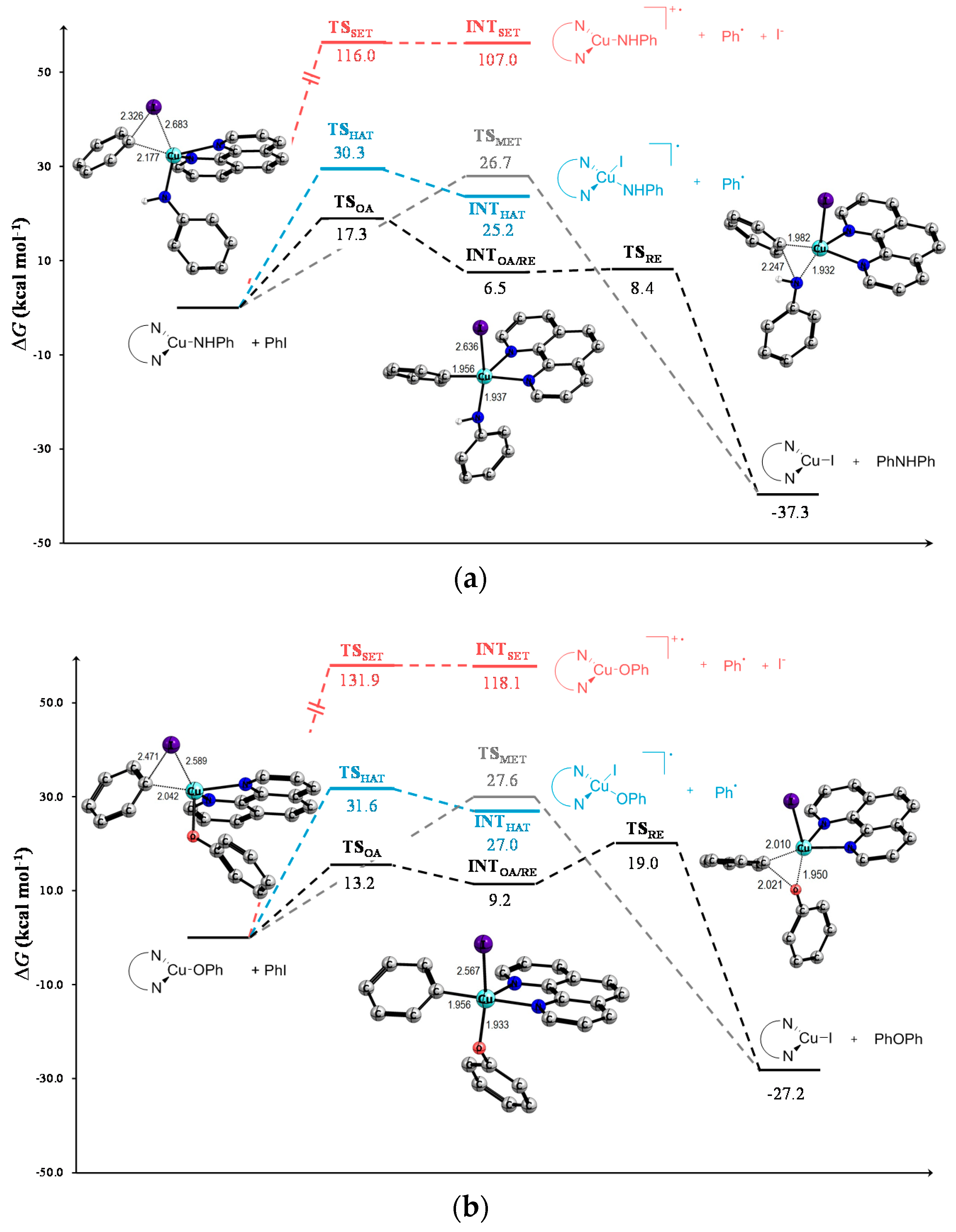
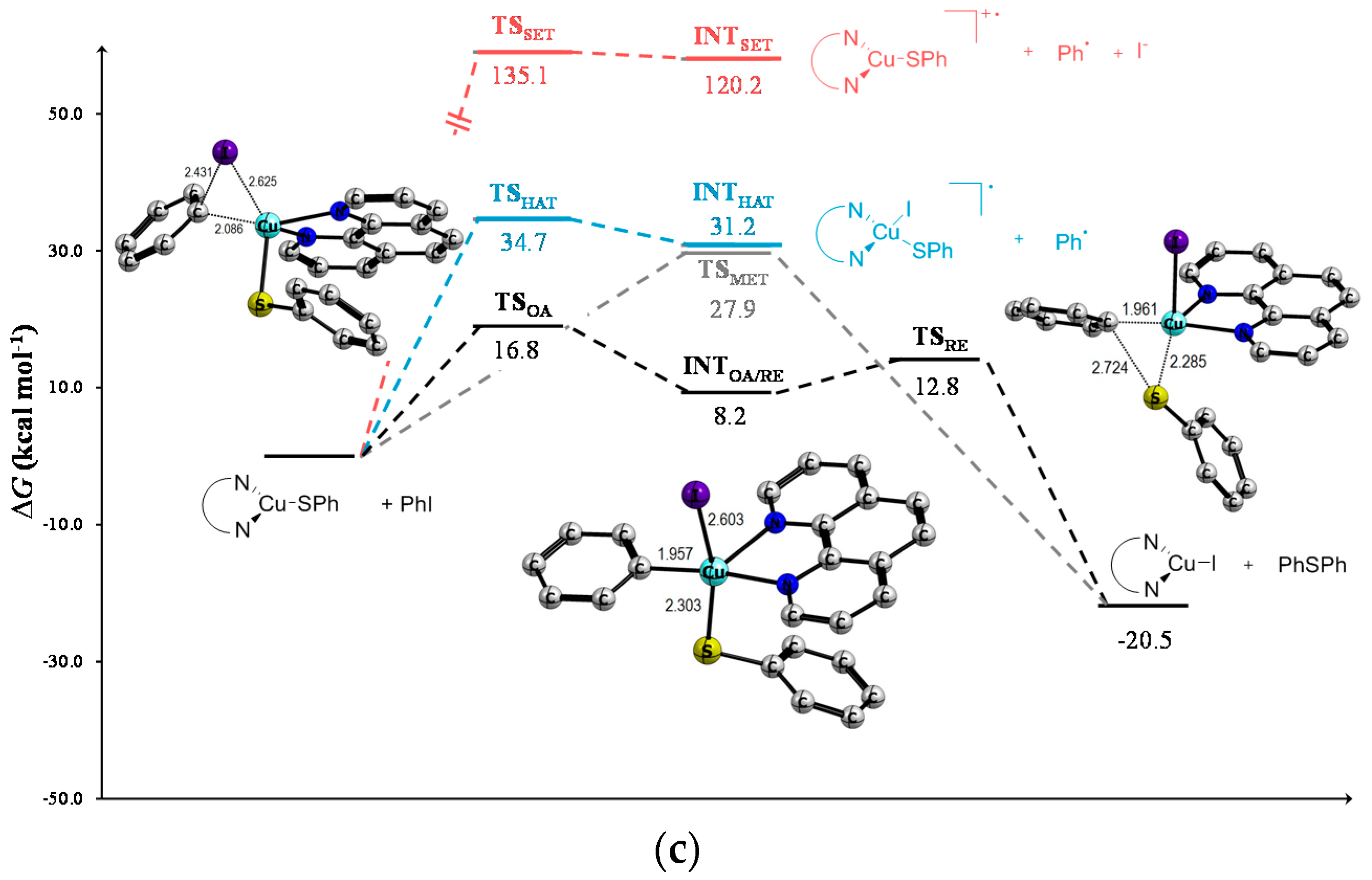
| 1 | TSOA | 17.3 | 18.9 | 20.4 | 13.2 | 15.5 | 17.0 | 16.8 | 19.0 | 20.5 |
| 2 | INTOA/RE | 6.5 | 7.5 | 8.5 | 9.2 | 11.4 | 13.2 | 8.2 | 9.3 | 10.0 |
| 3 | TSRE | 8.4 | 8.2 | 7.9 | 19.0 | 20.1 | 20.7 | 12.8 | 14.1 | 15.1 |
| 4 | TSMet | 26.7 | 28.0 | 29.2 | 27.6 | 30.0 | 32.0 | 27.9 | 29.7 | 31.6 |
| 5 | TSHAT | 30.3 | 29.5 | 32.6 | 31.6 | 31.8 | 35.1 | 34.7 | 34.6 | 37.7 |
| 6 | INTHAT | 25.2 | 23.7 | 22.2 | 27.0 | 27.0 | 26.3 | 31.2 | 30.8 | 30.3 |
| 7 | TSSET | 116.0 | 56.3 | 33.9 | 131.9 | 67.9 | 41.5 | 135.1 | 70.0 | 43.0 |
| 8 | INTSET | 107.0 | 56.2 | 24.3 | 118.1 | 67.8 | 35.6 | 120.2 | 69.7 | 37.7 |
| 9 | Prod | −37.3 | −39.6 | −40.9 | −27.2 | −28.2 | −28.5 | −20.5 | −21.8 | −22.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrada, D.M.; Soria-Castro, S.M.; Caminos, D.A.; Argüello, J.E.; Peñéñory, A.B. Understanding the Heteroatom Effect on the Ullmann Copper-Catalyzed Cross-Coupling of X-Arylation (X = NH, O, S) Mechanism. Catalysts 2017, 7, 388. https://doi.org/10.3390/catal7120388
Andrada DM, Soria-Castro SM, Caminos DA, Argüello JE, Peñéñory AB. Understanding the Heteroatom Effect on the Ullmann Copper-Catalyzed Cross-Coupling of X-Arylation (X = NH, O, S) Mechanism. Catalysts. 2017; 7(12):388. https://doi.org/10.3390/catal7120388
Chicago/Turabian StyleAndrada, Diego M., Silvia M. Soria-Castro, Daniel A. Caminos, Juan E. Argüello, and Alicia B. Peñéñory. 2017. "Understanding the Heteroatom Effect on the Ullmann Copper-Catalyzed Cross-Coupling of X-Arylation (X = NH, O, S) Mechanism" Catalysts 7, no. 12: 388. https://doi.org/10.3390/catal7120388