
Stereoselective Chemoenzymatic Synthesis of Optically Active Aryl-Substituted Oxygen-Containing Heterocycles †

 , , , ,
, , , ,
Abstract
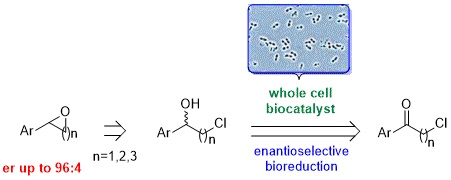
: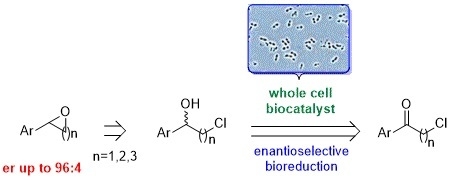
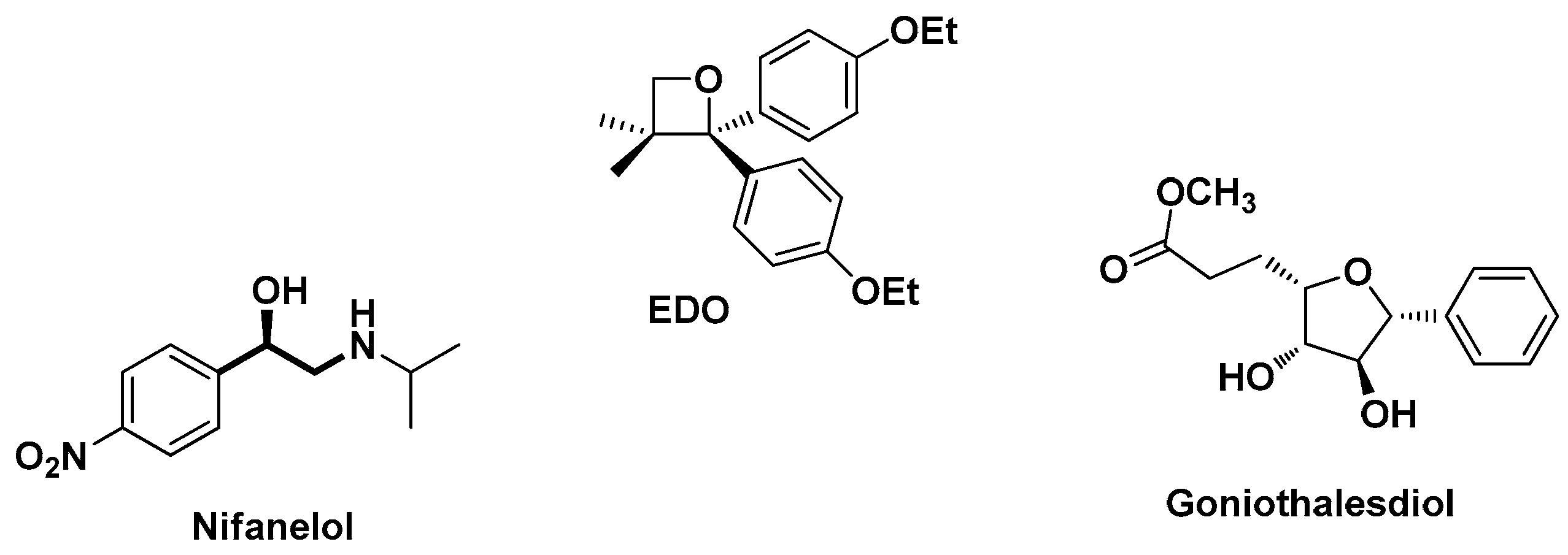
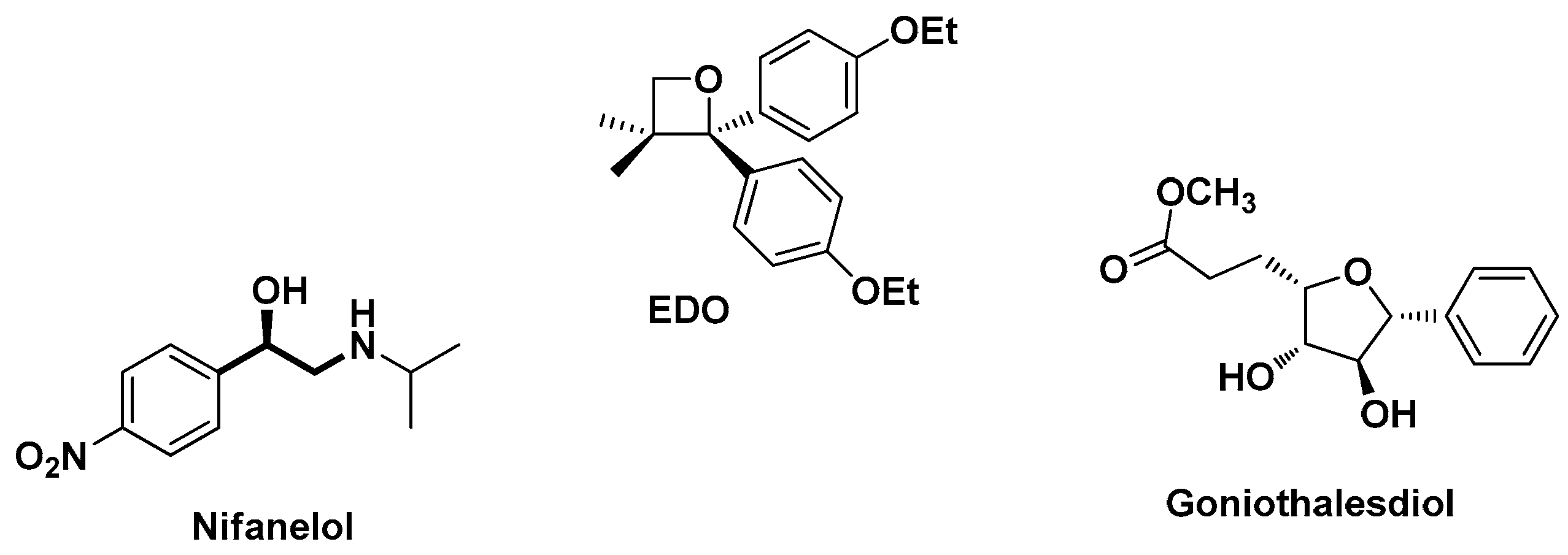
1. Introduction
2. Results
2.1. Screening of Biocatalysts for the Stereoselective Reduction of 3-Chloro-1-Arylpropanones
2.2. Screening of Biocatalysts for the Stereoselective Reduction of 4-Chloro-1-Aryl-1-Butanones
2.3. Screening of Biocatalysts for the Stereoselective Reduction of 2-Chloro-1-Acetophenones
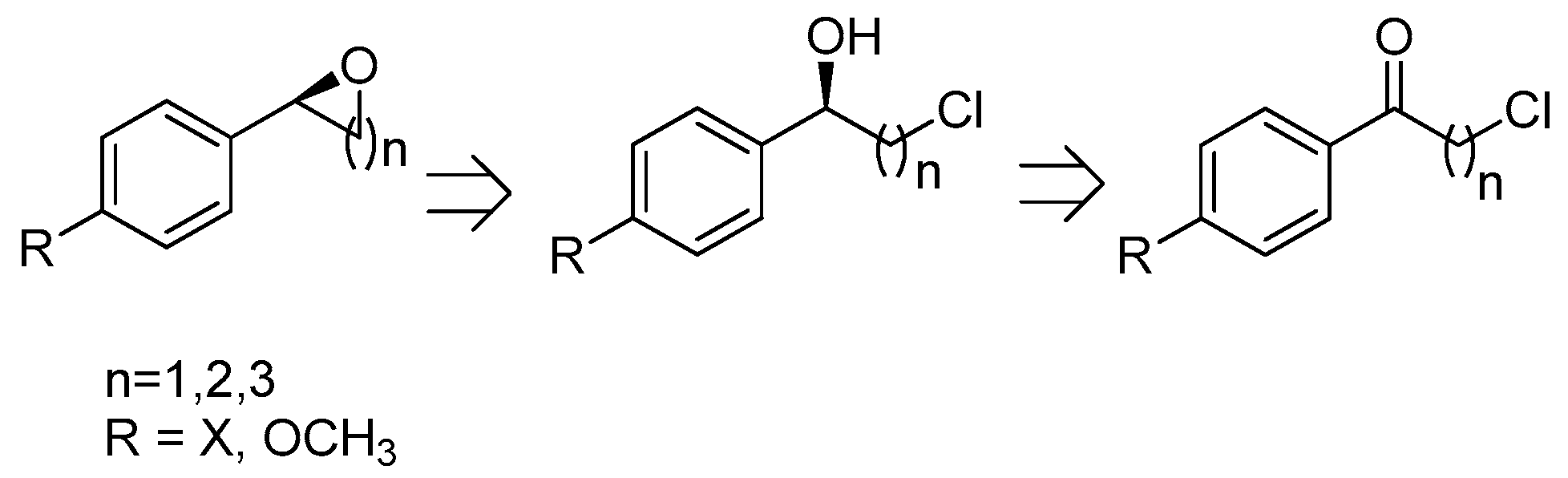
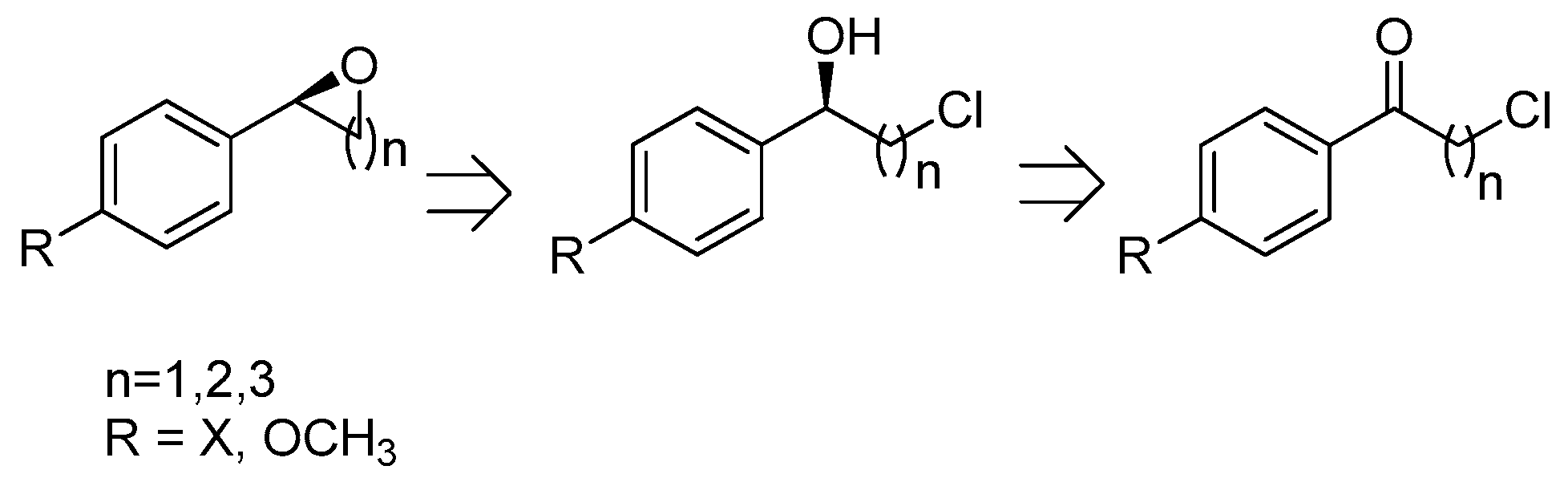
2.4. Synthesis of Optically Active 2-Aryloxetanes, 2-Phenyltetrahydrofurans, 2-Arylepoxides
3. Materials and Method
3.1. General Methods
3.2. Microorganism and Cultures
3.3. Blank Experiments
3.4. Bioreduction of Haloketones 1a,e by Yeasts Growing Cells: General Procedure
3.5. Baker’s Yeast Bioreductions General Procedure
3.6. Characterization Data of Compounds 2a-c,e,i,j and 3a,c,e,i,j
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Perna, F.M.; Salomone, A.; Capriati, V. Recent Developments in the Lithiation Reactions of Oxygen Heterocycles. In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Academic Press Inc.: Oxford, UK, 2016; Volume 118, pp. 91–127. [Google Scholar]
- Capriati, V.; Perna, F.M.; Salomone, A. “The Great Beauty” of organolithium chemistry: a land still worth exploring. Dalton Trans. 2014, 43, 14204–14210. [Google Scholar] [CrossRef] [PubMed]
- Florio, S.; Perna, F.M.; Salomone, A.; Vitale, P. Reduction of Epoxides. In Comprehensive Organic Synthesis, 2nd ed.; Molander, G.A., Knochel, P., Eds.; Elsevier: Oxford, UK, 2014; Volume 8, pp. 1086–1122. [Google Scholar]
- Salomone, A.; Perna, F.M.; Sassone, F.C.; Falcicchio, A.; Bezenšek, J.; Svete, J.; Stanovnik, B.; Florio, S.; Capriati, V. Preparation of Polysubstituted Isochromanes by Addition of ortho-Lithiated Aryloxiranes to Enaminones. J. Org. Chem. 2013, 78, 11059–11065. [Google Scholar] [CrossRef] [PubMed]
- Bayston, D.J.; Travers, C.B.; Polywka, M.E.C. Synthesis and evaluation of a chiral heterogeneous transfer hydrogenation catalyst. Tetrahedron Asymmetry 1998, 9, 2015–2018. [Google Scholar] [CrossRef]
- Cross, D.J.; Kenny, J.A.; Houson, I.; Campbell, L.; Walsgrove, T.; Wills, M. Rhodium versus ruthenium: Contrasting behaviour in the asymmetric transfer hydrogenation of α-substituted acetophenones. Tetrahedron Asymmetry 2001, 12, 1801–1806. [Google Scholar] [CrossRef]
- Morris, D.J.; Hayes, A.M.; Wills, M. The “Reverse-Tethered” Ruthenium (II) Catalyst for Asymmetric Transfer Hydrogenation: Further Applications. J. Org. Chem. 2006, 71, 7035–7044. [Google Scholar] [CrossRef]
- Perrone, M.G.; Santandrea, E.; Giorgio, E.; Bleve, L.; Scilimati, A.; Tortorella, P. A chemoenzymatic scalable route to optically active (R)-1-(pyridin-3-yl)-2-aminoethanol, valuable moiety of β3-adrenergic receptor agonists. Bioorg. Med. Chem. 2006, 14, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Soai, K.; Niwa, S.; Yamanoi, T.; Hikima, H.; Ishizaki, M. Asymmetric synthesis of 2-aryl substituted oxetanes by enantioselective reduction of β-halogenoketones using lithium borohydride modified with N,N′-dibenzoylcystine. J. Chem. Soc., Chem. Commun. 1986, 1018–1019. [Google Scholar] [CrossRef]
- Vitale, P.; Perna, F.M.; Agrimi, G.; Scilimati, A.; Salomone, A.; Cardellicchio, C.; Capriati, V. Asymmetric chemoenzymatic synthesis of 1,3-diols and 2,4-disubstituted aryloxetanes by using whole cell biocatalysts. Org. Biomol. Chem. 2016, 14, 11438–11445. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.M.; Fu, G.C. Applications of Planar-Chiral Heterocycles in Enantioselective Catalysis: Cu(I)/Bisazaferrocene-Catalyzed Asymmetric Ring Expansion of Oxetanes to Tetrahydrofurans. Tetrahedron 2001, 57, 2621–2634. [Google Scholar] [CrossRef]
- Malapit, C.A.; Howell, A.R. Pt-Catalyzed Rearrangement of Oxaspirohexanes to 3-Methylenetetrahydrofurans: Scope and Mechanism. J. Org. Chem. 2015, 80, 8489–8495. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Hasegawa, S.; Harada, T.; Tomisawa, T.; Fujii, A.; Takita, T. Oxetanocin, a novel nucleoside from bacteria. J. Antibiot. 1986, 39, 1623–1625. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Caggon, P.; McPhall, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Wuitschik, G.; Carreira, E.M.; Wagner, B.; Fischer, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Müller, K. Oxetanes in Drug Discovery: Structural and Synthetic Insights. J. Med. Chem. 2010, 53, 3227–3246. [Google Scholar] [CrossRef] [PubMed]
- Jalce, G.; Franck, X.; Figadère, B. Diastereoselective synthesis of 2,5-disubstituted tetrahydrofurans. Tetrahedron Asymmetry 2009, 20, 2537–2650. [Google Scholar] [CrossRef]
- Asano, K.; Matsubara, S. Asymmetric Catalytic Cycloetherification Mediated by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2011, 133, 16711–16713. [Google Scholar] [CrossRef] [PubMed]
- Murayama, H.; Nagao, K.; Ohmiya, H.; Sawamura, M. Copper(I)-Catalyzed Intramolecular Hydroalkoxylation of Unactivated Alkenes. Org. Lett. 2015, 17, 2039–2041. [Google Scholar] [CrossRef] [PubMed]
- Pauli, L.; Tannert, R.; Scheil, R.; Pfaltz, A. Asymmetric Hydrogenation of Furans and Benzofurans with Iridium-Pyridine-Phosphinite Catalysts. Chem. Eur. J. 2015, 21, 1482–1487. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Shibata, S.; Bakshi, R.K. An efficient and catalytically enantioselective route to (S)-(-)-phenyloxirane. J. Org. Chem. 1988, 53, 2861–2863. [Google Scholar] [CrossRef]
- Cordes, D.B.; Kwong, T.J.; Morgan, K.A.; Singaram, B. Chiral Styrene Oxides from α-haloacetophenones Using NaBH4 and TarB-NO2, a Chiral Lewis Acid. Tetrahedron Lett. 2006, 47, 349–351. [Google Scholar] [CrossRef]
- Hamada, T.; Torii, T.; Izawa, K.; Noyori, R.; Ikariya, T. Practical Synthesis of Optically Active Styrene Oxides via Reductive Transformation of 2-Chloroacetophenones with Chiral Rhodium Catalysts. Org. Lett. 2002, 4, 4373–4376. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Torii, T.; Onishi, T.; Izawa, K.; Ikariya, T. Asymmetric Transfer Hydrogenation of α-Aminoalkyl α’-Chloromethyl Ketones with Chiral Rh Complexes. J. Org. Chem. 2004, 69, 7391–7394. [Google Scholar] [CrossRef]
- Matharu, D.S.; Morris, D.J.; Kawamoto, A.M.; Clarkson, G.J.; Wills, M. A Stereochemically Well-Defined Rhodium(III) Catalyst for Asymmetric Transfer Hydrogenation of Ketones. Org. Lett. 2005, 7, 5489–5491. [Google Scholar] [CrossRef]
- Bevinakatti, S.; Banerji, A.A. Practical chemoenzymic synthesis of both enantiomers of propranolol. J. Org. Chem. 1991, 56, 5372–5375. [Google Scholar] [CrossRef]
- Ader, U.; Schneider, M.P. Enzyme assisted preparation of enantiomerically pure β-adrenergic blockers III. Optically active chlorohydrin derivatives and their conversion. Tetrahedron Asymmetry 1992, 3, 521–524. [Google Scholar] [CrossRef]
- Qin, F.; Qin, B.; Mori, T.; Wang, Y.; Meng, L.; Zhang, X.; Jia, X.; Abe, I.; You, S. Engineering of Candida glabrata Ketoreductase 1 for Asymmetric Reduction of α-Halo Ketones. ACS Catal. 2016, 6, 6135–6140. [Google Scholar] [CrossRef]
- De Miranda, A.S.; Simon, R.C.; Grischek, B.; de Paula, G.C.; Horta, B.A.C.; de Miranda, L.S.M.; Kroutil, W.; Kappe, C.O.; de Souza, R.O.M.A. Chiral Chlorohydrins from the Biocatalyzed Reduction of Chloroketones: Chiral Building Blocks for Antiretroviral Drugs. ChemCatChem 2015, 7, 984–992. [Google Scholar] [CrossRef]
- Wu, K.; Chen, L.; Fan, H.; Zhao, Z.; Wang, H.; Wei, D. Synthesis of enantiopure epoxide by ‘one pot’ chemoenzymatic approach using a highly enantioselective dehydrogenase. Tetrahedron Lett. 2016, 57, 899–904. [Google Scholar] [CrossRef]
- Guo, C.; Chen, Y.; Zheng, Y.; Zhang, W.; Tao, Y.; Feng, J.; Tang, L. Exploring the Enantioselective Mechanism of Halohydrin Dehalogenase from Agrobacterium radiobacter AD1 by Iterative Saturation Mutagenesis. Appl. Environ. Microbiol. 2015, 81, 2919–2926. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhu, X.; Zheng, H.; Jiang, R.; Majerić Elenkovb, M. Key Residues for Controlling Enantioselectivity of Halohydrin Dehalogenase from Arthrobacter sp. Strain AD2, Revealed by Structure-Guided Directed Evolution. Appl. Environ. Microbiol. 2012, 78, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Perna, F.M.; Ricci, M.A.; Scilimati, A.; Mena, M.C.; Pisano, I.; Palmieri, L.; Agrimi, G.; Vitale, P. Cheap and environmentally sustainable stereoselective arylketones reduction by Lactobacillus reuteri whole cells. J. Mol. Catal. B Enzym. 2016, 124, 29–37. [Google Scholar] [CrossRef]
- Vitale, P.; D’Introno, C.; Perna, F.M.; Perrone, M.G.; Scilimati, A. Kluyveromyces marxianus CBS 6556 growing cells as a new biocatalyst in the asymmetric reduction of substituted acetophenones. Tetrahedron Asymmetry 2013, 24, 389–394. [Google Scholar]
- Vitale, P.; Perna, F.M.; Perrone, M.G.; Scilimati, A. Screening On The Use Of Kluyveromyces Marxianus CBS 6556 Growing Cells As Enantioselective Biocatalyst For Ketones Reduction. Tetrahedron Asymmetry 2011, 22, 1985–1993. [Google Scholar] [CrossRef]
- Vitale, P.; Abbinante, V.M.; Perna, F.M.; Salomone, A.; Cardellicchio, C.; Capriati, V. Unveiling the Hidden Performance of Whole Cells in the Asymmetric Bioreduction of Aryl-containing Ketones in Aqueous Deep Eutectic Solvents. Adv. Synth. Catal. 2016. [Google Scholar] [CrossRef]
- Brenna, E. Synthetic Methods for Biologically Active Molecules: Exploring the Potential of Bioreductions; Brenna, E., Ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Faber, K. Biotransformations in Organic Chemistry: A Textbook, 6th ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Csuk, R.; Glanzer, I. Yeast-Mediated Stereoselective Biocatalysis; Patel, R.N., Ed.; Stereoselective Biocatalysis, Marcel Dekker: New York, NY, USA, 2000; pp. 527–578. [Google Scholar]
- Rodrigues, J.A.R.; Moran, P.J.S.; Conceicao, G.J.A.; Fardelone, L.C. Asymmetric Reduction of Carbonyl Compounds. Food Technol. Biotechnol. 2004, 42, 295–303. [Google Scholar]
- Utsukihara, T.; Okada, S.; Kato, N.; Horiuchi, C.A. Biotransformation of α-bromo and α,α′-dibromo alkanone to α-hydroxyketone and α-diketone by Spirulina platensis. J. Mol. Catal. B Enzym. 2007, 45, 68–72. [Google Scholar] [CrossRef]
- Wagner, P.J.; Lindstrom, M.J.; Sedon, J.H.; Ward, D.R. Photochemistry of delta-haloketones: Anchimeric assistance in triplet-state gamma-hydrogen abstraction and beta-elimination of halogen atoms from the resulting diradicals. J. Am. Chem. Soc. 1981, 103, 3842–3849. [Google Scholar] [CrossRef]
- Aleixo, L.M.; De Carvalho, M.; Moran, P.J.S.; Rodrigues, J.A.R. Hydride transfer versus electron transfer in the baker’s yeast reduction of α-haloacetophenones. Bioorg. Med. Chem. Lett. 1993, 3, 1637–1642. [Google Scholar] [CrossRef]
- Rocha, L.C.; Ferreira, H.V.; Pimenta, E.F.; Souza Berlinck, R.G.; Oliveira Rezende, M.O.; Landgraf, M.D.; Regali Seleghim, M.H.; Durães Sette, L.; Meleiro Porto, A.L. Biotransformation of α-bromoacetophenones by the marine fungus Aspergillus sydowii. Mar. Biotechnol. 2010, 12, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Barbasiewicz, M.; Brud, A.; Mąkosza, M. Synthesis of Substituted Tetrahydropyrans via Intermolecular Reactions of δ-Halocarbanions with Aldehydes. Synthesis 2007, 1209–1213. [Google Scholar] [CrossRef]
- De Carvalho, M.; Okamoto, M.T.; Moran, P.J.S.; Rodrigues, J.A.R. Baker’s yeast reduction of α-haloacetophenones. Tetrahedron 1991, 47, 2073–2080. [Google Scholar] [CrossRef]
- Wu, K.; Wang, H.; Chen, L.; Fan, H.; Zhao, Z.; Wei, D. Practical two-step synthesis of enantiopure styrene oxide through an optimized chemoenzymatic approach. Appl. Microbiol. Biotechnol. 2016, 100, 8757–8767. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.M.; Musa, M.M.; Rodriguez, L.; Sutton, D.A.; Popik, V.V.; Phillips, R.S. Mutation of Thermoanaerobacter ethanolicus secondary alcohol dehydrogenase at Trp-110 affects stereoselectivity of aromatic ketone reduction. Org. Biomol. Chem. 2014, 12, 5905–5910. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.A.; Russo, A.; Pisano, I.; Palmieri, L.; de Angelis, M.; Agrimi, G. Improved 1,3-Propanediol Synthesis from Glycerol by the Robust Lactobacillus reuteri Strain DSM 20016. J. Microbiol. Biotechnol. 2015, 6, 893–902. [Google Scholar] [CrossRef] [PubMed]
- De Man, J.C.; Rogosa, M.; Sharpe, M.E. A medium for the cultivation of lactobacilli. J. Appl. Bacteriol. 1960, 23, 130–135. [Google Scholar] [CrossRef]
- Zhou, J.-N.; Fang, Q.; Hu, Y.-H.; Yang, L.-Y.; Wu, F.-F.; Xie, L.-J.; Wu, J.; Li, S. Copper(II)-catalyzed enantioselective hydrosilylation of halo-substituted alkyl aryl and heteroaryl ketones: Asymmetric synthesis of (R)-fluoxetine and (S)-duloxetine. Org. Biomol. Chem. 2014, 12, 1009–1017. [Google Scholar] [CrossRef]
- Pop, L.A.; Czompa, A.; Paizs, C.; Toşa, M.I.; Vass, E.; Mátyus, P.; Irimie, F.-D. Lipase-Catalyzed Synthesis of Both Enantiomers of 3-Chloro-1-arylpropan-1-ols. Synthesis 2011, 18, 2921–2928. [Google Scholar]
- Janeczko, T.; Kostrzewa-Susłow, E. Enantioselective reduction of propiophenone formed from 3-chloropropiophenone and stereoinversion of the resulting alcohols in selected yeast cultures. Tetrahedron Asymmetry 2014, 25, 1264–1269. [Google Scholar] [CrossRef]
- Yu, F.; Zhou, J.-N.; Zhang, X.-C.; Sui, Y.-Z.; Wu, F.-F.; Xie, L.-J.; Chan, A.S.C.; Wu, J. Copper(II)-Catalyzed Hydrosilylation of Ketones Using Chiral Dipyridylphosphane Ligands: Highly Enantioselective Synthesis of Valuable Alcohols. Chem. Eur. J. 2011, 17, 14234–14240. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, B.; Xia, H.-F.; Ye, D.; Wu, J.; Shi, Y. Mesoporous silica KIT-6 supported superparamagnetic CuFe2O4 nanoparticles for catalytic asymmetric hydrosilylation of ketones in air. Green Chem. 2014, 16, 2680–2688. [Google Scholar] [CrossRef]
- Coppi, D.I.; Salomone, A.; Perna, F.M.; Capriati, V. 2-Lithiated-2-phenyloxetane: A new attractive synthon for the preparation of oxetane derivatives. Chem. Commun. 2011, 47, 9918–9920. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C. 2-Aryloxoetanes. I. Synthesis and NMR study. Bull. Soc. Chim. Fr. 1969, 10, 3648–3652. [Google Scholar]
- Schaal, C. Synthese. Extension des relations lineaires d’enthalpie libre aux parametres de resonance magnetique nucleaire. Bull. Soc. Chim. Fr. 1971, 8, 3064–3070. [Google Scholar]
- Chan, T.H.; Pellon, P. Chiral organosilicon compounds in synthesis. Highly enantioselective synthesis of arylcarbinols. J. Am. Chem. Soc. 1989, 111, 8737–8738. [Google Scholar] [CrossRef]
- Jokisaari, J.; Rahkamaa, E.; Malo, H. Studies on the PMR Spectra of Oxetanes: IV. 2-(4-Halophenyl) oxetanes. Zeitschrift für Naturforschung A 1971, 26, 973–978. [Google Scholar] [CrossRef]
- Zeng, X.H.; Miao, C.X.; Wang, S.F.; Xia, C.G.; Sun, W. Asymmetric 5-endo chloroetherification of homoallylic alcohols toward the synthesis of chiral β-chlorotetrahydrofurans. Chem. Commun. 2013, 49, 2418–2420. [Google Scholar] [CrossRef] [PubMed]
- Capriati, V.; Florio, S.; Luisi, R.; Salomone, A. Oxiranyl Anion-Mediated Synthesis of Highly Enantiomerically Enriched Styrene Oxide Derivatives. Org. Lett. 2002, 4, 2445–2448. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Wang, Z.; Li, Z. Preparation of (S)-2-, 3-, and 4-chlorostyrene oxides with the epoxide hydrolase from Sphingomonas sp. HXN-200. Tetrahedron Asymmetry 2008, 19, 407–415. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Biocatalyst | Ar | Ketone 1 | Product 2 (Yield %) b | Conversion % | er c | Abs. Conf. d |
| 1 | Baker’s yeast (RC) | C6H5 | 1a | 2a (42) | 50 | 94:6 | S |
| 2 | Saccharomyces cerevisiae (GC) e | C6H5 | 1a | 2a (48) | 55 | 75:25 | S |
| 3 | Kluyveromyces marxianus (GC) f | C6H5 | 1a | 2a (31) g | 70 | 58:42 | S |
| 4 | Baker’s yeast (RC) | 4-FC6H4 | 1b | 2b (13) | 15 | 63:37 | S |
| 5 | Baker’s yeast (RC) | 4-BrC6H4 | 1c | 2c (5) h | 85 | 95:5 | S |
| 6 | Baker’s yeast (RC) | 4-MeOC6H4 | 1d | 2d (–) | 12 | ND i | ND i |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Biocatayst | Ar | Substrate 1 | Product 2 (Yield %) b | Conversion (%) | er c | Abs. Conf. d |
| 1 | Baker’s yeast (RC) | C6H5 | 1e | 2e (44) | 49 | 95:5 | S |
| 2 | S. cerevisiae (GC) e | C6H5 | 1e | 2e (65) | 70 | 49:51 | S |
| 3 | K. marxianus (GC) f | C6H5 | 1e | 2e (4) | 7 | 42:58 | S |
| 4 | Baker’s yeast (RC) | 4-FC6H4 | 1f | 2f (–) g | 40 | ND h | ND h |
| 5 | Baker’s yeast (RC) | 4-BrC6H4 | 1g | 2g i | – i | ND h | ND h |
| 6 | Baker’s yeast (RC) | 4-CH3OC6H4 | 1h | 2h (–) j | 5 | ND h | ND h |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Biocatayst | Ar | Substrate | Chlorohydrin 2 (Yield %) a | Conversion (%) | Er b | Abs. Conf. c |
| 1 | Baker’s yeast d | C6H5 | 1i | 2i (53) | 55 | 90:10 | R |
| 2 | Baker’s yeast | 4-ClC6H4 | 1j | 2j (64) | 70 | 63:37 | R |
| 3 | L. reuteri (RC) e | 4-ClC6H4 | 1j | 2j (28) | 30 | 96:4 | S |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ar | Chlorohydrin 2 (er) | n | Product 3 (Yield %) b | er c | Abs. Conf. d |
| 1 | C6H5 | (S)-2a (94:6) | 2 | 3a (98) | 95:5 | S |
| 2 | 4-BrC6H4 | (S)-2c (95:5) | 2 | 3c (98) | 96:4 | S |
| 3 | C6H5 | (S)-2e (95:5) | 3 | 3e (98) | 95:5 | S |
| 4 | C6H5 | (R)-2i (90:10) | 1 e | 3i (95) | 90:10 | R |
| 5 | 4-ClC6H4 | (S)-2j (96:4) | 1 e | 3j (97) | 96:4 | S |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitale, P.; Digeo, A.; Perna, F.M.; Agrimi, G.; Salomone, A.; Scilimati, A.; Cardellicchio, C.; Capriati, V. Stereoselective Chemoenzymatic Synthesis of Optically Active Aryl-Substituted Oxygen-Containing Heterocycles. Catalysts 2017, 7, 37. https://doi.org/10.3390/catal7020037
Vitale P, Digeo A, Perna FM, Agrimi G, Salomone A, Scilimati A, Cardellicchio C, Capriati V. Stereoselective Chemoenzymatic Synthesis of Optically Active Aryl-Substituted Oxygen-Containing Heterocycles. Catalysts. 2017; 7(2):37. https://doi.org/10.3390/catal7020037
Chicago/Turabian StyleVitale, Paola, Antonia Digeo, Filippo Maria Perna, Gennaro Agrimi, Antonio Salomone, Antonio Scilimati, Cosimo Cardellicchio, and Vito Capriati. 2017. "Stereoselective Chemoenzymatic Synthesis of Optically Active Aryl-Substituted Oxygen-Containing Heterocycles" Catalysts 7, no. 2: 37. https://doi.org/10.3390/catal7020037