
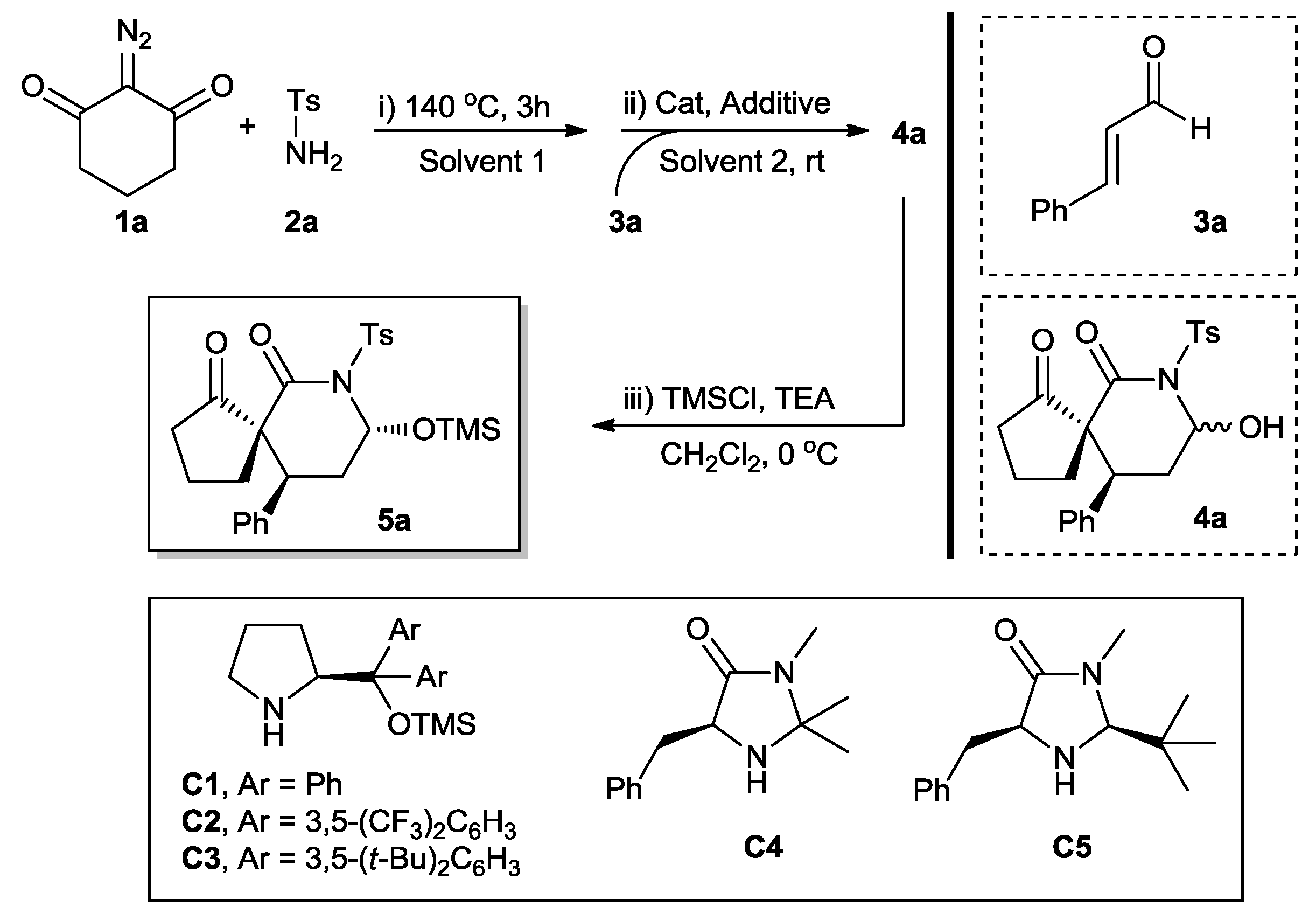
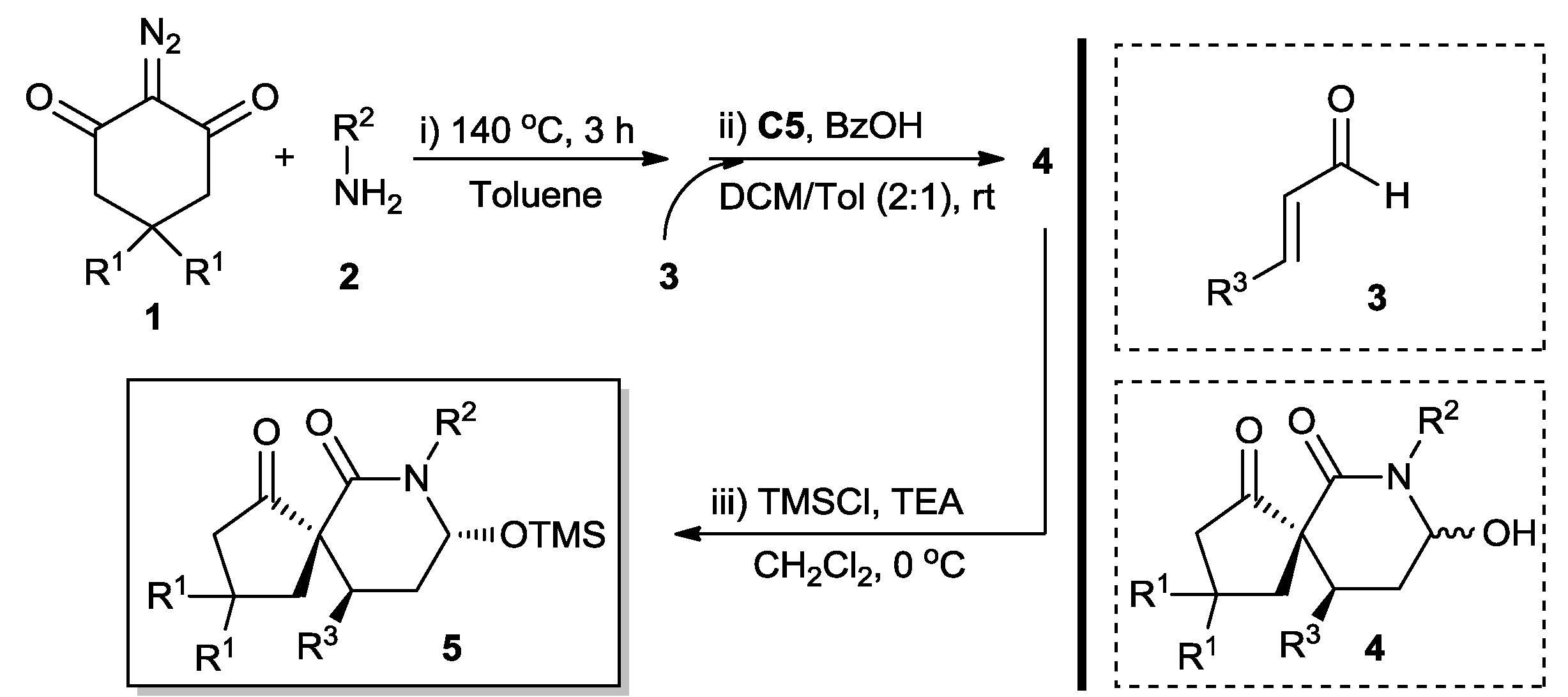
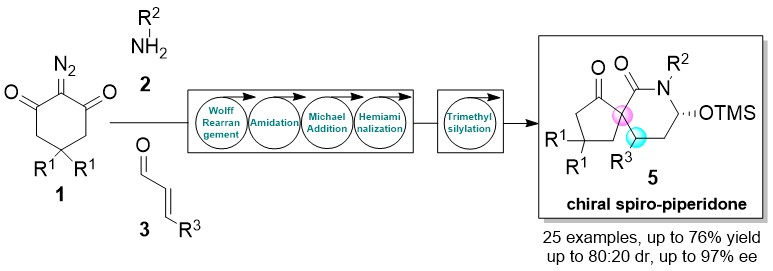
One-Pot Two-Step Organocatalytic Asymmetric Synthesis of Spirocyclic Piperidones via Wolff Rearrangement–Amidation–Michael–Hemiaminalization Sequence

 ,
,
Abstract
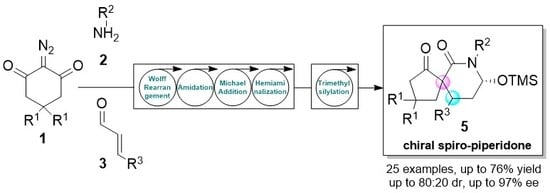
:
1. Introduction
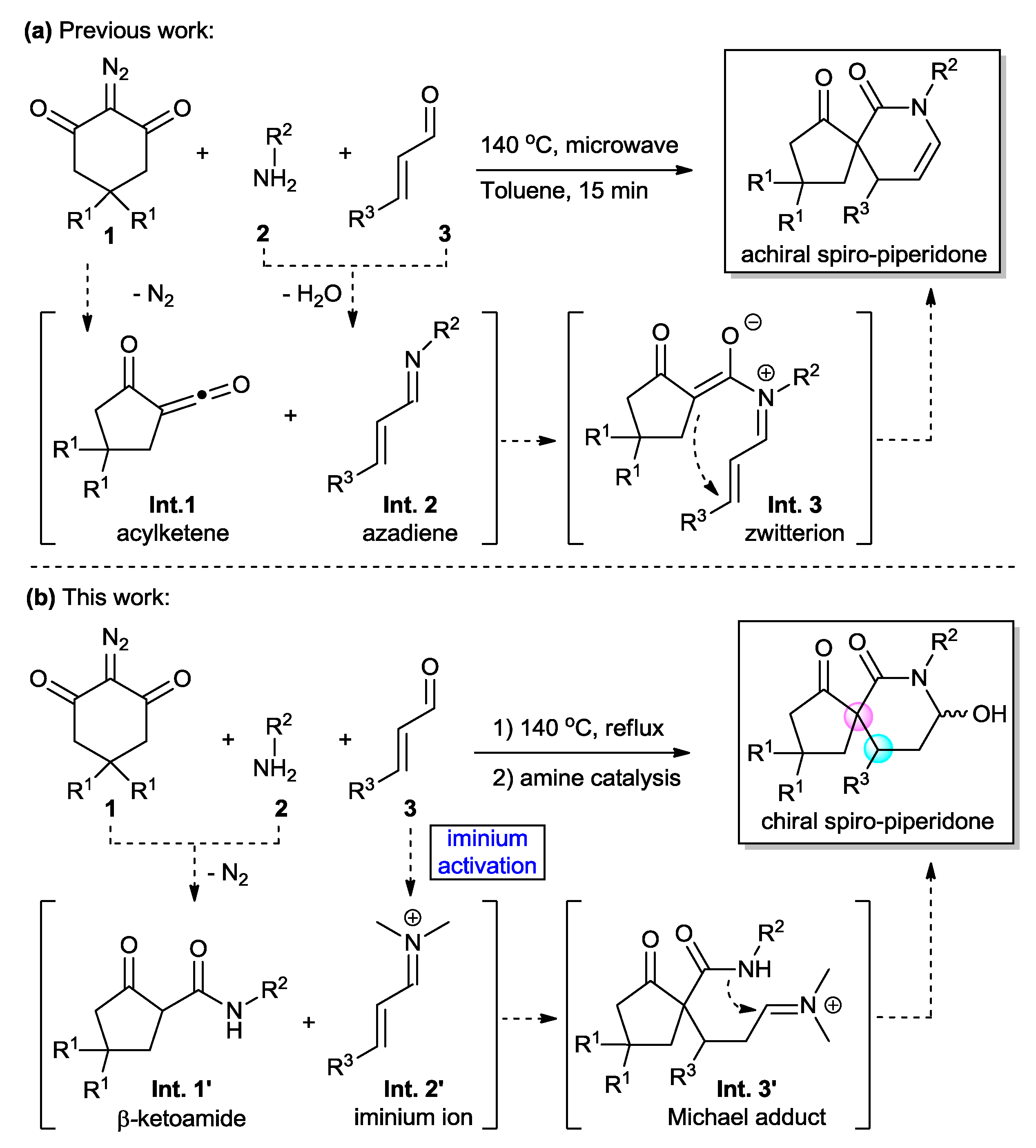
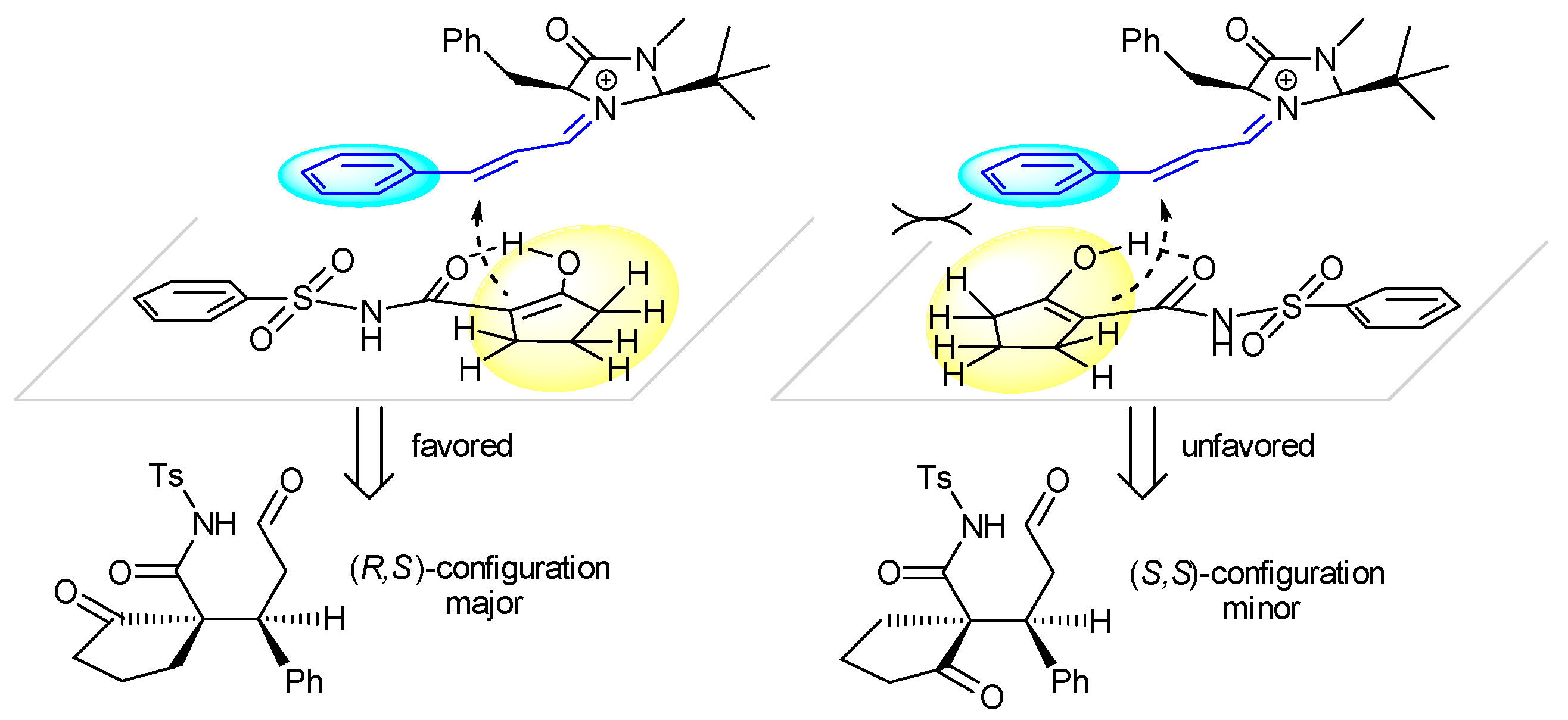
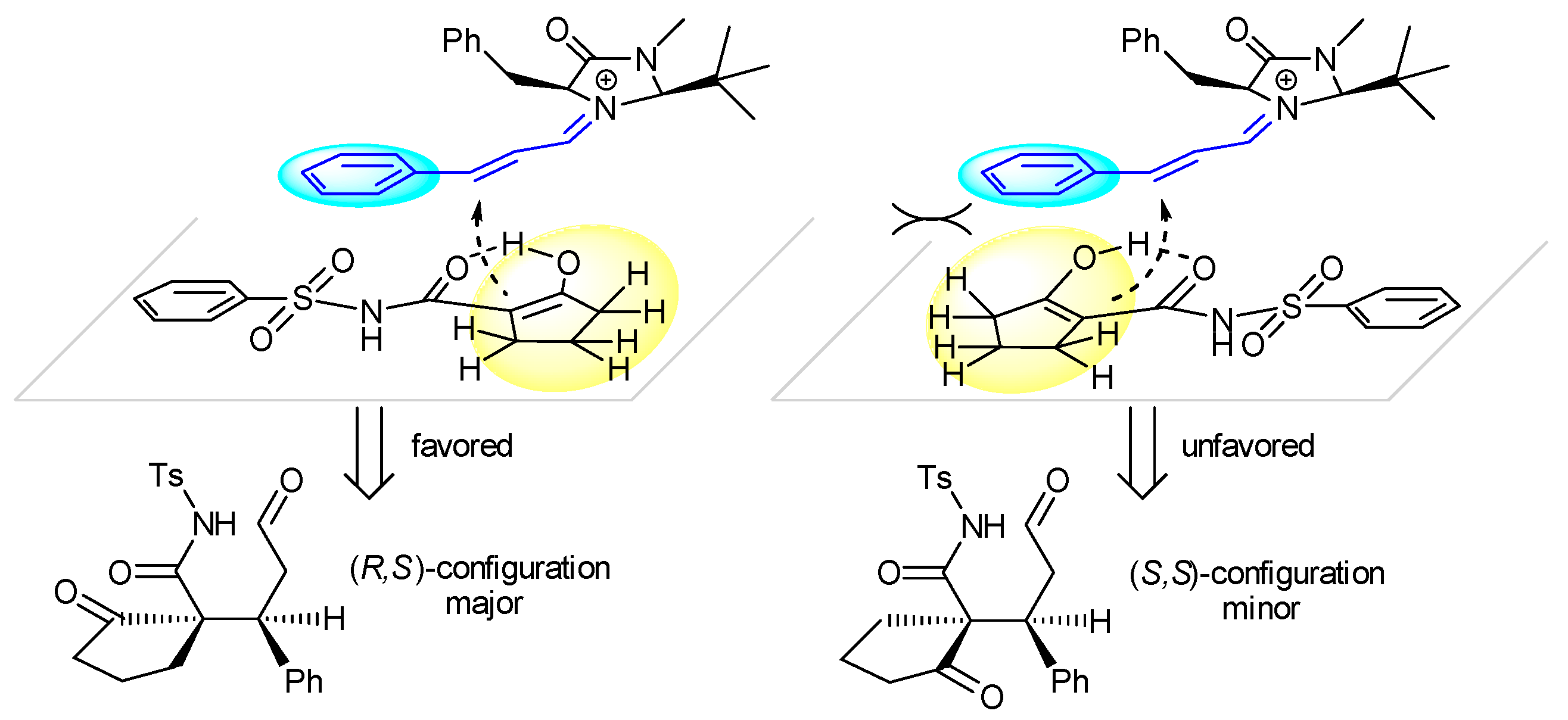
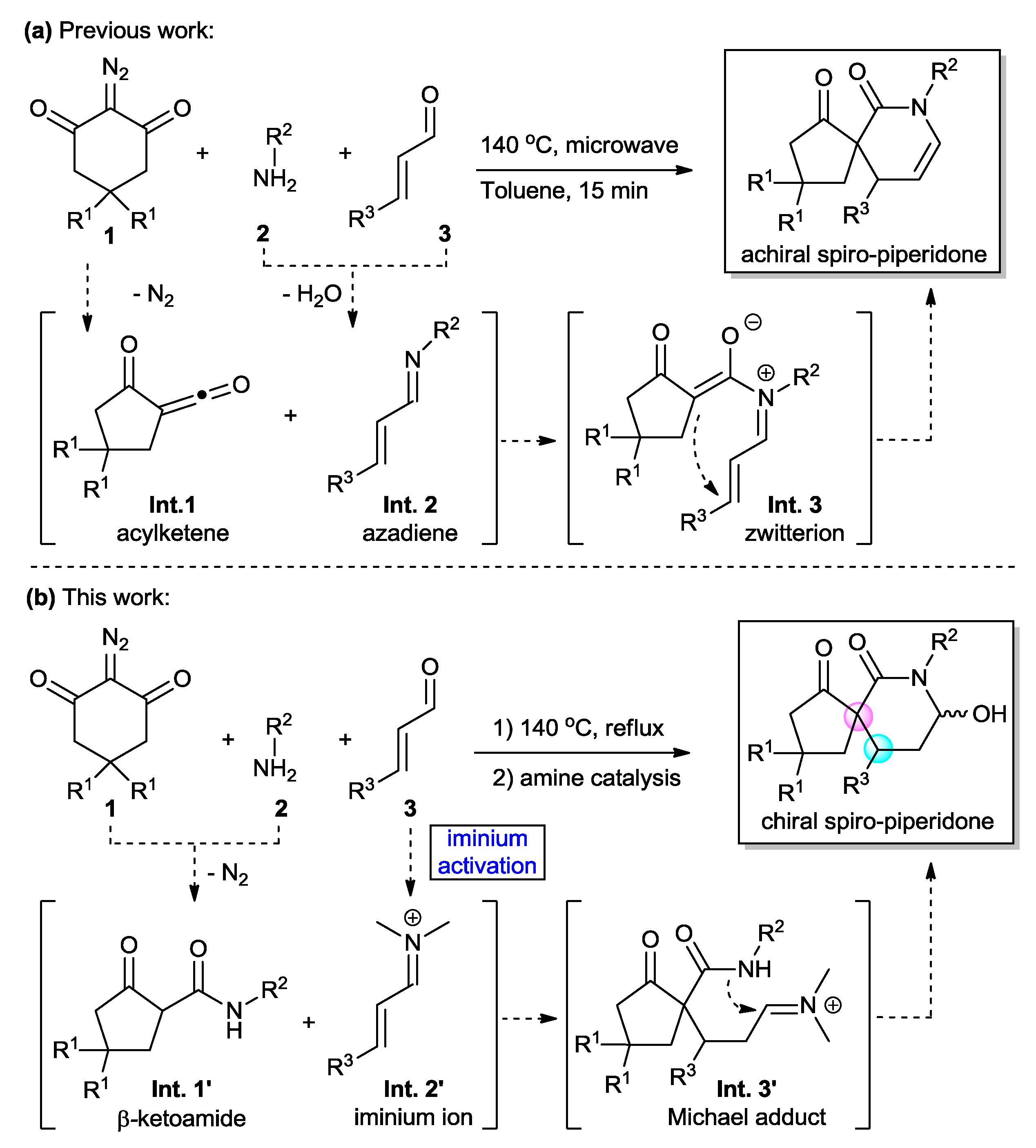
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Chiral Spirocyclic Piperidones 5
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rubiralta, M.; Giralt, E.; Diez, A. Piperidine: Structure, Preparation, Reactivity, and Synthetic Applications of Piperidine and Its Derivatives; Elsevier: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Albrecht, Ł.; Jiang, H.; Jørgensen, K.A. A simple recipe for sophisticated cocktails: Organocatalytic one-pot reactions—Concept, nomenclature, and future perspectives. Angew. Chem. Int. Ed. 2011, 50, 8492–8509. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K.A. Squaramides: Bridging from molecular recognition to bifunctional organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar]
- Bugaut, X.; Glorius, F. Organocatalytic umpolung: N-heterocyclic carbenes and beyond. Chem. Soc. Rev. 2012, 41, 3511–3522. [Google Scholar] [CrossRef] [PubMed]
- De Graaff, C.; Ruijter, E.; Orru, R.V.A. Recent developments in asymmetric multicomponent reactions. Chem. Soc. Rev. 2012, 41, 3969–4009. [Google Scholar] [CrossRef] [PubMed]
- Donslund, B.S.; Johansen, T.K.; Poulsen, P.H.; Halskov, K.S.; Jørgensen, K.A. The diarylprolinol silyl ethers: Ten years after. Angew. Chem. Int. Ed. 2015, 54, 13860–13874. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, F.; Gruttadauria, M.; Agrigento, P.; Noto, R. Low-loading asymmetric organocatalysis. Chem. Soc. Rev. 2012, 41, 2406–2447. [Google Scholar] [CrossRef] [PubMed]
- Chem, D.; Grossmann, A.; Enders, D. N-heterocyclic carbene catalyzed Domino reactions. Angew. Chem. Int. Ed. 2012, 51, 314–325. [Google Scholar]
- James, T.; van Gemmeren, M.; List, B. Development and applications of disulfonimides in enantioselective organocatalysis. Chem. Rev. 2015, 115, 9388–9409. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-L.; Liu, T.-Y.; Chen, Y.-C. Aminocatalytic asymmetric Diels-Alder reactions via HOMO activation. Acc. Chem. Res. 2012, 45, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-Y.; Xie, M.; Chen, Y.-C. Organocatalytic asymmetric transformations of modified Morita-Baylis-Hillman adducts. Chem. Soc. Rev. 2012, 41, 4101–4112. [Google Scholar] [CrossRef] [PubMed]
- Mahlau, M.; List, B. Asymmetric counteranion-directed catalysis: Concept, definition, and applications. Angew. Chem. Int. Ed. 2013, 52, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, P. Cinchona-based primary amine catalysis in the asymmetric functionalization of carbonyl compounds. Angew. Chem. Int. Ed. 2012, 51, 9748–9770. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, S.; Maruoka, K. Recent developments in asymmetric phase-transfer reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.E.; Bull, S.D.; Williams, J.M.J. Amidines, isothioureas, and guanidines as nucleophilic catalysts. Chem. Soc. Rev. 2012, 41, 2109–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volla, C.M.R.; Atodiresei, L.; Rueping, M. Catalytic C–C bond-forming multi-component cascade or domino reactions: Pushing the boundaries of complexity in asymmetric organocatalysis. Chem. Rev. 2014, 114, 2390–2431. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.D.; Millwood, P.A.; Smith, P.D. Asymmetric routes to substituted piperidines. Chem. Commun. 1998. [Google Scholar] [CrossRef]
- Felpin, F.-X.; Lebreton, J. Recent advances in the total synthesis of piperidine and pyrrolidine natural alkaloids with ring-closing metathesis as a key step. Eur. J. Org. Chem. 2003, 2003, 3693–3712. [Google Scholar] [CrossRef]
- Weintraub, P.M.; Sabol, J.S.; Kane, J.M.; Borcherding, D.R. Recent advances in the synthesis of piperidones and piperidines. Tetrahedron 2003, 59, 2953–2989. [Google Scholar] [CrossRef]
- Buffat, M.G.P. Synthesis of piperidines. Tetrahedron 2004, 60, 1701–1729. [Google Scholar] [CrossRef]
- Pearson, M.S.M.; Mathé-Allainmat, M.; Fargeas, V.; Lebreton, J. Recent advances in the total synthesis of piperidine azasugars. Eur. J. Org. Chem. 2005. [Google Scholar] [CrossRef]
- Terada, M.; Machioka, K.; Sorimachi, K. Chiral brønsted acid-catalyzed tandem aza-ene type reaction/cyclization cascade for a one-pot entry to enantioenriched piperidines. J. Am. Chem. Soc. 2007, 129, 10336–10337. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Gotoh, H.; Masui, R.; Ishikawa, H. Diphenylprolinol silyl ether as a catalyst in an enantioselective, catalytic, formal aza [3 + 3] cycloaddition reaction for the formation of enantioenriched piperidines. Angew. Chem. Int. Ed. 2008, 47, 4012–4015. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yu, J.; Sun, X.-X.; Rao, Q.-Q.; Gong, L.-Z. Organocatalytic asymmetric three-component cyclization of cinnamaldehydes and primary amines with 1,3-dicarbonyl compounds: Straightforward access to enantiomerically enriched dihydropyridines. Angew. Chem. Int. Ed. 2008, 47, 2458–2462. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Antonchick, A.P. A highly enantioselective brønsted acid catalyzed reaction cascade. Angew. Chem. Int. Ed. 2008, 47, 5836–5838. [Google Scholar] [CrossRef] [PubMed]
- Zu, L.; Xie, H.; Li, H.; Wang, J.; Yu, X.; Wang, W. Chiral amine-catalyzed enantioselective cascade aza-ene-type cyclization reactions. Chem. Eur. J. 2008, 14, 6333–6335. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Shi, F.; Gong, L.-Z. Brønsted-acid-catalyzed asymmetric multicomponent reactions for the facile synthesis of highly enantioenriched structurally diverse nitrogenous heterocycles. Acc. Chem. Res. 2011, 44, 1156–1171. [Google Scholar] [CrossRef] [PubMed]
- Dragutan, I.; Dragutan, V.; Demonceau, A. Targeted drugs by olefin metathesis: Piperidine-based iminosugars. RSC Adv. 2012, 2, 719–736. [Google Scholar] [CrossRef]
- Boddaert, T.; Coquerel, Y.; Rodriguez, J. Organocatalytic activity of N-heterocyclic carbenes in the Michael addition of 1,3-dicarbonyl compounds: Application to a stereoselective spirocyclization sequence. Adv. Synth. Catal. 2009, 351, 1744–1748. [Google Scholar] [CrossRef]
- Boddaert, T.; Coquerel, Y.; Rodriguez, J. Combination of rearrangement with metallic and organic catalyses—A step- and atom-economical approach to α-spiroactones and -lactams. Eur. J. Org. Chem. 2011, 43, 5061–5070. [Google Scholar] [CrossRef]
- Sanchez Duque, M.d.M.; Baslé, O.; Isambert, N.; Gaudel-Siri, A.; Génisson, Y.; Plaquevent, J.-C.; Rodriguez, J.; Constantieux, T. A cooperative participation of the amido group in the organocatalytic construction of all-carbon quaternary stereocenters by Michael addition with β-ketoamides. Org. Lett. 2011, 13, 3296–3299. [Google Scholar] [CrossRef] [PubMed]
- Bonne, D.; Constantieux, T.; Coquerel, Y.; Rodriguez, J. Stereoselective multiple bond-forming transformations (MBFTs): The power of 1,2- and 1,3-dicarbonyl compounds. Chem. Eur. J. 2013, 19, 2218–2231. [Google Scholar] [CrossRef] [PubMed]
- Galvez, J.; Castillo, J.-C.; Quiroga, J.; Rajzmann, M.; Rodriguez, J.; Coquerel, Y. Divergent chemo-, regio-, and diastereoselective normal electron-demand Povarov-type reactions with α-oxo-ketene dienophiles. Org. Lett. 2014, 16, 4126–4129. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Zhu, R.-Y.; Dai, W.; Wang, C.-S.; Tu, S.-J. Catalytic asymmetric formal [3 + 3] cycloaddition of an azomethine ylide with 3-indolylmethanol: Enantioselective construction of a six-membered piperidine framework. Chem. Eur. J. 2014, 20, 2597–2604. [Google Scholar] [CrossRef] [PubMed]
- Holmquist, M.; Blay, G.; Muñoz, M.C.; Pedro, J.R. Aza-Henry reaction of isatin ketimines with methyl 4-nrobutyrate en route to spiro[piperidine-3,3′-oxindoles]. Adv. Synth. Catal. 2015, 357, 3857–3862. [Google Scholar] [CrossRef]
- Hawner, C.; Alexakis, A. Metal-catalyzed asymmetric conjugate addition reaction: Formation of quaternary stereocenters. Chem. Commun. 2010, 46, 7295–7306. [Google Scholar] [CrossRef] [PubMed]
- Jautze, S.; Peters, R. Catalytic asymmetric Michael additions of α-cyanoacetates. Synthesis 2010. [Google Scholar] [CrossRef]
- Das, J.P.; Marek, I. Enantioselective synthesis of all-carbon quaternary stereogenic centers in acyclic systems. Chem. Commun. 2011, 47, 4593–4623. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Jia, Z.-J.; Chen, S.; Wu, L.; Chen, Y.-C. Organocatalytic tandem reaction to construct six-membered spirocyclic oxindoles with multiple chiral centres through a formal [2 + 2 + 2] annulation. Chem. Eur. J. 2010, 16, 2852–2856. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Shi, X.; Wang, S.; Sun, T.; Cao, Y.; Wang, R. Bifunctional organocatalytic strategy for inverse-electron-demand Diels-Alder reactions: Highly efficient in situ substrate generation and activation to construct azaspirocyclic skeletons. Angew. Chem. Int. Ed. 2012, 51, 2084–2087. [Google Scholar] [CrossRef] [PubMed]
- Goudedranche, S.; Bugaut, X.; Constantieux, T.; Bonne, D.; Rodriguez, J. α,β-Unsaturated acyl cyanides as new bis-electrophiles for enantioselective organocatalyzed formal [3 + 3] spiroannulation. Chem. Eur. J. 2014, 20, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Presset, M.; Coquerel, Y.; Rodriguez, J. Periselectivity switch of acylketenes in cycloadditions with 1-azadienes: Microwave-assisted diastereoselective domino three-component synthesis of α-spiro-δ-lactams. Org. Lett. 2010, 12, 4212–4215. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Peng, C.; He, G.; Leng, H.-J.; Wang, B.; Huang, W.; Han, B. Asymmetric synthesis of a structurally and stereochemically complex spirooxindole pyran scaffold through an organocatalytic multicomponent cascade reaction. Chem. Commun. 2012, 48, 10487–10489. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, L.; Peng, C.; Xie, X.; Leng, H.-J.; Wang, B.; Tang, Z.-W.; He, G.; Ouyang, L.; Huang, W.; et al. Organocatalytic tandem Morita-Baylis-Hillman-Michael reaction for asymmetric synthesis of a drug-like oxa-spirocyclic indanone scaffold. Chem. Commun. 2013, 49, 8692–8694. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Huang, W.; Ren, W.; He, G.; Wang, J.-H.; Peng, C. Asymmetric synthesis of cyclohexane-fused drug-like spirocyclic scaffolds containing six contiguous stereogenic centers via organocatalytic cascade reactions. Adv. Synth. Catal. 2015, 357, 561–568. [Google Scholar] [CrossRef]
- Zhou, R.; Wu, Q.; Guo, M.; Huang, W.; He, X.; Yang, L.; Peng, F.; He, G.; Han, B. Organocatalytic cascade reaction for the asymmetric synthesis of novel chroman-fused spirooxindoles that potently inhibit cancer cell proliferation. Chem. Commun. 2015, 51, 13113–13116. [Google Scholar] [CrossRef] [PubMed]
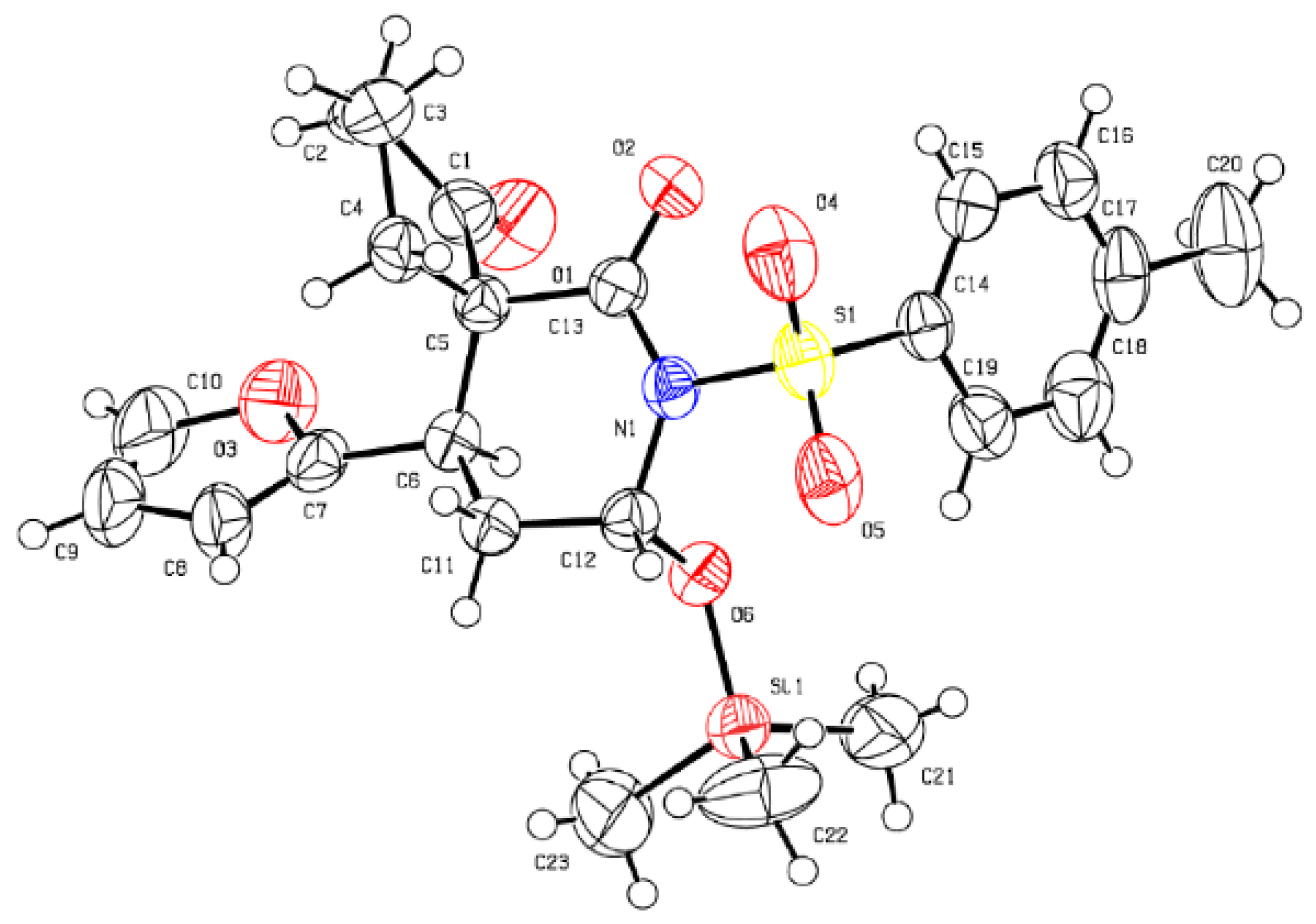
- CCDC 1499269 Contains The Supplementary Crystallographic Data of 5m. These Data Can be Obtained Free of Charge from the Cambridge Crystallographic Data Centre. Available online: www.ccdc.cam.ac.uk/data_request/cif (accessed on 16 August 2016).
- Austin, J.F.; MacMillan, D.W.C. Enantioselective organocatalytic indole alkylations. Design of a new and highly effective chiral amine for iminium catalysis. J. Am. Chem. Soc. 2002, 124, 1172–1173. [Google Scholar] [PubMed]
- Brown, S.P.; Goodwin, N.C.; MacMillan, D.W.C. The first enantioselective organocatalytic Mukaiyama-Michael reaction: A direct nethod for the synthesis of enantioenriched γ-butenolide architecture. J. Am. Chem. Soc. 2003, 125, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Presset, M.; Mailhol, D.; Coquerel, Y.; Rodriguez, J. Diazo-transfer reactions to 1,3-dicarbonyl compounds with tosyl azide. Synthesis 2011, 42, 2549–2552. [Google Scholar]

| Entry | Catalyst | Additive | Solvent 1 | Solvent 2 | Yield (%) b | dr c | ee (%) d |
|---|---|---|---|---|---|---|---|
| 1 | C1 | BzOH | Tol | Tol | 43 | 55:45 | 42 |
| 2 | C2 | BzOH | Tol | Tol | 45 | 58:42 | 48 |
| 3 | C3 | BzOH | Tol | Tol | 41 | 58:42 | 46 |
| 4 | C4 | TFA | Tol | Tol | 66 | 60:40 | 70 |
| 5 | C5 | TFA | Tol | Tol | 68 | 62:38 | 76 |
| 6 | C5 | TsOH | Tol | Tol | 65 | 60:40 | 74 |
| 7 | C5 | AcOH | Tol | Tol | 64 | 62:38 | 82 |
| 8 | C5 | BzOH | Tol | Tol | 64 | 65:35 | 84 |
| 9 | C5 | BzOH | Tol | MeCN | 73 | 62:38 | 80 |
| 10 | C5 | BzOH | Tol | THF | 70 | 60:40 | 78 |
| 11 | C5 | BzOH | Tol | DCM | 68 | 70:30 | 86 |
| 12 e | C5 | BzOH | Tol | DCM | 72 | 75:25 | 90 |
| 13 f | C5 | BzOH | Tol | DCM | 70 | 73:27 | 88 |
| Entry | R1 | R2 | R3 | Yield (5) (%) b | dr c | ee (%) d |
|---|---|---|---|---|---|---|
| 1 | H | Ts | Ph | 72 (5a) | 75:25 | 90 |
| 2 | H | Ts | 3-FC6H4 | 72 (5b) | 72:28 | 93 |
| 3 | H | Ts | 4-FC6H4 | 74 (5c) | 75:25 | 94 |
| 4 | H | Ts | 4-ClC6H4 | 74 (5d) | 73:27 | 95 |
| 5 | H | Ts | 4-BrC6H4 | 73 (5e) | 75:25 | 96 |
| 6 | H | Ts | 2-FC6H4 | 69 (5f) | 70:30 | 94 |
| 7 | H | Ts | 2-ClC6H4 | 68 (5g) | 70:30 | 91 |
| 8 | H | Ts | 2-NO2C6H4 | 70 (5h) | 72:28 | 93 |
| 9 | H | Ts | 4-NO2C6H4 | 76 (5i) | 78:22 | 97 |
| 10 | H | Ts | 2-MeOC6H4 | 64 (5j) | 70:30 | 91 |
| 11 | H | Ts | 4-MeC6H4 | 66 (5k) | 72:28 | 91 |
| 12 | H | Ts | 4-(Me)2NC6H4 | 62 (5l) | 68:32 | 90 |
| 13 | H | Ts | 2-furyl | 75 (5m) | 80:20 | 96 |
| 14 | H | Ts | Me | 53 (5n) | 64:36 | 50 |
| 15 | Me | Ts | Ph | 70 (5o) | 72:28 | 91 |
| 16 | Me | Ts | 4-BrC6H4 | 71 (5p) | 74:26 | 91 |
| 17 | Me | Ts | 2-furyl | 73 (5q) | 78:22 | 95 |
| 18 | H | PhSO2 | Ph | 76 (5r) | 76:24 | 94 |
| 19 | H | MeSO2 | Ph | 75 (5s) | 80:20 | 95 |
| 20 | H | Bn | Ph | 70 (5t) | 73:27 | 90 |
| 21 e | H | Ts | Ph | 70 (5u)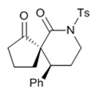 | 74:26 | 95 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Ouyang, L.; Tan, Y.; Tang, X.; Kang, J.; Wang, C.; Zhu, Y.; Peng, C.; Huang, W. One-Pot Two-Step Organocatalytic Asymmetric Synthesis of Spirocyclic Piperidones via Wolff Rearrangement–Amidation–Michael–Hemiaminalization Sequence. Catalysts 2017, 7, 46. https://doi.org/10.3390/catal7020046
Liu Y, Ouyang L, Tan Y, Tang X, Kang J, Wang C, Zhu Y, Peng C, Huang W. One-Pot Two-Step Organocatalytic Asymmetric Synthesis of Spirocyclic Piperidones via Wolff Rearrangement–Amidation–Michael–Hemiaminalization Sequence. Catalysts. 2017; 7(2):46. https://doi.org/10.3390/catal7020046
Chicago/Turabian StyleLiu, Yanqing, Liang Ouyang, Ying Tan, Xue Tang, Jingwen Kang, Chunting Wang, Yaning Zhu, Cheng Peng, and Wei Huang. 2017. "One-Pot Two-Step Organocatalytic Asymmetric Synthesis of Spirocyclic Piperidones via Wolff Rearrangement–Amidation–Michael–Hemiaminalization Sequence" Catalysts 7, no. 2: 46. https://doi.org/10.3390/catal7020046