Mechanism of Water Oxidation Catalyzed by a Dinuclear Ruthenium Complex Bridged by Anthraquinone

Abstract
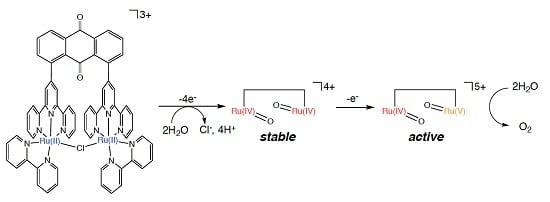
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
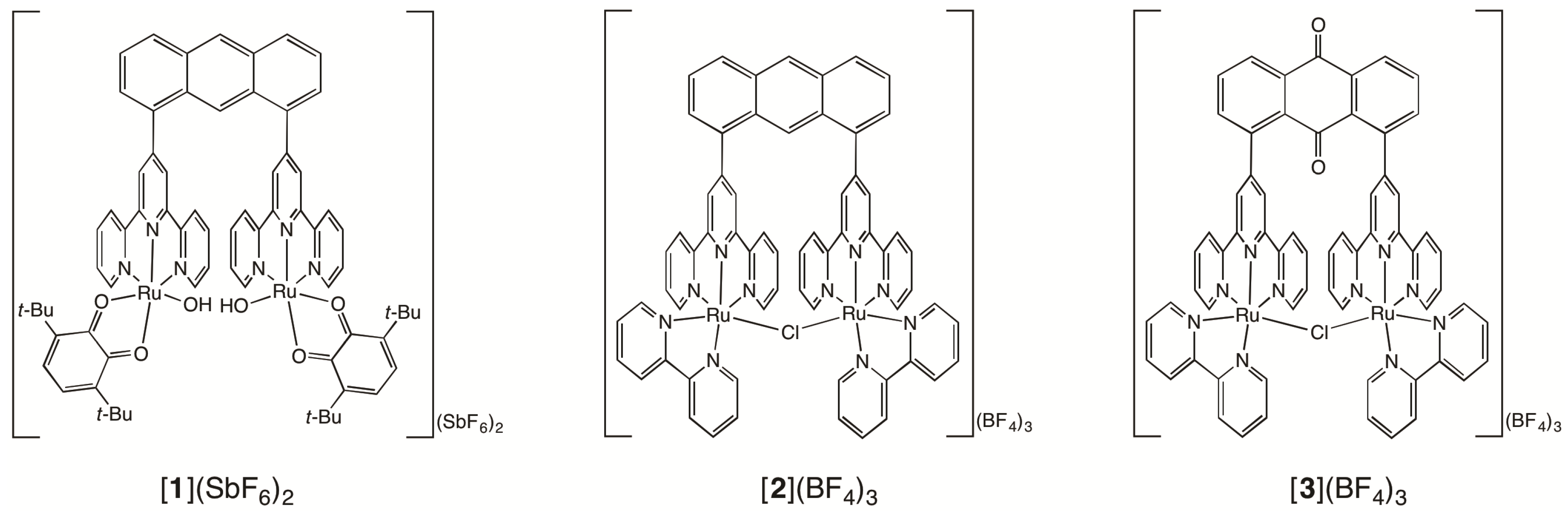
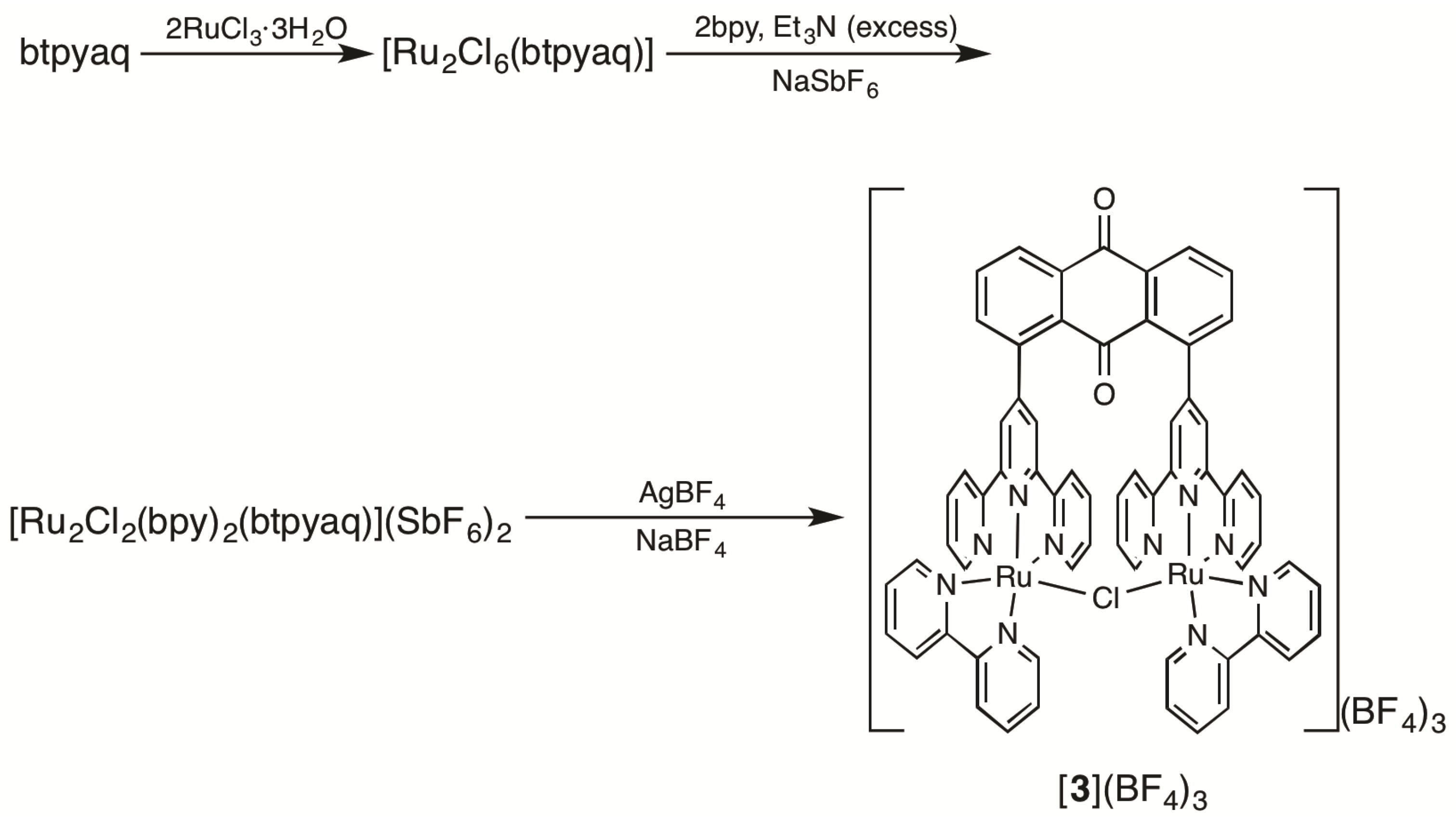
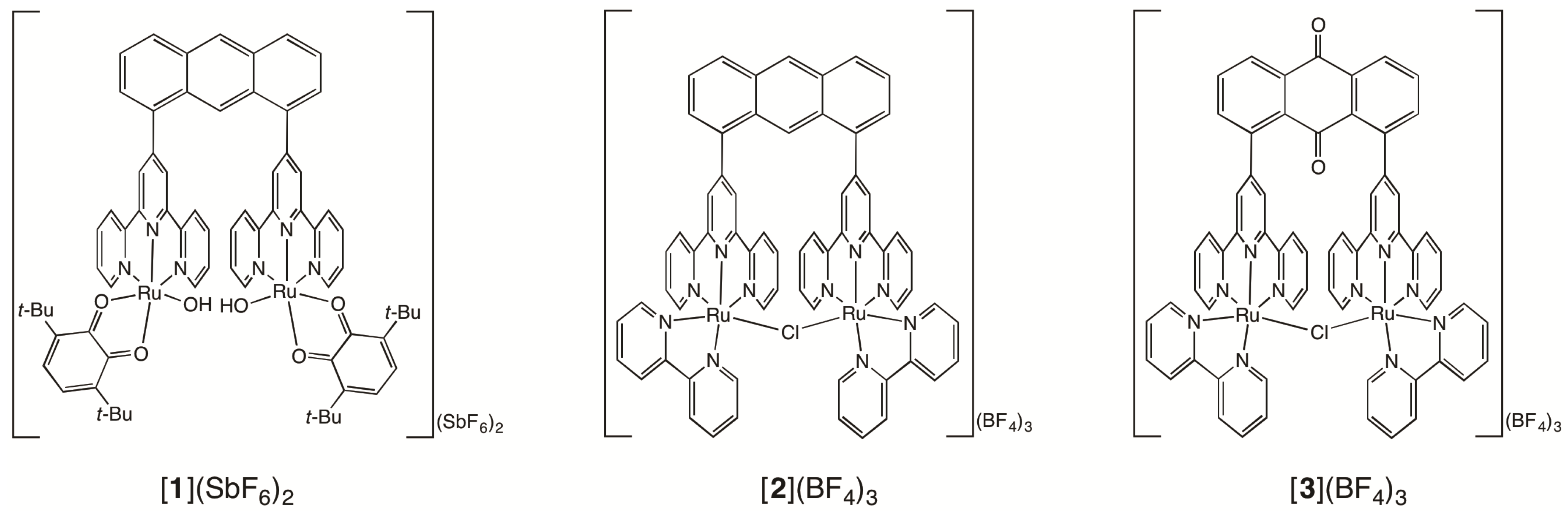
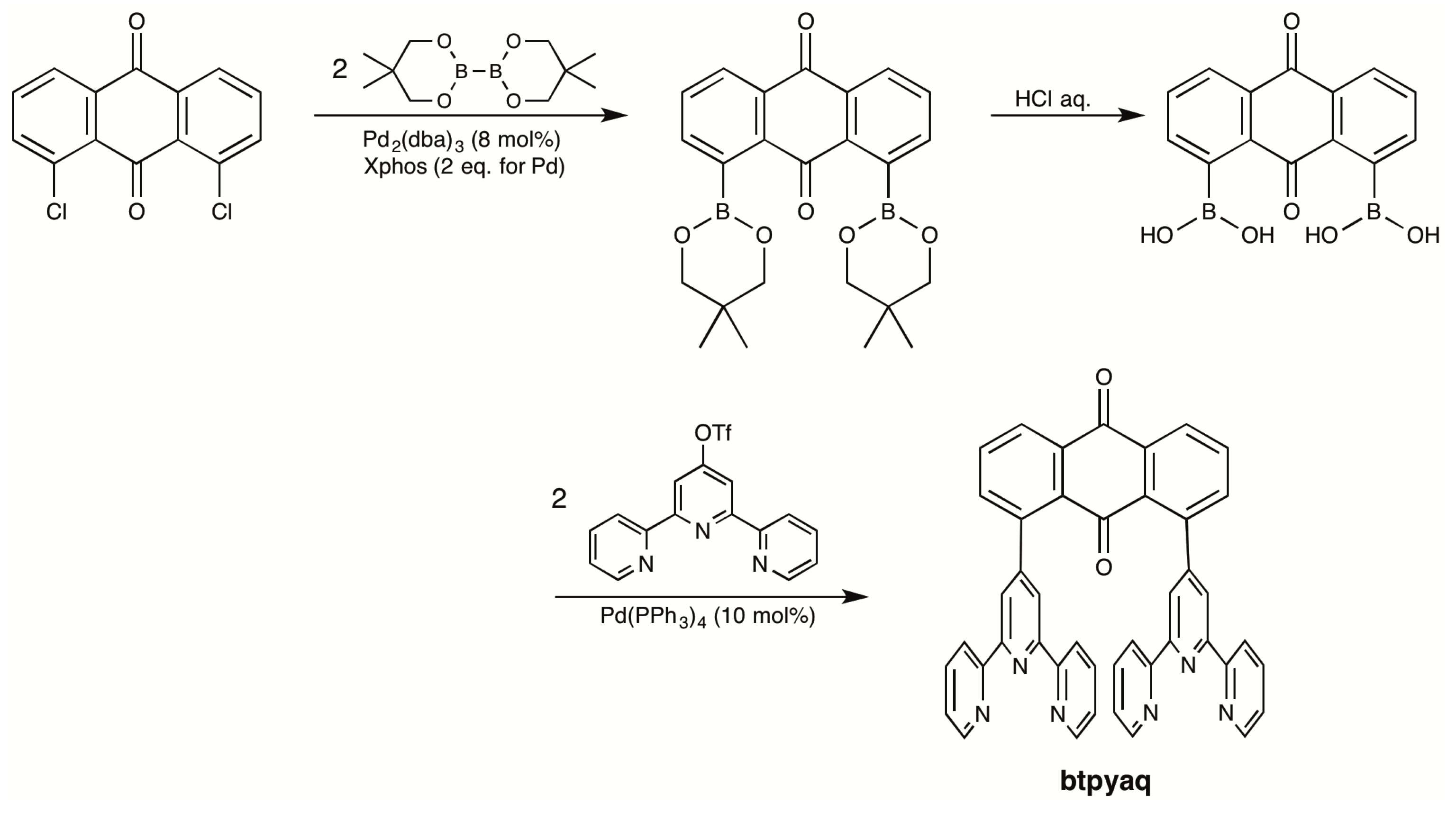
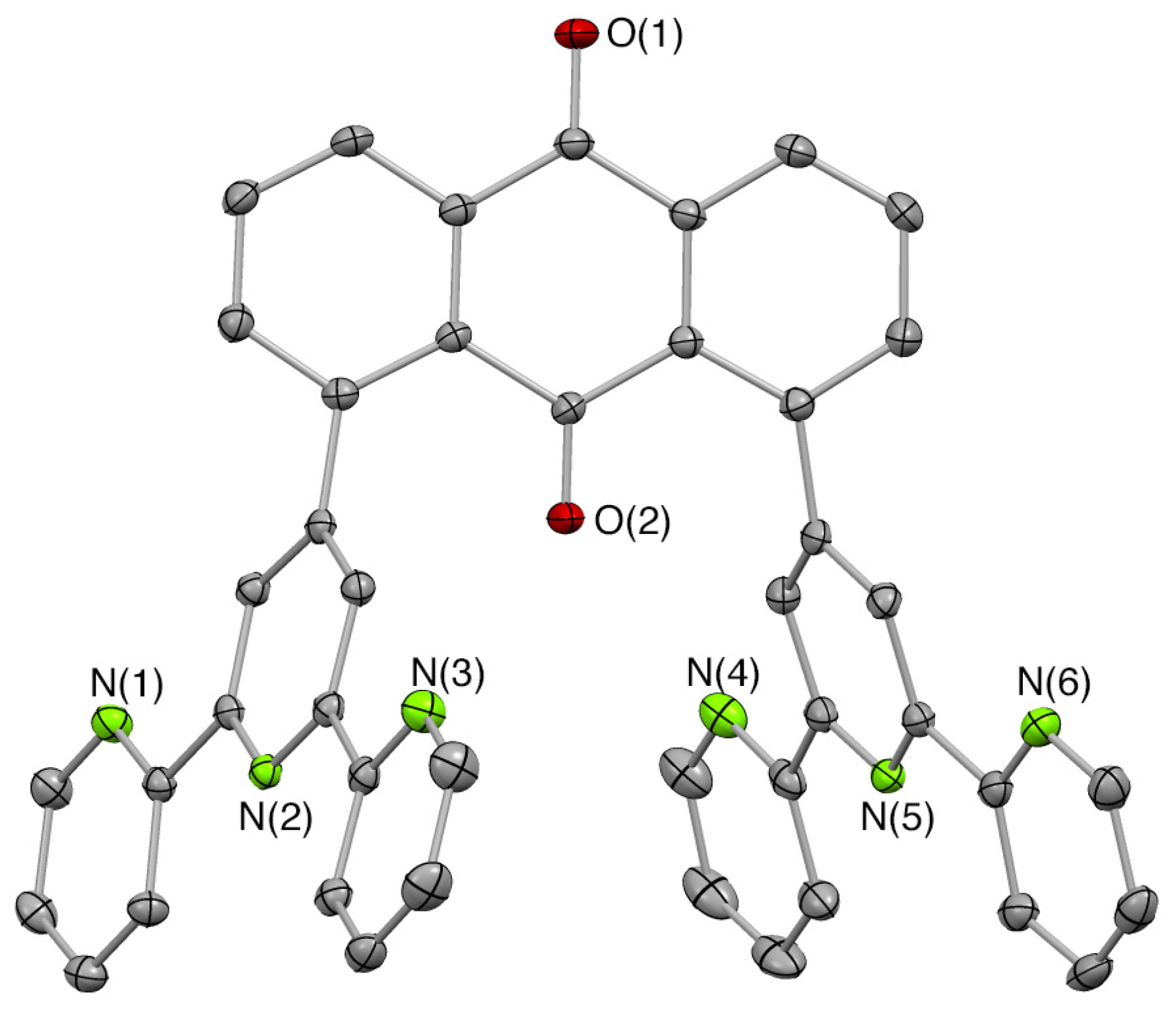
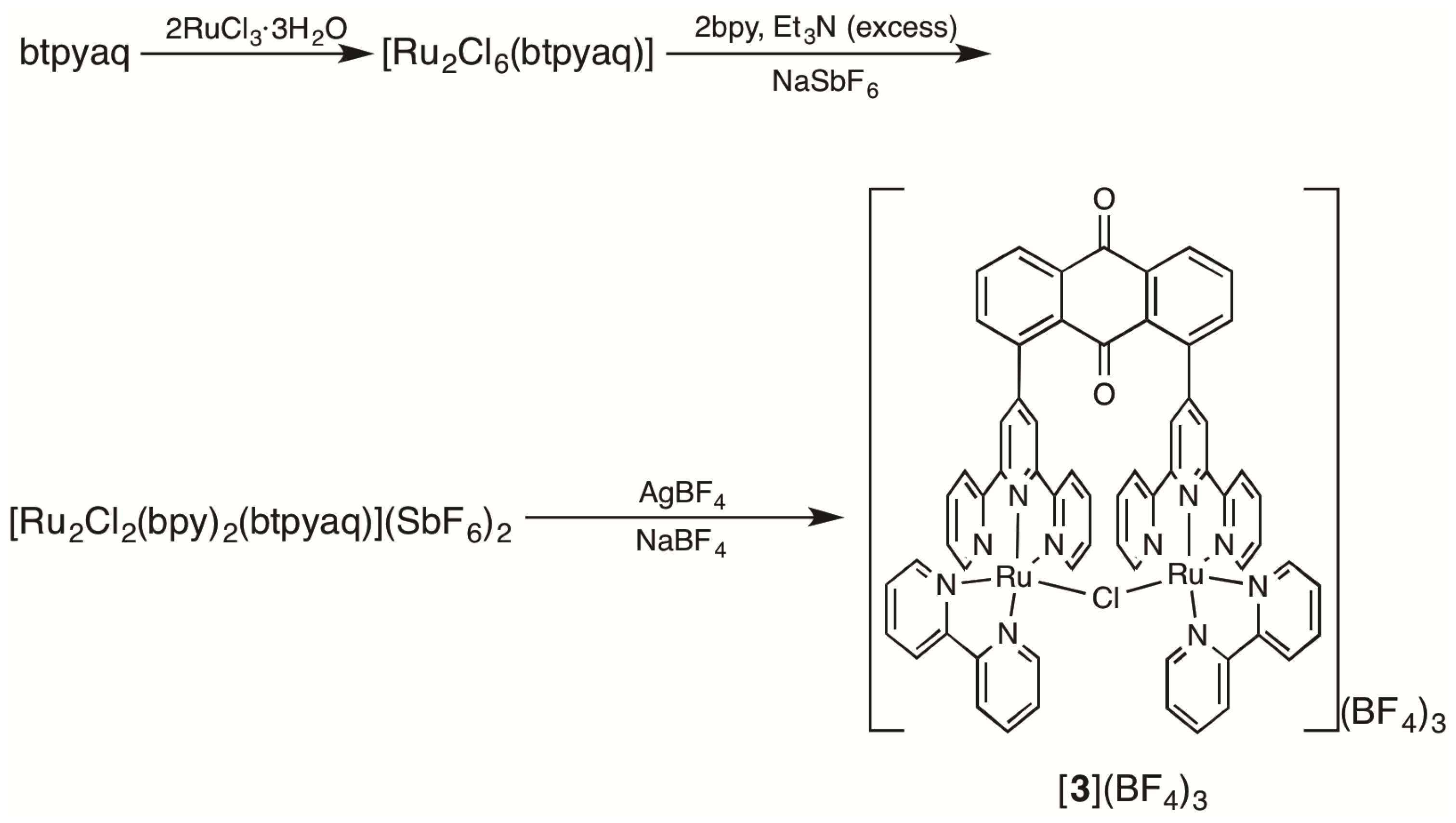
2.1. Syntheses of Bis(terpyridyl)anthraquinone and Dinuclear Ruthenium Complexes
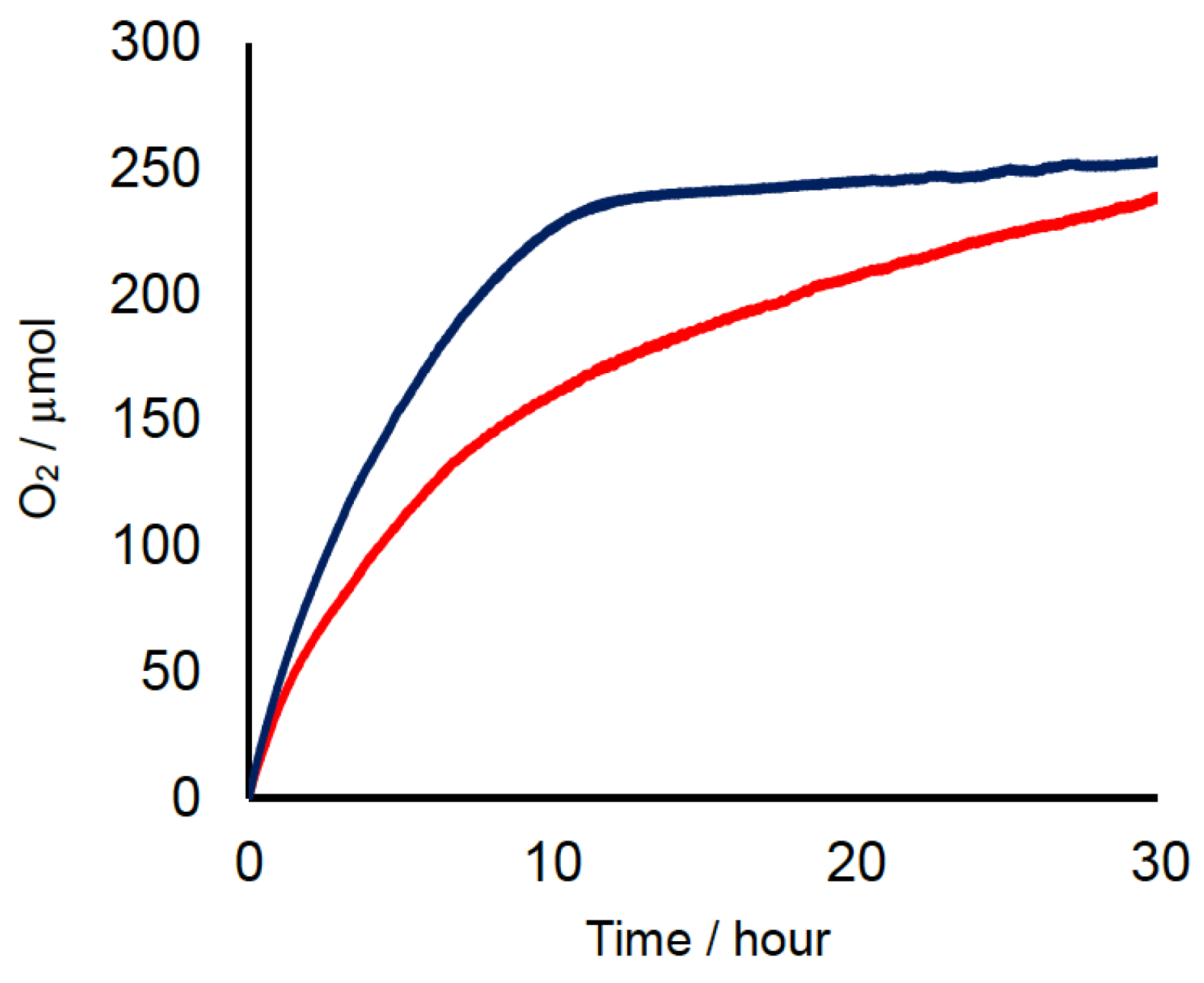
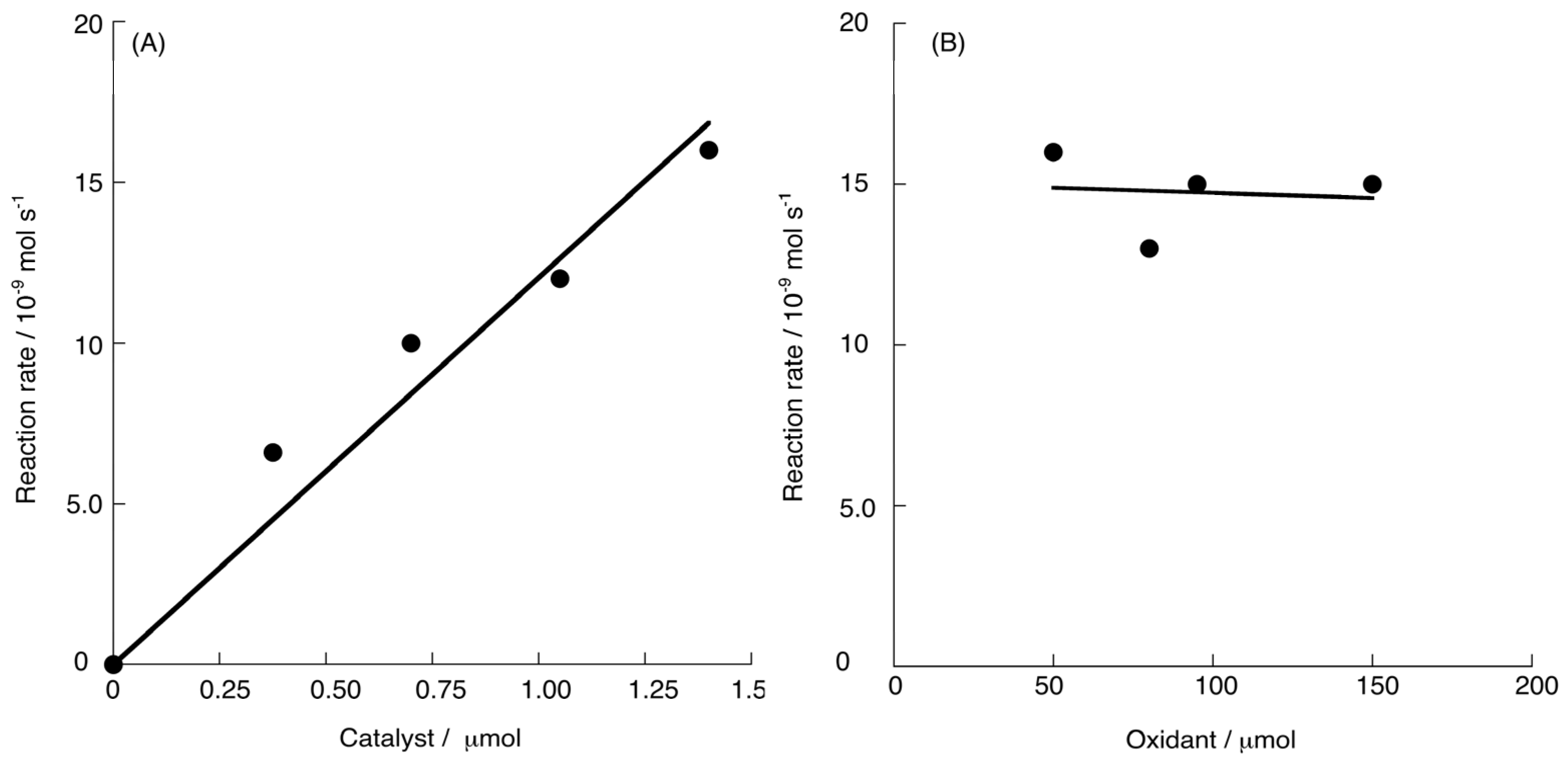
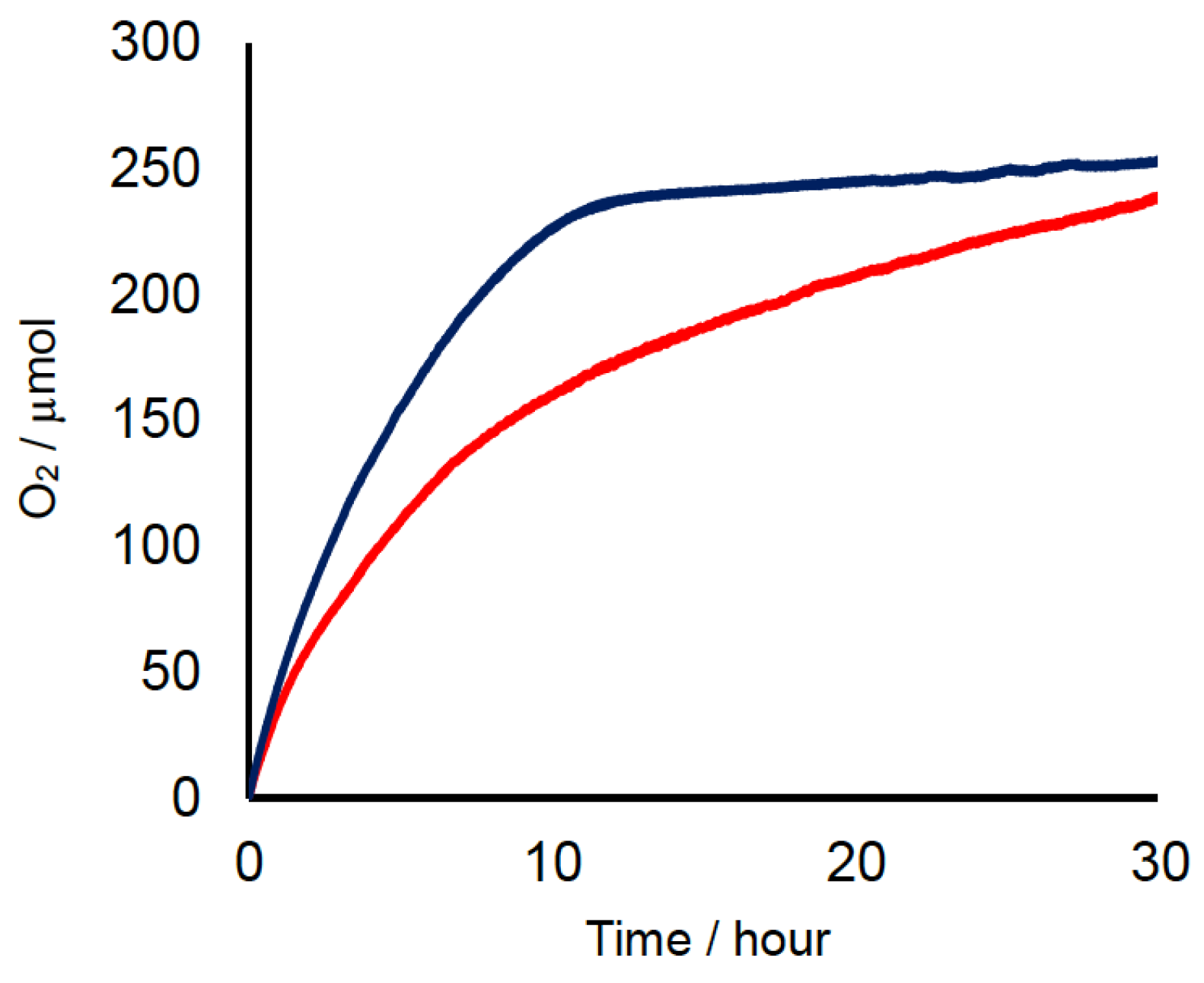
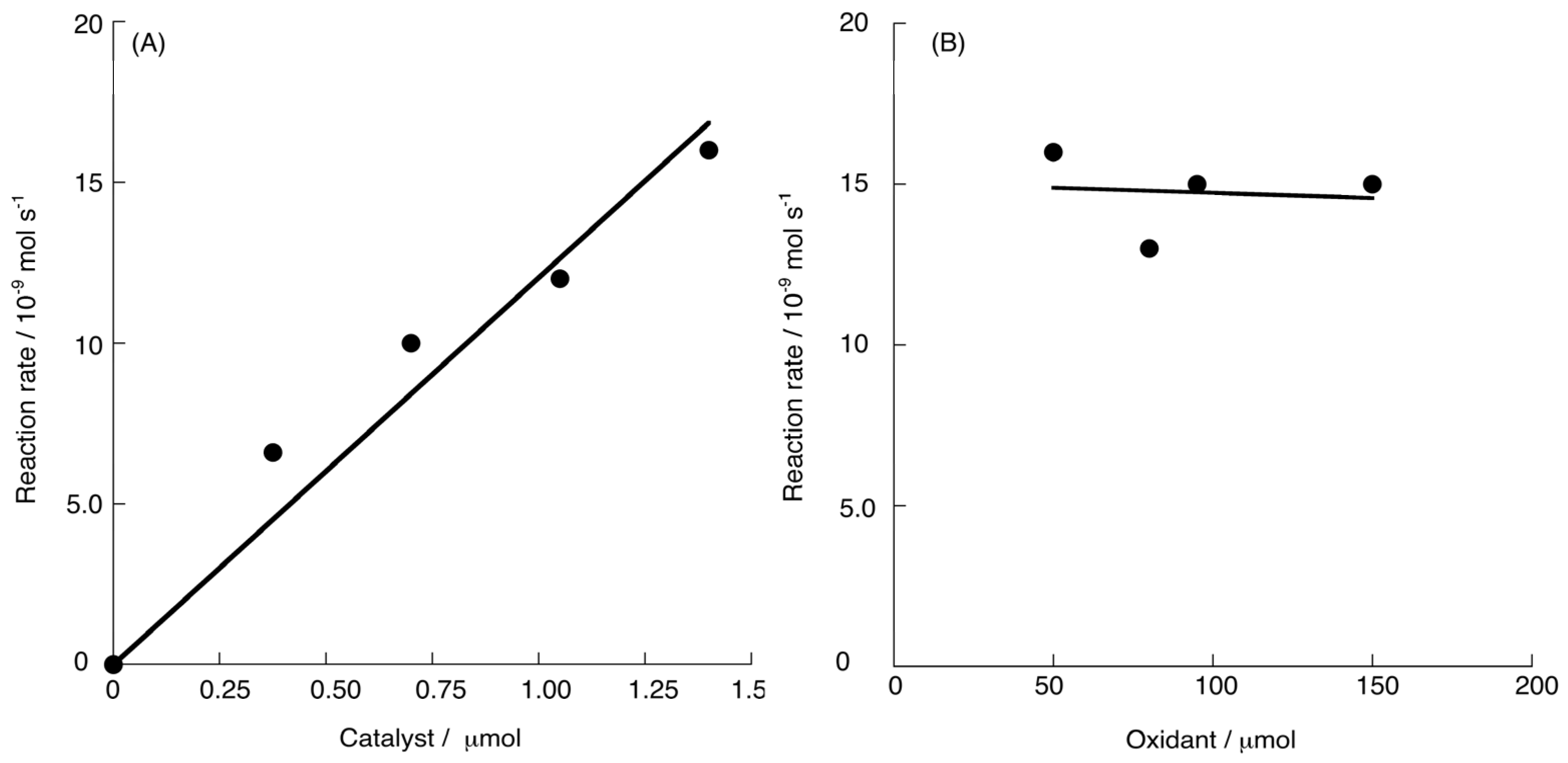
2.2. Chemical Water Oxidation
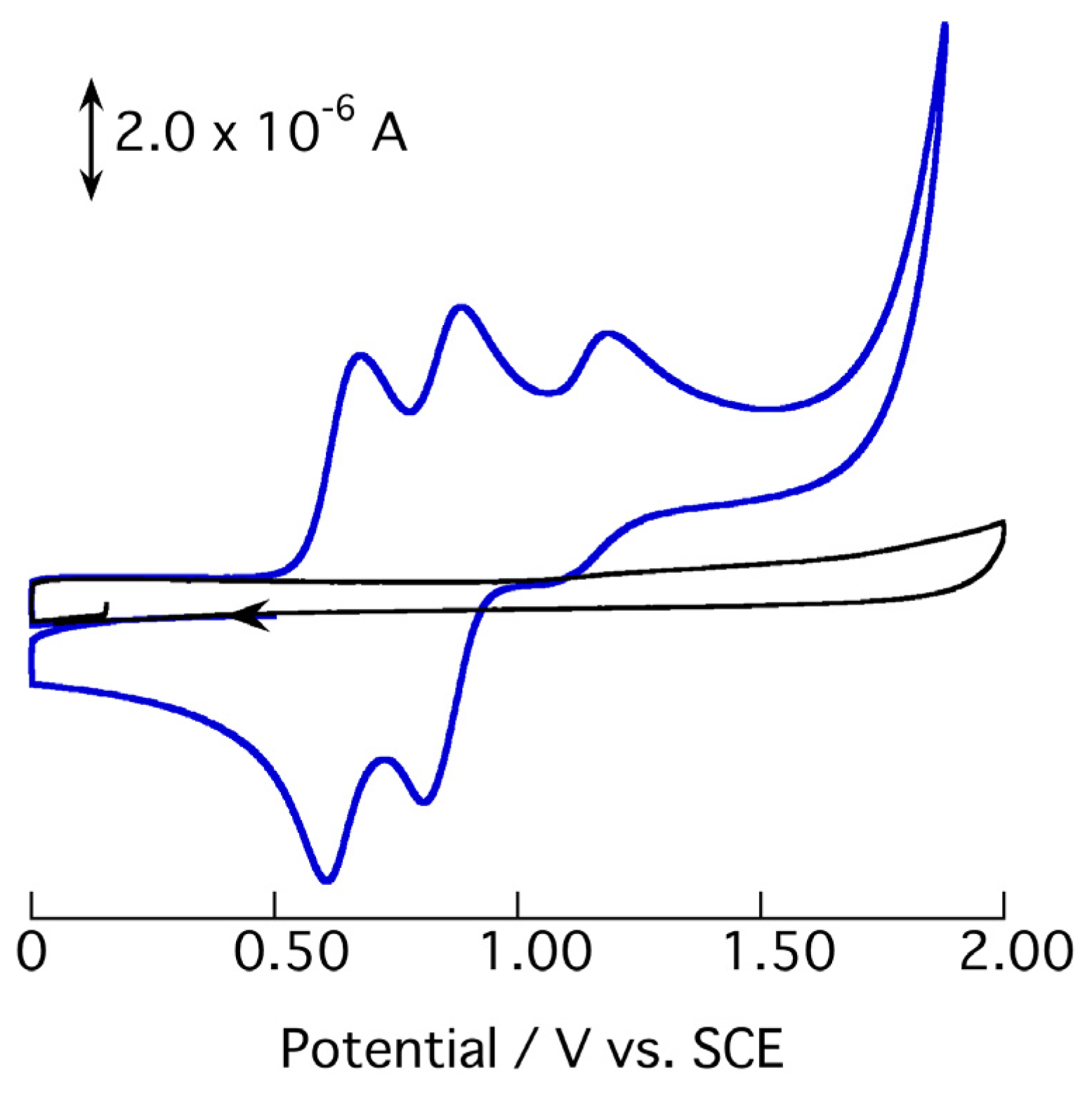
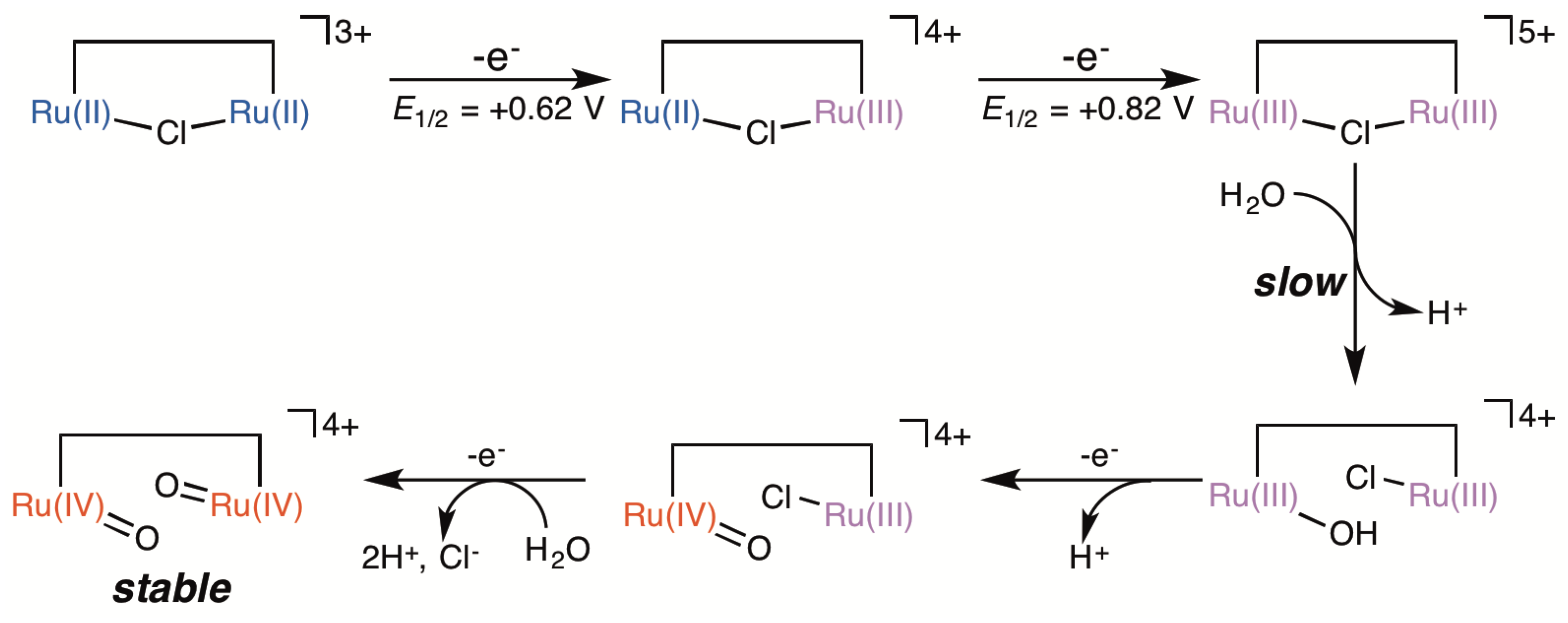
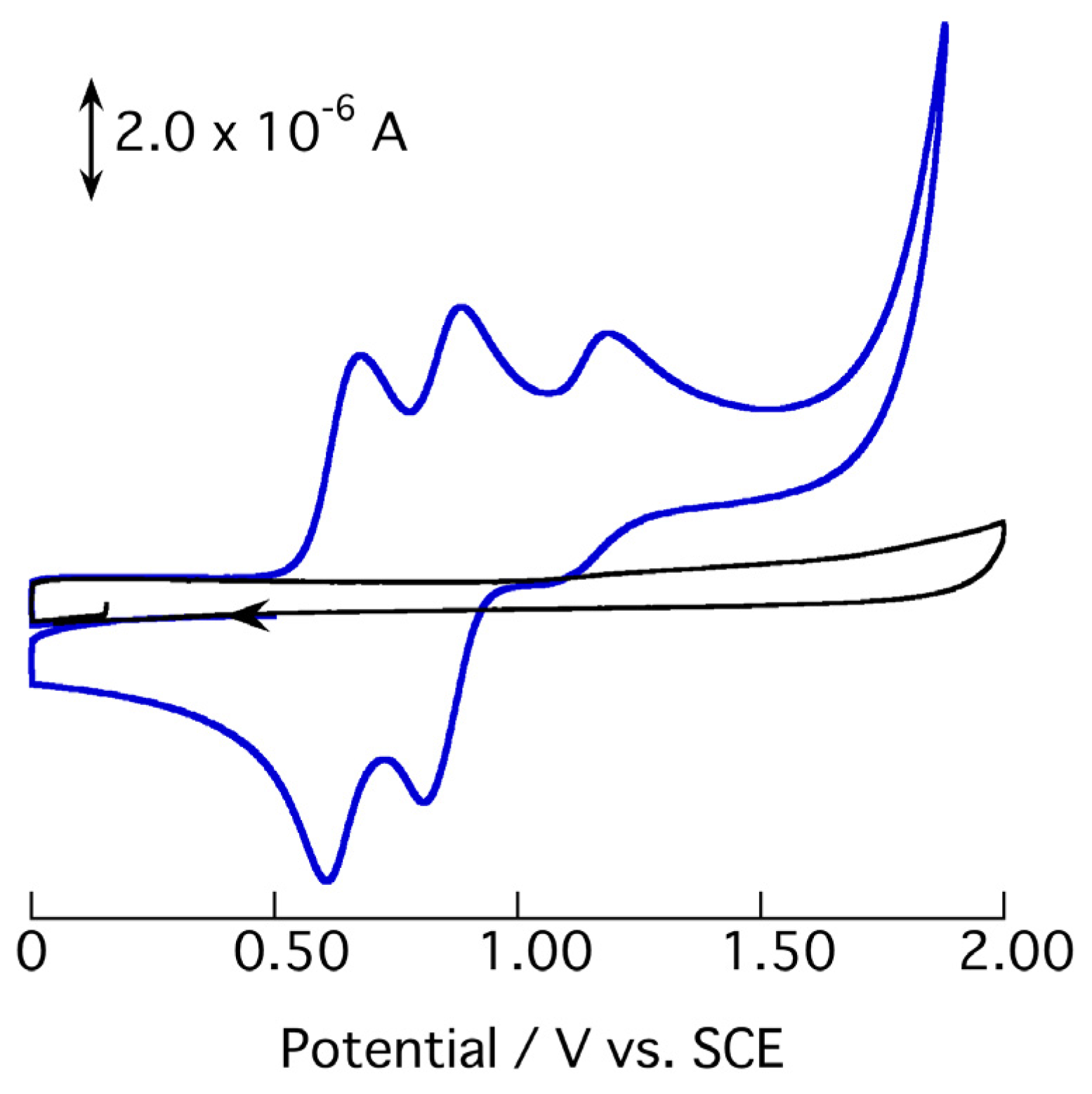
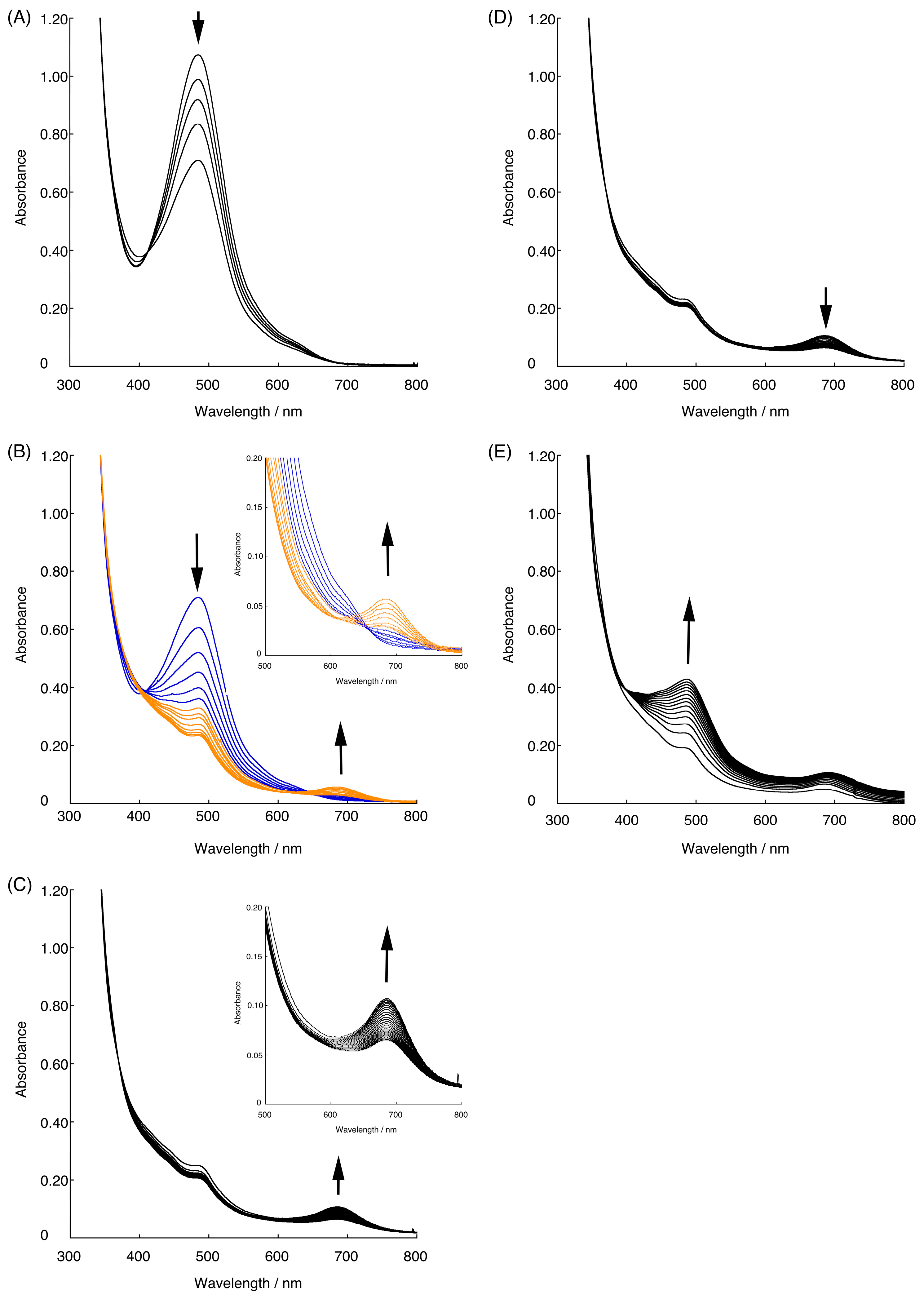
2.3. Redox Behavior of [3](BF4)3
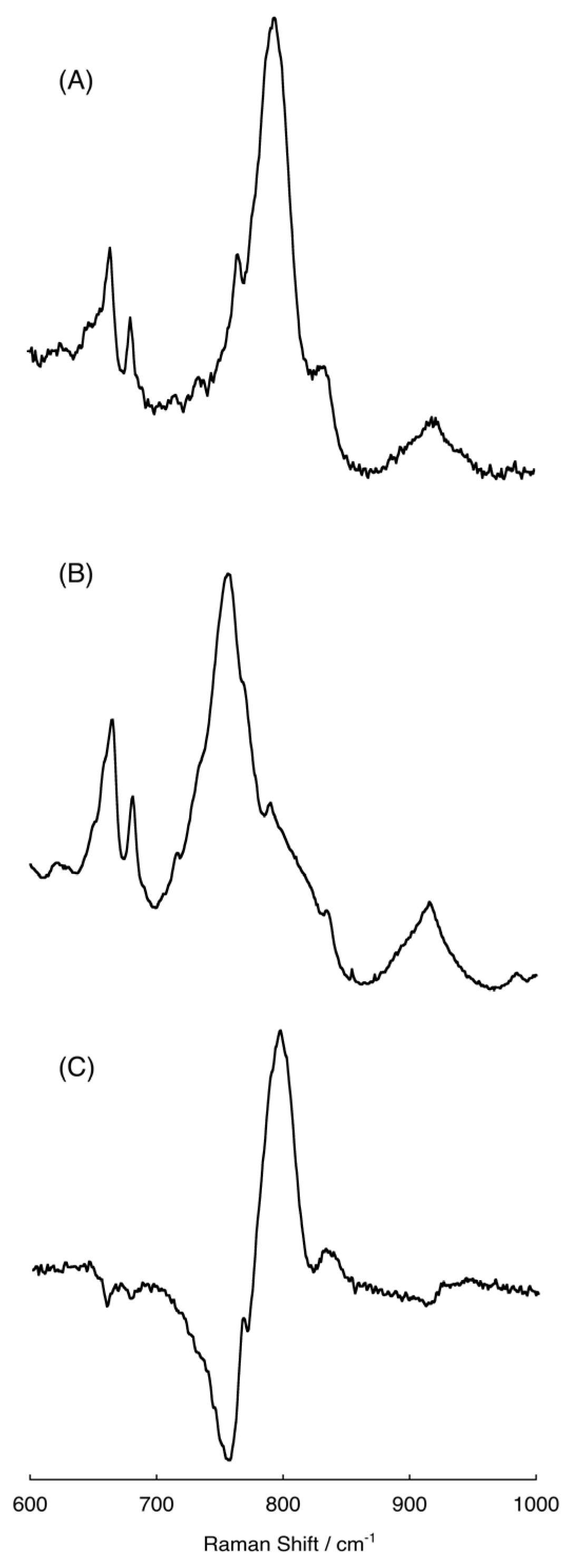
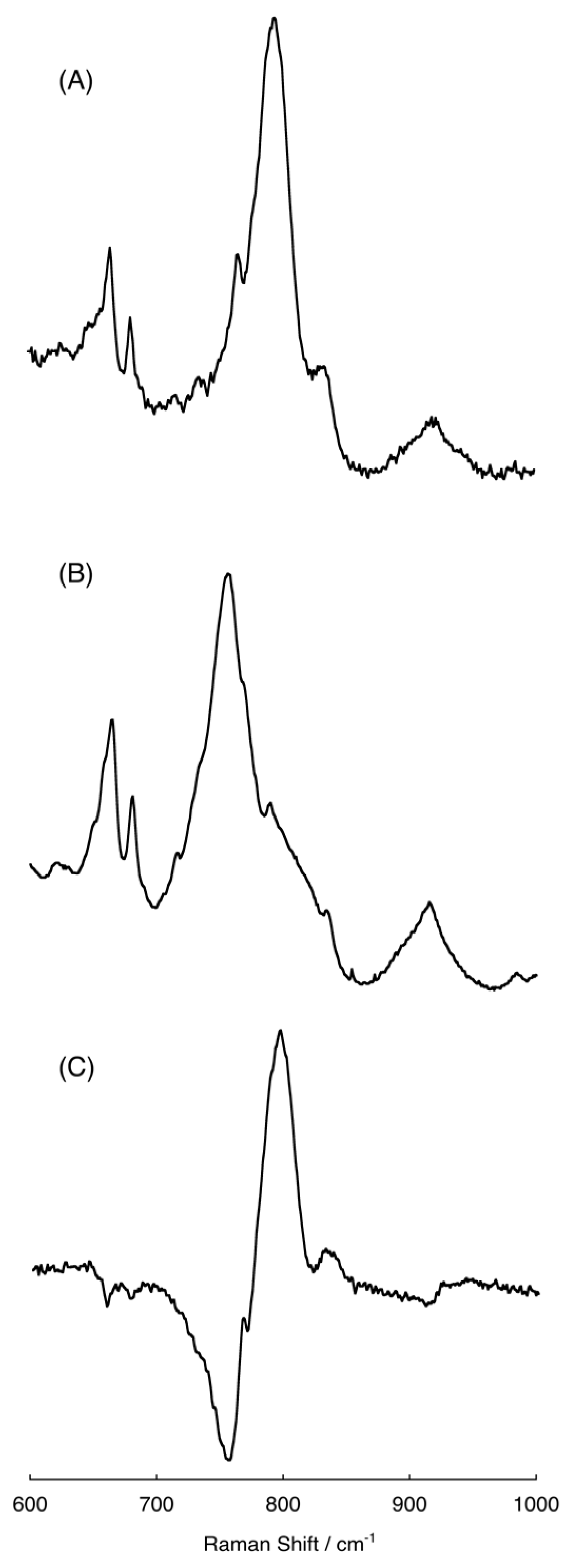
2.4. Raman Spectra of the Intermediate
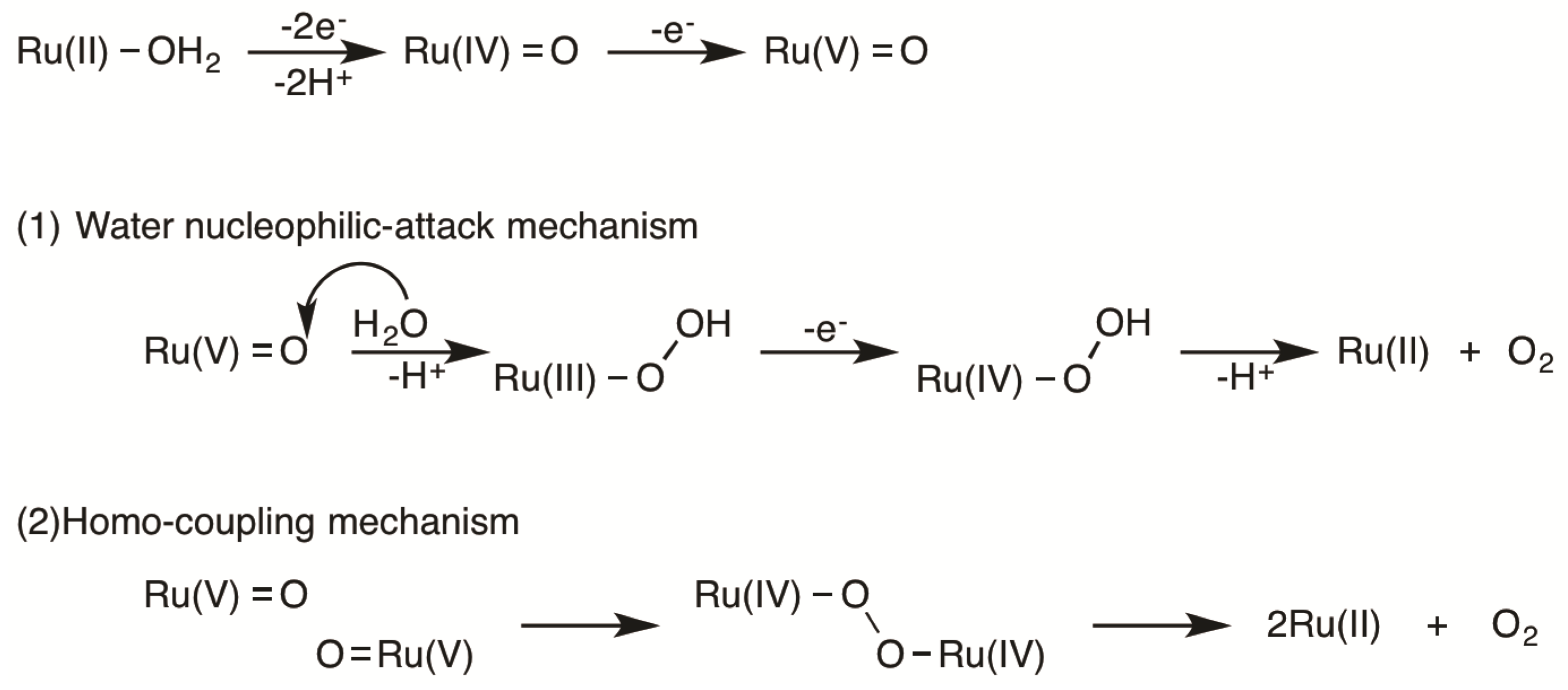
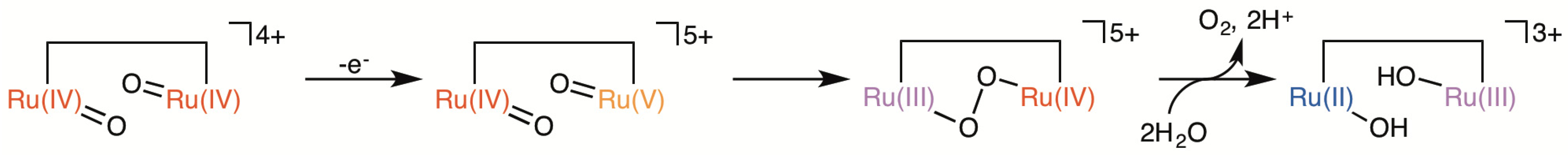
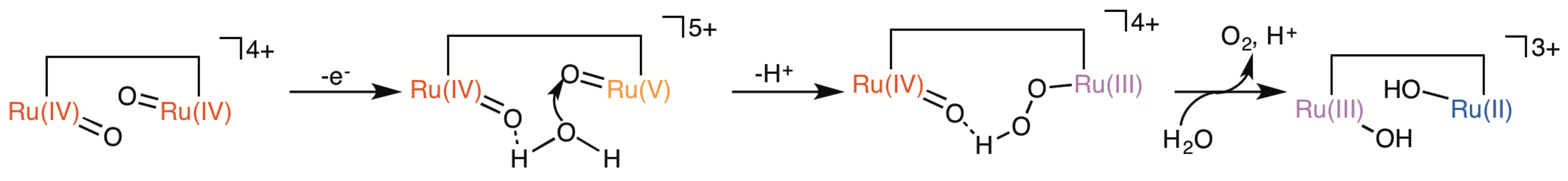
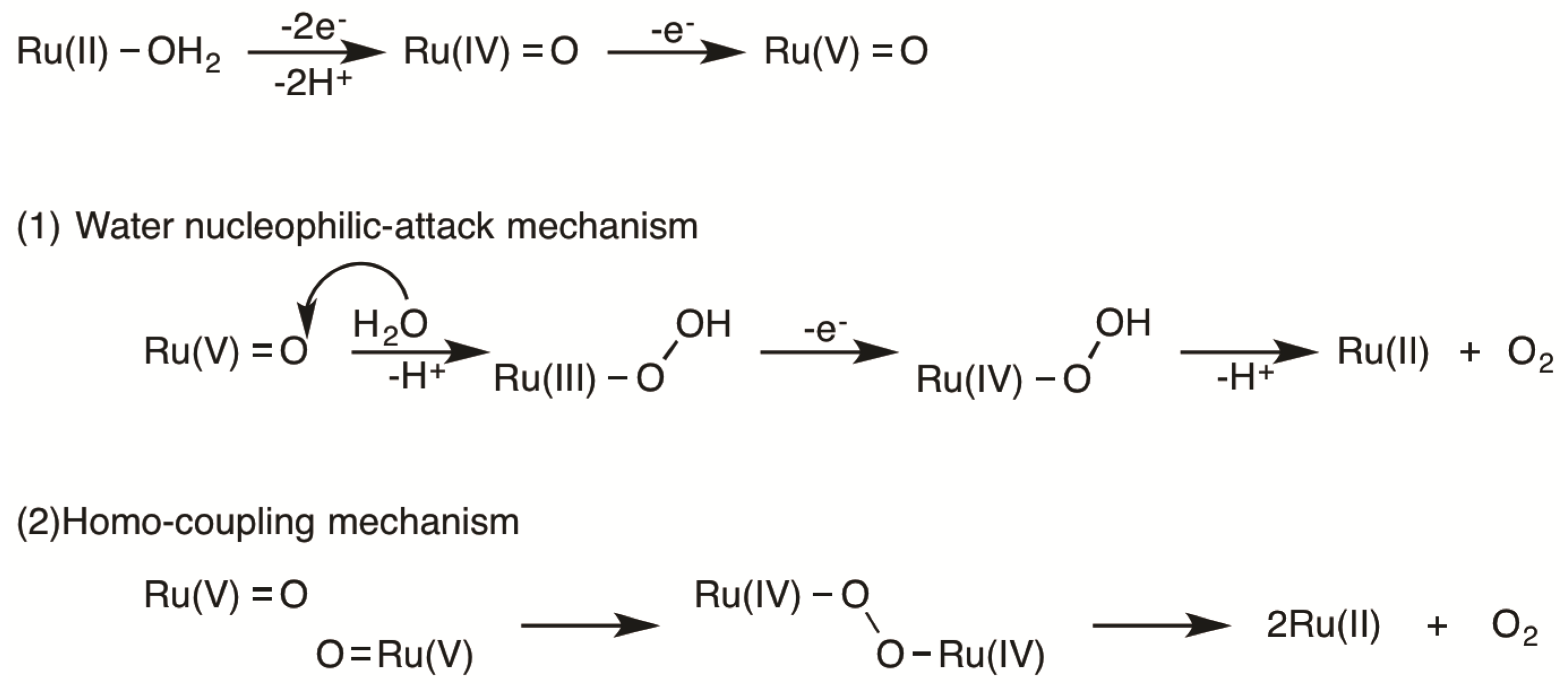
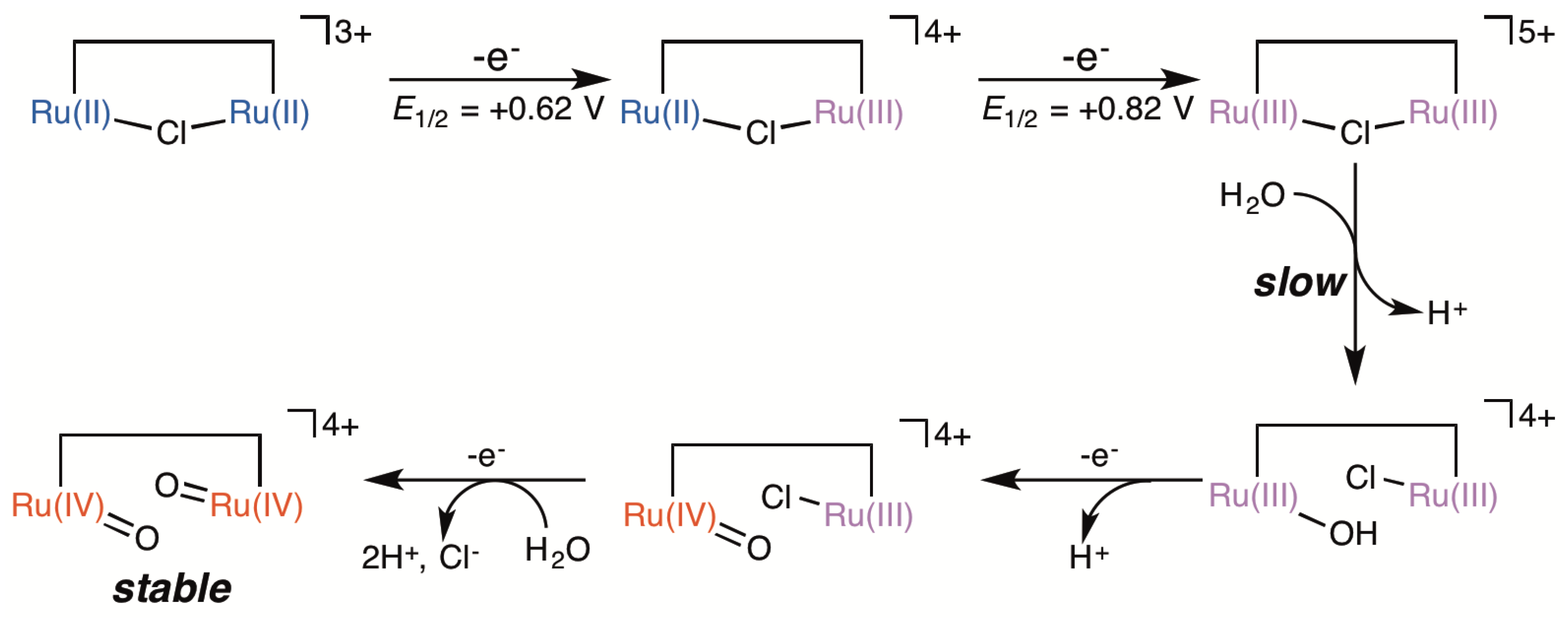
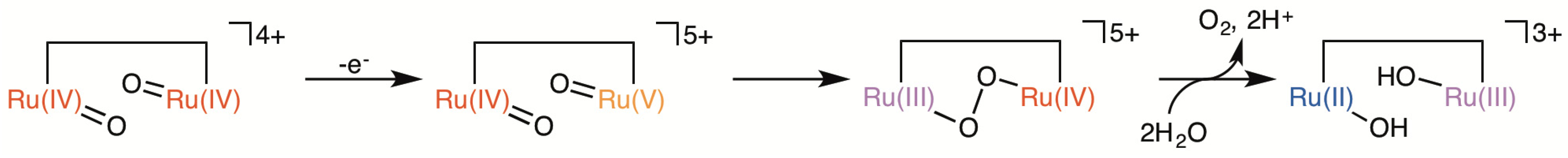
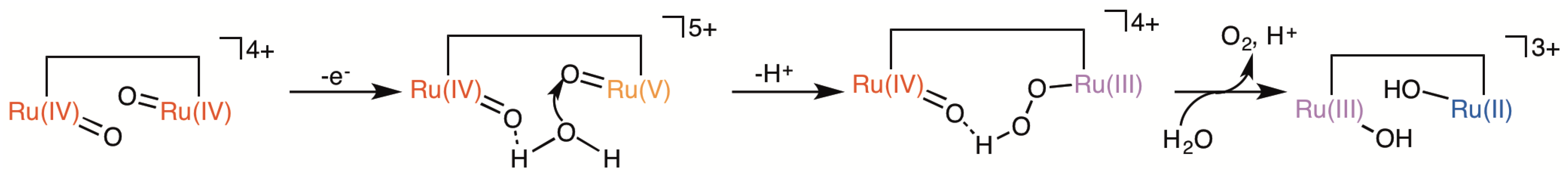
2.5. Catalytic Mechanism of Water Oxidation
3. Materials and Methods
3.1. Materials
3.2. Instrumentation
3.3. Syntheses
3.3.1. 1,8-Bis(neopentylglycolatoboryl)anthraquinone
3.3.2. Anthraquinone-1,8-diboronic Acid
3.3.3. 1,8-Bis(2,2′:6′,2″-terpyrid-4′-yl)anthraquinone (Btpyaq)
3.3.4. Ru2Cl6(btpyaq)
3.3.5. [Ru2Cl2(bpy)2(btpyaq)](SbF6)2
3.3.6. [Ru2(µ-Cl)(bpy)2(btpyaq)](BF4)3 ([3](BF4)3)
3.4. Water Oxidation
3.5. Measurements
3.5.1. Cyclic Voltammetry (CV) Measurements
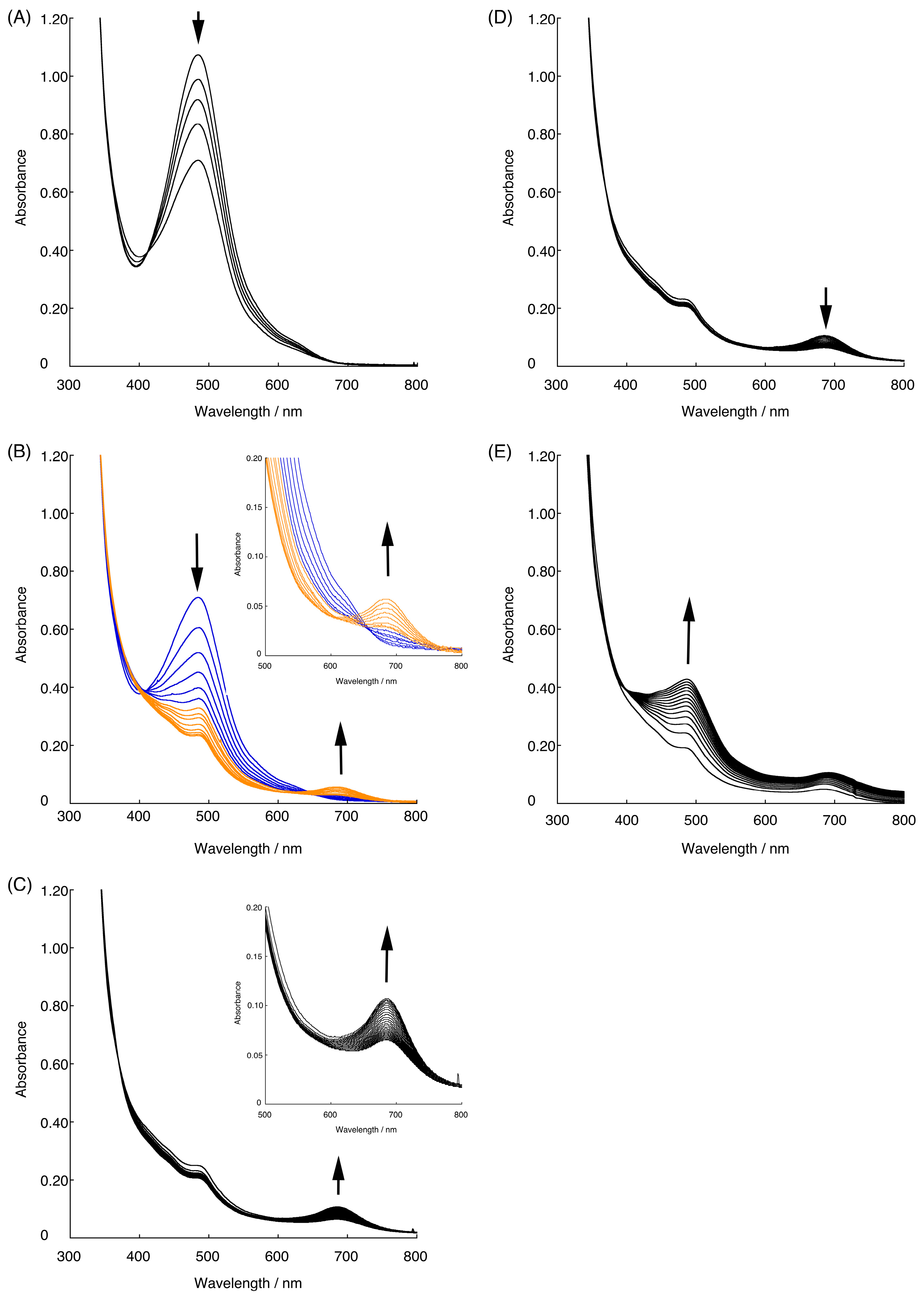
3.5.2. UV-Vis Spectral Measurements with Controlled-Potential Electrolysis
3.5.3. Raman Spectral Measurements with Controlled-Potential Electrolysis
3.5.4. Raman Spectral Measurements of the Chemically Oxidized Species
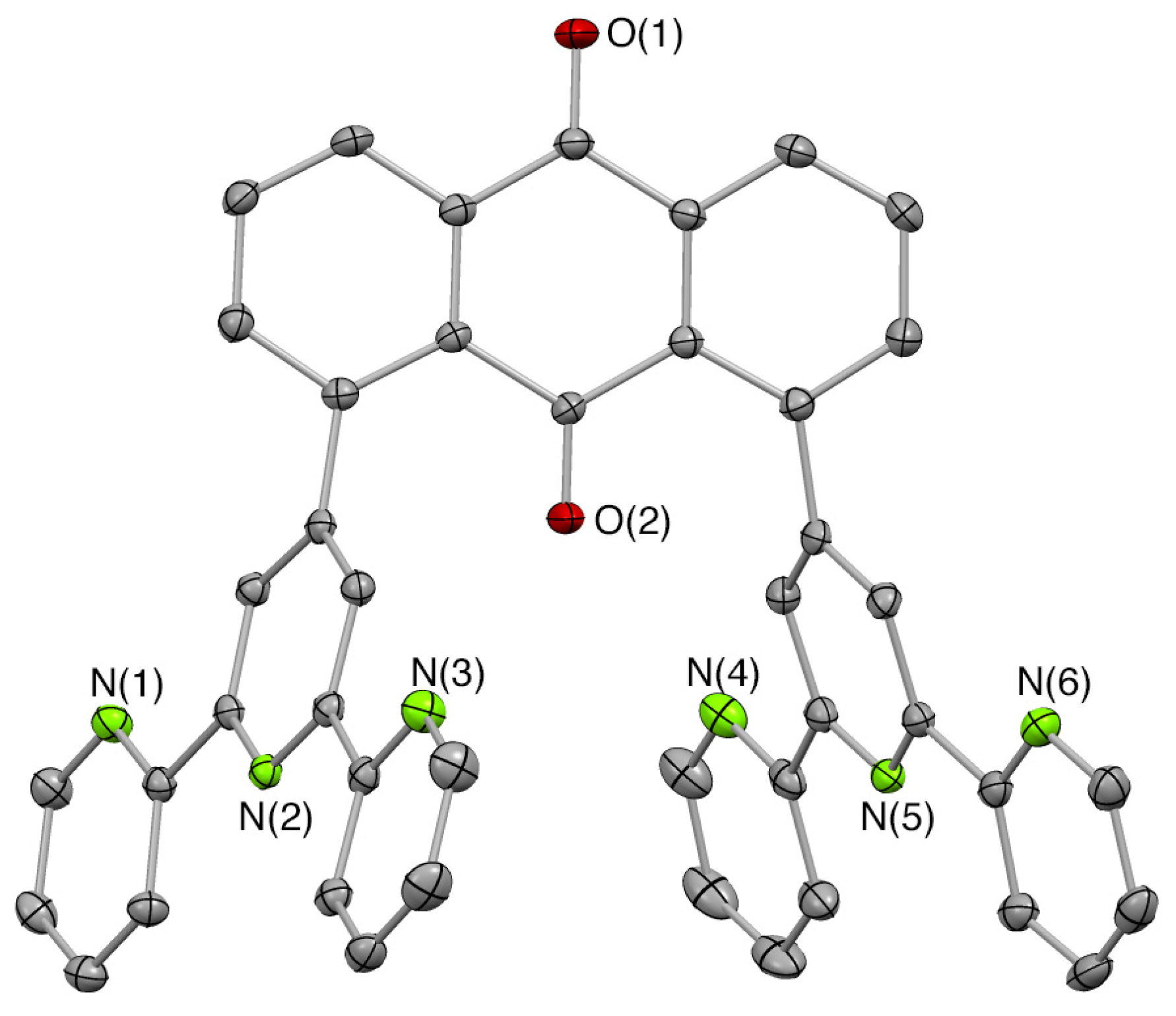
3.5.5. X-ray Crystallography
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, H.; Yan, R.; Zhang, D.; Fan, T. Challenges and perspectives in designing artificial photosynthetic systems. Chem. Eur. J. 2016, 22, 9870–9885. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Takeda, H.; Ishitani, O. Photocatalytic reduction of CO2 using metal complexes. J. Photochem. Photobiol. C Photochem. Rev. 2015, 25, 106–137. [Google Scholar] [CrossRef]
- Soman, S. Molecular systems for solar H2: Path to a renewable future. Comment Inorg. Chem. 2015, 35, 82–120. [Google Scholar] [CrossRef]
- House, R.L.; Iha, N.Y.M.; Coppo, R.L.; Alibabaei, L.; Sherman, B.D.; Kang, P.; Brennaman, M.K.; Hoertz, P.G.; Meyer, T.J. Artificial photosynthesis. J. Photochem. Photobiol. C Photochem. Rev. 2015, 25, 32–45. [Google Scholar] [CrossRef]
- Fukuzumi, S. Artificial photosynthetic systems for production of hydrogen. Curr. Opin. Chem. Biol. 2015, 25, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, A.Q.; Gregoire, J.M.; Luca, O.R. Electrocatalytic reduction of nitrogen and carbon dioxide to chemical fuels: Challenges and opportunities for a solar fuel device. J. Photochem. Photobiol. B Biol. 2015, 152, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Nocera, D.G. The artificial leaf. Acc. Chem. Res. 2012, 45, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, Z.; Brennaman, M.K.; Concepcion, J.J.; Patrocinio, A.O.T.; Iha, N.Y.M.; Meyer, T.J. Making solar fuels by artificial photosynthesis. Pure Appl. Chem. 2011, 83, 749–768. [Google Scholar] [CrossRef]
- Cook, T.R.; Dogutan, D.K.; Reece, S.Y.; Surendranath, Y.; Teets, T.S.; Nocera, D.G. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem. Rev. 2010, 110, 6474–6502. [Google Scholar] [CrossRef] [PubMed]
- Nocera, D.G. Chemistry of personalized solar energy. Inorg. Chem. 2009, 48, 10001–10017. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Shimada, T.; Kou, Y.; Nabetani, Y.; Masui, D.; Takagi, S.; Tachibana, H. The water oxidation bottleneck in artificial photosynthesis: How can we get through it? An alternative route involving a two-electron process. ChemSusChem 2011, 4, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Thummel, R.P. Mononuclear Ruthenium polypyridine complexes that catalyze water oxidation. Chem. Sci. 2016, 7, 6591–6603. [Google Scholar] [CrossRef]
- Xue, L.-X.; Meng, T.-T.; Yang, W.; Wang, K.-Z. Recent advances in Ruthenium complex-based light-driven water oxidation catalysts. J. Photochem. Photobiol. B Biol. 2015, 152, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, F.; Zhang, B.; Sun, L. Molecular complexes in water oxidation: Pre-catalysts or real catalysts. J. Photochem. Photobiol. C Photochem. Rev. 2015, 25, 71–89. [Google Scholar] [CrossRef]
- Duan, L.; Wang, L.; Li, F.; Li, F.; Sun, L. Highly fficient bioinspired molecules Ru water oxidation. Catalysts with negatively Charged backbone ligands. Acc. Chem. Res. 2015, 48, 2084–2096. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular catalysts for water oxidation. Chem. Rev. 2015, 115, 12947–13005. [Google Scholar] [CrossRef] [PubMed]
- Kaerkaes, M.D.; Verho, O.; Johnston, E.V.; Aakermark, B. Artificial photosynthesis: Molecular systems for catalytic water oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef] [PubMed]
- Kaerkaes, M.D.; Johnston, E.V.; Verho, O.; Aakermark, B. Artificial photosynthesis: From nanosecond electron transfer to catalytic water oxidation. Acc. Chem. Res. 2014, 47, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, M.; Shoji, A.; Yagi, M. Artificial manganese center models for photosynthetic oxygen evolution in photosystem II. Eur. J. Inorg. Chem. 2014, 2014, 595–606. [Google Scholar] [CrossRef]
- Francas, L.; Bofill, R.; Garcia-Anton, J.; Escriche, L.; Sala, X.; Llobet, A. Ru-based water oxidation catalysts. Mol. Water Oxid. Catal. 2014, 29–50. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Palmer, R.D.; Berlinguette, C.P. Homogeneous water oxidation catalysts containing a single metal site. Chem. Commun. 2013, 49, 218–227. [Google Scholar] [CrossRef] [PubMed]
- McAlpin, J.G.; Stich, T.A.; Casey, W.H.; Britt, R.D. Comparison of cobalt and manganese in the chemistry of water oxidation. Coord. Chem. Rev. 2012, 256, 2445–2452. [Google Scholar] [CrossRef]
- Artero, V.; Chavarot-Kerlidou, M.; Fontecave, M. Splitting water with cobalt. Angew. Chem. Int. Ed. 2011, 50, 7238–7266. [Google Scholar] [CrossRef] [PubMed]
- Yagi, M.; Syouji, A.; Yamada, S.; Komi, M.; Yamazaki, H.; Tajima, S. Molecular catalysts for water oxidation toward artificial photosynthesis. Photochem. Photobiol. Sci. 2009, 8, 139–147. [Google Scholar] [CrossRef]
- Yagi, M.; Kaneko, M. Molecular catalysts for water oxidation. Chem. Rev. 2001, 101, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Abe, T.; Kaneko, M. Historical overview and fundamental aspects of molecular catalysts for energy conversion. In Molecular Catalysts for Energy Conversion; Springer Series in Materials Science; Springer: Berlin/Heidelberg, Germany, 2009; Volume 111, pp. 1–36. [Google Scholar]
- Ruettinger, W.; Dismukes, G.C. Synthetic water-oxidation catalysts for artificial photosynthetic water oxidation. Chem. Rev. 1997, 97, 1–24. [Google Scholar] [CrossRef]
- Geletii Yurii, V.; Botar, B.; Kogerler, P.; Hillesheim Daniel, A.; Musaev Djamaladdin, G.; Hill Craig, L. An all-inorganic, stable, and highly active tetraRuthenium homogeneous catalyst for water oxidation. Angew. Chem. Int. Ed. 2008, 47, 3896–3899. [Google Scholar] [CrossRef] [PubMed]
- Sartorel, A.; Carraro, M.; Scorrano, G.; de Zorzi, R.; Geremia, S.; McDaniel, N.D.; Bernhard, S.; Bonchio, M. Polyoxometalate embedding of a tetraRuthenium(iv)-oxo-core by template-directed metalation of [γ-SiW10O36]8−: A totally inorganic oxygen-evolving catalyst. J. Am. Chem. Soc. 2008, 130, 5006–5007. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Lewis, F.W.; Harwood, L.M.; Hartl, F. Role of ligands in catalytic water oxidation by mononuclear Ruthenium complexes. Coord. Chem. Rev. 2015, 304–305, 88–101. [Google Scholar] [CrossRef]
- Sala, X.; Maji, S.; Bofill, R.; Garcia-Anton, J.; Escriche, L.; Llobet, A. Molecular water oxidation mechanisms followed by transition metals: State of the art. Acc. Chem. Res. 2014, 47, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Tanaka, K. Mechanistic approaches to molecular catalysts for water oxidation. Eur. J. Inorg. Chem. 2014, 2014, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Sala, X.; Romero, I.; Rodriguez, M.; Escriche, L.; Llobet, A. Molecular catalysts that oxidize water to dioxygen. Angew. Chem. Int. Ed. 2009, 48, 2842–2852. [Google Scholar] [CrossRef] [PubMed]
- Betley, T.A.; Wu, Q.; van Voorhis, T.; Nocera, D.G. Electronic design criteria for O–O bond formation via metal-oxo complexes. Inorg. Chem. 2008, 47, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, C.J.; Vannucci, A.K.; Concepcion, J.J.; Chen, Z.; Meyer, T.J. The role of proton coupled electron transfer in water oxidation. Energy Environ. Sci. 2012, 5, 7704–7717. [Google Scholar] [CrossRef]
- Meyer, T.J.; Huynh, M.H.V.; Thorp, H.H. The possible role of proton-coupled electron transfer (PCET) in water oxidation by photosystem II. Angew. Chem. Int. Ed. 2007, 46, 5284–5304. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.H.V.; Meyer, T.J. Proton-coupled electron transfer. Chem. Rev. 2007, 107, 5004–5064. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.J.; Huynh, M.H.V. The remarkable reactivity of high oxidation state Ruthenium and osmium polypyridyl complexes. Inorg. Chem. 2003, 42, 8140–8160. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, E.L.; Binstead, R.A.; Meyer, T.J. Mechanistic implications of proton transfer coupled to electron transfer. J. Am. Chem. Soc. 2001, 123, 10535–10544. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, J.J.; Tsai, K.; Muckerman, J.T.; Meyer, T.J. Mechanism of water oxidation by single-site Ruthenium complex catalysts. J. Am. Chem. Soc. 2010, 132, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Badiei, Y.M.; Polyansky, D.E.; Muckerman, J.T.; Szalda, D.J.; Haberdar, R.; Zong, R.; Thummel, R.P.; Fujita, E. Water oxidation with mononuclear Ruthenium(II) polypyridine complexes involving a direct Ru(IV)=O pathway in neutral and alkaline media. Inorg. Chem. 2013, 52, 8845–8850. [Google Scholar] [CrossRef] [PubMed]
- Romain, S.; Bozoglian, F.; Sala, X.; Llobet, A. Oxygen–oxygen bond formation by the Ru-Hbpp water oxidation catalyst occurs solely via an intramolecular reaction pathway. J. Am. Chem. Soc. 2009, 131, 2768–2769. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Duan, L.; Wang, Y.; Ahlquist, M.S.G.; Sun, L. Highly efficient and robust molecular water oxidation catalysts based on Ruthenium complexes. Chem. Commun. 2014, 50, 12947–12950. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular Ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Wang, L.; Inge, A.K.; Fischer, A.; Zou, X.; Sun, L. Insights into ru-based molecular water oxidation catalysts: Electronic and noncovalent-interaction effects on their catalytic activities. Inorg. Chem. 2013, 52, 7844–7852. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, Y.; Ishizuka, T.; Kotani, H.; Shiota, Y.; Yoshizawa, K.; Mieda, K.; Ogura, T.; Okajima, T.; Nozawa, S.; Kojima, T. A Ruthenium(III)-oxyl complex bearing strong radical character. Angew. Chem. Int. Ed. 2016, 55, 14041–14045. [Google Scholar] [CrossRef] [PubMed]
- Stull, J.A.; Stich, T.A.; Hurst, J.K.; Britt, R.D. Electron paramagnetic resonance analysis of a transient species formed during water oxidation catalyzed by the complex ion [(bpy)2Ru(OH2)]2O4+. Inorg. Chem. 2013, 52, 4578–4586. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Hiraide, T.; Miyazato, Y. Water oxidation catalyzed by a Ruthenium complex with an Ru–C bond. ChemistrySelect 2016, 1, 3045–3048. [Google Scholar] [CrossRef]
- Wada, T.; Tanaka, K.; Muckerman, J.T.; Fujita, E. Water oxidation by Ruthenium catalysts with non-innocent ligands. In Moecular Water Oxidation Catalysis, 1st ed.; Llobet, A., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2014; pp. 77–111. [Google Scholar]
- Wada, T.; Ohtsu, H.; Tanaka, K. Catalytic four-electron oxidation of water by intramolecular coupling of the oxo ligands of a bis(Ruthenium-bipyridine) complex. Chem. Eur. J. 2012, 18, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Muckerman, J.T.; Fujita, E.; Tanaka, K. Substituents dependent capability of bis(Ruthenium-dioxolene-terpyridine) complexes toward water oxidation. Dalton Trans. 2011, 40, 2225–2233. [Google Scholar] [CrossRef] [PubMed]
- Muckerman, J.T.; Polyansky, D.E.; Wada, T.; Tanaka, K.; Fujita, E. Water oxidation by a Ruthenium complex with noninnocent quinone ligands: Possible formation of an O–O bond at a low oxidation state of the metal. Inorg. Chem. 2008, 47, 1787–1802. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Tsuge, K.; Tanaka, K. Syntheses and redox properties of bis(hydroxoRuthenium) complexes with quinone and bipyridine ligands. Water-oxidation catalysis. Inorg. Chem. 2001, 40, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Tsuge, K.; Tanaka, K. Electrochemical oxidation of water to dioxygen catalyzed by the oxidized form of the bis(Ruthenium-hydroxo) complex in H2O. Angew. Chem. Int. Ed. 2000, 39, 1479–1482. [Google Scholar] [CrossRef]
- Isobe, H.; Tanaka, K.; Shen, J.-R.; Yamaguchi, K. Water oxidation chemistry of a synthetic dinuclear Ruthenium complex containing redox-active quinone ligands. Inorg. Chem. 2014, 53, 3973–3984. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yamanaka, S.; Isobe, H.; Tanaka, K.; Ueyama, N. Spin hamiltonian models for artificial and native water splitting systems revealed by hybrid dft calculations. Oxygen activation by high-valent mn and Ru ions. Int. J. Quantum Chem. 2012, 112, 3849–3866. [Google Scholar] [CrossRef]
- Tanaka, K.; Isobe, H.; Yamanaka, S.; Yamaguchi, K. Similarities of artificial photosystems by Ruthenium oxo complexes and native water splitting systems. Proc. Natl. Acad. Sci. USA 2012, 109, 15600–15605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Baik, M.-H. Redox properties of tanaka’s water oxidation catalyst: Redox noninnocent ligands dominate the electronic structure and reactivity. Inorg. Chem. 2011, 50, 5946–5957. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Masaoka, S. Cerium(IV)-driven oxidation of water catalyzed by mononuclear Ruthenium complexes. Res. Chem. Int. 2014, 40, 3169–3182. [Google Scholar] [CrossRef]
- Kimoto, A.; Yamauchi, K.; Yoshida, M.; Masaoka, S.; Sakai, K. Kinetics and dft studies on water oxidation by Ce4+ catalyzed by [Ru(terpy)(bpy)(OH2)]2+. Chem. Commun. 2012, 48, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Masaoka, S.; Abe, J.; Sakai, K. Catalysis of mononuclear aquaRuthenium complexes in oxygen evolution from water: A new radical coupling path using hydroxocerium(IV) species. Chem. Asian J. 2010, 5, 2369–2378. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Koike, T.; Hurst, J.K. Water exchange rates in the diRuthenium μ-oxo ion cis,cis-[(bpy)2Ru(OH2)]2O4+. J. Am. Chem. Soc. 2001, 123, 12775–12780. [Google Scholar] [CrossRef] [PubMed]
- Moonshiram, D.; Purohit, V.; Concepcion, J.J.; Meyer, T.J.; Pushkar, Y. Mechanism of catalytic water oxidation by the Ruthenium blue dimer catalyst: Comparative study in D2O versus H2O. Materials 2013, 6, 392–409. [Google Scholar] [CrossRef]
- Liu, F.; Concepcion, J.J.; Jurss, J.W.; Cardolaccia, T.; Templeton, J.L.; Meyer, T.J. Mechanisms of water oxidation from the blue dimer to photosystem II. Inorg. Chem. 2008, 47, 1727–1752. [Google Scholar] [CrossRef] [PubMed]
- Binstead, R.A.; Chronister, C.W.; Ni, J.; Hartshorn, C.M.; Meyer, T.J. Mechanism of water oxidation by the μ-oxo dimer [(bpy)2(H2O)RuIIIORuIII(OH2)(bpy)2]4+. J. Am. Chem. Soc. 2000, 122, 8464–8473. [Google Scholar] [CrossRef]
- Chronister, C.W.; Binstead, R.A.; Ni, J.; Meyer, T.J. Mechanism of water oxidation catalyzed by the μ-oxo dimer [(bpy)2(H2O)RuIIIORuIII(OH2)(bpy)2]4+. Inorg. Chem. 1997, 36, 3814–3815. [Google Scholar] [CrossRef]
- Ukai, T.; Kawazura, H.; Ishii, Y.; Bonnet, J.J.; Ibers, J.A. Chemistry of dibenzylideneacetone-palladium(0) complexes. I. Novel tris(dibenzylideneacetone)dipalladium(solvent) complexes and their reactions with quinones. J. Organomet. Chem. 1974, 65, 253–266. [Google Scholar] [CrossRef]
- Potts, K.T.; Konwar, D. Synthesis of 4′-vinyl-2,2′:6′,2″-terpyridine. J. Org. Chem. 1991, 56, 4815–4816. [Google Scholar] [CrossRef]
- APEX2, ver2014.11-0. Bruker-AXS: Madison, WI, USA, 2014.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Cryst. 2015, A71, 59–75. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wada, T.; Nishimura, S.; Mochizuki, T.; Ando, T.; Miyazato, Y. Mechanism of Water Oxidation Catalyzed by a Dinuclear Ruthenium Complex Bridged by Anthraquinone. Catalysts 2017, 7, 56. https://doi.org/10.3390/catal7020056
Wada T, Nishimura S, Mochizuki T, Ando T, Miyazato Y. Mechanism of Water Oxidation Catalyzed by a Dinuclear Ruthenium Complex Bridged by Anthraquinone. Catalysts. 2017; 7(2):56. https://doi.org/10.3390/catal7020056
Chicago/Turabian StyleWada, Tohru, Shunsuke Nishimura, Taro Mochizuki, Tomohiro Ando, and Yuji Miyazato. 2017. "Mechanism of Water Oxidation Catalyzed by a Dinuclear Ruthenium Complex Bridged by Anthraquinone" Catalysts 7, no. 2: 56. https://doi.org/10.3390/catal7020056