
Unprecedented Multifunctionality of Grubbs and Hoveyda–Grubbs Catalysts: Competitive Isomerization, Hydrogenation, Silylation and Metathesis Occurring in Solution and on Solid Phase


Abstract
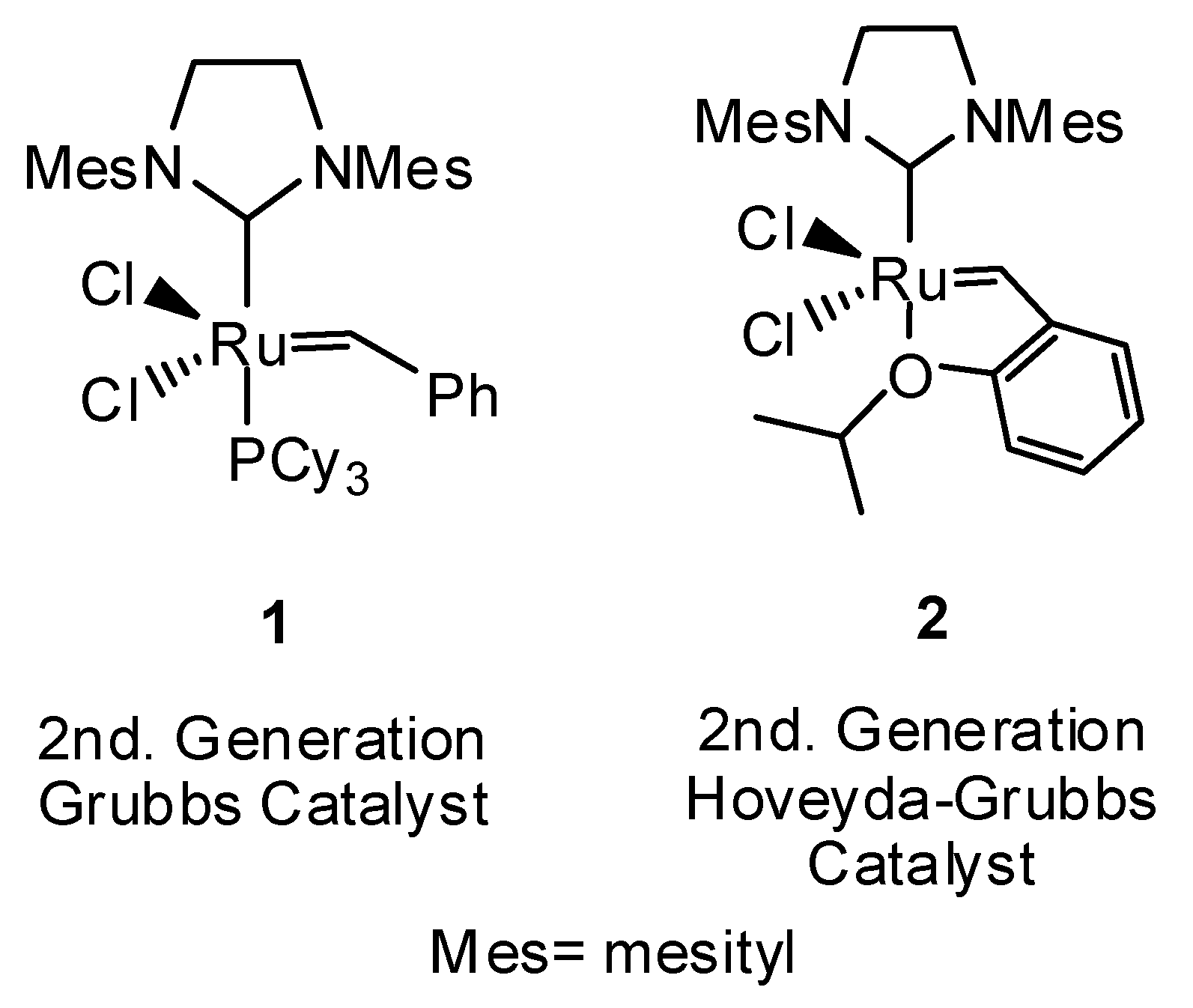
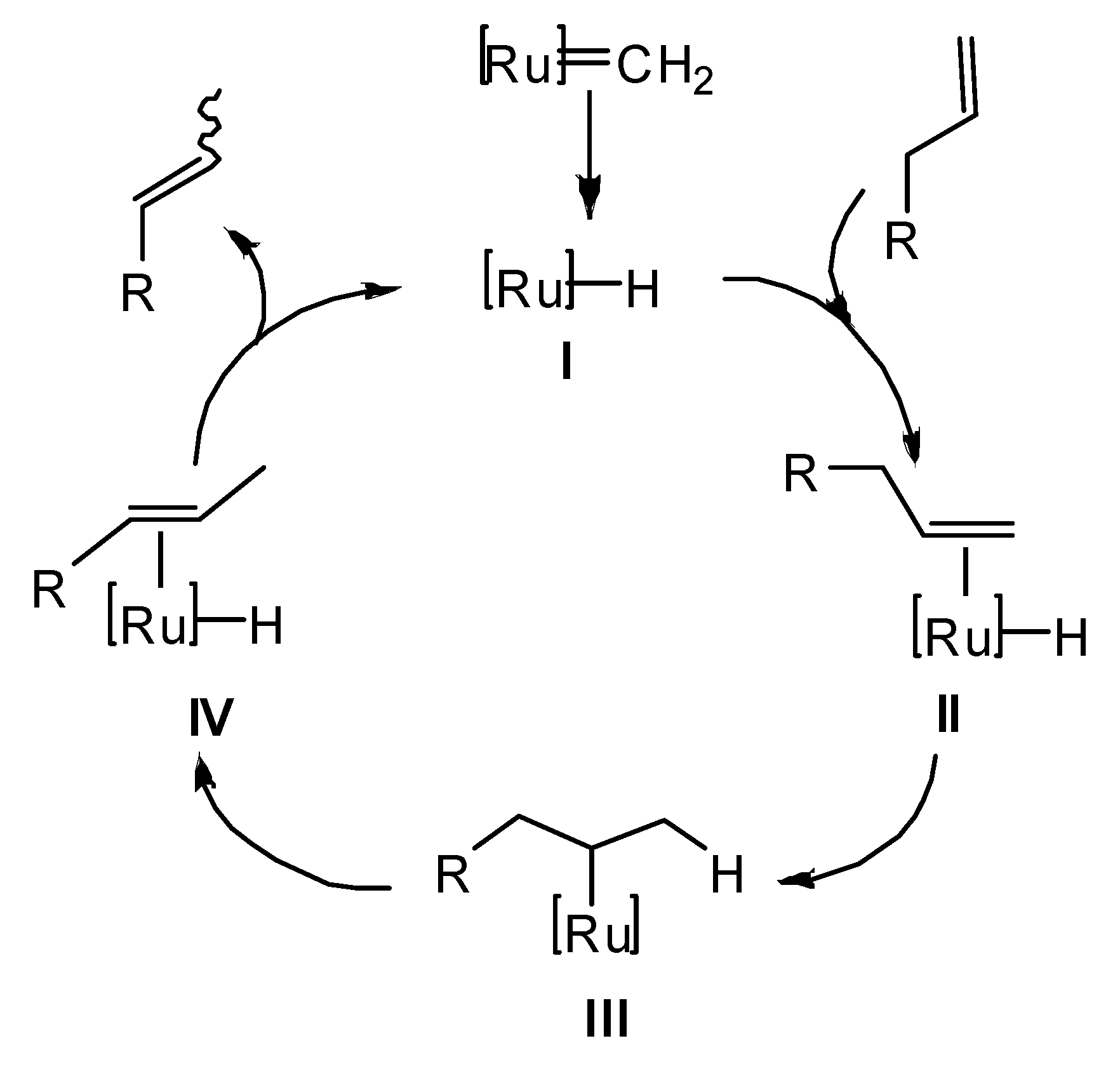
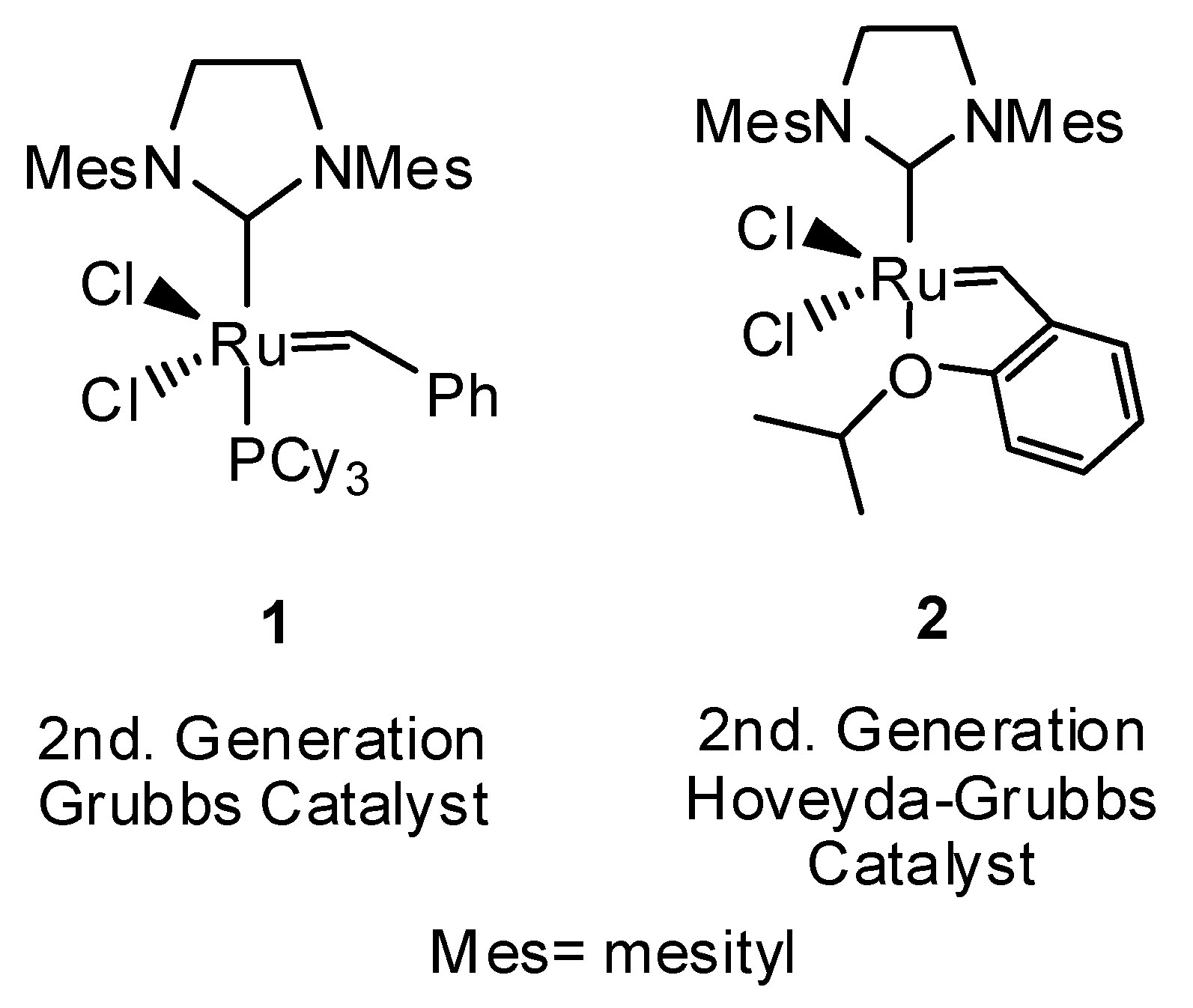
:1. Introduction
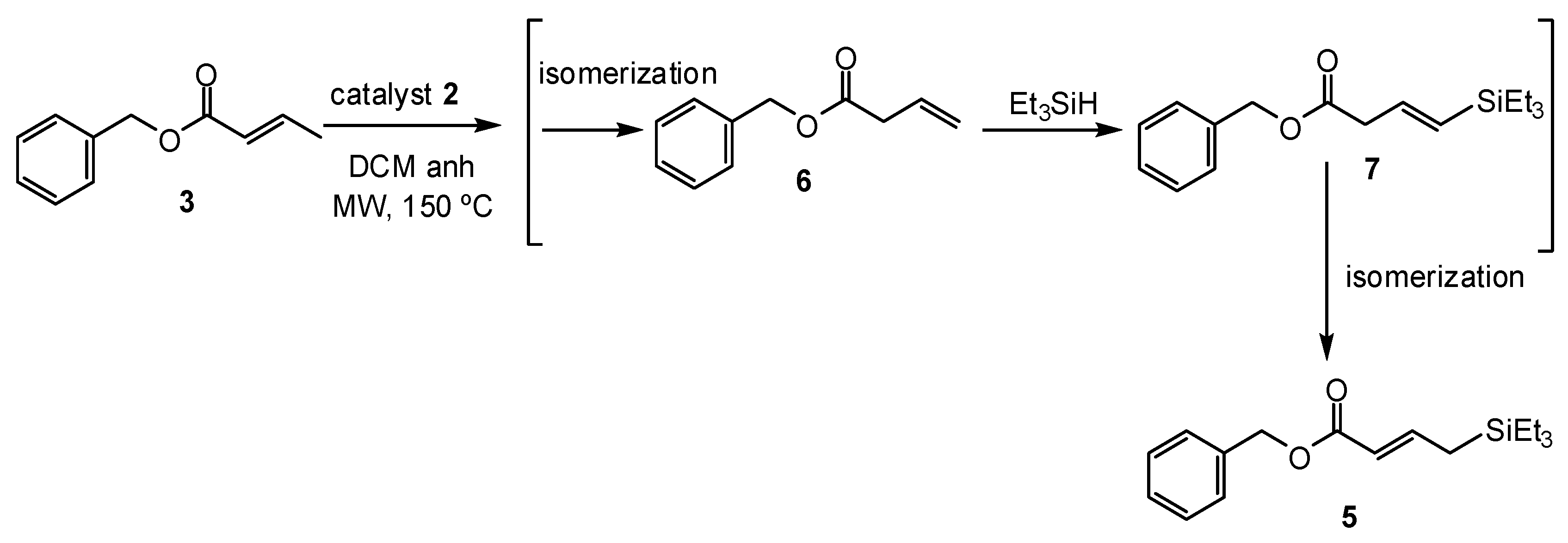
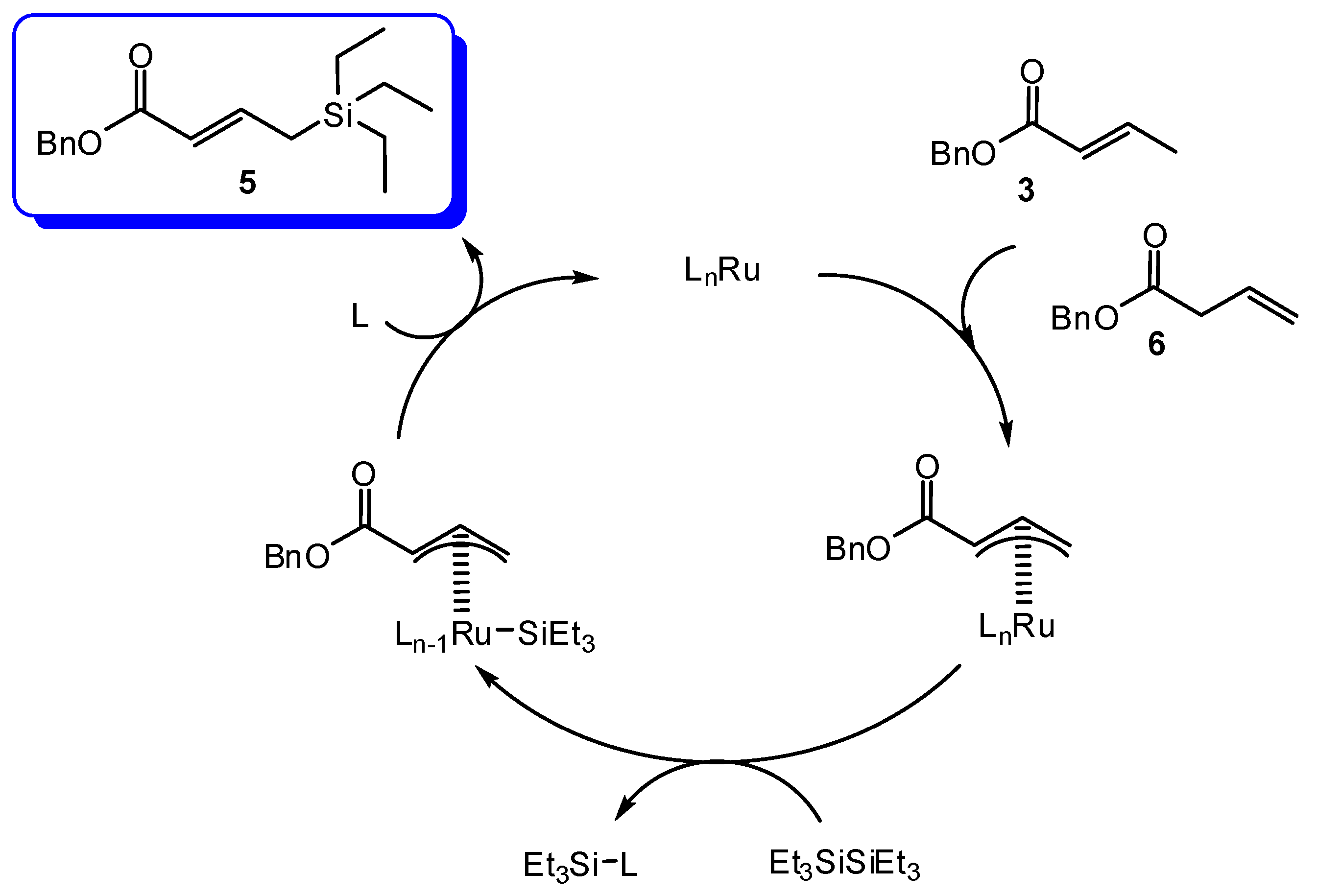
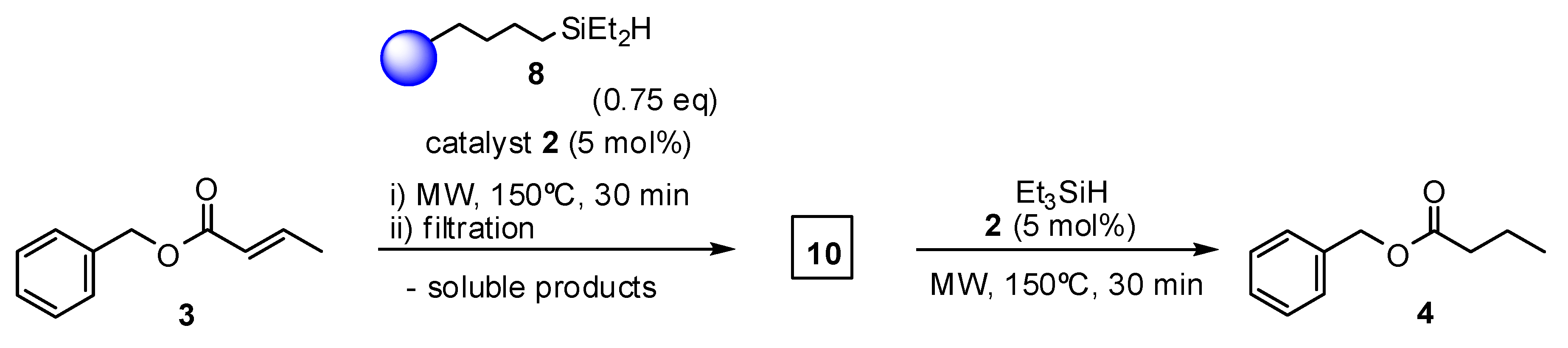
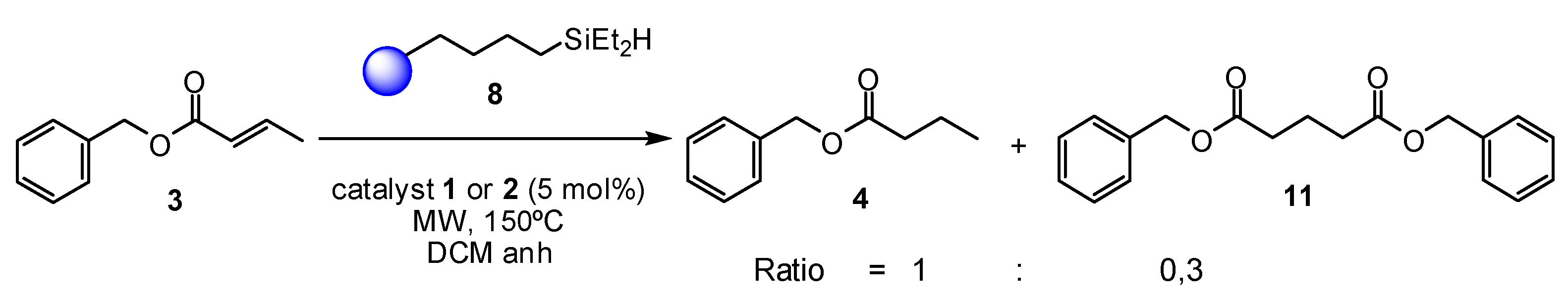
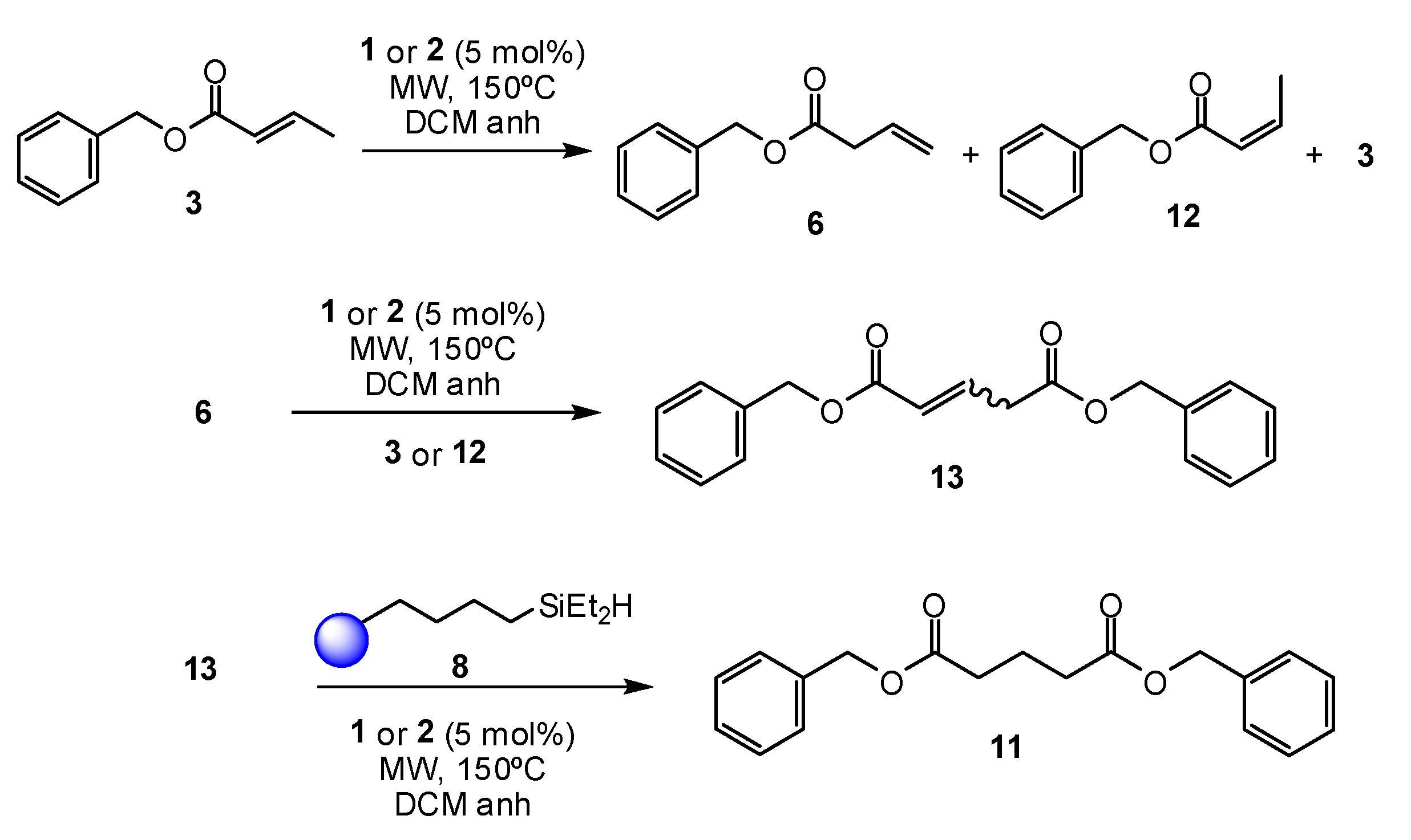
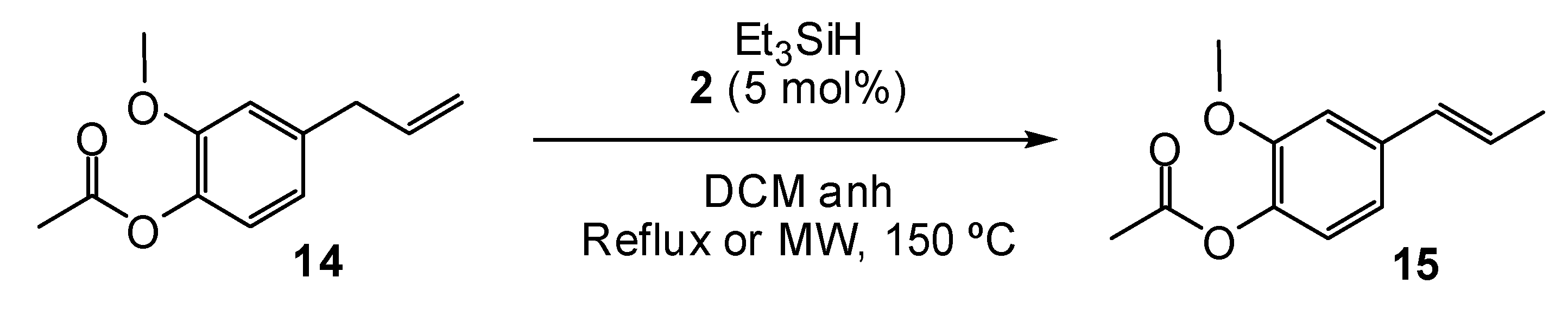
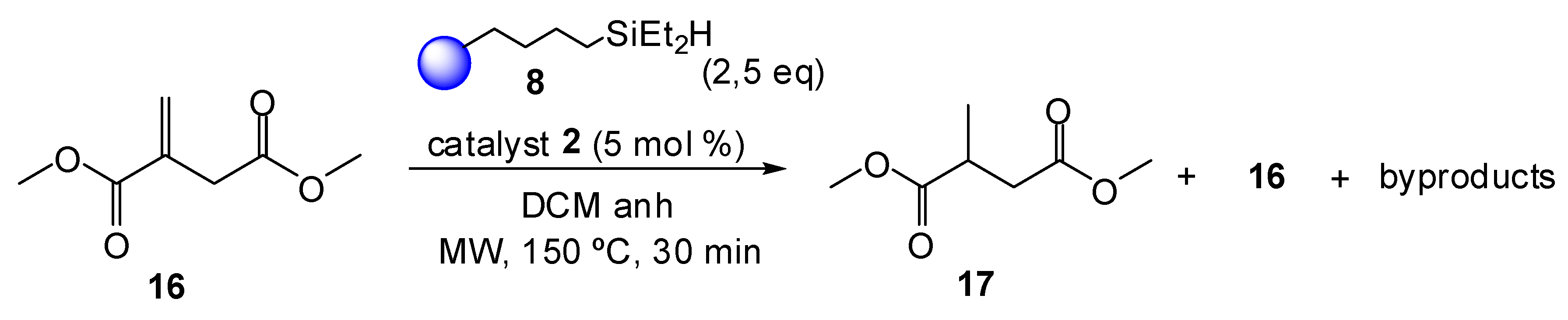
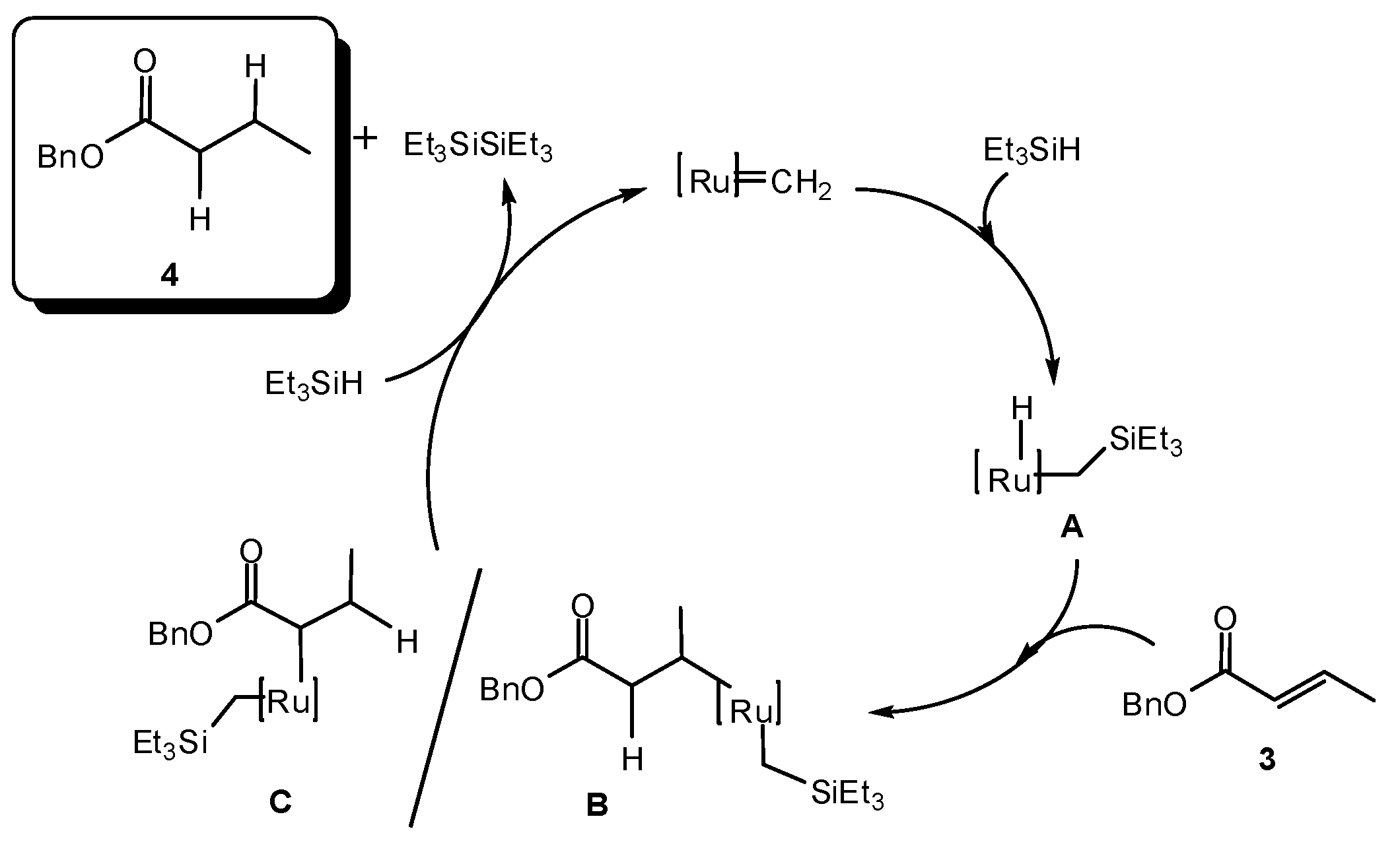
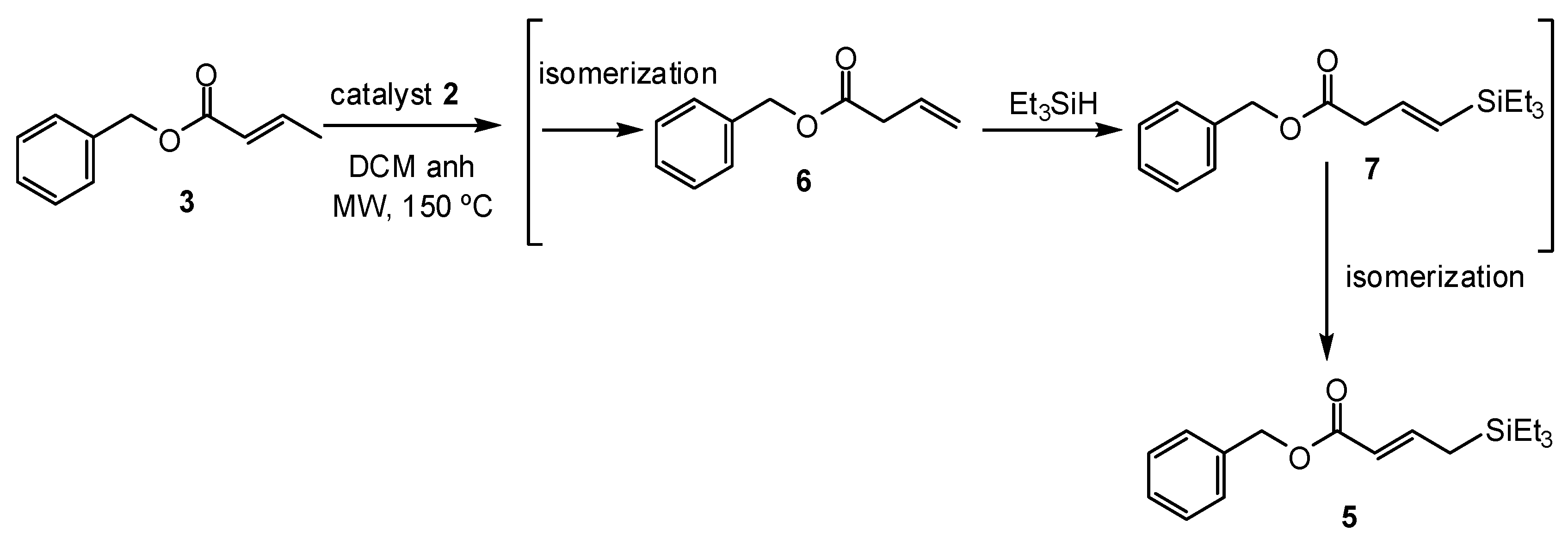
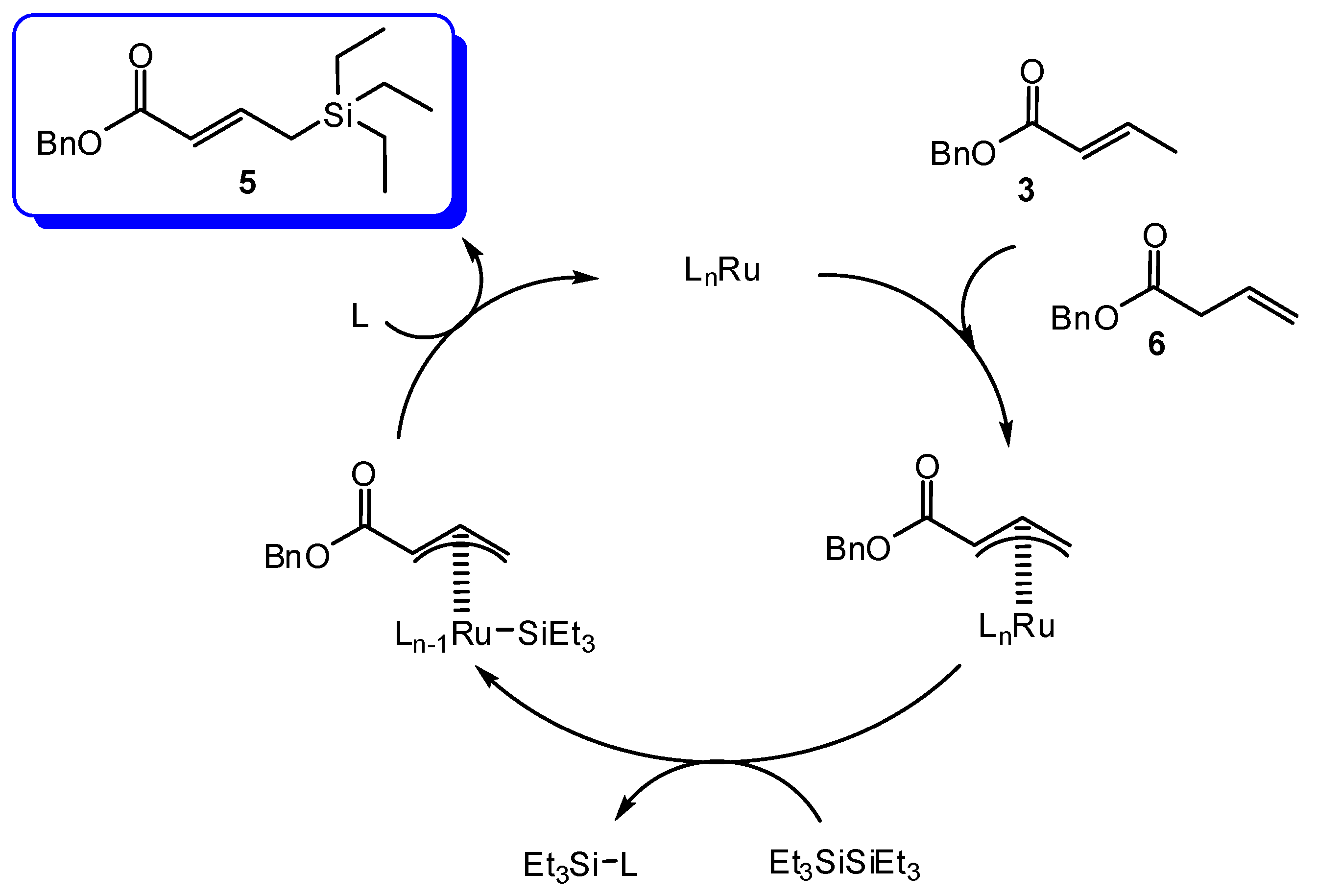
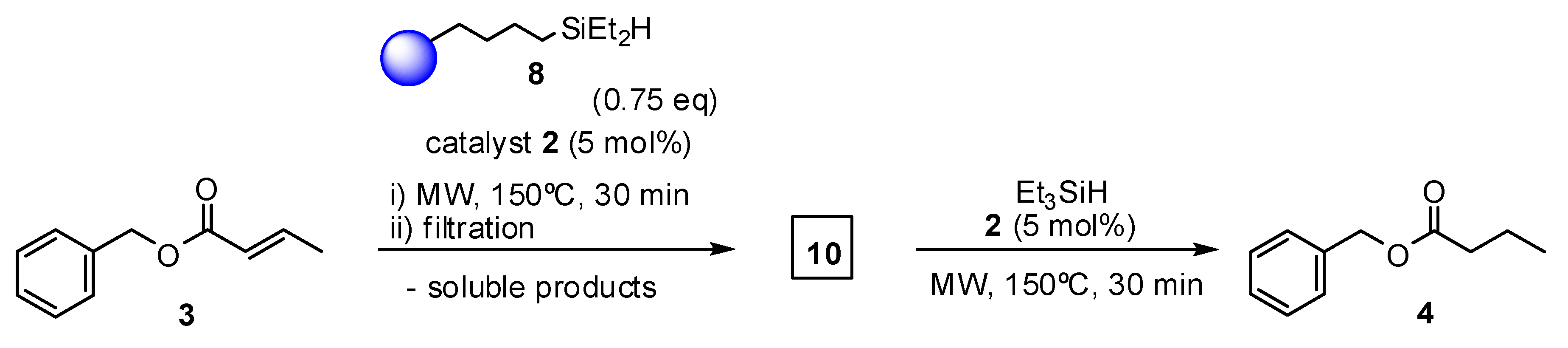
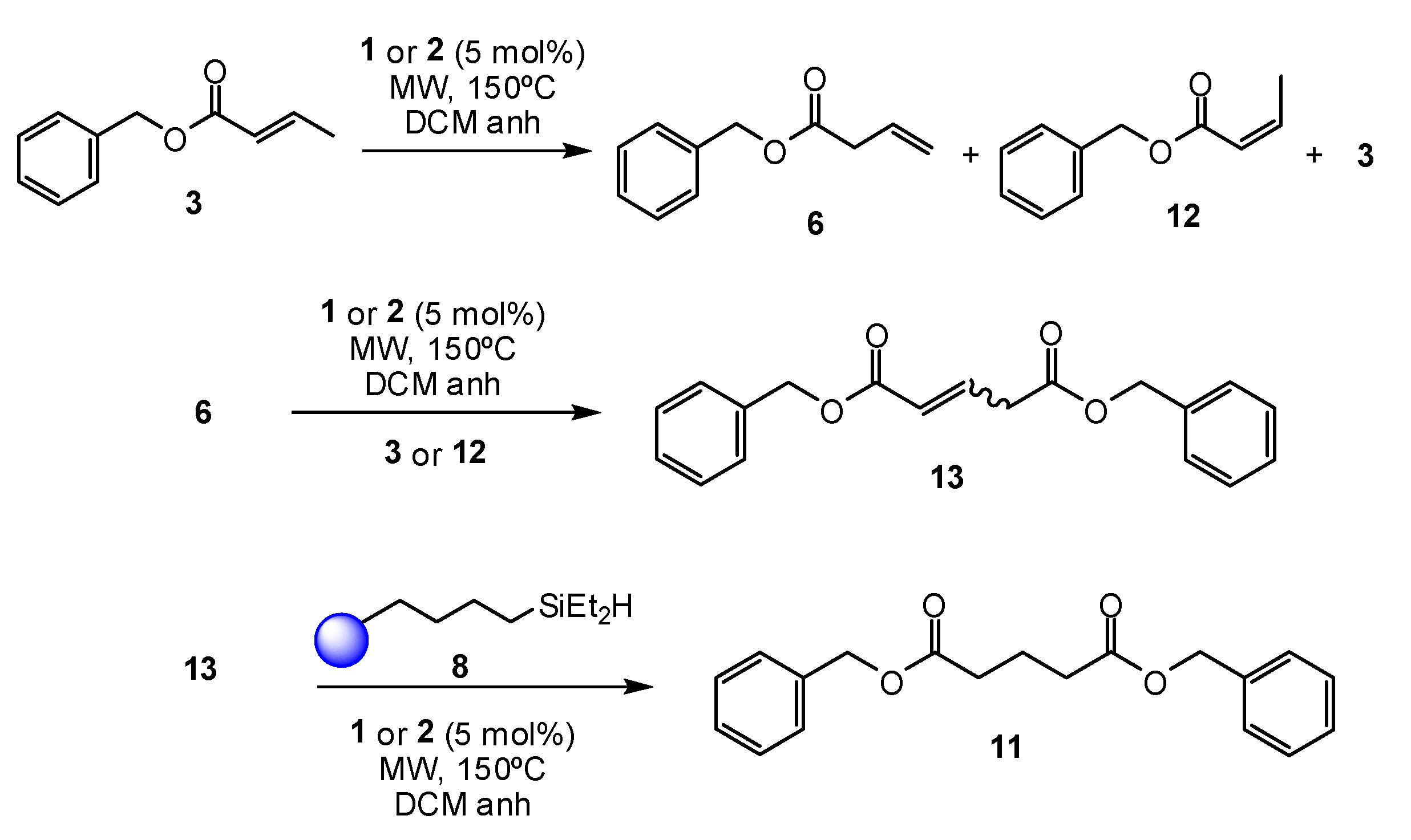
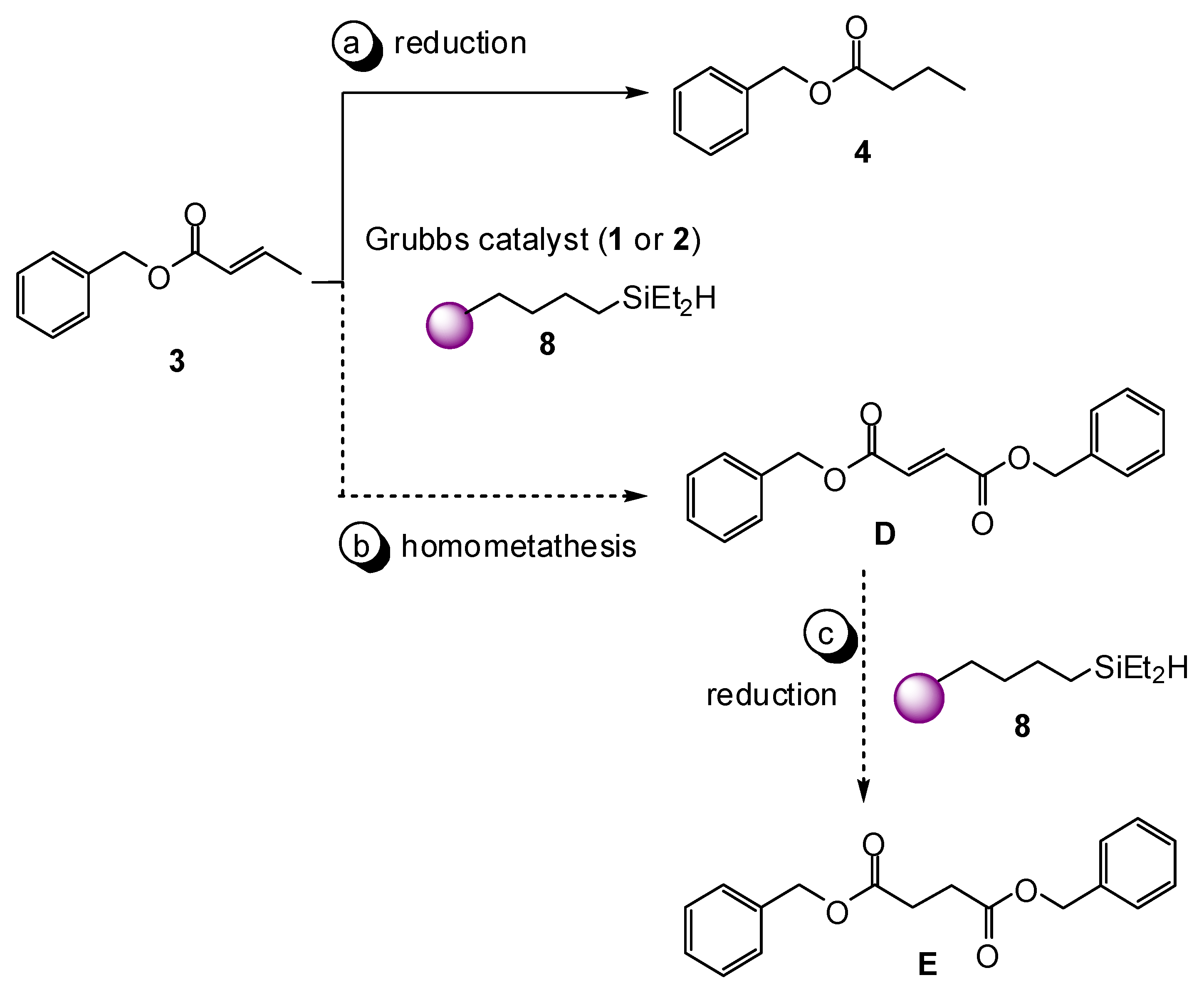
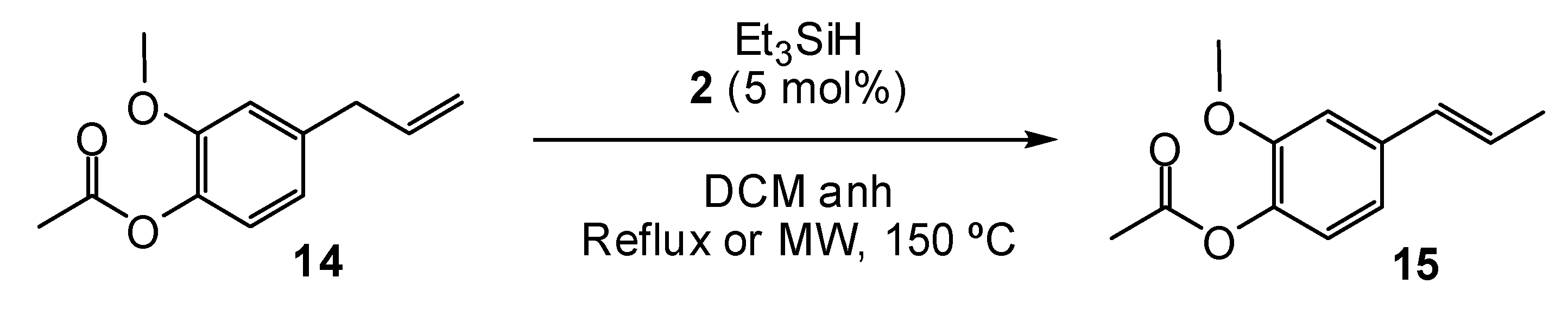
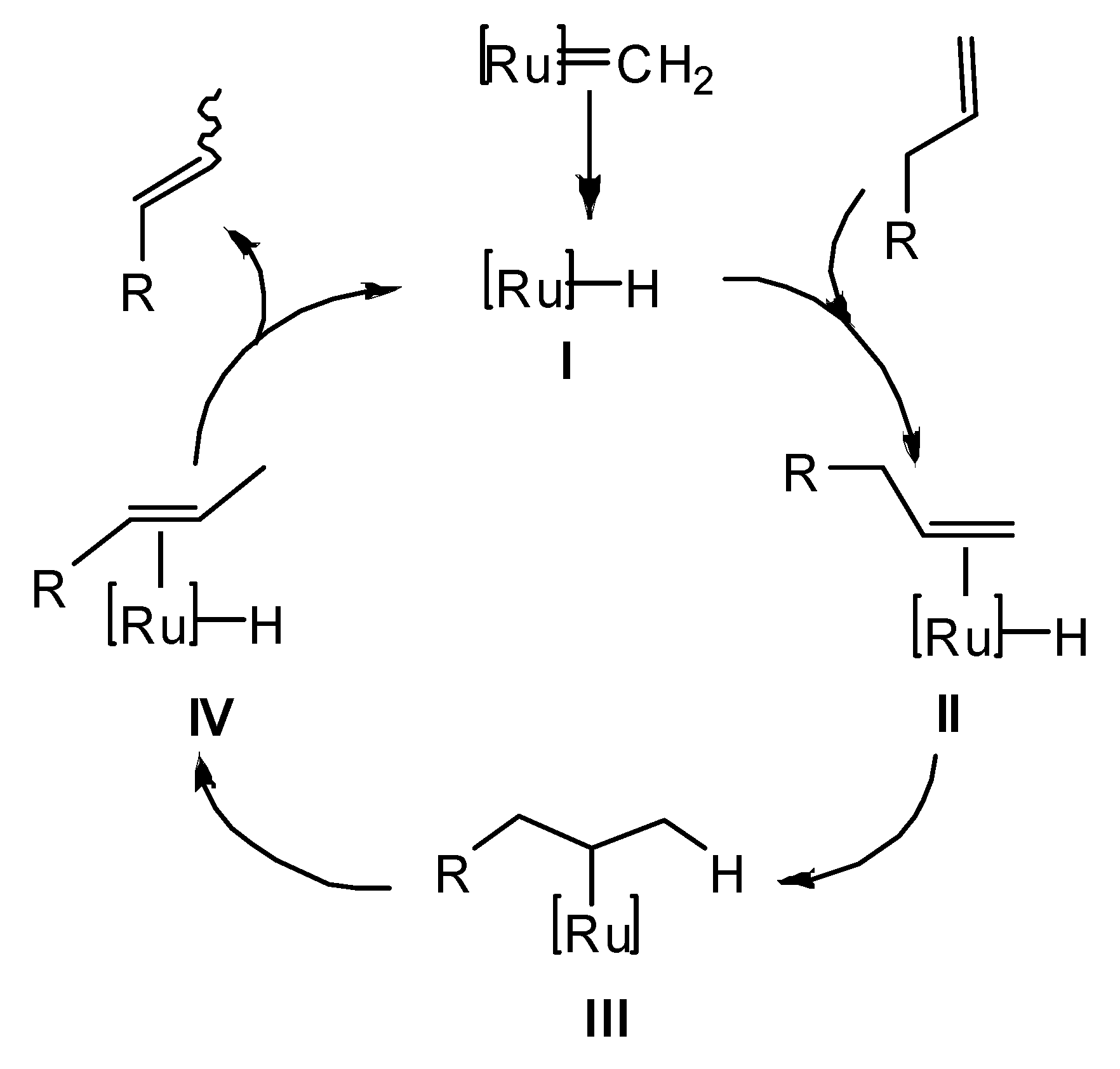
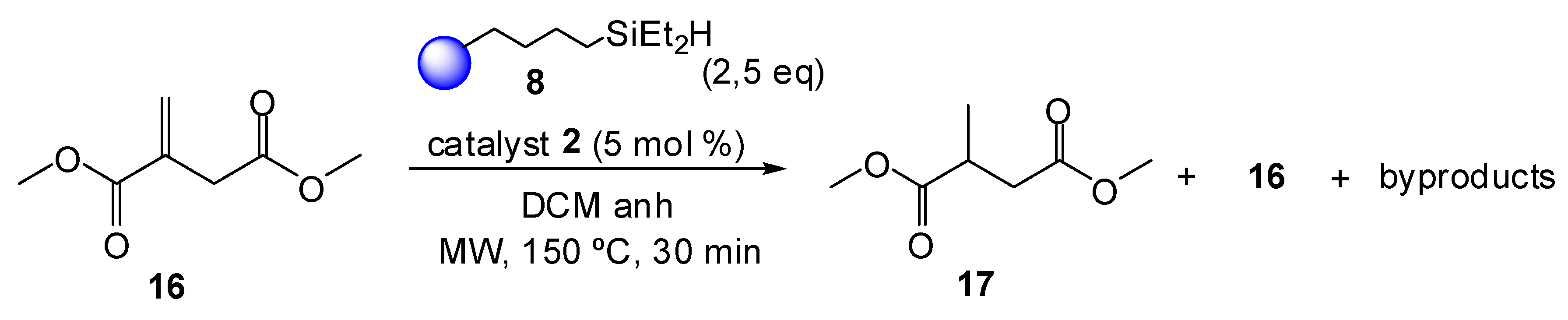
2. Results and Discussion
3. Materials and Methods
3.1. General Considerations
3.2. Instrumental and Physical Data
3.3. Synthetic Procedures
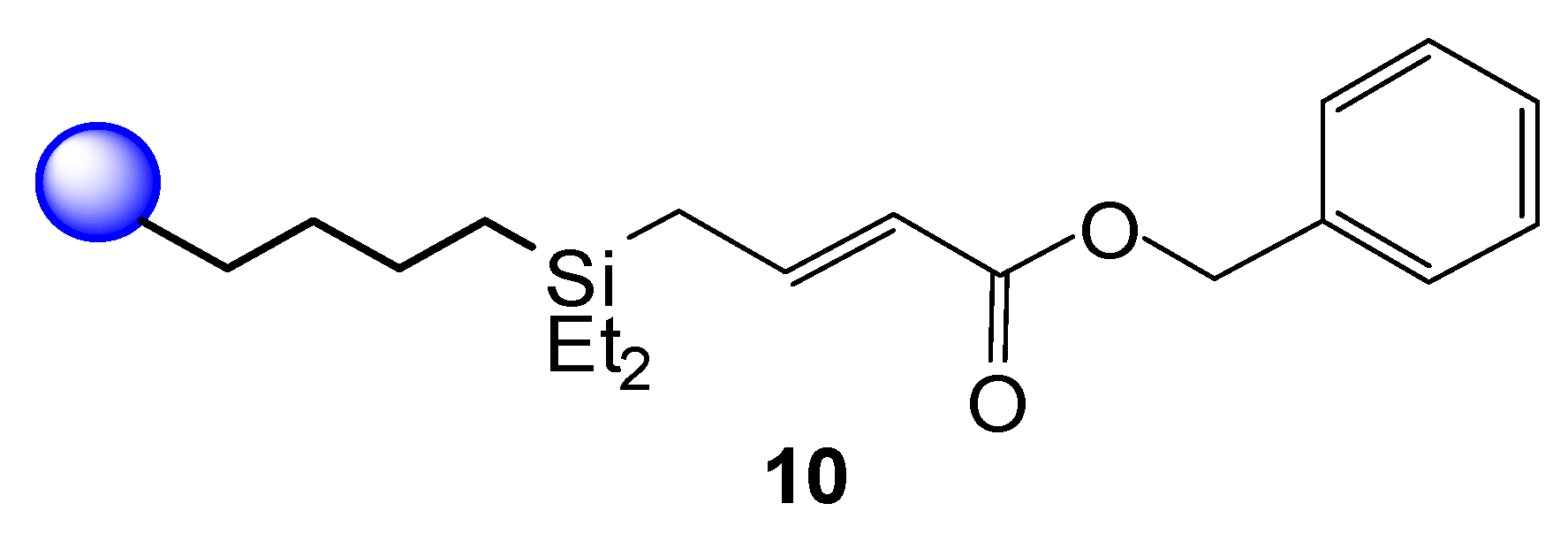
3.3.1. Synthesis of 4-(Diethylsilylbutyl) Polystyrene Resin (8)
3.3.2. General Procedure for Olefin Reduction in the Presence of Grubbs Catalysts and Triethylsilane
3.3.3. General Procedure for Olefin Reduction in the Presence of Grubbs Catalysts and Resin-Immobilized Silane (8)
3.4. Analytical Data





4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Kotha, S.; Misra, S.; Sreevani, G.; Babu, B.V. Non-metathetic behaviour of olefin metathesis catalysts. Curr. Org. Chem. 2013, 17, 2776–2795. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Luna, A. Grubbs’ ruthenium-carbenes beyond the metathesis reaction: Less conventional non-metathetic utility. Chem. Rev. 2009, 109, 3817–3858. [Google Scholar]
- Dragutan, V.; Dragutan, I. A resourceful new strategy in organic synthesis: Tandem and stepwise metathesis/non-metathesis catalytic processes. J. Organomet. Chem. 2006, 691, 5129–5147. [Google Scholar] [CrossRef]
- Schmidt, B. Catalysis at the interface of ruthenium carbene and ruthenium hydride chemistry: Organometallic aspects and applications to organic synthesis. Eur. J. Org. Chem. 2004, 2004, 1865–1880. [Google Scholar] [CrossRef]
- Schmidt, B. Connecting catalytic cycles by organometallic transformations in situ: Novel perspectives in the olefin metathesis field. Pure Appl. Chem. 2006, 78, 469–476. [Google Scholar] [CrossRef]
- Schmidt, B. Olefin metathesis and isomerization: From undesired side reactions to useful synthetic methodology. J. Mol. Catal. A 2006, 254, 53–57. [Google Scholar] [CrossRef]
- Schmidt, B.; Kunz, O. α,β-Unsaturated δ-valerolactones through RCM-isomerization sequence. Synlett 2012, 23, 851–854. [Google Scholar]
- Higman, C.S.; Lanterna, A.E.; Marin, M.L.; Scaiano, J.C.; Fogg, D.E. Catalyst decomposition during olefin metathesis yields: Isomerization-active ruthenium nanoparticles. ChemCatChem 2016, 8, 2446–2449. [Google Scholar] [CrossRef]
- For a previous publication about the reduction of olefins with Grubbs catalysts and Et3SiH, see e.g.: Menozzi, C.; Dalko, P.I.; Cossy, J. Reduction of olefins using ruthenium carbene catalysts and silanes. Synlett 2005, 2005, 2449–2452. [Google Scholar] [CrossRef]
- Moonen, K.; Dieltiens, N.; Stevens, C.V. Synthesis of 2-phosphonopyrroles via a one-pot RCM/oxidation sequence. J. Org. Chem. 2006, 71, 4006–4009. [Google Scholar] [CrossRef] [PubMed]
- Van Otterlo, W.A.L.; Coyanis, E.M.; Panaydes, J.-L.; de Koning, C.B.; Fernandes, M.A. Tandem catalysis: A ring-closing metathesis followed by dehydrogenative oxidation to afford substituted indenones. Synlett 2005, 2005, 501–505. [Google Scholar] [CrossRef]
- Tallarico, J.A.; Malnik, L.M.; Snapper, M.L. New reactivity from (PCy3)2Cl2 Ru=CHPh: A mild catalyst for Kharasch additions. J. Org. Chem. 1999, 64, 344–345. [Google Scholar] [CrossRef]
- Richel, A.; Delfosse, S.; Cremasco, C.; Delaude, L.; Demonceau, A.; Noels, A.F. Ruthenium catalysts bearing N-heterocyclic carbene ligands in atom transfer radical reactions. Tetrahedron Lett. 2003, 44, 6011–6015. [Google Scholar] [CrossRef]
- Schmidt, B.; Pohler, M. Ruthenium-catalyzed tandem ring closing metathesis (RCM)—Atom transfer radical cyclization (ATRC) sequences. J. Organomet. Chem. 2005, 690, 5552–5555. [Google Scholar] [CrossRef]
- Simal, F.; Delfosse, S.; Demonceau, A.; Noels, A.F.; Denk, K.; Kohl, F.J.; Weskamp, T.; Herrmann, W.A. Ruthenium alkylidenes: Modulation of a new class of catalysts for controlled radical polymerization of vinyl monomers. Chem. Eur. J. 2002, 8, 3047–3052. [Google Scholar] [CrossRef]
- Mallagaray, I.; Dominguez, G.; Gradillas, A.; Pérez-Castells, J. Tandem RCM—Isomerization—Cyclopropanation reactions. Org. Lett. 2008, 10, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Dragutan, I.; Dragutan, V.; Verpoort, F. Carbenoid transfer in competing reactions catalyzed by ruthenium complexes. Appl. Organomet. Chem. 2014, 28, 211–215. [Google Scholar] [CrossRef]
- Ding, F.; Sun, Y.; Verpoort, F.; Dragutan, V.; Dragutan, I. Catalytic activity and selectivity of a range of ruthenium complexes tested in the styrene/EDA reaction system. J. Mol. Catal. A 2014, 386, 86–94. [Google Scholar] [CrossRef]
- Méret, M.; Maj, A.M.; Demonceau, A.; Delaude, L. Ruthenium–arene catalysts bearing N-heterocyclic carbene ligands for olefin cyclopropanation and metathesis. Monatsh. Chem. 2015, 146, 1099–1105. [Google Scholar] [CrossRef]
- Mukai, C.; Itoh, R. Grubbs catalyst-mediated cycloisomerization of allenenes. Tetrahedron Lett. 2006, 47, 3971–3974. [Google Scholar] [CrossRef]
- Ashworth, I.W.; Hillier, I.H.; Nelson, D.J.; Percy, J.M.; Vincent, M.A. Searching for the hidden hydrides: The competition between alkene isomerization and metathesis with Grubbs catalysts. Eur. J. Org. Chem 2012, 2012, 5673–5677. [Google Scholar] [CrossRef]
- Higman, C.S.; Plais, L.; Fogg, D.E. Isomerization during olefin metathesis: An assessment of potential catalyst culprits. ChemCatChem 2013, 5, 3548–3551. [Google Scholar] [CrossRef]
- Hong, S.H.; Sanders, D.P.; Lee, C.W.; Grubbs, R.H. Prevention of undesirable isomerization during olefin metathesis. J. Am. Chem. Soc. 2005, 127, 17160–17161. [Google Scholar] [CrossRef] [PubMed]
- Czaban, J.; Schertzer, B.M.; Grela, K. Low catalyst loadings in self-metathesis of 1-dodecene. Adv. Synth. Catal. 2013, 355, 1997–2006. [Google Scholar] [CrossRef]
- Bourgeois, D.; Pancrazi, A.; Nolan, S.P.; Prunet, J. The Cl2(PCy3)(Imes)Ru(=CHPh) catalyst: Olefin metathesis versus olefin isomerization. J. Organomet. Chem. 2002, 643–644, 247–252. [Google Scholar] [CrossRef]
- Gimeno, N.; Formentín, P.; Steinke, J.H.G.; Vilar, R. Phenyl-phosphoric acid as a new additive to inhibit olefin isomerization in ruthenium-catalyzed metathesis reactions. Eur. J. Org. Chem. 2007, 2007, 918–924. [Google Scholar] [CrossRef]
- Kajetanowicz, A.; Milewski, M.; Rogińska, J.; Gajda, R.; Woźniak, K. Hoveyda-type quinone-containing complexes—Catalysts to prevent migration of the double bond under metathesis conditions. Eur. J. Org. Chem. 2017, 2017, 626–638. [Google Scholar] [CrossRef]
- Kadyrov, R. Olefin metathesis: Catalyst inhibition as a result of isomerization. ChemCatChem 2016, 8, 113–116. [Google Scholar] [CrossRef]
- Kaido, H.; Tupy, M.J.; Pederson, R.L.; Schrodi, Y. Metathesis Methods Involving Hydrogenation and Compositions Relating to Same. U.S. Patent 8,614,344, 24 December 2013. [Google Scholar]
- Nam, Y.H.; Snapper, M.L. Ruthenium-catalyzed tandem metathesis/non-metathesis processes. In Handbook of Metathesis, Applications in Organic Synthesis, 2nd ed.; Grubbs, R.H., O’Leary, D.J., Eds.; Wiley-VCH: Weinheim, Germany, 2015; Volume 2, pp. 311–380. [Google Scholar]
- Poeylaut-Palena, A.A.; Testero, S.A.; Mata, E.G. The non-metathetic role of Grubbs’ carbene complexes: From hydrogen-free reduction of α,β-unsaturated alkenes to solid-supported sequential cross-metathesis/reduction. Chem. Commun. 2011, 47, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Mendez, L.; Mata, E.G. Solid-supported cross-metathesis and a formal alkane metathesis for the generation of biologically relevant molecules. ACS Comb. Sci. 2015, 17, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Solid-Phase Organic Synthesis: Concepts, Strategies, and Applications; Toy, P.H.; Lam, Y. (Eds.) John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Testero, S.A.; Mata, E.G. Prospect of metal-catalyzed C–C forming cross-coupling reactions in modern solid-phase organic synthesis. J. Comb. Chem. 2008, 10, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Riveira, M.J.; Mata, E.G. Cross-metathesis on immobilized substrates—Application to the generation of synthetically and biologically relevant structures. Eur. J. Org. Chem. 2017, 1675–1693. [Google Scholar] [CrossRef]
- Jida, M.; Betti, C.; Schiller, P.W.; Tourwé, D.; Ballet, S. One-pot isomerization—Cross metathesis—Reduction (ICMR): Synthesis of lipophilic tetrapeptides. ACS Comb. Sci. 2014, 16, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Cherkupally, P.; Ramesh, S.; de la Torre, B.G.; Govender, T.; Kruger, H.G.; Albericio, F. Immobilized coupling reagents: Synthesis of amides/peptides. ACS Comb. Sci. 2014, 16, 579–601. [Google Scholar] [CrossRef] [PubMed]
- Pastre, J.C.; Browne, D.L.; Ley, S.V. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 2013, 42, 8849–8869. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Takeshita, K.; Kawamoto, K.; Murai, S.; Sonoda, N. Single-operation synthesis of vinylsilanes from alkenes and hydrosilanes with the aid of Ru3(CO)12. J. Org. Chem. 1986, 51, 3890–3895. [Google Scholar] [CrossRef]
- Bokka, A.; Jeon, J. Regio- and stereoselective dehydrogenative silylation and hydrosilylation of vinylarenes catalyzed by ruthenium alkylidenes. Org. Lett. 2016, 18, 5324–5327. [Google Scholar] [CrossRef] [PubMed]
- Hexaethyldisiloxane was obtained after performing an experiment using the conditions of Table 1, Entry 4, but in absence of the olefin.
- Sridhar, M.; Ramanaiah, B.C.; Narsaiah, C.; Swamy, M.K.; Mahesh, B.; Reddy, M.K.K. An efficient and simple method for the preparation of symmetrical disiloxanes from hydrosilanes by Lewis acid-catalyzed air oxidation. Tetrahedron Lett. 2009, 50, 7166–7168. [Google Scholar] [CrossRef]
- Delaude, L.; Demonceau, A.; Noels, A.F. Ruthenium-promoted radical processes toward fine chemistry. In Topics in Organometallic Chemistry; Bruneau, C., Dixneuf, P.H., Eds.; Springer: Berlin, Germany, 2004; Volume 11, pp. 155–171. [Google Scholar]
- Hu, Y.; Porco, J.A.; Labadie, J.W.; Gooding, O.W.; Trost, B.M. Novel polymer-supported trialkylsilanes and their use in solid-phase organic synthesis. J. Org. Chem. 1998, 63, 4518–4521. [Google Scholar] [CrossRef]
- Hanessian, S.; Giroux, S.; Larsson, A. Efficient allyl to propenyl isomerization in functionally diverse compounds with a thermally modified Grubbs second-generation catalyst. Org. Lett. 2006, 8, 5481–5484. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, T.J.; O’Riordan, T.J.C.; Rosa, C.P. Ruthenium-catalyzed isomerization of terminal olefins: Applications to synthesis. Angew. Chem. Int. Ed. 2009, 48, 1014–1017. [Google Scholar] [CrossRef] [PubMed]
- Giralt, E.; Rizo, J.; Pedroso, E. Application of gel-phase 13C-NMR to monitor solid phase peptide synthesis. Tetrahedron 1984, 40, 4141–4152. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Et3SiH (equiv.) | Products (3/4/5) b | |
|---|---|---|---|
| Molar Ratio | Percentage (%) | ||
| 1 | 0.5 | 10.5/1.0/1.4 | 81/8/11 |
| 2 | 1.0 | 4.4/1.0/0.4 | 76/17/7 |
| 3 | 2.5 | 0.9/1.0/0.3 | 41/46/13 |
| 4 | 5.0 | 0/1.0/0.1 | 0/91/9 |

| Entry | Olefin | Catalyst (5 mol %) | Silane (Equivalents) | Reaction Conditions a | Ratio 4:3 b | |
|---|---|---|---|---|---|---|
| Temp. (°C) | Time (h) | |||||
| 1 | 3 | 2 | 8 c, 3.0 | Reflux | 24 | 1:19 |
| 2 | 3 | 2 | 8 c, 2.5 | 55 (sealed system) | 16 | 1:5.6 |
| 3 | 3 | 2 | 8 c, 2.5 | 150 (MW) d | 0.5 | 1:0.3 e |
| 4 | 3 | 2 | - f | 150 (MW) d | 0.5 | 1:0.3 e |
| 5 | 3 | 1 | 8 c, 2.5 | 150 (MW) d | 0.75 | 1:3.2 e |
| 6 | 3 | 2 | 8 c, 0.75 | 150 (MW) d | 0.5 | ND g |
| 7 | Resin from Entry 6 | 2 | Et3SiH (2.5) | 150 (MW) d | 0.5 | 1:0.6 h |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Amezaga, M.; Delpiccolo, C.M.L.; Méndez, L.; Dragutan, I.; Dragutan, V.; Mata, E.G. Unprecedented Multifunctionality of Grubbs and Hoveyda–Grubbs Catalysts: Competitive Isomerization, Hydrogenation, Silylation and Metathesis Occurring in Solution and on Solid Phase. Catalysts 2017, 7, 111. https://doi.org/10.3390/catal7040111
Martinez-Amezaga M, Delpiccolo CML, Méndez L, Dragutan I, Dragutan V, Mata EG. Unprecedented Multifunctionality of Grubbs and Hoveyda–Grubbs Catalysts: Competitive Isomerization, Hydrogenation, Silylation and Metathesis Occurring in Solution and on Solid Phase. Catalysts. 2017; 7(4):111. https://doi.org/10.3390/catal7040111
Chicago/Turabian StyleMartinez-Amezaga, Maitena, Carina M. L. Delpiccolo, Luciana Méndez, Ileana Dragutan, Valerian Dragutan, and Ernesto G. Mata. 2017. "Unprecedented Multifunctionality of Grubbs and Hoveyda–Grubbs Catalysts: Competitive Isomerization, Hydrogenation, Silylation and Metathesis Occurring in Solution and on Solid Phase" Catalysts 7, no. 4: 111. https://doi.org/10.3390/catal7040111