
Perovskite Electrocatalysts for the Oxygen Reduction Reaction in Alkaline Media


Institute of Materials Physics, University of Goettingen, Friedrich-Hund Platz 1, 37077 Göttingen, Germany
Catalysts 2017, 7(5), 154; https://doi.org/10.3390/catal7050154
Submission received: 21 March 2017
/
Revised: 28 April 2017
/
Accepted: 10 May 2017
/
Published: 13 May 2017
(This article belongs to the Special Issue (Photo)Electrochemistry of Perovskites)
Abstract
:Oxygen reduction is considered a key reaction for electrochemical energy conversion but slow kinetics hamper application in fuel cells and metal-air batteries. In this review, the prospect of perovskite oxides for the oxygen reduction reaction (ORR) in alkaline media is reviewed with respect to fundamental insight into activity and possible mechanisms. For gaining these insights, special emphasis is placed on highly crystalline perovskite films that have only recently become available for electrochemical interrogation. The prospects for applications are evaluated based on recent progress in the synthesis of perovskite nanoparticles. The review concludes with the current understanding of oxygen reduction on perovskite oxides and a perspective on opportunities for future fundamental and applied research.

1. Introduction
The oxygen reduction reaction (ORR) [1] is a fundamental and ubiquitous reaction for energy conversion. It plays critical roles in the respiration of plants [2], in efficient operation of fuel cells [3,4,5,6,7,8,9,10], in discharge of primary and secondary metal-air batteries [11,12,13,14,15], as well as in corrosion processes of metals [16]. For application in fuel cells and metal-air batteries, the two main challenges are the slow kinetics of the ORR, hindering efficient utilization of chemically stored energy, and optimizing the product yield towards the reduction of oxygen to water. Fundamentally, the efficiency and product yield are both governed by the binding of oxygen and its reaction intermediates on the catalytic surface. The binding energy of oxygen depends on the structure, chemistry, and magnetism of the catalyst, which makes it complex to predict and ultimately control. In contrast to metal catalysts, the semiconducting properties of an oxide must also be considered as a prerequisite for a suitable electrode material for oxygen reduction (and evolution) [14,17,18,19,20]. Systematic empirical studies of the ORR on oxides have been performed since the 1970s (Table 1), where highly active oxides were characterized by amphoteric metallic conduction [17,21], sigma bands [17] with an occupation near unity of eg orbitals [22], high covalence [22], and paramagnetism [23,24] fostering a specific (‘side-on’) configuration of the likewise paramagnetic oxygen molecule on the surface. Oxides with perovskite or closely related structures played an important role in the discovery of these insights because their physicochemical properties can be systematically tuned [14,22,25,26,27,28].
This review is focused on non-precious perovskite oxides as electrocatalysts for the ORR in alkaline media. The focus on alkaline media was chosen as non-precious oxides, including perovskites, are not stable in acidic media [11,29,30,31]. A fundamental understanding of catalyst activity is central to the manuscript, which covers electrocatalysts spanning from thin films to nanoparticles. First, the perovskite structure is concisely introduced. Then, the effects of chemical substitutions on catalytic activity are discussed, followed by an introduction to common activity metrics. The microscopic origins of activity segue into efforts toward developments of an atomic picture of the catalytic mechanisms. Building on this foundation, the effects of structural, chemical, as well as magnetic properties on oxygen reduction are surveyed and their impact on activity is quantified. The review is concluded by summarizing the state-of-the-art understanding of the ORR on perovskites and a perspective for future research.
2. The Perovskite Structure
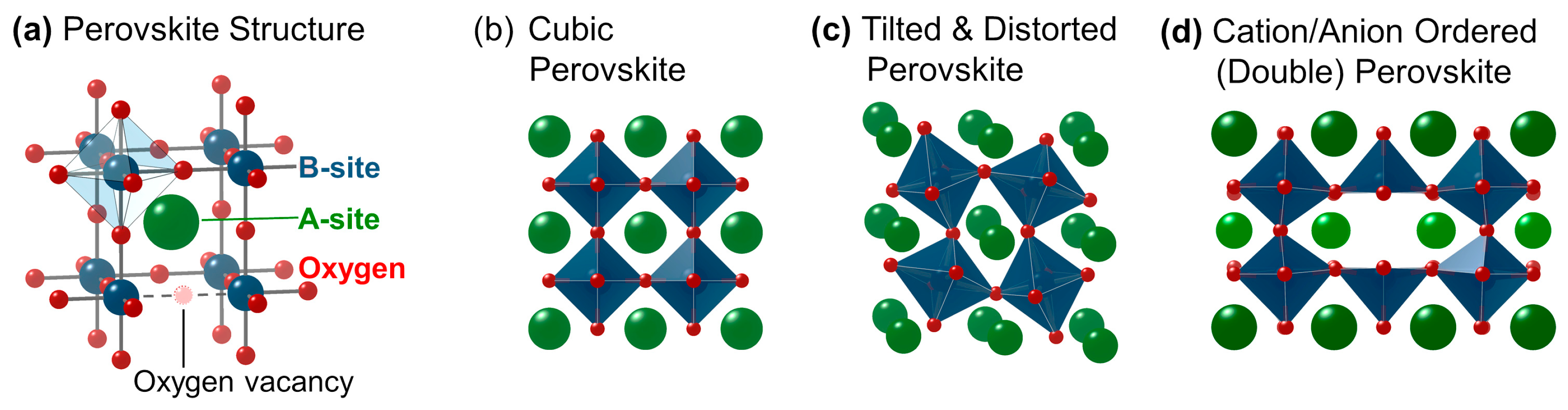
The mineral perovskite (CaTiO3) is the namesake for the crystal structure of oxides with general chemical formula of ABO3±δ (Figure 1) [32,33], where A is commonly a group II cation, a lanthanide cation, or a mix of both; B is one or more transition metals (here only d-block metals); and the oxygen non-stoichiometry δ is small for proper perovskites, i.e., there are few oxygen vacancies. A and B in the perovskite formula are commonly referred to as A-site and B-site. In a perfectly cubic perovskite without oxygen vacancies, the transition metals at the B-site are octahedrally coordinated to oxygen (Figure 1a), while the A-site is 12-fold coordinated to oxygen. Most combinations of A and B cations lead to finite δ and/or structural distortion, both of which lower the effective coordination of the cations. The degree of deviation from the cubic perovskite can be quantified using the Goldschmidt tolerance factor [34]
where rA, rB, and rO = 1.4 Å are the ionic radii of the A-site cation, B-site cation and oxygen, respectively. Most commonly, specific values are taken from Shannon’s Table [35] or linearly extrapolated from these tabulated values to the appropriate coordination. An alternative approach uses the average interatomic distances between the A-site cation and oxygen (|A—O|) and between the B-site cation and oxygen (|B—O|). These distances can be obtained from bond valence theory [36,37,38] (e.g., calculated using a web form [39] or SPuDS [40]) or from Rietveld refinement of experimental XRD data. In the latter case, t’ is also referred to as the observed tolerance factor [33]. Both computational approaches give comparable results as discussed, e.g., by Lufaso and Woodward [40].
The tolerance factor can be used to estimate the symmetry of the perovskite to some degree. A cubic perovskite (Figure 1b; space group, e.g., Pm-3m) has a tolerance factor near unity (t ≈ 1) and is thus likely when the A-O distance is about √2 times larger than the B—O distance. In other words, the B-site cations sit on the corners of a cube and the size of the A-site cation matches the diagonal. For smaller tolerance factors (t < 1), the size of the A-site cation is too small for the diagonal and orthorhombic (Figure 1c; space group, e.g., Pmna) or rhombohedral (space group, e.g., R-3c) distortions occur. This leads to tilt and rotation of the B-site octahedra, which changes the B—O bond angles and thereby strongly affects the hybridization of electronic orbitals. Additionally, the octahedron at the B-site can be distorted due to the Jahn–Teller effect [33,41,42] by which orbital degeneracy is removed upon elongation of one of the octahedral axes.
The stability range of the perovskite structure is constraint by two boundary conditions: (i) when t is 1/√2, A-site and B-site cations have equal size; and (ii) when t is larger than 1.0, the A-site cation is too large to fit on the diagonal of a (pseudo)cubic unit cell. Whether a perovskite structure is actually observed for a specific combination of A-site and B-site cations depends further on the ratio of the ionic radii of the B-site cation to that of oxygen, where ratios above 0.4 are required [43]. The multitude of possible chemical combinations within the perovskite structure results in a wide range of interesting electronic or magnetic properties and correlations [32].
Several crystal structures are closely related to the perovskite structure [44]. So-called hexagonal perovskites (space group, e.g., P63cm) can be observed on the cusp of the range of tolerance factors assigned to perovskites [32,45]. For larger deviations, various non-perovskite structures are likely found. When the requirements for the perovskite structure are met and two A-site cations with strongly differing sizes are used, additional complexity arises from ordering of A-sites and oxygen vacancies as in the double perovskite (AA’B2O5+δ) shown in Figure 1d (space group, e.g., P4/mmm). Ordering of oxygen vacancies without A-site ordering leads to structures related to brownmillerite (A2B2O5). Lastly, layering of perovskite slabs produces the Ruddlesden–Popper phases (An+1BnO3n+1). A detailed discussion of these perovskite-like structures is beyond the scope of this review and the reader is referred to other reviews and books [32,45]. They are introduced here as one can transition from a perovskite phase into these phases during systematic studies.
3. Effect of Chemical Substitution on Activity
The perovskite structure ABO3−δ has been chosen for many systematic studies of catalytic activity [25,46,47,48,49,50], including oxygen reduction and evolution (i.e., oxygen electrocatalysis) [14,22,25,26,27,28,51,52,53,54,55]. The key idea of this section is a demonstration of both intentional and unintentional effects of chemical substitution or changes in stoichiometry on the activity for oxygen reduction (Figure 2). A detailed analysis of correlations between intrinsic properties and catalytic activity requires discussing activity metrics (Section 4) and the mechanisms of ORR (Section 5). The focus on qualitative trends in this section has allowed including a wider range of studies to derive trends. Moreover, this focus facilitates a facile connection between activity for ORR and the components of the perovskite structure ABO3−δ as a foundation for the later discussion of the more universal structural, chemical, and magnetic origins of catalytic activity (Section 6).
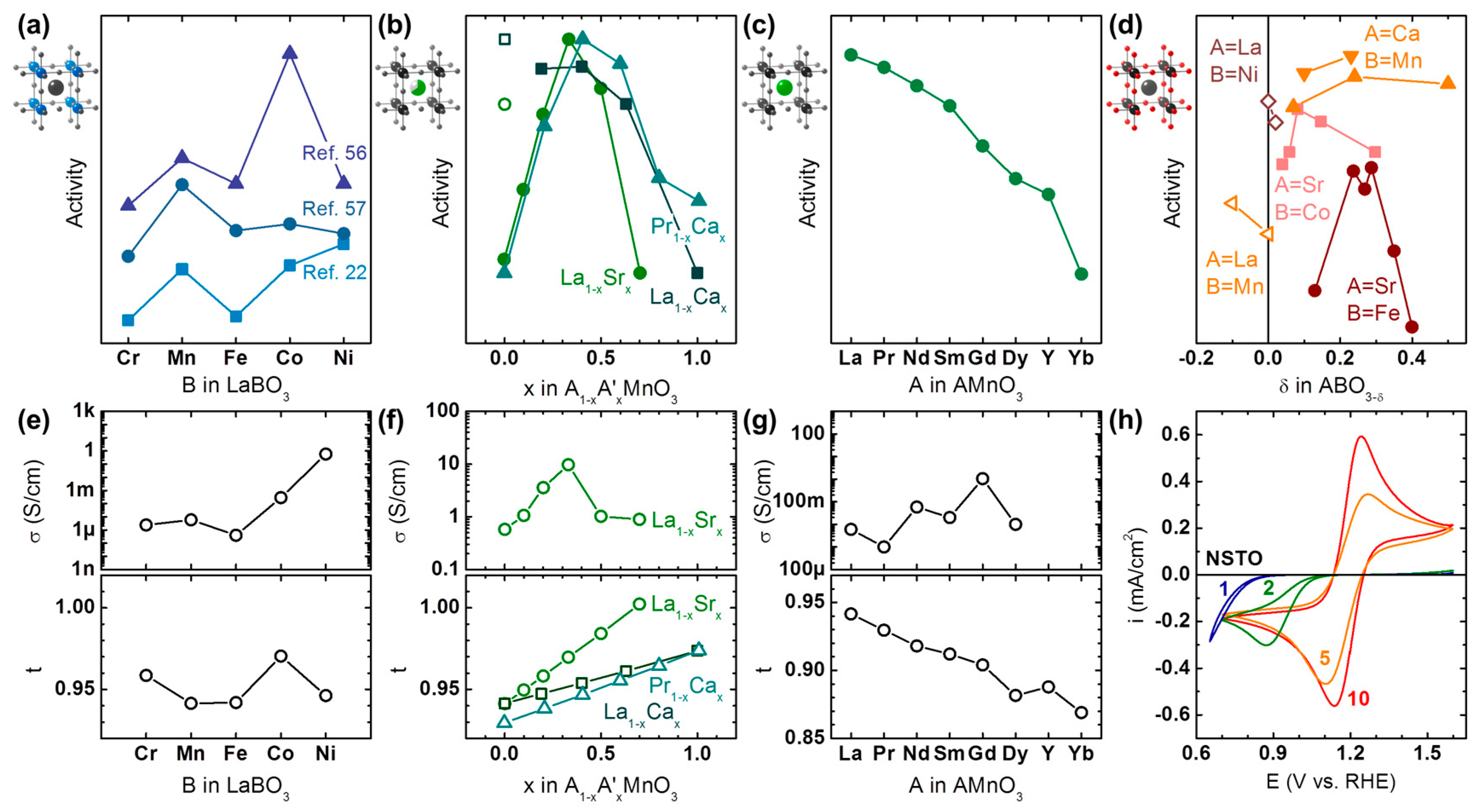
Catalytic trends with substitution of the B-site in LaBO3 were studied by Sunarso et al. [56], Celorrio et al. [57], and Suntivich et al. [22] in 0.1 M KOH for nominally trivalent 3d transition metals ranging from Cr to Ni in Figure 2a. In all three studies, Mn and Co at the B-site was more active than both Cr and Fe. On the other hand, relative differences between these elements and the most active B-site depended strongly on the study; possible reasons are: the used carbon additive, differences in surface roughness and area of the oxide (if not included in normalization), oxygen stoichiometry and phase purity. Ni-based perovskites are especially prone to phase segregation during synthesis [54,58] and ORR conditions [59] and it has been shown that X-ray absorption spectroscopy (XAS) is superior to XRD for verification of phase purity [58]. Even minor phase segregation can have great influence on the ORR activity [54], which likely explains the widely differing activity of Ni at the B-site relative to the baseline given by Cr and Fe. Interestingly, the carbon-free LaBO3 thin films studied by Stoerzinger et al. [60] show a local maximum of the ORR activity for Co as also observed by Sunarso et al. [56] and Celorrio et al. [57], which suggests that the used carbon did not affect the activity trends.
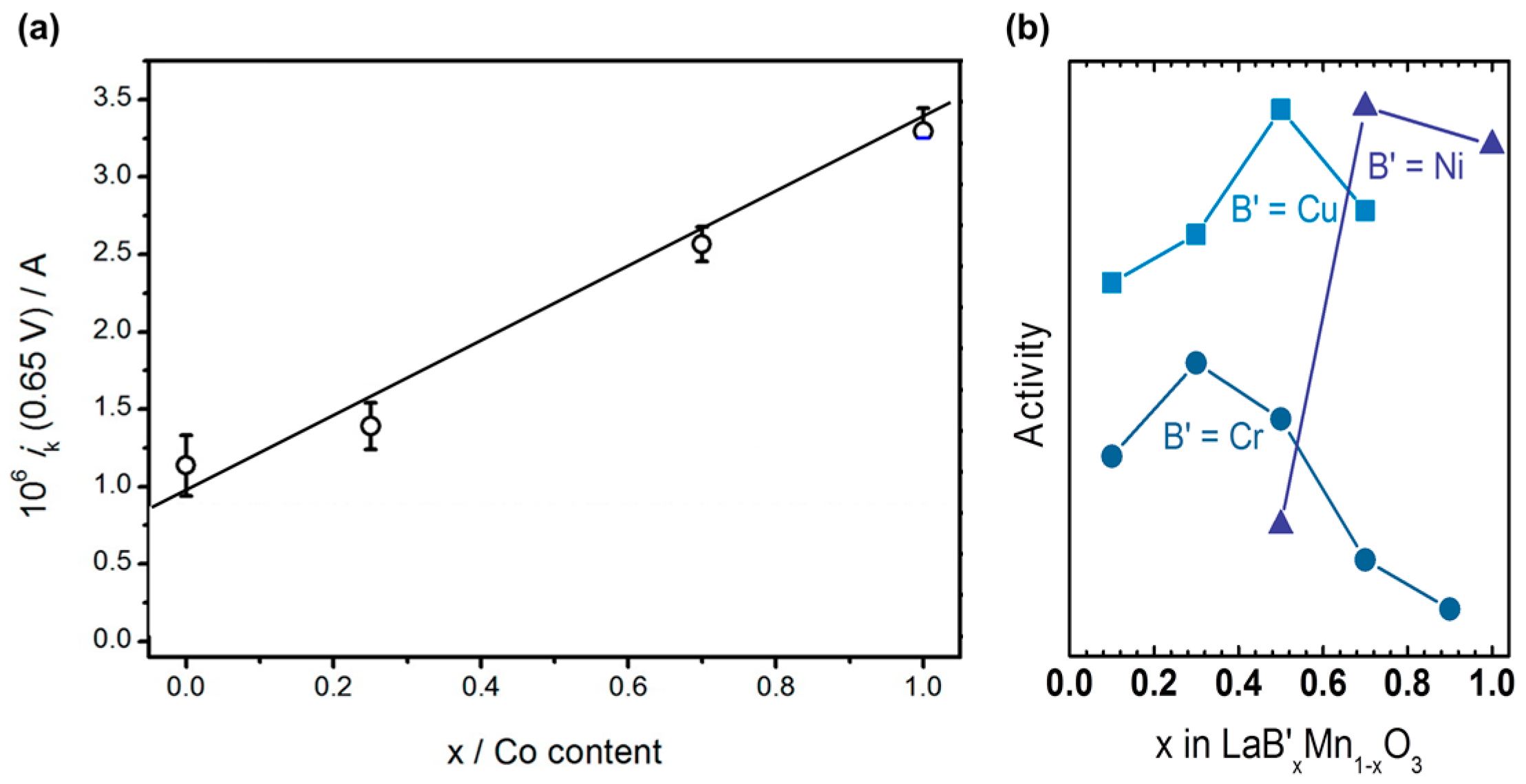
The composition of the B-site also influences activity as shown, e.g., by Suntivich et al. [22], Larsson & Johansson [61], and Celorrio et al. [62]. For many perovskites, it is also known that charge ordering effects lead to charge disproportionation [33]. Thus, understanding the observed changes requires complementary spectroscopic experiments. For LaNi0.5Mn0.5O3, Suntivich et al. [22] measured the ex situ valence of both Mn and Ni by XAS in the perovskite and concluded that Mn would be the more active element. Based on this argument, the activity should scale with the concentration of the active element as observed for Ba0.5Sr0.5Fe1−xCoxO3−δ (Figure A1a) [62]. However, linear trends were not observed for LaB’xMn1−xO3 (B’ = Cr, Ni, Cu) in the study of Larsson and Johansson (Figure A1b) [61], which strongly suggests that further effects significantly contribute to the observed activity as discussed in Section 6. Due to these complications, the composition at the B-site was not included in Figure 2, but can be found in Figure A1.
Trends with composition of the A-site were studied for AMnO3 by Hyodo et al. [65] (A = Pr1−xCax), Celorrio et al. [64] (A = La1−xCax) and Stoerzinger et al. [63] (A = La1−xSrx) as shown in Figure 2b. In the range of 10% to 100% of the divalent A-site, the maximum was found around 40% of the divalent A-site (formal valence Mn3.4+) in all studies. Similar trends with an activity maximum near 0.4 were also reported by Hammouche et al. [71] for La1−xCaxCoO3. Figure 2b further suggests that the maximum activity is found for lower concentrations of the larger Sr cation as compared to the smaller Ca cation. This is supported by a study of Hyodo et al. [66] where the size of the lanthanide at the A-site in AMnO3 was systematically varied (Figure 2c). Higher activity at nominally fixed Mn valence was observed for larger ionic radius of the lanthanide, which suggests that the tolerance factor affects the activity such that the activity is highest for a tolerance factor near unity.
The effect of oxygen stoichiometry on ORR activity was studied by Takeda et al. [67] for SrFeO3−δ and SrCoO3−δ as well as by Du et al. [68] for CaMnO3−δ (Figure 2d). The data of Suntivich et al. [22] (B = Mn) and Zhou & Sunarso [69] (B = Ni) was also included as a guide for LaBO3±δ. For divalent A-sites (solid symbols in Figure 2d), Mn- and Fe-based perovskites show maximum activity near δ = 0.25, while the maximum is closer to δ = 0.1 for Co-based perovskites. The data for trivalent A-sites (open symbols in Figure 2d) was very limited and should not be over-interpreted but it is clear that the oxygen stoichiometry also impacts the activity of those perovskites. Further studies are highly desirable for a more comprehensive understanding of the effect of oxygen stoichiometry on both activity and mechanism.
In a perfect experiment with an ideal material, the effect of changing a single property on catalytic activity could be studied. While the perovskite structure is fairly robust against chemical substitutions (Section 2), it is neither perfect nor ideal. Therefore, it must be expected that properties other than the intended ones, e.g., the number of d electrons at the B-site, are altered during the chemical substitutions in Figure 2a–c. The often unintended (estimated) changes in conductivity and tolerance factor are illustrated for example in Figure 2e–g.
Substitution of the B-site in LaBO3 drastically changes the expected bulk conductivity between ~1 µS/cm for Fe [72] to ~1 S/cm for Ni [73] (Figure 2e), while it has a clear maximum for La1−xSrxMnO3 [63] (Figure 2f). No discernable trend could be identified for substitution of the A-site from La to Dy [74,75,76,77,78,79,80] (Figure 2g) under the caveat that some of the data was only available for temperatures >300 K (Table A2). For the ORR data of Suntivich et al. [22], the activity trend matches that of the typical conductivity of the used LaBO3 perovskites. Moreover, the previously found local activity maximum of Mn relative to Cr and Fe in all data sets is also mirrored in the conductivity data [72,73,80,81]. The detrimental effect of insufficient conductivity is most pronounced in the thin film study of Stoerzinger et al. [60], where no activity was detected for LaCrO3 and LaFeO3 in the investigated voltage range. The series of (carbon-free) La1−xSrxMnO3 thin films of the same group [63] further show a clear correlation between measured conductivity and ORR activity (Figure 2f). Lastly, the oxygen stoichiometry also has a strong influence on conductivity, where the conductivity changes from 80 µS/cm for understoichiometric LaMnO3−δ [80] to 1.6 S/cm for overstoichiometric LaMnO3+δ [80]. In agreement with the above discussion, Suntivich et al. [22] report higher ORR activity for LaMnO3+δ than for LaMnO3. All these observations strongly support the early hypotheses of Meadowcroft [21] and Matsumoto et al. [17] (Table 1) that ORR catalysts must be conductive. The conductivity of LaBO3 and thus its activity can be improved by doping of the A-site with divalent cations such as Sr [54]. Therefore, it would be desirable to conduct systematic studies of the effect of B-site doping on activity using a suitably doped La1−xSrxBO3 series (e.g., x ≈ 0.4 for B = Mn; Figure 2f) rather than using the LaBO3 series and to report the conductivity of the material.
High bulk conductivity is an insufficient criterion for high ORR activity. Meadowcroft [21] proposed that amphoterism is additionally required (or in his words “having both p- and n-type conductivity”). An introduction to the semiconductor physics of oxides is beyond the scope of this review and the reader is referred to other reviews [20,82] and books [83,84,85] on the topic. Nonetheless, a very short introduction of relevant concepts is provided here. In contrast to metals, oxides can support locally charged regions (‘space charge layers’). As a consequence, transfer of either positive or negative charges across an interface with the oxide might be hindered by energy barriers if the Fermi levels of the isolated materials differ strongly. These effects are most strongly pronounced for highly crystalline thin films but they will also affect sufficiently crystalline particles. For thin films, they can be visualized and quantified using a fast-redox couple with suitable redox potential near the thermodynamic potential of oxygen reduction at 1.23 V vs. RHE (see Section 4). Figure 2h shows cyclic voltammetry of the ferri-/ferrocyanide redox on LaMnO3 thin films on Nb-doped SrTiO3 (LMO|NSTO) [70] as a metric for the perovskite oxide to transfer electrons across the electrode-electrolyte interface near 1.23 V vs. RHE. While the expected symmetric shape was observed for bulk-like 10 nm films, thinner films resulted in reduction of the peak currents and an asymmetry where currents are blocked above 1.23 V vs. RHE. Consequently, the ORR activity of the thinner films was drastically reduced compared to 10 nm LMO|NSTO. A similar correlation was also reported by Stoerzinger et al. [63] for thin films of the series La1−xSrxMnO3. It is also conceivable that a buried interface (e.g., between the support NSTO and another perovskite as the active catalyst) limits charge transfer. Therefore, all possible interfaces must be optimized when designing oxide catalysts.
The tolerance factor, t, also varies with B-site substitution, the composition at the A-site and A-site substitution (Figure 2e–g), which affects the activity. The influence of the tolerance factor on activity is known for high temperature oxygen reduction in solid oxide fuel cells (SOFC) [86,87]. Yet, its effect on low temperature oxygen reduction in alkaline electrolytes has not been comprehensively discussed. The relevance of the tolerance factor for the ORR in alkaline media is most clearly visible for substitution of the A-site in Figure 2g, where the highest activity is observed for tolerance factors closest to unity. The correlation between activity and tolerance factor might also explain the local activity maximum for LaCoO3 in the studies of Sunsarso et al. [56], Stoerzinger et al. [60], and Celorrio et al. [57] in the LaBO3 series (Figure 2e). Substituting divalent for trivalent cations leads to a linear increase in the predicted tolerance factor (Figure 2f), which could explain the asymmetry of the activity trend in Figure 2b. This analysis strongly suggests that the tolerance factor correlates with activity and it would be desirable to report it, preferably based on experimentally determined bond lengths.
In this section, the effect of the chemical composition on activity for oxygen reduction was discussed qualitatively. In order to achieve quantitative insight, activity metrics are introduced in the next section, which is followed by a discussion of possible oxygen reduction mechanisms on perovskite oxides in Section 5. This allows us to obtain microscopic insight into general structure–activity relationships, which complements the qualitative discussion above by a quantitative discussion in Section 6.
4. Activity Metrics
The overall reaction of a hydrogen fuel cell is
which is associated with a change in Gibbs free energy of −237 kJ/mol for the production of liquid water at 298 K and 101.3 kPa. [88] The free energy corresponds to the thermodynamic potential of the ORR of E0ORR = 1.229 V. The most meaningful reference potential for the ORR is the reversible hydrogen electrode (RHE), which is essentially the reversible potential of the hydrogen oxidation reaction at the counter electrode. The overpotential of the ORR is defined here as
where Emeasured is most commonly evaluated at a fixed current density. It is also common to report current densities at fixed overpotential to quantify the activity of the reaction on a given electrocatalyst.
H2 + ½O2 → H2Oaq
ηORR = E0ORR − Emeasured
Electrocatalysts can be included in a wide range of electrode assemblies, either in two-electrode setups as used in devices such as fuel cells and metal-air batteries or three-electrode setups as used in catalyst screening and fundamental research. In two-electrode setups, the electrocatalyst is loaded onto gas diffusion electrodes (GDE) [8,89,90], which typically contain a binder and carbon in addition to a catalyst with high surface area. While the electrodes can be used directly in devices, the quantification of the catalyst activity is hindered by an ambiguous electrochemically active surface area in these 10–100 micrometer thick films. More defined composite catalyst layers can be deposited on suitable support materials, commonly rotating disks that are measured in three-electrode setups. These electrodes are typically prepared by applying an ink or slurry of the catalyst particles that also contains a binder and carbon. The composite layer should be thinner than 100 nm to approximate a flat surface [91]. Single crystalline surfaces are best suited to evaluate the activity of a catalyst material and gain fundamental mechanistic insight but furthest removed from application in a device. These catalysts with low surface area do not include a binder or carbon.
The carbon added to composite electrodes serves several purposes. It disperses the catalyst particles, electrically connects the catalyst particles, enhances the conductivity locally and can act as a co-catalyst in some cases [92,93,94,95]. Most notably, the activity enhancement after Vulcan carbon addition to a conductive La0.8Sr0.2CoO3 strongly suggests a co-catalytic role of the used carbon [96], which is also likely on conductive LaCoO3 and La0.8Sr0.2MnO3 as supported by kinetic modeling [95,97]. In addition to activity, the carbon in composite electrodes may also affect the stability. Carbon may corrode or erode at oxidative potentials [98], which is a known problem for the stability of catalyst layers made from inks or slurries. The design of perovskite-carbon hybrid systems [98,99,100,101,102] is a possible avenue to make composite electrodes more robust so that catalyst stability is decoupled from catalyst activity. Synergetic effects between carbon supports and perovskite catalysts is a very active field of research and the reader is referred to a recent review for additional discussion [98].
Various metrics exist to quantify the catalytic effect of a material, i.e., its activity, for comparison with other electrocatalysts. The simplest metric is the current divided by the geometric area of the electrode, which is important on the device level. It will not be discussed here because it does not relate the current to a specific property of the electrocatalyst. Another straightforward metric is obtained by dividing the current by the active mass of the catalyst. It includes structural, chemical, and magnetic origins of catalytic activity as well as the surface area of the catalyst. For fundamental insight into the origin of catalytic activity, we could ideally measure the current associated with a specific product (e.g., hydroxide) and divide it by the number of active sites on the surface of the catalytic material. Unfortunately, neither the numerator nor denominator of this seemingly simple calculation is straightforward to obtain.
Oxygen concentration in alkaline media is of the order of 1 mM at room temperature [103]. It is therefore firstly necessary to separate the current limited by mass transport (im) from the current limited by the kinetics of oxygen reduction (ik). The reciprocal measured current is given by
which can be solved for ik if im was determined, e.g., as the limiting current in an experiment with forced convection. It should be noted here that both im and the apparent ik of composite electrodes depend on the dispersion and loading of the catalytic particles [91,93,94,104,105,106,107,108,109,110,111], which calls for optimization. Kinetic currents should be reported in the range of 10 to 80% of the limiting current and ideally below 50% [91]. It is highly recommended to obtain a measurement of background currents in an oxygen-free electrolyte (e.g., by purging with Ar) and to subsequently subtract these currents from the measurement in an oxygen-containing electrolyte to remove double-layer currents and possibly transition metal redox currents [14].
i−1 = im−1 + ik−1
Conceivable reduction products of oxygen (O2) in alkaline media are: superoxide (O2−, 1 e−), hydroperoxide (HO2−, 2 e−) and hydroxide (HO−, 4 e−) [112]. Hydroxide is the preferred product because the most electrons are transferred per reduced oxygen and due to the very reactive nature of the other products that may corrode cell components [113]. Product currents can be measured by forcing the product to convect to a second working electrode where it is detected using an electrochemical reaction. Most commonly, rotating ring disk electrodes (RRDE) [114,115,116,117] are employed where peroxide is oxidized at the ring. Currents can then be assigned to hydroxide and hydroperoxide production under the assumptions that (i) superoxide can be excluded; (ii) that there are no side reactions or (iii) transition metal redox; and (iv) that the determined collection efficiency is appropriate for the reaction products. The highly reactive superoxide radical is expected to disproportionate quickly in aqueous media so that it is unlikely to be detected in a conventional RRDE experiment. It was also not found below >6 M NaOH using a scanning microelectrode [118]. It is recommended to investigate the source of the measured currents by separate experiments to evaluate contributions due to side reactions, e.g., electrolyte decomposition, and due to B-site redox, which might not have been fully removed by background subtraction. Lastly, collection efficiencies are commonly obtained using fast redox couples such as ferri-/ferrocyanide. The latter undergoes ligand exchange in alkaline media [119], leading to degradation and possibly lowering of the measured collection efficiency, especially in the presence of UV light [120]. While hydroperoxide is miscible with water, it is also very reactive so that the time dependence should be considered when evaluating currents detected at the ring.
The measurement of kinetic currents, in particular using (R) RDEs, is well established for metals [1,91,117,121] and calculation of product-specific currents belonging to the hydroperoxide (2 e−) and hydroxide (4 e−) products is common. For the oxides discussed herein, product-specific currents [56,97] or kinetic constants [57,64] were exclusively reported for composite electrodes containing carbon. Therefore, the currents discussed in this review are not product-specific unless otherwise noted. As both hydroxide and hydroperoxide are likely products on composite electrodes of carbon and perovskite electrodes [57,92,95], it is highly desirable to obtain this information from Koutecký–Levich analysis [122] using an RDE or from the ring and disk currents of an RRDE [123,124].
As initially mentioned, the currents would ideally be normalized to the number of active sites to make measurements of different materials comparable. Unfortunately, this number is unknown and cannot be accessed directly for heterogeneous surfaces. Instead, currents are normalized by the entire surface area of the oxide [14,125], or by the number of redox active metal sites [57,62,64] to compare the specific activity of electrochemical surfaces [126]. Ideally, this is done for a single known surface facet so that the activity is representative of a specific chemical composition at the facet. In this respect, smooth epitaxial thin films and single crystals are ideally suited to investigate the specific activity of perovskite oxides (and any other material). A comprehensive summary of previous investigations of these materials can be found in Table 2, where their specific current densities are compared at 0.85 V vs. RHE (0.38 V overpotential). This overpotential was chosen as it is low enough to compare the kinetic currents of oxides and Pt surfaces, where the kinetic currents of both material classes are between 10 and 80% of the limiting current.
The studied B-sites of Mn, Co, and Ni match those identified in Section 3 as being most active in the LaBO3 series. All Mn-containing perovskites showed catalytic activity but the weak dependence on rotational speed in RDE measurements [127,128] suggests that oxygen is not directly reduced to water on these surfaces, which is in agreement with microkinetic analysis of composite electrodes [97]. LaCoO3 [60,129] and La0.64Ca0.35Co0.95O3 [130] were reported as active for oxygen reduction. La0.8Sr0.2CoO3 [96,131] showed no activity but could be activated by addition of carbon [96]. Due to the similarity in the chemistry of the Co-based perovskites, the discrepancy is unlikely to be caused by doping of divalent cations and requires further studies. Lastly, LaNiO3 on various substrates was found active for oxygen reduction [60,132]. In future work, it would be highly desirable to quantify the product yield of these surfaces in the absence of potential co-catalysts such as carbon.
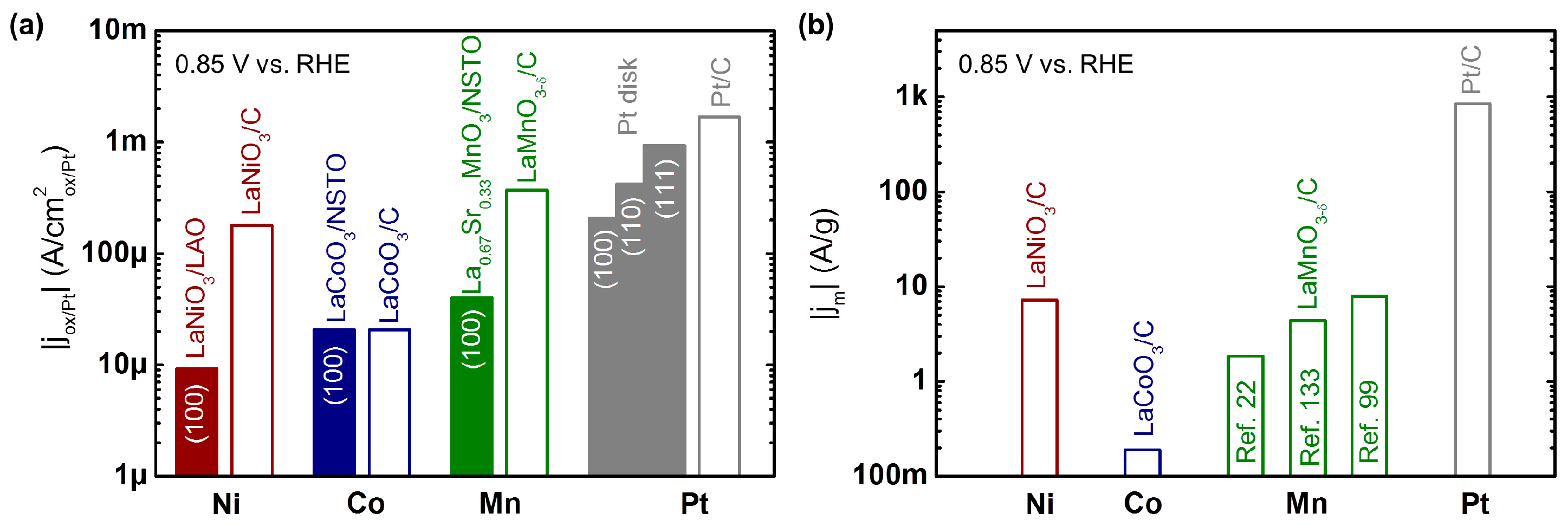
The activity for oxygen reduction of the most active Mn, Co, and Ni-based perovskite oxides are compared to those of low index Pt single crystals in Figure 3a. The current densities of the perovskite thin films increase from Ni [132] to Co [60] to Mn [63], where the most active oxide produced a factor 5 lower current than the least active Pt (100) [136] surface. However, the chemical, structural, and magnetic properties of the oxides are likely not fully optimized as will be argued in Section 6. In contrast to single crystalline Pt (Figure 3a), the activity of single crystalline perovskite oxides depends weakly on the surface facet (Table 2) in previous reports [63,96,131], so that the most commonly investigated (100) surfaces are compared. The most active composite electrodes [22] are also included for reference. The currents of the composites are equal or higher than those of the single crystalline surfaces because carbon can act as a co-catalyst [92,95,137,138,139] and disperses the catalyst [92,93]. The performance gain is particularly pronounced for Ni- and Mn-based perovskite, where LaMnO3−δ showed currents a factor 5 lower than Pt/C (46 wt% Pt on TKK carbon) [125]. Nonetheless, the higher elemental abundance by factors of factors of 3 × 105 and 1 × 104 (reference [140]) relative to Pt make these materials interesting candidate materials for optimization towards practical applications.
The mass activity of select composite electrodes of perovskite powders are compared to that of Pt/C (46 wt% Pt on TKK carbon) [125] in Figure 3b and Table 3. The mass of the oxide (or Pt) directly correlates with the weight and cost of a fuel cell cathode of a given size, which makes it a meaningful metric for the selection of catalyst materials. Further factors, such as stability and product yield, are also important for practical applications [113] but will not be covered here. Three main factors influence the mass activity: (i) the specific activity of the material per surface area, particularly of its majority facet; (ii) the size and microstructure of the particles; and (iii) the aforementioned interactions with the carbon support. The specific activities of the selected perovskites can be found in Figure 3 and Table 2. They are within one order of magnitude of Pt/C with the exception of LaCoO3 [22,57], which is less active. Tremendous progress has been achieved in making perovskite oxides with small particle size and large surface area. This is a great challenge due to the high formation energies of perovskites [141], which conventionally requires annealing at high temperatures, resulting in large particle sizes. Perovskite oxides with surface areas above 20 m2/g were investigated for oxygen reduction (Table 3). Overall, efforts to make nanoparticles with high surface area are focused on Mn-based perovskites.
The best perovskite oxide, LaMnO3 [22], reaches 7.9 A/g at 0.85 V vs. RHE, which is approximately two orders of magnitude lower than that of the Pt/C benchmark [125]. As the specific activity of LaMnO3 (Table 2) is within about an order of magnitude of Pt surfaces, this indicates that the surface area of the perovskite oxides needs further engineering to match that of Pt (and other metal particles or binary oxides). Additional activity could also be gained by careful optimization of the electrode composition, particularly considering the possible co-catalytic effect of carbon [92,137,143]. In practical terms, a 100-fold increase in loading of the oxides is currently necessary to match the activity of Pt with state-of-the-art perovskites [125]. In terms of cost, it was estimated composite electrodes of micron-sized LaMnO3+δ [22] and Pt have about equal activity per cost [26], so that the most active nanoparticle LaMnO3 composite in Table 3 would be about four times cheaper as compared to the commercial Pt/C benchmark in Figure 3b, albeit at significantly increased catalyst weight. Thus, perovskite oxides have been employed in many prototype devices of fuel cells [66,144,145] and Zn-air batteries [101,102,108], which is reviewed in detail elsewhere [11,89,90].
5. The Mechanisms of Oxygen Reduction on Perovskite Oxides
Surprisingly little is known conclusively about the mechanism of the oxygen reduction on perovskites and oxides in general. Even for the perhaps best-studied solid ORR catalyst, Pt, there is no generally accepted mechanism to date due to the elusiveness of reaction intermediates [10]. While the ORR on metal surfaces is clearly better understood experimentally [3,10,146,147] and theoretically [148,149,150,151,152,153,154], it is currently unknown if this knowledge can be transferred to oxides without significant adjustments due to properties inherent to oxides. As discussed in Section 2, perovskite oxides frequently exhibit strong electronic and/or magnetic correlations, band gaps, and bending, which may affect the mechanism. These properties have also thwarted insight by DFT calculations, which have been instrumental for a better understanding of the ORR on metals [148]. Nonetheless, select theoretical studies have been reported for various aspects of the ORR on perovskite oxides but exclusively in the LaBO3 series [30,59,155,156,157,158,159].
In this section, the mechanistic discussion of the ORR is prepared by firstly introducing the boundary conditions on perovskite oxides, which is followed by the definition of a minimalistic and universal set of elementary steps. These steps are then examined in the context of mechanistic sequences previously proposed for perovskite oxides.
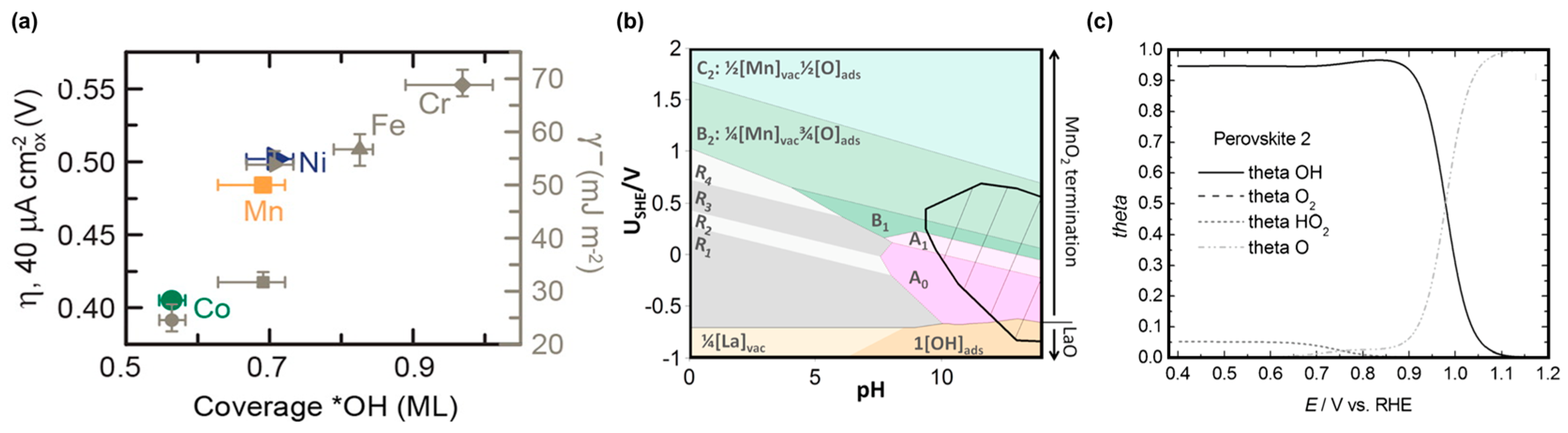
The surface of stoichiometric perovskites lacks terminal oxygen ions perpendicular to the surface, i.e., it is cleaved, to minimize surface energy. When brought in contact with an aqueous solution, the oxygen of water or hydroxide initially adsorbs on the surface and thus fully coordinates the surface metal sites. The adsorbed molecules engage in hydrogen bonding with each other and the solvent water/hydroxide molecules so that the electrochemical potentials of the perovskite and solution are in equilibrium [20]. The acid dissociation constant (pKa) of the surface site (s) or alternatively the point of zero charge (PZC) of the entire surface then determines the average surface protonation or surface charge. If the PZC is smaller than the pH of the solution, surface water will donate protons to the solvent, thus becoming hydroxide. Since common perovskites with B-sites of a first-row transition metal have PZCs in the range of 6–9 [52], their surfaces are covered by hydroxide in alkaline media. Stoerzinger et al. [60] found, by ambient pressure XPS, that lower hydroxide coverage of LaBO3 perovskites in a humid atmosphere correlated with lower voltage to support 40 µA/cm2 in 0.1 M KOH (Figure 4a). This correlation supports the importance of affinity for hydroxide coverage on the mechanisms of oxygen reduction.
The hydroxide coverage also depends on the applied voltage and solution pH. Thermodynamically stable (surface) phases can be visualized in an Eh-pH (or Pourbaix) diagram [29]. Rong and Kolpak [30] computed the most stable surface phases of (001)-oriented LaMnO3 in water using DFT (Figure 4b). In the commonly investigated alkaline conditions of pH 13 and 14, the surface traverses through the hydroxide covered phases (denoted B1, A1) to a cleaved surface (denoted A0) when decreasing the voltage from 0.4 V vs. SHE (no ORR overpotential; 1.23 V vs. RHE at pH 14) to −0.4 V vs. SHE (high ORR overpotential; 0.43 V vs. RHE at pH 14) [30]. A cleaved surface offers active sites for oxygen adsorption during the first step(s) of the oxygen reduction mechanisms. Poux et al. [97] calculated the voltage-dependence of a perovskite surface using the empiric rate constants of La0.8Sr0.2MnO3 at pH 14 (Figure 4c). In the investigated voltage range being typical for ORR, the calculated Langmuir-type coverage changed from predominantly O at low overpotential to predominantly OH at high overpotential, having equal coverages slightly below 1 V vs. RHE. Since the cleaved surface (A0) of the related LaMnO3 is thermodynamically favorable at high overpotential (Figure 4b), the increasing OH coverage with overpotential suggests OH displacement by O2 as the kinetically limiting step on the La0.8Sr0.2MnO3 surface.
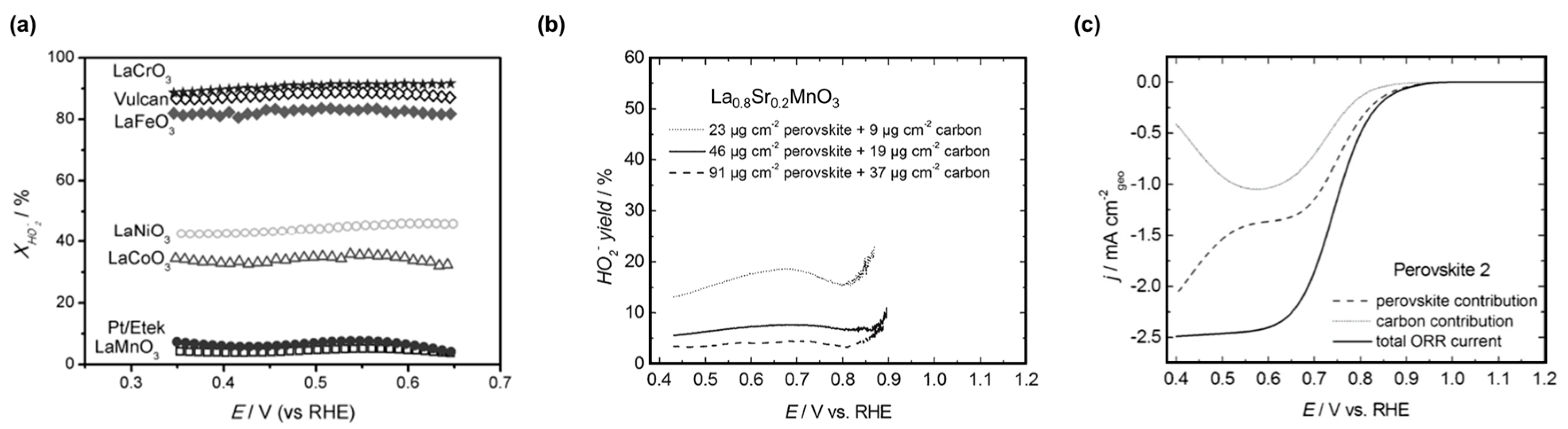
Hydroxide is the only desired product of the ORR in fuel cells and (aqueous) metal-air batteries but peroxide production is also frequently observed on composite electrodes of perovskite oxides [57,92,95]. Furthermore, perovskites containing Mn, Fe, and Co are known to chemically decompose peroxide [160,161,162]. Exemplary electrocatalytic hydroperoxide yields of composite electrodes containing LaBO3 perovskites and Vulcan carbon are shown in Figure 5a. The hydroperoxide yield was lowest for Mn at the B-site and comparable to the benchmark Pt/C, while it was in the range of 30—40% for Co and Ni B-sites [57]. For Fe and Cr B-sites, the yield was in excess of 80% and equal to Vulcan carbon, which supports negligible ORR activity of these badly conducting oxides. Hydroperoxide yields for composite electrodes are difficult to compare as they depend at least on (i) the total electrode loading (Figure 4b) [92,97]; (ii) the ratios of the perovskite and carbon loadings [92,95,143]; and (iii) the employed type of carbon [92,137]. None of these parameters are standardized in literature. Poux et al. [97] calculated the perovskite and carbon contributions using rate constants obtained from a composite electrode of La0.8Sr0.2MnO3 and Sibunit carbon (Figure 5c). While the total ORR current of the simulation is sigmoidal as frequently observed in experiments, the individual contributions have local minima between 0.5 and 0.6 V vs. RHE and diverge at lower voltages (higher overpotential). The ORR has also been studied on rotated carbon-free crystalline films of La1−xSrxMnO3 [127,128] (x = 0.33, 0.20) and La0.8Sr0.2CoO3 [96,128]. Cyclic voltammetry (CV) of these films does not show the expected sigmoidal shape and saturation currents in contrast to comparable measurements of composite electrodes containing these perovskites mixed with carbon [96]. Therefore, the coupling between the reactions on carbon and the perovskite must be considered when assigning a product yield to a perovskite oxide for discussions of the ORR mechanism.
Mn oxides containing Mn3+ in addition to Mn2+ or Mn4+ were identified as the most active for ORR [26], where the 3+/4+ redox is particularly important for Mn-based perovskites [163]. Direct evidence of the active valence by X-ray based methods is rare for the ORR in alkaline media. A notable exception is the recent XAS study at the Mn-K edge of Kan et al. [127] on (001)-oriented La0.67Sr0.33MnO3 in 0.1 M KOH, where Mn reduction of the thin film surface was found at 0.7 and 0.5 V vs. RHE relative to the film at open-circuit. While the spectra clearly support reduction, unfortunately, the analysis was not performed quantitatively. Operando experiments on non-perovskite Mn oxides showed reduction of Mn3+ to Mn2+ at high ORR overpotential [164]. Reduction of perovskites significantly below Mn3+ would require the formation of oxygen vacancies or change in the composition of the A-site for charge neutrality. Perovskite oxides such as LaMnO3 exhibit redox peaks at similar voltages to binary Mn oxides [163] and it was found that the most active sample in the series LaBO3 on carbon (i.e., LaMnO3) showed a redox wave slightly more positive than the onset of ORR [64]. Additionally, Ryabova et al. [163] found a correlation between the voltage of the Mn3+/4+ redox and ORR activity. The availability of Mn3+ was postulated as a requirement for O2 adsorption in their study, thus rationalizing the observed correlation.
Many of the above boundary conditions of surface coverage, product yield, and valence relate to one or more of the processes during catalytic oxygen reduction. Conceptually, the ORR should include the following basic processes
- P1
- Adsorption of molecular oxygen at a suitable site on the oxide surface
- P2
- Formation of intermediates to aid charge transfer and lower catalytic barriers
- P3
- Transfer of electrons/holes and protons/hydroxide to/from the catalyst
- P4
- Breaking of the O–O bond (if hydroxide is the product)
- P5
- Product release
where P1, P2, and P5 relate to coverage, P1, P3, and P5 relate to the valence and P4 and P5 relate to product yield. Much insight into these processes comes from DFT calculations of the reaction’s thermodynamics, in particular relating to intermediates (P2) and O–O bond breaking (P4), whose direct study is beyond current experimental capabilities for perovskite surfaces. A plethora of mechanisms are conceivable based on these five basic processes.
Hypothetical reaction pathways of the ORR are compared in Figure 6 where they are classified by the orientation of oxygen adsorption, which yields three prototypical idealized mechanisms. Oxygen can adsorb strongly through formation of a covalent bond with a single site (a2 in Figure 6a) which leads to an inner sphere electron transfer [165] that depends strongly on the electronic properties of the perovskite. On the other hand, oxygen can also adsorb weakly on multiple sites (b2 in Figure 6b) where the oxygen molecule might even be spatially separated by a monolayer of water/hydroxide. In the case of spatial separation, oxygen might be located in the outer Helmholtz layer of the electric double layer [166,167], which leads to an outer-sphere electron transfer that favors hydroperoxide production [165]. It was proposed that an outer sphere electron transfer is promoted by alkaline media on metals [166,167] and Fe–N–C [166,168] catalysts. In addition to materials properties such as magnetism to ensure proper alignment of the paramagnetic oxygen, electrolyte properties such as hydrogen bonding should be considered [149,150,166]. While an in-depth discussion is beyond the scope of this manuscript it should be noted that an outer-sphere electron transfer is conceivable for the mechanism in Figure 6b.
Further important distinctions of the idealized mechanisms are the nuclearity of the active site, which increases from Figure 6a to Figure 6c as well as the involvement of oxygen vacancies solely in Figure 6c. The shown mechanisms should be seen as idealized limiting cases. It is conceivable that those mechanisms occur simultaneously and compete on real surfaces, even to the extent that the mechanisms share intermediates, e.g., steps a1, b1, and c3.
The single site mechanism of Figure 6a is most commonly discussed in recent literature for perovskite surfaces. It can be traced back to work of the Goodenough group on the rutiles IrO2, RuO2; the pyrochlores Pb2Ru2−xPbxO7−y; and the perovskites Sr1−xNbO3−δ [169,170]. This mechanism is also assumed in DFT work of both metals [148] and metal oxides [30,59,155]. It starts with an OH covered surface (a1) and proceeds via displacement of an OH group for O2 (a2) that adsorbs in end-on. This is followed by protonation to an OOH group (a3), which might be released as hydroperoxide or remain an intermediate that undergoes O–O bond cleavage (a4) and is finally protonated to an OH group. In each step of this sequence, electron and proton transfers are thought to be coupled. Further coupling was proposed between the formation energy of the steps with a single bond between the metal and oxygen (a2,a4) and between the other two steps [171,172]. These so-called “scaling relations” [172,173,174,175,176] introduce a non-vanishing contribution to the overpotential [177,178,179,180], which could be a consequence of an isolated single active site that limits the available mechanistic pathways [181,182,183].
The mechanism in Figure 6b is based on the “pseudo-splitting” mechanism [184,185] on surface kinks adapted by the Tseung group for Li-doped NiO [23] and later pervoskites such as La0.5Sr0.5CoO3 [24] from an earlier proposal of Evans [185]. It was also discussed by Yeager who called it “bridge model” [55,186,187] and lifted the requirement of surface kinks (i.e., specific surface defects). The mechanism entails the participation of at least two metal sites and, therefore, could mitigate overpotentials caused by scaling between intermediates. This mechanism has been studied for Pt surfaces, where the first electron transfer (b2→b3 in Figure 6b) was proposed as the rate-limiting step [151]. The starting point is identical to the mechanism previously introduced, namely an OH-covered surface (a1 = b1). Then, two OH groups need to be displaced by O2, which is adsorbed in side-on configuration onto two metal sites on the OH-covered surface (b2), either to be released as hydroperoxide or the O–O bond is broken and two hydroxide molecules are released to a highly oxidized metal dimer on the surface (b3). Finally, the dimer is protonated to close the catalytic circle. The applicability of the mechanism depends on proper spacing of the participating metal sites. Furthermore, it was proposed that this mechanism requires magnetic ordering on oxides [23] (i.e., the experiment must be performed below the material’s Néel temperature at which an antiferromagnet becomes a paramagnet), which restricts the mechanism to paramagnetic oxides. It is expected that paramagnetism promotes “side-on” adsorption of the likewise paramagnetic O2 molecule [61]. While discussion of magnetism is scarce for ORR catalysts, many perovskite (-like) oxides can be paramagnetic in the temperature range of alkaline electrocatalysis (i.e., near 300 K) [61,188,189,190].
The mechanism in Figure 6c is motivated by recent reports of an active role of oxygen vacancies in the ORR, which is an example for the possible mechanistic role of surface defects. An in-depth discussion of the role of oxygen in perovskites for catalytic reactions, including the ORR, is reported by Yang and Grimaud [191] in this Special Issue. The shown mechanism is loosely based on high temperature work of mixed oxide ion/electronic conductor (MIEC) oxides [169]. Yet, related mechanisms have very recently been proposed for the reverse reaction of oxygen evolution on SrCoO3−δ perovskite oxides in alkaline media [192,193,194]. The starting point is an OH-covered surface as before but in this mechanism, one or more lattice oxygen vacancies (in the equatorial plane) are present additionally (c1). Next, one OH group near one of the vacancies is displaced and one oxygen of O2 fills the vacancy, while the other binds with the neighboring, previously undercoordinated, metal site (c2). Yeager [186] discussed a comparable configuration (albeit without oxygen vacancies), which would weaken the O–O bond so that a surface state similar to a4 in Figure 5a results. Alternatively, it is conceivable that the apical oxygen migrates on the surface to either recombine with another oxygen ion to yield molecular dioxygen or an oxygen vacancy, see e.g., the discussion in reference [169] (for high temperature electrocatalysis). The next step in the hypothetical mechanism of Figure 5c is protonation of the superoxide group to hydroxide (c3), which is identical to the initial steps (a1, b1) of the mechanism in Figure 5a,b. A route for recovery is protonation of the bridging oxygen (c4) as discussed by Grimaud et al. [194] (for oxygen evolution). The OH group in the bridging position is then discharged, thus recovering the catalyst. This mechanism also has special requirements, namely affinity for oxygen vacancies and structural stability in their presence, which restricts its applicability to a few materials classes among which are many perovskite (-like) oxides [195,196].
The discussed idealized mechanisms are expected to differ in their rate-limiting step and product yield. Under the common assumption of facile O–O bond breaking, the end-on mechanism in Figure 6a would produce hydroxide and be limited by either the initial hydroxide displacement (a1→a2) or recovery of the hydroxide surface (a4→a1). If the O–O bond breaking were limiting, which is commonly postulated for oxygen electrocatalysis using oxides, including perovskites, then hydroperoxide is the more likely a reaction product. The hydroperoxide could be subsequently catalyzed to hydroxide, resulting in an apparent transfer of four electrons per reduced O2. This is a likely scenario for many perovskite electrodes, particularly for composite electrodes containing carbon [92,95,96,137,138,139]. The side-on mechanism in Figure 6b requires that the O–O bond breaking in step b2 is not limiting. However, the interaction of oxygen with two metal sites is expected to weaken the O–O bond as compared to end-on adsorption on a single site of the same transition metal. The adsorption in this configuration (and preceding hydroxide displacement) should require a high degree of paramagnetism of the catalyst material to properly align the paramagnetic O2 molecule. It is thus expected to be limiting for non-magnetic materials. Furthermore, peroxide production seems less likely for side-on adsorption as compared to end-on adsorption as it would require the transition to a state similar to a2 for protonation. The O–O bond should be most weakened for bidentate adsorption in Figure 6c and thus the mechanism is least likely to produce hydroperoxide. The most likely limiting steps for bidentate adsorption at an oxygen vacancy are the chemical recovery of the vacancy or an electrochemical step having non-concerted electron and proton transfers [194]. In summary, the mechanistic pathway determines both the product yield of the ORR and the physical origin of the activity for the desired reduction of oxygen to hydroxide.
6. Derivation of Descriptors from Structure–Activity Relationships
Rational design of electrocatalysts requires fundamental insight into the microscopic origin of activity, which can be derived from structure–activity relationships. In oxygen electrocatalysis, this is frequently called the descriptor approach, where some physical property of the material serves as the descriptor for catalytic activity. Most commonly, chemical descriptors—such as eg occupancy [22] and valence [26]—are discussed and they have the greatest impact on the activity for oxygen reduction (Table 4) and also for the back reaction of oxygen evolution [197]. Further proposals include structural descriptors such as strain [129,132] and magnetic descriptors such as the magnetic moment [61].
Many descriptors can be varied in a parameter range that includes an optimal value as well as values where the parameter is either too weak/low or too strong/high so that a volcano-shaped trend naturally arises. The reason for the discontinuity of the trend at the tip of the volcano is a change of the limiting step in the mechanistic sequence (without changing the overall sequence). If the limiting step does not change in the studied parameter range, linear relations between activity and descriptors are frequently reported. In general, binding between catalyst and reactant should be neither too strong nor too weak for optimal catalysis, which is known as Sabatier’s principle.
A catalyst provides reaction pathways with energy barriers lower than that of the uncatalyzed reaction, which is achieved by the formation of reaction intermediates. Therefore, the Gibbs free energy of the formation of these intermediates is a conceptually obvious descriptor and it has proven very powerful and fruitful in theoretical investigations [30,59,148,150,155,198]. However, it is very challenging if not impossible to measure experimentally for oxide surfaces. As it has been postulated above that the recovery of the hydroxide surface is rate-limiting, the Gibbs free energy of formation of hydroxide is a suitable choice of an intermediate to use as a descriptor (Figure 7a). Lee et al. [59] calculated the Gibbs free energies of reactions a1→a2 and a4→a1 in Figure 6a as function of the Gibbs free energy of formation of metal hydroxide on LaBO3 perovskite surfaces with either La- or B-termination under 0.83 V vs. RHE polarization. For the intended structure-function relationship in Figure 7a, the y-axis should be a measure of the reaction kinetics. It can be obtained using the Eyring equation, which means that the y-axis is proportional to logarithmic current. Lee et al. [59] found Mn- and Co-terminated LaBO3 near optimal OH binding energy at the intersection of the grey lines, which is in agreement with the activity trends in Figure 2a and the assumed B-site termination in Figure 6. For LaNiO3, it was predicted that the La-terminated surface also shows near optimal binding, while the Ni-terminated surface is the least active. Aside from problems with phase purity in LaNiO3 [54], this might explain the diverging activity trends for LaNiO3 in the reported LaBO3 studies [22,56,57].
Another obvious choice for a descriptor is the chemisorption of the reactant, i.e., oxygen. Assuming proportionality between adsorption and desorption, temperature programmed desorption (TPD) can be used to measure desorption of O2 from perovskite surfaces for use as a descriptor. Suntivich and Shao-Horn [199] combined the TPD results of Yokoi and Uchida [200] with their activity measurements of the LaBO3 series (Figure 7b). In Figure 7b, the y-axis can be related to kinetics using the Butler–Volmer equation (and often the requirements for the simpler Tafel equation are met for ORR studies) so that the y-axis is again proportional to logarithmic current. The most active perovskites in the LaBO3 series exhibit temperatures of desorption between 120 °C (Ni) and 200 °C (Mn, Co). While few data are provided, the experimental descriptor seems promising, especially for comparison with DFT calculations. It would work with any of the mechanisms in Figure 6 with the caveats that solvent effects are excluded and that the onset of desorption might depend on the mode of adsorption (i.e., end-on vs. side-on vs. bidentate).
On a more fundamental level, the adsorption of oxygen could be rationalized by considering the hybridization of oxygen and frontier metal orbitals. For the mechanism in Figure 6a with end-on adsorption of oxygen, oxygen sp2-orbitals will form σ-bonds with metal eg orbitals in the octahedral coordination environment of perovskites. Following this rationale, Suntivich et al. [22] used the eg occupancy as a descriptor of oxygen reduction (Figure 7c), where the eg occupancy was either obtained from XAS or the chemical formula considering the spin state of the B-site. Again, B-sites having Mn, Ni, and Co are the most active and share a similar descriptor value of single occupancy of the eg orbital or slightly below. The most active sample in the study was oxygen-rich LaMnO3+δ with an eg occupancy of 0.8. While not included in the study of Suntivich et al. [22], manganite perovskites with La, Sr, Pr, and Ca doping [63,64,65] (Figure 2b) also show their maximum activity at estimated eg occupancies between 0.7 and 0.6. The optimum eg occupancy for Mn-based perovskites could thus fall below single occupancy. Yet, pinpointing the exact value requires challenging operando measurements of the eg occupancy.
No single descriptor can predict the optimal material for a given reaction, which was shown statistically for the reverse reaction of oxygen evolution [197]. Therefore, further activity gain can be achieved when a secondary descriptor is varied independently of a primary descriptor such as eg occupancy. These secondary descriptors can be chemical, structural, or magnetic.
The hybridization between oxygen and a metal site is a possible secondary chemical descriptor. At single eg occupancy, Suntivich et al. [22] discussed the M-O hybridization of adsorbed oxygen as a secondary chemical descriptor to explain differences in activity in the LaBO3 series (Figure 7d), where greater hybridization led to higher activity. For the back reaction of oxygen evolution, greater hybridization also led to higher activity in the LaBO3 series at single eg occupancy [51,201]. However, Abrishami et al. [202] report in this Special Issue that lower hybridization at an eg occupancy of 0.3 corresponds to higher activity and stability for first-order Ruddelsden–Popper phases that are related to the perovskite structure (see Section 2). This suggests that the details of the correlation between hybridization and activity might depend on the crystal structure and orbital filling. Further studies are required to fully understand the influence of hybridization on activity.
Strain is a possible secondary structural descriptor. Also at nominally single eg occupancy, Petrie et al. [132] grew epitaxial LaNiO3 on various substrates to vary the strain on LaNiO3 and ultimately the distortion of the Ni–O octahedron (Figure 7e). They found that compressive strain (negative values in Figure 7e) enhanced the activity. While strain also affected the activity of LaCoO3 at single eg occupancy, Stoerzinger et al. [129] found that the activity of LaCoO3 increased for tensile strain in contrast to LaNiO3. The origin of this discrepancy is unclear but a possible explanation for the differing trends might be the La0.67Sr0.33MnO3 buffer layer used in the study of Stoerzinger et al. [129]. While the catalytic effect of strain has been demonstrated, further studies are necessary to identify conclusive trends.
The magnetic moment is a possible secondary magnetic descriptor and was previously proposed by Larsson & Johansson [61]. Their data was used here after renormalizing by BET area to plot the intrinsic activity at fixed eg occupancy of Mn in the series LaB’0.5Mn0.5O3 (B’ = Cr, Ni, Cu). A Mn eg occupancy of ~0.3 was previously calculated from the valence measured by Suntivich et al. [22] using XAS for B’ = Ni and Cu, who argued that the valences of Ni and Cu should be close to each other having a value of ~2.3+ based on the greater Pauling electronegativity of Ni (1.91) and Cu (1.90) as compared to Mn (1.55) [203]. As the electronegativity of Cr (1.66) is also greater than that of Mn (1.55) [203], it is likely that Mn has very similar eg occupation for B’ = Cr, Ni, and Cu in the LaB’0.5Mn0.5O3 series but a direct experimental verification of this conjecture is missing. In the LaB’0.5Mn0.5O3 series in Figure 7f, a smaller magnetic moment yields a higher activity at similar eg occupancy. It is unknown if this trend is robust due to the scarcity of studies on the effect of magnetism on the ORR. Larsson & Johansson [61] could not identify obvious correlations between the magnetic moment and ORR activity. However, their analysis did not account for the chemistry of the materials in addition to magnetism.
Having introduced and discussed select descriptors in the previous paragraphs naturally raises the question which descriptor allows the greatest activity gain. To provide an answer, the descriptors previously proposed for perovskite oxides are ranked in Table 4 according to their activity range. Unfortunately, many activity metrics were used in these studies and activities were reported units of (over)potential or currents. The range was reproduced here as reported or shown in graphs in the original publications and calculated the other unit using the Tafel equation, Δη = a Δlog j, where a is the Tafel slope. Considering the mechanisms in Figure 6, the ORR will either be limited by a chemical step or an electrochemical step, for which Tafel slopes of 120 mV and 60 mV can be roughly estimated [205]. Thus, these Tafel slopes were picked to convert a potential range into a range of logarithmic current (Δlog j) and vice versa. It is noted that the reported activity range depends on the investigated parameter range and thus somewhat on the number of samples in the original study.
The first four descriptors in Table 4 are all related to chemistry and particularly occupation of orbitals, followed by structural and magnetic descriptors. For the top chemical descriptors, the reported activity changed by ~2 orders of magnitude (i.e., Δlog j ~ 2) for the eg occupancy of the B-site and the valence of the B-site accessed via the oxygen stoichiometry. The latter could also be a structural descriptor but too little is known about the role of oxygen vacancies in the ORR to separate structural and chemical effects. Activity changes of about one order of magnitude were reported for the valence accessed via the A-site composition, while the activity range reported for variation of the B—O covalence was below an order of magnitude and similar to the influence of the tolerance factor of the perovskite and the magnetic moment. Strain had the smallest reported activity range spanning about a factor of 3 (Δlog j ~ 0.5).
Based on the current knowledge of the ORR, an enhancement of the intrinsic activity of perovskite oxides by much more than an order of magnitude seems unlikely. In the best case, a secondary descriptor such as the covalence or the tolerance factor could improve the activity by about an order of magnitude at a fixed primary descriptor such as the eg occupancy. In practice, the gain should be lower the closer the primary descriptor gets to its optimum. Nonetheless, an increase by an order of magnitude through materials design would bring the activity of composite electrodes of perovskites and carbon to a level equal to that of composites of the benchmark Pt on carbon per metal/oxide area (Figure 3a). It is worth noting that the most active Pt alloys are currently about a factor 5 higher in activity as compared to pure Pt [206]. For practical applications, material utilization and weight are further challenges that need addressing as discussed above. However, the great advantage of perovskite oxides based on first row transition metals is their abundance, particularly for perovskites with Mn and Fe at the B-sites and Ca at the A-site [140]. This is highly relevant because the largest share (45%) of the produced Pt already has gone to automotive applications (i.e., catalytic converters) in 2014 [207] without commercial use of Pt-based fuel cells. Increased demand is expected due to more widespread use of fuel cells in automotive and stationary power applications. While it remains a great challenge to enhance the activity of perovskite oxides (and their composite electrodes) for oxygen reduction, the high abundance of their elements makes them a viable alternative to current Pt-based technology.
7. Concluding Remarks and Perspective
Correlating chemical, structural, and magnetic material properties to activity is the necessary first step towards a deep understanding of catalysis. From these correlations, fundamental guidelines can be distilled that allow predicting better catalysts. In this review, perovskite oxides are examined as electrocatalysts for the oxygen reduction in alkaline media. The understanding of the solid-state chemistry and oxide physics of perovskite oxides is very mature, which is particularly owed to advances in the preparation of epitaxial thin films and buried solid–solid interfaces since the late 1980s [208,209]. A similar revolution has yet to occur in understanding of the solid–liquid interface, where electrocatalysis takes place. Reaction intermediates at the solid–liquid interface remain elusive for the vast majority of catalysts for oxygen reduction and especially for perovskite oxides. A comprehensive knowledge of the reaction intermediates and the mechanistic sequence is paramount for fundamental insight into the origin of activity and product selectivity. The latter point is critical for the ORR where only reduction of oxygen to hydroxide is desired for alkaline fuel cells and metal-air batteries. Most of the current understanding draws from theoretical studies of the ORR on metals in the early 2000s, which have strongly influenced mechanistic studies on any catalyst. However, different mechanisms are also conceivable for perovskite oxides due to their magnetic and physicochemical properties.
The elusiveness of the intermediates and the mechanistic sequence calls for spectroscopic and microscopic operando measurements. While extremely challenging, it is desirable to perform these experiments on defect-free single crystals or epitaxial thin films for fundamental insight. Due to the limitation by the available signal, synchrotron-based experiments will be crucial for monitoring chemical and structural changes of the catalyst. Furthermore, surface-enhanced vibrational spectroscopy has also been used to track the formation of surface states with relevance to the ORR [210,211]. Changes in the morphology of a fuel cell material have been resolved by transmission electron microcopy (TEM) using a commercial liquid cell [212] but several challenges need to be overcome to provide the resolution required for catalytic insight [213]. The latter should be supported by rigorous electroanalytical analysis of the measured currents. Recent advances made it possible to perform RRDE measurements on epitaxial perovskite thin films [214,215], which is crucial for determining product-specific currents. Method development is thus an important cornerstone for the desired mechanistic insight.
Structure-function relationships (or catalytic descriptors) are an indispensable concept to derive fundamental guidelines for the design of more active catalysts after the mechanism is sufficiently understood. While chemical descriptors show the largest effect on catalysis of the ORR, magnetic and structural descriptors can have sizable influence on activity and they are clearly underrepresented in recent literature. Secondary descriptors are discussed to illustrate that it could be possible to further increase the activity near the optimum of one descriptor. Higher order descriptors should be considered in future material optimization for the ORR. Hong et al. [197] found, by statistical analysis, that a linear combination of nine descriptors was clearly superior to combining two descriptors for predicting the rate of the oxygen evolution reaction (i.e., the ORR back reaction) on perovskites. This illustrates the monumental challenge of optimizing materials for electrocatalysis. Due to the rich physics and chemistry of perovskites, systematic variation of chemical, structural, and magnetic properties could lead to new avenues to enhance the activity of perovskite oxides.
State-of-the-art electrocatalysts based on perovskite oxides still face both scientific and engineering challenges in competition with noble metal-based electrocatalysts. In addition to the aforementioned lack of mechanistic insight, further efforts are required to gain an improved understanding of the interplay between commonly employed carbon supports and perovskite oxides. It is still a matter of active debate whether and which perovskite oxides can independently perform the reduction of oxygen to hydroxide or whether they require a co-catalyst. While catalysts for acidic polymer electrolyte membrane (PEM) fuel cells are frequently tested with the established protocols [113], very little discussion can be found about the stability of perovskite oxides during oxygen reduction and about potential degradation mechanisms. Both stability and potential co-catalysis of the support must also be considered when engineering optimized composite electrodes containing perovskites. Furthermore, the surface area of perovskite oxides falls behind those of metals and simple metals. In addition to reducing the particle size, making perovskites with hierarchical porosity [216] is a promising approach to enhance mass activity. This could level their activity with that of the benchmark Pt, while preserving the advantage of large abundance, which would allow widespread deployment of technology based on perovskite oxides.
Acknowledgments
This work was supported by the DFG via CRC 1073. The author thanks V. Celorrio and D.J. Fermin for sharing their data as well as S. Mildner, J. Scholz, and J. Geppert for discussion.
Conflicts of Interest
The author declares no conflict of interest.
Appendix A. Effect of B-Site Composition on ORR Activity
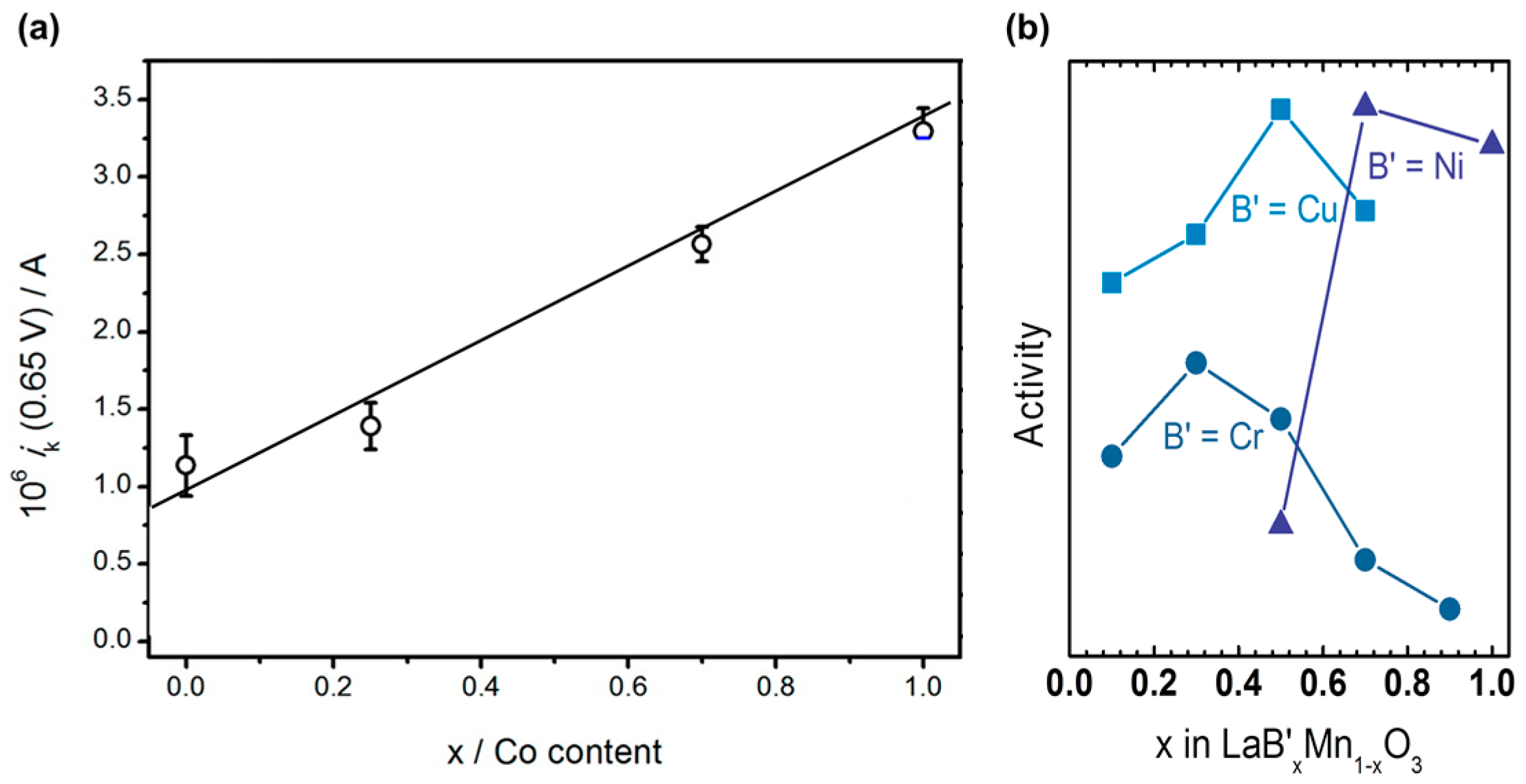
Figure A1.
(a) Kinetic currents of the system Ba0.5Sr0.5Fe1−xCoxO3−δ. At 0.65 V vs. RHE as function of Co content. Reproduced from [139]. Copyright 2016, American Chemical Society. (b) Trends of the activity for oxygen reduction of LaB’xMn1−xO3 (B’ = Cr, Ni, Cu). The data reported by Larsson & Johansson [61] was normalized by the BET area for the presentation in this figure.
Figure A1.
(a) Kinetic currents of the system Ba0.5Sr0.5Fe1−xCoxO3−δ. At 0.65 V vs. RHE as function of Co content. Reproduced from [139]. Copyright 2016, American Chemical Society. (b) Trends of the activity for oxygen reduction of LaB’xMn1−xO3 (B’ = Cr, Ni, Cu). The data reported by Larsson & Johansson [61] was normalized by the BET area for the presentation in this figure.
Appendix B. Bulk Conductivities of Select Perovskites

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
Reported conductivities and calculated tolerance factors of (stoichiometric) LaBO3 perovskites near room temperature.
Table A1.
Reported conductivities and calculated tolerance factors of (stoichiometric) LaBO3 perovskites near room temperature.
| Material | Space Group | t’ * | Conductivity (S/cm) | Temperature (K) | Reference |
|---|---|---|---|---|---|
| LaCrO3 | Pnma | 0.95863 | 2.4 × 10−3 | 298 | 81 |
| LaMnO3 | Pnma | 0.94151 | 6.0 × 10−3 | 298 | 80 |
| LaFeO3 | Pnma | 0.94198 | 4.0 × 10−4 | 280 | 72 |
| LaCoO3 | R-3c | 0.97039 | 2.8 × 10−1 | 298 | 72 |
| LaNiO3 | R-3c | 0.94621 | 5.7 × 10−3 | 298 | 73 |
* Tolerance factor, t’, Calculated using Equation (1) and data in Table A3.
Table A2.
Reported conductivities and calculated tolerance factors of (stoichiometric) AMnO3 perovskites near room temperature where known.
Table A2.
Reported conductivities and calculated tolerance factors of (stoichiometric) AMnO3 perovskites near room temperature where known.
| Material | Space Group | t’ * | Conductivity (S/cm) | Temperature (K) | Reference |
|---|---|---|---|---|---|
| YbMnO3 | Pbnm | 0.86893 | 2.1 × 103 | 603 | [74] |
| YMnO3 | P63cm | 0.88786 | 1.6 × 10−11 | 300 | [75] |
| DyMnO3 | Pbnm | 0.88155 | 1.0 × 10−2 | 600 | [76] |
| GdMnO3 | Pbnm | 0.90399 | 1.0 × 100 | 500 | [77] |
| SmMnO3 | Pbnm | 0.91206 | 2.0 × 10−2 | 600 | [76] |
| NdMnO3 | Pnma | 0.91802 | 5.8× 10−2 | 333 | [78] |
| PrMnO3 | Pbnm | 0.92959 | 1.0× 10−3 | 270 | [79] |
| LaMnO3 | Pnma | 0.94151 | 6.0 × 10−3 | 298 | [80] |
* Tolerance factor, t’, Calculated using Equation (1) and data in Table A3.
Appendix C. Bond Valence Parameters
Table A3.
Bond valence parameters used to calculate t’ using Equation (1).
| Ion | Coordination | M-O Distance (Å) | R0 (Å) | Reference |
|---|---|---|---|---|
| Cr3+ | 6 | 1.980 | 1.724 | [38] |
| Mn3+ | 6 | 2.016 | 1.760 | [38] |
| Mn4+ | 6 | 1.910 | 1.760 | [38] |
| Fe3+ | 6 | 2.015 | 1.759 | [38] |
| Fe3+ | 4 | 1.865 | 1.759 | [37] |
| Fe4+ | 6 | 1.909 | 1.759 | [37] |
| Co3+ | 6 | 1.956 | 1.700 | [37] |
| Co3+ | 4 | 1.806 | 1.700 | [37] |
| Co4+ | 6 | 1.850 | 1.700 | [37] |
| Ni3+ | 6 | 2.006 | 1.750 | [217] |
| Ca2+ | 12 | 2.630 | 1.967 | [38] |
| Sr2+ | 12 | 2.781 | 2.118 | [37] |
| La3+ | 12 | 2.685 | 2.172 | [38] |
| Pr3+ | 12 | 2.651 | 2.138 | [38] |
| Nd3+ | 12 | 2.618 | 2.105 | [38] |
| Sm3+ | 12 | 2.601 | 2.088 | [37] |
| Gd3+ | 12 | 2.578 | 2.065 | [37] |
| Dy3+ | 12 | 2.514 | 2.001 | [38] |
| Y3+ | 12 | 2.532 | 2.019 | [38] |
| Yb3+ | 12 | 2.478 | 1.965 | [38] |
b = 0.37.
References
- Song, C.; Zhang, J. Electrocatalytic Oxygen Reduction Reaction. In PEM Fuel Cell Electrocatalysts and Catalyst Layers; Springer: London, UK, 2008; pp. 89–134. [Google Scholar]
- Babcock, G.T. How Oxygen Is Activated and Reduced in Respiration. Proc. Natl. Acad. Sci. USA 1999, 96, 12971–12973. [Google Scholar] [CrossRef] [PubMed]
- Katsounaros, I.; Cherevko, S.; Zeradjanin, A.R.; Mayrhofer, K.J.J. Oxygen Electrochemistry as a Cornerstone for Sustainable Energy Conversion. Angew. Chem. Int. Ed. Engl. 2014, 53, 102–121. [Google Scholar] [CrossRef] [PubMed]
- Scherson, D.A.; Palencsr, A.; Tolmachev, Y.; Stefan, I. Transition Metal Macrocycles as Electrocatalysts for Dioxygen Reduction. In Electrochemical Surface Modification: Thin Films, Functionalization and Characterization; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 191–288. [Google Scholar]
- Zhang, Z.; Liu, J.; Gu, J.; Su, L.; Cheng, L. An Overview on Metal Oxide Materials as Electrocatalysts and Supports for Polymer Electrolyte Fuel Cells. Energy Environ. Sci. 2014, 7, 2535–2558. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity Benchmarks and Requirements for Pt, Pt-Alloy, and Non-Pt Oxygen Reduction Catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Cairns, E.J. Review—Recent Progress in Electrocatalysts for Oxygen Reduction Suitable for Alkaline Anion Exchange Membrane Fuel Cells. J. Electrochem. Soc. 2015, 162, F1504–F1539. [Google Scholar] [CrossRef]
- Nie, Y.; Li, L.; Wei, Z. Recent Advancements in Pt and Pt-Free Catalysts for Oxygen Reduction Reaction. Chem. Soc. Rev. 2015, 44, 2168–2201. [Google Scholar] [CrossRef] [PubMed]
- Gewirth, A.A.; Thorum, M.S. Electroreduction of Dioxygen for Fuel-Cell Applications: Materials and Challenges. Inorg. Chem. 2010, 49, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Jörissen, L. Bifunctional Oxygen/air Electrodes. J. Power Sources 2006, 155, 23–32. [Google Scholar] [CrossRef]
- Gupta, S.; Kellogg, W.; Xu, H.; Liu, X.; Cho, J.; Wu, G. Bifunctional Perovskite Oxide Catalysts for Oxygen Reduction and Evolution in Alkaline Media. Chem. Asian J. 2016, 11, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Sumboja, A.; Wuu, D.; An, T.; Li, B.; Goh, F.W.T.; Hor, T.S.A.; Zong, Y.; Liu, Z. Oxygen Reduction in Alkaline Media: From Mechanisms to Recent Advances of Catalysts. ACS Catal. 2015, 5, 4643–4667. [Google Scholar] [CrossRef]
- Hong, W.T.; Risch, M.; Stoerzinger, K.A.; Grimaud, A.J.L.; Suntivich, J.; Shao-Horn, Y. Toward the Rational Design of Non-Precious Transition Metal Oxides for Oxygen Electrocatalysis. Energy Environ. Sci. 2015, 8, 1404–1427. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Xu, D.; Xu, J.-J.; Zhang, X.-B. Oxygen Electrocatalysts in Metal–air Batteries: From Aqueous to Nonaqueous Electrolytes. Chem. Soc. Rev. 2014, 43, 7746–7786. [Google Scholar] [CrossRef] [PubMed]
- Sato, N. Basics of Corrosion Chemistry. In Green Corrosion Chemistry and Engineering; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 1–32. [Google Scholar]
- Matsumoto, Y.; Yoneyama, H.; Tamura, H. Influence of the Nature of the Conduction Band of Transition Metal Oxides on Catalytic Activity for Oxygen Reduction. J. Electroanal. Chem. Interfacial Electrochem. 1977, 83, 237–243. [Google Scholar] [CrossRef]
- Brenet, J.P. Electrochemical Behaviour of Metallic Oxides. J. Power Sources 1979, 4, 183–190. [Google Scholar] [CrossRef]
- Trasatti, S. Physical Electrochemistry of Ceramic Oxides. Electrochim. Acta 1991, 36, 225–241. [Google Scholar] [CrossRef]
- Gerischer, H. The Impact of Semiconductors on the Concepts of Electrochemistry. Electrochim. Acta 1990, 35, 1677–1699. [Google Scholar] [CrossRef]
- Meadowcroft, D.B. Low-Cost Oxygen Electrode Material. Nature 1970, 226, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Nakanishi, H.; Goodenough, J.B.; Shao-Horn, Y. Design Principles for Oxygen-Reduction Activity on Perovskite Oxide Catalysts for Fuel Cells and Metal-Air Batteries. Nat. Chem. 2011, 3, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Bevan, H.L.; Tseung, A.C.C. The Electrochemical Reduction of Oxygen on High Surface Area Lithium Doped Nickel Oxides. Electrochim. Acta 1974, 19, 201–206. [Google Scholar] [CrossRef]
- Hibbert, D.B.; Tseung, A.C.C. Homomolecular Oxygen Exchange and the Electrochemical Reduction of Oxygen on Semiconducting Oxides. J. Electrochem. Soc. 1978, 125, 74. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, P.; Dai, S. Recent Advances of Lanthanum-Based Perovskite Oxides for Catalysis. ACS Catal. 2015, 5, 6370–6385. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Risch, M.; Han, B.; Shao-Horn, Y. Recent Insights into Manganese Oxides in Catalyzing Oxygen Reduction Kinetics. ACS Catal. 2015, 5, 6021–6031. [Google Scholar] [CrossRef]
- Zhu, L.; Ran, R.; Tadé, M.; Wang, W.; Shao, Z. Perovskite Materials in Energy Storage and Conversion. Asia-Pac. J. Chem. Eng. 2016, 11, 338–369. [Google Scholar] [CrossRef]
- Chen, D.; Chen, C.; Baiyee, Z.M.; Shao, Z.; Ciucci, F. Nonstoichiometric Oxides as Low-Cost and Highly-Efficient Oxygen Reduction/Evolution Catalysts for Low-Temperature Electrochemical Devices. Chem. Rev. 2015, 115, 9869–9921. [Google Scholar] [CrossRef] [PubMed]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions; National Association of Corrosion Engineers: Houston, TX, USA, 1974. [Google Scholar]
- Rong, X.; Kolpak, A.M. Ab Initio Approach for Prediction of Oxide Surface Structure, Stoichiometry, and Electrocatalytic Activity in Aqueous Solution. J. Phys. Chem. Lett. 2015, 6, 1785–1789. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Si, W.; Luo, J.; Su, W.; Chang, H.; Li, J.; Hao, J.; Crittenden, J. Surface Tuning of La0.5Sr0.5CoO3 Perovskite Catalysts by Acetic Acid for NOX Storage and Reduction. Environ. Sci. Technol. 2016, 50, 6442–6448. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, N. ABO3-Type oxides—Their Structure and properties—A Bird’s Eye View. Mater. Sci. Eng. 1978, 36, 231–239. [Google Scholar] [CrossRef]
- Tilley, R.J.D. The ABX3 Perovskite Structure. In Perovskites; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 1–41. [Google Scholar]
- Goldschmidt, V.M. Die Gesetze der Krystallochemie. Naturwissenschaften 1926, 14, 477–485. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Tilley, R.J.D. Appendix A The Bond Valence Model for Perovskites. In Perovskites; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 303–306. [Google Scholar]
- Brese, N.E.; O’Keeffe, M. Bond-Valence Parameters for Solids. Acta Crystallogr. Sect. B Struct. Sci. 1991, 47, 192–197. [Google Scholar] [CrossRef]
- Brown, I.D.; Altermatt, D. Bond-Valence Parameters Obtained from a Systematic Analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. Sect. B Struct. Sci. 1985, 41, 244–247. [Google Scholar] [CrossRef]
- Faculty Innovation Center. Tolerance Factor Calculator v 0.2.1. Available online: http://www.me.utexas.edu/~benedekgroup/ToleranceFactorCalculator/ (accessed on 12 May 2017).
- Lufaso, M.W.; Woodward, P.M. Prediction of the Crystal Structures of Perovskites Using the Software Program SPuDS. Acta Crystallogr. Sect. B Struct. Sci. 2001, 57, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Jahn, H.A.; Teller, E. Stability of Polyatomic Molecules in Degenerate Electronic States. I. Orbital Degeneracy. Proc. R. Soc. A Math. Phys. Eng. Sci. 1937, 161, 220–235. [Google Scholar] [CrossRef]
- Englman, R. Jahn-Teller Effect in Molecules and Crystals; John Wiley & Sons, Ltd.: Chichester, UK, 1972. [Google Scholar]
- Li, C.; Soh, K.C.K.; Wu, P. Formability of ABO3 Perovskites. J. Alloy. Compd. 2004, 372, 40–48. [Google Scholar] [CrossRef]
- Tilley, R.J.D. ABX3—Related Structures. In Perovskites; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 42–78. [Google Scholar]
- Tilley, R.J.D. Hexagonal Perovskite-Related Structures. In Perovskites; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 79–122. [Google Scholar]
- Seiyama, T. Total Oxidation of Hydrocarbons on Perovskite Oxides. Catal. Rev. 1992, 34, 281–300. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Voorhoeve, R.J.H.; Johnson, D.W.; Remeika, J.P.; Gallagher, P.K. Perovskite Oxides: Materials Science in Catalysis. Science 1977, 195, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Tejuca, L.G.; Fierro, J.L.G.; Tascón, J.M.D. Structure and Reactivity of Perovskite-Type Oxides. Adv. Catal. 1989, 36, 237–328. [Google Scholar]
- Peña, M.A.; Fierro, J.L.G. Chemical Structures and Performance of Perovskite Oxides. Chem. Rev. 2001, 101, 1981–2017. [Google Scholar] [CrossRef] [PubMed]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Bockris, J.O. The Electrocatalysis of Oxygen Evolution on Perovskites. J. Electrochem. Soc. 1984, 131, 290–302. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Sato, E. Electrocatalytic Properties of Transition Metal Oxides for Oxygen Evolution Reaction. Mater. Chem. Phys. 1986, 14, 397–426. [Google Scholar] [CrossRef]
- Bockris, J.O.; Otagawa, T. Mechanism of Oxygen Evolution on Perovskites. J. Phys. Chem. 1983, 87, 2960–2971. [Google Scholar] [CrossRef]
- Tarasevich, M.R.; Sadkowski, A.; Yeager, E. Oxygen Electrochemistry. In Comprehensive Treatise of Electrochemistry; Springer: Boston, MA, USA, 1983; pp. 301–398. [Google Scholar]
- Sunarso, J.; Torriero, A.A.J.; Zhou, W.; Howlett, P.C.; Forsyth, M. Oxygen Reduction Reaction Activity of La-Based Perovskite Oxides in Alkaline Medium: A Thin-Film Rotating Ring-Disk Electrode Study. J. Phys. Chem. C 2012, 116, 5827–5834. [Google Scholar] [CrossRef]
- Celorrio, V.; Dann, E.; Calvillo, L.; Morgan, D.J.; Hall, S.R.; Fermin, D.J. Oxygen Reduction at Carbon-Supported Lanthanides: The Role of the B-Site. ChemElectroChem 2016, 3, 283–291. [Google Scholar] [CrossRef]
- Montini, T.; Bevilacqua, M.; Fonda, E.; Casula, M.F.; Lee, S.; Tavagnacco, C.; Gorte, R.J.; Fornasiero, P. Relationship between Electrical Behavior and Structural Characteristics in Sr-Doped LaNi0.6Fe0.4O3−δ Mixed Oxides. Chem. Mater. 2009, 21, 1768–1774. [Google Scholar] [CrossRef]
- Lee, Y.-L.; Gadre, M.J.; Shao-Horn, Y.; Morgan, D. Ab Initio GGA+U Study of Oxygen Evolution and Oxygen Reduction Electrocatalysis on the (001) Surfaces of Lanthanum Transition Metal Perovskites LaBO3 (B = Cr, Mn, Fe, Co and Ni). Phys. Chem. Chem. Phys. 2015, 17, 21643–21663. [Google Scholar] [CrossRef] [PubMed]
- Stoerzinger, K.A.; Hong, W.T.; Azimi, G.; Giordano, L.; Lee, Y.-L.; Crumlin, E.J.; Biegalski, M.D.; Bluhm, H.; Varanasi, K.K.; Shao-Horn, Y. Reactivity of Perovskites with Water: Role of Hydroxylation in Wetting and Implications for Oxygen Electrocatalysis. J. Phys. Chem. C 2015, 119, 18504–18512. [Google Scholar] [CrossRef]
- Larsson, R.; Johansson, L.Y. On the Catalytic Properties of Mixed Oxides for the Electrochemical Reduction of Oxygen. J. Power Sources 1990, 32, 253–260. [Google Scholar] [CrossRef]
- Celorrio, V.; Tiwari, D.; Fermin, D.J. Composition-Dependent Reactivity of Ba0.5Sr0.5CoxFe1−xO3−δ toward the Oxygen Reduction Reaction. J. Phys. Chem. C 2016, 120, 22291–22297. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Lü, W.; Li, C.; Ariando; Venkatesan, T.; Shao-Horn, Y. Highly Active Epitaxial La(1−x)SrxMnO3 Surfaces for the Oxygen Reduction Reaction: Role of Charge Transfer. J. Phys. Chem. Lett. 2015, 6, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Celorrio, V.; Calvillo, L.; Dann, E.; Granozzi, G.; Aguadero, A.; Kramer, D.; Russell, A.E.; Fermín, D.J. Oxygen Reduction Reaction at LaxCa1−xMnO3 Nanostructures: Interplay between A-Site Segregation and B-Site Valency. Catal. Sci. Technol. 2016, 6, 7231–7238. [Google Scholar] [CrossRef]
- Hyodo, T.; Hayashi, M.; Mitsutake, S.; Miura, N.; Yamazoe, N. Praseodymium–calcium Manganites (Pr1−xCaxMnO3) as Electrode Catalyst for Oxygen Reduction in Alkaline Solution. J. Appl. Electrochem. 1997, 27, 745. [Google Scholar] [CrossRef]
- Hyodo, T.; Hayashi, M.; Miura, N.; Yamazoe, N. Catalytic Activities of Rare-Earth Manganites for Cathodic Reduction of Oxygen in Alkaline Solution. J. Electrochem. Soc. 1996, 143, L266. [Google Scholar] [CrossRef]
- Takeda, Y.; Kanno, R.; Kondo, T.; Yamamoto, O.; Taguchi, H.; Shimada, M.; Koizumi, M. Properties of SrMO3−δ (M = Fe, Co) as Oxygen Electrodes in Alkaline Solution. J. Appl. Electrochem. 1982, 12, 275–280. [Google Scholar] [CrossRef]
- Du, J.; Zhang, T.; Cheng, F.; Chu, W.; Wu, Z.; Chen, J. Nonstoichiometric Perovskite CaMnO3−δ for Oxygen Electrocatalysis with High Activity. Inorg. Chem. 2014, 53, 9106–9114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Sunarso, J. Enhancing Bi-Functional Electrocatalytic Activity of Perovskite by Temperature Shock: A Case Study of LaNiO3−δ. J. Phys. Chem. Lett. 2013, 4, 2982–2988. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Risch, M.; Suntivich, J.; Lü, W.M.; Zhou, J.; Biegalski, M.D.; Christen, H.M.; Venkatesan, T.; Shao-Horn, Y.; Ariando; Venkatesan, T.; et al. Oxygen Electrocatalysis on (001)-Oriented Manganese Perovskite Films: Mn Valency and Charge Transfer at the Nanoscale. Energy Environ. Sci. 2013, 6, 1582–1588. [Google Scholar] [CrossRef]
- Hammouche, A.; Kahoul, A.; Sauer, D.U.; De Doncker, R.W. Influential Factors on Oxygen Reduction at La1−xCaxCoO3 Electrodes in Alkaline Electrolyte. J. Power Sources 2006, 153, 239–244. [Google Scholar] [CrossRef]
- Narasimhan, V.; Keer, H.V.; Chakrabarty, D.K. Structural and Electrical Properties of the LaCo1−xMnxO3 and LaCo1−xFexO3 Systems. Phys. Status Solidi 1985, 89, 65–71. [Google Scholar] [CrossRef]
- Tiwari, A.; Rajeev, K.P.; Narayan, J. Low Temperature Electrical Transport in La1−xNdxNiO3−δ. Solid State Commun. 2002, 121, 357–361. [Google Scholar] [CrossRef]
- Subba Rao, G.V.; Chandrashekhar, G.V.; Rao, C.N.R. Are Rare Earth Orthochromites Ferroelectric? Solid State Commun. 1968, 6, 177–179. [Google Scholar] [CrossRef]
- Sekhar, M.C.; Prasad, N.V. Dielectric, Impedance, Magnetic and Magnetoelectric Measurements on YMnO3. Ferroelectrics 2006, 345, 45–57. [Google Scholar] [CrossRef]
- Dabrowski, B.; Kolesnik, S.; Baszczuk, A.; Chmaissem, O.; Maxwell, T.; Mais, J. Structural, Transport, and Magnetic Properties of RMnO3 Perovskites (R = La, Pr, Nd, Sm, 153Eu, Dy). J. Solid State Chem. 2005, 178, 629–637. [Google Scholar] [CrossRef]
- Wandekar, R.V.; Wani, B.N.; Bharadwaj, S.R. High Temperature Thermal Expansion and Electrical Conductivity of Ln0.95MnO3+δ (Ln = La, Nd or Gd). J. Alloy. Compd. 2007, 433, 84–90. [Google Scholar] [CrossRef]
- Kamegashira, N.; Miyazaki, Y. Jahn-Teller Transition in NdMnO3. Phys. Status Solidi 1983, 76, K39–K42. [Google Scholar] [CrossRef]
- Lees, M.R.; Barratt, J.; Balakrishnan, G.; Paul, D.M.; Dewhurst, C.D. Low-Temperature Magnetoresistance and Magnetic Ordering in. J. Phys. Condens. Matter 1996, 8, 2967–2979. [Google Scholar] [CrossRef]
- Kamata, K.; Nakajima, T.; Hayashi, T.; Nakamura, T. Nonstoichiometric Behavior and Phase Stability of Rare Earth Manganites at 1200 °C: (1). LaMnO3. Mater. Res. Bull. 1978, 13, 49–54. [Google Scholar] [CrossRef]
- Li, G.; Kuang, X.; Tian, S.; Liao, F.; Jing, X.; Uesu, Y.; Kohn, K. Structure and Conductivity of Perovskites Sr1−xLaxTi1−xCrxO3. J. Solid State Chem. 2002, 165, 381–392. [Google Scholar] [CrossRef]
- Nozik, A.J.; Memming, R. Physical Chemistry of Semiconductor—Liquid Interfaces. J. Phys. Chem. 1996, 100, 13061–13078. [Google Scholar] [CrossRef]
- Memming, R. Semiconductor Electrochemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Krishnan Rajeshwar, K.; Rajeshwar, K. Fundamentals of Semiconductor Electrochemistry and Photoelectrochemistry. In Encyclopedia of Electrochemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. [Google Scholar]
- Memming, R. Processes at Semiconductor Electrodes. In Comprehensive Treatise of Electrochemistry; Springer: Boston, MA, USA, 1983; pp. 529–592. [Google Scholar]
- Hayashi, H.; Inaba, H.; Matsuyama, M.; Lan, N.G.; Dokiya, M.; Tagawa, H. Structural Consideration on the Ionic Conductivity of Perovskite-Type Oxides. Solid State Ion. 1999, 122, 1–15. [Google Scholar] [CrossRef]
- Richter, J.; Holtappels, P.; Graule, T.; Nakamura, T.; Gauckler, L.J. Materials Design for Perovskite SOFC Cathodes. Monatsh. Chem. Chem. Mon. 2009, 140, 985–999. [Google Scholar] [CrossRef]
- Vielstich, W. Ideal and Effective Efficiencies of Cell Reactions and Comparison to Carnot Cycles. In Handbook of Fuel Cells; John Wiley & Sons, Ltd.: Chichester, UK, 2010. [Google Scholar]
- Bidault, F.; Brett, D.J.L.; Middleton, P.H.; Brandon, N.P. Review of Gas Diffusion Cathodes for Alkaline Fuel Cells. J. Power Sources 2009, 187, 39–48. [Google Scholar] [CrossRef]
- Neburchilov, V.; Wang, H.; Martin, J.J.; Qu, W. A Review on Air Cathodes for Zinc—Air Fuel Cells. J. Power Sources 2010, 195, 1271–1291. [Google Scholar] [CrossRef]
- Mayrhofer, K.J.J.; Strmcnik, D.; Blizanac, B.B.; Stamenkovic, V.; Arenz, M.; Markovic, N.M. Measurement of Oxygen Reduction Activities via the Rotating Disc Electrode Method: From Pt Model Surfaces to Carbon-Supported High Surface Area Catalysts. Electrochim. Acta 2008, 53, 3181–3188. [Google Scholar] [CrossRef]
- Malkhandi, S.; Trinh, P.; Manohar, A.K.; Jayachandrababu, K.C.; Kindler, A.; Surya Prakash, G.K.; Narayanan, S.R. Electrocatalytic Activity of Transition Metal Oxide-Carbon Composites for Oxygen Reduction in Alkaline Batteries and Fuel Cells. J. Electrochem. Soc. 2013, 160, F943–F952. [Google Scholar] [CrossRef]
- Shinozaki, K.; Zack, J.W.; Pylypenko, S.; Pivovar, B.S.; Kocha, S.S. Oxygen Reduction Reaction Measurements on Platinum Electrocatalysts Utilizing Rotating Disk Electrode Technique-II. Influence of Ink Formulation, Catalyst Layer Uniformity and Thickness. J. Electrochem. Soc. 2015, 162, F1384–F1396. [Google Scholar] [CrossRef]
- Li, T.; Liu, J.; Jin, X.; Wang, F.; Song, Y. Composition-Dependent Electro-Catalytic Activities of Covalent Carbon-LaMnO3 Hybrids as Synergistic Catalysts for Oxygen Reduction Reaction. Electrochim. Acta 2016, 198, 115–126. [Google Scholar] [CrossRef]
- Poux, T.; Napolskiy, F.S.; Dintzer, T.; Kéranguéven, G.; Istomin, S.Y.; Tsirlina, G.A.; Antipov, E.V.; Savinova, E.R. Dual Role of Carbon in the Catalytic Layers of Perovskite/carbon Composites for the Electrocatalytic Oxygen Reduction Reaction. Catal. Today 2012, 189, 83–92. [Google Scholar] [CrossRef]
- Miyahara, Y.; Miyazaki, K.; Fukutsuka, T.; Abe, T. Influence of Surface Orientation on the Catalytic Activities of La0.8Sr0.2CoO3 Crystal Electrodes for Oxygen Reduction and Evolution Reactions. ChemElectroChem 2016, 3, 214–217. [Google Scholar] [CrossRef]
- Poux, T.; Bonnefont, A.; Kéranguéven, G.; Tsirlina, G.A.; Savinova, E.R. Electrocatalytic Oxygen Reduction Reaction on Perovskite Oxides: Series versus Direct Pathway. Chemphyschem 2014, 15, 2108–2120. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhou, W.; Shao, Z. Perovskite/Carbon Composites: Applications in Oxygen Electrocatalysis. Small 2017, 13, 1603793. [Google Scholar] [CrossRef] [PubMed]
- Kéranguéven, G.; Royer, S.; Savinova, E. Synthesis of Efficient Vulcan–LaMnO3 Perovskite Nanocomposite for the Oxygen Reduction Reaction. Electrochem. Commun. 2015, 50, 28–31. [Google Scholar] [CrossRef]
- Rincón, R.A.; Masa, J.; Mehrpour, S.; Tietz, F.; Schuhmann, W.; Muhler, M.; Schuhmann, W.; Chen, C.; Song, L.; Rauf, M.; et al. Activation of Oxygen Evolving Perovskites for Oxygen Reduction by Functionalization with Fe–Nx/C Groups. Chem. Commun. 2014, 50, 14760–14762. [Google Scholar] [CrossRef] [PubMed]
- Prabu, M.; Ramakrishnan, P.; Ganesan, P.; Manthiram, A.; Shanmugam, S. LaTi0.65Fe0.35O3−δ Nanoparticle-Decorated Nitrogen-Doped Carbon Nanorods as an Advanced Hierarchical Air Electrode for Rechargeable Metal-Air Batteries. Nano Energy 2015, 15, 92–103. [Google Scholar] [CrossRef]
- Lee, D.U.; Park, H.W.; Park, M.G.; Ismayilov, V.; Chen, Z. Synergistic Bifunctional Catalyst Design Based on Perovskite Oxide Nanoparticles and Intertwined Carbon Nanotubes for Rechargeable Zinc—Air Battery Applications. ACS Appl. Mater. Interfaces 2014. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Horvath, G.L.; Tobias, C.W. The Solubility and Diffusion Coefficient of Oxygen in Potassium Hydroxide Solutions. Electrochim. Acta 1967, 12, 287–297. [Google Scholar] [CrossRef]
- Speder, J.; Altmann, L.; Bäumer, M.; Kirkensgaard, J.J.K.; Mortensen, K.; Arenz, M. The Particle Proximity Effect: From Model to High Surface Area Fuel Cell Catalysts. RSC Adv. 2014, 4, 14971. [Google Scholar] [CrossRef]
- Nesselberger, M.; Roefzaad, M.; Fayçal Hamou, R.; Ulrich Biedermann, P.; Schweinberger, F.F.; Kunz, S.; Schloegl, K.; Wiberg, G.K.H.; Ashton, S.; Heiz, U.; et al. The Effect of Particle Proximity on the Oxygen Reduction Rate of Size-Selected Platinum Clusters. Nat. Mater. 2013, 12, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Ke, K.; Hiroshima, K.; Kamitaka, Y.; Hatanaka, T.; Morimoto, Y. An Accurate Evaluation for the Activity of Nano-Sized Electrocatalysts by a Thin-Film Rotating Disk Electrode: Oxygen Reduction on Pt/C. Electrochim. Acta 2012, 72, 120–128. [Google Scholar] [CrossRef]
- Masa, J.; Batchelor-McAuley, C.; Schuhmann, W.; Compton, R.G. Koutecky-Levich Analysis Applied to Nanoparticle Modified Rotating Disk Electrodes: Electrocatalysis or Misinterpretation. Nano Res. 2014, 7, 71–78. [Google Scholar] [CrossRef]
- Jung, J.-I.; Risch, M.; Park, S.; Kim, M.G.; Nam, G.; Jeong, H.-Y.; Shao-Horn, Y.; Cho, J. Optimizing Nanoparticle Perovskite for Bifunctional Oxygen Electrocatalysis. Energy Environ. Sci. 2016, 9, 176–183. [Google Scholar] [CrossRef]
- Fabbri, E.; Taylor, S.; Rabis, A.; Levecque, P.; Conrad, O.; Kötz, R.; Schmidt, T.J. The Effect of Platinum Nanoparticle Distribution on Oxygen Electroreduction Activity and Selectivity. ChemCatChem 2014, 6, 1410–1418. [Google Scholar] [CrossRef]
- Taylor, S.; Fabbri, E.; Levecque, P.; Schmidt, T.J.; Conrad, O. The Effect of Platinum Loading and Surface Morphology on Oxygen Reduction Activity. Electrocatalysis 2016, 7, 287–296. [Google Scholar] [CrossRef]
- Speder, J.; Spanos, I.; Zana, A.; Kirkensgaard, J.J.K.; Mortensen, K.; Altmann, L.; Bäumer, M.; Arenz, M. From Single Crystal Model Catalysts to Systematic Studies of Supported Nanoparticles. Surf. Sci. 2015, 631, 278–284. [Google Scholar] [CrossRef]
- Gattrell, M.; MacDougall, B. Reaction Mechanisms of the O2 Reduction/evolution Reaction. In Handbook of Fuel Cells; John Wiley & Sons, Ltd.: Chichester, UK, 2010. [Google Scholar]
- Borup, R.; Meyers, J.; Pivovar, B.; Kim, Y.S.; Mukundan, R.; Garland, N.; Myers, D.; Wilson, M.; Garzon, F.; Wood, D.; et al. Scientific Aspects of Polymer Electrolyte Fuel Cell Durability and Degradation. Chem. Rev. 2007, 107, 3904–3951. [Google Scholar] [CrossRef] [PubMed]
- Prater, K.B.; Bard, A.J. Rotating Ring-Disk Electrodes I. Fundamentals of the Digital Simulation Approach. Disk and Ring Transients and Collection Efficiencies. J. Electrochem. Soc. 1970, 117, 207–213. [Google Scholar] [CrossRef]
- Prater, K.B.; Bard, A.J. Rotating Ring-Disk Electrodes III. Catalytic and ECE Reactions. J. Electrochem. Soc. 1970, 117, 1517–1520. [Google Scholar] [CrossRef]
- Prater, K.B.; Bard, A.J. Rotating Ring-Disk Electrodes II. Digital Simulation of First and Second-Order Following Chemical Reactions. J. Electrochem. Soc. 1970, 117, 335–340. [Google Scholar] [CrossRef]
- Lee, S.-J.; Pyun, S.-I.; Lee, S.-K.; Kang, S.-J.L. Fundamentals of Rotating Disc and Ring-Disc Electrode Techniques and Their Applications to Study of the Oxygen Reduction Mechanism at Pt/C Electrode for Fuel Cells. Isr. J. Chem. 2008, 48, 215–228. [Google Scholar] [CrossRef]
- Zhang, C.; Fan, F.-R.F.; Bard, A.J. Electrochemistry of Oxygen in Concentrated NaOH Solutions: Solubility, Diffusion Coefficients, and Superoxide Formation. J. Am. Chem. Soc. 2009, 131, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.S.; Dzombak, D.A.; Luthy, R.G. Equilibrium Precipitation and Dissolution of Iron Cyanide Solids in Water. Environ. Eng. Sci. 1999, 16, 293–313. [Google Scholar] [CrossRef]
- Arellano, C.A.P.; Martínez, S.S. Effects of pH on the Degradation of Aqueous Ferricyanide by Photolysis and Photocatalysis under Solar Radiation. Sol. Energy Mater. Sol. Cells 2010, 94, 327–332. [Google Scholar] [CrossRef]
- Paulus, U.A.; Schmidt, T.J.; Gasteiger, H.A.; Behm, R.J. Oxygen Reduction on a High-Surface Area Pt/Vulcan Carbon Catalyst: A Thin-Film Rotating Ring-Disk Electrode Study. J. Electroanal. Chem. 2001, 495, 134–145. [Google Scholar] [CrossRef]
- Treimer, S.; Tang, A.; Johnson, D.C. A Consideration of the Application of Koutecky-Levich Plots in the Diagnoses of Charge-Transfer Mechanisms at Rotated Disk Electrodes. Electroanalysis 2002, 14, 165–171. [Google Scholar] [CrossRef]
- Damjanovic, A.; Genshaw, M.A.; Bockris, J.O. Distinction between Intermediates Produced in Main and Side Electrodic Reactions. J. Chem. Phys. 1966, 45, 4057–4059. [Google Scholar] [CrossRef]
- Appleby, A.J.; Savy, M. Kinetics of Oxygen Reduction Reactions Involving Catalytic Decomposition of Hydrogen Peroxide: Application to Porous and Rotating Ring-Disk Electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1978, 92, 15–30. [Google Scholar] [CrossRef]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Shao-Horn, Y. Electrocatalytic Measurement Methodology of Oxide Catalysts Using a Thin-Film Rotating Disk Electrode. J. Electrochem. Soc. 2010, 157, B1263–B1268. [Google Scholar] [CrossRef]
- Trasatti, S.; Petrii, O.A. Real Surface Area Measurements in Electrochemistry. J. Electroanal. Chem. 1992, 327, 353–376. [Google Scholar] [CrossRef]
- Kan, D.; Orikasa, Y.; Nitta, K.; Tanida, H.; Kurosaki, R.; Nishimura, T.; Sasaki, T.; Guo, H.; Ozaki, Y.; Uchimoto, Y.; et al. Overpotential-Induced Introduction of Oxygen Vacancy in La0.67Sr0.33MnO3 Surface and Its Impact on Oxygen Reduction Reaction Catalytic Activity in Alkaline Solution. J. Phys. Chem. C 2016, 120, 6006–6010. [Google Scholar] [CrossRef]
- Miyahara, Y.; Miyazaki, K.; Fukutsuka, T.; Abe, T. Catalytic Roles of Perovskite Oxides in Electrochemical Oxygen Reactions in Alkaline Media. J. Electrochem. Soc. 2014, 161, F694–F697. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Choi, W.S.; Jeen, H.; Lee, H.N.; Shao-Horn, Y. Role of Strain and Conductivity in Oxygen Electrocatalysis on LaCoO3 Thin Films. J. Phys. Chem. Lett. 2015, 6, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Lippert, T.; Montenegro, M.J.; Döbeli, M.; Müller, S.; Willmott, P.R.; Wokaun, A. Perovskite Thin Films Deposited by Pulsed Laser Ablation as Model Systems for Electrochemical Applications. Prog. Solid State Chem. 2007, 35, 221–231. [Google Scholar] [CrossRef]
- Komo, M.; Hagiwara, A.; Taminato, S.; Hirayama, M.; Kanno, R. Oxygen Evolution and Reduction Reactions on La0.8Sr0.2CoO3 (001), (110), and (111) Surfaces in an Alkaline Solution. Electrochemistry 2012, 80, 834–838. [Google Scholar] [CrossRef]
- Petrie, J.R.; Cooper, V.R.; Freeland, J.W.; Meyer, T.L.; Zhang, Z.; Lutterman, D.A.; Lee, H.N. Enhanced Bifunctional Oxygen Catalysis in Strained LaNiO3 Perovskites. J. Am. Chem. Soc. 2016, 138, 2488–2491. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Anoop, G.; Lee, H.J.; Lee, S.S.; Kwak, J.H.; Lee, H.J.; Jo, J.Y. Facile Synthesis of Perovskite LaMnO3+δ Nanoparticles for the Oxygen Reduction Reaction. J. Catal. 2016, 344, 578–582. [Google Scholar] [CrossRef]
- Risch, M.; Stoerzinger, K.A.; Maruyama, S.; Hong, W.T.; Takeuchi, I.; Shao-Horn, Y. La0.8Sr0.2MnO3−δ Decorated with Ba0.5Sr0.5Co0.8Fe0.2O3−δ: A Bi-Functional Surface for Oxygen Electrocatalysis with Enhanced Stability and Activity. J. Am. Chem. Soc. 2014, 136, 5229–5232. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Sato, E. Electrochemical Properties of the Single Crystal La0.7Pb0.3MnO3 Electrode. Electrochim. Acta 1980, 25, 585–589. [Google Scholar] [CrossRef]
- Markovic, N.M.; Gasteiger, H.A.; Ross, P.N. Oxygen Reduction on Platinum Low-Index Single-Crystal Surfaces in Alkaline Solution: Rotating Ring DiskPt(hkl) Studies. J. Phys. Chem. 1996, 100, 6715–6721. [Google Scholar]
- Molina-García, M.A.; Rees, N.V. Effect of Catalyst Carbon Supports on the Oxygen Reduction Reaction in Alkaline Media: A Comparative Study. RSC Adv. 2016, 6, 94669–94681. [Google Scholar] [CrossRef]
- Mohamed, R.; Fabbri, E.; Levecque, P.; Kötz, R.; Schmidt, T.J.; Conrad, O. Understanding the Influence of Carbon on the Oxygen Reduction and Evolution Activities of BSCF/Carbon Composite Electrodes in Alkaline Electrolyte. ECS Trans. 2014, 58, 9–18. [Google Scholar] [CrossRef]
- Fabbri, E.; Mohamed, R.; Levecque, P.; Conrad, O.; Kötz, R.; Schmidt, T.J. Composite Electrode Boosts the Activity of Ba0.5Sr0.5Co0.8Fe0.2O3−δ Perovskite and Carbon toward Oxygen Reduction in Alkaline Media. ACS Catal. 2014, 4, 1061–1070. [Google Scholar] [CrossRef]
- Vesborg, P.C.K.; Jaramillo, T.F. Addressing the Terawatt Challenge: Scalability in the Supply of Chemical Elements for Renewable Energy. RSC Adv. 2012, 2, 7933–7947. [Google Scholar] [CrossRef]
- Zeng, Z.; Calle-Vallejo, F.; Mogensen, M.B.; Rossmeisl, J. Generalized Trends in the Formation Energies of Perovskite Oxides. Phys. Chem. Chem. Phys. 2013, 15, 7526–7533. [Google Scholar] [CrossRef] [PubMed]
- Christ, J.; Ngo, C.; Batson, T.; Cadigan, C.; Tong, J.; Richards, R.; O’Hayre, R.; Pylypenko, S. Synthesis of High Surface Area CaxLa(1−X)Al(1−X)MnxO(3−δ) Perovskite Oxides for Oxygen Reduction Electrocatalysis in Alkaline Media. Catal. Sci. Technol. 2016, 6, 7744–7751. [Google Scholar] [CrossRef]
- Fabbri, E.; Mohamed, R.; Levecque, P.; Conrad, O.; Kötz, R.; Schmidt, T.J. Ba0.5Sr0.5Co0.8Fe0.2O3−δ Perovskite Activity towards the Oxygen Reduction Reaction in Alkaline Media. ChemElectroChem 2014, 1, 338–342. [Google Scholar] [CrossRef]
- Shimizu, Y.; Uemura, K.; Miura, N.; Yamazoe, N. Gas-Diffusion Electrodes for Oxygen Reduction Loaded with Large Surface Area La1−xCaxMO3 (M = Co, Mn). Chem. Lett. 1988, 17, 1979–1982. [Google Scholar] [CrossRef]
- Yuasa, M.; Shimanoe, K.; Teraoka, Y.; Yamazoe, N. High-Performance Oxygen Reduction Catalyst Using Carbon-Supported La-Mn-Based Perovskite-Type Oxide. Electrochem. Solid-State Lett. 2011, 14, A67. [Google Scholar] [CrossRef]
- Hsueh, K.-L.; Chin, D.-T.; Srinivasan, S. Electrode Kinetics of Oxygen Reduction: A Theoretical and Experimental Analysis of the Rotating Ring-Disc Electrode Method. J. Electroanal. Chem. Interfacial Electrochem. 1983, 153, 79–95. [Google Scholar] [CrossRef]
- Adzic, R.R.; Wang, J.X. Configuration and Site of O2 Adsorption on the Pt(111) Electrode Surface. J. Phys. Chem. B 1998, 102, 8988–8993. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Anderson, A.B. Insights into Electrocatalysis. Phys. Chem. Chem. Phys. 2012, 14, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.B.; Albu, T.V. Ab Initio Determination of Reversible Potentials and Activation Energies for Outer-Sphere Oxygen Reduction to Water and the Reverse Oxidation Reaction. J. Am. Chem. Soc. 1999, 121, 11855–11863. [Google Scholar] [CrossRef]
- Sidik, R.A.; Anderson, A.B. Density Functional Theory Study of O2 Electroreduction When Bonded to a Pt Dual Site. J. Electroanal. Chem. 2002, 528, 69–76. [Google Scholar] [CrossRef]
- Anderson, A.B.; Albu, T.V. Catalytic Effect of Platinum on Oxygen Reduction An Ab Initio Model Including Electrode Potential Dependence. J. Electrochem. Soc. 2000, 147, 4229–4238. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Nørskov, J.K.; Taylor, C.D.; Janik, M.J.; Neurock, M. Calculated Phase Diagrams for the Electrochemical Oxidation and Reduction of Water over Pt(111). J. Phys. Chem. B 2006, 110, 21833–21839. [Google Scholar] [CrossRef] [PubMed]
- Janik, M.J.; Taylor, C.D.; Neurock, M. First-Principles Analysis of the Initial Electroreduction Steps of Oxygen over Pt(111). J. Electrochem. Soc. 2009, 156, B126–B135. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, H.-P. Oxygen Reduction Activity on Perovskite Oxide Surfaces: A Comparative First-Principles Study of LaMnO3, LaFeO3, and LaCrO3. J. Phys. Chem. C 2013, 117, 2106–2112. [Google Scholar] [CrossRef]
- Ahmad, E.A.; Tileli, V.; Kramer, D.; Mallia, G.; Stoerzinger, K.A.; Shao-Horn, Y.; Kucernak, A.R.; Harrison, N.M. Optimizing Oxygen Reduction Catalyst Morphologies from First Principles. J. Phys. Chem. C 2015, 119, 16804–16810. [Google Scholar] [CrossRef]
- Ahmad, E.A.; Mallia, G.; Kramer, D.; Kucernak, A.R.; Harrison, N.M. The Stability of LaMnO3 Surfaces: A Hybrid Exchange Density Functional Theory Study of an Alkaline Fuel Cell Catalyst. J. Mater. Chem. A 2013, 1, 11152–11162. [Google Scholar] [CrossRef]
- Zhou, Y.; Lü, Z.; Wei, B.; Xu, S.; Xu, D.; Yang, Z. The Comparative Theoretical Study of the LaBO3 (B = Mn, Fe, Co, and Ni) Surface Properties and Oxygen Adsorption Mechanisms. Ionics 2016, 22, 1153–1158. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Tripković, V.; Tritsaris, G.A.; Calle-Vallejo, F. Electro-Catalysis of Oxygen Reduction Reaction. ECS Trans. 2010, 33, 43–50. [Google Scholar]
- Carbonio, R.E.; Fierro, C.; Tryk, D.; Scherson, D.; Yeager, E. Perovskite-Type Oxides: Oxygen Electrocatalysis and Bulk Structure. J. Power Sources 1988, 22, 387–398. [Google Scholar] [CrossRef]
- Lee, Y.N.; Lago, R.M.; Fierro, J.L.G.; González, J. Hydrogen Peroxide Decomposition over Ln1−xAxMnO3 (Ln = La or Nd and A = K or Sr) Perovskites. Appl. Catal. A Gen. 2001, 215, 245–256. [Google Scholar] [CrossRef]
- Zhang, H. Oxygen Sorption and Catalytic Properties of La1−xSrxCo1−yFeyO3 Perovskite-Type Oxides. J. Catal. 1990, 121, 432–440. [Google Scholar] [CrossRef]
- Ryabova, A.S.; Napolskiy, F.S.; Poux, T.; Istomin, S.Y.; Bonnefont, A.; Antipin, D.M.; Baranchikov, A.Y.; Levin, E.E.; Abakumov, A.M.; Kéranguéven, G.; et al. Rationalizing the Influence of the Mn(IV)/Mn(III) Red-Ox Transition on the Electrocatalytic Activity of Manganese Oxides in the Oxygen Reduction Reaction. Electrochim. Acta 2016, 187, 161–172. [Google Scholar] [CrossRef]
- Gorlin, Y.; Lassalle-Kaiser, B.; Benck, J.D.; Gul, S.; Webb, S.M.; Yachandra, V.K.; Yano, J.; Jaramillo, T.F. In Situ X-Ray Absorption Spectroscopy Investigation of a Bifunctional Manganese Oxide Catalyst with High Activity for Electrochemical Water Oxidation and Oxygen Reduction. J. Am. Chem. Soc. 2013, 135, 8525–8534. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.M.; Gil, A.F.; Galicia, L.; González, I. Understanding the Difference between Inner- and Outer-Sphere Mechanisms: An Electrochemical Experiment. J. Chem. Educ. 1996, 73, 808. [Google Scholar] [CrossRef]
- Ramaswamy, N.; Mukerjee, S. Fundamental Mechanistic Understanding of Electrocatalysis of Oxygen Reduction on Pt and Non-Pt Surfaces: Acid versus Alkaline Media. Adv. Phys. Chem. 2012, 2012, 1–17. [Google Scholar] [CrossRef]
- Ramaswamy, N.; Mukerjee, S. Influence of Inner-and Outer-Sphere Electron Transfer Mechanisms during Electrocatalysis of Oxygen Reduction in Alkaline Media. J. Phys. Chem. C 2011, 115, 18015–18026. [Google Scholar] [CrossRef]
- Vezzù, K.; Bach Delpeuch, A.; Negro, E.; Polizzi, S.; Nawn, G.; Bertasi, F.; Pagot, G.; Artyushkova, K.; Atanassov, P.; Di Noto, V. Fe-Carbon Nitride “Core-Shell” Electrocatalysts for the Oxygen Reduction Reaction. Electrochim. Acta 2016, 222, 1778–1791. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Cushing, B.L.; Goodenough, J.B.; Cushing, B.L. Oxide-Based ORR Catalysts. In Handbook of Fuel Cells; John Wiley & Sons, Ltd.: Chichester, UK, 2010. [Google Scholar]
- Goodenough, J.B.; Manoharan, R.; Paranthaman, M. Surface Protonation and Electrochemical Activity of Oxides in Aqueous Solution. J. Am. Chem. Soc. 1990, 112, 2076–2082. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of Water on (Oxidized) Metal Surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Martínez, J.I.; García-Lastra, J.M.; Rossmeisl, J.; Koper, M.T.M. Physical and Chemical Nature of the Scaling Relations between Adsorption Energies of Atoms on Metal Surfaces. Phys. Rev. Lett. 2012, 108, 116103. [Google Scholar] [CrossRef] [PubMed]
- Fernández, E.M.; Moses, P.G.; Toftelund, A.; Hansen, H.A.; Martínez, J.I.; Abild-Pedersen, F.; Kleis, J.; Hinnemann, B.; Rossmeisl, J.; Bligaard, T.; et al. Scaling Relationships for Adsorption Energies on Transition Metal Oxide, Sulfide, and Nitride Surfaces. Angew. Chem. Int. Ed. Engl. 2008, 47, 4683–4686. [Google Scholar] [CrossRef] [PubMed]
- Montemore, M.M.; Medlin, J.W. Scaling Relations between Adsorption Energies for Computational Screening and Design of Catalysts. Catal. Sci. Technol. 2014, 4, 3748–3761. [Google Scholar] [CrossRef]
- Abild-Pedersen, F.; Greeley, J.; Studt, F.; Rossmeisl, J.; Munter, T.R.; Moses, P.G.; Skúlason, E.; Bligaard, T.; Nørskov, J.K. Scaling Properties of Adsorption Energies for Hydrogen-Containing Molecules on Transition-Metal Surfaces. Phys. Rev. Lett. 2007, 99, 16105. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, J.; Qu, Z.-W.; Zhu, H.; Kroes, G.-J.; Nørskov, J.K. Electrolysis of Water on Oxide Surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Koper, M.T.M. Thermodynamic Theory of Multi-Electron Transfer Reactions: Implications for Electrocatalysis. J. Electroanal. Chem. 2011, 660, 254–260. [Google Scholar] [CrossRef]
- Koper, M.T.M. Theory of Multiple Proton–electron Transfer Reactions and Its Implications for Electrocatalysis. Chem. Sci. 2013, 4, 2710–2723. [Google Scholar] [CrossRef]
- Viswanathan, V.; Hansen, H.A. Unifying Solution and Surface Electrochemistry: Limitations and Opportunities in Surface Electrocatalysis. Top. Catal. 2013, 57, 215–221. [Google Scholar] [CrossRef]
- Dau, H.; Limberg, C.; Reier, T.; Risch, M.; Roggan, S.; Strasser, P. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem 2010, 2, 724–761. [Google Scholar] [CrossRef]
- Halck, N.B.; Petrykin, V.; Krtil, P.; Rossmeisl, J. Beyond the Volcano Limitations in Electrocatalysis—Oxygen Evolution Reaction. Phys. Chem. Chem. Phys. 2014, 16, 13682–13688. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Halck, N.B.; Kramm, U.; Siahrostami, S.; Krtil, P.; Rossmeisl, J. Beyond the Top of the Volcano?—A Unified Approach to Electrocatalytic Oxygen Reduction and Oxygen Evolution. Nano Energy 2016, 29, 126–135. [Google Scholar] [CrossRef]
- Vojvodic, A.; Nørskov, J.K. New Design Paradigm for Heterogeneous Catalysts. Natl. Sci. Rev. 2015, 2, 140–149. [Google Scholar] [CrossRef]
- Goldstein, J.R.; Tseung, A.C.C. A Joint Pseudo-splitting/Peroxide Mechanism for Oxygen Reduction at Fuel Cell Cathodes. Nature 1969, 222, 869–870. [Google Scholar] [CrossRef]
- Evans, U.R. Cathodic Reduction of Oxygen in Fuel Cells and Corrosion Cells. Nature 1968, 218, 602–603. [Google Scholar] [CrossRef]
- Yeager, E. Mechanisms of Electrochemical Reactions on Non-Metallic Surfaces. In Cathode Catalysts for Oxygen Reduction; National Bureau of Standards Special Publication 455: Gaithersburg, MD, USA, 1976. [Google Scholar]
- Yeager, E. Electrocatalysts for O2 Reduction. Electrochim. Acta 1984, 29, 1527–1537. [Google Scholar] [CrossRef]
- Tilley, R.J.D. Magnetic Properties. In Perovskites; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 217–246. [Google Scholar]
- Paraskevopoulos, M.; Mayr, F.; Hemberger, J.; Loidl, A.; Heichele, R.; Maurer, D.; Müller, V.; Mukhin, A.A.; Balbashov, A.M. Magnetic Properties and the Phase Diagram of La1−xSrxMnO3 for X ≤ 0.2. J. Phys. Condens. Matter 2000, 12, 3993–4011. [Google Scholar] [CrossRef]
- Jirák, Z.; Krupička, S.; Šimša, Z.; Dlouhá, M.; Vratislav, S. Neutron Diffraction Study of Pr1−xCaxMnO3 Perovskites. J. Magn. Magn. Mater. 1985, 53, 153–166. [Google Scholar] [CrossRef]
- Yang, C.; Grimaud, A. Factors Controlling the Redox Activity of Oxygen in Perovskites: From Theory to Application for Catalytic Reactions. Catalysts 2017, 7, 149. [Google Scholar] [CrossRef]
- Mefford, J.T.; Rong, X.; Abakumov, A.M.; Hardin, W.G.; Dai, S.; Kolpak, A.M.; Johnston, K.P.; Stevenson, K.J. Water Electrolysis on La1−xSrxCoO3−δ Perovskite Electrocatalysts. Nat. Commun. 2016, 7, 11053. [Google Scholar] [CrossRef] [PubMed]
- Grimaud, A.; Hong, W.T.; Shao-Horn, Y.; Tarascon, J.-M. Anionic Redox Processes for Electrochemical Devices. Nat. Mater. 2016, 15, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Grimaud, A.; Diaz-Morales, O.; Han, B.; Hong, W.T.; Lee, Y.-L.; Giordano, L.; Stoerzinger, K.A.; Koper, M.T.M.; Shao-Horn, Y. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 2017, 9, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Nemudry, A.; Goldberg, E.L.; Aguirre, M.; Alario-Franco, M.Á. Electrochemical Topotactic Oxidation of Nonstoichiometric Perovskites at Ambient Temperature. Solid State Sci. 2002, 4, 677–690. [Google Scholar] [CrossRef]
- Grenier, J.C.; Wattiaux, A.; Doumerc, J.P.; Dordor, P.; Fournes, L.; Chaminade, J.P.; Pouchard, M. Electrochemical Oxygen Intercalation into Oxide Networks. J. Solid State Chem. 1992, 96, 20–30. [Google Scholar] [CrossRef]
- Hong, W.T.; Welsch, R.E.; Shao-Horn, Y. Descriptors of Oxygen-Evolution Activity for Oxides: A Statistical Evaluation. J. Phys. Chem. C 2016, 120, 78–86. [Google Scholar] [CrossRef]
- Viswanathan, V.; Hansen, H.A.; Rossmeisl, J.; Nørskov, J.K. Unifying the 2e− and 4e− Reduction of Oxygen on Metal Surfaces. J. Phys. Chem. Lett. 2012, 3, 2948–2951. [Google Scholar] [CrossRef] [PubMed]
- Suntivich, J.; Shao-Horn, Y. Trend in Oxygen Reduction Reaction on Transition Metal Oxide Surfaces. ECS Trans. 2013, 58, 715–726. [Google Scholar] [CrossRef]
- Yokoi, Y.; Uchida, H. Catalytic Activity of Perovskite-Type Oxide Catalysts for Direct Decomposition of NO: Correlation between Cluster Model Calculations and Temperature-Programmed Desorption Experiments. Catal. Today 1998, 42, 167–174. [Google Scholar] [CrossRef]
- Suntivich, J.; Hong, W.T.; Lee, Y.-L.; Rondinelli, J.M.; Yang, W.; Goodenough, J.B.; Dabrowski, B.; Freeland, J.W.; Shao-Horn, Y. Estimating Hybridization of Transition Metal and Oxygen States in Perovskites from O K -Edge X-Ray Absorption Spectroscopy. J. Phys. Chem. C 2014, 118, 1856–1863. [Google Scholar] [CrossRef]
- Ebrahimizadeh Abrishami, M.; Risch, M.; Scholz, J.; Roddatis, V.; Osterthun, N.; Jooss, C. Oxygen Evolution at Manganite Perovskite Ruddlesden-Popper Type Particles: Trends of Activity on Structure, Valence and Covalence. Materials 2016, 9, 921. [Google Scholar] [CrossRef]
- Allred, A.L. Electronegativity Values from Thermochemical Data. J. Inorg. Nucl. Chem. 1961, 17, 215–221. [Google Scholar] [CrossRef]
- Bronoel, G.; Grenier, J.C.; Reby, J. Comparative Behaviour of Various Oxides in the Electrochemical Reactions of Oxygen Evolution and Reduction in Alkaline Medium. Electrochim. Acta 1980, 25, 1015–1018. [Google Scholar] [CrossRef]
- Fletcher, S. Tafel Slopes from First Principles. J. Solid State Electrochem. 2008, 13, 537–549. [Google Scholar] [CrossRef]
- Escudero-Escribano, M.; Malacrida, P.; Hansen, M.H.; Vej-Hansen, U.G.; Velázquez-Palenzuela, A.; Tripkovic, V.; Schiøtz, J.; Rossmeisl, J.; Stephens, I.E.L.; Chorkendorff, I. Tuning the Activity of Pt Alloy Electrocatalysts by Means of the Lanthanide Contraction. Science 2016, 352, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Loferski, P. 2014 Minerals Yearbook—Platinum Group Metals; United States Geological Survey: Reston, VA, USA, 2016.
- Dagotto, E. When Oxides Meet Face to Face. Science 2007, 318, 1076–1077. [Google Scholar] [CrossRef] [PubMed]
- Mannhart, J.; Schlom, D.G. Oxide Interfaces—An Opportunity for Electronics. Science 2010, 327, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; de Respinis, M.; Frei, H. Time-Resolved Observations of Water Oxidation Intermediates on a Cobalt Oxide Nanoparticle Catalyst. Nat. Chem. 2014, 6, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-F.; Kooyman, P.J.; Koper, M.T.M.; Bodappa, N.; Wandlowski, T. Intermediate Stages of Electrochemical Oxidation of Single-Crystalline Platinum Revealed by in Situ Raman Spectroscopy. Nat. Commun. 2016, 7, 12440. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-Z.; Prabhudev, S.; Yang, J.; Gabardo, C.M.; Botton, G.A.; Soleymani, L. In Situ Liquid Cell TEM Study of Morphological Evolution and Degradation of Pt–Fe Nanocatalysts During Potential Cycling. J. Phys. Chem. C 2014, 118, 22111–22119. [Google Scholar] [CrossRef]
- Hodnik, N.; Dehm, G.; Mayrhofer, K.J.J. Importance and Challenges of Electrochemical in Situ Liquid Cell Electron Microscopy for Energy Conversion Research. Acc. Chem. Res. 2016, 49, 2015–2022. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Risch, M.; Stoerzinger, K.A.; Wartner, G.; Shao-Horn, Y.; Jooss, C. Rotating Ring–Disk Electrode Study of Oxygen Evolution at a Perovskite Surface: Correlating Activity to Manganese Concentration. J. Phys. Chem. C 2016, 120, 27746–27756. [Google Scholar] [CrossRef]
- Scholz, J.; Risch, M.; Wartner, G.; Luderer, C.; Roddatis, V.; Jooss, C. Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemsitry. Catalysts 2017, 7, 139. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, L.; Mai, L.; Han, C.; An, Q.; Xu, X.; Liu, X.; Zhang, Q. Hierarchical Mesoporous Perovskite La0.5Sr0.5CoO2.91 Nanowires with Ultrahigh Capacity for Li-Air Batteries. Proc. Natl. Acad. Sci. USA 2012, 109, 19569–19574. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I. Accumulated Table of Bond Valence Parameters. Available online: http://www.iucr.org/__data/assets/file/0007/126574/bvparm2016.cif (accessed on 12 May 2017).
Figure 1.
(a) Illustration of a unit cell of the perovskite structure. (b) A cubic perovskite. (c) A perovskite with tilted and distorted octahedral and (d) A perovskite with both cation and anion ordering (a double perovskite is shown). View along y-direction in (b–d).
Figure 1.
(a) Illustration of a unit cell of the perovskite structure. (b) A cubic perovskite. (c) A perovskite with tilted and distorted octahedral and (d) A perovskite with both cation and anion ordering (a double perovskite is shown). View along y-direction in (b–d).

Figure 2.
Trends in catalytic activity towards oxygen reduction with (a) substitution of the B-site (data extracted from references [22,56,57]), (b) composition of the A-site (data extracted from references [63,64,65]), (c) substitution of the A-site (data extracted from reference [66]), and (d) oxygen non-stoichiometry (data extracted from references [22,67,68,69]). LaMnO3 (open symbols in panel b) was excluded from the trend as it is prone to oxygen non-stoichiometry. Chemical substitution also influences the bulk conductivity and tolerance factor (Table A1 and Table A2), as shown in examples for (e) substitution of the B-site and (f) composition of the A-site (conductivity data extracted from reference [62]) and (g) substitution of the A-site. Tolerance factors were calculated using the bond valence parameters in Table A3. (h) Cyclic voltammetry of the ferri-/ferrocyanide redox on LaMnO3 on Nb-doped STO (LMO|NSTO) showing rectifying behavior with thinning the LMO layer (numbers are LMO thickness in nm). Reproduced from reference [70]. Copyright 2013, Royal Society of Chemistry.
Figure 2.
Trends in catalytic activity towards oxygen reduction with (a) substitution of the B-site (data extracted from references [22,56,57]), (b) composition of the A-site (data extracted from references [63,64,65]), (c) substitution of the A-site (data extracted from reference [66]), and (d) oxygen non-stoichiometry (data extracted from references [22,67,68,69]). LaMnO3 (open symbols in panel b) was excluded from the trend as it is prone to oxygen non-stoichiometry. Chemical substitution also influences the bulk conductivity and tolerance factor (Table A1 and Table A2), as shown in examples for (e) substitution of the B-site and (f) composition of the A-site (conductivity data extracted from reference [62]) and (g) substitution of the A-site. Tolerance factors were calculated using the bond valence parameters in Table A3. (h) Cyclic voltammetry of the ferri-/ferrocyanide redox on LaMnO3 on Nb-doped STO (LMO|NSTO) showing rectifying behavior with thinning the LMO layer (numbers are LMO thickness in nm). Reproduced from reference [70]. Copyright 2013, Royal Society of Chemistry.
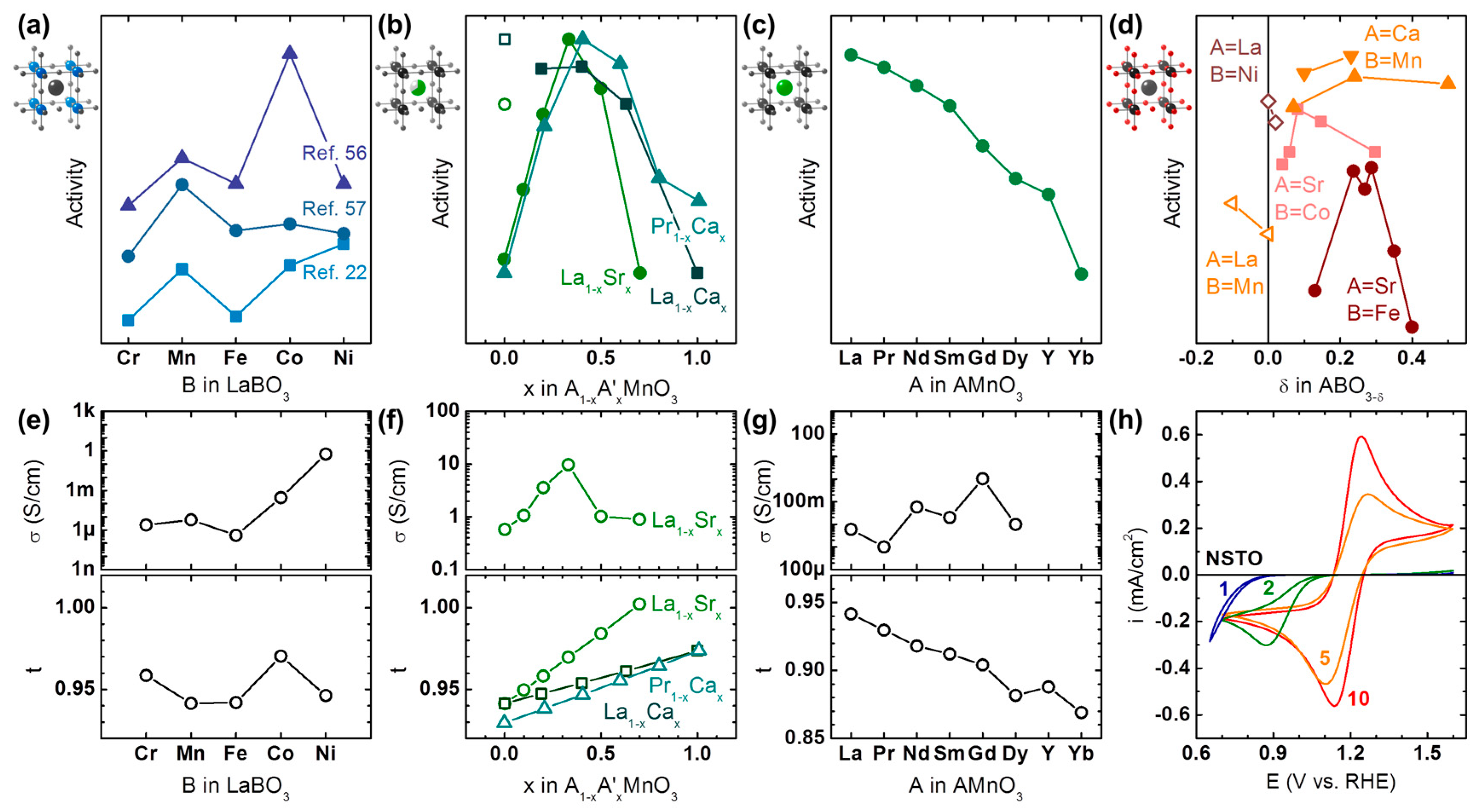
Figure 3.
Activity metrics for the oxygen reduction reaction at 0.85 V vs. RHE (380 mV overpotential) in alkaline media normalized by (a) the surface area of the oxides or Pt (data extracted from references [22,60,63,125,132,136]) and (b) by the mass loading of the oxides or Pt (data extracted from references [22,99,125,133]).
Figure 3.
Activity metrics for the oxygen reduction reaction at 0.85 V vs. RHE (380 mV overpotential) in alkaline media normalized by (a) the surface area of the oxides or Pt (data extracted from references [22,60,63,125,132,136]) and (b) by the mass loading of the oxides or Pt (data extracted from references [22,99,125,133]).
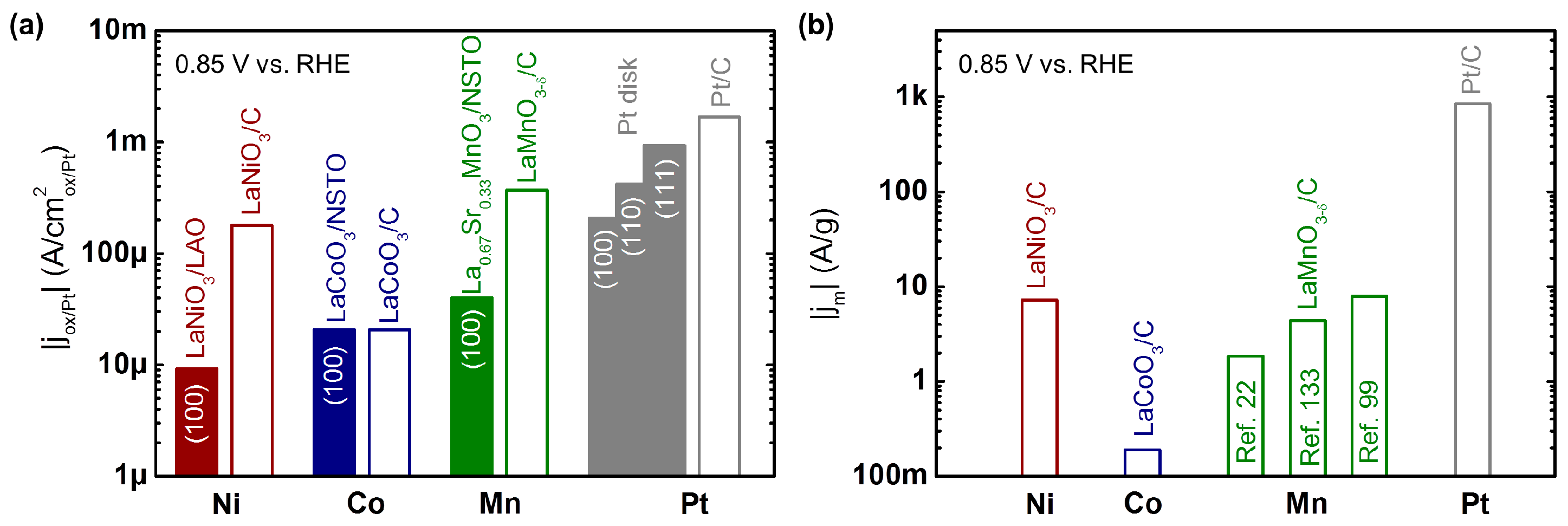
Figure 4.
The surface before and during the oxygen reduction reaction. (a) Influence of experimental hydroxide coverage on ORR activity of LaBO3 perovskites. Reproduced from reference [60]. Copyright 2015, American Chemical Society. (b) Surface Eh-pH (Pourbaix) diagram of the La-Mn-O-H system obtained by DFT calculations. Reproduced from reference [30]. Copyright 2015, American Chemical Society. (c) Variation of the surface coverage obtained from the rate constants of La0.8Sr0.2MnO3 in 1 M NaOH. Reproduced from reference [97]. Copyright 2014, Wiley and Sons.
Figure 4.
The surface before and during the oxygen reduction reaction. (a) Influence of experimental hydroxide coverage on ORR activity of LaBO3 perovskites. Reproduced from reference [60]. Copyright 2015, American Chemical Society. (b) Surface Eh-pH (Pourbaix) diagram of the La-Mn-O-H system obtained by DFT calculations. Reproduced from reference [30]. Copyright 2015, American Chemical Society. (c) Variation of the surface coverage obtained from the rate constants of La0.8Sr0.2MnO3 in 1 M NaOH. Reproduced from reference [97]. Copyright 2014, Wiley and Sons.
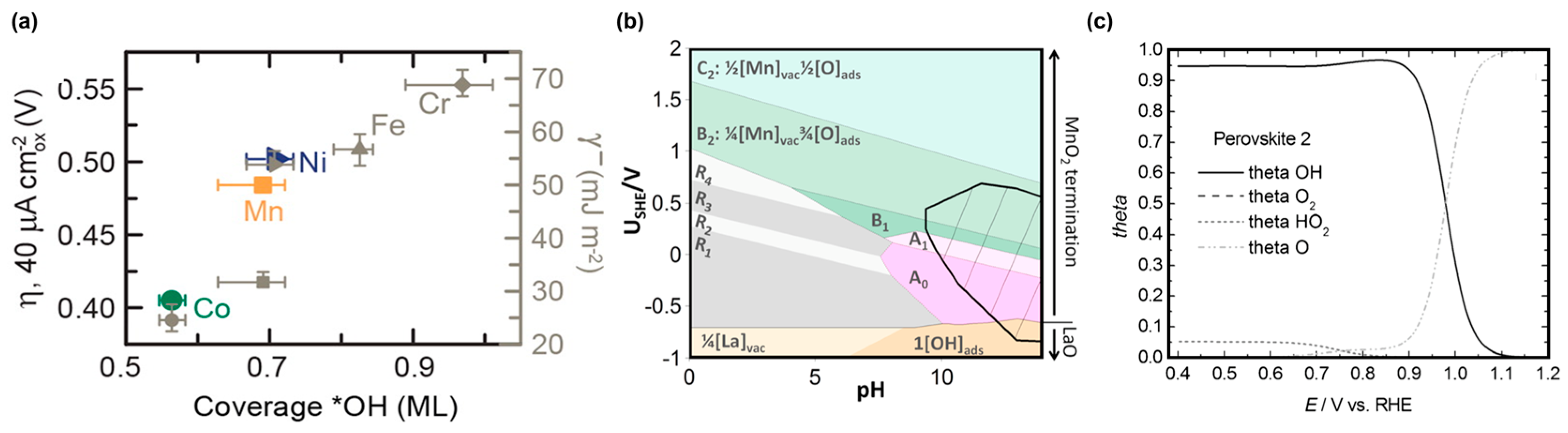
Figure 5.
Product yield of oxygen reduction on select perovskites. (a) Measurement of produced peroxide on composite LaBO3 electrodes containing Vulcan carbon. Reproduced from reference [57]. Copyright 2016, Wiley and Sons. (b) Dependence of the experimental peroxide yield on the loading of La0.8Sr0.2MnO3 and Sibunit carbon. Reproduced from reference [97]. Copyright 2014, Wiley and Sons. (c) Simulation of the contributions of La0.8Sr0.2MnO3 and Sibunit carbon to the total ORR current based on empirical rate constants. Reproduced from reference [97]. Copyright 2014, Wiley and Sons.
Figure 5.
Product yield of oxygen reduction on select perovskites. (a) Measurement of produced peroxide on composite LaBO3 electrodes containing Vulcan carbon. Reproduced from reference [57]. Copyright 2016, Wiley and Sons. (b) Dependence of the experimental peroxide yield on the loading of La0.8Sr0.2MnO3 and Sibunit carbon. Reproduced from reference [97]. Copyright 2014, Wiley and Sons. (c) Simulation of the contributions of La0.8Sr0.2MnO3 and Sibunit carbon to the total ORR current based on empirical rate constants. Reproduced from reference [97]. Copyright 2014, Wiley and Sons.
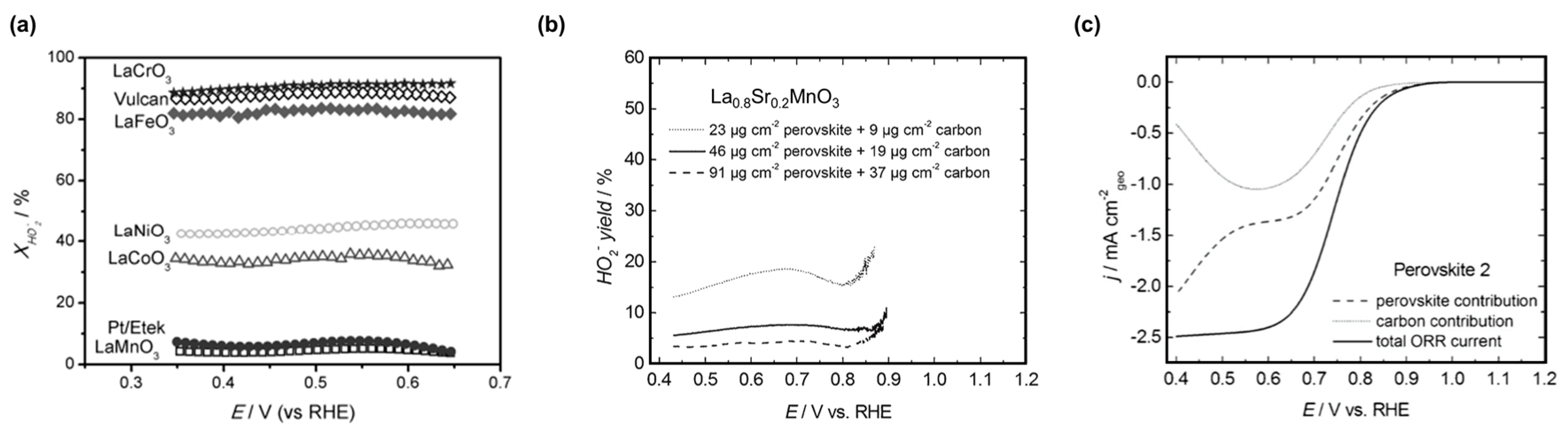
Figure 6.
Hypothetical proposals for electrolytic reduction of oxygen to hydroxide in alkaline media. Prototypical O2 adsorption in (a) end-on configuration, (b) side-on configuration and (c) bidentate configuration with participation of a lattice oxygen vacancy.
Figure 6.
Hypothetical proposals for electrolytic reduction of oxygen to hydroxide in alkaline media. Prototypical O2 adsorption in (a) end-on configuration, (b) side-on configuration and (c) bidentate configuration with participation of a lattice oxygen vacancy.
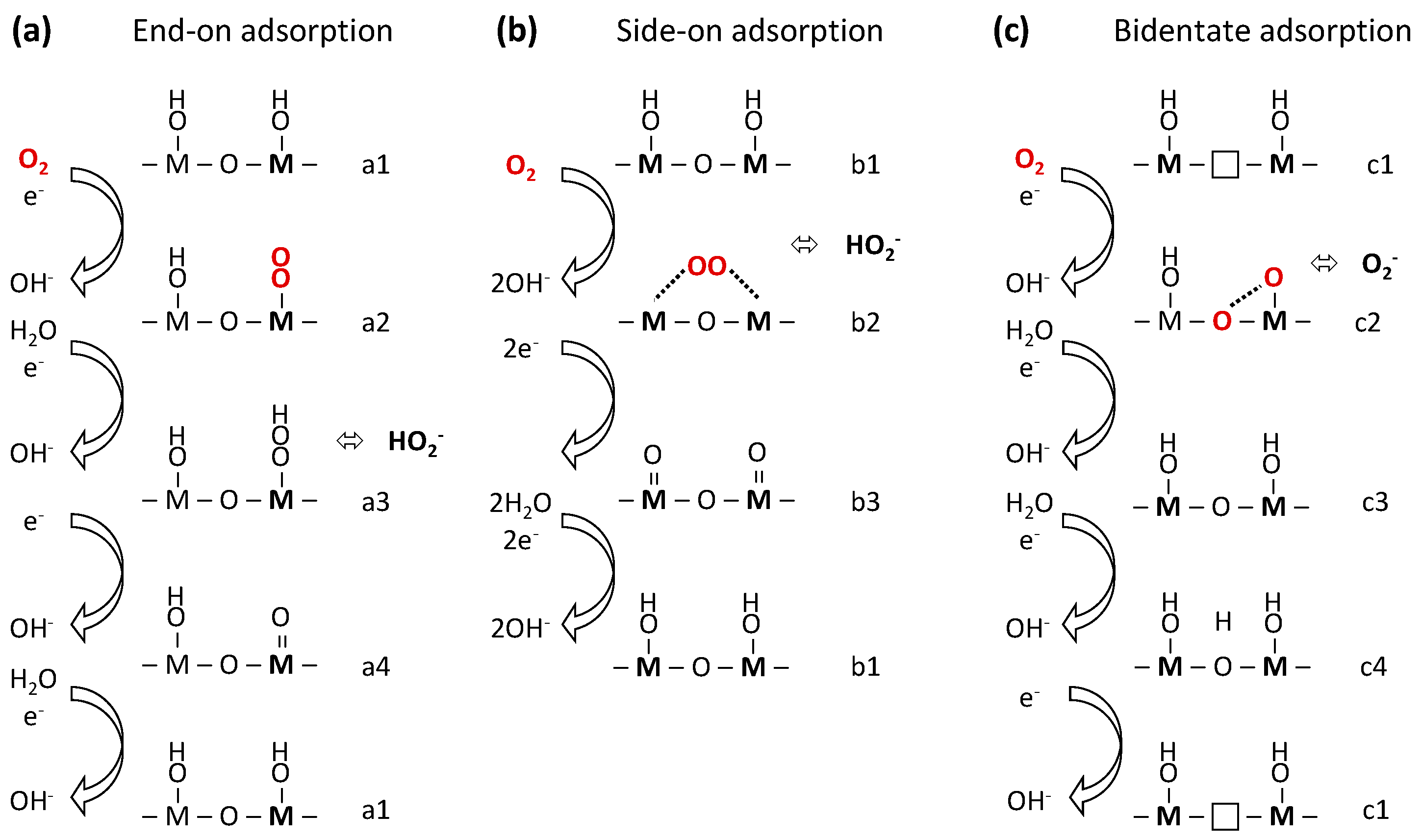
Figure 7.
Select structure–activity relationships of perovskites catalyzing the ORR. (a) The theoretical overpotential of oxygen reduction (ΔG) for the LaBO3 series terminated either by LaO (A-LaBO3) or BO2 (B-LaBO3) calculated as function of the formation of OH intermediates using the GGA + Ueff method. Reproduced from reference [59]. Copyright 2015, Royal Society of Chemistry. (b) Experimental voltage at 50 µA/cm2ox for powders of the LaBO3 perovskite series as function of experimental oxygen desorption temperature (solid circles) and an estimate for LaFeO3 (open circle). Reproduced from reference [199]. Copyright 2013, Electrochemical Society. (c) Experimental voltage at 50 µA/cm2ox as function of estimated eg occupancy of the LaBO3 series (solid circles) and other relevant perovskites (open circles). Reproduced from reference [22]. Copyright 2011, Nature Chemistry. (d) Experimental voltage at 50 µA/cm2ox as function of measured absorbance as a proxy of metal-oxygen covalence. Reproduced from reference [22]. Copyright 2011, Nature Chemistry. (e) The current at 0.4 V overpotential of epitaxial LaNiO3 thin films strained by growth on various substrates. Reprinted from reference [132]. Copyright 2016, American Chemical Society. (f) Current logarithm at −20 mV vs. Hg/HgO (in 6 M KOH) of perovskites in the series LaB’0.5Mn0.5O3 as function of their magnetic moment (data extracted from reference [61]).
Figure 7.
Select structure–activity relationships of perovskites catalyzing the ORR. (a) The theoretical overpotential of oxygen reduction (ΔG) for the LaBO3 series terminated either by LaO (A-LaBO3) or BO2 (B-LaBO3) calculated as function of the formation of OH intermediates using the GGA + Ueff method. Reproduced from reference [59]. Copyright 2015, Royal Society of Chemistry. (b) Experimental voltage at 50 µA/cm2ox for powders of the LaBO3 perovskite series as function of experimental oxygen desorption temperature (solid circles) and an estimate for LaFeO3 (open circle). Reproduced from reference [199]. Copyright 2013, Electrochemical Society. (c) Experimental voltage at 50 µA/cm2ox as function of estimated eg occupancy of the LaBO3 series (solid circles) and other relevant perovskites (open circles). Reproduced from reference [22]. Copyright 2011, Nature Chemistry. (d) Experimental voltage at 50 µA/cm2ox as function of measured absorbance as a proxy of metal-oxygen covalence. Reproduced from reference [22]. Copyright 2011, Nature Chemistry. (e) The current at 0.4 V overpotential of epitaxial LaNiO3 thin films strained by growth on various substrates. Reprinted from reference [132]. Copyright 2016, American Chemical Society. (f) Current logarithm at −20 mV vs. Hg/HgO (in 6 M KOH) of perovskites in the series LaB’0.5Mn0.5O3 as function of their magnetic moment (data extracted from reference [61]).
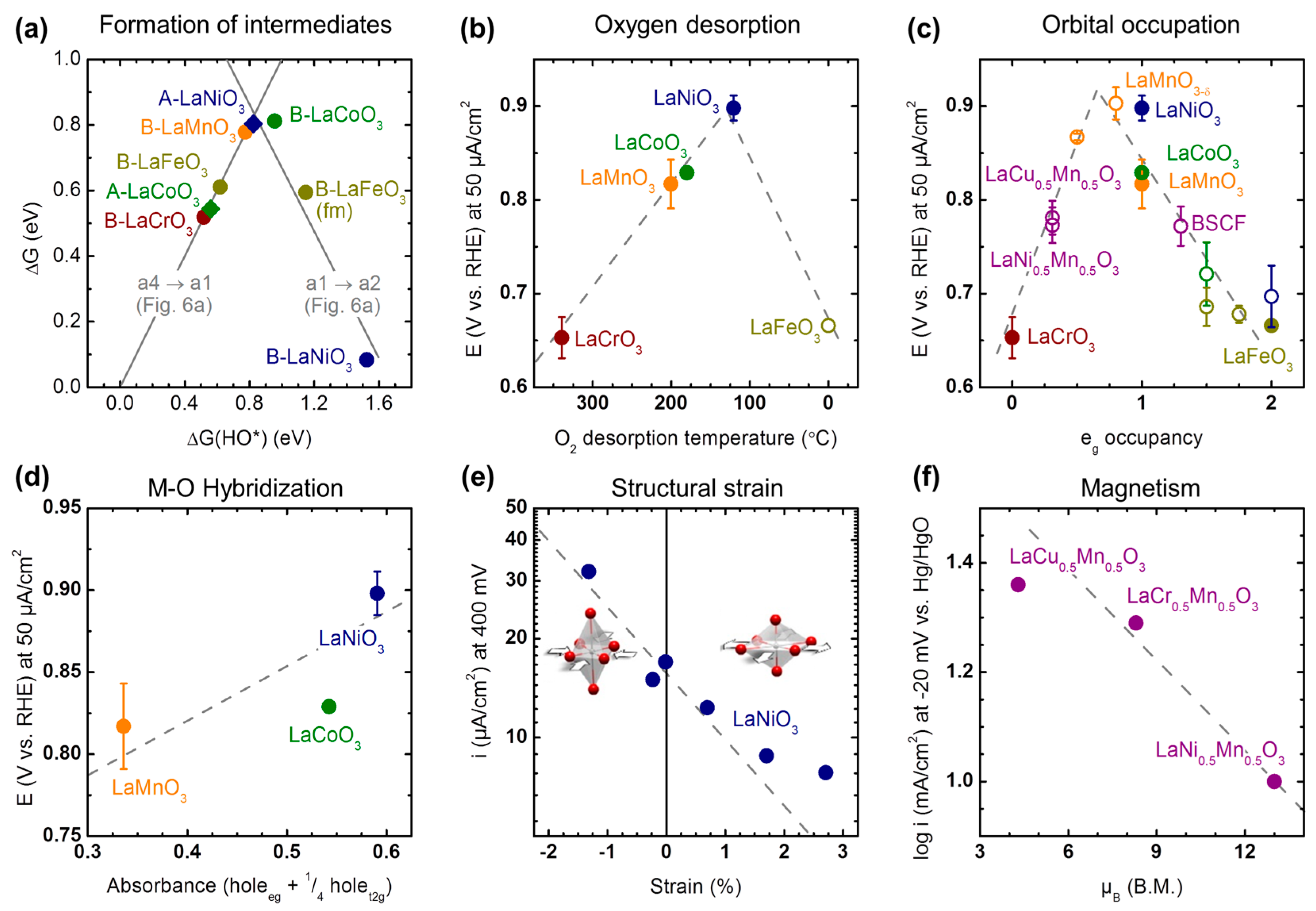
Table 1.
Requirements for oxides with high activity towards oxygen reduction.
| Year | Requirement | Type | Reference |
|---|---|---|---|
| 1970 | both n- and p-type conductivity (amphoterism) | C | [21] |
| 1970 | small band gap | C | [21] |
| 1973 | paramagnetism | M | [23,24] |
| 1977 | metallic conductivity | C | [17] |
| 1977 | occupied sigma band | C | [17] |
| 2011 | eg occupancy close to one | C | [22] |
| 2011 | high M-O covalence | C | [22] |
Types: C = chemical; M = magnetic.
Table 2.
Activity of perovskite thin films and select references normalized by the surface area of the oxide obtained by various common methods.
Table 2.
Activity of perovskite thin films and select references normalized by the surface area of the oxide obtained by various common methods.
| Material | Substrate/Support | Type | Orientation | |j| at 0.85 V vs. RHE a (µA/cm2ox) | Method b | Reference |
|---|---|---|---|---|---|---|
| LaMnO3 (10 nm) | Nb:SrTiO3 | Thin film | (100) | 15 | CV, 0 rpm | [70] |
| LaMnO3 | Pt | Thin film | poly | 33 *,c | CV, 1600 rpm | [128] |
| LaMnO3 | AB carbon | Composite | poly | 21 | CV, kinetic | [22] |
| LaMnO3+δ | AB carbon | Composite | poly | 350 | CV, kinetic | [22] |
| LaMnO3+δ | Vulcan carbon | Composite | poly | 20 | CV, kinetic | [133] |
| LaMnO3 | Nb:SrTiO3 | Thin film | (100) | 18 | CV, 0 rpm | [63] |
| LaMnO3 | Nb:SrTiO3 | Thin film | (100) | 6 | CV, 0 rpm | [63] |
| La0.9Sr0.1MnO3 | Nb:SrTiO3 | Thin film | (100) | 7 | CV, 0 rpm | [63] |
| La0.8Sr0.2MnO3 | Nb:SrTiO3 | Thin film | (100) | 16 | CV, 0 rpm | [63] |
| La0.8Sr0.2MnO3 | Pt | Thin film | poly | 71 *,c | CV, 1600 rpm | [128] |
| La0.8Sr0.2MnO3 | Nb:SrTiO3 | Thin film | (100) | 8 | CV, 0 rpm | [134] |
| Ba0.5Sr0.5Co0.8Fe0.2O3 on La0.8Sr0.2MnO3 d | Nb:SrTiO3 | Thin film | (100) | 28 | CV, 0 rpm | [134] |
| La0.67Sr0.33MnO3 | Nb:SrTiO3 | Thin film | (100) | 40 | CV, 0 rpm | [63] |
| La0.67Sr0.33MnO3 | Nb:SrTiO3 | Thin film | (100) | 37 | CA, 0 rpm | [63] |
| La0.67Sr0.33MnO3 | Nb:SrTiO3 | Thin film | (110) | 19 | CA, 0 rpm | [63] |
| La0.67Sr0.33MnO3 | Nb:SrTiO3 | Thin film | (111) | 20 | CA, 0 rpm | [63] |
| La0.5Sr0.5MnO3 | Nb:SrTiO3 | Thin film | (100) | 21 | CV, 0 rpm | [63] |
| La0.5Sr0.5MnO3 | Nb:SrTiO3 | Thin film | (100) | 21 | CA, 0 rpm | [63] |
| La0.5Sr0.5MnO3 | Nb:SrTiO3 | Thin film | (110) | 21 | CA, 0 rpm | [63] |
| La0.5Sr0.5MnO3 | Nb:SrTiO3 | Thin film | (111) | 23 | CA, 0 rpm | [63] |
| La0.3Sr0.7MnO3 | Nb:SrTiO3 | Thin film | (100) | 4 | CV, 0 rpm | [63] |
| La0.7Pb0.3MnO3 e | n/a | Single cryst | (100) | 28 * | CA, 0 rpm | [135] |
| LaCoO3 | Nb:SrTiO3 | Thin film | (100) | 21 | CV, 0 rpm | [60] |
| LaCoO3 | La0.67Sr0.33MnO3 on SrTiO3 | Thin film | (100) | 20 | CV, 0 rpm | [129] |
| LaCoO3 | La0.67Sr0.33MnO3 on LaAlO3 | Thin film | (100) | 13 | CV, 0 rpm | [129] |
| LaCoO3 | La0.67Sr0.33MnO3 on (LaAlO3)0.3 (Sr2AlTaO6)0.7 | Thin film | (100) | 33 | CV, 0 rpm | [129] |
| LaCoO3 | AB carbon | Composite | poly | 27 | CV, kinetic | [22] |
| La0.8Sr0.2CoO3 | Pt | Thin film | poly | 38 *,c | CV, 1600 rpm | [128] |
| La0.8Sr0.2CoO3 | SrTiO3 | Thin film | (100) | n/a *,f | n/a | [131] |
| La0.8Sr0.2CoO3 | SrTiO3 | Thin film | (110) | n/a *,f | n/a | [131] |
| La0.8Sr0.2CoO3 | SrTiO3 | Thin film | (111) | n/a *,f | n/a | [131] |
| La0.8Sr0.2CoO3 | n/a | Single cryst | (100) | ~0 * | CV, 1600 rpm | [96] |
| Vulcan carbon | La0.8Sr0.2CoO3 | Composite | (100) | 760 * | CV, 1600 rpm | [96] |
| La0.8Sr0.2CoO3 | n/a. | Single cryst | (110) | ~0 * | CV, 1600 rpm | [96] |
| La0.8Sr0.2CoO3 | n/a. | Single cryst | (111) | ~0 * | CV, 1600 rpm | [96] |
| La0.64Ca0.35Co0.95O3 | MgO | Thin film | (100) | 3 * | CV, 0 rpm | [130] |
| LaNiO3 | Nb:SrTiO3 | Thin film | (100) | 6 | CV, 0 rpm | [60] |
| LaNiO3 | AB carbon | Composite | poly | 185 | CV, kinetic | [22,125] |
| LaNiO3 | LaAlO3 | Thin film | (100) | 9 | CV, 0 rpm | [132] |
| LaNiO3 (relaxed) | LaAlO3 | Thin film | (100) | 6 | CV, 0 rpm | [132] |
| LaNiO3 (relaxed) | LaSrAlO4 | Thin film | (100) | 5 | CV, 0 rpm | [132] |
| LaNiO3 | (LaAlO3)0.3 (Sr2AlTaO6)0.7 | Thin film | (100) | 4 | CV, 0 rpm | [132] |
| LaNiO3 | SrTiO3 | Thin film | (100) | 3 | CV, 0 rpm | [132] |
| LaNiO3 | DyScO3 | Thin film | (100) | 2 | CV, 0 rpm | [132] |
Digitized from published data and linearly interpolated near 0.85 V vs. RHE. Only activities obtained by an identical method can be compared directly. a Electrolyte was 0.1 M KOH if not noted otherwise. * 1 M KOH. b rotation rate of electrode indicated or kinetic current if reported. The current can be considered kinetically limited below ~40 µA/cm2ox [70], so that the effect of rotation should be negligible for most of the thin films. c capacitive background subtracted. Surface roughness not quantified. d 50% BSCF coverage. e after eight voltage cycles. f a Pt pseudo reference was used and it is unclear if activity above background was observed. Abbreviations: RHE = reversible hydrogen electrode; CV = cyclic voltammetry; CA = chronoamperometry; rpm = revolutions per minute; AB carbon = acetylene black carbon.
Table 3.
Activity of perovskite thin films and select references normalized by the mass of the oxide.
Table 3.
Activity of perovskite thin films and select references normalized by the mass of the oxide.
| Material | Support | Specific Surface Area (m2/g) | |j| at 0.85 V vs. RHE a (A/g) | Method | Reference |
|---|---|---|---|---|---|
| LaMnO3 | Vulcan carbon | n/a | 7.9 * | CV, 900 rpm | [99] |
| LaMnO3 | Vulcan carbon | 26.8 | 1.4 | CV, 1600 rpm | [57] |
| LaMnO3+δ | Vulcan carbon | 22.1 | 4.3 | CV, kinetic | [133] |
| LaMnO3+δ | AB carbon | 0.5 | 1.9 | CV, kinetic | [22] |
| La1−xCaxAl1−xMnxO3−δ | Vulcan carbon | 6.1 | 3.5 | CV, 900 rpm b | [142] |
| LaCoO3 | AB carbon | 0.7 | 0.2 | CV, kinetic | [22] |
| LaCoO3 | Super P carbon | n/a | 0.2 | CV, 1600 rpm | [56] |
| LaNiO3 | AB carbon | 3.5 | 7.0 | CV, kinetic | [125] |
| LaNiO3−δ | Carbon black | ~3.8 | 0.2 | CV, 1600 rpm | [69] |
Digitized from published data linearly interpolation near 0.85 V vs. RHE. a Electrolyte was 0.1 M KOH if not noted otherwise. * 1 M NaOH. b rotation rate estimated from limiting current of Vulcan carbon as not reported.
Table 4.
Ranking and classification of descriptors proposed for the ORR.
| Rank | Type | Parameter | Range of Activity | Parameter Range | Reference |
|---|---|---|---|---|---|
| 1 | C | eg occupancy | 250 mV (Δlog j = 2.1 to 4.2) | 1 unit (eg 1 to eg 0 or 2) | [22] |
| 2 | C, S | Valence via oxygen stoichiometry | Δlog j = 2.2 (132 to 264 mV) | SrFeO3−δ, 0.1 < δ < 0.4 | [67] |
| Δlog j = 0.8 (48 to 96 mV) | SrCoO3−δ, 0.0 < δ < 0.3 | [67] | |||
| 86 mV (Δlog j = 0.7 to 1.4) | LaMnO3+δ, 0.0 < δ < 0.1 | [22] | |||
| Δlog j = 0.3 (19 to 38 mV) | CaMnO3+δ, 0.1 < δ < 0.5 | [68] | |||
| 60 mV (Δlog j = 0.5 to 1.0) | LaNiO3−δ, δ = 0.00, 0.02 | [69] | |||
| Δlog j = 0.3 (20 to 41 mV) | La0.33Sr0.66O3−δ, 0.2 < δ < 0.3 | [204] | |||
| 3 | C | Valence via A-site composition | 115 mV (Δlog j = 1.0 to 1.9) | La1−xSrxMnO3, 0.0 < x < 0.7 | [63] |
| Δlog j = 0.9 (54 to 108 mV) | La1−xCaxMnO3, 0.0 < x < 1.0 | [64] | |||
| Δlog j = 0.2 (12 to 24 mV) | Pr1−xSrxMnO3, 0.0 < x < 1.0 | [65] | |||
| 4 | C | Covalence | 81 mV (Δlog j = 0.7 to 1.4) | 0.25 absorbance units | [22] |
| 5 | S | Tolerance factor | 80 mV (Δlog j = 0.7 to 1.3) | LaNiO3 a, 1.00 < t < 1.01 | [69] |
| Δlog j = 0.4 (24 to 48 mV) | AMnO3 b, 0.87 < t < 0.94 | [66] | |||
| 6 | M | Magnetic moment | Δlog j = 0.7 (42 to 84 mV) | LaMn0.5M0.5O3, 4 < µ < 13 | [61] |
| 7 | S | Strain (in-plane) | 49 mV (Δlog j = 0.4 to 0.8) | LaCoO3, −0.5% < ε < 2.6% | [129] |
| Δlog j = 0.6 (24 to 48 mV) | LaNiO3, −2.2% < ε < 2.7% | [132] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Risch, M. Perovskite Electrocatalysts for the Oxygen Reduction Reaction in Alkaline Media. Catalysts 2017, 7, 154. https://doi.org/10.3390/catal7050154
AMA Style
Risch M. Perovskite Electrocatalysts for the Oxygen Reduction Reaction in Alkaline Media. Catalysts. 2017; 7(5):154. https://doi.org/10.3390/catal7050154
Chicago/Turabian StyleRisch, Marcel. 2017. "Perovskite Electrocatalysts for the Oxygen Reduction Reaction in Alkaline Media" Catalysts 7, no. 5: 154. https://doi.org/10.3390/catal7050154
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.