
Reactivity of Copper Electrodes towards Functional Groups and Small Molecules in the Context of CO2 Electro-Reductions

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
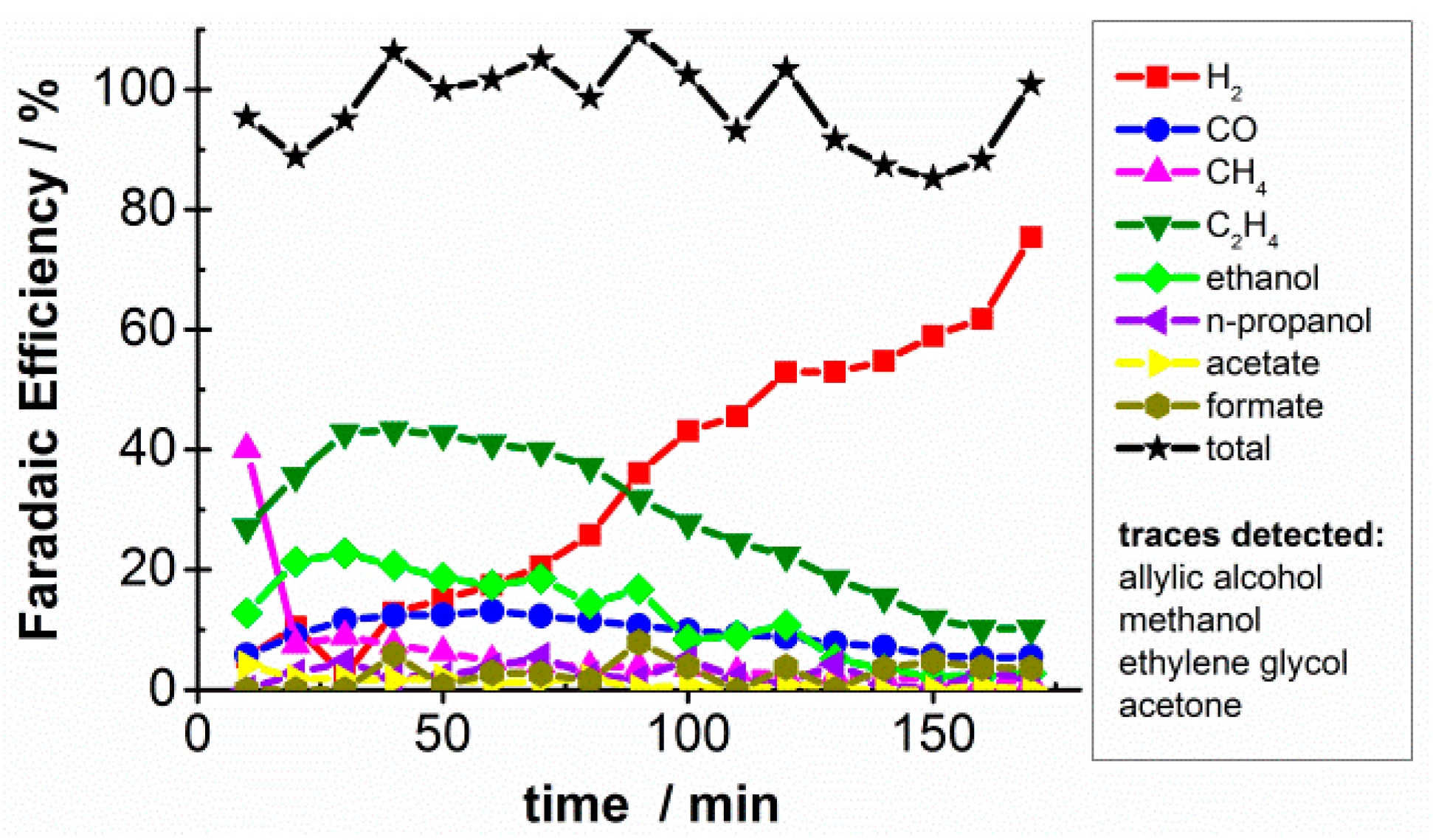
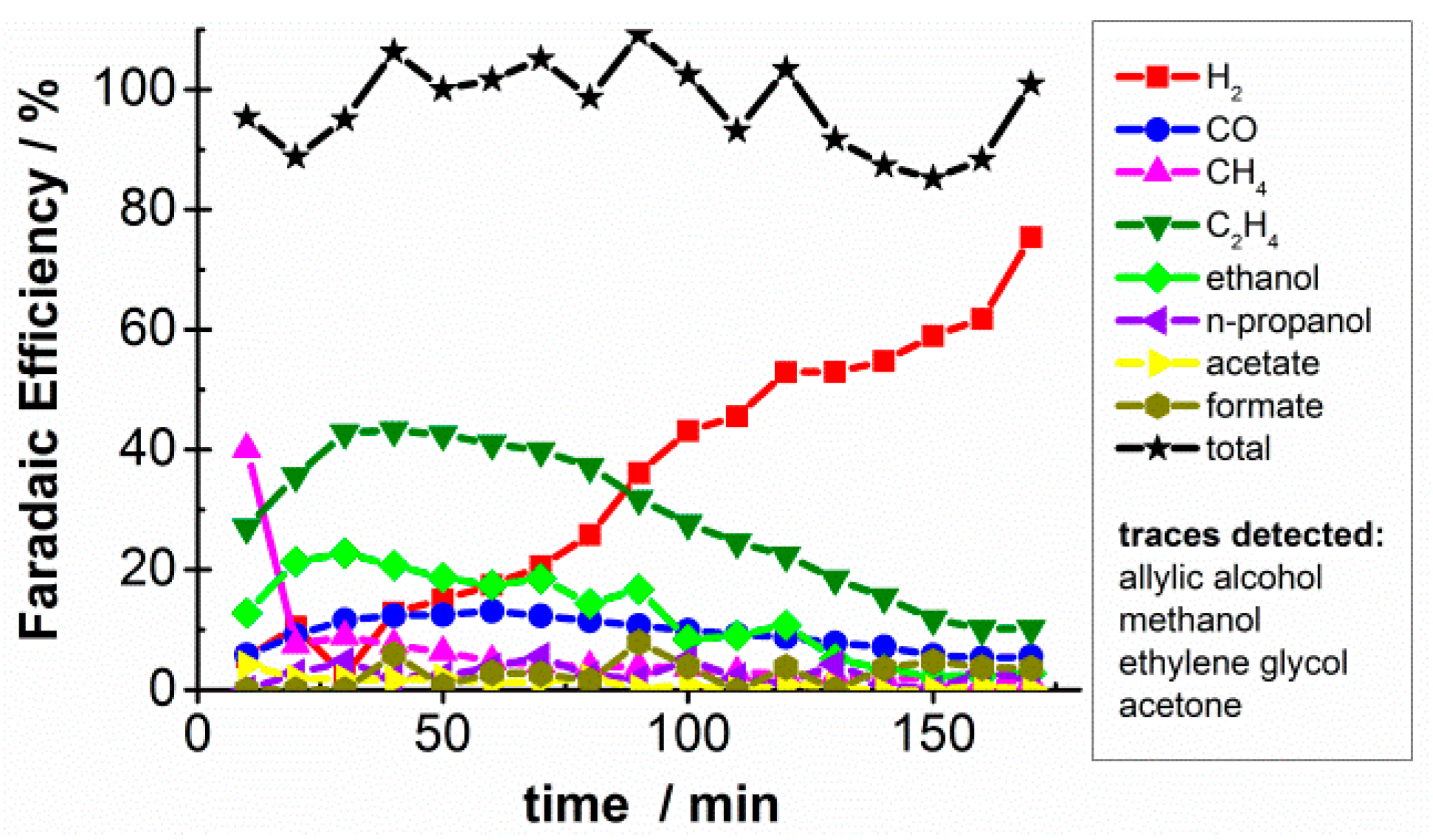
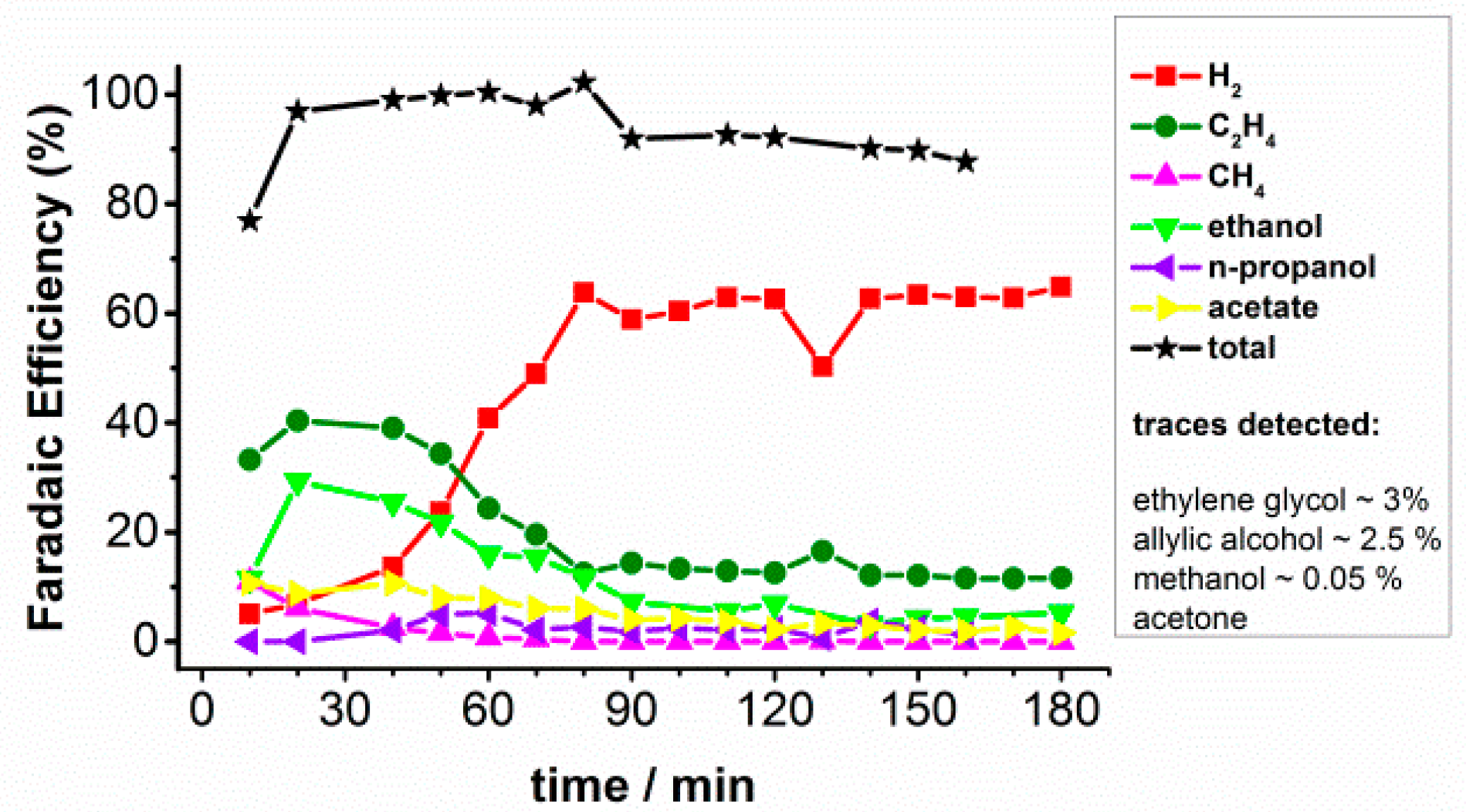
2.1. CO2 Bulk Electrolysis Benchmark
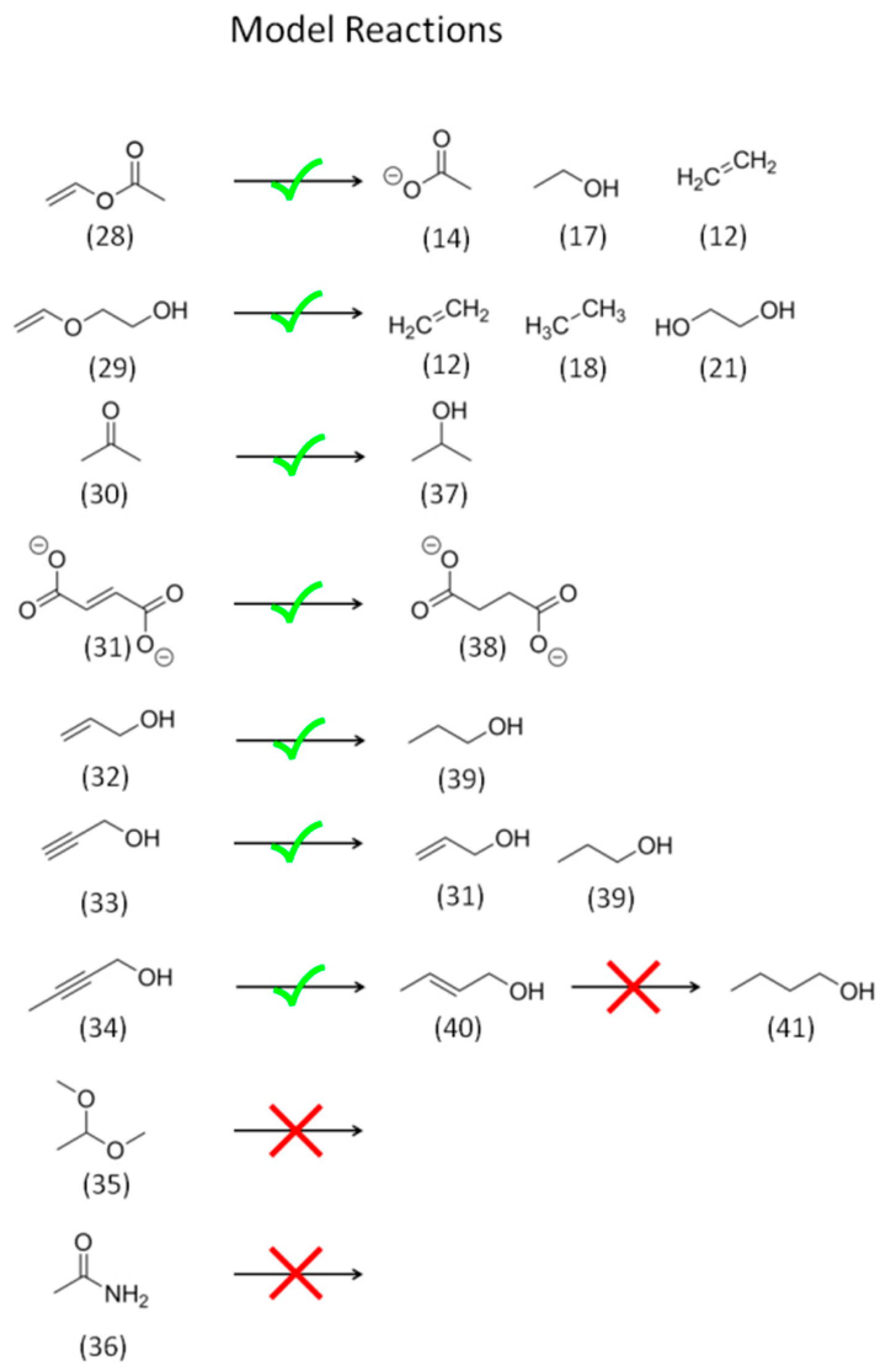
2.2. Model Reagent Screening
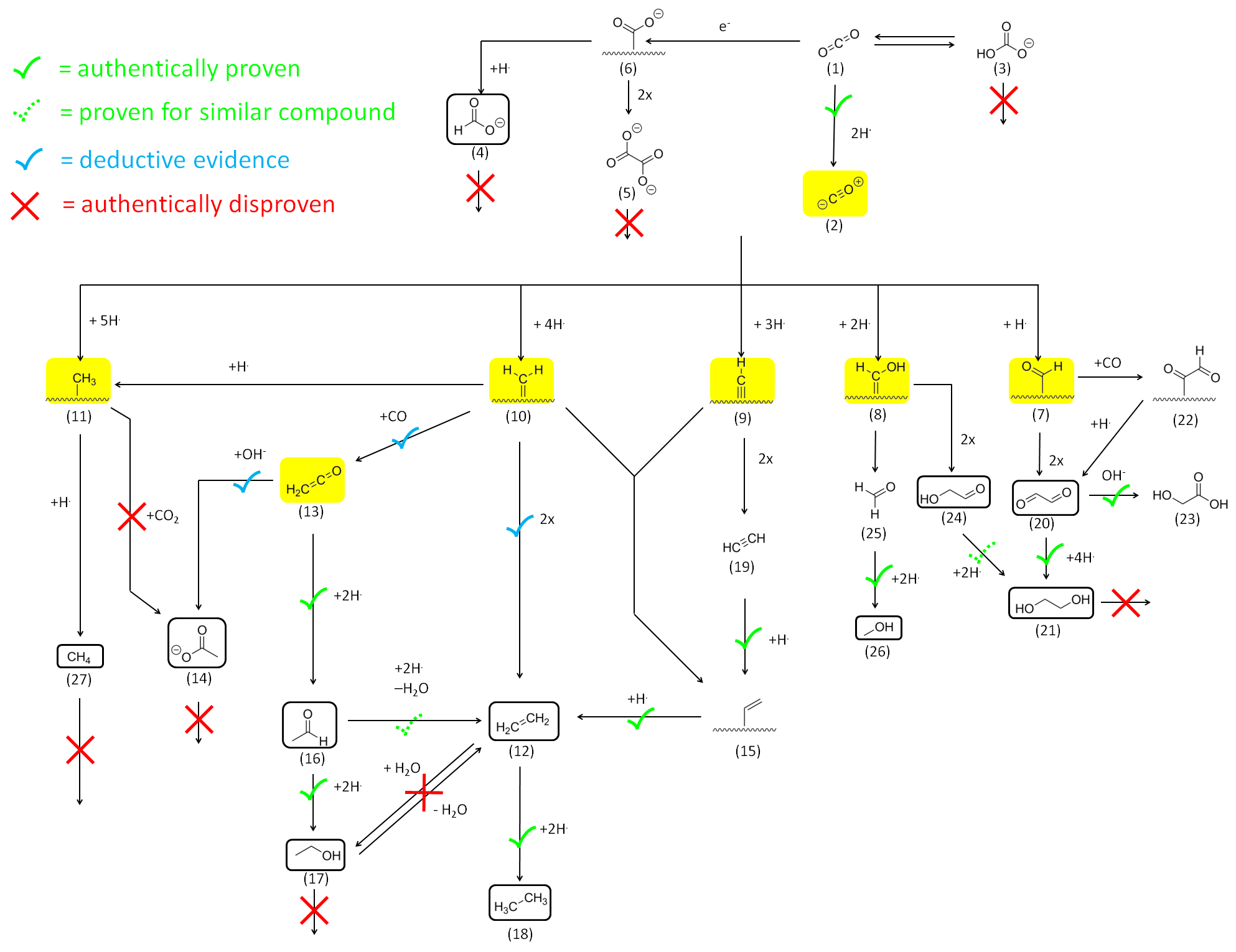
2.2.1. Reactivity of CO2 and HCO3−
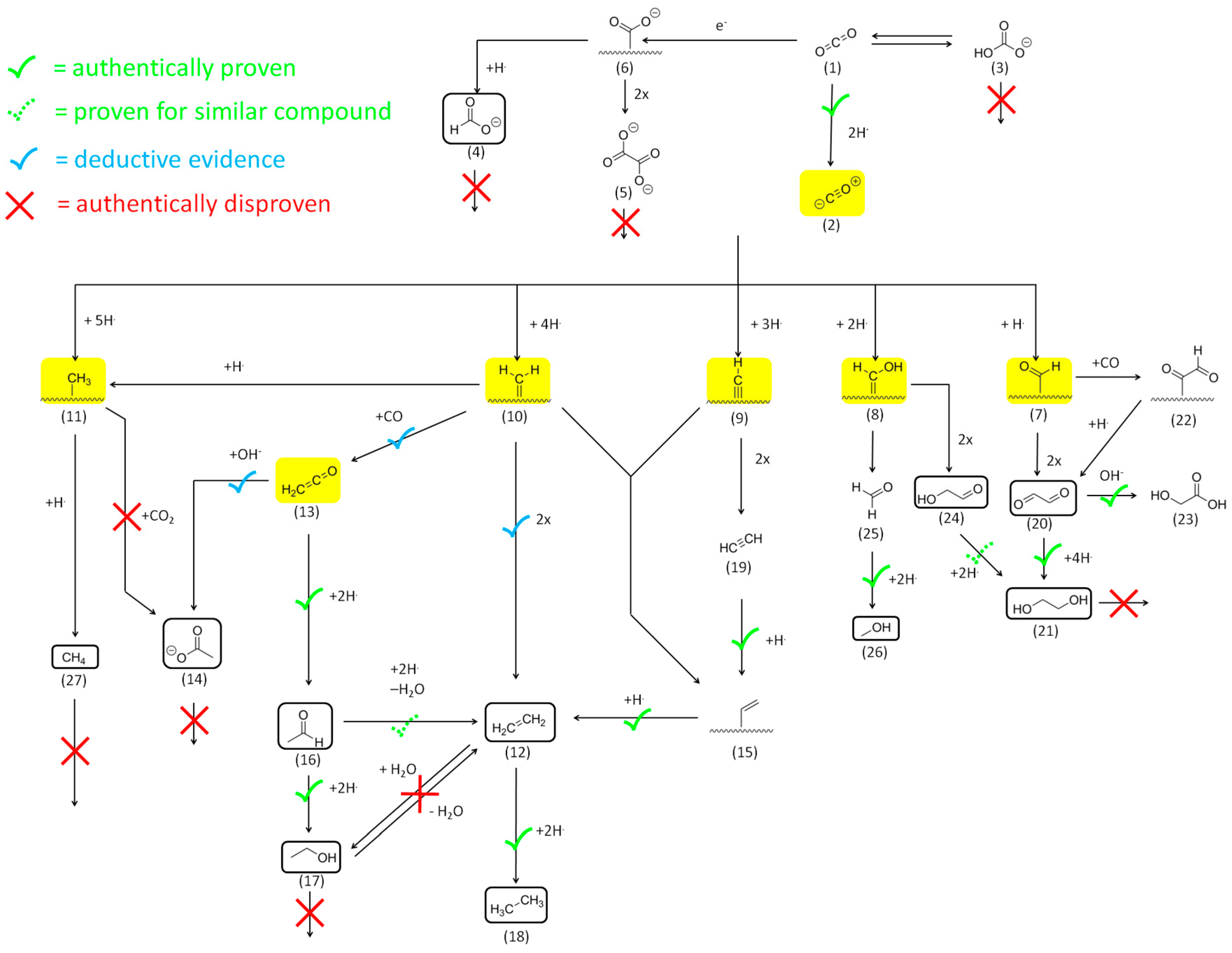
2.2.2. Reactivity and Role of 2e− Carboxylate Species
2.2.3. Reactivity of Higher Carboxylates
2.2.4. The Role of Acetaldehyde Acyl-Species and Enolates
2.2.5. Reactivity of C=O Double Bonds
2.2.6. Reactivity of Alcohols and Ethers
2.2.7. Reactivity of Alkenes
2.2.8. Reactivity of Alkynes
2.3. CO Bulk Electrolysis at High Current Density
Mechanistic Conclusions: Acetate and Ketene
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- DENA Grid Study II. Available online: http://www.dena.de/fileadmin/user_upload/Projekte/Erneuerbare/Dokumente/dena_Grid_Study_II_-_final_report.pdf (accessed on 17 March 2016).
- DENA Verteilernetzstudie. Available online: http://www.dena.de/fileadmin/user_upload/Projekte/Energiesysteme/Dokumente/denaVNS_Abschlussbericht.pdf (accessed on 17 March 2016).
- Sridhar, N.; Agarwal, A.; Rode, E. Electrochemical Production of Chemicals—Applicability of CO2 Conversion; Det Norse Veritas AS: Oslo, Norway, 2012. [Google Scholar]
- Schlögl, R. The Role of Chemistry in the Energy Challenge. ChemSusChem 2010, 3, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Liu, Y.; Hong, F.; Zang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y. Modern Aspects of Electrochemistry; Vayenas, C.G., White, R.E., Eds.; Springer: New York, NY, USA, 2008; p. 16. ISSN 0076-9924. [Google Scholar]
- Hori, Y. Handbook of Fuel Cells—Fundamentals, Technology and Applications; Vielstich, W., Lamm, A., Gasteiger, H.A., Eds.; Wiley VCH: Weinheim, Germany, 2003; ISBN 047-1-49-9269. [Google Scholar]
- Paik, W.; Andersen, T.N.; Eyring, H. Kinetic studies of the electrolytic reduction of carbon dioxide on the mercury electrode. Electrochim. Acta 1969, 14, 1217–1232. [Google Scholar] [CrossRef]
- Hori, Y.; Suzuki, S. Electrolytic Reduction of Carbon Dioxide at Mercury Electrode in Aqueous Solution. Bull. Chem. Soc. Jpn. 1982, 55, 660–665. [Google Scholar] [CrossRef]
- Hori, Y.; Kikuchi, K.; Shin Suzuki, K. Production of CO and CH4 in electrochemical reduction of CO2 at Metal Electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14, 1695–1698. [Google Scholar] [CrossRef]
- Cook, R.L.; MacDuff, R.C.; Sammels, A.F. On the Electrochemical Reduction of Carbon Dioxide at In Situ electrodeposited Copper. J. Electrochem. Soc. 1988, 135, 1320–1326. [Google Scholar] [CrossRef]
- Cook, R.L.; MacDuff, R.C.; Sammels, A.F. High Rate Gas Phase CO2 Reduction to Ethylene and Methane using Gas Diffusion Electrodes. J. Electrochem. Soc. 1990, 137, 607–608. [Google Scholar] [CrossRef]
- Hori, Y.; Murata, A.; Takahashi, R. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at copper electrodes in aqueous solution. J. Chem. Soc. Faraday Trans. 1989, 85, 2309–2326. [Google Scholar] [CrossRef]
- Jermann, B.; Augustynski, J. Long-term activation of the copper cathode in the course of CO2 reduction. Electrochim. Acta 1994, 39, 1891–1896. [Google Scholar] [CrossRef]
- Ogura, K.; Yano, H.; Shirai, F. Catalytic Reduction of CO2 to Ethylene by Electrolysis at a Three-Phase Interface. J. Electrochem. Soc. 2003, 150, D163–D168. [Google Scholar] [CrossRef]
- Verela, A.S.; Kroschel, M.; Reier, P.; Strasser, P. Controlling the selectivity of CO2 electroreduction on copper: The effect of the electrolyte concentration and the importance of the local pH. Catal. Today 2016, 260, 8–13. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Li, C.W.; Kanan, M.W. CO2 Reduction at Low Overpotential on Cu Electrodes Resulting from the reduction of Thick Cu2O Films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef] [PubMed]
- Chi, D.; Yang, H.; Du, Y.; Lu, T.; Sui, G.; Wang, H.; Lu, J. Morphology-controlled CuO nanoparticles for the electroreduction of CO2 to ethanol. RCS Adv. 2014, 4, 37329–37332. [Google Scholar]
- Kas, R.; Kortlever, R.; Milbrat, A.; Koper, M.T.M.; Maul, G.; Baltrisaitis, J. Electrochemical CO2 reduction on Cu2O-derived copper nanoparticles: Controlling the selectivity of hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Varela, A.S.; Bonifacio, C.S.; Zegkinoglou, I.; Sinev, I.; Choi, Y.-W.; Kisslinger, K.; Stach, E.A.; Yang, J.C.; Strasser, P.; et al. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 2016, 7, 12123. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Cook, R.L.; Kehoe, V.M.; MacDuff, R.C.; Patel, J.; Sammells, A.F. Carbon Dioxide Reduction to Alcohols using Perovskite-Type Electrocatalysts. J. Electrochem. Soc. 1993, 140, 614–618. [Google Scholar] [CrossRef]
- Jermann, B. Réduction Electrochimique de Longue Durée du Dioxyde de Carbone sur des Cathodes en Cuivre; Université de Genéve: Geneva, Switzerland, 1995. [Google Scholar]
- Chen, C.S.; Handoko, A.D.; Wan, J.H.; Ma, L.; Ren, D.; Yeo, B.S. Stable and selective electrochemical reduction of carbon dioxide to ethylene on copper mesocrystals. Catal. Sci. Technol. 2015, 5, 161–168. [Google Scholar] [CrossRef]
- Yano, H.; Tanaka, T.; Nakayama, M.; Ogura, K. Selective electrochemical reduction of CO2 to ethylene at a three-phase interface on copper(I) halide-confined Cu-mesh electrodes in acidic solutions of potassium halides. J. Electroanal Chem. 2004, 565, 287–293. [Google Scholar] [CrossRef]
- Ogura, K.; Oohara, R.; Kudo, Y. Reduction of CO2 to Ethylene at Three-Phase Interface Effects of Electrode Substrate and Catalytic Coating. J. Electrochem. Soc. 2005, 152, D213–D219. [Google Scholar] [CrossRef]
- Albo, J.; Sáez, A.; Solla-Gullón, J.; Montiel, V.; Irabien, A. Production of methanol from CO2 electroreduction at Cu2O and Cu2O/ZnO-based electrodes in aqueous solution. Appl. Catal. Environ. 2015, 176, 709–717. [Google Scholar] [CrossRef]
- Grote, J.-P.; Zeradjanin, A.R.; Cherevko, S.; Savan, A.; Breitbach, B.; Ludwig, A.; Mayrhofer, K.J.J. Screening of material libraries of electrochemical CO2 reduction catalysts—Improving the selectivity of Cu by mixing with Co. J. Catal. 2016, 343, 248–256. [Google Scholar] [CrossRef]
- Albo, J.; Vallejo, D.; Beobide, G.; Catillo, O.; Irabien, A. Copper-Based Metal-Organic Porous Materials for CO2 Electrocatalytic Reduction to Alcohols. ChemSusChem 2017, 6, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.N.; Andreiadis, E.S.; Heidkamp, J.; Simon, P.M.; Derat, E.; Cobo, S.; Royal, G.; Bergmann, A.; Strasser, P.; Dau, H.; et al. From molecular copper complexes to composite electrocatalytic materials for selective reduction of CO2 to formic acid. J. Mater. Chem. A 2015, 3, 3901–3907. [Google Scholar] [CrossRef]
- Gatrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons on copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Jitaru, M. Electrochemical Carbon Dioxide Reduction—Fundamental and Applied Topics. J. Univ. Chem. Technol. Metall. 2007, 42, 333–344. [Google Scholar]
- Oloman, C.; Lui, H. Electrochemical Processing of Carbon Dioxide. ChemSusChem 2008, 1, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Whipple, D.T.; Kenis, P.J.A. Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. [Google Scholar] [CrossRef]
- Neubauer, S.S.; Krause, R.K.; Schmid, B.; Guldi, D.M.; Schmid, G. Overpotentials and Faraday Efficiencies in CO2 Electrocatalysis—The Impact of 1-Ethyl-3-Methylimidazolium Trifluormethanesulfonate. Adv. Energy Mater. 2016, 6, 1502231. [Google Scholar] [CrossRef]
- Seshadri, G.; Lin, C.; Bocarsly, A.B. A new homogeneous electrocatalyst for the reduction of carbon dioxide. J. Electroanal. Chem. 1994, 372, 145–150. [Google Scholar] [CrossRef]
- Albo, J.; Beobide, G.; Castano, P.; Irabien, A. Methanol Synthesis from CO2 at Cu2O/ZnO promoted by pyridine-based aqueous solutions. J. CO2 Util. 2017, 18, 164–172. [Google Scholar] [CrossRef]
- Torelli, D.A.; Francis, S.A.; Crompton, J.C.; Javiert, A.; Thompson, J.R.; Brunschwig, B.S.; Soriaga, M.P.; Lewis, N.S. Nickel-Gallium-Catalyzed Electrochemical Reduction of CO2 to Highly Reduced Products at Low Overpotentials. ACS Catal. 2016, 6, 2100–2104. [Google Scholar] [CrossRef]
- Shen, J.; Kortlever, R.; Kas, R.; Birdja, Y.Y.; Diaz-Morales, O.; Kwon, Y.; Ledezma-Yanez, I.; Schouten, K.J.P.; Mul, G.; Koper, M.T.M. Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoprophyrin. Nat. Commun. 2016, 6, 8177. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Durand, W.J.; Peterson, A.A.; Peterson, F.; Studt, F.; Abild-Pedersen, F.; Nørskov, J.K. Structure effects on the energetics of the electrochemical reduction of CO2 by copper surfaces. Surf. Sci. 2011, 605, 1254–1359. [Google Scholar] [CrossRef]
- Schouten, K.J.P.; Kwon, Y.; van der Ham, C.J.M.; Qim, Z.; Koper, M.T.M. An new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes. Chem. Sci. 2011, 2, 1902–1909. [Google Scholar] [CrossRef]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Calle-Vallejo, F.; Koper, M.T.M. Theoretical Conciderations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes. Angew. Chem. Int. Ed. 2013, 52, 7282–7285. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Esopi, M.R.; Janik, M.J.; Asthagiri, A. Selectivity of CO2 Reduction on Copper Electrodes: The Role of the Kinetics of Elemetentary Steps. Angew. Chem. Int. Ed. 2013, 52, 2459–2462. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Deng, Y.; Handoko, A.D.; Chem, C.S.; Malkhandi, S.; Yeo, B.S. Selective Electrochemical Reduction of Carbon Dioxide to Ethylene and Ethanol of Copper(I)Oxide Catalysts. ACS Catal. 2015, 5, 2814–2821. [Google Scholar] [CrossRef]
- Kim, D.; Lee, S.; Ocon, J.D.; Jeong, B.; Lee, J.K.; Lee, J. Insights into an autonomously formed oxygen-evacuated Cu2O electrode for the selective production of C2H4 from CO2. Phys. Chem. Chem. Phys. 2015, 17, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Schouten, K.J.P.; Qin, Z.; Gallent, E.P.; Koper, M.T.M. Two Pathways for the formation of Ethylene in CO Reduction on Single-Crystal Copper Electrodes. J. Am. Chem. Soc. 2012, 134, 9864–9867. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Peterson, A.A.; Varela, A.S.; Jovanov, Z.P.; Bech, L.; Durand, W.J.; Dahl, S.; Nørskov, J.K.; Chrokendorff, I. The importance of surface morphology in contolling the selectivity of polycrystalline copper for CO2 electroreduction. Phys. Chem. Chem. Phys. 2012, 14, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Bertheussen, E.; Verdaguer-Casadevall, A.; Ravasio, D.; Montoya, J.H.; Trimarco, D.B.; Roy, C.; Meier, S.; Wendland, J.; Nørskov, J.K.; Stephens, I.E.L.; et al. Acetaldehyde as an intermediate in the Electroreduction of Carbon Monoxide to Ethanol on Oxide-Derived Copper. Angew. Chem. Int. Ed. 2016, 55, 1450–1454. [Google Scholar] [CrossRef] [PubMed]
- Reller, C.; Krause, R.K.; Neubauer, S.S.; Schmid, G.; Fleischer, M. CO2-to-value direct electrocatalytic reduction of CO2 towards chemical feedstock. In Proceedings of the 48 Jahrestreffen Deutscher Katalytiker, Congress Centrum Neue Weimarhalle, Weimar, Germany, 11–13 March 2015. [Google Scholar]
- Reller, C.; Krause, R.; Volkova, E.; Schmid, B.; Neubauer, S.; Rucki, A.; Schuster, M.; Schmid, G. Selective Electroreduction of CO2 toward Ethylene on Nano Dendritic Copper Catalysts at High Current Density. Adv. Energy Mater. 2017. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Javier, A.; Baricuatro, J.H.; Soriaga, M.P. Regulating the Product Distribution of CO Reduction by the Atomic Level Structural Modification of the Cu Electrode Surface. Electrocatalysis 2016, 7, 391–399. [Google Scholar] [CrossRef]
- Zinola, C.F. Electrocatalyis: Computational, Experimental and Industrial Aspects; CRC Press: Boca Raton, FL, USA, 2010; ISBN 978142004544. [Google Scholar]
- Ma, S.; Sadakiyo, M.; Luo, R.; Heima, M.; Yamauchi, M.; Kenis, P.J.A. One-step electrosynthesis of ethylene and ethanol from CO2 in an alkaline electrolyzer. J. Power Sources 2016, 301, 219–228. [Google Scholar] [CrossRef]
- Li, C.W.; Ciston, J.; Kanan, M.W. Electroreduction of Carbon Monoxide to Liquid Fuels on Oxide-Derived Nanocrystalline Copper. Nature 2014, 508, 504–520. [Google Scholar] [CrossRef] [PubMed]
- Wesselbaum, S.; Moha, V.; Meuresch, M.; Brosinski, S.; Thenert, K.M.; vom Stein, T.; Englert, U.; Hölscher, M.; Klankermeyer, J.; Leitner, W. Hydrogenation of carbon dioxide to methanol using a homogeneous ruthenium-Triphos catalyst: From mechanistic investigations to multiphase catalysis. Chem. Sci. 2015, 6, 693–704. [Google Scholar] [CrossRef]
- Murata, Y.; Hori, Y. Product Selectivity Affected by Cationic Species in Electrochemical Reduction of CO2 and CO at a Cu Electrode. Bull. Chem. Soc. Jpn. 1991, 64, 123–127. [Google Scholar] [CrossRef]
- Wuttig, A.; Liu, C.; Peng, Q.; Yaguchi, M.; Hendon, C.H.; Motobayashi, K.; Ye, S.; Osawa, M.; Surendranath, Y. Tracking a Common Surface-Bound Intermediate during CO2-to-Fuels Catalysis. ACS Cent. Sci. 2016, 2, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Ledezma-Yanes, I.; Pérez Gallent, E.; Koper, M.T.M.; Calle-Vallejo, F. Structure-sensitive electroreduction of acetaldehyde to ethanol on copper and its mechanistic implications for CO and CO2 reduction. Catal. Today 2016, 262, 90–94. [Google Scholar] [CrossRef]
- Loeffler, K.W.; Koehler, C.A.; Paul, N.M.; De Haan, D.O. Oligomer formation in evaporating aqueous glyoxal and methyl glyocal solutions. Environ. Sci. Technol. 2006, 40, 6318–6323. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Gattrell, M.; MacDougall, B. Calculations for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J. Appl. Electrochem. 2006, 36, 161–172. [Google Scholar] [CrossRef]
- Montoya, J.H.; Peterson, A.A.; Nørskov, J.K. Insights into C–C coupling in CO2 Electroreduction on Copper Electrodes. ChemCatChem 2013, 5, 737–742. [Google Scholar] [CrossRef]
- Kropp, K.; Skibbe, V.; Erker, G.; Krueger, C. Fischer-Tropsch intermediates: Tris[(.eta.2-formaldehyde)zirconocene] from the carbonylation of a zirconium hydride. J. Am. Chem. Soc. 1983, 105, 3353–3354. [Google Scholar] [CrossRef]
- Suess, D.L.M.; Peters, J.C. A CO-Derived Iron Dicarbyne That Releases Olefin upon Hydrogenation. J. Am. Chem. Soc. 2013, 135, 12580–12583. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Du, H.; Liang, Z.; Zhu, Z.; Duan, D.; Liu, S. Paired Electrochemical Synthesis of Ethylene and Oxalic Acid from Acetylene. Int. J. Electrochem. Sci. 2013, 8, 6566–6573. [Google Scholar]
- Rüchardt, C.; Schrauzer, G.N. Über die Carbonylierung von Carbenen und die katalytische Zersetzung von Diazoalkanen mit Nickeltetracarbonyl. Chem. Ber. 1960, 93, 1840–1848. [Google Scholar] [CrossRef]
- Mankad, N.P.; Peters, J.C. Diazoalkanes react with a bis(phosphino)borate copper(I) source to generate [Ph2BPtbu2]Cu(η1-N2CR2),[Ph2BPtBu2]Cu(CPh2), and [Ph2BPtbu2]Cu-N(CPh2)(NCPh2). Chem. Commun. 2008, 1061–1063. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Warren, T.H. Discrete Bridging and Terminal Copper Carbenes in Copper-Catalyzed Cyclopropanation. J. Am. Chem. Soc. 2004, 126, 10085–10094. [Google Scholar] [CrossRef] [PubMed]
- Grotjahn, D.B.; Bikzhanova, G.B.; Collins, L.S.B.; Concolino, T.; Lam, K.-C.; Rheingold, A.L. Controlled, Reversible Conversion of a Ketene Ligand to Carbene and CO Ligands on a Single Metal Center. J. Am. Chem. Soc. 2000, 122, 5222–5223. [Google Scholar] [CrossRef]
- Bodnar, T.W.; Cutler, A.R. Formation of a stable (.eta.2-C,C) ketene compound dicarbonyl(cyclopentadienyl)keteneiron hexafluorophosphate [(C5H5)Fe(CO)2(CH2CO)+ PF6-] by carbonylation of an iron methylidene complex. A novel entry into carbonyl-derived C2 chemistry. J. Am. Chem. Soc. 1983, 105, 5926–5928. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmid, B.; Reller, C.; Neubauer, S.S.; Fleischer, M.; Dorta, R.; Schmid, G. Reactivity of Copper Electrodes towards Functional Groups and Small Molecules in the Context of CO2 Electro-Reductions. Catalysts 2017, 7, 161. https://doi.org/10.3390/catal7050161
Schmid B, Reller C, Neubauer SS, Fleischer M, Dorta R, Schmid G. Reactivity of Copper Electrodes towards Functional Groups and Small Molecules in the Context of CO2 Electro-Reductions. Catalysts. 2017; 7(5):161. https://doi.org/10.3390/catal7050161
Chicago/Turabian StyleSchmid, Bernhard, Christian Reller, Sebastian S. Neubauer, Maximilian Fleischer, Romano Dorta, and Guenter Schmid. 2017. "Reactivity of Copper Electrodes towards Functional Groups and Small Molecules in the Context of CO2 Electro-Reductions" Catalysts 7, no. 5: 161. https://doi.org/10.3390/catal7050161