
Synthesis, Structural Characterization and Catalytic Evaluation of Anionic Phosphinoferrocene Amidosulfonate Ligands


Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 40 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(6), 167; https://doi.org/10.3390/catal7060167
Submission received: 20 April 2017
/
Revised: 19 May 2017
/
Accepted: 19 May 2017
/
Published: 24 May 2017
(This article belongs to the Special Issue Suzuki–Miyaura Cross-Coupling Reaction and Potential Applications)
Abstract
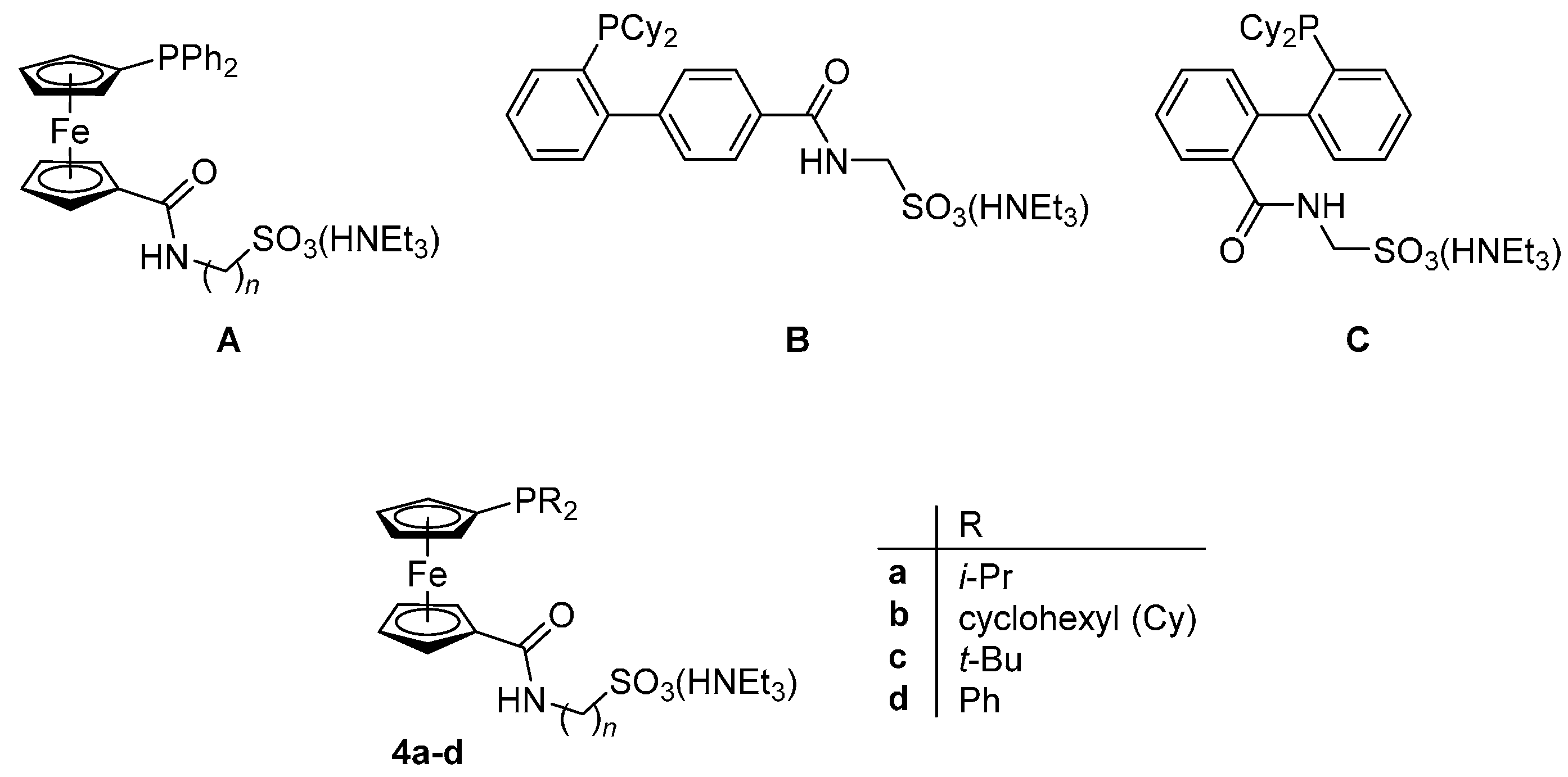
:Triethylammonium salts of phosphinoferrocene amidosulfonates with electron-rich dialkyphosphino substituents, R2PfcCONHCH2SO3(HNEt3) (4a–c), where fc = ferrocene-1,1′-diyl, and R = i-Pr (a), cyclohexyl (Cy; b), and t-butyl (c), were synthesized from the corresponding phosphinocarboxylic acids-borane adducts, R2PfcCO2H·BH3 (1a–c), via esters R2PfcCO2C6F5·BH3 (2a–c) and adducts R2PfcCONHCH2SO3(HNEt3)·BH3 (3a–c). Compound 4b was shown to react with [Pd(μ-Cl)(η-C3H5)]2 and AgClO4 to afford the zwitterionic complex [Pd(η3-C3H5)(Cy2PfcCONHCH2SO3-κ2O,P)] (5b), in which the amidosulfonate ligand coordinates as a chelating donor making use of its phosphine moiety and amide oxygen. The structures of 3b·CH2Cl2, 4b and 5b·CH2Cl2 were determined by single-crystal X-ray diffraction analysis. Compounds 4a–c and their known diphenylphosphino analogue, Ph2PfcCONHCH2SO3(HNEt3) (4d), were studied as supporting ligands in Pd-catalyzed cyanation of aryl bromides with K4[Fe(CN)6] and in Suzuki–Miyaura biaryl cross-coupling performed in aqueous reaction media under mild reaction conditions. In the former reaction, the best results were achieved with a catalyst generated from [PdCl2(cod)] (cod = η2:η2-cycloocta-1,5-diene) and 2 equiv. of the least electron-rich ligand 4d in dioxane–water as a solvent. In contrast, the biaryl coupling was advantageously performed with a catalyst resulting from palladium(II) acetate and ligand 4a (1 equiv.) in the same solvent.
1. Introduction
Sulfonation of phosphines represents an efficient and time-tested approach toward hydrophilic ligands. Introduction of a single sulfonate moiety into a molecule of a phosphine donor usually increases polarity and hydrophilicity to such an extent that the resulting derivatives can be used as donors to prepare highly hydrophilic coordination compounds as well as supporting ligands for transition metal-catalyzed reactions performed in aqueous and water-organic biphase media [1,2,3]. Phosphinosulfonate Donors including the archetypal and widely used trisodium tris(sulfonatophenyl)phosphine (TPPTS) [4,5,6] are typically obtained by direct sulfonation of the parent phosphines, which somewhat limits the scope of the accessible compounds, mainly due to problems associated with a high reactivity of the sulfonation agents and their compatibility with other functional groups.
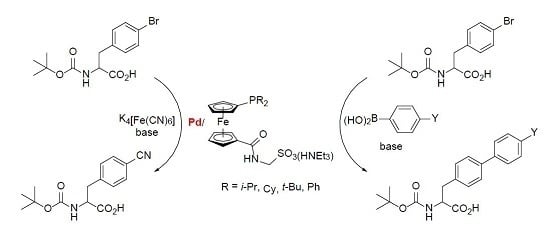
We have recently demonstrated that phosphinosulfonate donors can also be accessed via amidation of phosphinocarboxylic acids with ω-aminosulfonic acids [7]. This synthetic approach, which inherently leads to a simultaneous incorporation of the polar amide linking group, is modular and allows the synthesis of libraries of donors with modified structures and also eliminates some limitations of the direct-sulfonation approach. So far, we have utilized this method to prepare a series of phosphinoferrocene amidosulfonates differing in the length of the aliphatic spacer between the amide and sulfonate groups, compounds A (n = 1–3) [7], and a pair of isomeric phosphinobiphenyl donors B and C [8,9] (Scheme 1). These compounds were successfully tested as supporting ligands in Pd-catalyzed cyanation of aryl bromides and in Suzuki–Miyaura cross-coupling [7,8,9].
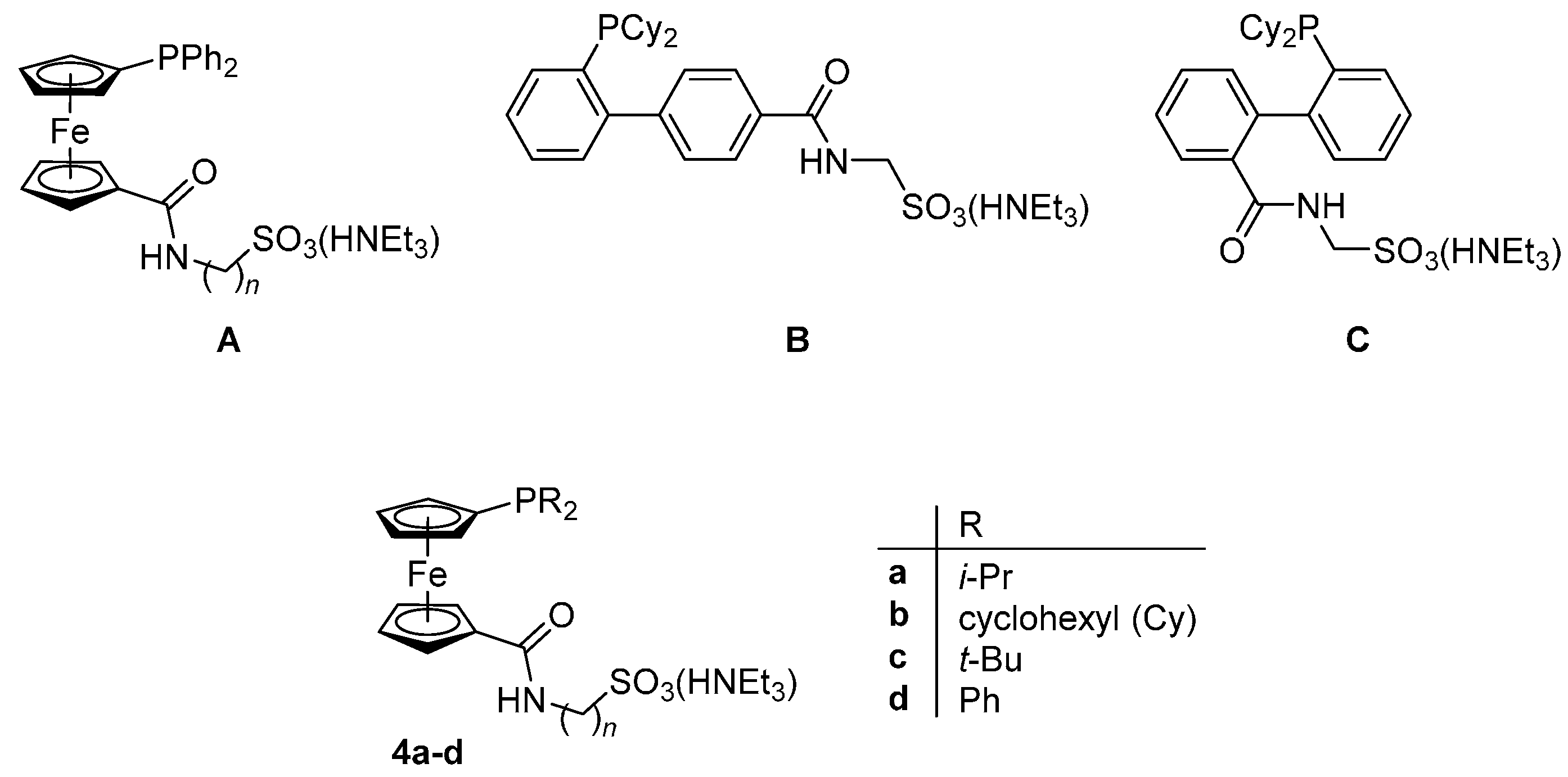
Having recently established a reliable synthetic route to new (dialkylphosphino)ferrocenecarboxylic acids in their stable, phosphine-protected form (viz. compounds 1a–c) [10], we decided to use these compounds further in the preparation of phosphinoferrocene amidosulfonate donors with varied, electron-rich phosphine substituents, compounds 4a–c (Scheme 1). The synthesis, structural characterization and an evaluation of these compounds as supporting ligands in Pd-catalyzed cyanation of aryl bromides to the corresponding nitriles [11,12] and Suzuki–Miyaura cross-coupling of aryl bromides with arylboronic acids [13,14,15,16,17] performed in aqueous reaction media are reported in this contribution.
2. Results and Discussion
2.1. Synthesis of the Ligands
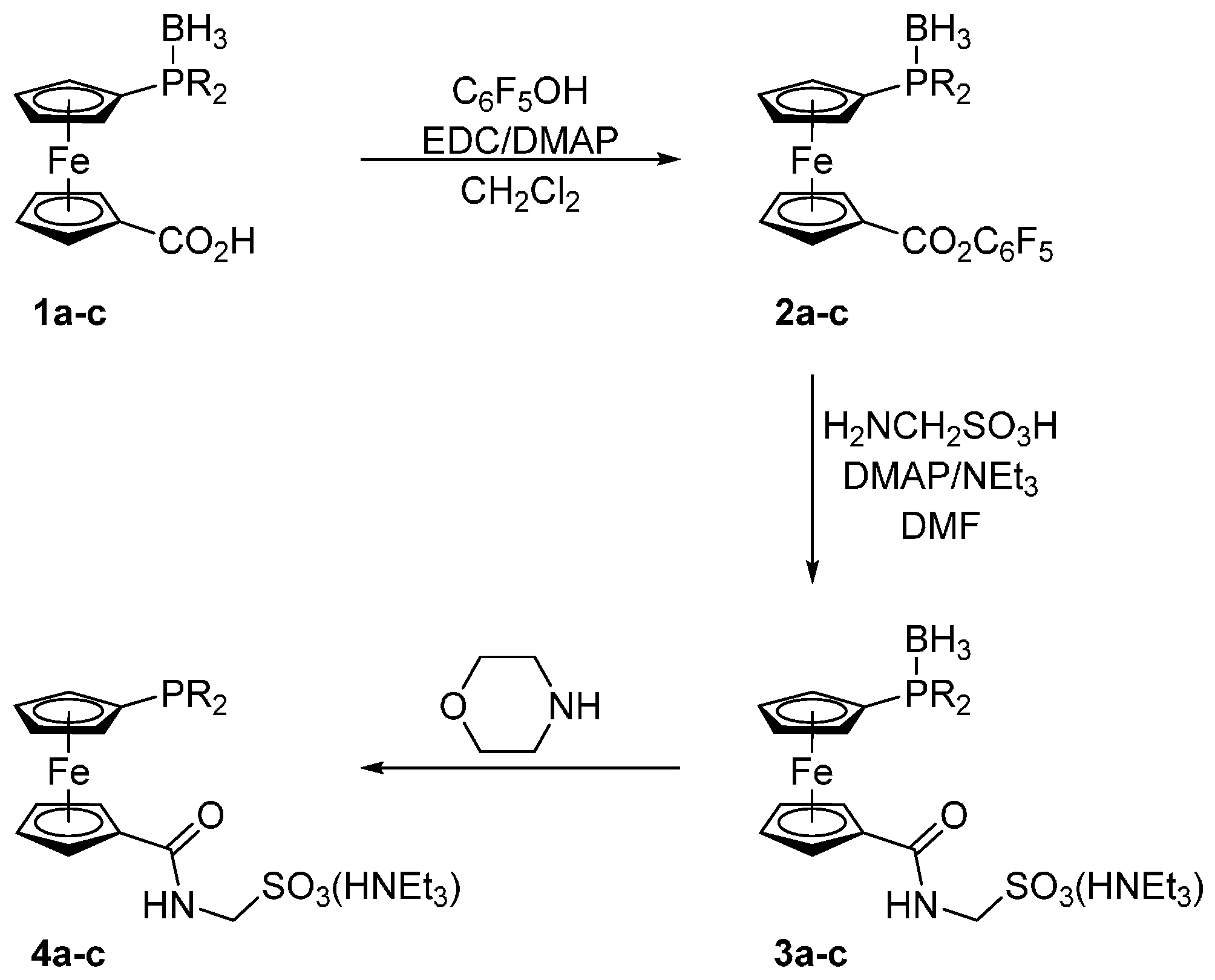
Compounds 1a–c were prepared in analogy to the amidosulfonate donors A–C mentioned in the Introduction except that their oxidation-sensitive phosphine moieties were protected in the form of BH3 adducts [18] during the synthesis (Scheme 2). In the first step, phosphinocarboxylic acid-borane adducts 1a–c were reacted with pentafluorophenol in the presence of 1-ethyl- -3-[3-(dimethylamino)propyl]carbodiimide (EDC) and 4-(dimethylamino)pyridine (DMAP) to afford the corresponding esters 2a–c. These active esters were, in turn, reacted with aminomethanesulfonic acid in a mixture of N,N-dimethylformamide (DMF) and triethylamine to give the target ligands in their P-protected form, compound 3a–c. In the last step, the borane protecting group was removed by heating these borane adducts in neat morpholine (65 °C/16 h) and the resulting free phosphines 4a–c were isolated by column chromatography using a NEt3-pretreated silica gel column to avoid cation exchange. The target ligands 4a–c as well as all reaction intermediates were characterized by NMR and IR spectroscopy, electrospray ionization (ESI) mass spectrometry and by elemental analysis.
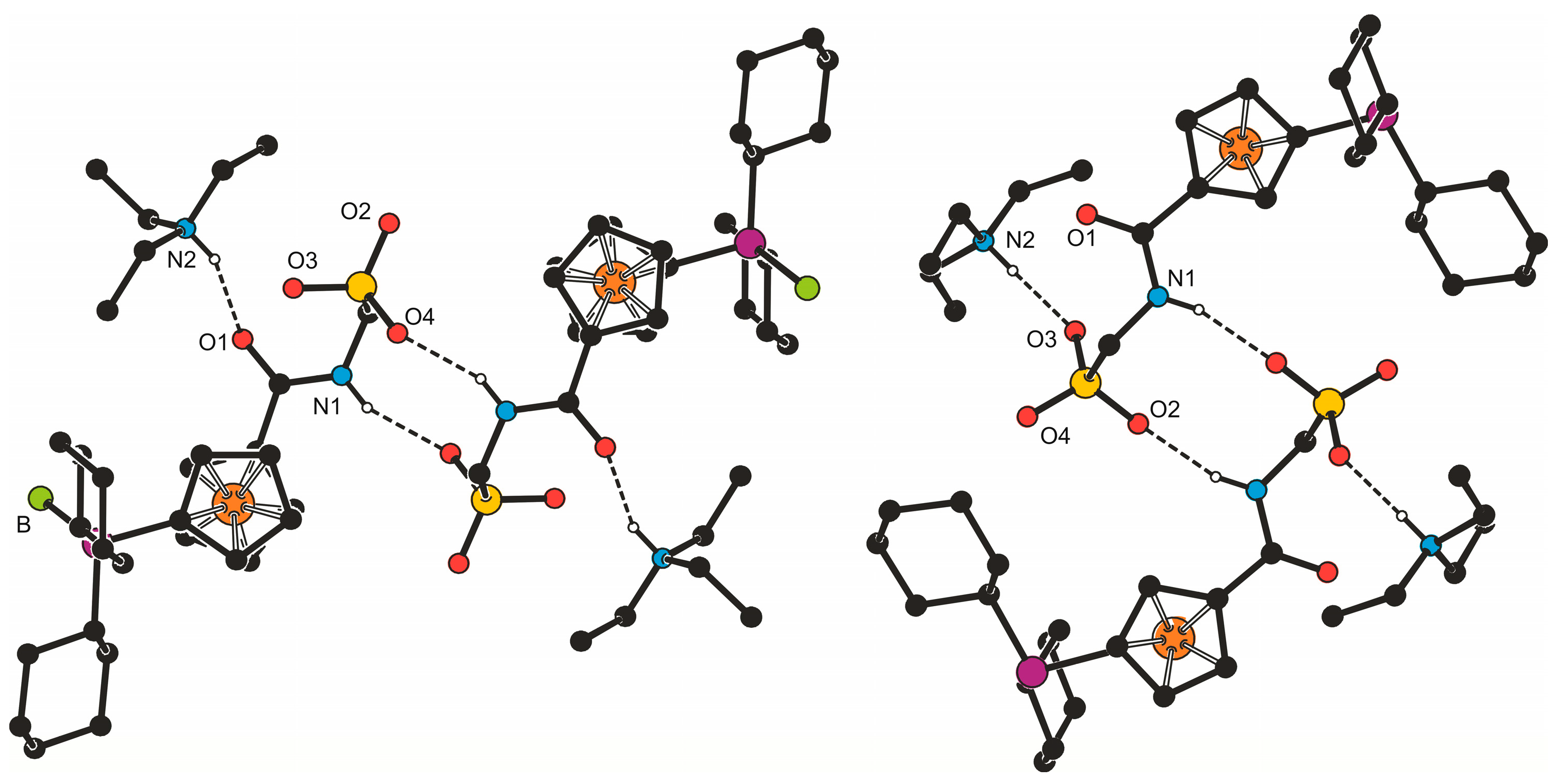
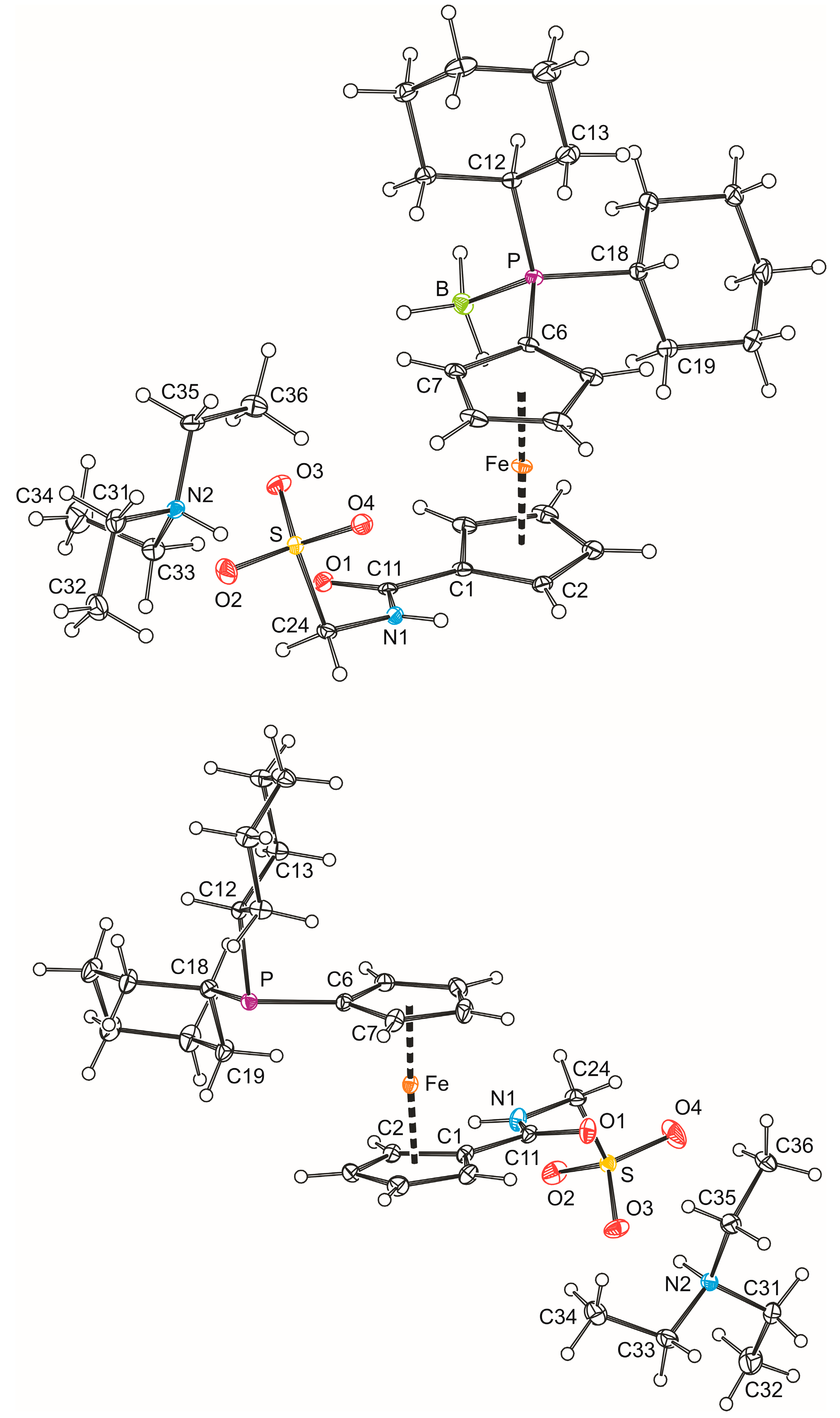
In addition, solid-state structures of 3b·CH2Cl2 and 4b were determined by single-crystal X-ray diffraction analysis. Views of the molecular structures are shown in Figure 1 and the relevant structural parameters for the phosphinoferrocene amidosulfonate anions are presented in Table 1. The ferrocene units in both structures adopt their usual geometry with similar Fe–C distances (cf. Fe–C(1–10): 2.036(2)–2.066(2) Å for 3b·CH2Cl2 and 2.032(2)–2.070(1) Å for 4b) and tilt angles not exceeding 5°. The substituents attached in the positions 1 and 1′ are diverted from each other, so that the ferrocene cyclopentadienyls assume conformations near to anticlinal eclipsed (compare τ angles with the ideal value of 144° [19]). Generally, the individual geometric parameters compare well with the values reported previously for compound 4d [7] and acid 1b [10]. In the pair, the structures of the anions differ mainly by the orientation of their CH2SO3 pendant groups. In the structure of 3b·CH2Cl2, this moiety extends above the amide plane toward the ferrocene unit, whereas in 4b, it is directed away from the ferrocene moiety (compare the dihedral angles C11-N1-C24-S of −88.5(2)° and 105.3(1)° in 3b·CH2Cl2 and 4b, respectively). Slight differences are observed also in the overall molecular conformation (compare angles τ and φ in Table 1) and in the lengths of the P–C bonds, which are longer in the free phosphine 4b than in the corresponding BH3 adduct 3b·CH2Cl2 [3b·CH2Cl2: P–C6 1.798(2) Å, P–C12 1.843(2) Å, and P–C18 1.840(2) Å; 4b: P–C6 1.825(1) Å, P–C12 1.873(1) Å, and P–C18 1.862(1) Å]. The cyclohexyl substituents assume chair conformations, which is clearly indicated by the ring puckering parameter θ being 0.0(2)° [2.2(2)] and 178.0(2)° [175.3(2)°] for the rings C(12–17) and C(18–23) in 3b·CH2Cl2 [4b], respectively (N.B. ideal chair requires θ = 0/180°, see ref. [20]). In all rings, the P–C bonds occupy equatorial positions.
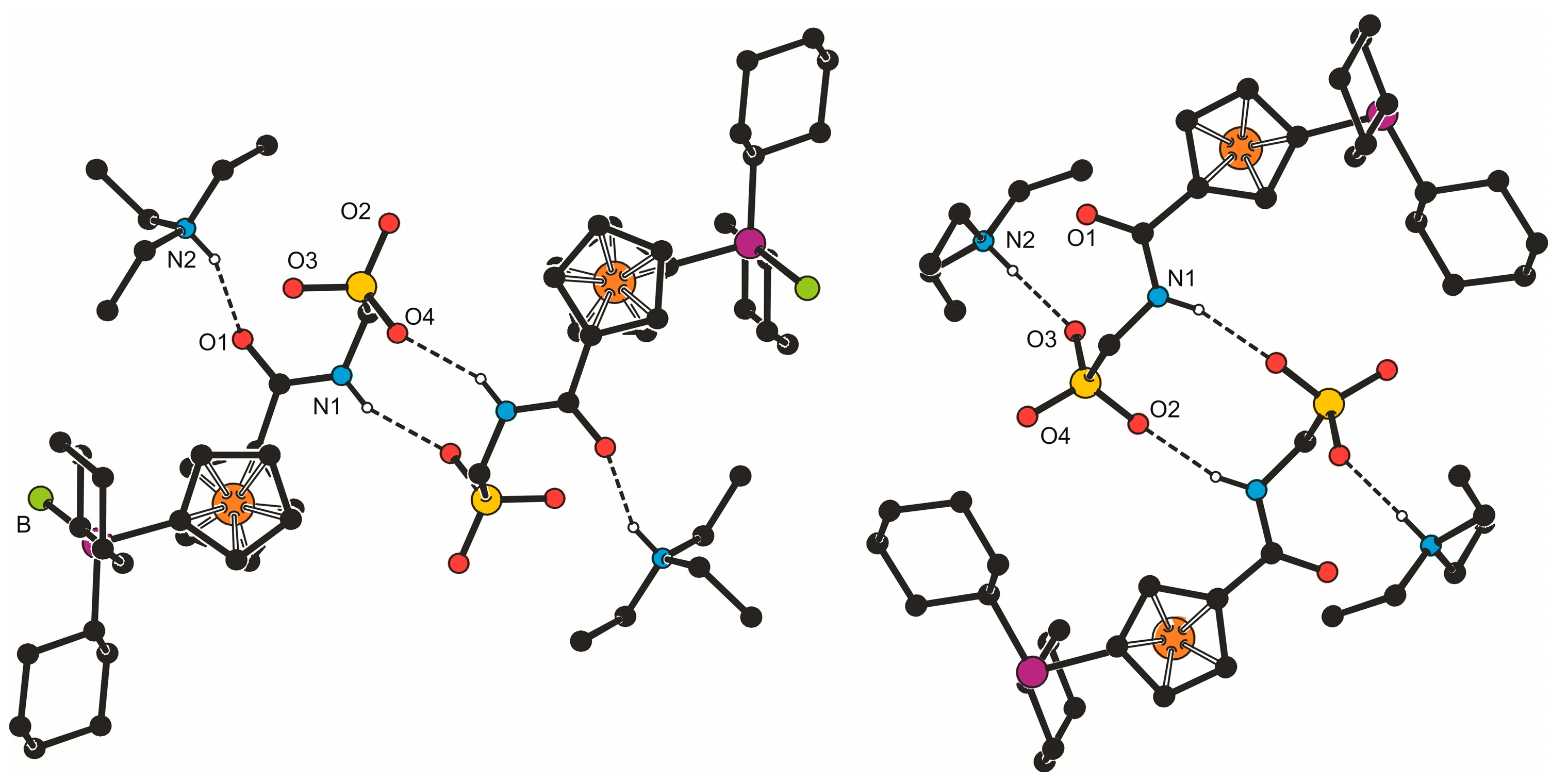
Despite their obvious structural similarity, compounds 3b·CH2Cl2 and 4b constitute different solid-state assemblies in their crystals (Figure 2). The amidosulfonate anions in the structure of 3b·CH2Cl2 interact with their inversion-related counterparts through a pair of N1–H1N···O4 hydrogen bonds (N1···O4 = 2.811(2) Å) to form a closed dimeric motif, which further binds two adjacent Et3NH+ cations via N2–H2N···O1 hydrogen bonds involving the amide oxygen (N2···O1 = 2.794(2) Å). A similar central unit formed from a pair of inversion-related amidosulfonate anions is found also in the structure of 4b (N1···O2 = and 2.813(2) Å) and this dimeric moiety even interacts with a pair of the triethylammonium cations though via hydrogen bonds toward the sulfonate oxygen O3 (N2–H2N ··· O3 = 2.717(2) Å).
The reaction of [Pd(μ-Cl)(η-C3H5)]2 with ligand 4b chosen as a representative and then with silver(I) perchlorate proceeded under cleavage of the chloride bridges in the dimeric Pd(II) precursor and removal of the Pd-bound chloride to afford the zwitterionic (η3-C3H5)Pd(II) complex 5b wherein the phosphinosulfonate anion Cy2PfcCONHCH2SO3− coordinates through its phosphine substituent and the amide oxygen, forming an O,P-chelate ring (Scheme 3). The coordination of the phosphine group in 5b was manifested through a shift of the 31P NMR resonance to a lower field (the 31P NMR coordination shift, ΔP = δP(complex) − δP(free ligand), was 36.0 ppm), whereas the shift of the amide C=O vibration in the IR spectrum by 65 cm−1 to lower energies suggested that the amide oxygen is involved in coordination to the (η3-C3H5)Pd fragment rather that the anionic sulfonate moiety [7].
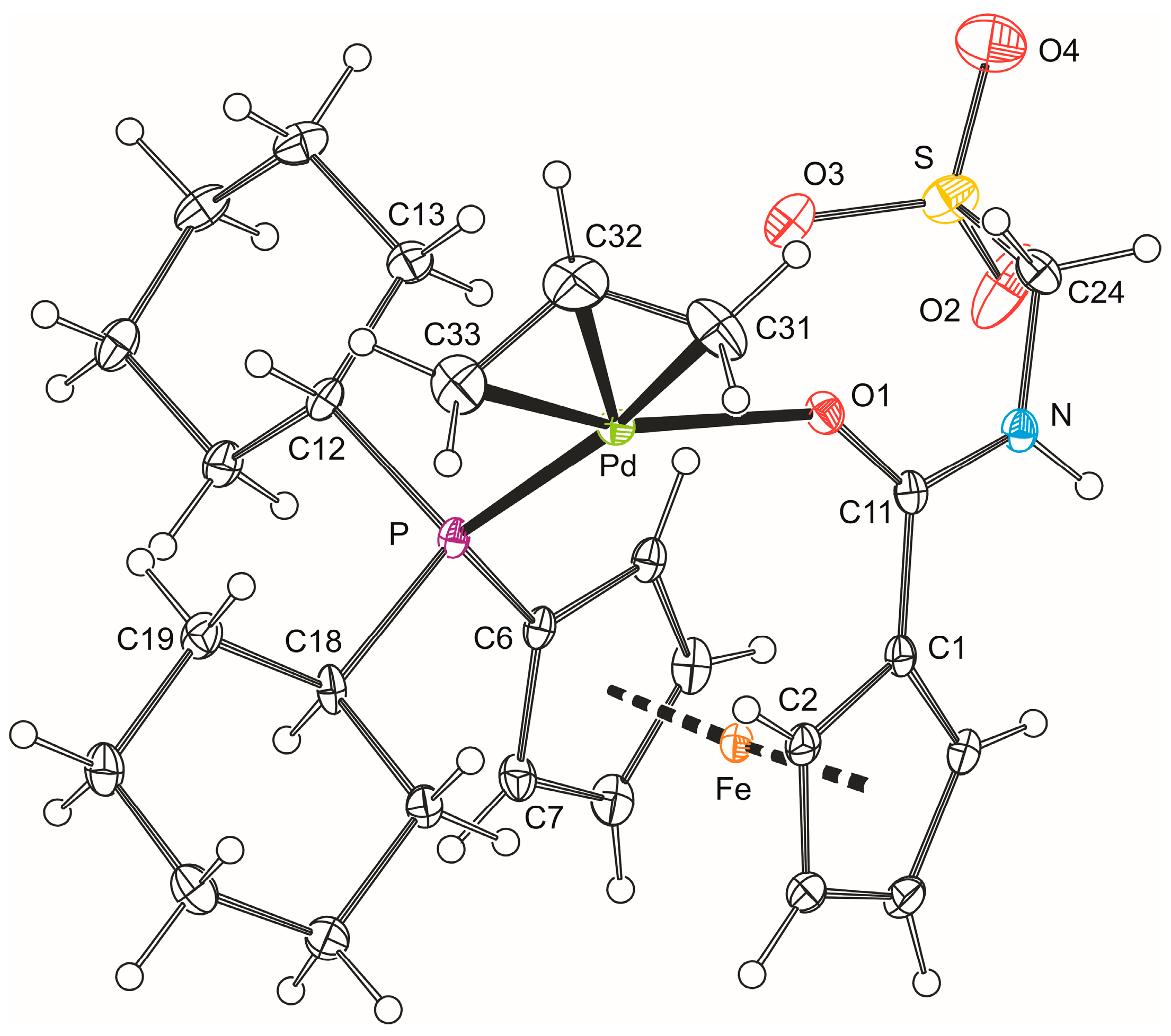
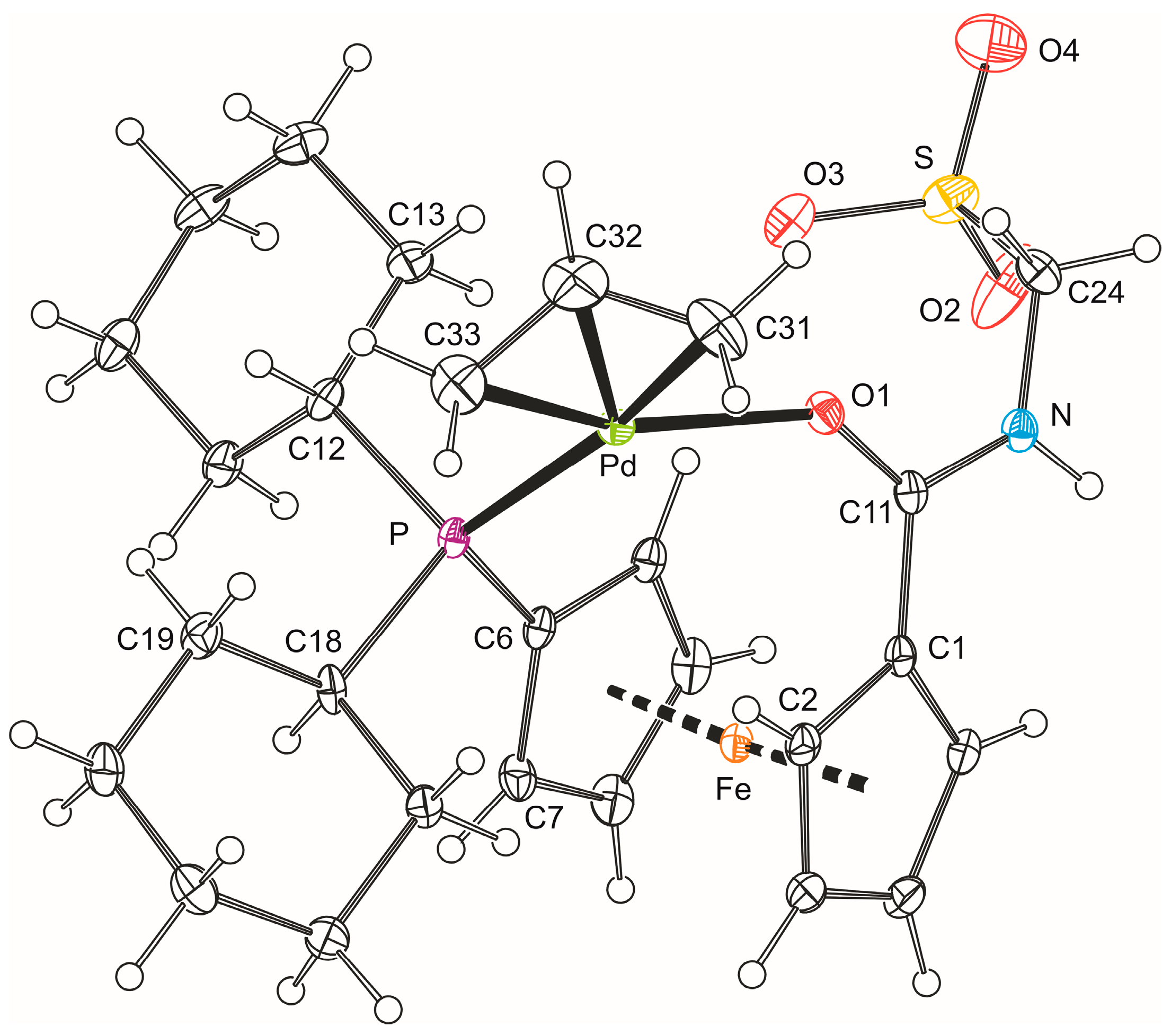
The formulation of 5b was unequivocally corroborated by X-ray diffraction analysis on the stoichiometric solvate 5b·CH2Cl2. A view of the complex molecule is shown in Figure 3, and the selected geometric parameters are given in Table 2. The allyl moiety {C31, C32, C33} in the structure of 5b is rotated by 69.7(4)° with respect to the plane defined by the palladium atom and the remaining ligating atoms P and O1, so that the carbon atom C32 in the meso position is diverted from the ferrocene ligand [21]. The individual Pd–C(allyl) distances decrease gradually from C31 to C33, reflecting the trans-influence of the donor atoms located opposite the allyl moiety (P > O) [22,23].
Apparently because of chelate coordination, the 1,1′-disubtituted ferrocene unit is less open than in the structure of 4b, adopting a near synclinal eclipsed conformation as evidenced by the torsion angle C1–Cg1–Cg2–C6 (τ) of −65.1(2)° (cf. the ideal value: 72°). The ferrocene moiety is somewhat tilted (dihedral angle: 6.5(2)°) with the C1 and C10 atoms that reside above each other in the ferrocene unit forming the shortest bonds toward the central iron atom (N.B. the individual Fe–C bonds span the range 2.011(2)–2.074(2) Å). More importantly, the distortion propagates to the amide unit, which appears twisted by 15.3(3)° with respect to its parent cyclopentadienyl ring Cp1 and the pivotal atom C11 is displaced from the Cp1 plane inward of the ferrocene unit by as much as 0.214(2) Å. All this aids in bringing the amide oxygen O1 into a position suitable for chelate ring formation. The CH2SO3 arm is oriented above the amide unit and to the side of the ferrocene unit so that the angle subtended by the C24–S bond and the axis of the ferrocene unit (Cg1···Cg2) is 21.75(6)°. In contrast, the phosphine phosphorus lies in the plane of its bonding cyclopentadienyl ring (perpendicular distance from the Cp2 plane is only 0.047(1) Å). However, because of the proximity of one of the cyclohexyl substituents, the C7–C6–P angle is 3.6° less acute than the C10–C6–P angle opening to the less sterically encumbered side of the ferrocene moiety (N.B. the difference between the C2–C1–C11 and C5–C1–C11 angles is only 2.0°).
In the crystal, the molecules of complex 5b associate into dimers via hydrogen bonds between their amide NH groups and sulfonate oxygen O2 in a proximal molecule, N–H1N···O2 (N1···O2 = 2.779(3) Å, angle at H1N = 155°). Additional soft C–H···O3S interactions interconnect these dimers into a three-dimensional array.
2.2. Catalytic Experiments
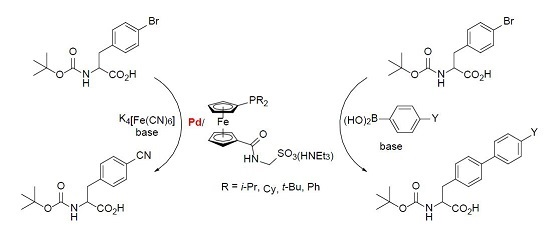
The series of phosphinoferrocene amidosulfonates 4a–d bearing different substituents at the phosphorus atom, which can be regarded as the primary coordination site for the catalytically active soft metal ions, was firstly evaluated in Pd-catalyzed cyanation of aryl bromides leading to the corresponding nitriles [11,12] using potassium hexacyanoferrate(II) as a practically non-toxic and environmentally benign cyanide source [24]. In particular, we chose the cyanation of N-Boc protected 4-bromophenylalanine 6 (Scheme 4) performed in dioxane–water (1:1) mixture at 100 °C in the presence of potassium carbonate as a base. The results collected in Table 3 indicate that the yield of the coupling product 7 depends strongly on the Pd source. At a Pd loading of 2 mol. % and with the model ligand 4d, the best catalyst performance resulted from [PdCl2(cod)] (cod = η2:η2-cycloocta-1,5-diene) and 2 equiv. of the phosphine ligand, which reached full conversion of 6 to 7 within 3 h. An analogous catalyst prepared at a 1:1 [PdCl2(cod)]:4d molar ratio ensued in only 30% conversion. Similar catalysts resulting from 4d and palladium(II) acetate, which is commonly used as a Pd-precursor in cross-coupling reactions [25] and even afforded very good yields of the coupling products in similar cyanation reactions [7,26], performed considerably worse (conversions <5%). Poor results were also obtained when [Pd(μ-Cl)(η-C3H5)]2 was employed as the Pd source, whereas the reaction performed in the presence of [PdCl2(cod)] (2 mol. %) without any supporting ligand did not proceed in any appreciable extent. Notably, the yields of the coupling product 7 markedly decreased when the supporting ligand 4d, used during the screening experiments, was replaced with its more electron-rich dialkylphosphino analogues 4a–c (see Table 3).
Next, we turned to Suzuki–Miyaura biaryl coupling, which is one of the most widely utilized cross-coupling reactions [14,15,16,17]. We chose the reactions of bromobenzoic (8o/8m/8p) and (bromophenyl)acetic (9o/9m/9p) acids with 4-fluorophenyl (10a) and 4-tolylboronic (10b) acids (Scheme 5) that can be advantageously performed in aqueous solvents, and used the most stable and accessible ligand 4d for the initial screening experiments. The reaction conditions were optimized for the coupling of 9p and 10a yielding biphenyl 12pa, which can be easily monitored by 1H and 19F NMR spectroscopy. The results are summarized in Table 4.
The first reaction tests were aimed at finding a suitable solvent (Table 4, entries 1–5). When performed in water at 40 °C and with a catalyst formed from [PdCl2(cod)] and 4d (1:2 ratio, 1 mol. %), the model reaction produced the coupling product 12pa in a decent 60% yield after 2 h and a 72% yield after 6 h. Better results were obtained in ethanol and, in particular, an ethanol–water 1:1 mixture, where the yield was 81% after 6 h. Reaction in pure dioxane proceeded to only a negligible extent (10% yield after 6 h), presumably for solubility reasons. However, the addition of water to the system markedly improved the reaction outcome. Thus, the yields of 12pa achieved in a 1:1 dioxane–water mixture were 82% after 2 h and quantitative after 6 h at 40 °C.
Evaluation of different Pd(II) precursors performed next (Table 4, entries 5–13) revealed that the best yield of the coupling product (85%) is obtained with a catalyst resulting in situ from palladium(II) acetate and 1 equiv. of the amidosulfonate ligand 4d. Addition of another equivalent of 4d slightly reduced the yield of 12pa. A different trend was noted for [PdCl2(cod)] in which case the catalyst obtained after the addition of 2 equiv. of 4d achieved a higher yield than the analogous catalyst after the addition of only 1 equiv. of the supporting ligand. The catalyst generated from [Pd(μ-Cl)(η-C3H5)]2 and 4d (1 equiv. per Pd atom) proved to be inactive. It is also noteworthy that unsupported palladium(II) acetate also catalyzed the reaction but the yield was substantially lower than for both tested Pd(OAc)2/4d catalysts.
In the last step, we have compared catalysts resulting from palladium(II) acetate and different ligands 4. For this purpose, the metal loading was reduced to 0.5 mol. % and the reaction was monitored after 2 h and 6 h to check whether the catalysts retain their activity. In the case of the Pd(OAc)2/4d catalyst, the yields achieved after 2 and 6 h were 85% and 89%, respectively. Analogous catalyst resulting from 4b showed a similar yield after 6 h (namely 88%) but a significantly lower yield after 2 h (only 74%), which may suggest a slower catalyst activation and/or slower reaction rate [6]. A similar situation was noted in the case of ligand 4a possessing diisopropylphosphino substituent, except that the yield of 12pa after 6 h was the highest among the catalysts tested (nearly quantitative). In contrast, catalysts resulting from the most bulky and electron-rich ligand 4c acquired the lowest conversions in the entire series, presumably due to rapid deactivation [10]. Based on these results, the Pd(OAc)2/4a catalyst (0.5 mol. % Pd) was chosen for the following reaction scope tests, which were limited to aryl bromides because the reaction with (4-chlorophenyl)acetic acid and 10a did not yield any coupling product (0.5 mol. % Pd(OAc)2/4a, dioxane–water, 40 or 80 °C/6 h).
The results collected in Table 5 reveal several trends. First of all, both 2-bromobenzoic and (2-bromophenyl)acetic acid reacted only sluggishly with arylboronic acids 10a/b, which can be ascribed to a steric hindrance. All other isomeric aryl bromide substrates reacted well, achieving very good to practically complete conversions. In all cases, the reactions with 10a bearing the electron-withdrawing substituent proceeded with higher conversions than those with 10b. The differences in pairs of the analogous reactions involving 10a and 10b were considerably larger for ortho- and para-substituted aryl bromides than for their meta-substituted counterparts, which in turn points to a dominant electronic influence (I- and M-effect).
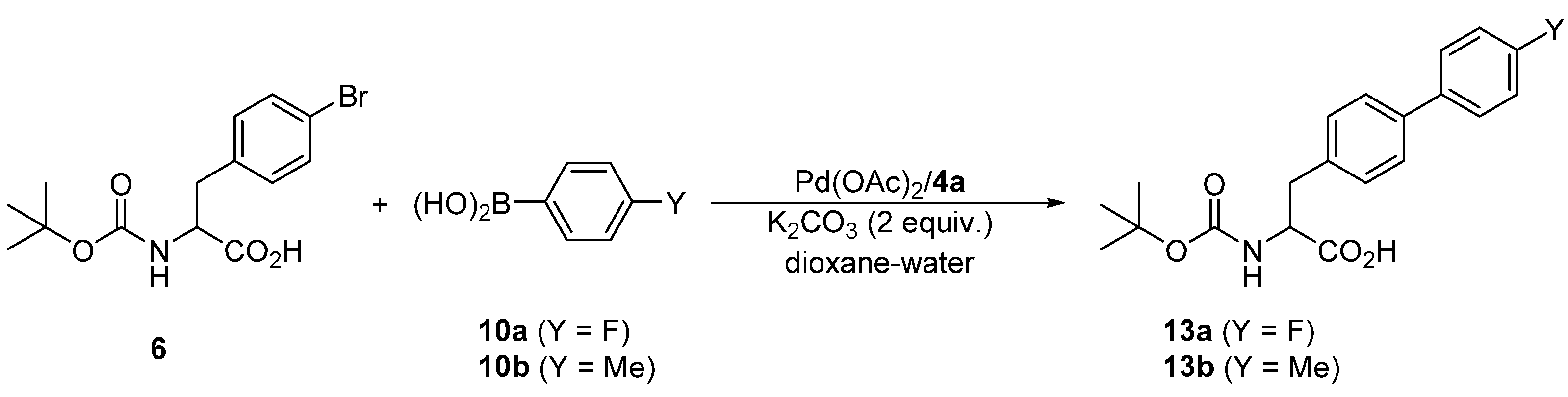
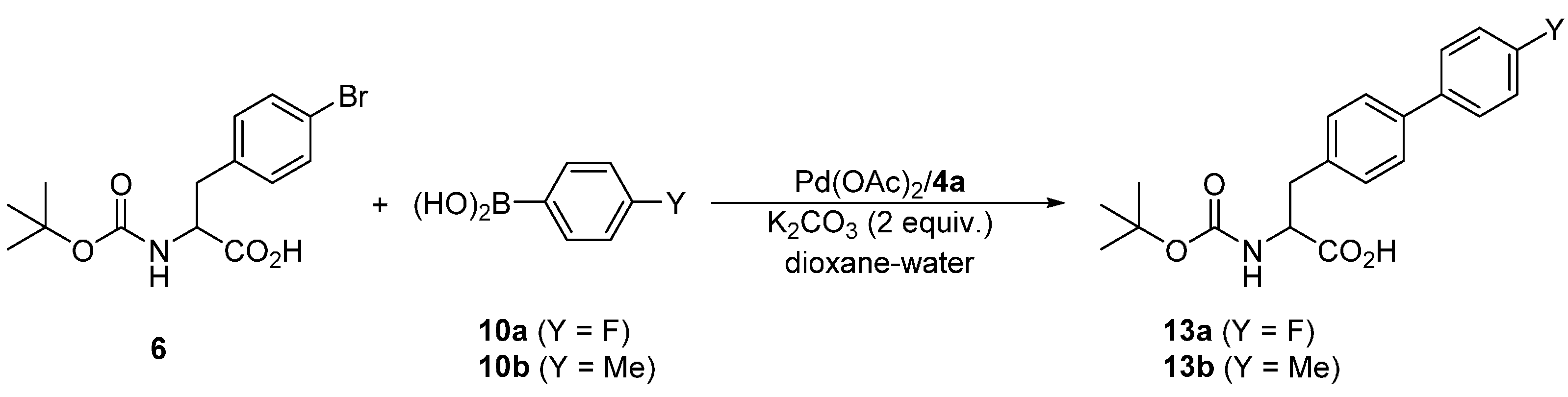
Attempted coupling of 4-bromophenyl alanine substrate 6 with 10a and 10b (Scheme 6) under similar conditions proceeded satisfactorily, producing the biphenyl amino acids 13a and 13b, respectively, in approximately 90% yields (Table 6). Unfortunately, these products could not be separated from unreacted 6a either by chromatography or crystallization due to a similar retention characteristic and reluctance to crystallize, respectively. Upon increasing the amount of the catalyst to 1 mol. %, the reaction achieved complete conversions within 6 h at 40 °C, which in turn allowed for the isolation of the pure products 13 in good yields. Notably, NMR analysis of the crude reaction mixtures revealed that the Boc-protecting group is stable under the reaction conditions, which is indeed manifested in the good yields.
3. Experimental
3.1. Materials and Methods
Compounds 1a–c [10] and 4d [7] were prepared according to the literature methods. Anhydrous dichloromethane was obtained from a PureSolv MD5 Solvent Purification System (Innovative Technology Inc., Amesbury, MA, USA). Triethylamine and dioxane were distilled from sodium metal. Other chemicals (Alfa-Aesar or Sigma-Aldrich, Ward Hill, MA, USA; Saint Louis, MO, USA), anhydrous N,N-dimethylformamide over molecular sieves (Sigma-Aldrich), anhydrous ethanol (Penta, Prague, Czech Republic) and all solvents (reagent grade from Lach-Ner, Neratovice, Czech Republic) used for workup, column chromatography and crystallizations were without any additional purification.
NMR spectra were recorded at 298 K on a Varian Unity Inova 400 spectrometer (1H, 399.95 MHz; 13C, 100.58 MHz; and 31P, 161.90 MHz) or a Bruker AVANCE III 400 spectrometer (1H, 400.13 MHz; 13C{1H}, 100.62 MHz; 19F, 376.46 MHz; and 31P, 161.97 MHz). Chemical shifts (δ/ppm) are given relative to internal tetramethylsilane (1H and 13C NMR), to external 85% aqueous H3PO4 (31P NMR), and to external neat CFCl3 (19F NMR), respectively. In addition to the standard notation of NMR signals, vt and vq are used to distinguish virtual triplets and quartets arising from the C=O and phosphine-substituted cyclopentadienyl rings, respectively. Conventional low-resolution electrospray ionization mass spectra (ESI MS) were recorded with an Esquire 3000 (Bruker). High-resolution (HR) analyses were performed with a compact Q-TOF (Bruker Daltonik) instrument. The samples were dissolved in HPLC-grade methanol. Infrared spectra were collected in Nujol mulls on a FTIR Thermo Fisher Nicolet 760 instrument in the range 400–4000 cm−1.
3.2. General Procedure for the Synthesis of Esters H3B·R2PfcCO2C6F5 (2a–c)
The respective acids H3B·R2PfcCO2H (1a–c, 5.0 mmol), 1-ethyl-3-[3-(dimethylamino)propyl]-carbodiimide hydrochloride (1.15 g, 6.0 mmol), pentafluorophenol (1.10 g, 6.0 mmol) and 4-(dimethylamino)pyridine (122 mg, 1.0 mmol) were dissolved in dichloromethane (50 mL). After stirring overnight, brine (50 mL) was added to the reaction mixture and stirring was continued for another 10 min. Then, the organic layer was separated and washed with brine (50 mL). The combined aqueous phases were back-extracted with dichloromethane (20 mL). The combined organic layers were dried over anhydrous MgSO4 and evaporated to dryness. The crude product was purified by flash column chromatography over silica gel as described below.
3.2.1. Preparation of iPr2PfcCO2C6F5·BH3 (2a)
Ester 2a was synthesized from acid 1a (1.80 g, 5.0 mmol) following the general procedure and isolated as an orange solid. Ethyl acetate-hexane (1:8) was used during the chromatography. Only the first band from the product was collected. Yield: 2.46 g (94%).
1H NMR (CDCl3): δ 0.15–1.10 (br m, 3 H, BH3), 1.16 (dd, 3JHH = 7.1 Hz, 3JPH = 2.7 Hz, 6 H, CHMe2), 1.19 (dd, 3JHH = 7.1 Hz, 3JPH = 3.5 Hz, 6 H, CHMe2), 2.16 (d of sept, 2JPH = 10.1 Hz, 3JHH = 7.1 Hz, 2 H, CHMe2), 4.52 (vq, J′ = 1.8 Hz, 2 H, fc), 4.61 (d of vt, J’ ≈ 0.9, 1.9 Hz, 2 H, fc), 4.80 (vt, J′ = 2.0 Hz, 2 H, fc), 5.06 (vt, J′ = 2.0 Hz, 2 H, fc). 13C{1H} NMR (CDCl3): δ 17.16 (s, 2 C, CHMe2), 17.47 (d, 2JPC = 2 Hz, 2 C, CHMe2), 22.58 (d, 1JPC = 35 Hz, 2 C, CHMe2), 68.05 (s, Cipso-CO of fc), 70.65 (d, 1JPC = 53 Hz, Cipso-P of fc), 72.32 (s, 2 C, CH of fc), 73.44 (d, JPC = 7 Hz, 2 C, CH of fc), 73.59 (d, JPC = 6 Hz, 2 C, CH of fc), 75.43 (s, 2 C, CH of fc), 125.16 (s, Cipso of C6F5), 137.96 (dm, 1JFC = 253 Hz, Cmeta of C6F5), 139.45 (dm, 1JFC = 253 Hz, Cpara of C6F5), 141.48 (dm, 1JFC = 250 Hz, Cortho of C6F5), 167.45 (s, C=O). 19F{1H} NMR (CDCl3): δ = −162.60 (m, Fmeta of C6F5), −158.34 (t, 3JFF = 22 Hz, Fpara of C6F5), −153.08 (m, Fortho of C6F5) ppm. 31P{1H} NMR (CDCl3): δ 31.9 (br d). IR (Nujol): 2373 m, 2342 m, 1768 s, 1522 s, 1305 w, 1261 (s), 1203 w, 1169 w, 1142 w, 1072 s, 1058 m, 1023 m, 1011 m, 974 m, 885 m, 870 w, 852 w, 833 w, 753 w, 639 w, 611 w, 531 w, 505 w cm−1. MS (ESI+): m/z 549.1 ([M + Na]+), 565.1 ([M + K]+). Anal. Calc. for C23H25BF5FeO2P (526.06): C 52.51, H 4.79%. Found: C 52.50, H 4.69%.
3.2.2. Preparation of Cy2PfcCO2C6F5·BH3 (2b)
Ester 2b was obtained from acid 1b (2.20 g, 5.0 mmol) as described above and isolated as an orange solid. Ethyl acetate-hexane (1:10) was used during the chromatography. Only the first band from the product was collected. Yield: 2.54 g (84%).
1H NMR (CDCl3): δ 0.15–1.05 (br m, 3 H, BH3), 1.10–1.40 (m, 10 H, Cy), 1.65–1.73 (m, 2 H, Cy), 1.75–2.01 (m, 10 H, Cy), 4.49 (vq, J′ = 1.8 Hz, 2 H, fc), 4.60 (d of vt, J′ ≈ 0.9, 1.8 Hz, 2 H, fc), 4.78 (vt, J′ = 2.0 Hz, 2 H, fc), 5.04 (vt, J′ = 2.0 Hz, 2 H, fc). 13C{1H} NMR (CDCl3): δ 25.89 (d, JPC = 1 Hz, 2 C, CH2 of Cy), 26.77 (d, JPC = 2 Hz, 2 C, CH2 of Cy), 26.88 (d, JPC = 1 Hz, 2 C, CH2 of Cy), 26.95 (s, 2 C, CH2 of Cy), 27.27 (d, JPC = 2 Hz, 2 C, CH2 of Cy), 32.33 (d, 1JPC = 34 Hz, 2 C, CH of Cy), 68.03 (s, Cipso-CO of fc), 71.31 (d, 1JPC = 53 Hz, Cipso-P of fc), 72.34 (s, 2 C, CH of fc), 73.42 (d, JPC = 6 Hz, 2 C, CH of fc), 73.58 (d, JPC = 7 Hz, 2 C, CH of fc), 75.39 (s, 2 C, CH of fc), 125.18 (m, Cipso of C6F5), 137.95 (dm, 1JFC = 252 Hz, Cmeta of C6F5), 139.44 (dm, 1JFC = 253 Hz, Cpara of C6F5), 141.38 (dm, 1JFC = 251 Hz, Cortho of C6F5), 167.46 (s, C=O). 19F{1H} NMR (CDCl3): δ = −162.62 (m, Fmeta of C6F5), −158.33 (t, 3JFF = 22 Hz, Fpara of C6F5), −153.10 (m, Fortho of C6F5) ppm. 31P{1H} NMR (CDCl3): δ 24.3 (br d). IR (Nujol): 2373 m, 2342 m, 1768 s, 1522 s, 1305 w, 1261 m, 1203 w, 1169 w, 1142 w, 1072 s, 1058 m, 1023 m, 1011 s, 974 m, 885 m, 870 w, 852 w, 833 w, 753 w, 639 w, 611 w, 531 w, 505 w cm−1. MS (ESI+): m/z 629.2 ([M + Na]+), 645.1 ([M + K]+). Anal. Calc. for C29H33BF5FeO2P (606.18): C 57.50, H 5.50%. Found: C 57.46, H 5.49%.
3.2.3. Preparation of H3B·tBu2PfcCO2C6F5 (2c)
Ester 2c was synthesized from 1c (1.94 g, 5.0 mmol) by using the general procedure and isolated as an orange solid. Ethyl acetate-hexane (1:8) was used during the column chromatography. Only the first band from the product was collected. Yield: 2.06 g (93%).
1H NMR (CDCl3): δ 0.20–1.20 (br m, 3 H, BH3), 1.30 (d, 3JPH = 12.9 Hz, 18 H, CMe3), 4.62–4.65 (m, 4 H, fc), 4.80 (vt, J′ = 2.0 Hz, 2 H, fc), 5.03 (vt, J′ = 2.0 Hz, 2 H, fc). 13C{1H} NMR (CDCl3): δ 28.64 (d, 2JPC = 2 Hz, 6 C, CMe3), 33.42 (d, 1JPC = 27 Hz, 2 C, CMe3), 67.95 (s, Cipso-CO of fc), 72.46 (s, 2 C, CH of fc), 72.76 (d, 1JPC = 48 Hz, Cipso-P of fc), 73.51 (d, JPC = 6 Hz, 2 C, CH of fc), 74.91 (d, JPC = 6 Hz, 2 C, CH of fc), 75.92 (s, 2 C, CH of fc), 125.18 (m, Cipso of C6F5), 137.92 (dm, 1JFC = 252 Hz, Cmeta of C6F5), 139.44 (dm, 1JFC = 253 Hz, Cpara of C6F5), 143.37 (dm, 1JFC = 251 Hz, Cortho of C6F5), 167.45 (s, C=O). 19F{1H} NMR (CDCl3): δ = −162.60 (m, Fmeta of C6F5), −158.35 (t, 3JFF = 22 Hz, Fpara of C6F5), −153.01 (m, Fortho of C6F5) ppm. 31P{1H} NMR (CDCl3): δ 45.5 (br d). IR (Nujol): 2391 m, 2363 m, 2344 m, 1752 s, 1521 s, 1304 w, 1267 s, 1162 w, 1144 w, 1080 s, 1057 w, 1026 m, 1006 w, 995 m, 980 w, 910 w, 895 m, 874 w, 852 w, 833 m, 815 w, 755 w, 647 w, 626 w, 573 w, 531 w, 514 w cm−1. MS (ESI+): m/z 577.2 ([M + Na]+), 593.1 ([M + K]+). Anal. Calc. for C25H29BF5FeO2P (554.11): C 54.19, H 5.28%. Found: C 54.18, H 5.22%.
3.3. General Procedure for the Synthesis of Amides R2PfcCONHCH2SO3(HNEt3)·BH3 (3a–c)
The respective ester 1 (3.0 mmol), aminomethanesulfonic acid (0.51 g, 4.5 mmol) and 4-(dimethylamino)pyridine (122 mg, 1.0 mmol) were dissolved in a mixture of N,N-dimethylformamide (10 mL) and triethylamine (2 mL). After stirring overnight, the solvents were evaporated under vacuum and the residual DMF was removed by trituration with diethyl ether (50 mL). The crude product was purified by flash column chromatography over silica gel. The column was packed in CH2Cl2–MeOH–Et3N (100:5:5) and the product was eluted with CH2Cl2–MeOH (100:5). The first orange band was collected and evaporated. The solid residue was triturated with diethyl ether (3 × 20 mL) to remove residual triethylamine and, finally, dried under vacuum.
3.3.1. Preparation of iPr2PfcCONHCH2SO3(HNEt3)·BH3 (3a)
Amide 3a was synthesized from ester 2a (1.58 g, 3.0 mmol) following the general procedure described above and isolated as an orange crystalline solid after crystallization from hot ethyl acetate. Yield: 1.32 g (79%).
1H NMR (CD2Cl2): δ 0.10–1.10 (br m, 3 H, BH3), 1.11 (dd, 3JHH = 7.1 Hz, 3JPH = 1.4 Hz, 6 H, CHMe2), 1.15 (d, 3JHH = 7.1 Hz, 6 H, CHMe2), 1.31 (t, 3JHH = 7.3 Hz, 9 H, CH3 of Et3NH+), 2.13 (d of sept, 2JPH = 10.1 Hz, 3JHH = 7.1 Hz, 2 H, CHMe2), 3.10 (q, 3JHH = 7.3 Hz, 6 H, CH2 of Et3NH+), 4.40 (vq, J′ = 1.7 Hz, 2 H, fc), 4.43 (d, 3JHH = 6.6 Hz, 2 H, CH2N), 4.50 (vt, J′ = 2.0 Hz, 2 H, fc), 4.65 (d of vt, J′ ≈ 0.9, 1.9 Hz, 2 H, fc), 4.83 (vt, J′ = 2.0 Hz, 2 H, fc), 6.92 (t, 3JHH = 6.5 Hz, 1 H, NH). 13C{1H} NMR (CD2Cl2): δ 8.85 (s, 3 C, CH3 of Et3NH+), 17.30 (s, 2 C, CHMe2), 17.62 (d, 2JPC = 2 Hz, 2 C, CHMe2), 22.82 (d, 1JPC = 35 Hz, 2 C, CHMe2), 46.46 (s, 3 C, CH2 of Et3NH+), 56.05 (s, CH2N), 69.36 (d, 1JPC = 55 Hz, Cipso-P of fc), 70.09 (s, 2 C, CH of fc), 73.35 (s, 2 C, CH of fc), 73.59 (s, 2 C, CH of fc), 73.66 (d, JPC = 1 Hz, 2 C, CH of fc), 76.96 (s, Cipso-CO of fc), 169.54 (s, C=O). 31P{1H} NMR (CD2Cl2): δ 31.6 (br d). IR (Nujol): 3312 m, 2376 w, 2360 m, 2332 m, 1654 s, 1533 s, 1499 m, 1366 m, 1313 w, 1285 w, 1245 m, 1216 m, 1165 s, 1070 m, 1035 m, 1012 m, 980 m, 899 w, 888 w, 868 w, 852 w, 840 m, 831 m, 812 w, 797 w, 754 m, 692 w, 667 w, 632 w, 615 m, 585 w, 535 w, 515 m cm−1. MS (ESI+): m/z 555.2 ([M + H]+), 656.4 ([M + HNEt3]+); MS (ESI−): m/z 451.9 ([M − HNEt3]+). Anal. Calc. for C24H44BFeN2O4PS (554.30): C 52.00, H 8.00, N 5.06%. Found: C 51.90, H 8.03, N 4.93%.
3.3.2. Preparation of Cy2PfcCONHCH2SO3(HNEt3)·BH3 (3b)
Amide 3b was synthesized from 2b (1.82 g, 3.0 mmol) according to the general procedure and was isolated as an orange crystalline solid after crystallization from hot ethyl acetate. Yield: 1.67 g (88%).
1H NMR (CD2Cl2): δ 0.04–1.04 (br m, 3 H, BH3), 1.08–1.38 (m, 10 H, Cy), 1.31 (t, 3JHH = 7.3 Hz, 9 H, CH3 of Et3NH+), 1.64–1.71 (m, 2 H, Cy), 1.74–1.99 (m, 10 H, Cy), 3.11 (q, 3JHH = 7.3 Hz, 6 H, CH2 of Et3NH+), 4.37 (vq, J′ = 1.8 Hz, 2 H, fc), 4.42 (d, 3JHH = 6.6 Hz, 2 H, CH2N), 4.47 (vt, J′ = 2.0 Hz, 2 H, fc), 4.64 (d of vt, J′ ≈ 0.9, 1.8 Hz, 2 H, fc), 4.80 (vt, J′ = 1.9 Hz, 2 H, fc), 6.85 (t, 3JHH = 6.6 Hz, 1 H, NH). 13C{1H} NMR (CD2Cl2): δ 8.87 (s, 3 C, CH3 of Et3NH+), 26.39 (d, JPC = 2 Hz, 2 C, CH2 of Cy), 27.15–27.30 (m, 6 C, CH2 of Cy), 27.57 (d, JPC = 2 Hz, 2 C, CH2 of Cy), 32.49 (d, 1JPC = 35 Hz, 2 C, CH of Cy), 46.50 (s, 3 C, CH2 of Et3NH+), 56.02 (s, CH2N), 69.99 (d, 1JPC = 55 Hz, Cipso-P of fc), 70.04 (s, 2 C, CH of fc), 73.35 (s, 2 C, CH of fc), 73.48 (d, JPC = 7 Hz, 2 C, CH of fc), 73.70 (d, JPC = 7 Hz, 2 C, CH of fc), 76.98 (s, Cipso-CO of fc), 169.44 (s, C=O). 31P{1H} NMR (CD2Cl2): δ 24.1 (br d). IR (Nujol): 3531 m, 3456 m, 3337 m, 2378 m, 2334 m, 1648 s, 1523 m, 1296 w, 1253 w, 1211 m, 1168 s, 1053 m, 890 w, 853 w, 826 w, 765 w, 635 w, 616 w, 528 w, 506 w cm−1. MS (ESI+): m/z 635.2 ([M + H]+), 736.5 ([M + HNEt3]+); MS (ESI−): m/z 532.0 ([M − HNEt3]+). Anal. Calc. for C30H52BFeN2O4PS (634.43): C 56.79, H 8.26, N 4.42%. Found: C 56.64, H 8.31, N 4.29%.
3.3.3. Preparation of tBu2PfcCONHCH2SO3(HNEt3)·BH3 (3c)
Amide 3c was synthesized from 2c (1.66 g, 3.0 mmol) according to the general procedure and isolated as an orange solid. The compound decomposes upon attempted crystallization. Yield: 1.62 g (92%).
1H NMR (CD2Cl2): δ 0.15–1.15 (br m, 3 H, BH3), 1.27 (d, 3JPH = 12.8 Hz, 18 H, CMe3), 1.31 (t, 3JHH = 7.3 Hz, 9 H, CH3 of Et3NH+), 3.10 (q, 3JHH = 7.3 Hz, 6 H, CH2 of Et3NH+), 4.43 (d, 3JHH = 6.6 Hz, 2 H, CH2N), 4.48 (vt, J′ = 2.0 Hz, 2 H, fc), 4.50 (vq, J′ = 1.7 Hz, 2 H, fc), 4.69 (d of vt, J′ ≈ 0.9, 1.9 Hz, 2 H, fc), 4.80 (vt, J′ = 2.0 Hz, 2 H, fc), 6.91 (t, 3JHH = 6.5 Hz, 1 H, NH). 13C{1H} NMR (CD2Cl2): δ 8.86 (s, 3 C, CH3 of Et3NH+), 28.84 (d, 2JPC = 2 Hz, 6 C, CMe3), 33.60 (d, 1JPC = 28 Hz, 2 C, CMe3), 46.44 (s, 3 C, CH2 of Et3NH+), 56.03 (s, CH2N), 70.23 (s, 2 C, CH of fc), 71.66 (d, 1JPC = 49 Hz, Cipso-P of fc), 73.49 (d, JPC = 6 Hz, 2 C, CH of fc), 73.85 (s, 2 C, CH of fc), 75.16 (d, JPC = 7 Hz, 2 C, CH of fc), 76.93 (s, Cipso-CO of fc), 169.42 (s, C=O). 31P{1H} NMR (CD2Cl2): δ 45.1 (br m). IR (Nujol): 3296 m, 2382 m, 2363 m, 2343 m, 1656 s, 1517 m, 1498 m, 1316 w, 1299 w, 1251 w, 1211 m, 1161 s, 1068 w, 1039 s, 1014 m, 983 m, 895 w, 858 w, 838 w, 814 w, 751 w, 647 w, 613 w, 598 w, 516 w cm−1. MS (ESI+): m/z 684.4 ([M + HNEt3]+), 605.3 ([M + Na]+); MS (ESI−): m/z 479.9 ([M − HNEt3]+). Anal. Calc. for C26H48BFeN2O4PS·0.1CH2Cl2 (590.84): C 53.05, H 8.22, N 4.74%. Found: C 52.98, H 7.88, N 4.49%.
3.4. General Procedure for Deprotection of Borane Aducts 3
A Schlenk flask was charged with a borane adduct 3 (1.5 mmol) and freshly distilled morpholine (10 mL). The reaction mixture was thoroughly degassed by five freeze–pump–thaw cycles and then heated at 65 °C for 16 h. Next, the morpholine was removed under vacuum and the crude product was purified by flash column chromatography over silica gel. The chromatographic column was packed in CH2Cl2–MeOH–Et3N (100:5:5) and the product was eluted with CH2Cl2–MeOH (100:5). The first orange band was collected and evaporated. The residue was triturated with diethyl ether (3 × 20 mL) to remove residual triethylamine.
3.4.1. Preparation of iPr2PfcCONHCH2SO3(HNEt3) (4a)
Amide 4a was prepared from 3a (0.831 g, 1.5 mmol) as outlined above and isolated as an orange crystalline solid after crystallization from hot ethyl acetate. Yield: 0.657 g (81%).
1H NMR (CD2Cl2): δ 1.05 (dd, 3JHH = 7.0 Hz, 3JPH = 2.1 Hz, 6 H, CHMe2), 1.08 (dd, 3JHH = 7.0 Hz, 3JPH = 4.2 Hz, 6 H, CHMe2), 1.32 (t, 3JHH = 7.3 Hz, 9 H, CH3 of Et3NH+), 1.91 (d of sept, 2JPH = 2.0 Hz, 3JHH = 7.3 Hz, 2 H, CHMe2), 3.12 (q, 3JHH = 7.3 Hz, 6 H, CH2 of Et3NH+), 4.25 (vq, J′ = 1.6 Hz, 2 H, fc), 4.31 (vt, J′ = 1.9 Hz, 2 H, fc), 4.41 (d, 3JHH = 6.5 Hz, 2 H, CH2N), 4.47 (vt, J′ = 1.8 Hz, 2 H, fc), 4.68 (vt, J′ = 1.9 Hz, 2 H, fc), 6.77 (t, 3JHH = 5.8 Hz, 1 H, NH). 13C{1H} NMR (CD2Cl2): δ 8.90 (s, 3 C, CH3 of Et3NH+), 20.03 (d, 2JPC = 11 Hz, 2 C, CHMe2), 20.25 (d, 2JPC = 15 Hz, 2 C, CHMe2), 23.76 (d, 1JPC = 12 Hz, 2 C, CHMe2), 46.61 (s, 2 C, CH2 of Et3NH+), 56.08 (s, CH2N), 69.66 (s, 2 C, CH of fc), 72.21 (d, JPC = 2 Hz, 2 C, CH of fc), 72.75 (s, 2 C, CH of fc), 73.13 (d, JPC = 10 Hz, 2 C, CH of fc), 76.28 (s, Cipso-CO of fc), 76.28 (d, 1JPC = 10 Hz, Cipso-P of fc), 170.09 (s, C=O). 31P{1H} NMR (CD2Cl2): δ − 0.1 (s). IR (Nujol): 3509 s, 3448 s, 3285 s, 1659 s, 1637 s, 1540 s, 1342 w, 1315 m, 1286 m, 1243 m, 1209 m, 1157 s, 1073 w, 1038 s, 964 w, 893 w, 834 m, 769 w, 658 w, 635 w, 616 m, 595 w, 541 w, 525 w, 517 w cm−1. MS (ESI+): m/z 440.0 ([M + H − NEt3]+), 541.2 ([M + H]+); MS (ESI−): m/z 437.9 ([M − HNEt3]+). Anal. Calc. for C24H41BFeN2O4PS (540.47): C 53.33, H 7.65, N 5.18%. Found: C 53.07, H 7.69, 5.24%.
3.4.2. Preparation of Cy2PfcCONHCH2SO3(HNEt3) (4b)
Amide 4b was synthesized from 3b (0.952 g, 1.5 mmol) by following the general procedure and isolated as an orange crystalline solid after crystallization from hot ethyl acetate. Yield: 0.78 g (84%).
1H NMR (CD2Cl2): δ 0.96–1.37 (m, 10 H, Cy), 1.32 (t, 3JHH = 7.3 Hz, 9 H, CH3 of Et3NH+), 1.61–1.82 (m, 10 H, Cy), 1.86–1.95 (m, 2 H, Cy), 3.12 (q, 3JHH = 7.3 Hz, 6 H, CH2 of Et3NH+), 4.22 (vq, J′ = 1.6 Hz, 2 H, fc), 4.29 (vt, J′ = 1.9 Hz, 2 H, fc), 4.42 (d, 3JHH = 6.5 Hz, 2 H, CH2N), 4.46 (vt, J′ = 1.8 Hz, 2 H, fc), 4.67 (vt, J′ = 1.9 Hz, 2 H, fc), 6.86 (t, 3JHH = 6.3 Hz, 1 H, NH). 13C{1H} NMR (CD2Cl2): δ 8.91 (s, 3 C, CH3 of Et3NH+), 26.87 (d, JPC = 1 Hz, 2 C, CH2 of Cy), 27.65 (d, JPC = 9 Hz, 2 C, CH2 of Cy), 27.78 (d, JPC = 11 Hz, 2 C, CH2 of Cy), 30.48 (d, JPC = 2 Hz, 2 C, CH2 of Cy), 30.60 (s, 2 C, CH2 of Cy), 33.73 (d, JPC = 12 Hz, 2 C, CH of Cy), 46.63 (s, 3 C, CH2 of Et3NH+), 56.09 (s, CH2N), 69.67 (s, 2 C, CH of fc), 72.16 (d, JPC = 3 Hz, 2 C, CH of fc), 72.76 (d, JPC = 1 Hz, 2 C, CH of fc), 73.32 (d, JPC = 10 Hz, 2 C, CH of fc), 76.23 (s, Cipso-CO of fc), 170.21 (s, C=O). One ferrocene resonance (Cipso-P) was not identified, presumably due to overlaps. 31P{1H} NMR (CD2Cl2): δ − 8.3 (s). IR (Nujol): 3309 s, 1653 s, 1533 s, 1314 m, 1284 m, 1247 w, 1217 m, 1169 s, 1077 w, 1044 s, 966 w, 891 w, 847 w, 831 w, 771 w, 613 m, 547 w, 533 w, 516 w cm−1. MS (ESI+): m/z 621.3 ([M + H]+); MS (ESI−): m/z 518.0 ([M − HNEt3]+). Anal. Calc. for C30H49BFeN2O4PS (620.59): C 58.06, H 7.96, N 4.52%. Found: C 57.81, H 7.82, N 4.50%.
3.4.3. Preparation of tBu2PfcCONHCH2SO3(HNEt3) (4c)
Amide 4c was synthesized from 3c (0.874 g, 1.5 mmol) as described above and isolated as an orange solid. Yield: 0.605 g (71%).
1H NMR (CD2Cl2): δ 1.18 (d, 3JPH = 11.2 Hz, 18 H, CMe3), 1.32 (t, 3JHH = 6.0 Hz, 9 H, CH3 of Et3NH+), 3.12 (q, 3JHH = 6.4 Hz, 6 H, CH2 of Et3NH+), 4.32–4.45 (m, 4 H, fc), 4.42 (d, 3JHH = 6.5 Hz, 2 H, CH2N), 4.54 (vt, J′ = 1.8 Hz, 2 H, fc), 4.71 (vt, J′ = 1.9 Hz, 2 H, fc), 6.76 (t, 3JHH = 5.4 Hz, 1 H, NH). 13C{1H} NMR (CD2Cl2): δ 8.92 (s, 3 C, CH3 of Et3NH+), 30.92 (d, 2JPC = 13 Hz, 6 C, CMe3), 33.00 (d, 1JPC = 20 Hz, 2 C, CMe3), 46.58 (s, 3 C, CH2 of Et3NH+), 56.10 (s, CH2N), 69.72 (s, 2 C, CH of fc), 72.17 (d, JPC = 3 Hz, 2 C, CH of fc), 73.34 (s, 2 C, CH of fc), 74.65 (d, JPC = 12 Hz, 2 C, CH of fc), 76.11 (s, Cipso-CO of fc), 170.00 (s, C=O). One ferrocene resonance (Cipso-P) was not identified, presumably due to overlaps. 31P{1H} NMR (CD2Cl2): δ 27.3 (s). IR (Nujol): 3448 s, 3324 s, 1644 s, 1541 s, 1317 m, 1292 m, 1186 s, 1072 w, 1039 s, 936 w, 896 w, 837 m, 812 m, 756 w, 734 w, 617 m, 522 m cm−1. MS (ESI+): m/z 468.1 ([M + H − NEt3]+), 569.2 ([M + H]+); MS (ESI−): m/z 465.9 ([M − HNEt3]+). Anal. Calc. for C26H45FeN2O4PS·0.3CH2Cl2 (594.00): C 53.18, H 7.74, N 4.72%. Found: C 53.16, H 7.75, N 4.53%.
3.4.4. Preparation of [Pd(Cy2PfcCONHCH2SO3-κ2O,P)(η3-C3H5)] (5b)
Solid [Pd(μ-Cl)(η3-C3H5)]2 (18.3 mg, 0.05 mmol) was added to a solution of 4b (62.1 mg, 0.1 mmol) in dichloromethane (5 mL). After stirring for 30 min, a solution of AgClO4 (21.0 mg, 0.1 mmol) in benzene (1 mL) was added causing immediate precipitation of AgCl. The resulting mixture was stirred for 1 h and filtered through a syringe filter (PTFE, 0.45 μm pore size). The filtrate was evaporated under vacuum to afford a crude product, which was purified by two subsequent crystallizations by liquid-phase diffusion of diethyl ether into a chloroform solution of the complex to fully remove triethylammonium perchlorate. Yield after two crystallizations: 41 mg (62%).
1H NMR (CDCl3): δ 1.11–1.44 (m, 11 H, Cy), 1.67–2.09 (m, 11 H, Cy), 4.41 (br m, 2 H, fc), 4.53 (br m, 2 H, fc), 4.60 (br d, 3JHH = 5.4 Hz, 2 H, CH2N), 4.76 (br m, 2 H, fc), 4.87 (br m, 2 H, fc), 5.67 (quint, 3JHH = 9.9 Hz, 1 H, CH of C3H5), 7.14 (br s, 1 H, NH). 31P{1H} NMR (CDCl3): δ 27.7 (s). IR (Nujol): 3240 s, 1588 s, 1566 s, 1329 w, 1297 m, 1258 m, 1249 w, 1226 w, 1204 s, 1175 s, 1091 s, 1039 s, 1007 m, 976 w, 959 w, 937 w, 921 w, 846 m, 836 w, 771 w, 743 m, 619 m, 604 w, 594 w, 548 w, 529 w, 500 w. MS (ESI+): m/z 688.1 ([M + Na]+). Anal. Calc. for C27H38FeNO4PPdS (665.86): C 48.70, H 5.75, N 2.10%. Found: C 48.52, H 5.68, N 2.04%.
3.5. Catalytic Tests in Pd-Catalyzed Cyanation
A solution of ligand (2, 4 or 8 mol. % with respect to the aryl bromide 6) in dry dichloromethane (3 mL) was added to the respective palladium source (2 or 4 mol. %) placed in the reaction vessel; the obtained mixture was stirred for 5 min and then evaporated under vacuum. Anhydrous potassium hexacyanoferrate(II) (184 mg, 0.5 mmol), potassium carbonate (138 mg, 1.0 mmol) and 6 (172 mg, 0.5 mmol) were added to the pre-formed catalyst. The flask was equipped with a magnetic stirring bar, flushed with argon, and sealed with a septum. Solvent (1,4-dioxane/water, 1:1; 4 mL) was introduced, the septum was replaced with a glass stopper, and the reaction flask was transferred to an oil bath maintained at 100 °C. After stirring for 3 h, the reaction mixture was cooled to room temperature and diluted with water (10 mL), ethyl acetate (5 mL) and 3 M HCl (3 mL). The organic layer was separated and washed with brine (10 mL). The aqueous layer was back-extracted with ethyl acetate (3 × 5 mL). The combined organic layers were dried over anhydrous magnesium sulfate and evaporated under reduced pressure. The conversion was determined by integration of 1H NMR spectrum.
Characterization data for the coupling product 7. 1H NMR (CDCl3): δ 1.35 (s, 9 H, CH3), 2.95–3.38 (m, 2 H, CH2), 4.25–5.10 (m, 2 H, CH and NH), 7.32 and 7.60 (2 × d, JHH = 8.0 Hz, 2 H, C6H4). The NMR data are in accordance with the literature [27].
3.6. Catalytic Tests in Pd-Catalyzed Suzuki–Miyaura Cross-Coupling
The procedure was modified from ref. [28]. Thus, palladium(II) acetate (1.0 or 0.5 mol. %) and the respective ligand 4 (1 or 2 equiv. with respect to Pd) were placed into a Schlenk tube and dissolved in dichloromethane (2 mL) under argon. The mixture was stirred for 5 min and then evaporated under vacuum. Next, aryl halide (1.00 mmol), boronic acid (1.15 mmol) and K2CO3 (2.00 mmol) were added to the Schlenk tube and the reaction vessel was filled with argon and sealed with a rubber septum. Degassed water (2 mL) and dioxane (2 mL) were introduced and the reaction flask was placed into a preheated oil bath (40 °C). After stirring for 6 h, the reaction was terminated by cooling on ice and the simultaneous addition of 3 M aqueous HCl (3 mL), water (7 mL) and ethyl acetate (10 mL). The aqueous layer was separated and extracted with ethyl acetate (3 × 15 mL). The organic layers were combined, washed with brine (30 mL), dried over MgSO4 and evaporated under reduced pressure. Conversion was determined by 1H NMR spectroscopy.
If appropriate, the coupling products were isolated as follows. K2CO3 (ca. 300 mg) was added to the crude product and the mixture was dissolved in water (10 mL). Brine (10 mL) and NaCl (ca. 300 mg) were added to the solution and the resulting precipitate was filtered. The collected precipitate was dissolved in boiling water (100 mL) and filtered immediately by suction to remove the by-product resulting from self-coupling of aryl boronic acids. The filtrate was cooled to laboratory temperature and acidified with dilute hydrochloric acid until pH 3–4 was reached whereupon a white precipitate formed. The separated product was filtered off, washed with distilled water (20 mL) and then taken up with ethyl acetate (30 mL). The solution was dried over MgSO4 and evaporated under reduced pressure to afford pure coupling product, which was analyzed by 1H NMR spectroscopy. Characterization data of the coupling products were as follows.
2-(4-Fluorophenyl)benzoic acid (11oa). 1H NMR (CDCl3): δ 7.13–7.01 (m, 2 H), 7.31–7.25 (m, 2 H), 7.33 (dd, J = 7.8, 1.1 Hz, 1H), 7.43 (td, J = 7.6, 1.4 Hz, 1H), 7.57 (td, J = 7.6, 1.4 Hz), 7.96 (dd, J = 7.8, 1.1 Hz, 1H), (all aromatics). The collected data correspond to those in the literature; see ref. [29].
3-(4-Fluorophenyl)benzoic acid (11ma). 1H NMR (dmso-d6): δ 7.28–8.17 (m, 8 H, aromatics), 13.16 (br s, 1 H, CO2H). The NMR data are in line with those in the literature (see ref. [27]).
4-(4-Fluorophenyl)benzoic acid (11pa). 1H NMR (dmso-d6): δ 7.30–7.37 (m, 2 H, aromatics), 7.75–7.83 (m, 4 H, aromatics), 7.98–8.04 (m, 2 H, aromatics), 13.02 (br s, 1 H, CO2H). The data are in accordance with those in the literature; see ref. [27].
2-(4-Tolyl)benzoic acid (11ob). 1H NMR (CDCl3): δ 2.38 (s, 3 H, CH3), 7.26–7.14 (m, 4 H, aromatics), 7.35 (dd, J = 7.7, 0.9 Hz, 1 H, aromatics), 7.39 (td, J = 7.6, 1.4 Hz, 1 H, aromatics), 7.53 (td, J = 7.6, 1.4 Hz, 1 H, aromatics), 7.92 (dd, J = 7.8, 1.1 Hz, 1 H, aromatics). The analytical data are in agreement with those in the literature, see ref. [28].
3-(4-Tolyl)benzoic acid (11mb). 1H NMR (dmso-d6): δ 2.35 (s, 3 H, CH3), 7.27–8.19 (m, 8 H, aromatics). The NMR data are in agreement with those in the literature; see ref. [27].
4-(4-Tolyl)benzoic acid (11pb). 1H NMR (CDCl3): δ 2.42 (s, 3 H, CH3) 7.27–7.32 (m, 2 H, aromatics), 7.52–7.57 (m, 2 H, aromatics), 7.66–7.71 (m, 2 H, aromatics), 8.13–8.18 (m, 2 H, aromatics). These data correspond with those in the literature; see ref. [27].
2-(4-Fluorophenyl)phenylacetic acid (12oa). 1H NMR (dmso-d6): δ 3.50 (s, 2 H, CH2), 7.21–7.38 (m, 8 H, aromatics), 12.27 (br s, 1 H, CO2H). 13C{1H} NMR (dmso-d6): δ 38.49 (s, CH2), 115.06 (d, 2JCF = 21 Hz, 2 C, aromatic CH), 126.91 (s, CH), 127.45 (s, CH) 129.76 (s, aromatic CH), 130.79 (s, CH), 130.83 (d, 3JCF = 7.9 Hz, 2 C, aromatic CH), 132.53 (s, aromatic C), 137.06 (d, 4JCF = 3 Hz, aromatic C), 140.76 (s, aromatic C), 161.42 (d, 1JCF = 244 Hz, aromatic C), 172.67 (s, CO2H). HRMS (ESI−) calc. for C14H10FO2 ([M − H]−): 229.0665, found 229.0670.
3-(4-Fluorophenyl)phenylacetic acid (12ma). 1H NMR (dmso-d6): δ 3.65 (s, 2 H, CH2), 2.24–2.33 (m, 3 H, aromatics), 7.37–7.43 (m, 1 H, aromatics), 7.50–7.55 (m, 2 H, aromatics), 7.65–7.71 (m, 2 H, aromatics), 12.35 (br s, 1 H, CO2H). 13C{1H} NMR (dmso-d6): δ 40.61 (s, CH2), 115.68 (d, 2JCF = 21 Hz, 2 C aromatic CH), 124.88 (s, aromatic CH), 127.79 (s, aromatic CH), 128.56 (s, aromatic CH), 128.60 (d, 3JCF = 7.9 Hz, 2 C aromatic CH) 128.83 (s, aromatic CH) 135.72 (s, aromatic C), 136.50 (d, 4JCF = 3 Hz, aromatic C), 139.04 (s, aromatic C), 161.82 (d, 1JCF = 244 Hz, aromatic C), 172.62 (s, CO2H). HRMS (ESI−) calc. for C14H10FO2 ([M − H]−): 229.0665, found 229.0669.
4-(4-Fluorophenyl)phenylacetic acid (12pa). 1H NMR (dmso-d6): δ 3.61 (s, 2 H, CH2), 7.24–7.74 (m, 8 H, aromatics), 12.38 (br s, 1 H, CO2H). The data are in accordance with those in the literature; see ref. [30].
2-(4-Tolyl)phenylacetic acid (12ob). 1H NMR (CDCl3): δ 2.39 (s, 3 H, CH3), 3.59 (s, 2 H, CH2), 7.25–7.55 (m, 8 H, aromatics). The NMR parameters are in agreement with those in the literature; see ref. [31].
3-(4-Tolyl)phenylacetic acid (12mb). 1H NMR (dmso-d6): δ 2.34 (s, 3 H, CH3), 3.64 (s, 2 H, CH2), 7.20–7.56 (m, 8 H, aromatics), 12.35 (br s, 1 H, CO2H). 13C{1H} NMR (dmso-d6): δ 20.67 (s, CH3), 40.66 (s, CH2), 124.68 (s, aromatic CH), 126.45 (s, 2 C, aromatic CH), 127.56 (s, aromatic CH), 128.17(s, aromatic CH), 128.78(s, aromatic CH), 129.51 (s, 2 C, aromatic CH), 135.62 (s, aromatic C), 136.72 (s, aromatic C), 137.15(s, aromatic C), 140.013 (s, aromatic C), 172.69 (s, CO2H). HRMS (ESI−) calc. for C15H13O2 ([M − H]−): 225.0916, found 225.0957.
4-(4-Tolyl)phenylacetic acid (12pb). 1H NMR (CDCl3): δ 2.39 (s, 3 H, CH3), 3.59 (s, 2 H, CH2), 7.25–7.55 (m, 8 H, aromatics). The data correspond with those in the literature (ref. [27]).
Rac-2-{[(tert-butyloxy)carbonyl]amino}-3-(4′-fluorobiphenyl-4-yl)propionic acid (13a). 1H NMR (dmso-d6): δ 1.32 (s, 9 H, CMe3) 2.83–2.89 (m, 1 H, CH2), 3.03–3.08 (m, 1 H, CH2), 4.01–4.15 (m, 1 H, CH), 7.16–7.09 (m, 1 H), 7.13 (d, 3JHH = 8.3 Hz, 1 H, NH), 7.24–7.30 (m, 2 H, aromatics), 7.33 (d, J = 8.1 Hz, 2 H, aromatics), 7.56 (d, J = 8.2 Hz, 2 H, aromatics), 12.65 (br s, 1 H, CO2H). The data are in accordance with those reported in ref. [32].
Rac-2-{[(tert-butyloxy)carbonyl]amino}-3-(4′-methylbiphenyl-4-yl)propionic acid (13b). 1H NMR (dmso-d6): δ 1.32 (s, 9 H, CMe3), 2.33 (s, 3 H, CH3), 2.76–3.08 (m, 2 H, CH2), 4.01‑4.16 (m, 1 H, CH), 7.12 (d,3JHH = 8.3 Hz, 1 H, NH), 7.24–7.26 (m, 2 H, aromatics), 7.30–7.32 (m, 2 H, aromatics), 7.51–7.56 (m, 4 H, aromatics), 12.63 (br s, 1 H, CO2H). The analytical data are in agreement with those in the literature (see ref. [8]).
3.7. X-ray Crystallography
The crystals of 3b·CH2Cl2 (orange prism, 0.17 × 0.20 × 0.25 mm3) and 4b (orange plate, 0.07 × 0.17 × 0.23 mm3) were grown from dichloromethane–hexane and ethyl acetate–heptane, respectively. Full-set diffraction data for both compounds (±h ± k ± l, θmax = 27.5°) were collected on a Bruker D8 VENTURE Kappa Duo PHOTON100 diffractometer equipped with IµS micro-focus sealed tube (MoKα radiation, λ = 0.71073 Å) and a Cryostream cooling device (Oxford Cryosystems) at 150(2) K.
Selected crystallographic data for 3b·CH2Cl2: C31H54BCl2FeN2O4PS·CH2Cl2 (M = 719.35 g mol−1), triclinic, space group P − 1 (no. 2); a = 8.3583(3) Å, b = 11.0812(4) Å, c = 20.6164(8) Å, α = 99.276(1)°, β = 100.551(1)°, γ = 102.947(1)°; V = 1788.3(1) Å3, Z = 2, Dcalc = 1.336 g mL−1, F(000) = 764, μ(MoKα) = 0.711 mm−1. A total of 37,660 diffractions were collected, of which were 8211 unique (Rint = 2.93%) and 7117 observed according to the Io > 2σ(Io) criterion.
Selected crystallographic data for 4b: C30H49FeN2O4PS (M = 620.59 g mol−1), monoclinic, space group P21/c (no. 14); a = 17.3904(6) Å, b = 9.1114(3) Å, c = 19.7014(7) Å, β = 97.552(1)°; V = 3094.6(2) Å3, Z = 4, Dcalc = 1.332 g mL−1, F(000) = 1328, μ(MoKα) = 0.643 mm−1. A total of 82,146 diffractions were collected, of which were 7125 unique (Rint = 4.12%) and 3275 observed according to the Io > 2σ(Io) criterion.
The structures of 3b·CH2Cl2 and 4b were solved by direct methods (XT2014 [33]) and refined by unrestricted least-squares against F2 (SHELXL-97 [34] or SHELXL-2014 [35]). All non-hydrogen atoms were refined with anisotropic displacement parameters. The amide hydrogens H1N were identified on an electron density map and refined as riding atom with Uiso(H1N) set to 1.2Ueq(N). Hydrogen atoms in the CHn groups were included in their theoretical positions using the standard HFIX instructions in SHELXL-2014 and refined as riding atoms. A recent version of the PLATON program [36] was used to perform all geometric calculations and prepare the structural diagrams.
In the case of 3b·CH2Cl2, the refinement converged (Δ/σ ≤ 0.002, 391 parameters) to R = 2.96% and wR = 7.02% for the observed diffractions and to R = 3.84% and wR = 7.54% for all diffractions. Extremes on the final difference electron density map were: Δρmax = 0.72, Δρmin = −0.79 e Å−3. CCDC deposition number: 1545049.
For 4b, the refinement converged (Δ/σ ≤ 0.002, 355 parameters) to R = 2.68% and wR = 6.53% for the observed diffractions and to R = 3.31% and wR = 6.83% for all diffractions. Extremes on the final difference electron density map were: Δρmax = 0.36, Δρmin = −0.36 e Å−3. CCDC deposition number: 1545048.
An orange, bar-like crystal of 5b·CH2Cl2 with approximate dimensions of 0.14 × 0.25 × 0.36 mm3 was obtained by recrystallization of the complex from dichloromethane–diethyl ether. Full-diffraction data (±h ± k ± l, θmax = 27.56°) were collected at 150(2) K with a Nonius Kappa CCD diffractometer equipped with a Bruker APEX-II image plate detector and a Cryostream cooling device (Oxford Cryosystems) using MoKα radiation (λ = 0.71073 Å). A total of 26,856 diffractions were collected, of which 7049 were independent (Rint = 2.98%) and 5970 observed according to the Io > 2σ(Io) criterion. Selected crystallographic data: C27H38FeNO4PPdS·CH2Cl2 (M = 750.79 g mol−1), monoclinic, space group P2/c (no. 13); a = 17.257(1) Å, b = 10.4304(6) Å, c = 17.2618(8) Å, β = 100.882(2)°; V = 3051.2(3) Å3, Z = 4, Dcalc = 1.634 g mL−1, F(000) = 1535, μ(MoKα) = 1.395 mm−1.
The structure was solved and refined as described above for 3b·CH2Cl2 and 4b. The refinement converged (Δ/σ ≤ 0.002, 352 parameters) to R = 3.30% and wR = 7.74% for the observed diffractions and to R = 4.26% and wR = 8.22% for all diffractions. The final difference electron density map showed peaks of no chemical significance (Δρmax = 1.51, Δρmin = −0.75 e Å−3). CCDC deposition number: 1544852.
4. Conclusions
In summary, several new phosphinoferrocene amidosulfonate donors with different dialkylphosphino substituents were synthesized and characterized. Together with their diphenylphosphino-substituted counterpart, these compounds were used as a ligand in Pd-mediated cyanation of aryl bromides and Suzuki–Miyaura cross-coupling of bromo-substituted benzoic and phenylacetic acids with boronic acids to the corresponding biphenyls. The testing reactions—aimed mainly at comparing different ligands and catalysts resulting thereof, rather than at obtaining the highest yields—revealed differences between the two types of reactions. Namely, ligand 4d bearing the least electron-donating phosphine moiety and [PdCl2(cod)] as a palladium source provided the best results in the cyanation reaction, while the biaryl coupling reactions provided the best yields with a catalyst resulting from the electron-rich donor 4a and palladium(II) acetate. In a wider perspective, the collected results confirm that careful optimization of the catalytic system and reaction conditions is needed in each particular case to obtain good results in Pd-catalyzed C–C bond forming reactions.
Acknowledgments
The research leading to these results has received funding from the Norwegian Financial Mechanism 2009–2014 and the Ministry of Education, Youth and Sports of the Czech Republic under Project Contract no. MSMT-23681/2015-2.
Author Contributions
Jiří Schulz prepared and characterized the ligands evaluated in this study and Pd-complex 5b; Filip Horký performed all catalytic tests; Ivana Císařová collected the diffraction data; Petr Štěpnička conceived the experiments and analyzed the collected results. All co-authors participated in writing the paper.
Conflicts of Interest
The authors declare no conflict of interest. The funding agency had no role in the design of the present study; in the collection, analyses, or interpretation of the data; in the preparation of the manuscript and in the decision to publish the results.
References
- Herrmann, W.A.; Kohlpainter, C.W. Water-soluble ligands, metal complexes, and catalysts: Synergism of homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. Engl. 1993, 32, 1524–1544. [Google Scholar] [CrossRef]
- Pinault, N.; Bruce, D.W. Homogeneous catalysts based on water-soluble phosphines. Coord. Chem. Rev. 2003, 241, 1–25. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Cornils, B. Aqueous-Phase Organometallic Catalysis, 2nd ed.; Wiley-VCH: Weinheim, Geramny, 2004. [Google Scholar]
- Herrmann, W.A.; Kulpe, J.A.; Kellner, J.; Riepl, H.; Bahrmann, H.; Konkol, W. Water-Soluble Metal Complexes of the Sulfonated Triphenylphosphane TPPTS: Preparation of the Pure Compounds and their Use in Catalysis. Angew. Chem. Int. Ed. Engl. 1990, 29, 391–393. [Google Scholar] [CrossRef]
- Cornils, B.; Kuntz, E.G. Introducing TPPTS and related ligands for industrial biphasic processes. J. Organomet. Chem. 1995, 502, 177–186. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Albanese, G.P.; Manetsberger, R.B.; Lappe, P.; Bahrmann, H. New Process for the Sulfonation of Phosphane Ligands for Catalysts. Angew. Chem. Int. Ed. Engl. 1995, 34, 811–813. [Google Scholar] [CrossRef]
- Schulz, J.; Císařová, I.; Štěpnička, P. Phosphinoferrocene Amidosulfonates: Synthesis, Palladium Complexes, and Catalytic Use in Pd-Catalyzed Cyanation of Aryl Bromides in an Aqueous Reaction Medium. Organometallics 2012, 31, 729–738. [Google Scholar] [CrossRef]
- Schulz, J.; Císařová, I.; Štěpnička, P. Synthesis of an amidosulfonate-tagged biphenyl phosphine and its application in the Suzuki-Miyaura reaction affording biphenyl-substituted amino acids in water. J. Organomet. Chem. 2015, 796, 65–72. [Google Scholar] [CrossRef]
- Schulz, J.; Horký, F.; Štěpnička, P. Different Performance of Two Isomeric Phosphinobiphenyl Amidosulfonates in Pd-Catalyzed Cyanation of Aryl Bromides. Catalysts 2016, 6, 182. [Google Scholar] [CrossRef]
- Schulz, J.; Vosáhlo, P.; Uhlík, F.; Císařová, I.; Štěpnička, P. Probing the Influence of Phosphine Substituents on the Donor and Catalytic Properties of Phosphinoferrocene Carboxamides: A Combined Experimental and Theoretical Study. Organometallics 2017, 36, 1828–1841. [Google Scholar] [CrossRef]
- Anbarasan, P.; Schareina, T.; Beller, M. Recent developments and perspectives in palladium-catalyzed cyanation of aryl halides: Synthesis of benzonitriles. Chem. Soc. Rev. 2011, 40, 5049–5067. [Google Scholar] [CrossRef] [PubMed]
- Vafaeezadeh, M.; Hashemi, M.M.; Karbalaie-Reza, M. The possibilities of palladium-catalyzed aromatic cyanation in aqueous media. Inorg. Chem. Commun. 2016, 72, 86–90. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Miyaura, N. Organoboron compounds. Top. Curr. Chem. 2002, 219, 11–59. [Google Scholar]
- Miyaura, N. Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; De Meijere, A., Diederich, F., Eds.; Wiley-VCH: Weinheim, Germany, 2004; Volume 1, Chapter 2; pp. 41–123. [Google Scholar]
- Molnár, Á. Palladium-Catalyzed Coupling Reactions; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Maluenda, I.; Navarro, O. Recent developments in the Suzuki-Miyaura reaction: 2010–2014. Molecules 2015, 20, 7528–7557. [Google Scholar] [CrossRef] [PubMed]
- Brunel, J.M.; Faure, B.; Maffei, M. Phosphane–boranes: Synthesis, characterization and synthetic applications. Coord. Chem. Rev. 1998, 178–180, 665–698. [Google Scholar] [CrossRef]
- Gan, K.-S.; Hor, T.S.A. Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science; Togni, A., Hayashi, T., Eds.; VCH: Weinheim, Germany, 1995; Chapter 1; pp. 18–35. [Google Scholar]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Redhouse, A.D. The chemistry of the metal-carbon bond. In The Structure, Preparation, Thermochemistry and Characterization of Organometallic Compounds; Hartley, F.R., Patai, S., Eds.; John Wiley: New York, NY, USA, 1982; Volume 1, Chapter 1; pp. 20–22. [Google Scholar]
- Appleton, T.G.; Clark, H.C.; Manzer, L.E. The trans-Influence. Its measurement and significance. Coord. Chem. Rev. 1973, 10, 335–422. [Google Scholar] [CrossRef]
- Hartley, F.R. Cis- and trans-effects of ligands. Chem. Soc. Rev. 1973, 2, 163–179. [Google Scholar] [CrossRef]
- Schareina, T.; Zapf, A.; Beller, M. Potassium hexacyanoferrate(II)—A new cyanating agent for the palladium-catalyzed cyanation of aryl halides. Chem. Commun. 2004, 1388–1389. [Google Scholar] [CrossRef] [PubMed]
- Carole, W.A.; Colacot, T.J. Understanding Palladium Acetate from a User Perspective. Chem. Eur. J. 2016, 22, 7686–7695. [Google Scholar] [CrossRef] [PubMed]
- Škoch, K.; Císařová, I.; Štěpnička, P. Phosphinoferrocene ureas: Synthesis, structural characterization and catalytic use in Pd-catalyzed cyanation of aryl bromides. Organometallics 2015, 34, 1942–1956. [Google Scholar] [CrossRef]
- Kuroki, Y.; Ueno, H.; Tanaka, M.; Takata, K.; Motoyama, T.; Baba, K. N-acylamino Acid Amide Compounds and Intermediates for Preparation Thereof. U.S. Patent 6265418, 24 July 2001. [Google Scholar]
- Shiwen, L.; Meiyun, L.; Daoan, X.; Xiaogang, L.; Xiuling, Z.; Mengping, G. A highly efficient catalyst of a nitrogen-based ligand for the Suzuki coupling reaction at room temperature under air in neat water. Org. Biomol. Chem. 2014, 12, 4511–4516. [Google Scholar]
- Ramirez, N.P.; Bosque, I.; Gonzalez-Gomez, J.C. Photocatalytic dehydrogenative lactonization of 2-arylbenzoic acids. Org. Lett. 2015, 17, 4550–4553. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, E.; Zinzi, L.; Cantore, M.; Contino, M.; Perrone, M.G.; Luurtsema, G.; Berardi, F.; Perrone, R.; Colabufo, N.A. SAR studies on tetrahydroisoquinoline derivatives: The Role of flexibility and bioisosterism to raise potency and selectivity toward P-glycoprotein. J. Med. Chem. 2014, 57, 9983–9994. [Google Scholar] [CrossRef] [PubMed]
- Dastbaravardeh, N.; Toba, T.; Farmer, M.E.; Jin-Quan, Y. Monoselective o-C-H functionalizations of mandelic acid and α-phenylglycine. J. Am. Chem. Soc. 2015, 137, 9877–9884. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.T.; Parmeggiani, F.; Weise, N.J.; Flitsch, S.L.; Turner, N.J. Chemoenzymatic synthesis of optically pure and l- and d-biarylalanines through biocatalytic asymmetric amination and palladium-catalyzed arylation. ACS Catal. 2015, 5, 5410–5413. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Examples of phosphino-amidosulfonate donors.
Scheme 2.
Synthesis of ligands 4a–c. Legend: R = i-Pr (a), cyclohexyl (Cy; b), and t-Bu (c); EDC = 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide, DMAP = 4-(dimethylamino)pyridine, and DMF = N,N-dimethylformamide.
Scheme 2.
Synthesis of ligands 4a–c. Legend: R = i-Pr (a), cyclohexyl (Cy; b), and t-Bu (c); EDC = 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide, DMAP = 4-(dimethylamino)pyridine, and DMF = N,N-dimethylformamide.
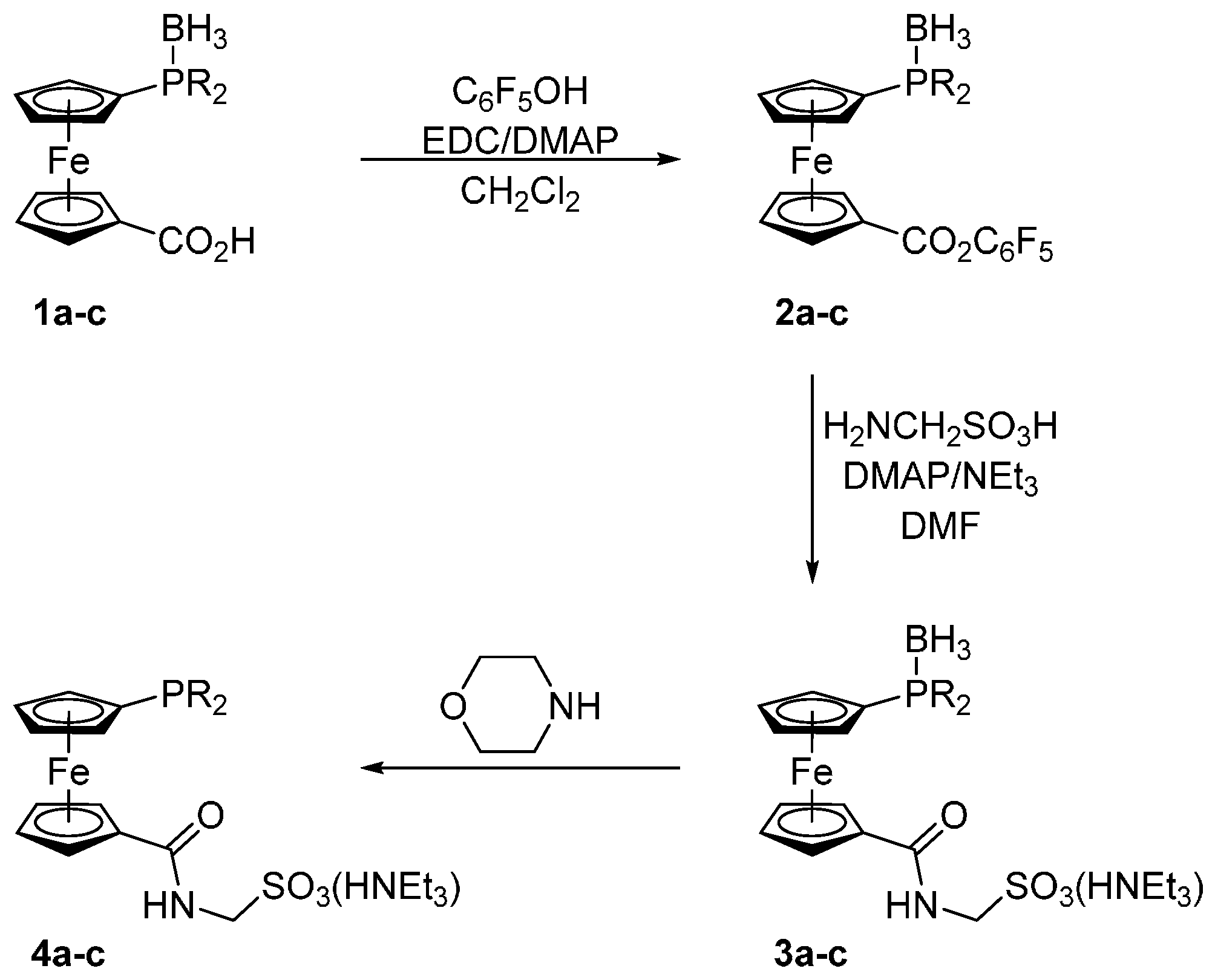
Figure 1.
PLATON plots of the molecular structures of 3b·CH2Cl2 (top) and 4b (bottom). The displacement ellipsoids are scaled to the 30% probability level. Note: all rings are numbered consecutively and, hence, only the labels of the pivotal and the adjacent atom are shown to avoid complicating the Figure. The molecule of solvent is also omitted.
Figure 1.
PLATON plots of the molecular structures of 3b·CH2Cl2 (top) and 4b (bottom). The displacement ellipsoids are scaled to the 30% probability level. Note: all rings are numbered consecutively and, hence, only the labels of the pivotal and the adjacent atom are shown to avoid complicating the Figure. The molecule of solvent is also omitted.
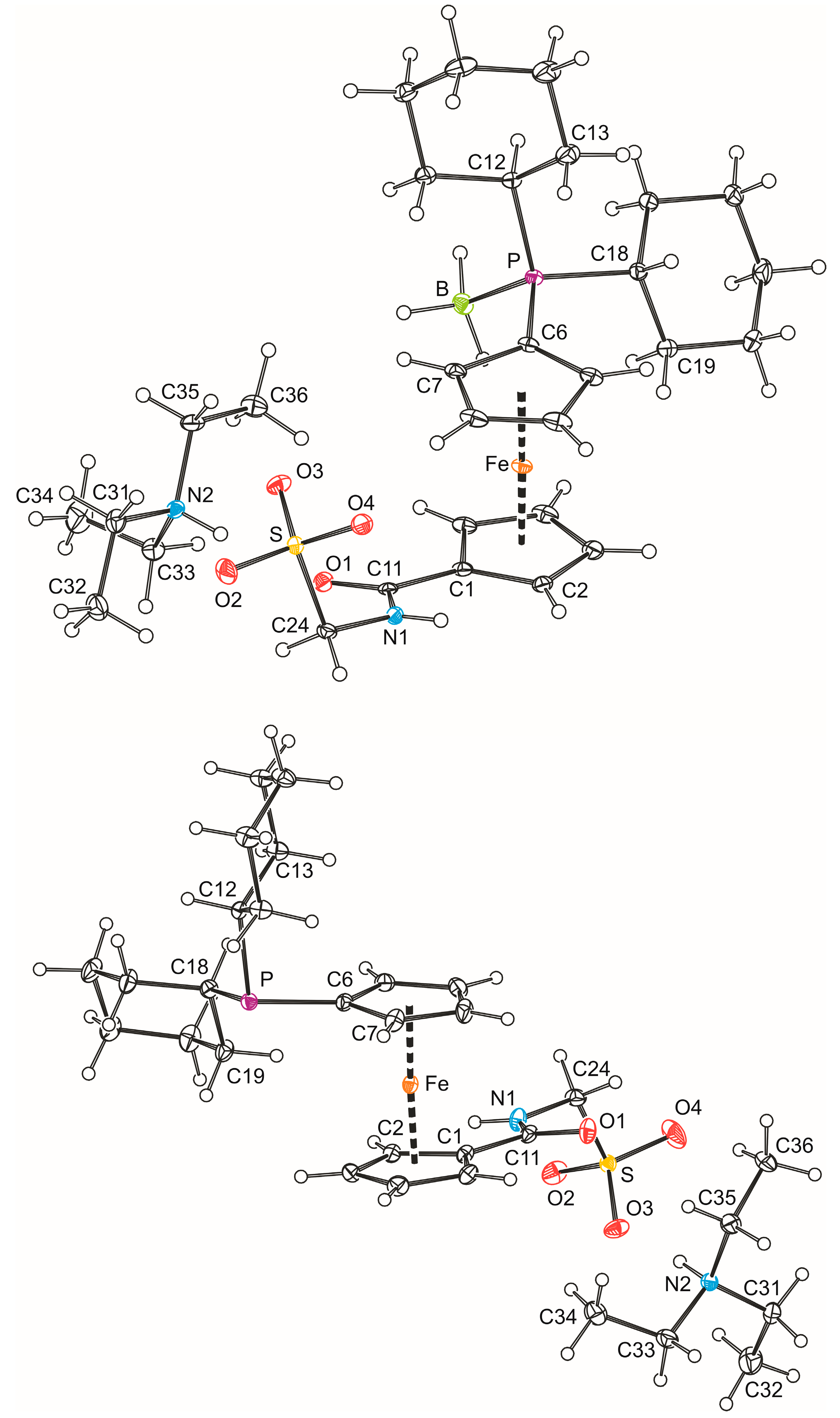
Figure 2.
Simplified packing diagrams for 3b·CH2Cl2 (left) and 4b (right).
Scheme 3.
Synthesis of complex 5b.
Figure 3.
PLATON plot of the complex molecule in the structure of 5b·CH2Cl2 showing displacement ellipsoids at the 30% probability level.
Figure 3.
PLATON plot of the complex molecule in the structure of 5b·CH2Cl2 showing displacement ellipsoids at the 30% probability level.
Scheme 4.
Pd-catalyzed cyanation of N-protected (4-bromophenyl)alanine 6.
Scheme 5.
Pd-catalyzed Suzuki–Miyaura cross-coupling of aromatic acids 8/9 with boronic acids 10a and 10b.
Scheme 5.
Pd-catalyzed Suzuki–Miyaura cross-coupling of aromatic acids 8/9 with boronic acids 10a and 10b.
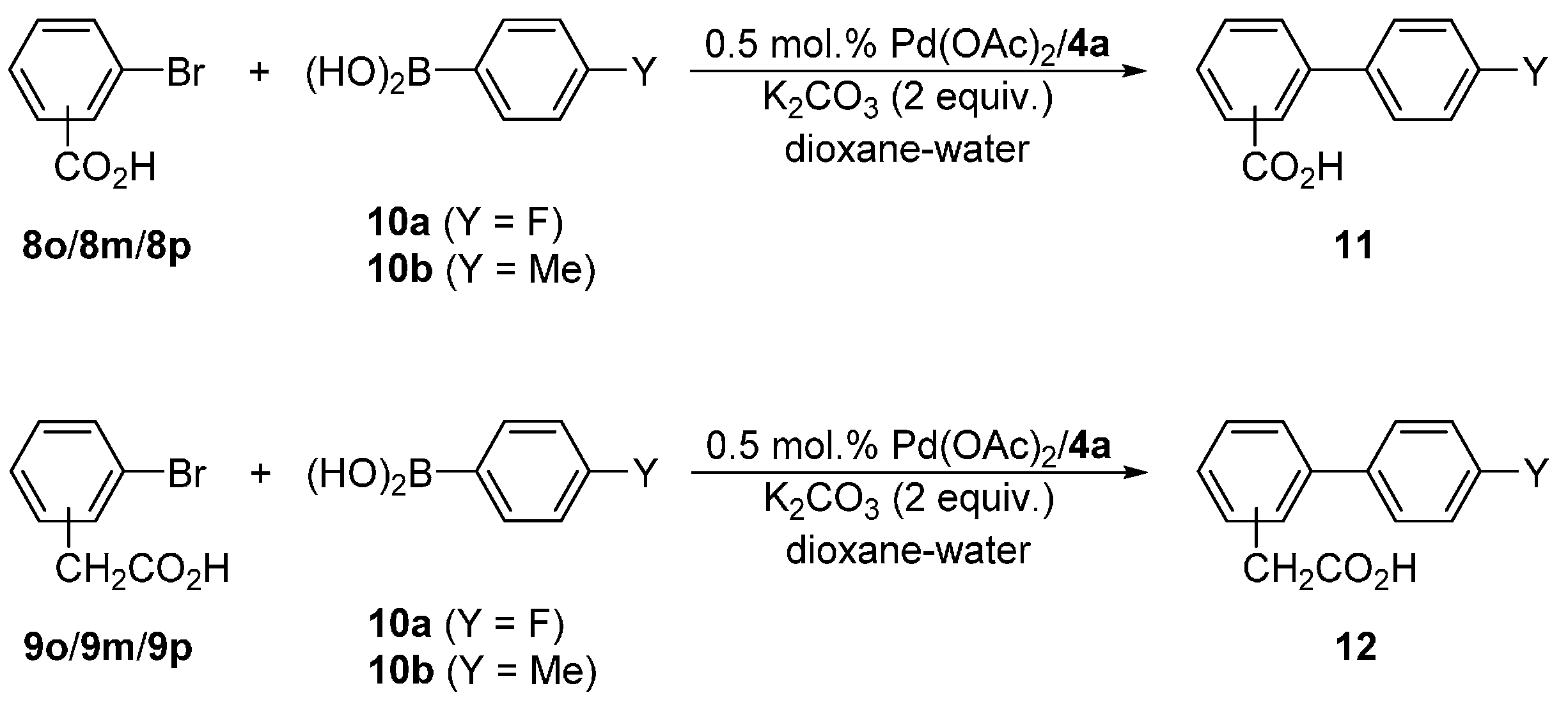
Scheme 6.
Pd-catalyzed Suzuki–Miyaura biaryl coupling involving substrate 6.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
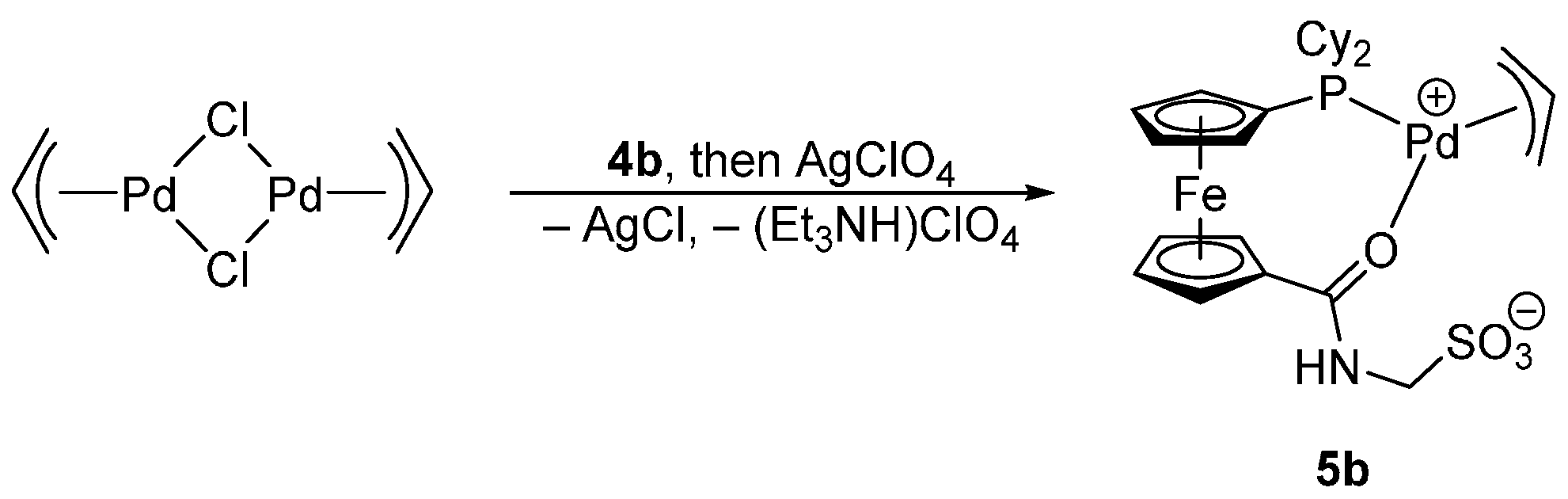{kind=link}
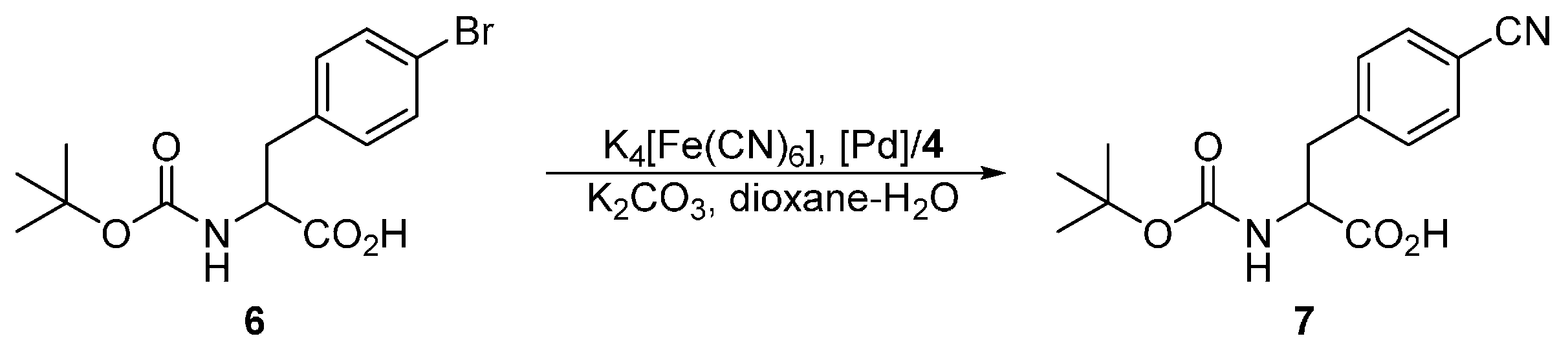{kind=link}
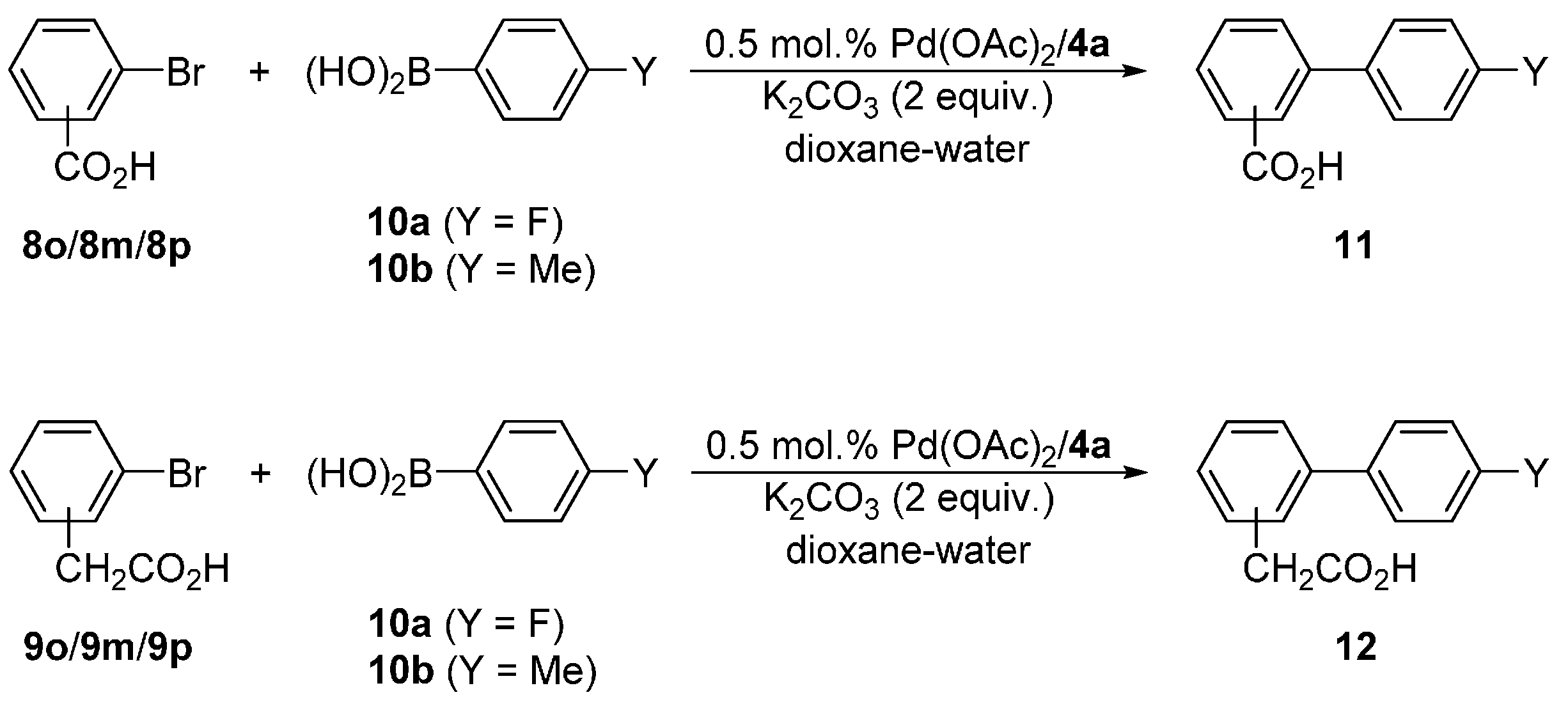{kind=link}
{kind=link}
Table 1.
Selected distances and angles for the anions in the structures of 3b·CH2Cl2 and 4b (in Å and deg) a.
Table 1.
Selected distances and angles for the anions in the structures of 3b·CH2Cl2 and 4b (in Å and deg) a.
| Parameter | 3b·CH2Cl2 | 4b |
|---|---|---|
| Fe–Cg1 | 1.6506(8) | 1.6452(7) |
| Fe–Cg2 | 1.6490(8) | 1.6490(8) |
| ∠Cp1, Cp2 | 4.8(1) | 4.50(9) |
| τ | −130.3(1) | 146.1(1) |
| P–C6 | 1.798(2) | 1.825(1) |
| P–B | 1.931(2) | n.a. |
| C1–C11 | 1.483(2) | 1.477(2) |
| C11–O1 | 1.239(2) | 1.230(2) |
| C11–N1 | 1.346(2) | 1.353(2) |
| O–C11–N1 | 123.0(1) | 122.6(1) |
| φ | 11.9(2) | 5.9(2) |
| N1–C24 | 1.443(2) | 1.434(2) |
| C24–S | 1.804(2) | 1.791(2) |
| S–O2 | 1.453(1) | 1.445(1) |
| S–O3 | 1.454(1) | 1.458(1) |
| S–O4 | 1.460(1) | 1.442(1) |
| N1–C24-S | 112.3(1) | 115.0(1) |
| C24–S–O b | 104.59(7)–106.20(7) | 103.06(8)–107.16(7) |
| O2–S–O3 | 114.07(7) | 110.89(7) |
| O2–S–O4 | 113.27(8) | 114.73(8) |
| O3–S–O4 | 112.16(7) | 113.57(8) |
a Definition of the parameters: Cp1 and Cp2 are the cyclopentadienyl rings C(1–5) and C(6–10), respectively. Cg1 and Cg2 are the respective centroids. τ denotes the dihedral angle C1–Cg1–Cg2–Cg, and φ is the angle subtended by the planes of the ring Cp1 and the amide group {C11, O1 N}. n.a. = not applicable. b The range of C24–S–O(2,3,4) angles.
Table 2.
Selected distances and angles for 5b·CH2Cl2 (in Å and deg) a.
| Pd–P | 2.3161(7) | P–Pd–O1 | 105.07(5) |
|---|---|---|---|
| Pd–O1 | 2.131(2) | C31–Pd–C33 | 67.1(1) |
| Pd–C31 | 2.225(3) | P–Pd–C33 | 97.00(9) |
| Pd–C32 | 2.140(3) | O1–Pd–C31 | 91.1(1) |
| Pd–C33 | 2.082(3) | C31–C32–C33 | 121.4(3) |
| Fe–Cg1 | 1.650(1) | ∠Cp1,Cp2 | 6.5(2) |
| Fe–Cg2 | 1.652(1) | τ | −65.1(2) |
| C1–C11 | 1.467(3) | O1–C11–N | 120.6(2) |
| C11–O | 1.249(3) | φ | 15.3(3) |
| C11–N | 1.342(3) | C11–N–C24 | 123.2(2) |
| N–C24 | 1.436(4) | N–C24–S | 112.5(2) |
| C24–S | 1.793(3) | C24–S–O b | 103.0(2)–106.8(2) |
| S–O2 | 1.442(3) | O2–S–O3 | 111.1(2) |
| S–O3 | 1.460(2) | O2–S–O4 | 116.9(2) |
| S–O4 | 1.443(3) | O3–S–O4 | 111.8(2) |
| C6–P | 1.813(3) | C6–P–C12 | 104.6(1) |
| P–C12 | 1.844(3) | C6–P–C18 | 101.2(1) |
| P–C18 | 1.854(3) | C12–P–C18 | 104.9(1) |
a Definition of the parameters: Cp1 and Cp2 are the cyclopentadienyl rings C(1–5) and C(6–10), respectively. Cg1 and Cg2 denote their respective centroids. τ stands for the dihedral angle C1–Cg1–Cg2–Cg, and φ is the angle subtended by the planes of the ring Cp1 and the amide moiety {C11, O1 N}. b The range of C24–S–O(2,3,4) angles.
Table 3.
Summary of the catalytic results in Pd-catalyzed cyanation of substrate 6 a.
| Entry | Pd Source (Loading) | Ligand (Amount) | NMR Yield of 7 [%] |
|---|---|---|---|
| 1 | [PdCl2(cod)] (2 mol. %) | 4d (4 mol. %) | 100 (95 c) |
| 2 | [PdCl2(cod)] (2 mol. %) | 4d (2 mol. %) | 30 |
| 3 | Pd(OAc)2 (2 mol. %) | 4d (4 mol. %) | <5% |
| 4 | Pd(OAc)2 (2 mol. %) | 4d (2 mol. %) | <5% |
| 5 | [Pd(μ-Cl)(η-C3H5)]2 (2 mol. %) b | 4d (2 mol. %) | <5% |
| 6 | [PdCl2(cod)] (2 mol. %) | none | 0 |
| 7 | [PdCl2(cod)] (2 mol. %) | 4a (2 mol. %) | 6 |
| 8 | [PdCl2(cod)] (2 mol. %) | 4b (2 mol. %) | 14 |
| 9 | [PdCl2(cod)] (2 mol. %) | 4c (2 mol. %) | 12 |
a Conditions: Substrate 6 (0.50 mmol), K2CO3 (1 mmol) and K4[Fe(CN)6] (0.25 mmol) were reacted in the presence of in situ generated catalysts in dioxane–water (1:1, 4 mL) at 100 °C for 3 h. b 2 mol. % of Pd. c Isolated yield.
Table 4.
Summary of the optimization experiments for the model coupling of 8p and 10a.
| Entry | Pd Source (Loading) | Ligand | Solvent | NMR Yield of 12pa [%] after | |
|---|---|---|---|---|---|
| 2 h | 6 h | ||||
| 1 | [PdCl2(cod)] (1 mol. %) | 4d (2 mol. %) | water | 60 | 72 |
| 2 | [PdCl2(cod)] (1 mol. %) | 4d (2 mol. %) | ethanol | 72 | 75 |
| 3 | [PdCl2(cod)] (1 mol. %) | 4d (2 mol. %) | ethanol–water (1:1) | 72 | 81 |
| 4 | [PdCl2(cod)] (1 mol. %) | 4d (2 mol. %) | dioxane | 0 | 10 |
| 5 | [PdCl2(cod)] (1 mol. %) | 4d (2 mol. %) | dioxane–water (1:1) | 82 | quant. |
| 6 | [PdCl2(cod)] (1 mol. %) | 4d (1 mol. %) | dioxane–water (1:1) | 41 | n.a. |
| 7 | Pd(OAc)2 (1 mol. %) | 4d (2 mol. %) | dioxane–water (1:1) | 78 | n.a. |
| 8 | Pd(OAc)2 (1 mol. %) | 4d (1 mol. %) | dioxane–water (1:1) | 85 | n.a. |
| 9 | [Pd(μ-Cl)(η-C3H5)]2 (1 mol. %) b | 4d (1 mol. %) | dioxane–water (1:1) | 0 | n.a. |
| 10 | Pd(OAc)2 (1 mol. %) | none | dioxane–water (1:1) | 56 | n.a. |
| 11 | Pd(OAc)2 (0.5 mol. %) | 4a (0.5 mol. %) | dioxane–water (1:1) | 79 | 98 |
| 12 | Pd(OAc)2 (0.5 mol. %) | 4b (0.5 mol. %) | dioxane–water (1:1) | 74 | 88 |
| 13 | Pd(OAc)2 (0.5 mol. %) | 4c (0.5 mol. %) | dioxane–water (1:1) | 43 | 67 |
| 14 | Pd(OAc)2 (0.5 mol. %) | 4d (0.5 mol. %) | dioxane–water (1:1) | 85 | 89 |
a Conditions: substrates 8p (1.0 mmol) and 10a (1.15 mmol) were reacted in the presence of in situ generated catalysts and K2CO3 (2.0 mmol) in 4 mL of the respective solvent at 40 °C. n.a. = not available. b 1 mol. % of Pd.
Table 5.
Summary of the reaction scope tests a.
| Entry | Aryl Bromide | Boronic Acid | Product | NMR Yield [%] | Isolated Yield [%] |
|---|---|---|---|---|---|
| 1 | 8o | 10a | 11oa | 19 | n.d. |
| 2 | 8m | 10a | 11ma | 89 | 67 |
| 3 | 8p | 10a | 11pa | 92 | 77 |
| 4 | 8o | 10b | 11ob | 24 | n.d. |
| 5 | 8m | 10b | 11mb | 88 | 77 |
| 6 | 8p | 10b | 11pb | 84 | 64 |
| 7 | 9o | 10a | 12oa | <5 | n.d. |
| 8 | 9m | 10a | 12ma | 97 | 78 |
| 9 | 9p | 10a | 12pa | 98 | 78 |
| 10 | 9o | 10b | 12ob | 12 | n.d. |
| 11 | 9m | 10b | 12mb | 93 | 90 |
| 12 | 9p | 10b | 12pb | 80 | 71 |
a Conditions: the respective substrates 8/9 (1.0 mmol) and 10 (1.15 mmol) were reacted in the presence of a catalyst generated from palladium(II) acetate (0.5 mol.%) and ligand 4a (1 equiv. with respect to Pd) and K2CO3 (2.0 mmol) as the base in 4 mL of dioxane–water (1:1) at 40 °C. n.d. = not determined.
Table 6.
Reactions of 10a/b with amino acid 6 a.
| Entry | Boronic Acid | Product | Pd Loading | NMR Yield [%] | Isolated Yield [%] |
|---|---|---|---|---|---|
| 1 | 10a | 13a | 0.5 mol. % | 88 | n.d. |
| 2 | 10b | 13b | 0.5 mol. % | 91 | n.d. |
| 3 | 10a | 13a | 1.0 mol. % | 100 | 76 |
| 4 | 10b | 13b | 1.0 mol. % | 100 | 83 |
a Conditions: compound 6 (1.0 mmol) and 10a or 10b (1.15 mmol) were reacted in the presence of a catalyst generated from palladium(II) acetate (0.5 or 1.0 mol. %) and ligand 4a (1 equiv.) and K2CO3 (2.0 mmol) as the base in 4 mL of dioxane–water (1:1) at 40 °C for 6 h. n.d. = not determined.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schulz, J.; Horký, F.; Císařová, I.; Štěpnička, P. Synthesis, Structural Characterization and Catalytic Evaluation of Anionic Phosphinoferrocene Amidosulfonate Ligands. Catalysts 2017, 7, 167. https://doi.org/10.3390/catal7060167
AMA Style
Schulz J, Horký F, Císařová I, Štěpnička P. Synthesis, Structural Characterization and Catalytic Evaluation of Anionic Phosphinoferrocene Amidosulfonate Ligands. Catalysts. 2017; 7(6):167. https://doi.org/10.3390/catal7060167
Chicago/Turabian StyleSchulz, Jiří, Filip Horký, Ivana Císařová, and Petr Štěpnička. 2017. "Synthesis, Structural Characterization and Catalytic Evaluation of Anionic Phosphinoferrocene Amidosulfonate Ligands" Catalysts 7, no. 6: 167. https://doi.org/10.3390/catal7060167
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.