
Chiral Catalyst Deactivation during the Asymmetric Hydrogenation of Acetophenone


1
Department of Process Engineering, Universidad Autónoma Metropolitana—Iztapalapa, San Rafael Atlixco # 186, Ciudad de México C.P. 09340, Mexico
2
Facultad de Ciencias Químico Biológicas, Universidad Autónoma de Sinaloa, Josefa Ortiz de Domínguez S/N, Ciudad Universitaria, Culiacán Sinaloa C.P. 80013, Mexico
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(7), 193; https://doi.org/10.3390/catal7070193
Submission received: 18 May 2017
/
Revised: 14 June 2017
/
Accepted: 20 June 2017
/
Published: 23 June 2017
(This article belongs to the Special Issue Commemorative Issue in Honor of Professor Emeritus Calvin H. Bartholomew in Anticipation of His 75th Birthday)
Abstract
:Asymmetric hydrogenation in solution catalyzed by chiral catalysts is a powerful tool to obtain chiral secondary alcohols. It is possible to reach conversions and enantiomeric excesses close to 99%, but that frequently requires the use of non-optimal amounts of catalysts or long reaction times. That is in part caused by the lack of kinetic information needed for the design of large-scale reactors, including few reported details about catalyst deactivation. In this work, we present a kinetic model for the asymmetric hydrogenation in solution of acetophenone, a prochiral substrate, catalyzed by different bisphosphine-diamine Ru complexes. The experimental data was fitted with a first order model that includes first order deactivation of the catalyst and the presence of residual activity. The fit of the experimental data is very good, and an analysis of the kinetic and deactivation parameters gives further insight into the role of each ligand present in the Ru catalysts. This is the first report of a kinetic analysis of homogenous complexes’ catalysis including an analysis of their deactivation.

1. Introduction
Asymmetric hydrogenation of prochiral ketones is important to obtain chiral secondary alcohols [1], which are intermediates in the production of pharmaceuticals, agrochemicals and perfumes [2]. In the hydrogenation of the carbonyl group it is customary to use a homogeneous chiral catalyst to facilitate the reaction and to avoid the formation of undesired enantiomers [3]. In this area, Noyori and his group [4] have developed a series of chiral Ru complexes used as catalysts that have an excellent performance during the asymmetric hydrogenation of several prochiral substrates. Unfortunately, that is not always the case for all the complexes studied in solution. There are reports of experiments in which the catalyst activity decreased drastically or even ceased completely. In some cases, this can be explained by the inhibition of the catalyst by the product. Another possibility, probably the most common, is that the catalyst deactivates. This may occur by the loss of a ligand, the reduction of the central metal and ripening, or formation of dimers, etc. Despite its relevance, there has not been much interest in the open literature in understanding what occurs to the catalysts [5,6]. The research effort has been focused upon the trial and error development of more efficient catalysts to attain high conversion and/or enantioselectivity.
There are few reports in the literature where a mention is made of deactivation and its possible causes. Grasa et al. [7] stated that deactivation occurred during the hydrogenation of acetophenone with a bisphosphine/diamine-Ru catalyst, and suggested that a Ru-hydride active center was formed initially but that it decomposed during reaction. A limited set of experimental data, conversion and enantiomeric excess (ee), was included, which makes an adequate comparison between catalysts more difficult. Abdur-Rashid et al. [8] conducted several experiments with the same reaction and observed decay in the activity of the catalyst, also a bisphosphine/diamine-Ru complex. They presented a kinetic model to adjust the experimental data, but there was no attempt to relate it with the structure of the complex.
Deactivation is not limited to the bisphosphine/diamine-Ru catalysts. It has also been documented with other catalysts and reactions in solution, as in the case of a [Rh(DIPAMP)(MeOH)2]BF4 complex in solution used to catalyze the asymmetric hydrogenations of C–C double bonds [9]. Deactivation has also been reported during C–C coupling with a [(PtBu3)-PdBr]2 catalyst [10], and olefin hydroamination catalyzed by PtBr2/Br− [11]. A recent study of the dehydrogenation of formic acid catalyzed by a TfDPEN-derived iridium complex explains the conversion-time results in terms of deactivation stemming from the irreversible transformation of the active complex to two inactive iridacycles [12].
As the few references above indicate, in the field of organometallic complex catalysts there has been little use of kinetics models, and much less of models that include deactivation. It is apparent that understanding the nature of the phenomena that cause the loss in activity of the catalysts developed is relevant in order to design more efficient processes. In this work, we present results for a series of Ru complexes used to catalyze the asymmetric hydrogenation of the prochiral ketone acetophenone. During reaction, a loss in the activity of the catalyst was observed and to adjust our data a kinetic model that accounts for deactivation was developed. The conversion-time data was fitted successfully, and by analyzing the kinetic and deactivation parameters we could differentiate the effect of each type of ligand in the Ru complexes used as catalysts.
2. Results and Discussion
2.1. Synthesis of the Complexes
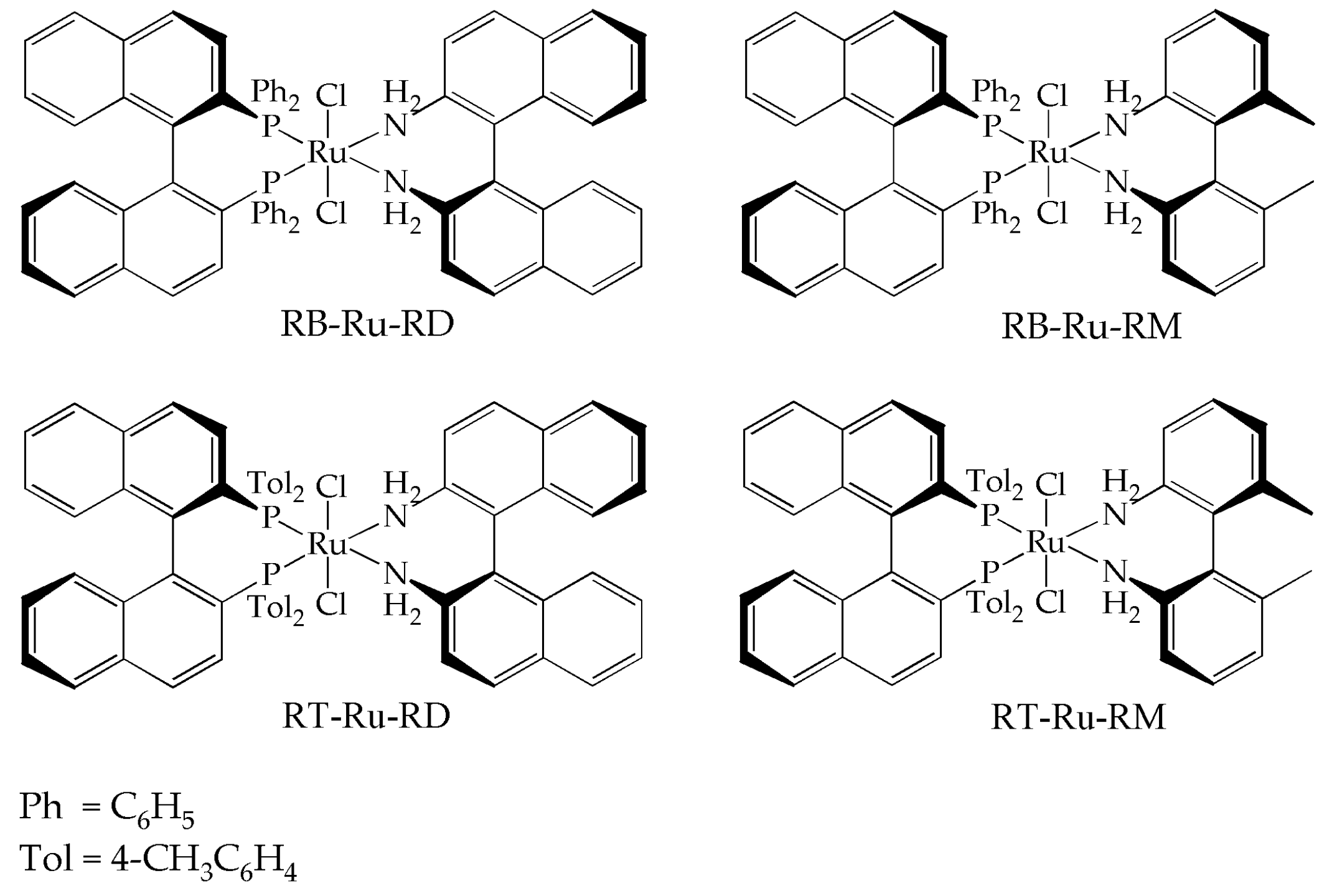
The Ru complexes were synthesized following standard Schlenk techniques. The ligands used were (R)-BINAP ((R)-2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene), (R)-Tol-BINAP ((R)-2,2′-Bis(di-p-tolylphosphino)-1,1′-binaphthyl), (R)-DABN ((R)-(+)-1-1′-Bi(2-naphthylamine)) and (R)-MAB ((R)-6,6′-dimethyl-2,2′-diaminobiphenyl). The details, as well as the spectrocopic characterization of the Ru complexes (31P Nuclear Magnetic Resonance and Infrared) can be found in a previous work [13]. The complex structures are shown in Figure 1.
2.2. Catalytic Performance
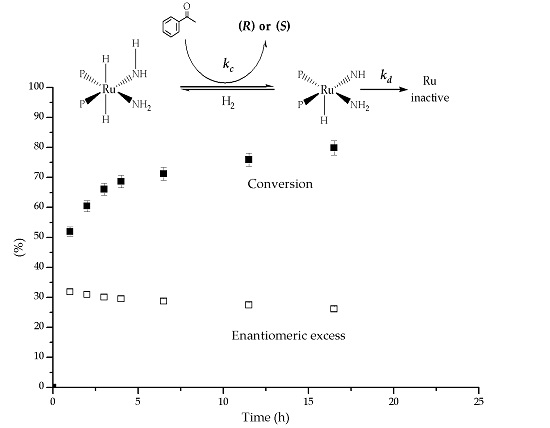
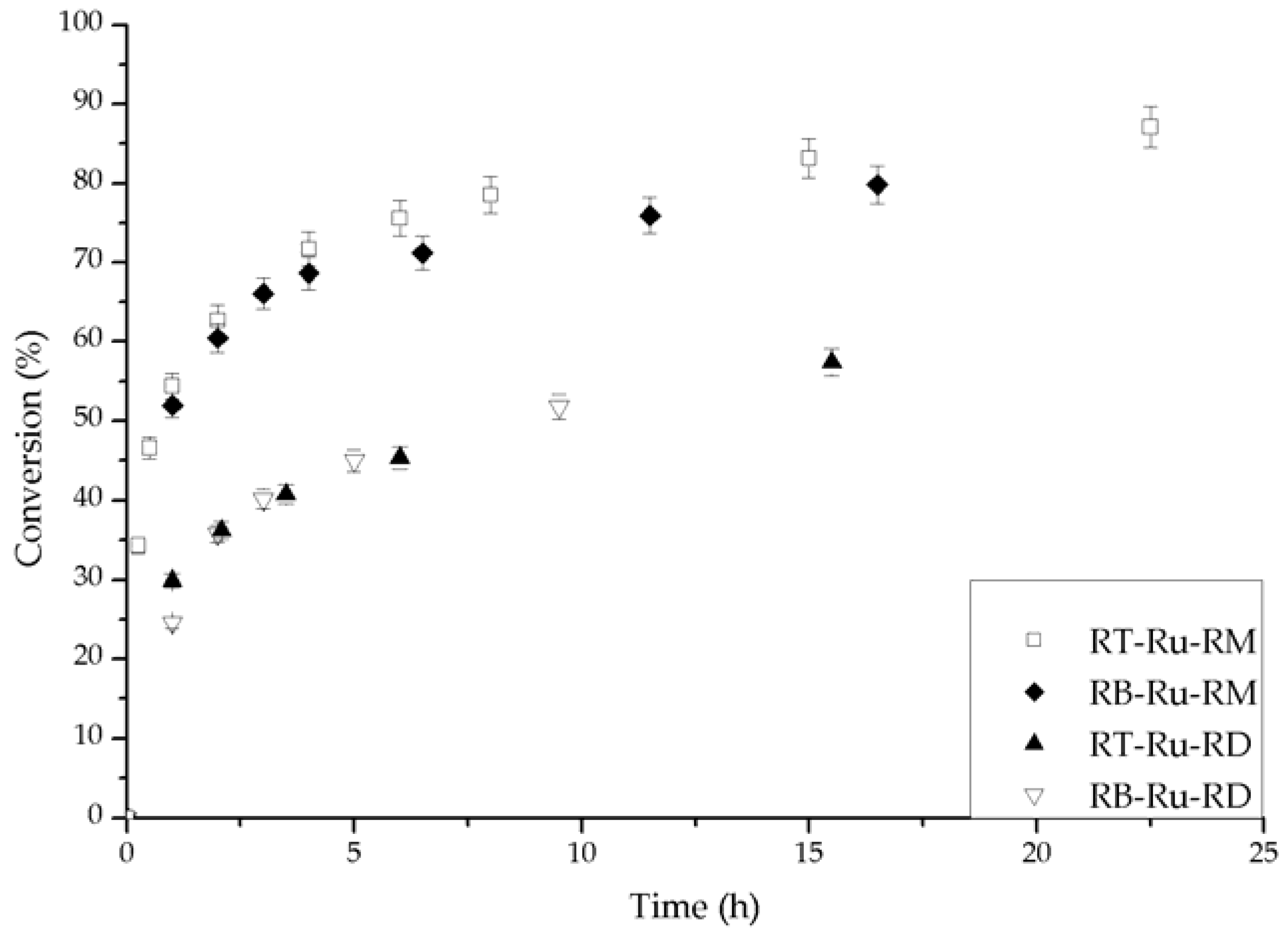
During catalytic experiments in a batch reactor, Figure 2, all the Ru complexes hydrogenated acetophenone asymmetrically. The only product was 1-phenylethanol and (R) was the preferred enantiomer, with ee up to 43%. In all cases, the initial ee was the largest, and then there was decay with time. The conversion (X) vs. time behavior generally consisted of an initial period with fast reaction, followed by a slow rate region. As we discuss later, we attributed the changes to catalyst deactivation.
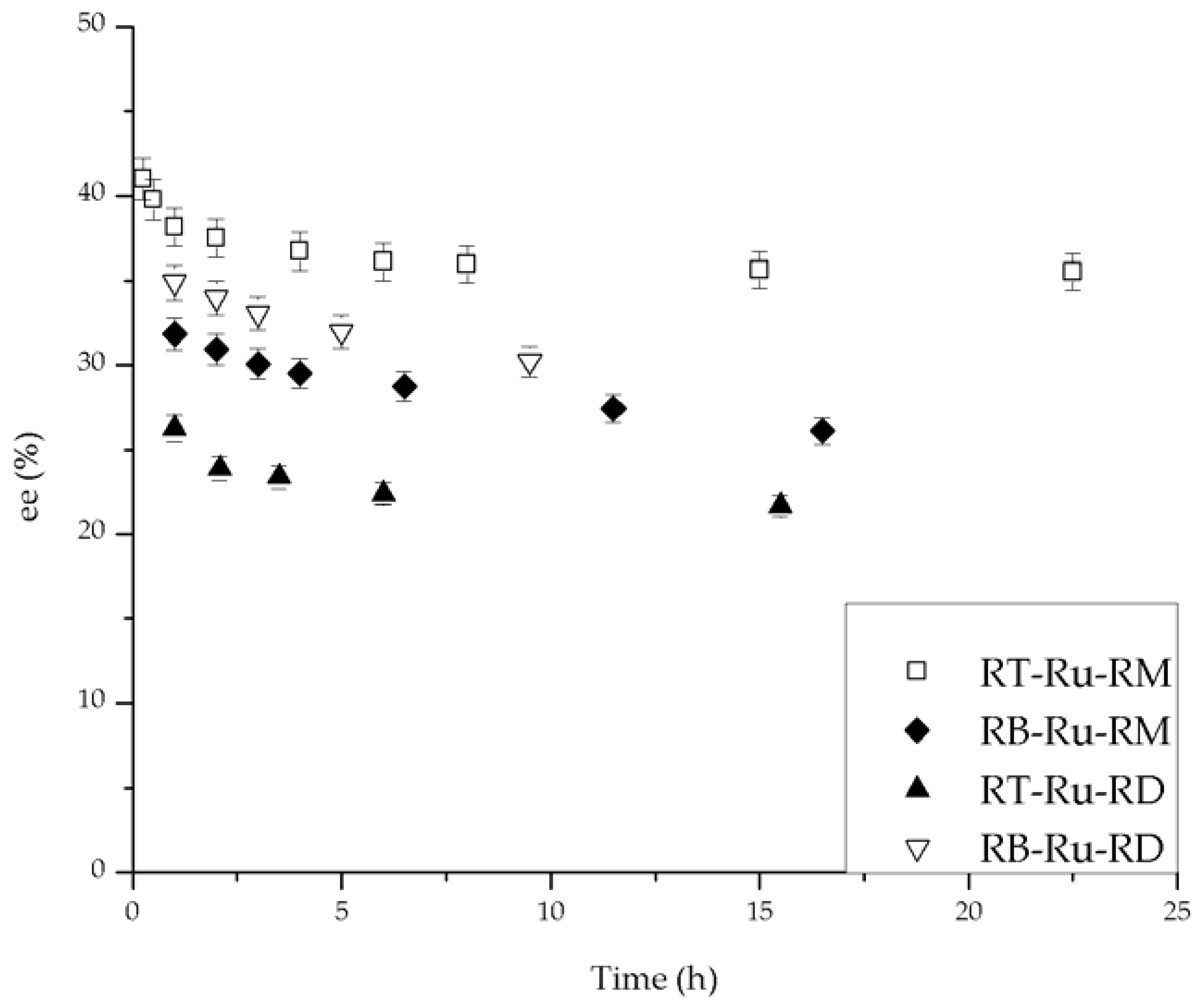
The conversion and ee (%) vs. time behavior for the different Ru complexes is given in Figure 3 and Figure 4, respectively. The effect of each ligand is different. First, the complexes bearing (R)-MAB (RB-Ru-RM and RT-Ru-RM) convert over 80% of the substrate and have a similar trend with time (Figure 3). The complexes having (R)-DABN (RB-Ru-RD and RT-Ru-RD) are not as active, with conversions below 60%. This underscores the role of the MAB ligand in favoring a higher activity. As for the bisphosphine ligand, its effect over the performance of the catalyst can be examined on Figure 4. There is at least a 7% difference in the ee between the RT-Ru-RM and the RB-Ru-RM complexes as a result of a difference in the bisphosphine ligand. The difference increases with reaction time. In the case of the RT-Ru-RD and RB-Ru-RD complexes there is also a difference in the ee, about 9%. Hence, a variation in the bisphosphine ligand appears to have a larger effect upon the enantioselectivity.
There is evidence in the literature about the effect of the bisphosphine ligand on the ee, but the role of the diamine ligand is more difficult to understand, although it is essential for these catalysts in order to be functional. Grasa et al. [7] synthesized several Ru complexes with different 1,4-diamine ligands, active in the hydrogenation of acetophenone. The ee reached with (R)-P-Phos in the catalyst was 75% for the (R) product, but with a more sterically bulky bisphosphine, (R)-Xyl-P-Phos and the same diamine, the ee was 55% towards the (S) configuration of the product. The conversion reported for each complex was the same. This data supports our conclusions, because the diamine affects the conversion, whereas the bisphosphine ligand modifies the ee.
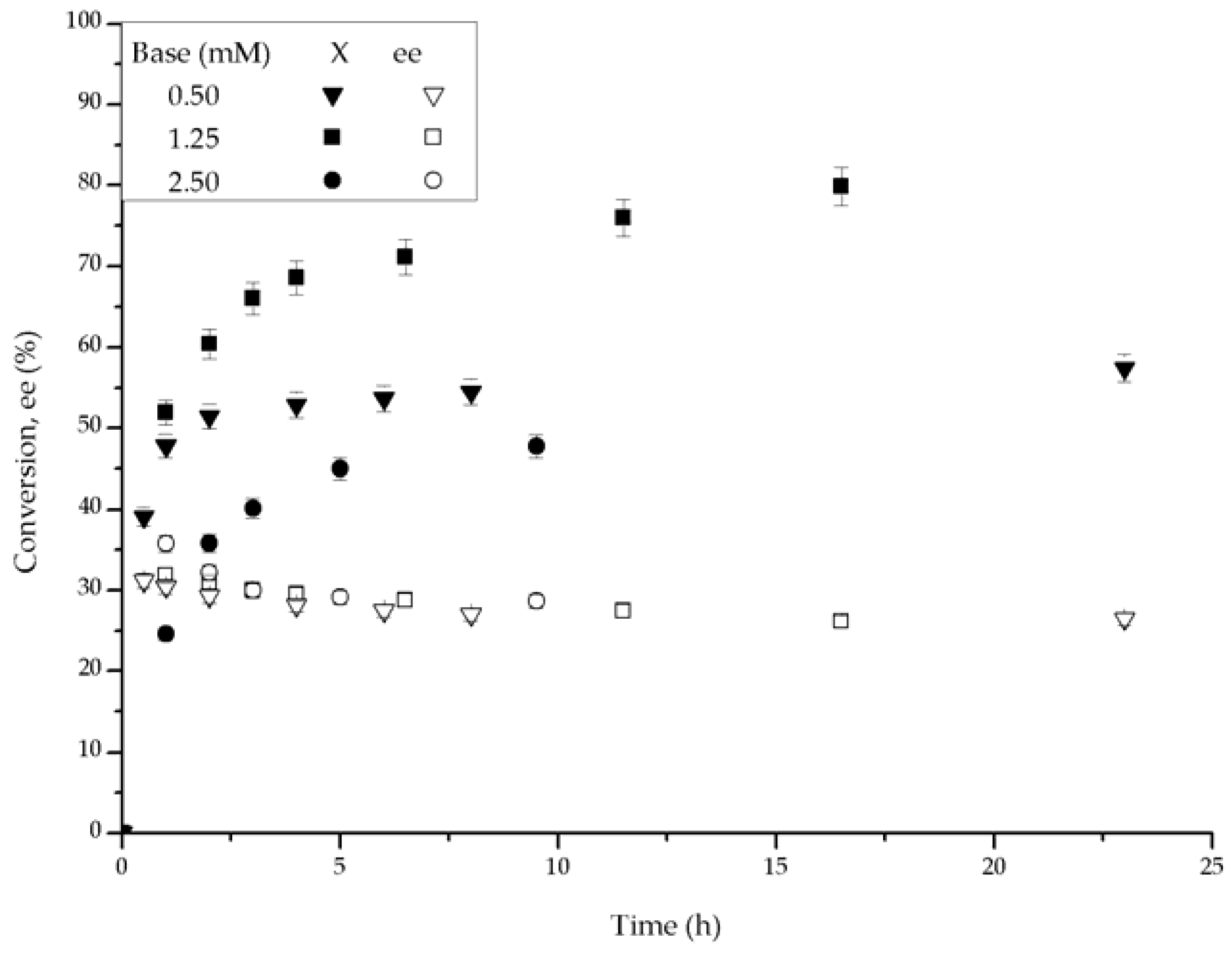
After comparing the activity of the complexes under the same conditions, we studied the effect of the base concentration upon the conversion and ee. In these experiments, we used RB-Ru-RM, Figure 5, and RT-Ru-RM, Figure 6, because of their better catalytic performance. With RB-Ru-RM, the largest product yield occurred with a base concentration equal to 1.25 mM, which corresponds to a base/catalyst molar ratio (B/C) of 2.5. With other base concentrations, the production of the secondary alcohol was lower. Interestingly, the initial rates with base concentrations of 0.50 and 1.25 mM were similar, but in the first case the reaction stopped almost completely after about 2 h. The difference between the initial and final ee values increased with the base concentration. It was the largest when 2.50 mM of base were used.
To address the origin of the loss in activity of the catalysts, an additional experiment was conducted. After the last sampling at 23 h with the RB-Ru-RM complex and 1.25 mM of base, acetophenone (1 mmol) was added again to the reactor. After another 5 h of reaction the analysis of the reaction media showed that there had been no further reaction. This result led us to conclude that there was catalyst deactivation rather than product inhibition. If the latter were occurring, there would have been some extra production of the secondary alcohol. An example of product inhibition was reported during the oxidation of alcohol to aldehyde catalyzed by Bis(pyridyl)siloxane Pd(II) complex [14].
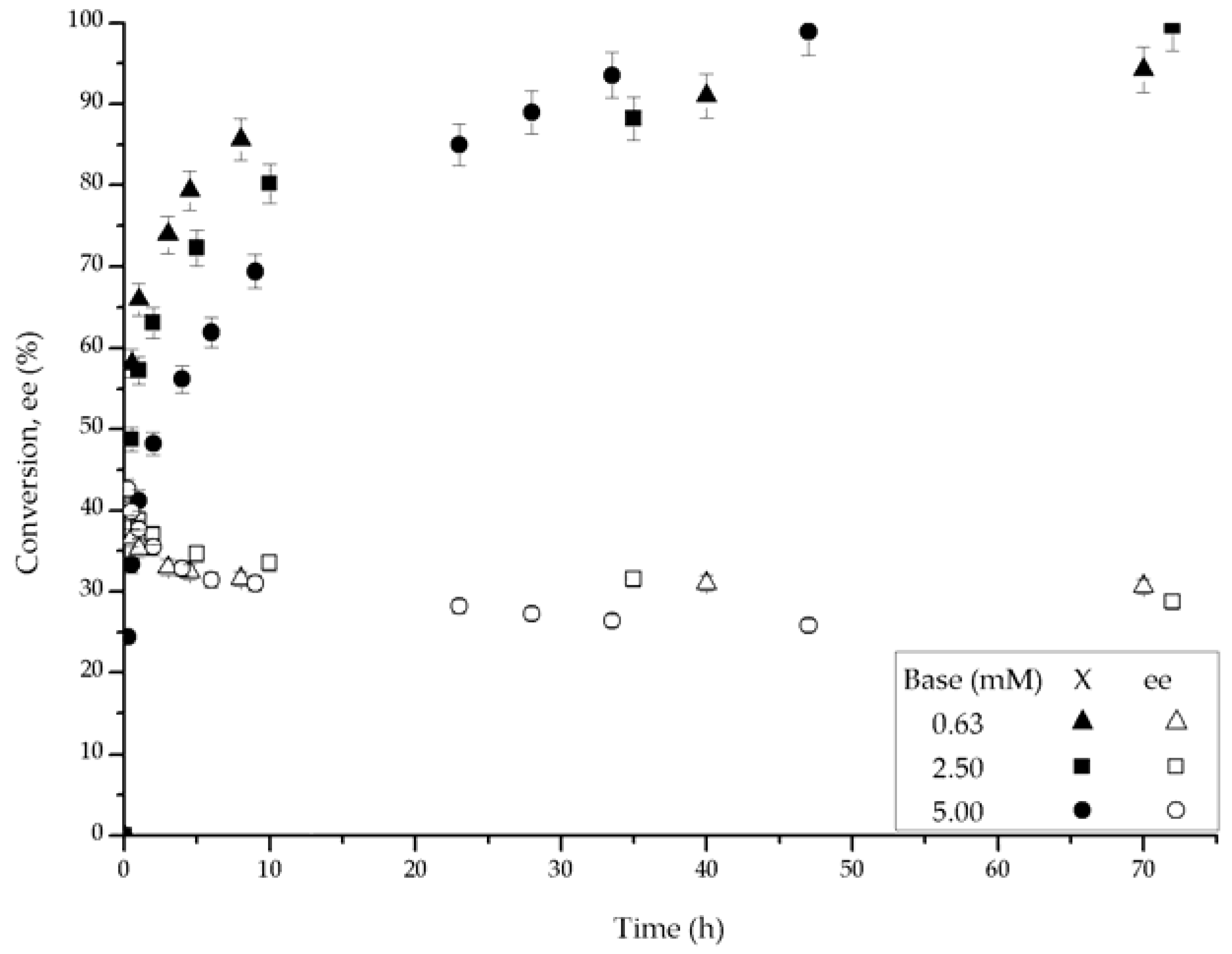
With the RT-Ru-RM complex, shown in Figure 6, the effect of the base concentration can be broadly divided in two zones. For reaction times smaller than 20 h, the use of high base concentrations resulted in lower initial reaction rates, but at larger reaction times there was a crossover in the conversion-time behavior, which made the use of higher base concentrations more favorable. As to the ee values, they varied according to the base concentration, the larger the base concentration, the larger the initial ee and its decay. These results stress the importance of doing a full analysis of the time evolution of this type of reaction. Unfortunately, it is customary to reach conclusions based on one-point samplings. In this case, as shown in Figure 6, the opposite conclusions can be derived depending on whether the results are analyzed before or after 20 h of reaction.
Sandoval et al. [15] report that the initial reaction rate depends on the base concentration and that there is an optimum, but in their case the ee remains constant. In our case, the base concentration seems to influence not only the initial reaction rate but also the final conversion level and the decay in ee. This implies that the deactivation of the catalysts depends upon the base concentration. In a recent review of the enantioselective reduction of ketones catalyzed by Noyori and Noyori-Ikariya bifunctional catalysts [16], it is stated that the base concentration can affect the formation of the major product originated by a diastereomeric transition state that is stabilized either by N-H···π or N-K···π ligand-aromatic ring non-covalent attractive interactions in the catalysts-substrate complex and/or the absence of steric constrains. A dependence of the ee on the base concentration has been reported by other authors. Abdur-Rashid et al. [8] found that by adding t-BuOK in excess the ee values increased but no explanation was given. In a more recent work, Abbel et al. [17] conducted several experiments to address the variation in ee observed during reaction. The authors varied the initial proportion of different Ru complex isomers (t,c-3a, Λc,c-3a and Δc,c-3a, nomenclature used in their work) in the asymmetric hydrogenation of acetophenone. They observed that by having a larger proportion of the t,c-3a isomer, the conversion towards 1-phenylethanol increased and the ee was constant at 63% (S). However, if they continued the same experiment for longer times, the additional isomers (Λc,c-3a and Δc,c-3a) began to contribute to the reaction, and that led to higher conversions of the secondary alcohol but to a decrease in ee values (50–30% (S)). This suggests that the Λc,c-3a and Δc,c-3a isomers were active for the hydrogenation of the substrate, but their capability to do it asymmetrically was lower than that of the t,c-3a complex.
To rationalize our results, it is plausible to assume there is isomerization of the Ru complex during reaction and that would affect the ee and result in deactivation, providing an explanation of the experimental observations. To test this assumption, the experimental results were fitted to a kinetic + deactivation model in the next section.
2.3. Kinetic Model with Deactivation
There has been much study of deactivation in the case of solid catalysts as shown in recent reviews [18,19], but that is not the case for homogeneous catalysts in solution. In the present study, an analysis of the conversion-time results indicates that there is a different degree of residual activity after the initial fast decay. We tested a kinetic model that includes deactivation with residual activity [20] and found that first order kinetics for the substrate, coupled with first order deactivation, was the best model for fitting our experimental results. The equations representing our experimental setup (batch reactor at constant volume and pressure) are:
Here, CA is the concentration of acetophenone, kc is the rate constant, kd is the deactivation constant, a is the activity of the catalyst and ass is a residual activity. Equations (1) and (2) are a case of the so-called separable deactivation kinetics and their solution, using as initial conditions that CA = CAo and ao = 1 at t = 0, is given as Equation (3):
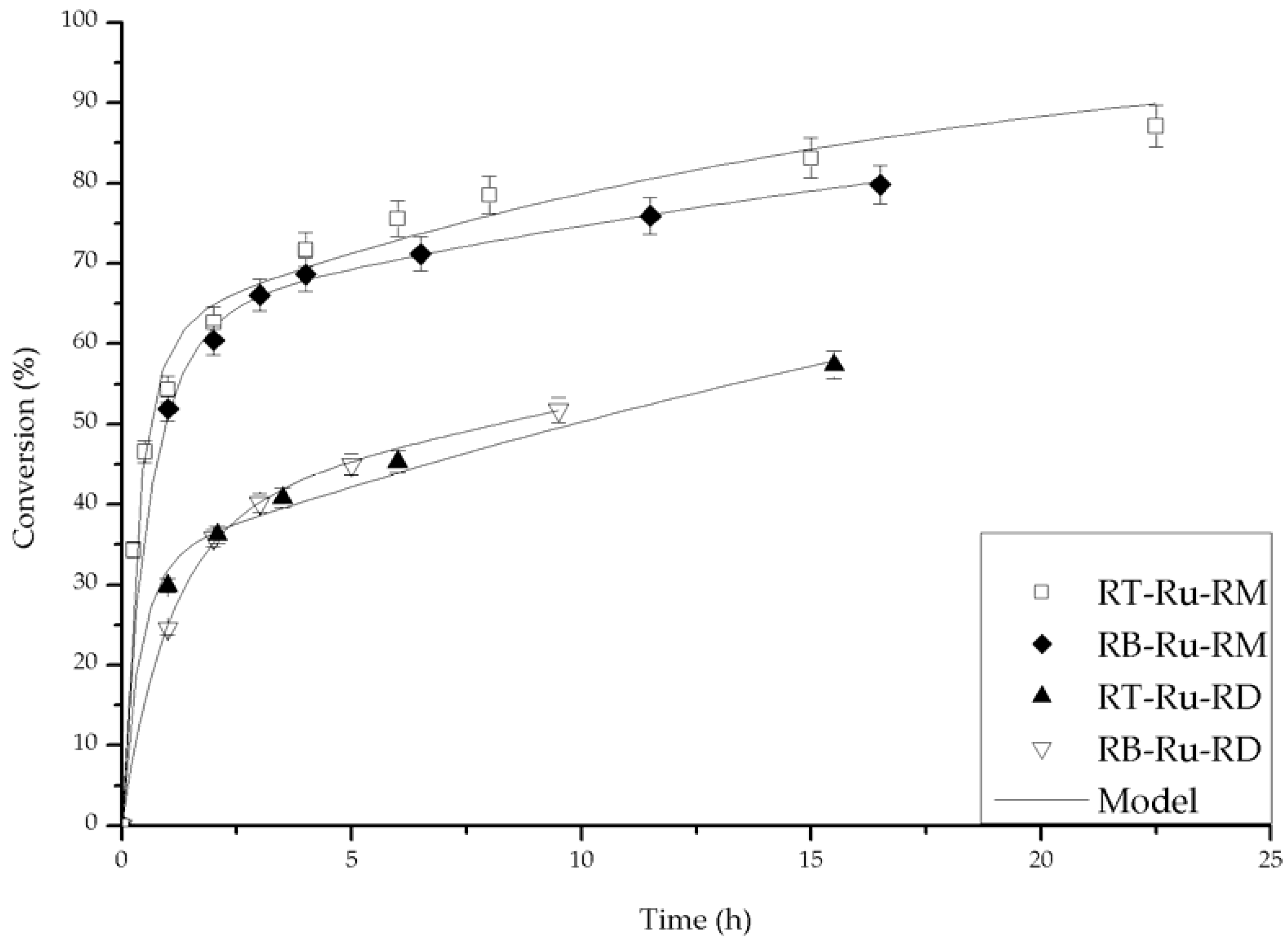
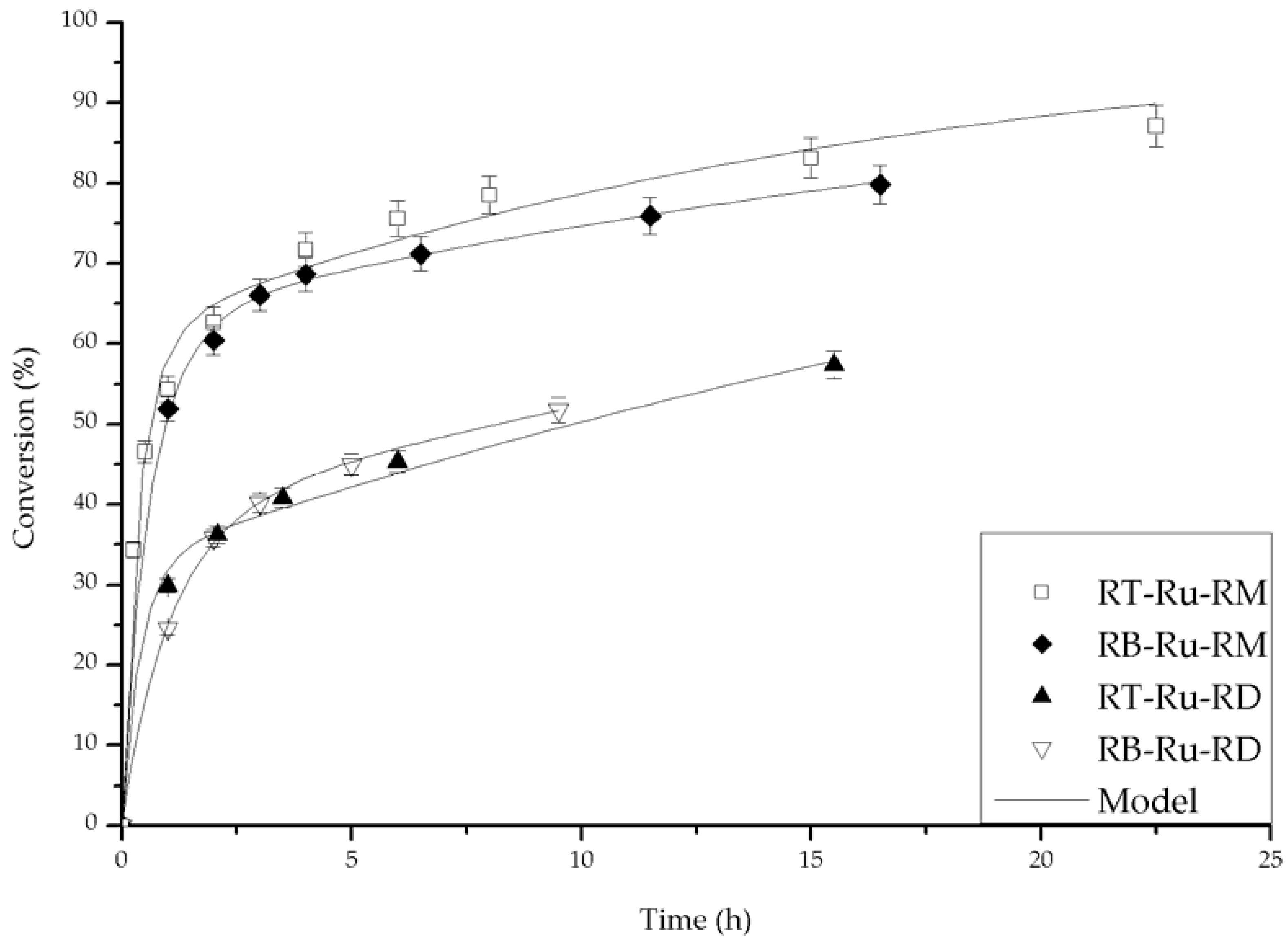
The parameters kc, kd and ass of Equation (3) were determined from a non-linear least squares fit (Mathematica, version 10, Wolfram Research, Champaign, IL, USA). The adjusted curves are plotted against the experimental values in Figure 7. It is evident that there is excellent agreement. The fitted parameters for the different experimental condition are summarized in Table 1. A direct comparison of the fitted parameters under the same experimental conditions for all the catalysts can be analyzed on entries 1, 2, 3 and 4. The complexes bearing the MAB ligand were the best catalysts to produce the secondary alcohol, as their kc values were the highest. In addition, the dynamics of the main reaction and of the complex deactivation were of the same order of magnitude, as can be appreciated from the kc and kd values. On the other hand, the catalysts with the ligand DABN resulted in lower acetophenone conversions, and their kc values were smaller when compared to the complexes with MAB.
The fitted parameters are a function of the base concentration for the RB-Ru-RM catalyst, as seen in entries 3, 5 and 6. Under entry 3 conditions, there was an optimum base concentration for which kc was slightly larger than kd and ass was the largest. This corresponds to the higher conversion profiles observed in Figure 5. The base concentration affected the performance of the RT-Ru-RM catalyst and that can be concluded from the analysis of the fitted parameters in entries 7, 8 and 9. When the base concentration increased, deactivation was more noticeable, i.e., high kd and low kc values. Interestingly, the presence of higher kd leads to a faster approach to the residual activity, and since the ass for the entry 9 was the largest, the conversion reached was the highest at shorter reaction time (Figure 6).
2.4. Mechanistic Considerations
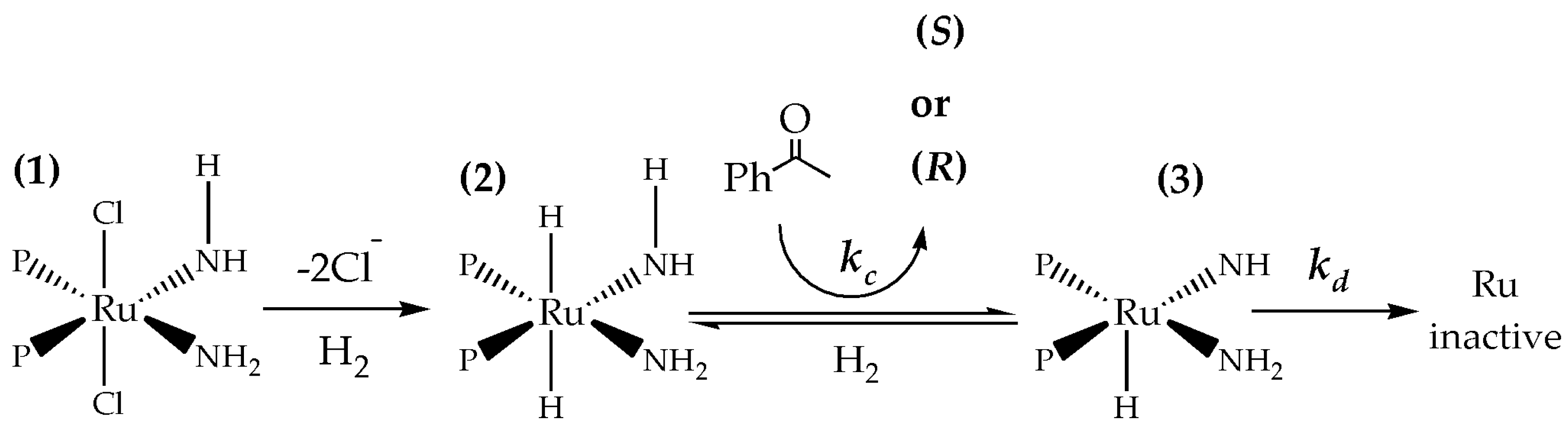
A representation of the different steps occurring during reaction is shown in Scheme 1 (more details of the catalyzed pathway can be found in reference [21]). Initially, the pre-catalyst 1 is transformed into the catalyst 2. Then, the catalyst hydrogenates the substrate and is transformed into 3. The product of the reaction is mainly the R enantiomer but as the time of reaction increases, the structure-directing ability of the catalyst to produce the R enantiomer decreases. At the end of the reaction cycle, part of the species 3 is regenerated by hydrogen and another fraction is converted to an inactive species. The species 3 has a square based pyramidal geometry and it can form an isomer with trigonal bipyramidal geometry. The latter species is probably difficult to be regenerated with H2, hence leading to overall deactivation. Such isomerization can proceed via the Berry pseudorotation [22].
Although the mechanism proposed cannot be ascertained based only on kinetics, it is based on reasonably well established ideas in the field. On the other hand, the kinetic model developed here allows to compare the interplay between reaction and deactivation through the analysis of the values for kc, kd and ass. These are sensitive to the structure of the catalyst and to the experimental conditions used. Besides the use of novel complexes as catalysts, this is the first report of the modeling of deactivation for asymmetric hydrogenation catalyzed by Ru complexes in solution.
3. Materials and Methods
All the manipulations of the reagents were carried out using Schlenk techniques inside a glove bag filled several times with N2. (R)-BINAP, (R)-Tol-BINAP ((R)-2,2′-Bis(di-p-tolylphosphino)-1,1′-binaphthyl), RuCl2(η6-C6H6) dimer, (R)-DABN ((R)-(+)-1-1′-Bi(2-naphthylamine)), acetophenone, potassium terbutoxide (t-BuOK), dimethylformamide (DMF) and anhydrous isopropyl alcohol (IPA) were purchased from Sigma-Aldrich Química (Toluca, MX, Mexico). (R)-MAB ((R)-6,6′-dimethyl-2,2′-diaminobiphenyl) was synthesized according to previous reports [23,24]. 31P Nuclear Magnetic Resonance in the liquid state (CDCl3 as solvent) was performed in a Bruker spectrometer (Avance III 500, resonance frequency of 1H 500 MHz, Bruker Mexicana, Ciudad de México, Mexico). The IR spectra were acquired in a Bruker FTIR spectrometer (Tensor 27, Bruker Mexicana, Ciudad de México, Mexico) from KBr pellets. The products of reaction were analyzed by Gas Chromatography (HP 5890, Agilent Technologies Mexico, Ciodad de Mexico, Mexico) with a chiral column (cp-chirasildex-cb 7502). The final configuration was determined by comparison with standards ((R) and (S) 1-phenylethanol also purchased from Sigma-Aldrich Química).
3.1. Synthesis of the Complexes
Preparation of (R)-BINAP-Ru-(R)-DABN complex (RB-Ru-RD): We followed the procedures reported by Ohkuma et al. [25] and Grasa et al. [7] with some minor modifications. (R)-BINAP (0.3 mmol) and of RuCl2(η6-C6H6) dimer (0.15 mmol) were mixed in a Schlenk flask and DMF (12 mL) was then poured in. The solution was heated to 115 °C during 3 h and then cooled to room temperature. To this mixture (R)-DABN (0.3 mmol) was added, and the resulting solution was left under stirring overnight. DMF was removed under vacuum while keeping the temperature constant at 40 °C.
3.2. Procedure for the Hydrogenation of Acetophenone
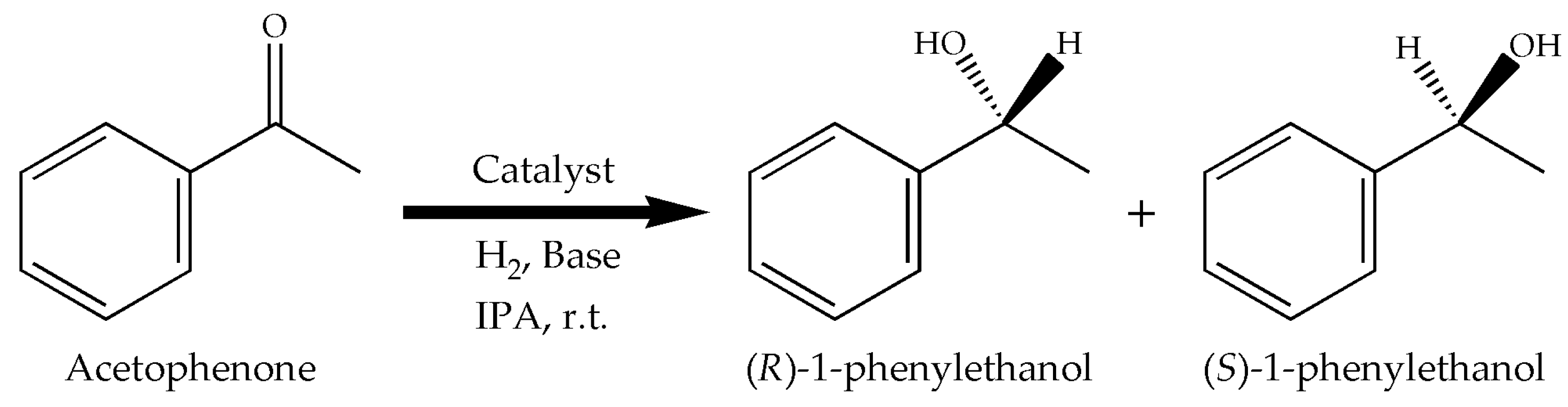
The model reaction used to test the complexes was the hydrogenation of acetophenone (Figure 2). The expected product, 1-phenylethanol, is employed in the pharmaceutical and fragrances industries [26,27]. Typically, the desired amounts of complex (0.01 mmol) and of t-BuOK were poured in the reactor, followed by IPA (20 or 40 mL) and then acetophenone (1 mmol). The reactor was flushed 5 times with pure H2 before the pressure was set at 689.5 kPa (100 psi) to begin the reaction at room temperature. Samples were withdrawn during the reaction to obtain the conversion vs. reaction time data.
3.3. Parameter Estimation for the Kinetic Model with Catalyst Deactivation
We used the integral method to analyze the kinetics of acetophenone hydrogenation. As the deactivation Equation (2) is independent, it was first integrated and simplified to give Equation (4) for the activity
Equation (4) was substituted in Equation (1) to give Equation (5):
Integration of Equation (5) resulted in:
Applying the definition of conversion, Equation (3) was obtained:
The kc, kd and ass parameters were determined from a non-linear least-squares fit of Equation (3) using the Levenberg-Marquardt algorithm option in Mathematica (version 10, Wolfram Research). As this involves an iterative process, we used different seed values for the parameters in order to assure that the converged solution was optimal.
4. Conclusions
Four complexes of the type bisphosphine/diamine-Ru were used as catalysts for the asymmetric hydrogenation of acetophenone. The diamine ligands employed have axial chirality similar to the bisphosphine ligands normally employed. The role of each ligand in the catalyst was assigned according to our experimental observations. The diamine ligand had a major influence on the conversion reached, while the bisphosphine ligand affected the ee values. The catalysts behave in a singular way because the base concentration affects the initial reaction rates and subsequent deactivation. A first order kinetic model that includes deactivation was proposed, and it fitted the experimental observations with very good agreement. Deactivation was modeled as a first order process with residual activity. This is one of the few studies where deactivation is modeled using this type of catalyst. A scheme for the structural changes in the complexes during reaction, based on generally accepted knowledge in the field and the deactivation model, is proposed to explain the transient variations in ee and conversion during the asymmetric hydrogenation of acetophenone in solution catalyzed by bisphosphine/diamine Ru complexes.
Acknowledgments
J.P.R.-L. thanks Consejo Nacional de Ciencia y Tecnología (CONACyT) for the graduate scholarships granted. The authors acknowledge the financial support of UAS and UAM-Iztapalapa during the development of this work.
Author Contribution
J.P.R.-L. performed experiments and contributed to the numerical analysis and writing of the manuscript. G.A.F. directed the research and contributed to the modeling and writing of the manuscript. Both authors have approved the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Noyori, R. Facts are the enemy of truth-reflections on serendipitous discovery and unforeseen developments in asymmetric catalysis. Angew. Chem. Int. Ed. 2013, 52, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Ohkuma, T. Asymmetric catalysis by architectural and functional molecular engineering: Pactical chemo- and stereoselective hydrogenation of ketones. Angew. Chem. Int. Ed. 2001, 40, 40–73. [Google Scholar] [CrossRef]
- Hayashi, T.; Yamasaki, K. Rhodium-catalyzed asymmetric 1,4-addition and its related asymmetric reactions. Chem. Rev. 2003, 103, 2829–2844. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R. Asymmetric catalysis: Science and opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar]
- Jones, C.W. On the stability and recyclability of supported metal-ligand complex catalysts: Myths, misconceptions and critical research needs. Top. Catal. 2010, 53, 942–952. [Google Scholar]
- Van Leeuwen, P.W.N.M. Decomposition pathways of homogeneous catalysts. Appl. Catal. A-Gen. 2001, 212, 61–81. [Google Scholar] [CrossRef]
- Grasa, G.A.; Zanotti-Gerosa, A.; Medlock, J.A.; Hems, W.P. Asymmetric hydrogenation of isobutyrophenone using a [(diphosphine) RuCl2 (1,4-diamine)] catalyst. Org. Lett. 2005, 7, 1449–1451. [Google Scholar] [CrossRef] [PubMed]
- Abdur-Rashid, K.; Clapham, S.E.; Harvey, J.N.; Lough, A.J.; Morris, R.H. Mechanism of the hydrogenation of ketones catalyzed by trans-dihydrido(diamine)ruthenium(II) complexes. J. Am. Chem. Soc. 2002, 124, 15104–15118. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Baumann, W.; Drexler, H.J.; Heller, D. Unusual deactivation in the asymmetric hydrogenation of itaconic acid. J. Organomet. Chem. 2011, 696, 1760–1767. [Google Scholar] [CrossRef]
- Proutiere, F.; Aufiero, M.; Schoenebeck, F. Reactivity and stability of dinuclear Pd(I) complexes: Studies on the active catalytic species, insights into precatalyst activation and deactivation, and application in highly selective cross-coupling reactions. J. Am. Chem. Soc. 2012, 134, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Dub, P.A.; Béthegnies, A.; Poli, R. DFT and experimental studies on the PtX2/X–catalyzed olefin hydroamination: Effect of halogen, amine basicity, and olefin on activity, regioselectivity, and catalyst deactivation. Organometallics 2012, 31, 294–305. [Google Scholar] [CrossRef]
- Matsunami, A.; Kuwata, S.; Kayaki, Y. A bifunctional iridium catalyst modified for persistent hydrogen generation from formic acid: Understanding deactivation via cyclometalation of a 1,2-Diphenylethylenediamine motif. ACS Catal. 2017, 7, 4479–4484. [Google Scholar] [CrossRef]
- Rivera, V.M.; Ruelas-Leyva, J.P.; Fuentes, G.A. Pd and Ru complexes bearing axially chiral ligands for the asymmetric hydrogenation of C=C and C=O double bonds. Catal. Today 2013, 213, 109–114. [Google Scholar] [CrossRef]
- Missaghi, M.N.; Galloway, J.M.; Kung, H.H. Bis(pyridyl)siloxane-Pd(II) complex catalyzed oxidation of alcohol to aldehyde: Effect of ligand tethering on catalytic activity and deactivation behavior. Appl. Catal. A-Gen. 2011, 391, 297–304. [Google Scholar] [CrossRef]
- Sandoval, C.A.; Ohkuma, T.; Muñiz, K.; Noyori, R. Mechanism of asymmetric hydrogenation of ketones catalyzed by BINAP/1,2-diamine-Ruthenium(II) complexes. J. Am. Chem. Soc. 2003, 125, 13490–13503. [Google Scholar] [CrossRef] [PubMed]
- Dub, P.A.; Gordon, J.C. The mechanism of enantioselective ketone reduction with Noyori and Noyori-Ikariya bifunctional catalysts. Dalton Trans. 2016, 45, 6756–6781. [Google Scholar] [CrossRef] [PubMed]
- Abbel, R.; Abdur-Rashid, K.; Faatz, M.; Hadzovic, A.; Lough, A.J.; Morris, R.H. A succession of isomers of Ruthenium dihydride complexes. Which one is the ketone hydrogenation catalyst? J. Am. Chem. Soc. 2005, 127, 1870–1882. [Google Scholar] [CrossRef] [PubMed]
- Argyle, M.; Bartholomew, C.H. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Argyle, M. Advances in catalyst deactivation and regeneration. Catalysts 2015, 5, 949–954. [Google Scholar] [CrossRef]
- Fuentes, G.A. Catalyst deactivation and steady state activity: A generalized power-law equation. Appl. Catal. 1985, 15, 33–40. [Google Scholar] [CrossRef]
- Li, L.; Pan, Y.; Lei, M. The enantioselectivity in asymmetric ketone hydrogenation catalyzed by RuH2(diphosphine)(diamine) complexes: Insights from a 3D-QSSR and DFT study. Catal. Sci. Technol. 2016, 6, 4450–4457. [Google Scholar] [CrossRef]
- Ugi, I.; Marquarding, D.; Klusacek, H.; Gillespie, P.; Ramirez, F. Berry pseudorotation and turnstile rotation. Acc. Chem. Res. 1971, 4, 288–296. [Google Scholar] [CrossRef]
- Pérez, C.; Pérez, S.; Fuentes, G.A.; Corma, A. Preparation and use of a chiral amine Ruthenium hydrogenation catalyst supported on mesoporous silica. J. Mol. Catal. A-Chem. 2003, 197, 275–281. [Google Scholar] [CrossRef]
- Uehara, A.; Kubota, T.; Tsuchiya, R. New atropisomeric chiral bisphosphine, (S)-6-6′-dimethyl-2-2′-bis(diphenylphosphinamino)biphenyl, and asymmetric hydrogenation using the Rh(I) complex thereof. Chem. Lett. 1983, 13, 441–444. [Google Scholar] [CrossRef]
- Ohkuma, T.; Hattori, T.; Hirohito, O.; Inoue, T.; Noyori, R. BINAP/1,4-diamine-Ruthenium(II) complexes for efficient asymmetric hydrogenation of 1-tetralones and analogues. Org. Lett. 2004, 6, 2681–2683. [Google Scholar] [CrossRef] [PubMed]
- Bertero, N.M.; Apesteguía, C.R.; Marchi, A.J. Catalytic and kinetic study of the liquid-phase hydrogenation of acetophenone over Cu/SiO2 catalysts. Appl. Catal. A-Gen. 2008, 349, 100–109. [Google Scholar] [CrossRef]
- Yadav, G.D.; Mewada, R.K. Selective hydrogenation of acetophenone to 1-phenyl ethanol over nanofibrous Ag-OMS-2 catalysts. Catal. Today 2012, 198, 330–337. [Google Scholar] [CrossRef]
Figure 1.
Structure and nomenclature of the complexes used in this work. (R)-BINAP-RuCl2-(R)-DABN = RB-Ru-RD; (R)-BINAP-RuCl2-(R)-MAB = RB-Ru-RM; (R)-Tol-BINAP-RuCl2-(R)-DABN = RT-Ru-RD; (R)-Tol-BINAP-RuCl2-(R)-MAB = RT-Ru-RM.
Figure 1.
Structure and nomenclature of the complexes used in this work. (R)-BINAP-RuCl2-(R)-DABN = RB-Ru-RD; (R)-BINAP-RuCl2-(R)-MAB = RB-Ru-RM; (R)-Tol-BINAP-RuCl2-(R)-DABN = RT-Ru-RD; (R)-Tol-BINAP-RuCl2-(R)-MAB = RT-Ru-RM.
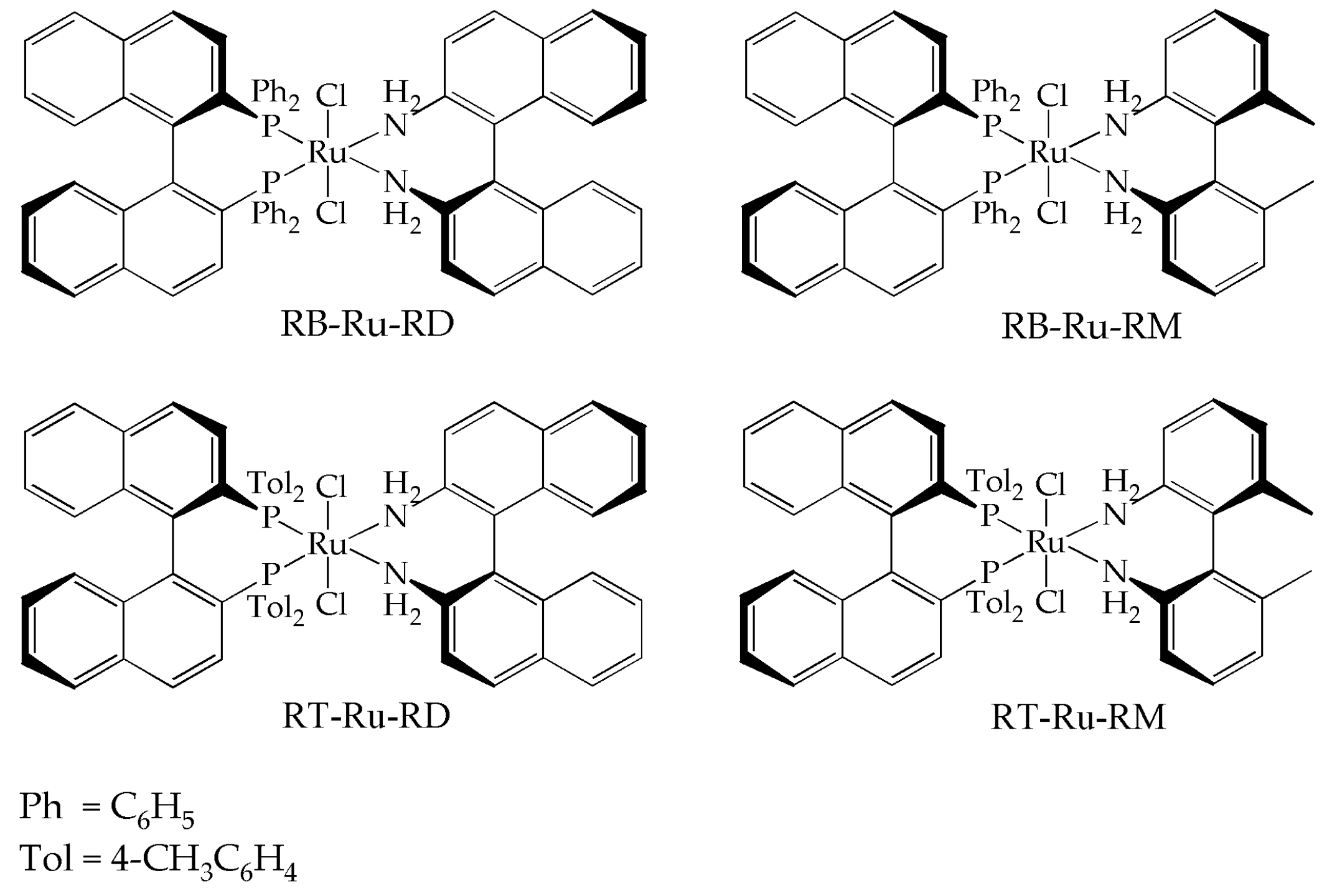
Figure 2.
Hydrogenation of acetophenone with the Ru complexes as catalysts.
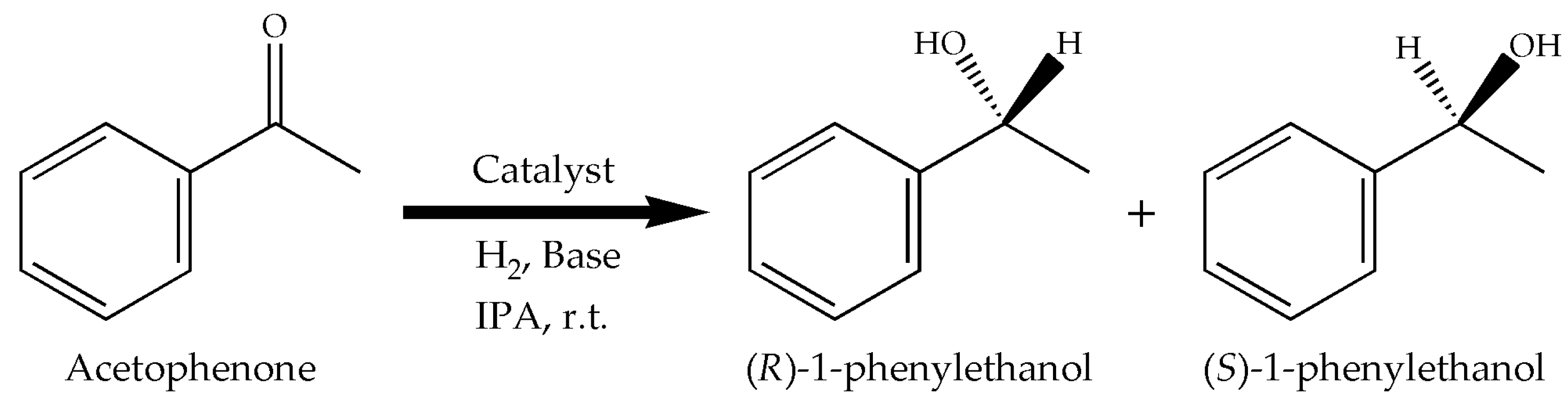
Figure 3.
Conversion during the hydrogenation of acetophenone catalyzed by the different Ru-complexes. Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 20 mL isopropanol (IPA), 1.25 mM t-BuOK and 0.01 mmol of catalyst.
Figure 3.
Conversion during the hydrogenation of acetophenone catalyzed by the different Ru-complexes. Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 20 mL isopropanol (IPA), 1.25 mM t-BuOK and 0.01 mmol of catalyst.
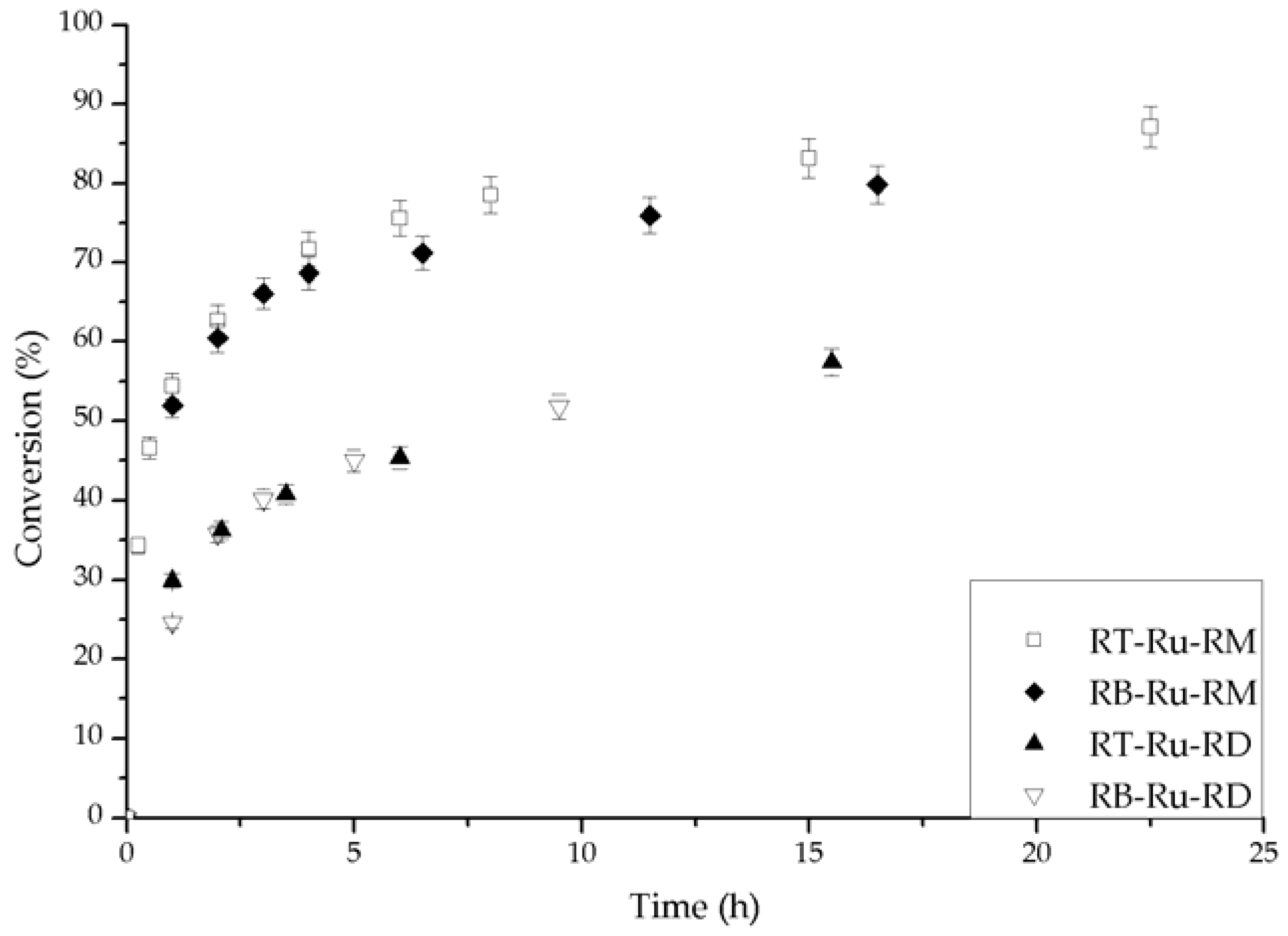
Figure 4.
Enantioselectivity during hydrogenation of acetophenone catalyzed by Ru-complexes. Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 20 mL isopropanol (IPA), 1.25 mM t-BuOK and 0.01 mmol of catalyst.
Figure 4.
Enantioselectivity during hydrogenation of acetophenone catalyzed by Ru-complexes. Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 20 mL isopropanol (IPA), 1.25 mM t-BuOK and 0.01 mmol of catalyst.
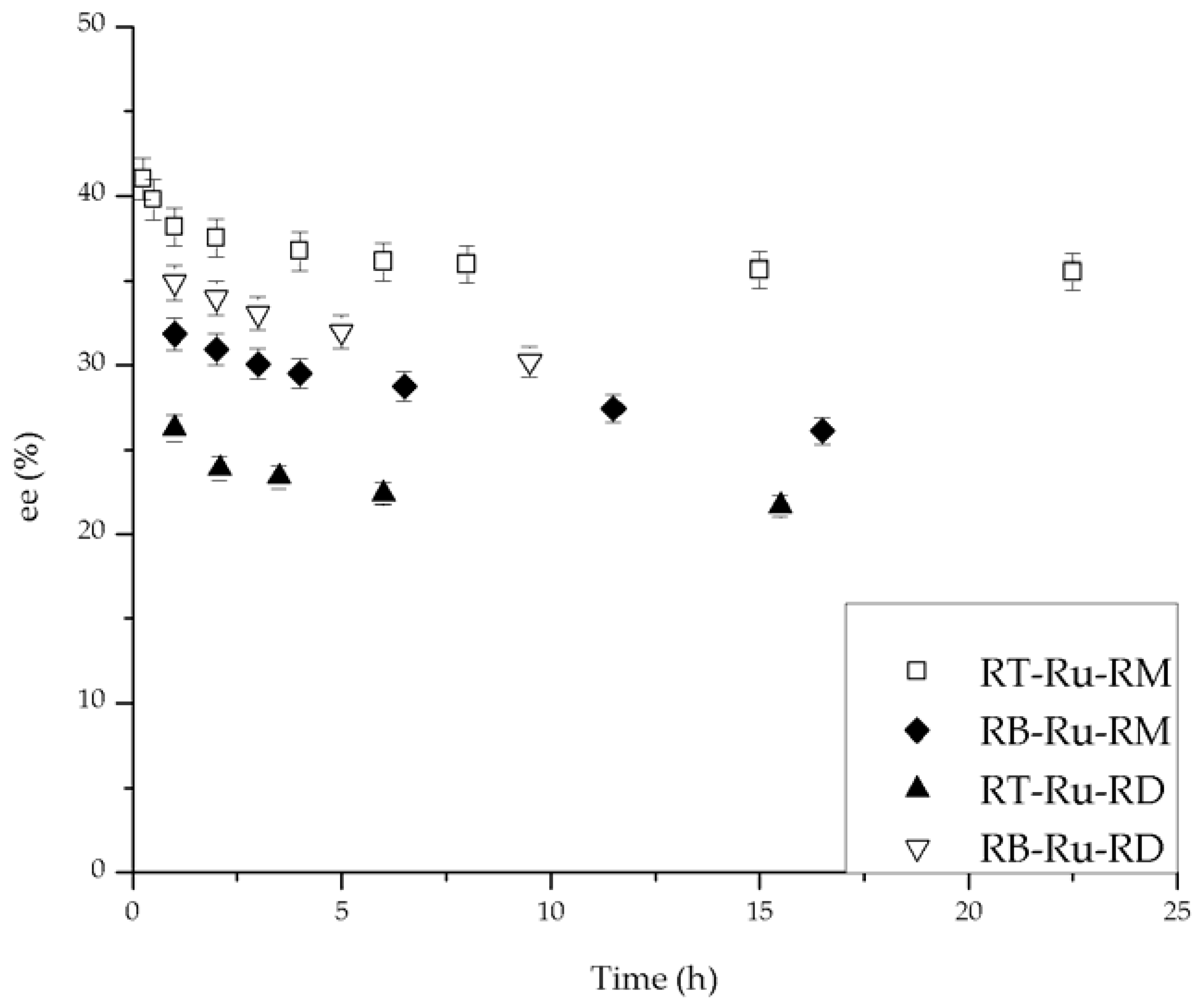
Figure 5.
Conversion and ee vs. time plots for different base concentrations with the RB-Ru-RM complex as catalyst. Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 20 mL IPA, base t-BuOK and 0.01 mmol of catalyst.
Figure 5.
Conversion and ee vs. time plots for different base concentrations with the RB-Ru-RM complex as catalyst. Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 20 mL IPA, base t-BuOK and 0.01 mmol of catalyst.

Figure 6.
Effect of the base concentration upon conversion and ee with the RR-Ru-RM catalyst. Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 40 mL IPA, base t-BuOK and 0.01 mmol of catalyst.
Figure 6.
Effect of the base concentration upon conversion and ee with the RR-Ru-RM catalyst. Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 40 mL IPA, base t-BuOK and 0.01 mmol of catalyst.

Figure 7.
Fit of the experimental data with the deactivation model (Equation (3)). Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 20 mL isopropanol (IPA), 1.25 mM t-BuOK and 0.01 mmol of catalyst.
Figure 7.
Fit of the experimental data with the deactivation model (Equation (3)). Conditions: 100 psi of H2, room temperature, 1 mmol acetophenone, 20 mL isopropanol (IPA), 1.25 mM t-BuOK and 0.01 mmol of catalyst.
Scheme 1.
Proposed steps involved in asymmetric hydrogenation considering one active site and deactivation. For clarity, the complete structure of the ligands is omitted.
Scheme 1.
Proposed steps involved in asymmetric hydrogenation considering one active site and deactivation. For clarity, the complete structure of the ligands is omitted.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the experimental condition and the resulting parameters for the kinetic model with deactivation.
Table 1.
Summary of the experimental condition and the resulting parameters for the kinetic model with deactivation.
| Entry | Complex | IPA mL | Base (mM) | kc (h−1) | kd (h−1) | ass | TOF (h−1) 1 |
|---|---|---|---|---|---|---|---|
| 1 | RB-Ru-RD | 20 | 1.250 | 0.402 | 0.773 | 0.063 | 40.2 |
| 2 | RT-Ru-RD | 20 | 1.250 | 0.913 | 2.22 | 0.033 | 91.3 |
| 3 | RB-Ru-RM | 20 | 1.250 | 1.183 | 1.151 | 0.032 | 118.3 |
| 4 | RT-Ru-RM | 20 | 1.250 | 1.935 | 1.98 | 0.031 | 193.5 |
| 5 | RB-Ru-RM | 20 | 0.500 | 1.627 | 2.222 | 0.003 | 162.7 |
| 6 | RB-Ru-RM | 20 | 2.500 | 0.383 | 0.644 | 0.019 | 38.3 |
| 7 | RT-Ru-RM | 40 | 0.625 | 2.422 | 1.697 | 0.012 | 242.2 |
| 8 | RT-Ru-RM | 40 | 2.500 | 1.907 | 1.806 | 0.021 | 190.7 |
| 9 | RT-Ru-RM | 40 | 5.000 | 1.213 | 2.047 | 0.052 | 121.3 |
1 Calculated with TOF initial = (kc·NA0)/NRu; NA0 = initial moles of acetophenone and NRu = initial moles of Ru. In all cases 1 mmol of acetophenone and 0.01 mmol of catalyst were used, together with the proper amount of t-BuOK and IPA. ass is dimensionless.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ruelas-Leyva, J.P.; Fuentes, G.A. Chiral Catalyst Deactivation during the Asymmetric Hydrogenation of Acetophenone. Catalysts 2017, 7, 193. https://doi.org/10.3390/catal7070193
AMA Style
Ruelas-Leyva JP, Fuentes GA. Chiral Catalyst Deactivation during the Asymmetric Hydrogenation of Acetophenone. Catalysts. 2017; 7(7):193. https://doi.org/10.3390/catal7070193
Chicago/Turabian StyleRuelas-Leyva, Jose P., and Gustavo A. Fuentes. 2017. "Chiral Catalyst Deactivation during the Asymmetric Hydrogenation of Acetophenone" Catalysts 7, no. 7: 193. https://doi.org/10.3390/catal7070193
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.