1. Introduction
Suzuki–Miyaura coupling reaction is a powerful protocol for the synthesis of polyarylenes containing π-conjugated polymers [
1]. We have found that
t-Bu
3PPd(Ar)Br [
2,
3] initiates chain-growth Suzuki–Miyaura coupling polymerization of haloarylboronic acid (ester) as an AB type monomer to afford well-defined polyfluorene [
4], poly(
p-phenylene) [
5], and poly(hexylthiophene) [
6], and other researchers have obtained well-defined poly(phenanthrene) [
7] and poly(fluorene-
alt-benzothiadiazole) [
8]. The chain-growth polymerization progresses via intramolecular transfer of the Pd catalyst to the terminal C–X (X = halogen) bond after reductive elimination of polymer–Pd–ArX. Therefore, these types of chain-growth polymerizations, including Kumada–Tamao and other coupling polymerizations, are known as catalyst-transfer condensation polymerizations (CTCPs) [
9,
10].
When this
t-Bu
3PPd(0) catalyst, which has a propensity for intramolecular catalyst transfer on a π-electron face, was used for Suzuki–Miyaura coupling polymerization of dibromoarene and arenyldiboronic acid ester (AA + BB polycondensation), high-molecular-weight π-conjugated polymer with a boronate moiety at both ends was obtained, even though excess dibromoarene was used [
11]. This unstochiometric polycondensation behavior is accounted for by successive substitution of the bromides in dibromoarene with arenyldiboronic acid ester or oligomers having boronate moieties at both ends through intramolecular transfer of the Pd catalyst on the π face of dibromoarene. However, Suzuki–Miyaura coupling reaction of 4,4’-dibromostilbene with phenylboronic acid in the presence of
t-Bu
3PPd(0) catalyst did not selectively afford diphenyl-substituted stilbene, implying that the Pd catalyst did not walk from one benzene ring to the other through the carbon–carbon double bond (C=C) in the stilbene after the first substitution of 4,4’-dibromostilbene with phenylboronic acid [
12]. We found that this failure of intramolecular catalyst transfer is due to bimolecular intermolecular transfer of
t-Bu
3PPd(0) on the C=C of dibromostilbene to the C=C of another dibromostilbene. However, this intermolecular transfer of the catalyst could be suppressed by introduction of alkoxy groups at the ortho positions of the C=C.
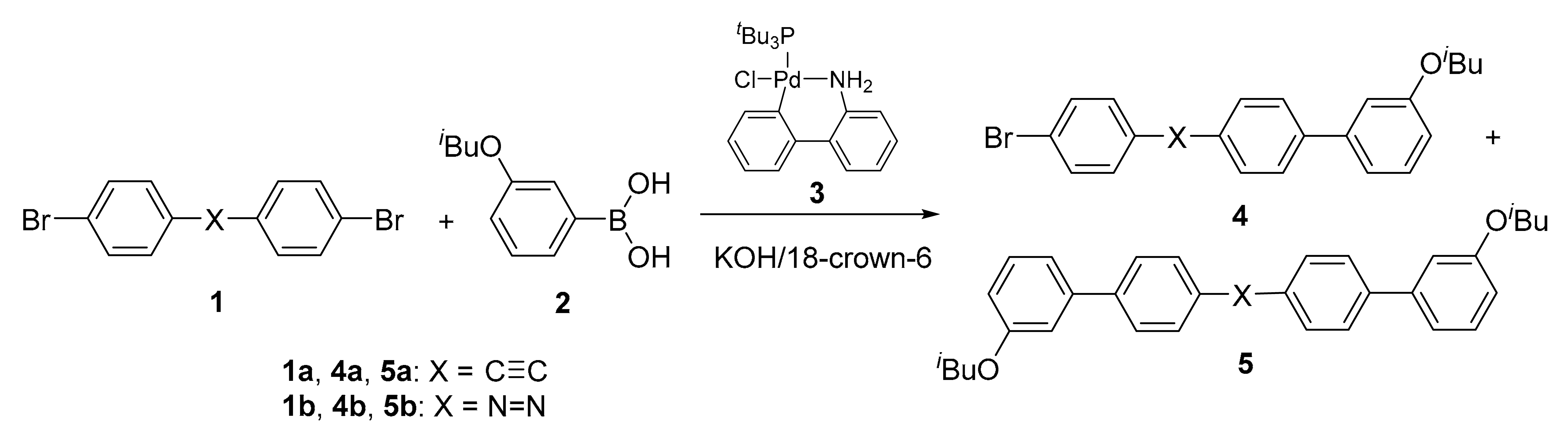
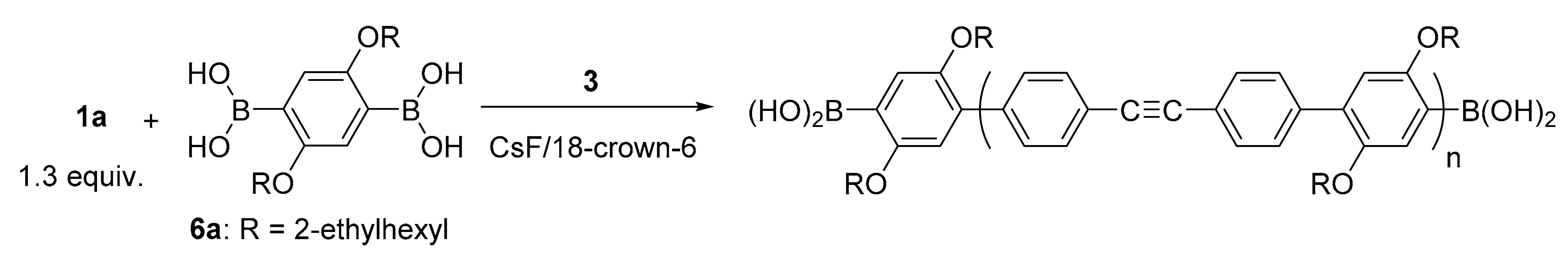
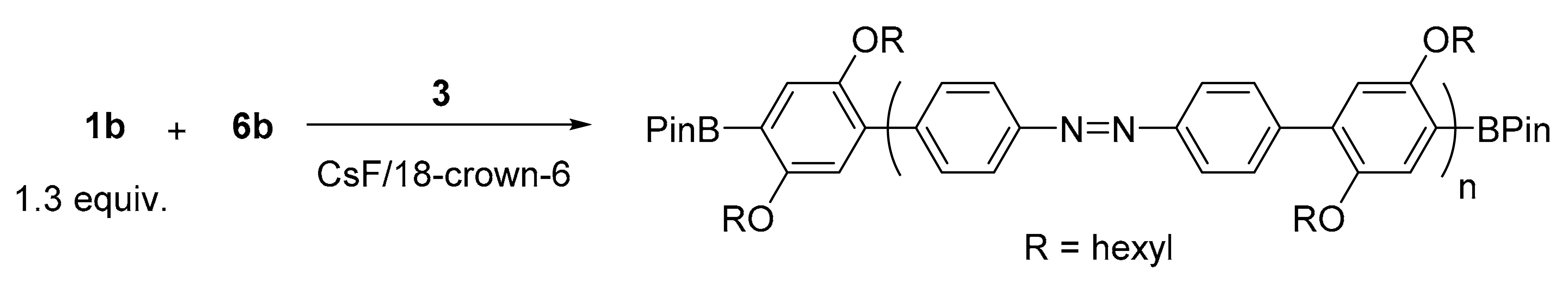
We were next interested in whether or not the Pd catalyst undergoes intramolecular catalyst transfer on other multiple bonds. In the present study, we investigated catalyst transfer on a carbon–carbon triple bond (C≡C) and nitrogen–nitrogen double bond (N=N) by means of Suzuki–Miyaura coupling reaction of 4,4’-dibromotolan (
1a) or 4,4’-dibromoazobenzene (
1b) with phenylboronic acid
2 in the presence of
t-Bu
3PPd(0) precatalyst
3 [
13] (
Scheme 1). If the catalyst undergoes intramolecular transfer on the multiple bond X in
1 and then inserts itself into the C–Br bond after the first substitution, the main product would be disubstituted
5. On the other hand, if the catalyst diffuses into the reaction mixture after the first substitution, the main product would be monosubstituted
4. Bielawski and coworkers have demonstrated intramolecular catalyst transfer of
t-Bu
3PPd(0) catalyst on C≡C in Stille CTCP of 4-(tributylstannylethynyl)-2,5-dialkoxyiodobenzene as an AB monomer [
14]. However, this catalyst transfer on C≡C might be dependent on the steric effect of the tributylstannyl and/or alkoxy groups, as in the case of catalyst transfer on C=C in
o-alkoxy-substituted stilbene. In the present work, we found that
t-Bu
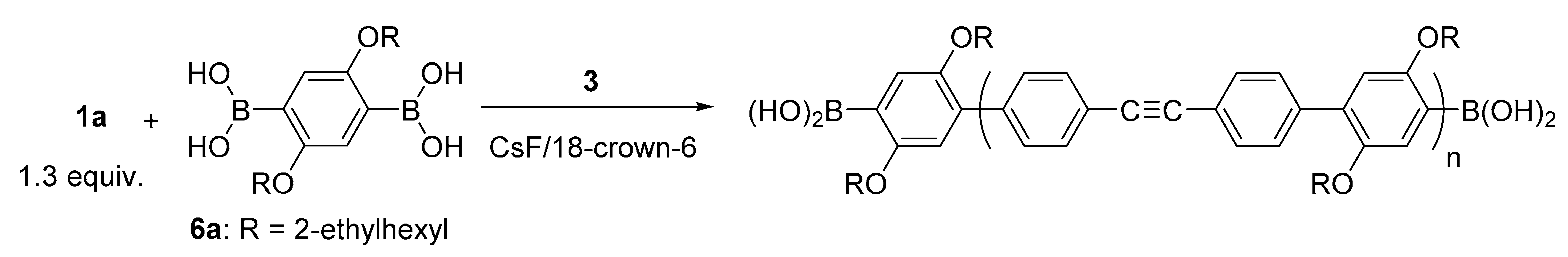
3PPd(0) catalyst undergoes intramolecular catalyst transfer on C≡C and N=N even if the benzene rings adjacent to these multiple bonds are unsubstituted, in contrast to the case of catalyst transfer on C=C. To demonstrate effective catalyst transfer on C≡C and N=N, we further conducted unstoichiometric Suzuki–Miyaura polycondensation of excess
1 and 1.0 equiv. of phenylenediboronic acid (ester) in the presence of
3, obtaining π-conjugated polymer with a boronate moiety at both ends.
3. Materials and Methods
3.1. Materials
All starting materials were purchased from commercial suppliers (TCI, Aldrich, Wako, and Kanto, Tokyo, Japan) and used without further purification. Dry tetrahydrofuran (THF, stabilizer-free, Kanto, Tokyo, Japan) and distilled water (Wako, Tokyo, Japan) were used as received. Dibromoazobenzene
1b [
18] and phenylenediboronic acid
6a [
11] were prepared according to the literature.
3.2. Measurements
1H and 13C NMR spectra were obtained on JEOL ECA-500 (JEOL, Tokyo, Japan and ECA-600 spectrometers (JEOL, Tokyo, Japan). The internal standard for 1H NMR spectra in CDCl3 was tetramethylsilane (0.00 ppm) and the internal standard for 13C NMR spectra in CDCl3 was the midpoint of CDCl3 (77.0 ppm). IR spectra were recorded on a JASCO FT/IR-410 (JASCO, Tokyo, Japan). All melting points were measured with a Yanagimoto hot stage melting point apparatus (Yanaco, Tokyo, Japan) without correction. GC was performed on a Shimadzu GC-14B gas chromatograph (Shimazu, Kyoto, Japan) equipped with a Shimadzu fused silica capillary column CBP1-W12–100 (12 m length, 0.53 mm i.d.) and a flame ionization detector (FID). Isolation of 4b and 5b was carried out on a Japan Analytical Industry LC908-C60 recycling preparative HPLC (eluent, CHCl3) (JAI, Tokyo, Japan) with two JAIGEL columns (1H-40 and 2H-40). Column chromatography was performed on silica gel (Kieselgel 60, 230–400 mesh, Merck, Darmstadt, Germany) with a specified solvent. The Mn and Mw/Mn values of polymer were measured on a Tosoh HLC-8020 gel permeation chromatography (GPC) unit (eluent, THF; calibration, polystyrene standards) (Tosoh, Yamaguchi, Japan) with two TSK-gel columns (2 × Multipore HXL-M). MALDI-TOF mass spectra were recorded on a Shimadzu/Kratos AXIMA-CFR plus (Shimadzu, Kyoto, Japan) in the reflectron ion mode (laser λ = 337 nm). DCTB (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile) was used as the matrix for the MALDI-TOF mass measurements.
3.3. Synthesis of Monosubstituted Tolan 4a
Transfer of reagents and withdrawal of small aliquots of the reaction mixture for analysis were carried out via a syringe from a three-way stopcock under a stream of nitrogen. A two-necked round-bottomed flask was equipped with a three-way stopcock and a dimroth condenser. Dibromotolan 1a (134.9 mg, 0.40 mmol), phenylboronic acid 2 (81.4 mg, 0.42 mmol), (PPh3)4Pd (23.3 mg, 0.020 mmol), K3PO4 (370.9 mg, 1.75 mmol), and 18-crown-6 (846 mg, 3.26 mmol) were placed in the flask, and the flask was flushed with argon. Dry THF (10.0 mL) and distilled water (0.60 mL) were added to the flask via a syringe, and the reaction mixture was refluxed for 3 h. The reaction was quenched with 1 M hydrochloric acid, and the mixture was extracted with CHCl3. The organic layer was washed with water and dried over anhydrous MgSO4. The solvent was removed under reduced pressure, and the residue was purified by means of column chromatography (SiO2, hexane) to afford 4a as a pale yellow viscous liquid (111.3 mg, 69%); mp 96–99 °C.
1H NMR (500 MHz, CDCl3) δ 7.58 (s, 4H), 7.50 (d, J = 8.6 Hz, 2H), 7.40 (d, J = 8.6 Hz, 2H), 7.35 (t, J = 8.0 Hz, 1H), 7.18 (d, J = 6.9 Hz, 1H), 7.13 (s, 1H), 6.90 (dd, J = 5.7 Hz and 2.3 Hz, 1H), 3.78 (d, J = 6.9 Hz, 2H), 2.14-2.10 (m, 1H), 1.05 (d, J = 6.3 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 159.7, 141.8, 140.9, 132.0, 129.8, 127.3, 122.3, 119.3, 113.4, 90.0, 75.6, 30.9, 28.3, 19.3; IR (KBr) 3460, 2908, 1606, 1471, 1350, 1206, 1105, 965, 776, 690, 513 cm−1.
3.4. Synthesis of Disubstituted Tolan 5a
Transfer of reagents and withdrawal of small aliquots of the reaction mixture for analysis were carried out via a syringe from a three-way stopcock under a stream of nitrogen. Dibromotolan 1a (134.3 mg, 0.40 mmol), 2 (157.7 mg, 0.81 mmol), 3 (10.2 mg, 0.020 mmol), KOH (105.4 mg, 1.88 mmol), and 18-crown-6 (861.0 mg, 3.26 mmol) were placed in a flask equipped with a three-way stop cock, and the atmosphere in the flask was replaced with argon. Dry THF (10.0 mL) and distilled water (0.60 mL) were added to the flask via a syringe, and the mixture was degassed under reduced pressure and filled with argon. The reaction mixture was stirred at room temperature for 2 h. The reaction was quenched with hydrochloric acid, and the mixture was extracted with CHCl3. The organic layer was washed with water and dried over anhydrous MgSO4. The solvent was removed under reduced pressure, and the residue was purified by means of column chromatography (SiO2, hexane) to afford 5a as a dark yellow viscous solid (184.1 mg, 96%); mp 130–134 °C.
1H NMR (500 MHz, CDCl3) δ 7.60 (s, 8H), 7.35 (t, J = 7.7 Hz, 2H), 7.18 (d, J = 7.7 Hz, 2H), 7.15 (s, 2H), 6.90 (dd, J = 6.0 and 2.0 Hz, 2H), 3.79 (d, J = 6.6 Hz, 4H), 2.13-2.10 (m, 2H), 1.04 (d, J = 6.9 Hz, 12H); 13C NMR (126 MHz, CDCl3) δ 167.7, 152.2, 147.2, 144.7, 130.8, 124.5, 115.5, 114.2, 83.9, 69.8, 29.7, 24.8; IR (KBr) 3461, 2907, 1606, 1522, 1295, 1206, 1050, 1013, 873, 830, 776, 690, 538 cm−1.
3.5. Synthesis of Mono- and Disubstituted Azobenzene 4b and 5b
Transfer of reagents and withdrawal of small aliquots of the reaction mixture for analysis were carried out via a syringe from a three-way stopcock under a stream of nitrogen. Dibromoazobenzene 1b (50.9 mg, 0.15 mmol), 2 (30.6 mg, 0.16 mmol), 3 (4.2 mg, 0.016 mmol), KOH (37.8 mg, 0.68 mmol), and 18-crown-6 (318.4 mg, 1.2 mmol) were placed in a flask equipped with a three-way stop cock, and the atmosphere in the flask was replaced with argon. Dry THF (6.0 mL) and distilled water (0.36 mL) were added to the flask via a syringe, and the mixture was degassed under reduced pressure and filled with argon. The reaction mixture was stirred at room temperature for 24 h. The reaction was quenched with hydrochloric acid, and the mixture was extracted with CHCl3. The organic layer was washed with water and dried over anhydrous MgSO4. The solvent was removed under reduced pressure, and the residue was purified by means of HPLC to afford 4b as a pale yellow viscous solid (10.4 mg, 17%) and 5b as a pale yellow viscous solid (22.3 mg, 31%).
4b: mp 89–95 °C; 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.6 Hz, 2H), 7.82 (d, J = 8.9 Hz, 2H), 7.74 (d, J = 8.6 Hz, 2H), 7.66 (d, J = 8.6 Hz, 2H), 7.37 (t, J = 8.0 Hz, 1H), 7.24 (d, J = 7.7 Hz, 1H), 7.20 (s, 1H), 6.94 (dd, J = 7.7 and 1.7 Hz, 1H), 3.80 (d, J = 6.6 Hz, 1H), 2.16–2.10 (m, 1H), 1.06 (d, J = 6.6 Hz, 6H), 13C NMR (126 MHz, CDCl3) δ 159.7, 151.9, 143.6, 141.6, 129.8, 127.8, 123.3, 119.4, 113.8, 113.6, 74.5, 28.3, 19.3, IR (KBr) 3460, 2911, 1599, 1469, 1397, 1217, 1108, 965, 840, 610, 552 cm−1.
5b: mp 195–198 °C; 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 8.6 Hz, 4H), 7.75 (d, J = 8.3 Hz, 4H), 7.37 (t, J = 8.0 Hz, 2H), 7.24 (d, J = 8.3 Hz, 2H), 7.20 (s, 2H), 6.93 (dd, J = 6.3 and 2.0 Hz, 2H), 3.81 (d, J = 6.3 Hz, 4H), 2.17–2.11 (m, 2H), 1.06 (d, J = 6.6 Hz, 12H), 13C NMR (126 MHz, CDCl3) δ 159.7, 151.6, 132.3, 129.9, 128.3, 127.8, 124.3, 119.4, 113.9, 113.6, 77.3, 28.3, 19.3, IR (KBr) 3448, 2924, 2362, 1719, 1571, 1473, 1281, 1065, 1006, 835, 657, 577 cm−1.
3.6. Suzuki–Miyaura Coupling Reaction of 1a with 2
Transfer of reagents and withdrawal of small aliquots of the reaction mixture for analysis were carried out via a syringe from a three-way stopcock under a stream of nitrogen. Dibromotolan 1a (0.100 mmol), 2 (0.105 mmol), 3 (0.005 mmol), KOH (0.5 mmol), 18-crown-6 (0.80 mmol), and 1,4-bis(hexyloxy)benzene (0.040 mmol) as an internal standard substance were placed in the flask, and the atmosphere in the flask was replaced with argon. Dry THF (4.0 mL), distilled water (0.40 mL), and additive (none or 0.100 mmol) were added to the flask via a syringe, and the mixture was degassed under reduced pressure and filled with argon. The reaction mixture was stirred at room temperature for 2 h, and the reaction was quenched with 1 M hydrochloric acid. The mixture was extracted with CHCl3, and the organic layer was subjected to GC analysis for estimation of conversion of 1a and the product ratio of 4a to 5a.
3.7. Suzuki–Miyaura Coupling Reaction of 1b with 2
Transfer of reagents and withdrawal of small aliquots of the reaction mixture for analysis were carried out via a syringe from a three-way stopcock under a stream of nitrogen. Dibromoazobenzene 1b (50.9 mg, 0.15 mmol), 2 (30.6 mg, 0.16 mmol), 3 (4.2 mg, 0.008 mmol), KOH (37.8 mg, 0.68 mmol), 18-crown-6 (318.4 mg, 1.2 mmol), and 1,4-bis(hexyloxy)benzene (27.2 mg, 0.016 mmol) as an internal standard substance were placed in the flask, and the atmosphere in the flask was replaced with argon. Dry THF (6.0 mL) and distilled water (0.36 mL) were added to the flask via a syringe, and the mixture was degassed under reduced pressure and filled with argon. The reaction mixture was stirred at room temperature for 24 h, and the reaction was quenched with 1 M hydrochloric acid. The mixture was extracted with CHCl3 and dried over MgSO4. The solvent was removed under reduced pressure. The whole mixture was dissolved in CDCl3 and then subjected to 1H NMR analysis to determine conversion of 1b and the product ratio of 4b to 5b.
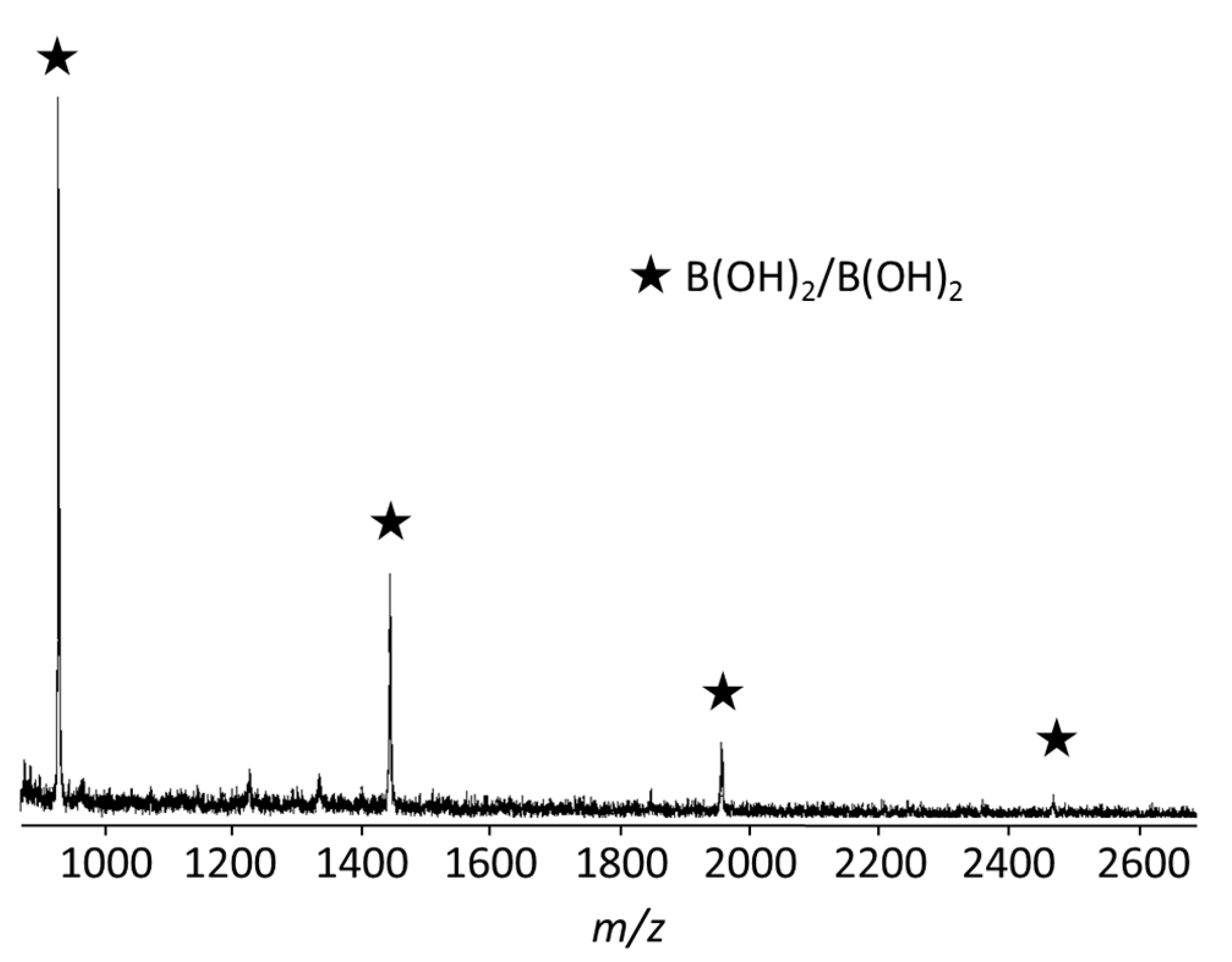
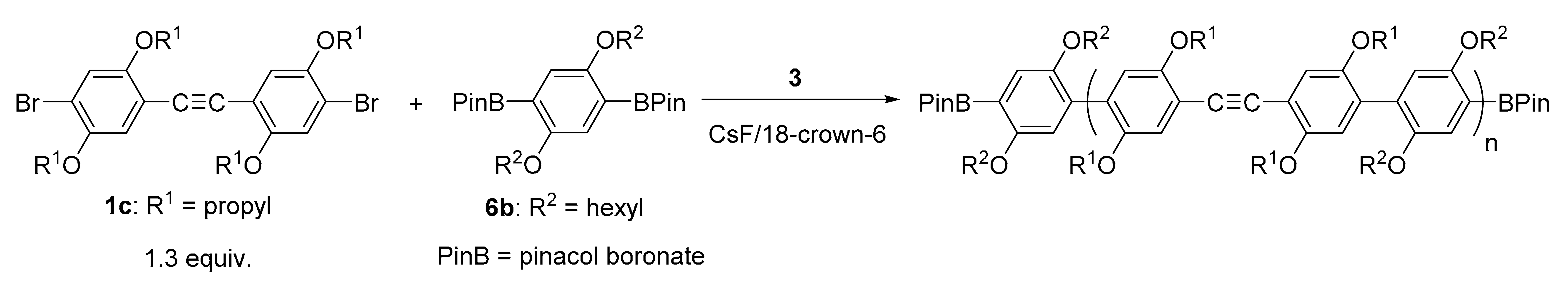
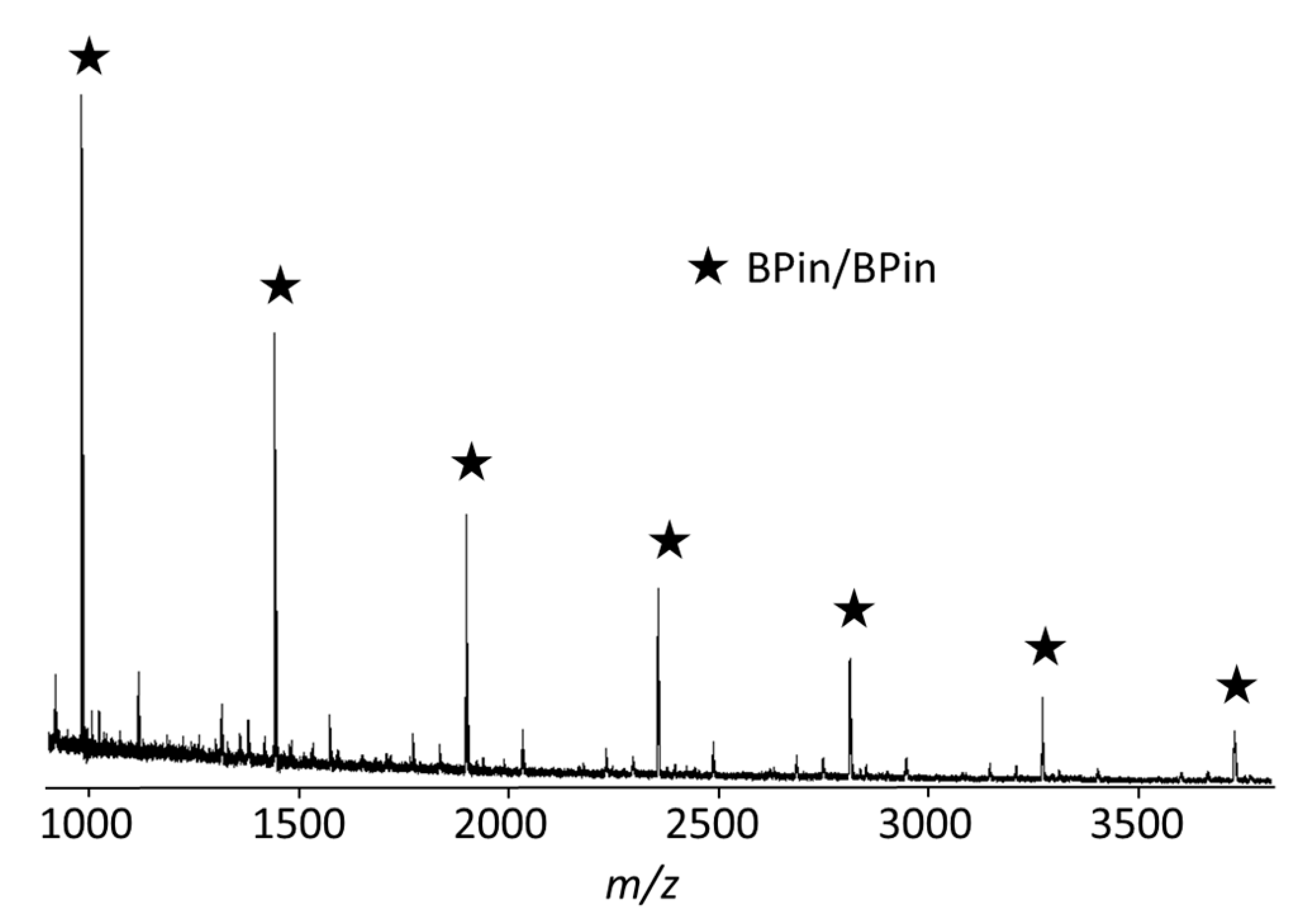
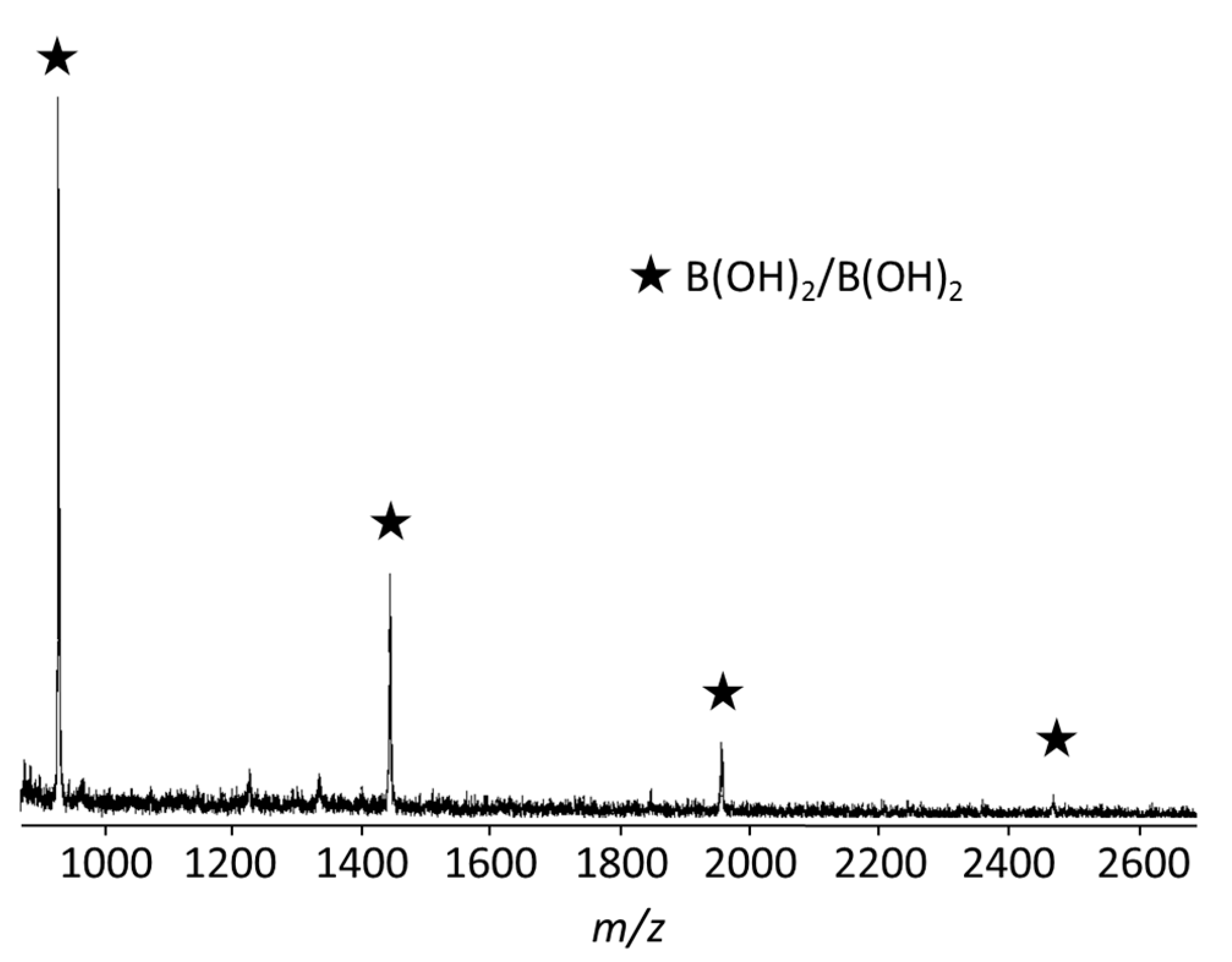
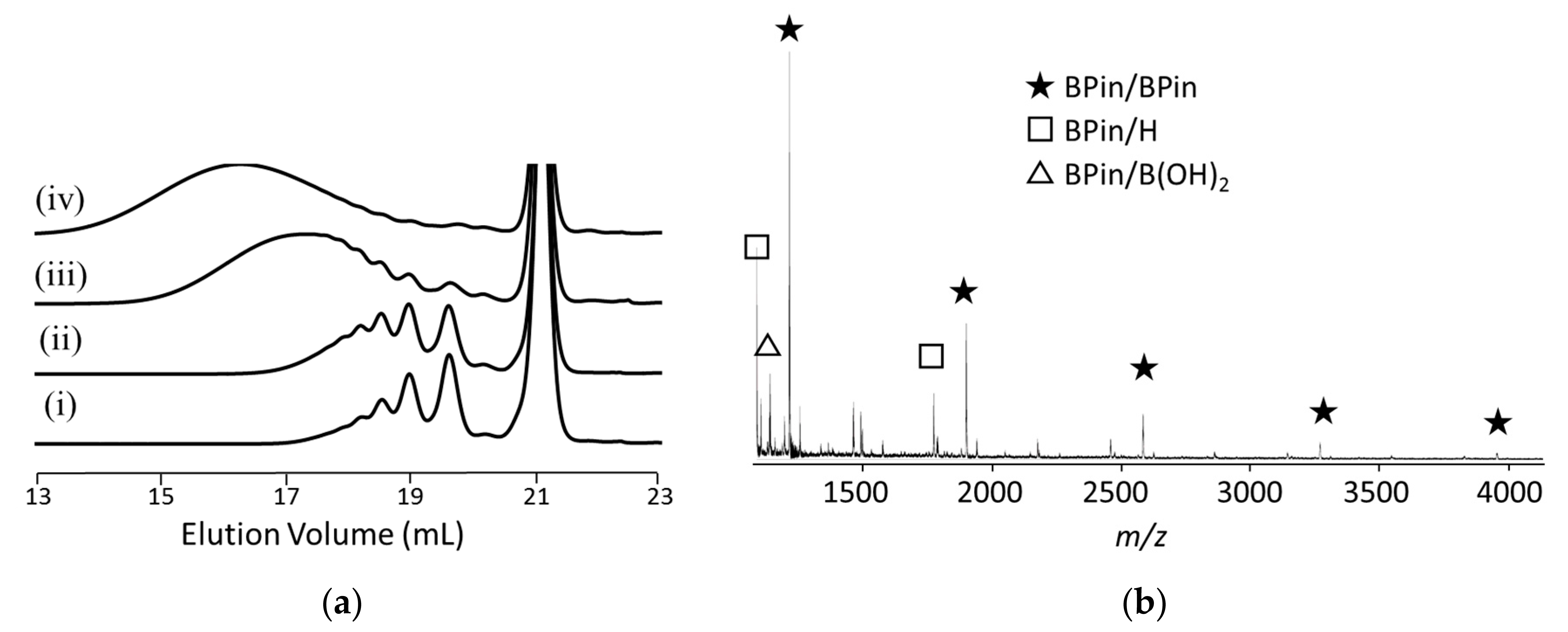
3.8. Polycondensation of 1c and 6b
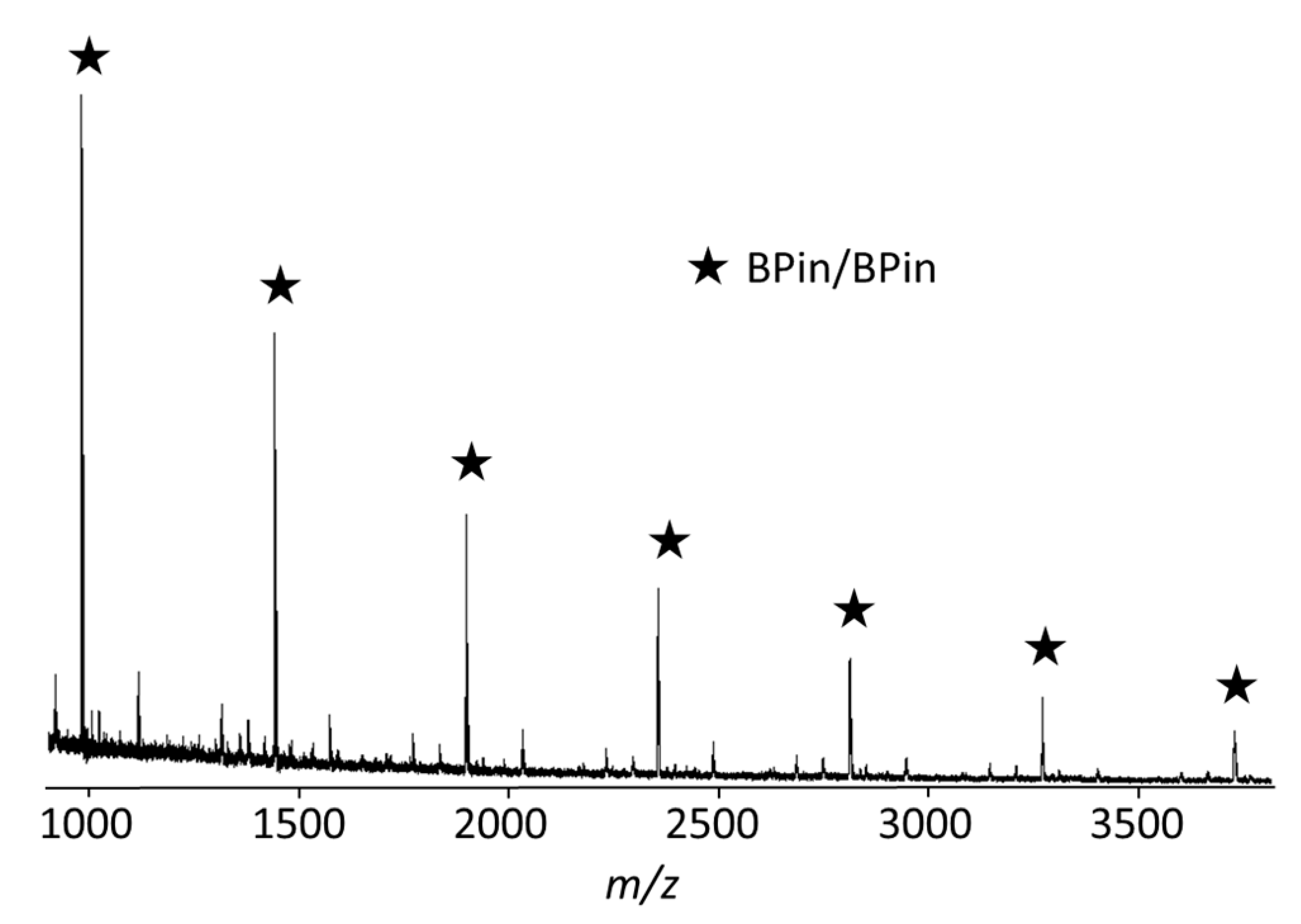
Transfer of reagents and withdrawal of small aliquots of the reaction mixture for analysis was carried out via a syringe from a three-way stopcock under a stream of nitrogen. Dibromo monomer 1c (36.89 mg, 0.065 mmol), phenylenediboronic acid pinacol ester 6b (26.91 mg, 0.051 mmol), CsF (34.1 mg, 0.22 mmol), 18-crown-6 (119.1 mg, 0.45 mmol), and 3 (1.4 mg, 0.027 mmol) were placed in the flask, and the atmosphere in the flask was replaced with argon. Dry THF (6.0 mL) and distilled water (0.2 mL) were added to the flask via a syringe, and the mixture was degassed under reduced pressure and filled with argon. The reaction mixture was stirred at room temperature for two days, and the reaction was quenched with 1 M hydrochloric acid. The mixture was extracted with CHCl3, and the organic layer was washed with water, then dried over anhydrous MgSO4. Concentration under reduced pressure gave residue, which was purified by preparative HPLC to give polymer (32.7 mg, 94%).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}