Surface Oxidation of Supported Ni Particles and Its Impact on the Catalytic Performance during Dynamically Operated Methanation of CO2



Abstract
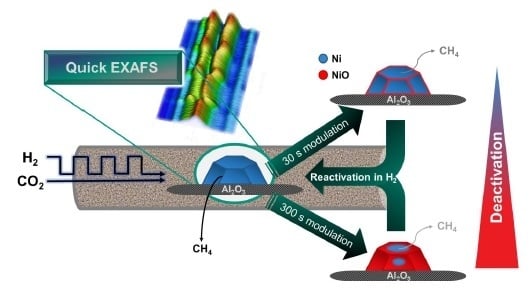
:
1. Introduction
2. Results and Discussion
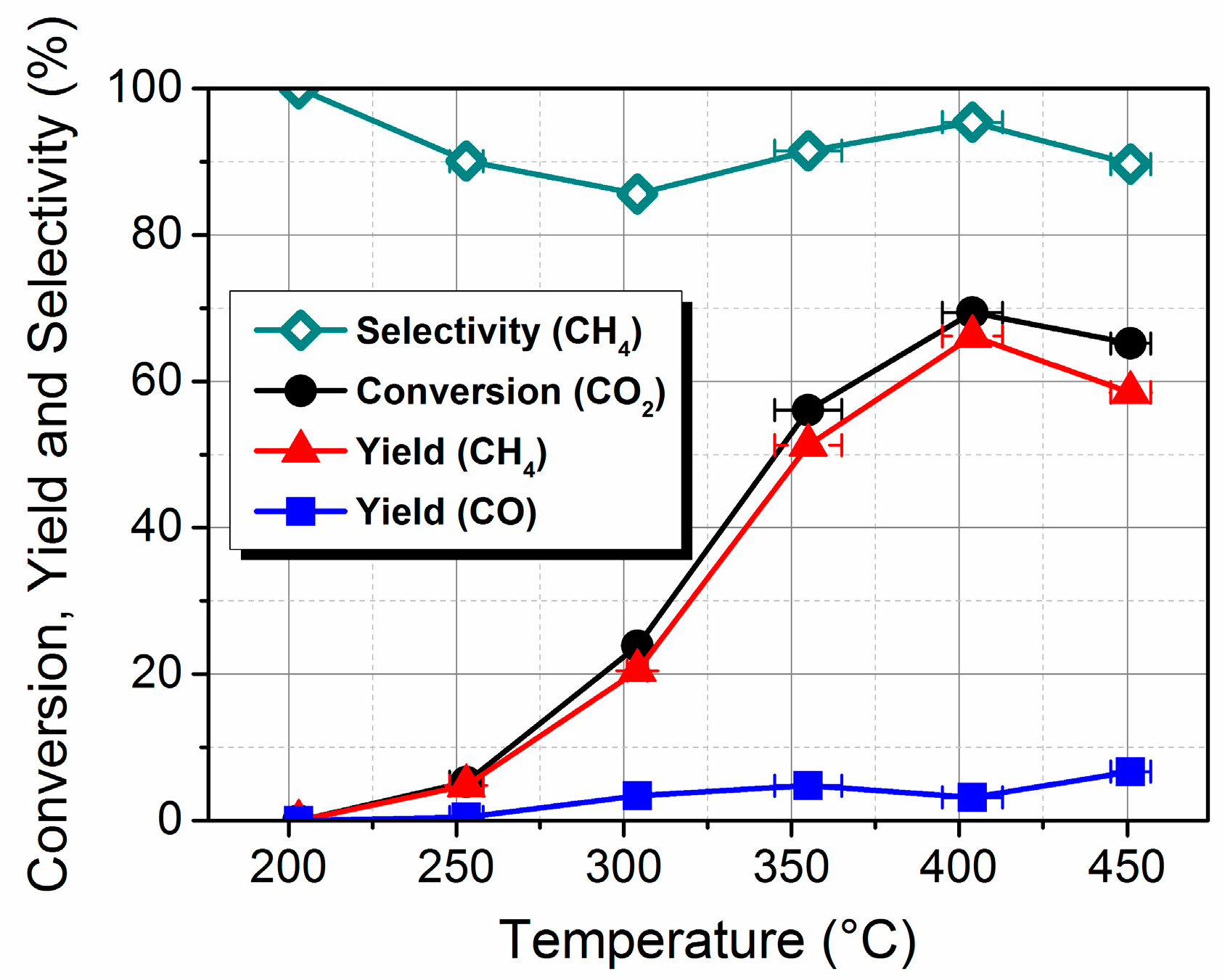
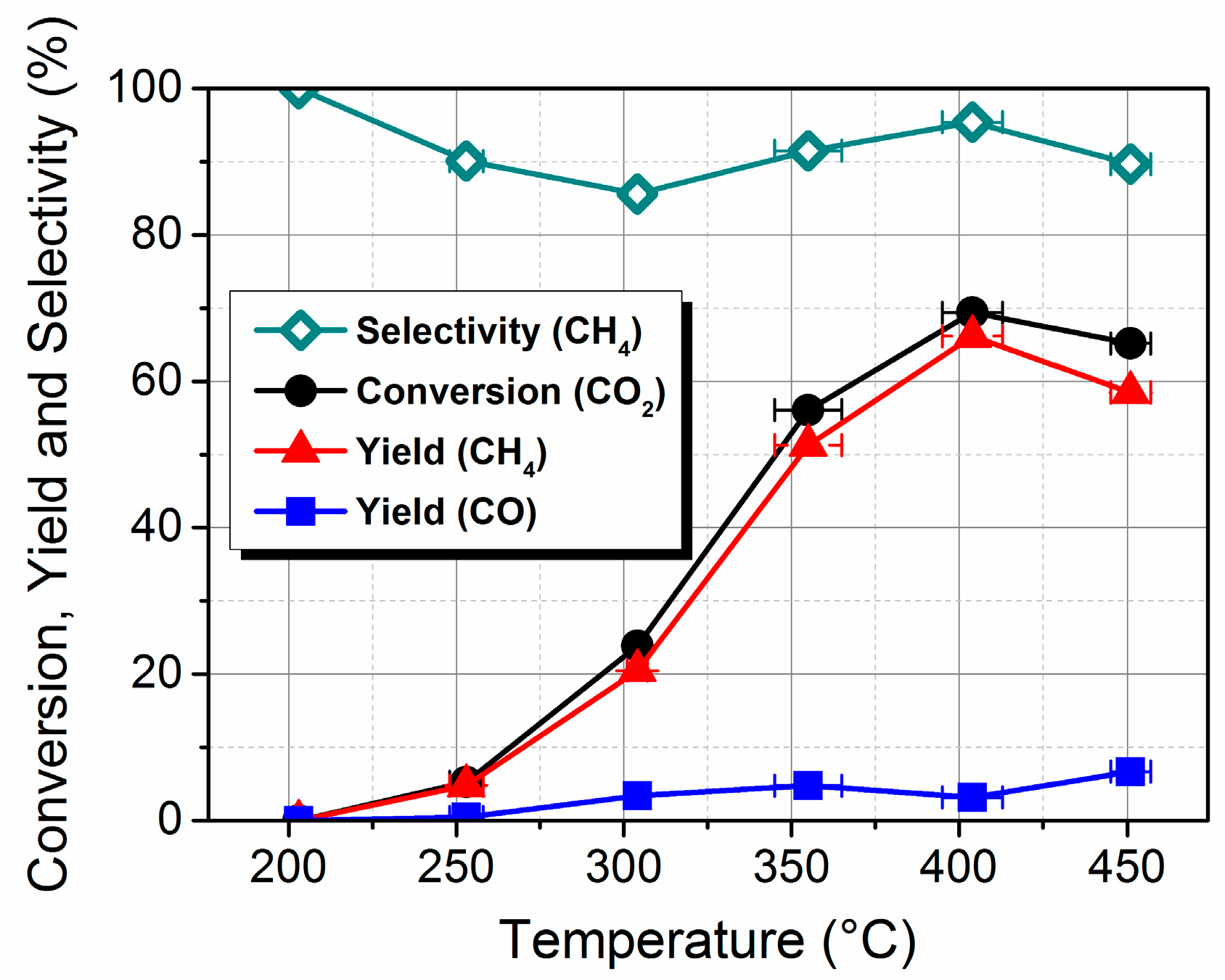
2.1. Preparation, Characterization and Catalytic Performance
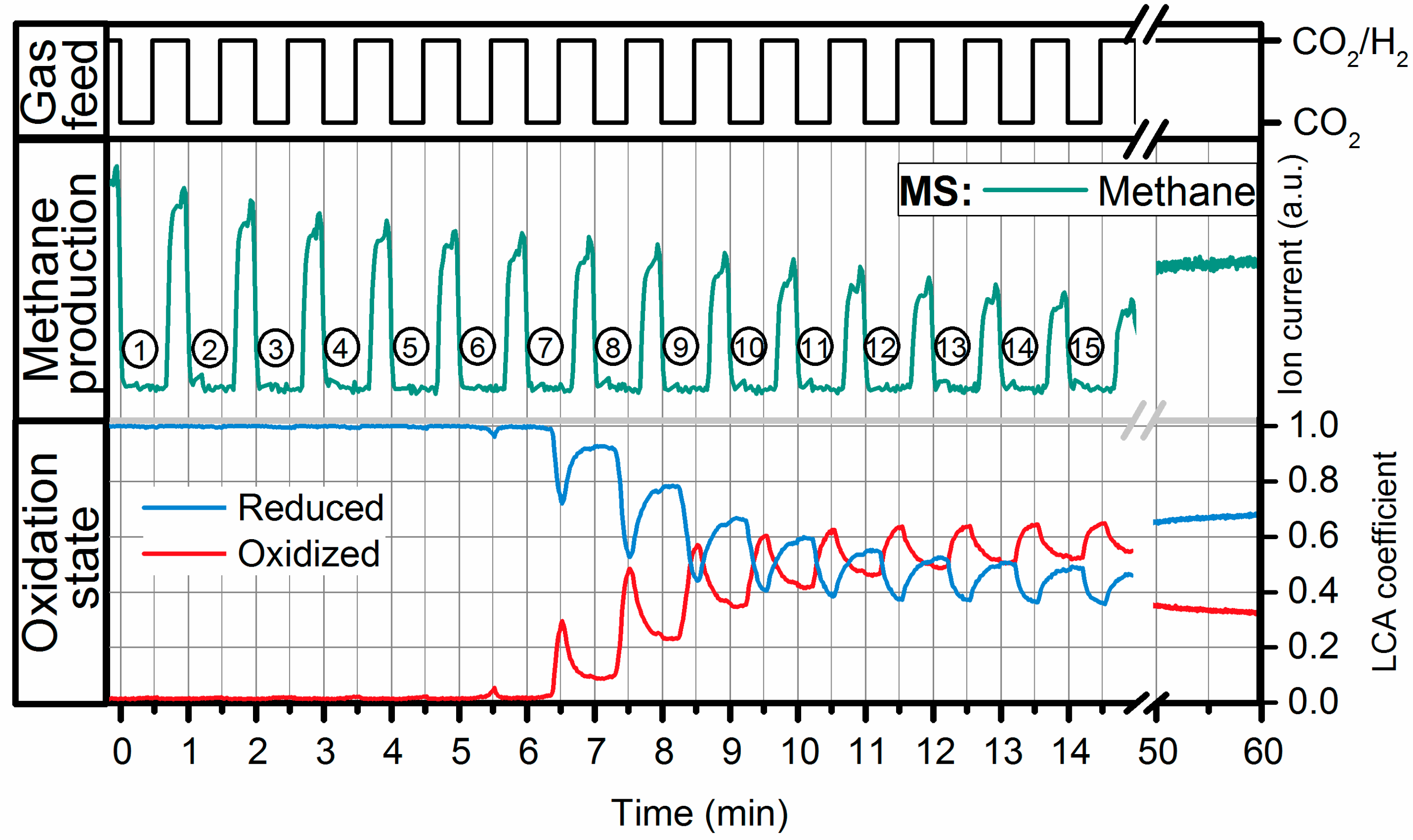
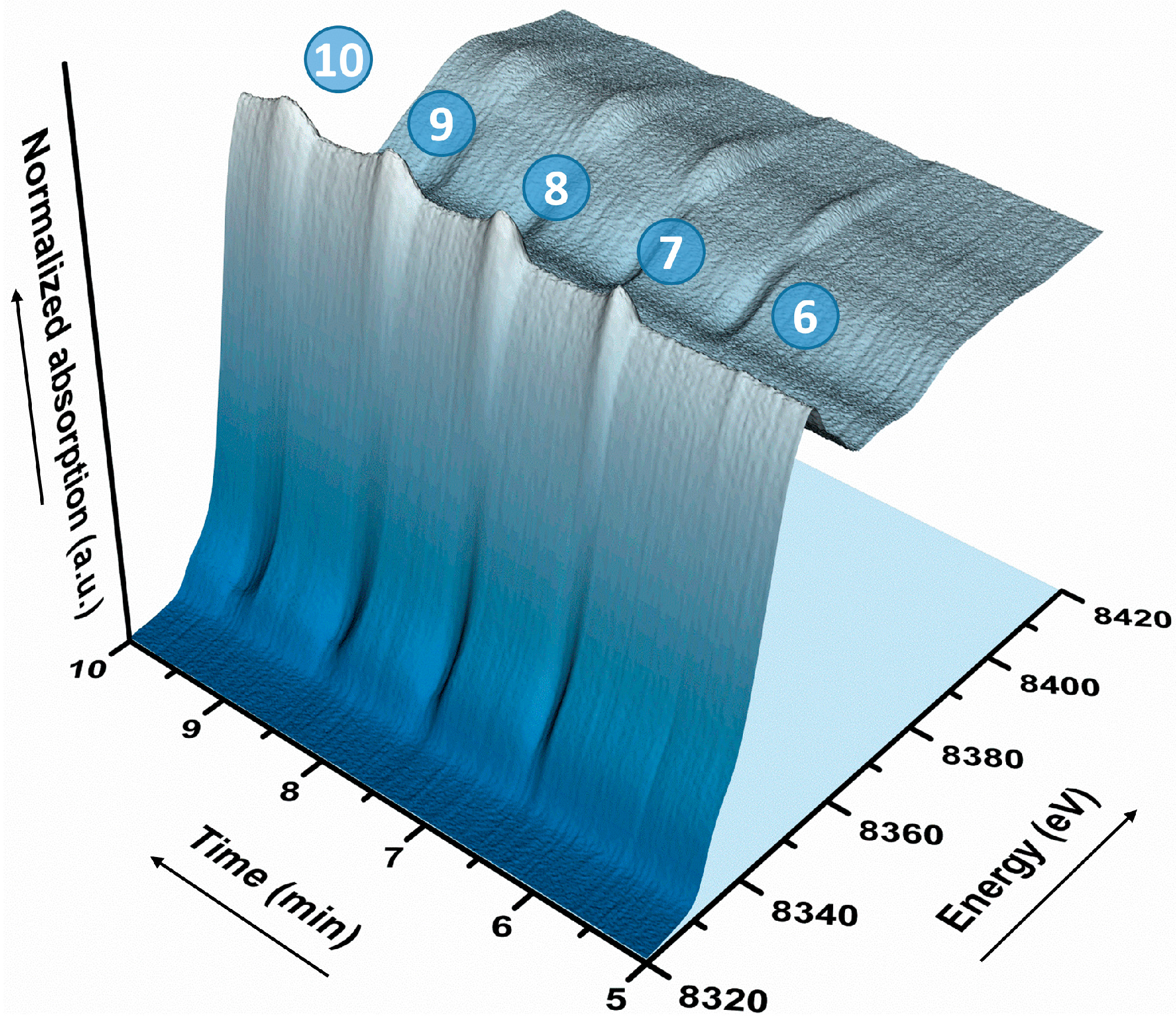
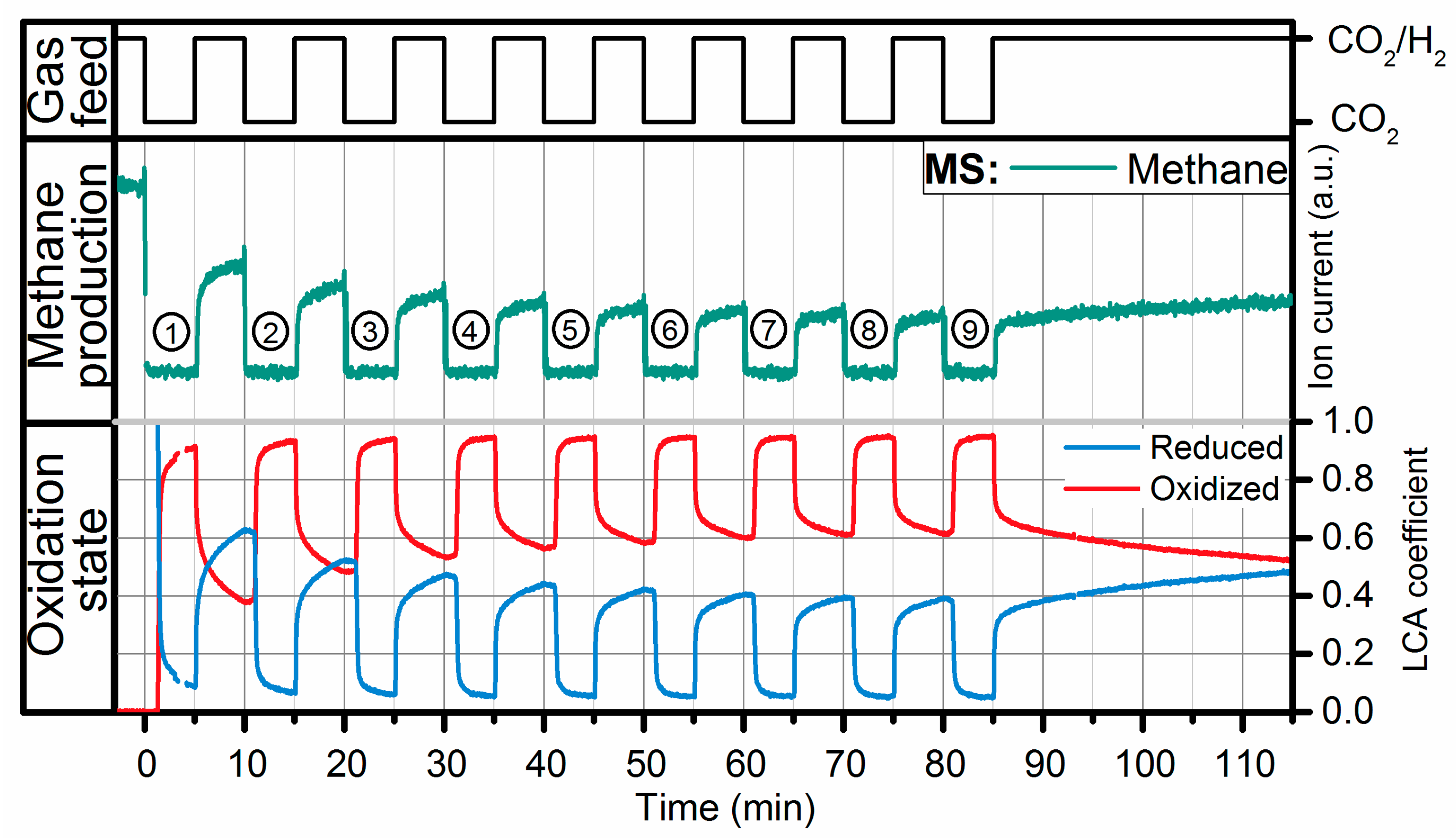
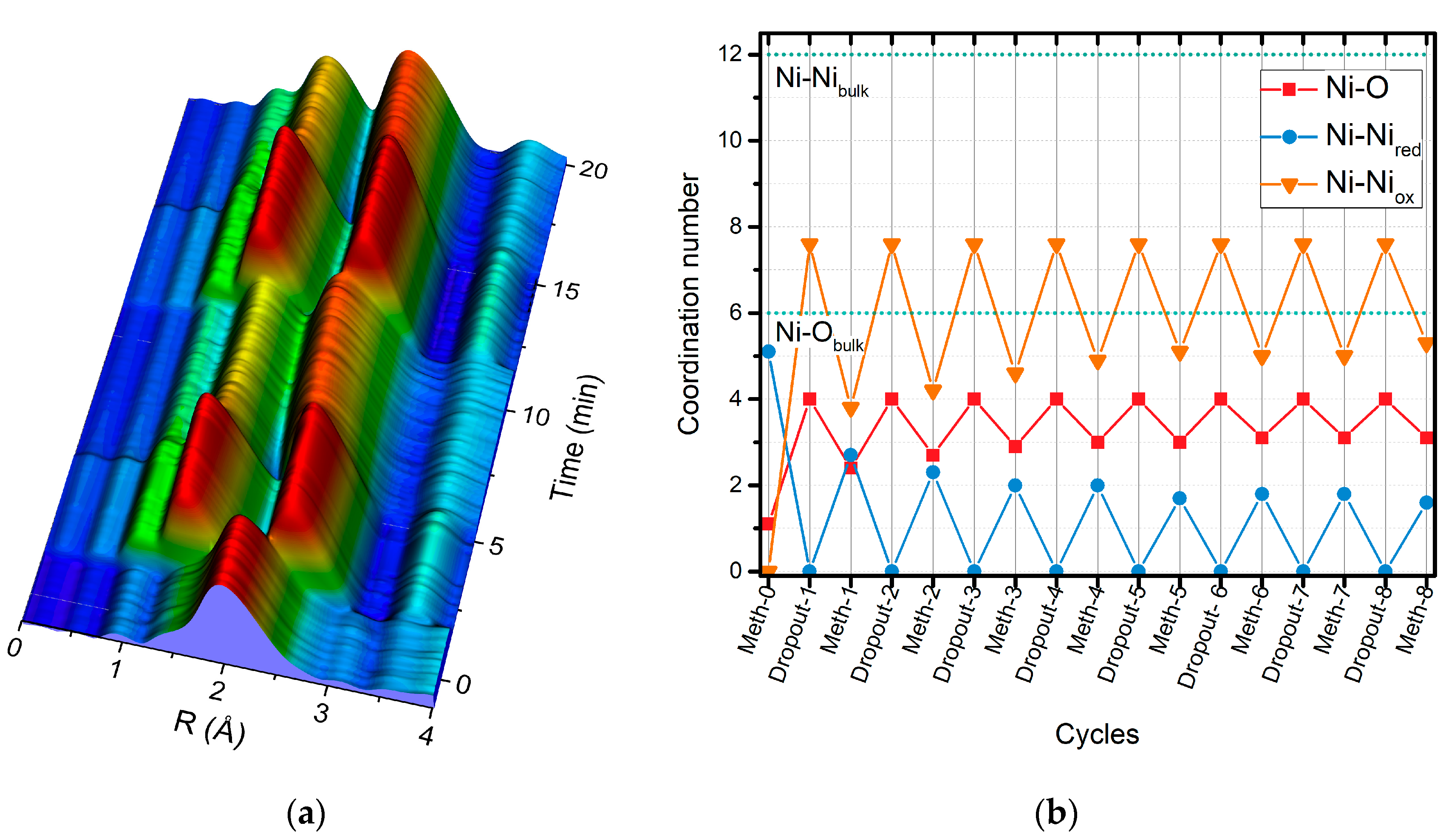
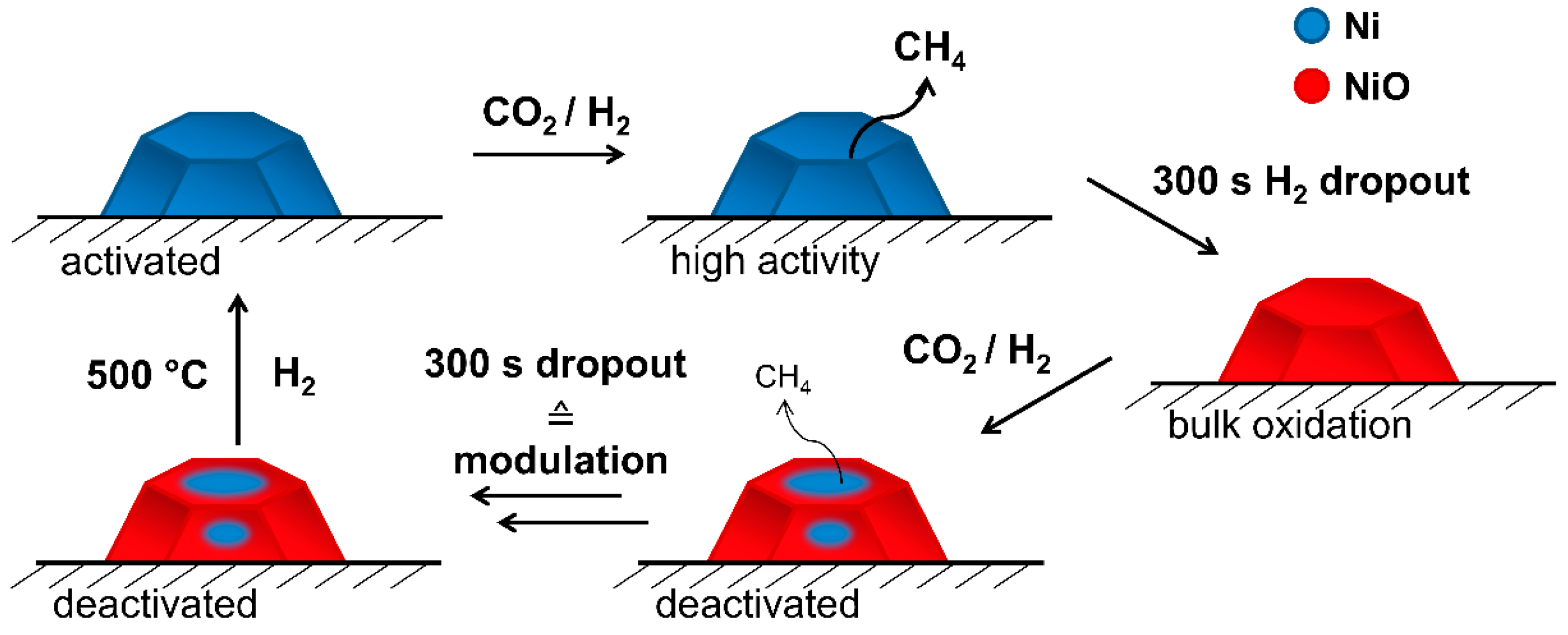
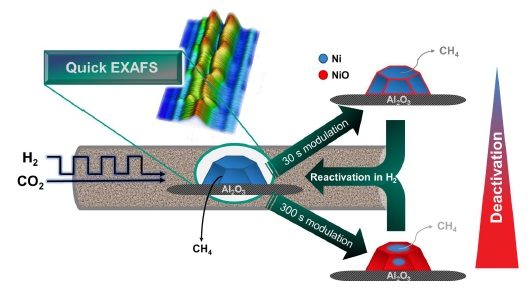
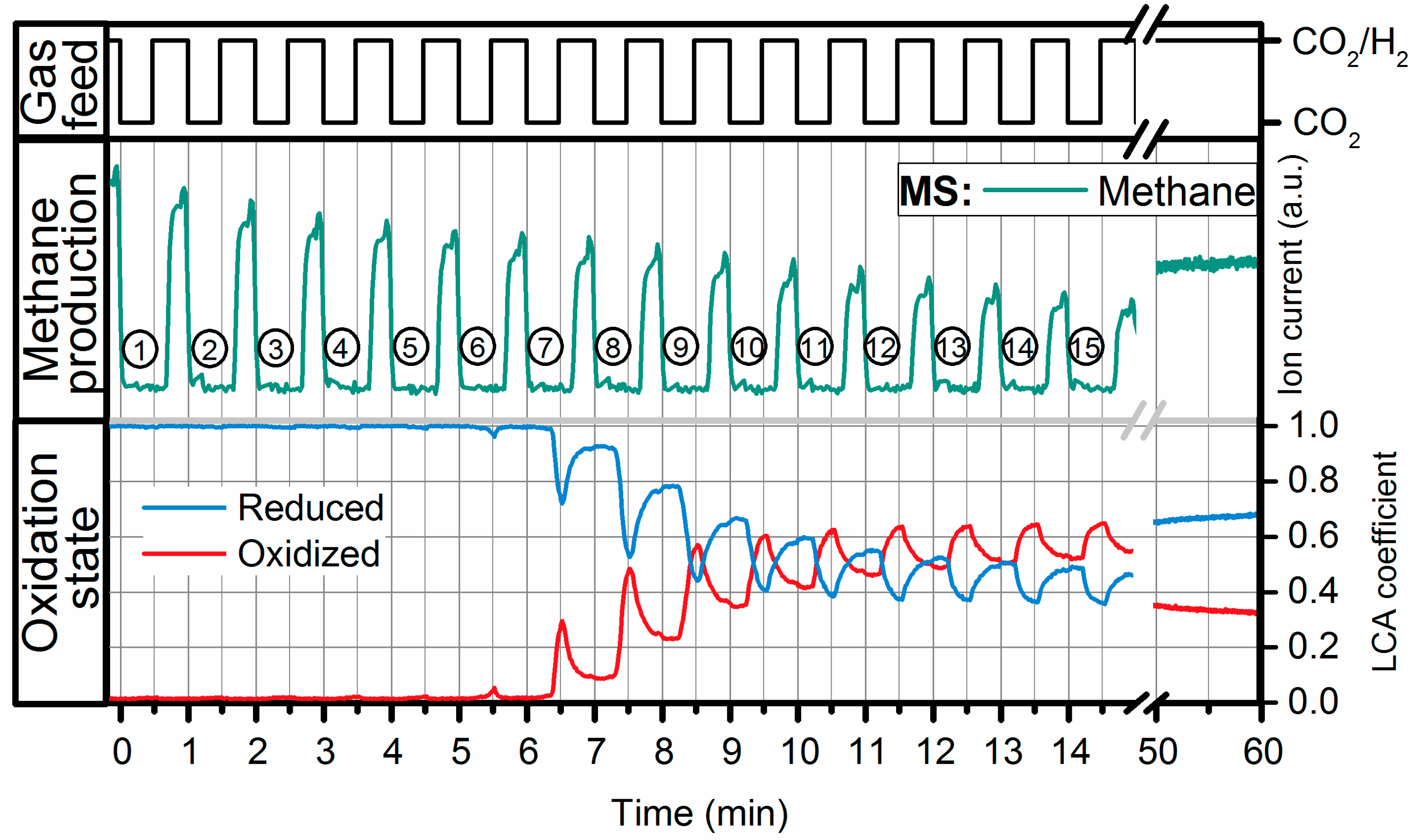
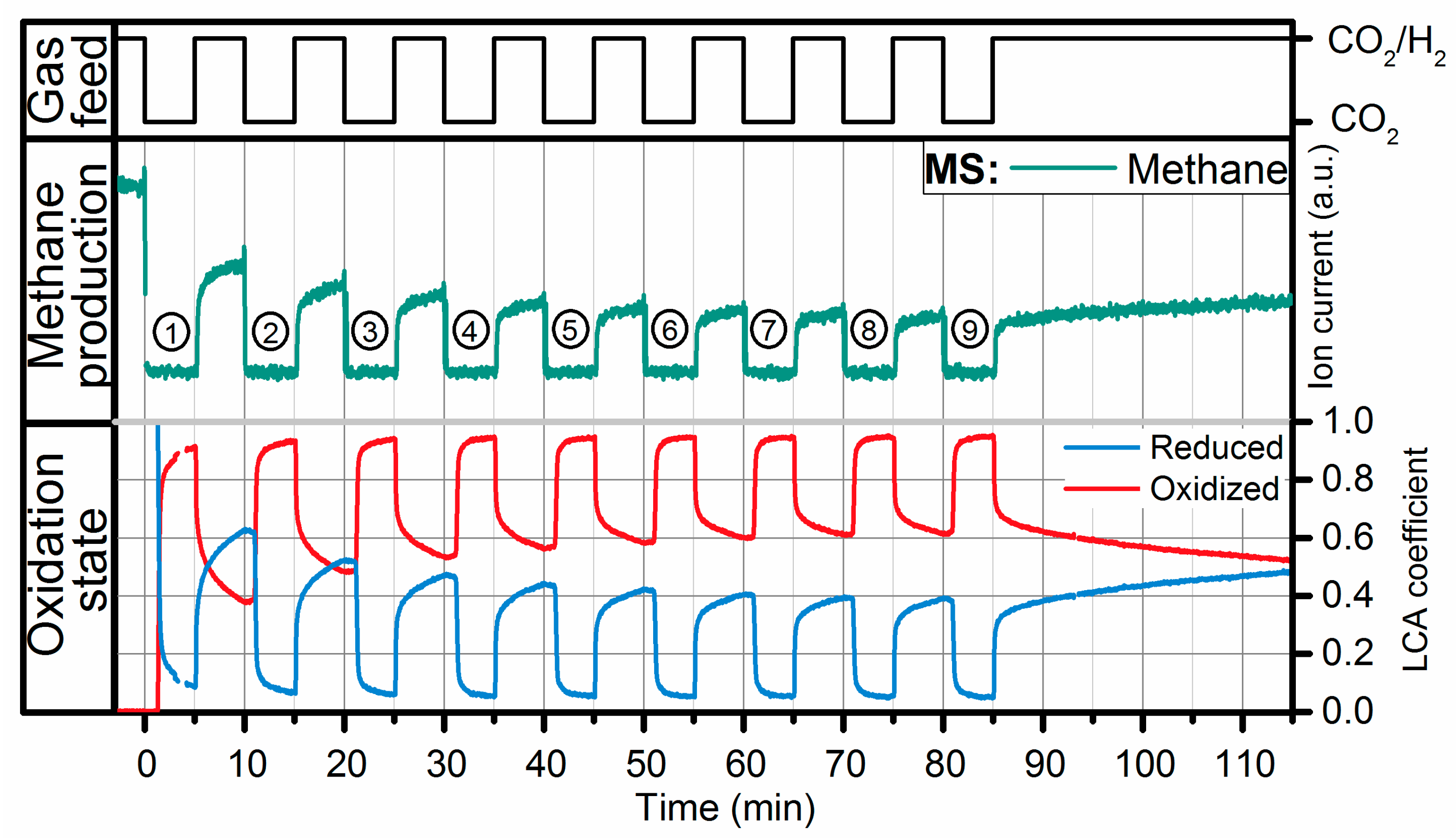
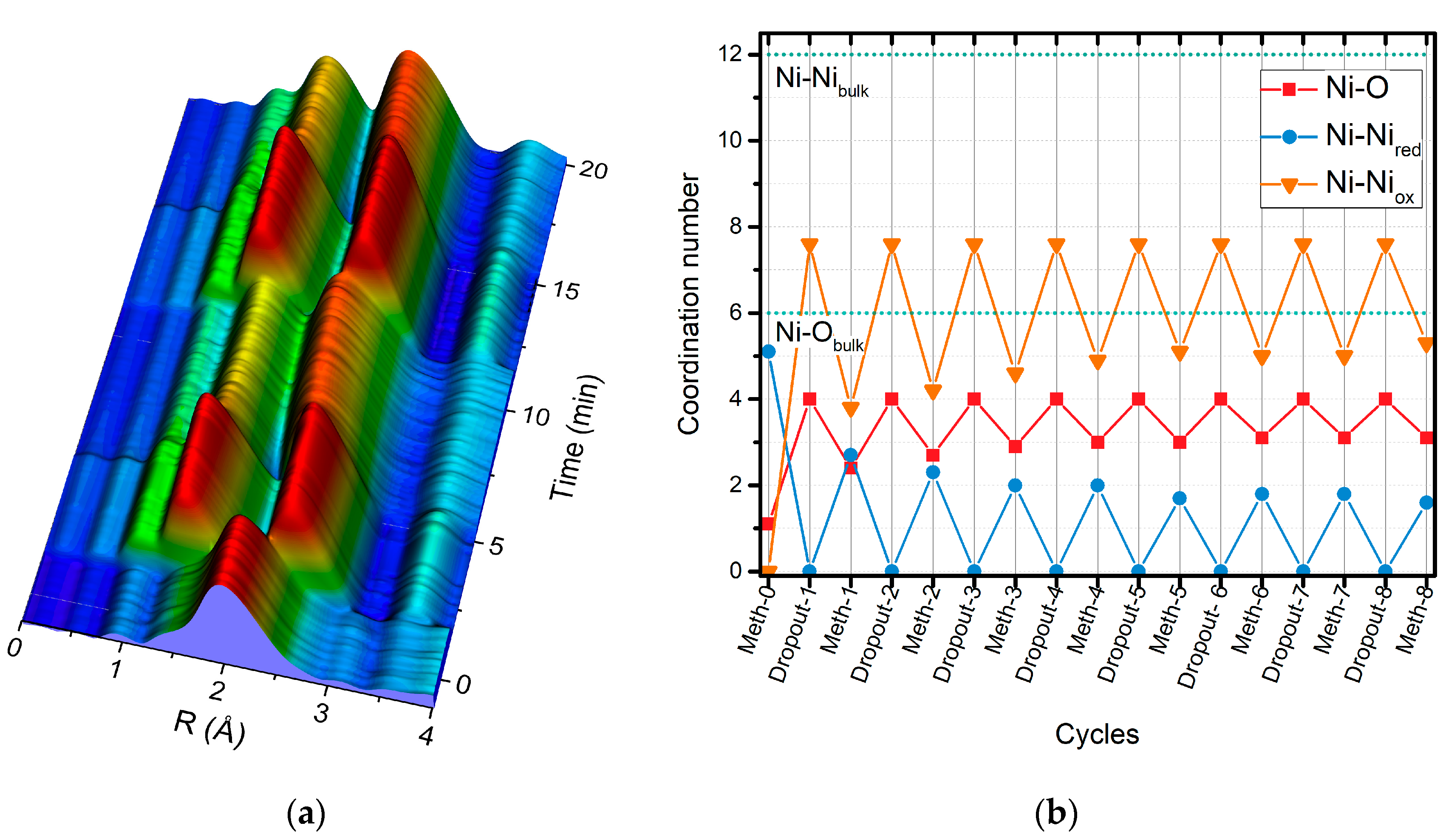
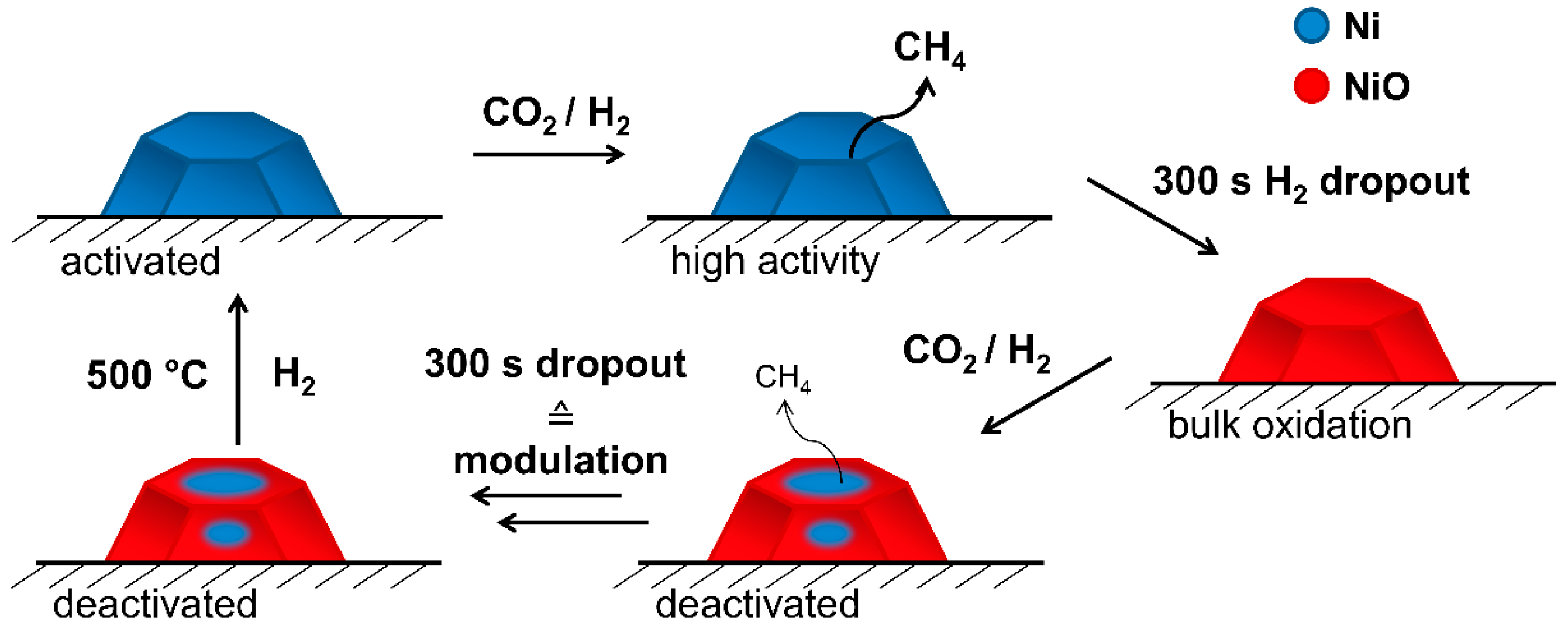
2.2. Operando QEXAFS Studies under Transient Reaction Conditions
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Characterization
3.3. Catalytic Performance
3.4. Operando XAS Experiments
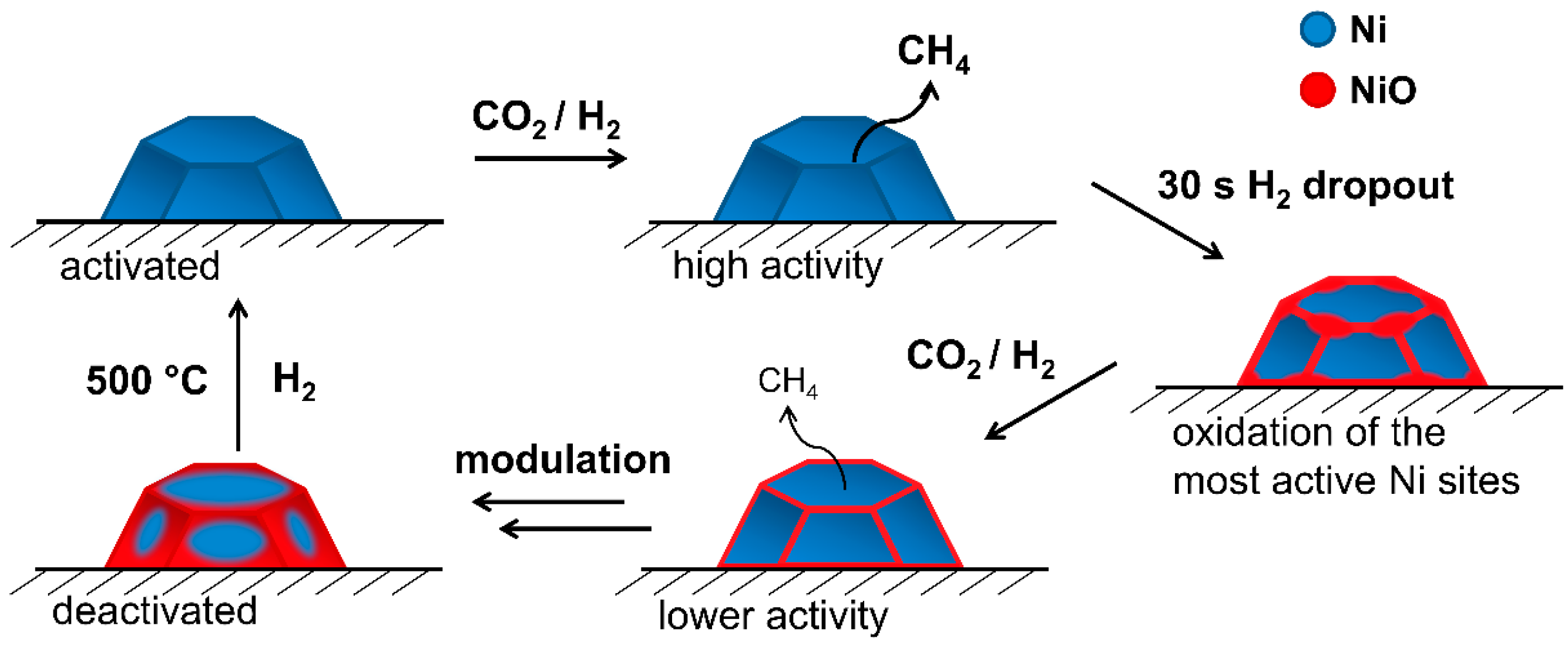
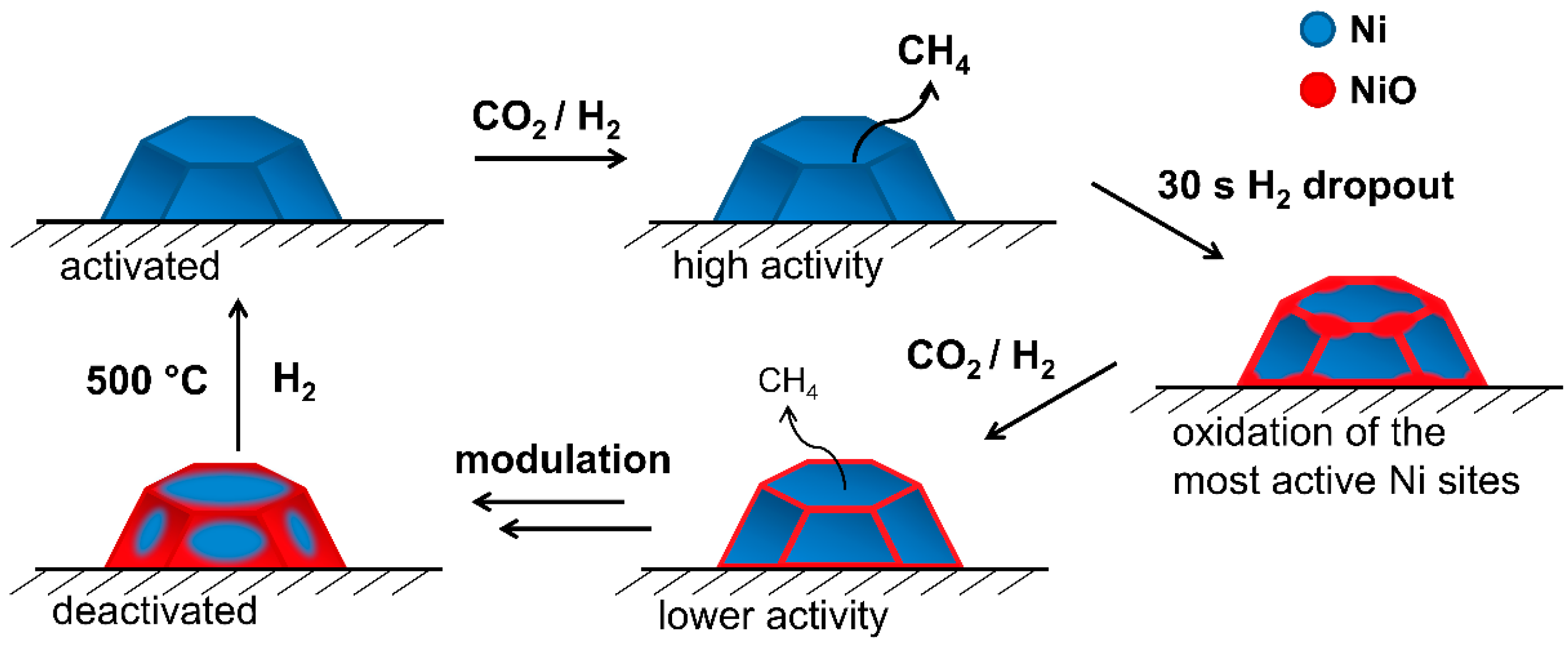
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schüth, F. Chemical Compounds for Energy Storage. Chem. Ing. Technol. 2011, 83, 1984–1993. [Google Scholar] [CrossRef]
- Schlögl, R. Chemical Energy Storage; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2013. [Google Scholar]
- Sterner, M. Erneuerbare Energien und Energieeffizienz—Renewable Energies and Energy Efficiency; Kassel University Press GmbH: Kassel, Germany, 2009; Volume 14. [Google Scholar]
- Hashimoto, K.; Yamasaki, M.; Fujimura, K.; Matsui, T.; Izumiya, K.; Komori, M.; El-Moneim, A.A.; Akiyama, E.; Habazaki, H.; Kumagai, N.; et al. Global CO2 recycling—Novel materials and prospect for prevention of global warming and abundant energy supply. Mater. Sci. Eng. A 1999, 267, 200–206. [Google Scholar] [CrossRef]
- Kalz, K.F.; Kraehnert, R.; Dvoyashkin, M.; Dittmeyer, R.; Gläser, R.; Krewer, U.; Reuter, K.; Grunwaldt, J.-D. Future Challenges in Heterogeneous Catalysis: Understanding Catalysts under Dynamic Reaction Conditions. Chem. Cat. Chem. 2017, 9, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Götz, M.; Lefebvre, J.; Mörs, F.; McDaniel Koch, A.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Iglesias González, M.; Schaub, G. Fischer-Tropsch Synthesis with H2/CO2—Catalyst Behavior under Transient Conditions. Chem. Ing. Technol. 2015, 87, 848–854. [Google Scholar] [CrossRef]
- Iglesias González, M.; Eilers, H.; Schaub, G. Flexible Operation of Fixed-Bed Reactors for a Catalytic Fuel Synthesis–CO2 Hydrogenation as Example Reaction. Energy Technol. 2016, 4, 90–103. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Q.; Gu, F.; Liu, B.; Zhong, Z.; Su, F. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Mukti, R.R.; Taufiq-Yap, Y.H.; Sazegar, M.R. Highly active Ni-promoted mesostructured silica nanoparticles for CO2 methanation. Appl. Catal. B 2014, 147, 359–368. [Google Scholar] [CrossRef]
- Bian, L.; Zhang, L.; Xia, R.; Li, Z. Enhanced low-temperature CO2 methanation activity on plasma-prepared Ni-based catalyst. J. Nat. Gas Sci. Eng. 2015, 27, 1189–1194. [Google Scholar] [CrossRef]
- Liu, J.; Li, C.; Wang, F.; He, S.; Chen, H.; Zhao, Y.; Wei, M.; Evans, D.G.; Duan, X. Enhanced low-temperature activity of CO2 methanation over highly-dispersed Ni/TiO2 catalyst. Catal. Sci. Technol. 2013, 3, 2627–2633. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Roger, A.-C. Methanation of carbon dioxide over nickel-based Ce0.72Zr0.28O2 mixed oxide catalysts prepared by sol-gel method. Appl. Catal. A 2009, 369, 90–96. [Google Scholar] [CrossRef]
- Rahmani, S.; Rezaei, M.; Meshkani, F. Preparation of highly active nickel catalysts supported on mesoporous nanocrystalline γ-Al2O3 for CO2 methanation. J. Ind. Eng. Chem. 2014, 20, 1346–1352. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrogen Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- He, S.; Li, C.; Chen, H.; Su, D.; Zhang, B.; Cao, X.; Wang, B.; Wei, M.; Evans, D.G.; Duan, X. A Surface Defect-Promoted Ni Nanocatalyst with Simultaneously Enhanced Activity and Stability. Chem. Mater. 2013, 25, 1040–1046. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Chai, R.; Zhao, G.; Liu, Y.; Lu, Y.; Cao, F. Ni-Al2O3/Ni-foam catalyst with enhanced heat transfer for hydrogenation of CO2 to methane. AlChE J. 2015, 61, 4323–4331. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Riani, P.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrogen Energy 2015, 40, 9171–9182. [Google Scholar] [CrossRef]
- Abe, T.; Tanizawa, M.; Watanabe, K.; Taguchi, A. CO2 methanation property of Ru nanoparticle-loaded TiO2 prepared by a polygonal barrel-sputtering method. Energy Environ. Sci. 2009, 2, 315–321. [Google Scholar] [CrossRef]
- Hoekman, S.K.; Broch, A.; Robbins, C.; Purcell, R. CO2 recycling by reaction with renewably-generated hydrogen. Int. J. Greenh. Gas Control 2010, 4, 44–50. [Google Scholar] [CrossRef]
- Mutz, B.; Carvalho, H.W.P.; Mangold, S.; Kleist, W.; Grunwaldt, J.-D. Methanation of CO2: Structural response of a Ni-based catalyst under fluctuating reaction conditions unraveled by operando spectroscopy. J. Catal. 2015, 327, 48–53. [Google Scholar] [CrossRef]
- Aldana, P.A.U.; Ocampo, F.; Kobl, K.; Louis, B.; Thibault-Starzyk, F.; Daturi, M.; Bazin, P.; Thomas, S.; Roger, A.C. Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy. Catal. Today 2013, 215, 201–207. [Google Scholar] [CrossRef]
- Zarfl, J.; Ferri, D.; Schildhauer, T.J.; Wambach, J.; Wokaun, A. DRIFTS study of a commercial Ni/γ-Al2O3 CO methanation catalyst. Appl. Catal. A 2015, 495, 104–114. [Google Scholar] [CrossRef]
- Rönsch, S.; Köchermann, J.; Schneider, J.; Matthischke, S. Global Reaction Kinetics of CO and CO2 Methanation for Dynamic Process Modeling. Chem. Eng. Technol. 2016, 39, 208–218. [Google Scholar] [CrossRef]
- Rönsch, S.; Matthischke, S.; Müller, M.; Eichler, P. Dynamische Simulation von Reaktoren zur Festbettmethanisierung—Dynamic Simulation of Fixed-Bed Methanation Reactors. Chem. Ing. Technol. 2014, 86, 1198–1204. [Google Scholar] [CrossRef]
- Eilers, H.; Schaub, G. Fischer-Tropsch-Synthese unter instationären Bedingungen im Suspensionsreaktor: Experimentelle und rechnerische Studien—Fischer-Tropsch Synthesis under Transient Conditions in a Slurry Reactor: Experimental and Mathematical Investigations. Chem. Ing. Technol. 2015, 87, 837–842. [Google Scholar] [CrossRef]
- Newton, M.A. Dynamic adsorbate/reaction induced structural change of supported metal nanoparticles: Heterogeneous catalysis and beyond. Chem. Soc. Rev. 2008, 37, 2644–2657. [Google Scholar] [CrossRef] [PubMed]
- Stötzel, J.; Frahm, R.; Kimmerle, B.; Nachtegaal, M.; Grunwaldt, J.-D. Oscillatory Behavior during the Catalytic Partial Oxidation of Methane: Following Dynamic Structural Changes of Palladium Using the QEXAFS Technique. J. Phys. Chem. C 2012, 116, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Grunwaldt, J.-D.; Molenbroek, A.M.; Topsøe, N.Y.; Topsøe, H.; Clausen, B.S. In Situ Investigations of Structural Changes in Cu/ZnO Catalysts. J. Catal. 2000, 194, 452–460. [Google Scholar] [CrossRef]
- Hansen, P.L.; Wagner, J.B.; Helveg, S.; Rostrup-Nielsen, J.R.; Clausen, B.S.; Topsøe, H. Atom-resolved imaging of dynamic shape changes in supported copper nanocrystals. Science 2002, 295, 2053–2055. [Google Scholar] [CrossRef] [PubMed]
- Gänzler, A.M.; Casapu, M.; Boubnov, A.; Müller, O.; Conrad, S.; Lichtenberg, H.; Frahm, R.; Grunwaldt, J.-D. Operando spatially and time-resolved X-ray absorption spectroscopy and infrared thermography during oscillatory CO oxidation. J. Catal. 2015, 328, 216–224. [Google Scholar] [CrossRef]
- Eslava, J.L.; Iglesias-Juez, A.; Agostini, G.; Fernández-García, M.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Time-Resolved XAS Investigation of the Local Environment and Evolution of Oxidation States of a Fischer–Tropsch Ru–Cs/C Catalyst. ACS Catal. 2016, 6, 1437–1445. [Google Scholar] [CrossRef]
- Mutz, B.; Carvalho, H.W.P.; Kleist, W.; Grunwaldt, J.-D. Dynamic transformation of small Ni particles during methanation of CO2 under fluctuating reaction conditions monitored by operando X-ray absorption spectroscopy. J. Phys. Conf. Ser. 2016, 712, 012050. [Google Scholar] [CrossRef]
- Abbas, Z.; Mezher, T.; Abu-Zahra, M.R.M. Evaluation of CO2 Purification Requirements and the Selection of Processes for Impurities Deep Removal from the CO2 Product Stream. Energy Procedia 2013, 37, 2389–2396. [Google Scholar] [CrossRef]
- Köppel, W.; Götz, M.; Graf, F. Biogas upgrading for injection into the gas grid. Gwf-Gas Erdgas. 2009, 150, 26–35. [Google Scholar]
- Burattin, P.; Che, M.; Louis, C. Metal Particle Size in Ni/SiO2 Materials Prepared by Deposition−Precipitation: Influence of the Nature of the Ni(II) Phase and of Its Interaction with the Support. J. Phys. Chem. B 1999, 103, 6171–6178. [Google Scholar] [CrossRef]
- Bitter, J.H.; van der Lee, M.K.; Slotboom, A.G.T.; van Dillen, A.J.; de Jong, K.P. Synthesis of Highly Loaded Highly Dispersed Nickel on Carbon Nanofibers by Homogeneous Deposition–Precipitation. Catal. Lett. 2003, 89, 139–142. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Magistri, L.; Busca, G. A study of the methanation of carbon dioxide on Ni/Al2O3 catalysts at atmospheric pressure. Int. J. Hydrogen Energy 2014, 39, 11557–11565. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Yoon, W.-L.; Lee, T.-W.; Rhee, Y.-W.; Seo, Y.-S. A highly active Ni-Al2O3 catalyst prepared by homogeneous precipitation using urea for internal reforming in a molten carbonate fuel cell (MCFC): Effect of the synthesis temperature. Int. J. Hydrogen Energy 2010, 35, 11237–11244. [Google Scholar] [CrossRef]
- Seo, J.G.; Youn, M.H.; Park, D.R.; Nam, I.; Song, I.K. Hydrogen production by steam reforming of liquefied natural gas (LNG) over Ni–Al2O3 catalysts prepared by a sequential precipitation method: Effect of precipitation agent. Int. J. Hydrogen Energy 2009, 34, 8053–8060. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Xu, L.; Wang, F.; Chen, M.; Zhang, J.; Yuan, K.; Wang, L.; Wu, K.; Xu, G.; Chen, W. CO2 methanation over a Ni based ordered mesoporous catalyst for the production of synthetic natural gas. RSC Adv. 2016, 6, 28489–28499. [Google Scholar] [CrossRef]
- Frahm, R. Quick scanning EXAFS: First experiments. Nucl. Instrum. Methods Phys. Res. Sect. A 1988, 270, 578–581. [Google Scholar] [CrossRef]
- Frahm, R. New method for time dependent X-ray absorption studies. Rev. Sci. Instrum. 1989, 60, 2515–2518. [Google Scholar] [CrossRef]
- Nachtegaal, M.; Müller, O.; König, C.; Frahm, R. QEXAFS: Techniques and Scientific Applications for Time-Resolved XAS. In X-ray Absorption and X-ray Emission Spectroscopy; van Bokhoven, J.A., Lamberti, C., Eds.; Wiley-VCH: Weinheim, Germany, 2016; pp. 155–183. [Google Scholar]
- Doronkin, D.E.; Casapu, M.; Günter, T.; Müller, O.; Frahm, R.; Grunwaldt, J.-D. Operando Spatially- and Time-Resolved XAS Study on Zeolite Catalysts for Selective Catalytic Reduction of NOX by NH3. J. Phys. Chem. C 2014, 118, 10204–10212. [Google Scholar] [CrossRef]
- Abild-Pedersen, F.; Lytken, O.; Engbæk, J.; Nielsen, G.; Chorkendorff, I.; Nørskov, J.K. Methane activation on Ni(111): Effects of poisons and step defects. Surf. Sci. 2005, 590, 127–137. [Google Scholar] [CrossRef]
- Molenbroek, A.M.; Nørskov, J.K.; Clausen, B.S. Structure and Reactivity of Ni−Au Nanoparticle Catalysts. J. Phys. Chem. B 2001, 105, 5450–5458. [Google Scholar] [CrossRef]
- Vang, R.T.; Honkala, K.; Dahl, S.; Vestergaard, E.K.; Schnadt, J.; Laegsgaard, E.; Clausen, B.S.; Norskov, J.K.; Besenbacher, F. Controlling the catalytic bond-breaking selectivity of Ni surfaces by step blocking. Nat. Mater. 2005, 4, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Bengaard, H.S.; Nørskov, J.K.; Sehested, J.; Clausen, B.S.; Nielsen, L.P.; Molenbroek, A.M.; Rostrup-Nielsen, J.R. Steam Reforming and Graphite Formation on Ni Catalysts. J. Catal. 2002, 209, 365–384. [Google Scholar] [CrossRef]
- Kirstein, W.; Petraki, I.; Thieme, F. A study on oxygen adsorption and coadsorption with carbonmonoxide on a stepped nickel surface. Surf. Sci. 1995, 331, 162–167. [Google Scholar] [CrossRef]
- Marwood, M.; Doepper, R.; Renken, A. In-situ surface and gas phase analysis for kinetic studies under transient conditions The catalytic hydrogenation of CO2. Appl. Catal. A 1997, 151, 223–246. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S. Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Andersson, M.P.; Abild-Pedersen, F.; Remediakis, I.N.; Bligaard, T.; Jones, G.; Engbæk, J.; Lytken, O.; Horch, S.; Nielsen, J.H.; Sehested, J.; et al. Structure sensitivity of the methanation reaction: H2-induced CO dissociation on nickel surfaces. J. Catal. 2008, 255, 6–19. [Google Scholar] [CrossRef]
- Holloway, P.H. Chemisorption and oxide formation on metals: Oxygen-nickel reaction. J. Vac. Sci. Technol. 1981, 18, 653–659. [Google Scholar] [CrossRef]
- Besenbacher, F.; Nørskov, J.K. Oxygen chemisorption on metal surfaces: General trends for Cu, Ni and Ag. Prog. Surf. Sci. 1993, 44, 5–66. [Google Scholar] [CrossRef]
- Dahl, S.; Logadottir, A.; Egeberg, R.; Larsen, J.; Chorkendorff, I.; Törnqvist, E.; Nørskov, J.K. Role of steps in N2 activation on Ru(0001). Phys. Rev. Lett. 1999, 83, 1814. [Google Scholar] [CrossRef]
- Gracia, F.J.; Bollmann, L.; Wolf, E.E.; Miller, J.T.; Kropf, A.J. In situ FTIR, EXAFS, and activity studies of the effect of crystallite size on silica-supported Pt oxidation catalysts. J. Catal. 2003, 220, 382–391. [Google Scholar] [CrossRef]
- Kale, M.J.; Christopher, P. Utilizing Quantitative in Situ FTIR Spectroscopy To Identify Well-Coordinated Pt Atoms as the Active Site for CO Oxidation on Al2O3-Supported Pt Catalysts. ACS Catal. 2016, 6, 5599–5609. [Google Scholar] [CrossRef]
- Boubnov, A.; Gänzler, A.; Conrad, S.; Casapu, M.; Grunwaldt, J.-D. Oscillatory CO Oxidation Over Pt/Al2O3 Catalysts Studied by In situ XAS and DRIFTS. Top. Catal. 2013, 56, 333–338. [Google Scholar] [CrossRef]
- Jacobsen, C.J.H.; Dahl, S.; Hansen, P.L.; Törnqvist, E.; Jensen, L.; Topsøe, H.; Prip, D.V.; Møenshaug, P.B.; Chorkendorff, I. Structure sensitivity of supported ruthenium catalysts for ammonia synthesis. J. Mol. Catal. A Chem. 2000, 163, 19–26. [Google Scholar] [CrossRef]
- Van Hardeveld, R.; van Montfoort, A. The influence of crystallite size on the adsorption of molecular nitrogen on nickel, palladium and platinum. Surf. Sci. 1966, 4, 396–430. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.-D.; Jensen, P.A.; Jensen, A.D. Influence on nickel particle size on the hydrodeoxygenation of phenol over Ni/SiO2. Catal. Today 2016, 259, 277–284. [Google Scholar] [CrossRef]
- Benfield, R.E. Mean coordination numbers and the non-metal-metal transition in clusters. J. Chem. Soc. Faraday Trans. 1992, 88, 1107–1110. [Google Scholar] [CrossRef]
- Geus, J.W.; van Dillen, A.J. Preparation of Supported Catalysts by Deposition–Precipitation. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 428–467. [Google Scholar]
- De Jong, K.P. Deposition Precipitation. In Synthesis of Solid Catalysts; de Jong, K.P., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 111–134. [Google Scholar]
- Van der Lee, M.K.; van Dillen, J.; Bitter, J.H.; de Jong, K.P. Deposition Precipitation for the Preparation of Carbon Nanofiber Supported Nickel Catalysts. JACS 2005, 127, 13573–13582. [Google Scholar] [CrossRef] [PubMed]
- Bergeret, G.; Gallezot, P. Particle Size and Dispersion Measurements. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 738–765. [Google Scholar]
- Müller, O.; Nachtegaal, M.; Just, J.; Lutzenkirchen-Hecht, D.; Frahm, R. Quick-EXAFS setup at the SuperXAS beamline for in situ X-ray absorption spectroscopy with 10 ms time resolution. J. Synchrotron. Radiat. 2016, 23, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Müller, O.; Lützenkirchen-Hecht, D.; Frahm, R. Quick scanning monochromator for millisecond in situ and in operando X-ray absorption spectroscopy. Rev. Sci. Instrum. 2015, 86, 093905. [Google Scholar] [CrossRef] [PubMed]
- Grunwaldt, J.-D.; van Vegten, N.; Baiker, A. Insight into the structure of supported palladium catalysts during the total oxidation of methane. Chem. Commun. 2007, 4635–4637. [Google Scholar] [CrossRef] [PubMed]
- Grunwaldt, J.-D.; Caravati, M.; Hannemann, S.; Baiker, A. X-ray absorption spectroscopy under reaction conditions: Suitability of different reaction cells for combined catalyst characterization and time-resolved studies. Phys. Chem. Chem. Phys. 2004, 6, 3037–3047. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. Athena, Artemis, Hephaestus: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron. Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Rehr, J.J.; Albers, R. Theoretical approaches to X-ray absorption fine structure. Rev. Mod. Phys. 2000, 72, 621. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
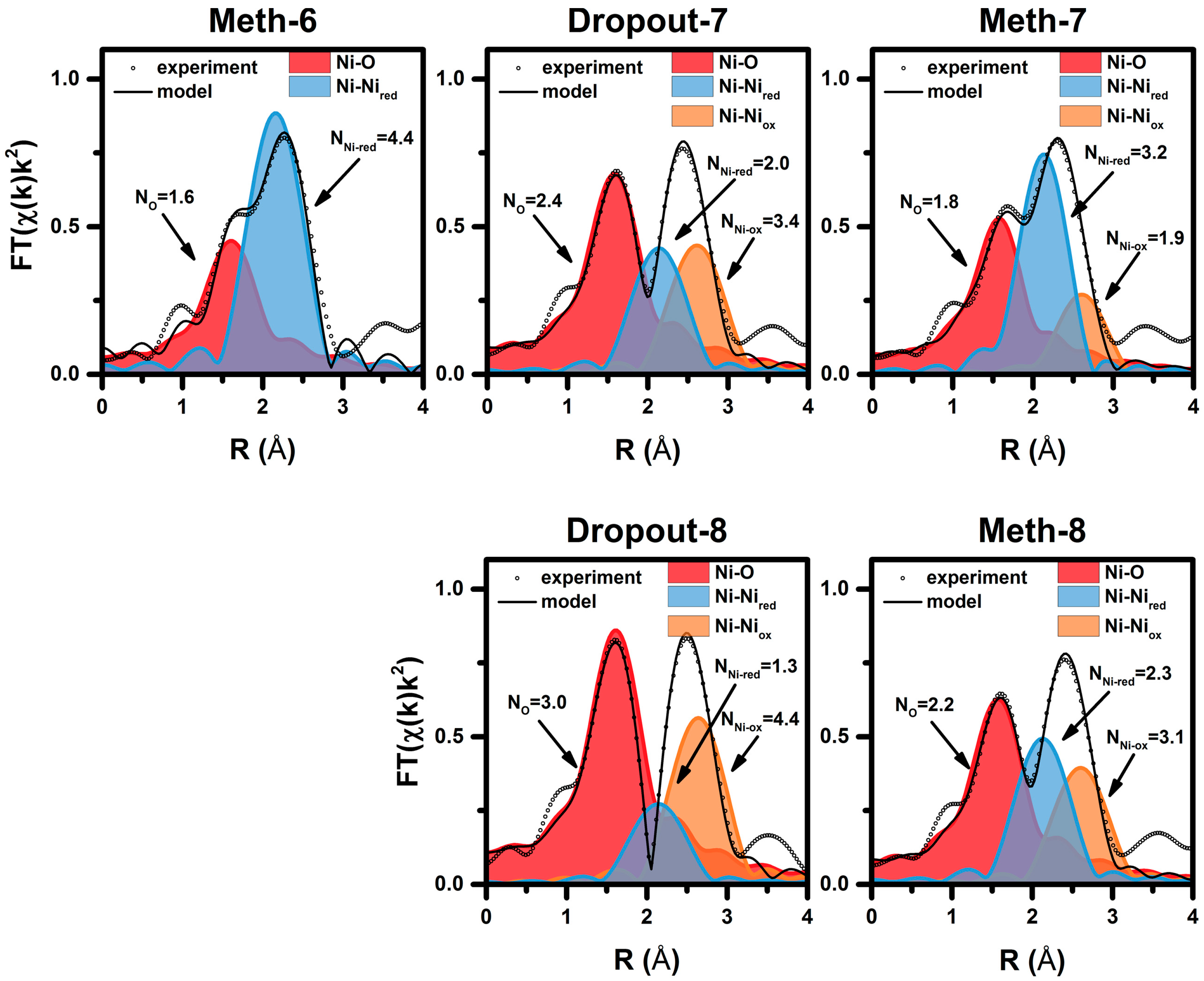
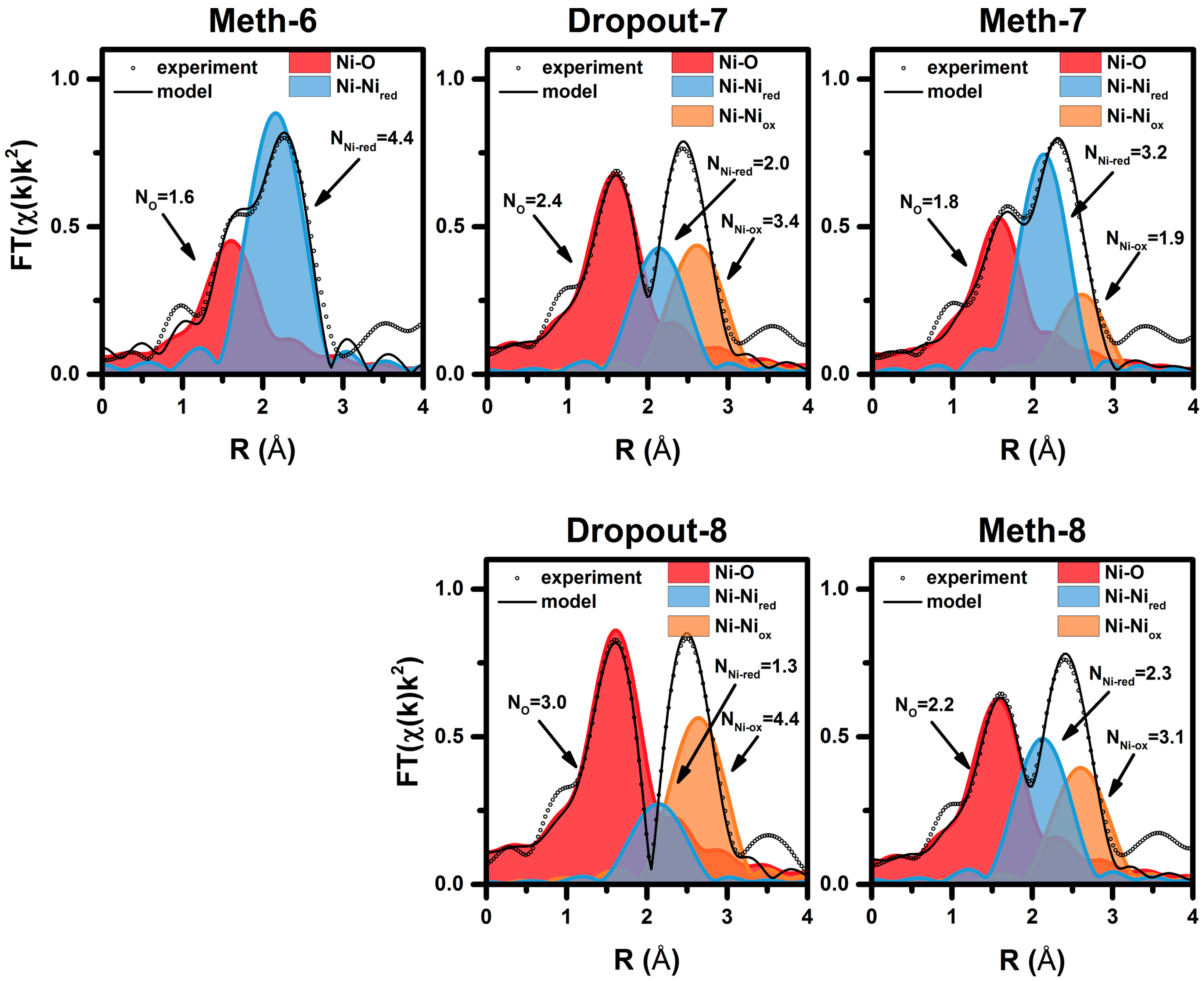
| Cycle | Atom | N | R (Å)a | σ2·10−3 (Å2) | E0 (eV) | R Factor (%) |
|---|---|---|---|---|---|---|
| Meth-6 | O | 1.6 ± 0.2 | 2.03 ± 0.2 | 8.3 f | 9.0 ± 1.5 | 0.27 |
| Ni | 4.4 ± 0.3 | 2.49 ± 0.1 | 11.8 f | |||
| Dropout-7 | O | 2.4 ± 0.2 | 2.02 ± 0.03 | 8.3 f | 7.6 ± 2.9 | 0.25 |
| Ni | 2.0 ± 0.6 | 2.48 ± 0.02 | 11.4 f | |||
| Ni | 3.4 ± 0.9 | 2.96 ± 0.03 | 11.8 f | |||
| Meth-7 | O | 1.8 ± 0.2 | 2.01 ± 0.2 | 8.3 f | 6.4 ± 2.2 | 0.23 |
| Ni | 3.2 ± 0.4 | 2.48 ± 0.1 | 11.4 f | |||
| Ni | 1.9 ± 0.6 | 2.95 ± 0.3 | 11.8 f | |||
| Dropout-8 | O | 3.0 ± 0.2 | 2.04 ± 0.02 | 8.3 f | 8.9 ± 2.1 | 0.14 |
| Ni | 1.3 ± 0.6 | 2.48 ± 0.03 | 11.4 f | |||
| Ni | 4.4 ± 0.7 | 2.97 ± 0.03 | 11.8 f | |||
| Meth-8 | O | 2.2 ± 0.2 | 2.02 ± 0.03 | 8.3 f | 6.8 ± 2.9 | 0.23 |
| Ni | 2.3 ± 0.6 | 2.48 ± 0.01 | 11.4 f | |||
| Ni | 3.1 ± 0.9 | 2.96 ± 0.03 | 11.8 f |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mutz, B.; Gänzler, A.M.; Nachtegaal, M.; Müller, O.; Frahm, R.; Kleist, W.; Grunwaldt, J.-D. Surface Oxidation of Supported Ni Particles and Its Impact on the Catalytic Performance during Dynamically Operated Methanation of CO2. Catalysts 2017, 7, 279. https://doi.org/10.3390/catal7090279
Mutz B, Gänzler AM, Nachtegaal M, Müller O, Frahm R, Kleist W, Grunwaldt J-D. Surface Oxidation of Supported Ni Particles and Its Impact on the Catalytic Performance during Dynamically Operated Methanation of CO2. Catalysts. 2017; 7(9):279. https://doi.org/10.3390/catal7090279
Chicago/Turabian StyleMutz, Benjamin, Andreas Martin Gänzler, Maarten Nachtegaal, Oliver Müller, Ronald Frahm, Wolfgang Kleist, and Jan-Dierk Grunwaldt. 2017. "Surface Oxidation of Supported Ni Particles and Its Impact on the Catalytic Performance during Dynamically Operated Methanation of CO2" Catalysts 7, no. 9: 279. https://doi.org/10.3390/catal7090279