Hydrogen-Etched TiO2−x as Efficient Support of Gold Catalysts for Water–Gas Shift Reaction


College of Biological, Chemical Sciences and Engineering, Jiaxing University, 118 Jiahang Road, Jiaxing 314001, China
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(1), 26; https://doi.org/10.3390/catal8010026
Submission received: 30 December 2017
/
Revised: 8 January 2018
/
Accepted: 9 January 2018
/
Published: 15 January 2018
(This article belongs to the Special Issue Catalysis in Steam Reforming)
Abstract
:Hydrogen-etching technology was used to prepare TiO2−x nanoribbons with abundant stable surface oxygen vacancies. Compared with traditional Au-TiO2, gold supported on hydrogen-etched TiO2−x nanoribbons had been proven to be efficient and stable water–gas shift (WGS) catalysts. The disorder layer and abundant stable surface oxygen vacancies of hydrogen-etched TiO2−x nanoribbons lead to higher microstrain and more metallic Au0 species, respectively, which all facilitate the improvement of WGS catalytic activities. Furthermore, we successfully correlated the WGS thermocatalytic activities with their optoelectronic properties, and then tried to understand WGS pathways from the view of electron flow process. Hereinto, the narrowed forbidden band gap leads to the decreased Ohmic barrier, which enhances the transmission efficiency of “hot-electron flow”. Meanwhile, the abundant surface oxygen vacancies are considered as electron traps, thus promoting the flow of “hot-electron” and reduction reaction of H2O. As a result, the WGS catalytic activity was enhanced. The concept involved hydrogen-etching technology leading to abundant surface oxygen vacancies can be attempted on other supported catalysts for WGS reaction or other thermocatalytic reactions.
1. Introduction
Hydrogen is a promising clean, efficient and sustainable energy source and can be a suitable candidate to reduce the dependence on fossil fuels. In this sense, the water–gas shift (WGS) reaction (CO + H2O = CO2 + H2) is crucial in industrially producing pure hydrogen, because the reaction provides not only hydrogen generation but also CO cleanup. However, the traditional WGS catalysts (e.g., Cu-Zn-Al, Fe-Cr and Co-Mo based catalysts) usually need long and tedious pretreatment procedures or are easily pyrophoric [1]. To develop new and more efficient WGS catalysts, supported catalysts have been believed to be good candidate. For example, both precious metals (e.g., Pt, Au, Pd, Rh, Ir or Ru) [2,3] and cheap transition metals (e.g., Cu or Ni) [4,5] were deposited on various supports including TiO2 [6,7,8,9,10,11,12,13,14,15,16,17], CeO2 [18,19,20,21,22,23], Mo2C [24,25,26], FeOx [27,28,29], Co3O4 [30], Al2O3 [31,32], ZrO2 [33,34], CeO2-TiO2 [35], CeO2-ZrO2 [36,37] and CeO2-La2O3 [38]. Au-TiO2 catalysts have been given considerable attentions for WGS activities, due to the high activity and low side reactions of dispersed Au [38], and some advantages of TiO2 supports (e.g., low price, easy preparation, adjustable properties and strong interaction with active metal).
Au-TiO2 has been considered as a bifunctional supported catalyst [39,40,41], suggesting that the WGS activity can be strongly affected by not only the properties of active gold species but also the structural and surface properties of appropriate supports. For example, the nature of the support has an important effect on the activation and stabilization of the gold nanoparticles [39]. Similarly, the surface structures of ceria supports ((100) surface versus (111) surface) have a strong effect on the morphology, size, and atomic interface structures of gold [42]. Furthermore, the support can directly participate by activating H2O molecules [12], which dissociate into H2 and O atom on the surface oxygen vacancies of CeO2 support during the WGS reaction [43]. Notably, in terms of Pt/TiO2 (110) and Au/TiO2 model catalysts, the oxygen vacancies of TiO2 supports have played a critical role in the dissociation of H2O, which determines the WGS catalytic activity [41,44,45]. Also, the H2O dissociation on the surface oxygen vacancy is generally considered as the rate determining step [44,45]. From the above, in order to design an effective Au-TiO2 WGS catalyst, it is essential to develop appropriate supports, which prefer to TiO2 supports with abundant surface oxygen vacancies. Generally, most scientists devoted to increase the oxygen vacancies by adding metal or nonmetal impurities [7,15,46]. However, the amount of introduced dopants is generally not enough to form desired oxygen vacancies, and the introduction of dopants will lead to a complicated catalyst system and will confuse us with respect to the real reaction mechanism.
Recently, a dopant-free black TiO2 (i.e., pure TiO2 phase) was obtained through high-pressure hydrogenation [47]. The black TiO2 presents the surface disorder layer with Ti3+ ions, surface oxygen vacancies and point defects, which are exactly required to show high WGS activities. For instance, Panagiotopoulou et al. [48] reported that partial reduction of TiO2 support results in the creation of new active sites for CO adsorption, probably located at metallic Pt in contact with Ti3+ ions. However, to date, black TiO2 is mainly considered for photocatalysis [49,50,51,52,53], as well as supercapacitors [54], lithium and Al-ion batteries [55]. To our best knowledge, the black TiO2 based catalyst has not been reported for the WGS reaction, or even for other thermocatalytic reactions.
In addition, metal–support interactions are known to influence the catalytic activity. Recently, it has been reported that “hot-electron flow” can be formed between metal-oxide interfaces during exothermic chemical processes, and the “hot-electron flow” has strongly influenced the catalytic activity of CO oxidation [46,56,57,58]. It is well-known that the WGS reaction is a typical exothermic redox reaction with electron exchange. Therefore, it is interesting, based on electron flow processes, to investigate the effect of black TiO2 surface properties (e.g., oxygen vacancies) on their WGS catalytic activity.
Here, a blue black TiO2−x nanoribbon rich in stable surface oxygen vacancies was prepared by hydrogen-etching technology. Blue black TiO2−x nanoribbon had been proven to be efficient and stable support of gold catalyst for WGS reaction. According to the in-depth characterization results, we try to understand WGS pathway from the view of the electron flow process. Hereinto, the reduction of the forbidden band gap leads to the decrease in Ohmic barrier; thus, the transmission efficiency of “hot-electron flow” is improved. Meanwhile, the abundant surface oxygen vacancies become electron traps, thus promoting the flow of “hot-electron” and reduction reaction of H2O, so as to enhance the WGS catalytic activity.
2. Results
2.1. Catalytic Activities and Stabilities
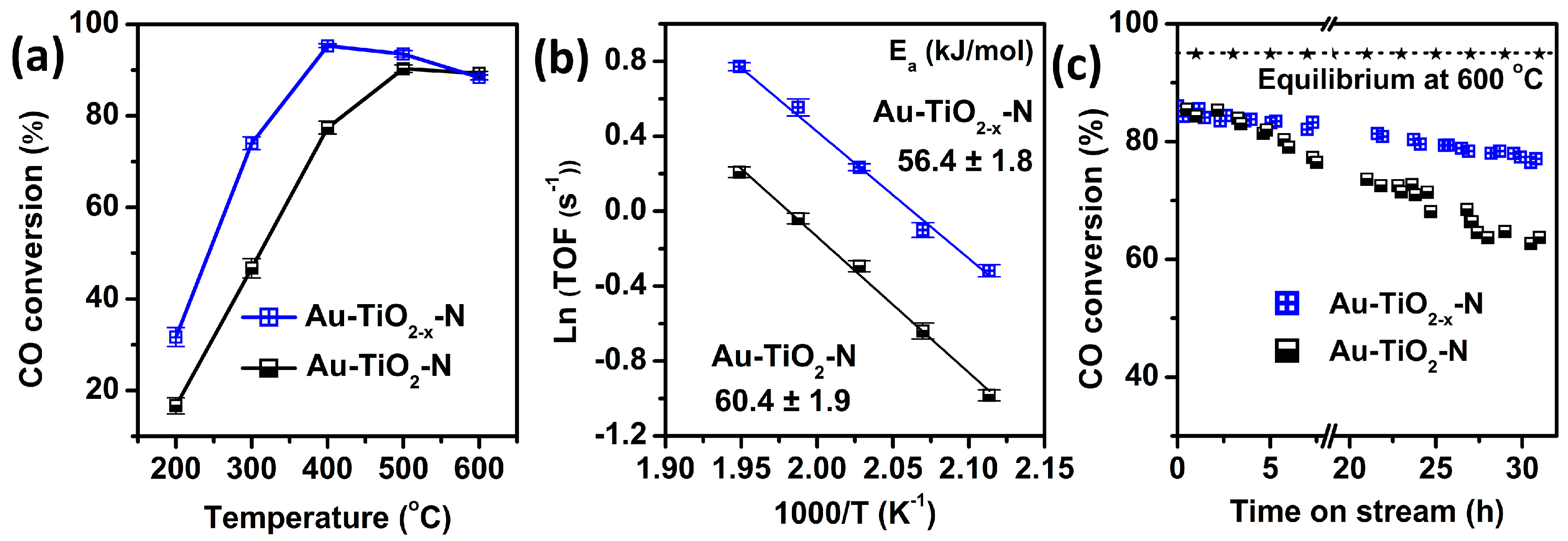
As shown in Figure 1a, Au-TiO2−x-N catalyst shows higher CO conversion than the corresponding Au-TiO2-N catalyst from 200 to 400 °C, indicating that the TiO2−x-N support can obviously improve WGS catalytic activities compared with the white TiO2-N support. For example, compared with Au-TiO2-N catalyst, the CO conversion of Au-TiO2−x-N catalyst increase by 59% (i.e., from 46.7% to 74% at 300 °C). Therefore, H2 atmosphere leads to higher catalytic activity, because the only difference between the two Au-TiO2 catalysts is calcination atmosphere (H2 or air) of supports.
To make a more realistic comparison of catalytic activities, turnover frequencies (TOFs) were measured in the kinetic region with negligible heat and mass-transfer effects (keeping CO conversion below 12%), as shown in Figure 1b. Au-TiO2−x-N catalyst presents higher TOF and slight smaller active energy (Ea) than Au-TiO2-N catalyst. For example, the TOF of Au-TiO2−x-N catalyst arrives 0.73 s−1 at 200 °C (Table S1), nearly double that of Au-TiO2-N catalyst (0.37 s−1 at 200 °C). A slight decrease in Ea from 60.4 ± 1.9 kJ/mol (Au-TiO2-N) to 56.4 ± 1.8 kJ/mol (Au-TiO2−x-N) is also observed. As shown in Table S2, compared with other Au or Pt supported on TiO2 catalysts reported in literatures, our Au-TiO2−x-N catalyst presented higher TOF. Even comparing with the other candidate catalysts, the activity of our Au-TiO2−x-N catalyst is still ranked at the top.
Figure 1c shows high-temperature stabilities of the Au-TiO2 catalysts for WGS at 600 °C. The CO conversion of Au-TiO2−x-N catalyst only decrease by 19.1% (from 86% to 69.6%) over a 32-h run, which obviously presents higher stability than Au-TiO2-N catalyst (decrease by 36.8% from 85.5% to 54.7%). Meanwhile, our Au-TiO2−x-N catalyst has a comparable or even higher stability than the Au-TiO2 and Au/CeZrO4 catalysts reported in the literature [14,37].
Therefore, the colors of TiO2 supports seemingly have an important effect on the WGS catalytic activity and stability; thus, H2 atmosphere has an indelible function.
2.2. Structural Properties
As shown in Figure 2a,b, TiO2 precursors were calcined in air and high-purity H2 atmospheres, which produce white TiO2-N and blue black TiO2−x-N supports, respectively. As shown in Figure 2b,c, both TiO2−x-N and TiO2-N presents the nanoribbon morphology. Thus, H2 atmosphere has little effect on their morphology. However, H2 atmosphere indeed has significant effect on their colors and catalytic activities, which must be attributed to other reasons. For this reason, the structural and optoelectronic properties of various TiO2 supports and Au catalysts should be further and deeply investigated with the aid of various characterization techniques.
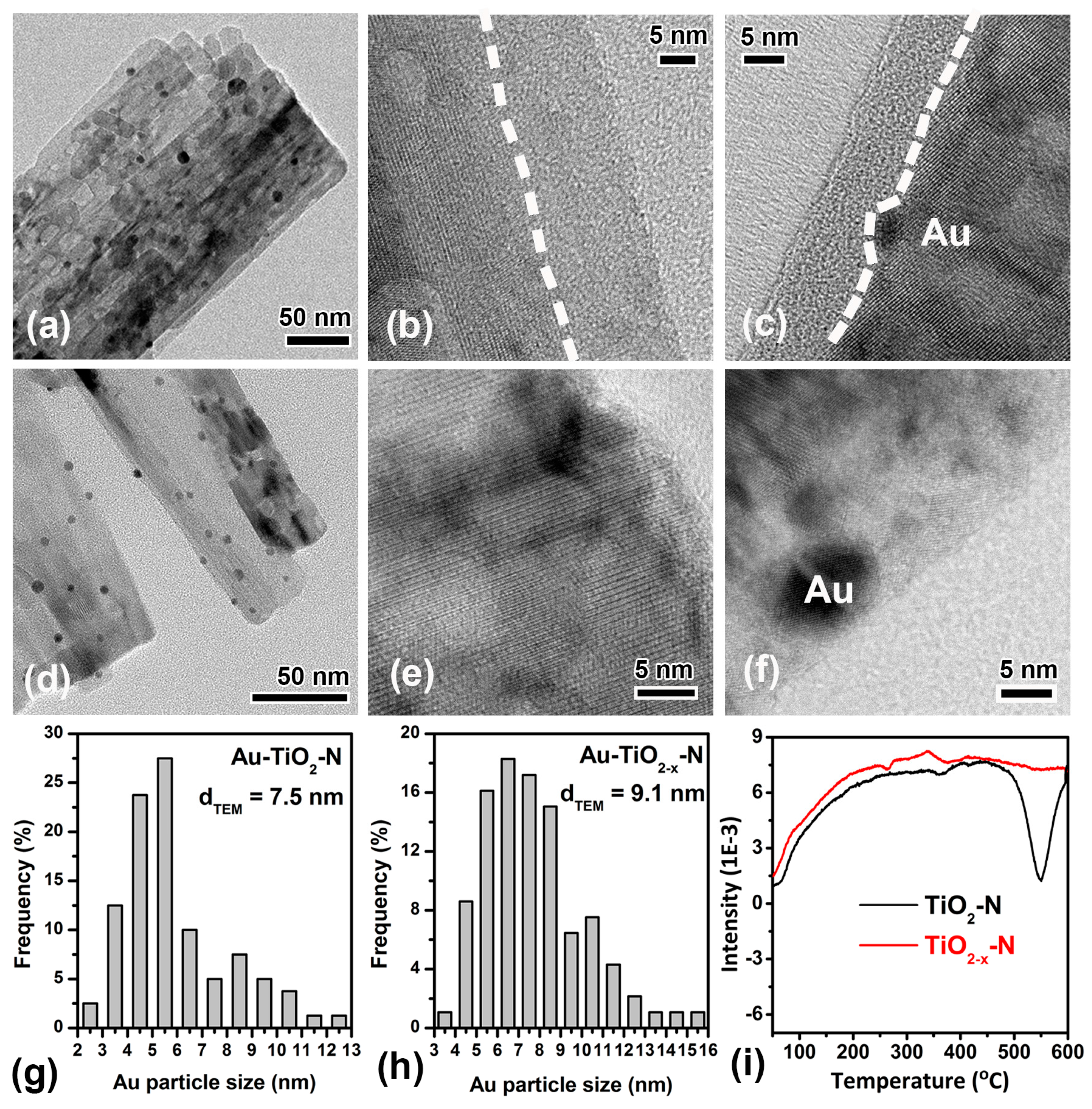
As shown in Figure 3a,d, TiO2−x-N and TiO2-N supports were decorated with Au particles (dark dots). The surface-averaged Au cluster diameters (dTEM) of Au-TiO2−x-N and Au-TiO2-N are 9.1 nm and 7.5 nm, respectively (Figure 3g,h). Impressively, the high resolution transmission electron microscopy (HRTEM) images were further investigated in detail. As shown in Figure 3b, the surface of TiO2−x-N nanocrystals became disordered where the disordered outer layer surrounds a crystalline core. After Au was deposited on TiO2 supports, Au-TiO2−x-N retains the microstructure composed of crystalline core and disordered shell, in spite of the fact that the thickness of the disordered surface layer has been reduced. However, the HRTEM images of TiO2-N and Au-TiO2-N show no obvious disordered shell near the surface region (Figure 3e,f). Thus, the results indicate that H2 atmosphere resulted in the surface structure rearrangement and disordered shell, in agreement with the reported results [47,49], so the calcination in H2 atmosphere was chosen as the hydrogen-etching technology.
Furthermore, the evolution of surface structure was also confirmed by the H2-TPR results (Figure 3i). As shown in Figure 3i, their reduction peaks occur at 550 °C, in very good agreement with the temperature of hydrogen-etching. The reducible capacity of TiO2−x-N is smaller than that of TiO2-N, indicating that the surface oxygen species of TiO2−x-N are smaller than those of TiO2-N. In other words, disordered shell should stem from the decrease of oxygen species (TiO2 + xH2 = TiO2−x + xH2O) [48] due to H2 reduction during the hydrogen-etching technology.
As shown in Figure 4, TiO2-N and TiO2−x-N present two sets of diffraction peaks: anatase TiO2 marked with “$” (JCPDS file No. 71-1166), and monoclinic TiO2 marked with “#” (JCPDS file No. 74-1940). After Au was deposited on the above-mentioned TiO2 supports, the all diffraction peaks remain unchanged and the diffraction peaks of any Au species cannot be observed. Furthermore, compared with TiO2-N and Au-TiO2-N, diffraction peaks of TiO2−x-N and Au-TiO2−x-N became broader, embodying that the FWHM of the diffraction peak at 25.3° increased from 0.564° to 0.597° and from 0.602° to 0.609°, respectively (Table 1). Usually, the broadening of diffraction peaks may be due to the increase of microstrain (Δd/d) in anatase [18,59] (i.e., increase from 0.600 to 0.617 and from 0.615 to 0.625, Table 1) and monoclinic TiO2 (the increase from 0.702 to 0.720 and from 0.802 to 0.812, Table S3). The above increase of microstrain might stem from lattice distortion including two aspects: (1) The reduction of d-spacing. Compared with air atmosphere, hydrogen-etching leads to the decrease of d-spacing in anatase (decrease from 3.519 to 3.508 Å and from 3.525 to 3.521 Å, Table 1) and monoclinic TiO2 (decrease from 6.246 to 6.212 Å and from 6.264 to 6.250 Å, Table S3). Meanwhile, hydrogen-etching also gives rise to the positive shift of 2θ, which conforms to the inverse correlation between d and θ according to Bragg equation (nλ = 2dsinθ). (2) The decrease in cell parameters. For example, both cell volume (V) of anatase (136.33 → 136.27 Å3 and 136.09 → 135.99 Å3, Table 1) and monoclinic TiO2 (285.466 → 285.141 Å3 and 285.006 →284.252 Å3, Table S3) decreased. Hydrogen-etching resulted in the reduction of cell parameters (i.e., the lattice contraction), which is responsible for the reduction of d-spacing. Thus, these lattice distortions (i.e., the reduction of cell parameters and d-spacing) finally lead to the increase of microstrain. As reported in our previous studies [60,61], the larger the microstrain is, the stronger the interaction between active metals and supports is, and then the higher the surface energy is. Obviously, the stronger interaction and higher surface energy are all in favor of higher catalytic activities. Therefore, hydrogen-etching technology resulted in surface lattice distortion of TiO2−x-N support, reflected by a larger microstrain value, which gives rise to higher surface energy and stronger interaction between Au and TiO2−x-N supports and, as a consequence, higher catalytic activities of Au-TiO2−x-N.
The increase of microstrain can also be proven by the blue-shift and asymmetrical broadening of Raman bands, as presented in Figure 5. These spectra display two sets of Raman bands: (a) anatase TiO2 presents six (3Eg + 2B1g + A1g) bands at about 144, 197, 399, 515, 519 (superimposed with the 515 cm−1 band), and 639 cm−1, respectively [47]; and (b) monoclinic TiO2 presents the most intense mode at 123 cm−1, and other bands superimposed with those of anatase TiO2 (i.e., 145, 161, 172, and 196 cm−1) [62]. For the strongest band at about 144 cm−1 (external vibration of Ti-O bond), compared with TiO2-N and Au-TiO2-N, the bands of TiO2−x-N and Au-TiO2−x-N shift to higher wavenumber (i.e., blue-shift from 142.5 to 146.1 cm−1 and from 148.6 to 149.6 cm−1, respectively). Raman shift (Δν) is the wavenumber difference between Stokes scattered light (νs) and incident light (ν0). To explain these phenomena, the calculation of the Raman band is simplified by a physical model-harmonic oscillator. The wavenumber (ν) are expressed by Hooke’s law as follows:
where c is the velocity of light, k is the bond force constant, and μ is the reduced mass. On the basis of the equation, the blue-shift of Raman bands at 144 cm−1 should be ascribed to the increase of the Ti-O bond force constant (k), which arises from the decrease of the length of Ti-O bond. It shall result in the decrease of cell parameters, which is in very good agreement with XRD Rietveld analysis results (Table 1).
ν (cm−1) = (k/μ)1/2/2πc
In addition, as far as asymmetrical broadening is concerned, the TiO2−x-N and Au-TiO2−x-N presented asymmetrical broadening bands compared with TiO2-N and Au-TiO2-N. It has been reported that structural disorder (localized defects associated with oxygen vacancies) was rather responsible for the broadening of the bands observed in black TiO2 [51]. The results indicate that H2 atmosphere resulted in the surface oxygen vacancies due to the reduction of the surface oxygen species, agreeing with the results of H2-TPR (Figure 3i).
In other words, during the hydrogen-etching procedure, the reduction of the surface oxygen species resulted in the decrease of the length of Ti-O bond. As a consequence, cell parameters and d-spacing are reduced and lattice distortions (i.e., increased microstrain) arise; finally, disorder layers and stable surface oxygen vacancies are formed (Figure S2). Then, Au particles are bound more strongly to surface oxygen vacancies [63], which facilitate the WGS reaction happening at the interface between Au and oxygen vacancies [41]. Usually, the decrease of surface oxygen species should lead to more surface oxygen vacancies and Ti3+ ions, because of the charge balance (2Ti4+ + O2− = 2Ti3+ + VӦ, VӦ being a doubly ionized oxygen vacancy). The existence of oxygen vacancies and Ti3+ ions can be confirmed by the results of electron paramagnetic resonance (EPR) and X-ray photoelectron spectroscopy (XPS).
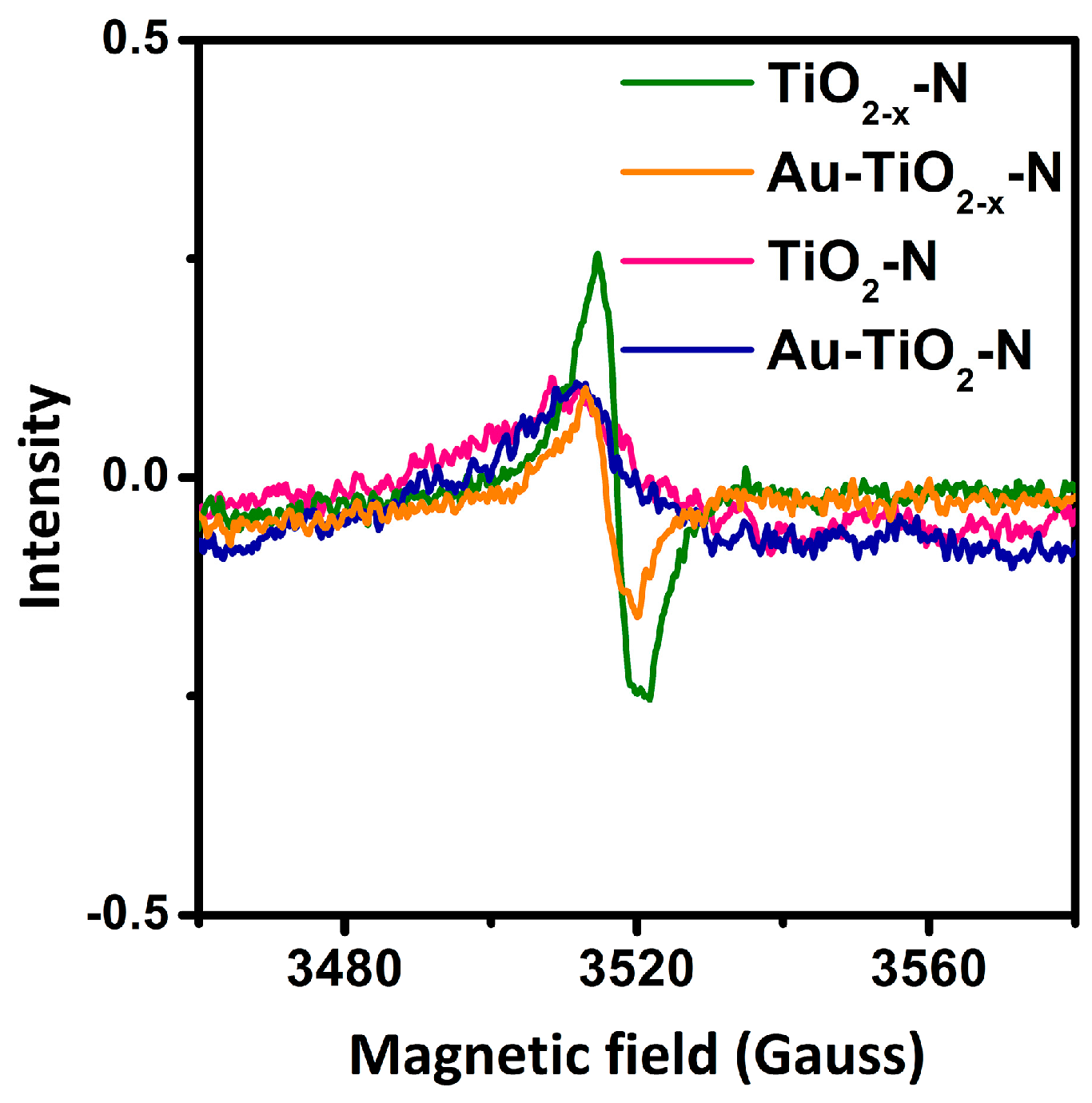
EPR measurements were carried out to determine the possible unpaired electrons from hydrogen-etching processes. As shown in Figure 6, TiO2−x-N give rise to a EPR signal at g-value of 2.004, which should be attributed to surface Ti3+ [49,51] and single electron O2− radical trapped by O2 (from air) adsorbed at oxygen vacancy [64]. However, a very weak signal could be found for TiO2-N. The results indicate that hydrogen-etching technology results in more surface Ti3+ and oxygen vacancies of TiO2−x-N. Moreover, Au-TiO2−x-N presents stronger EPR signals than Au-TiO2-N. Thus, surface Ti3+ and oxygen vacancies should be highly stable, in spite of immersion in water during the preparation process of Au catalysts. Furthermore, it was found that the color of blue black TiO2−x-N did not change in air over several months.
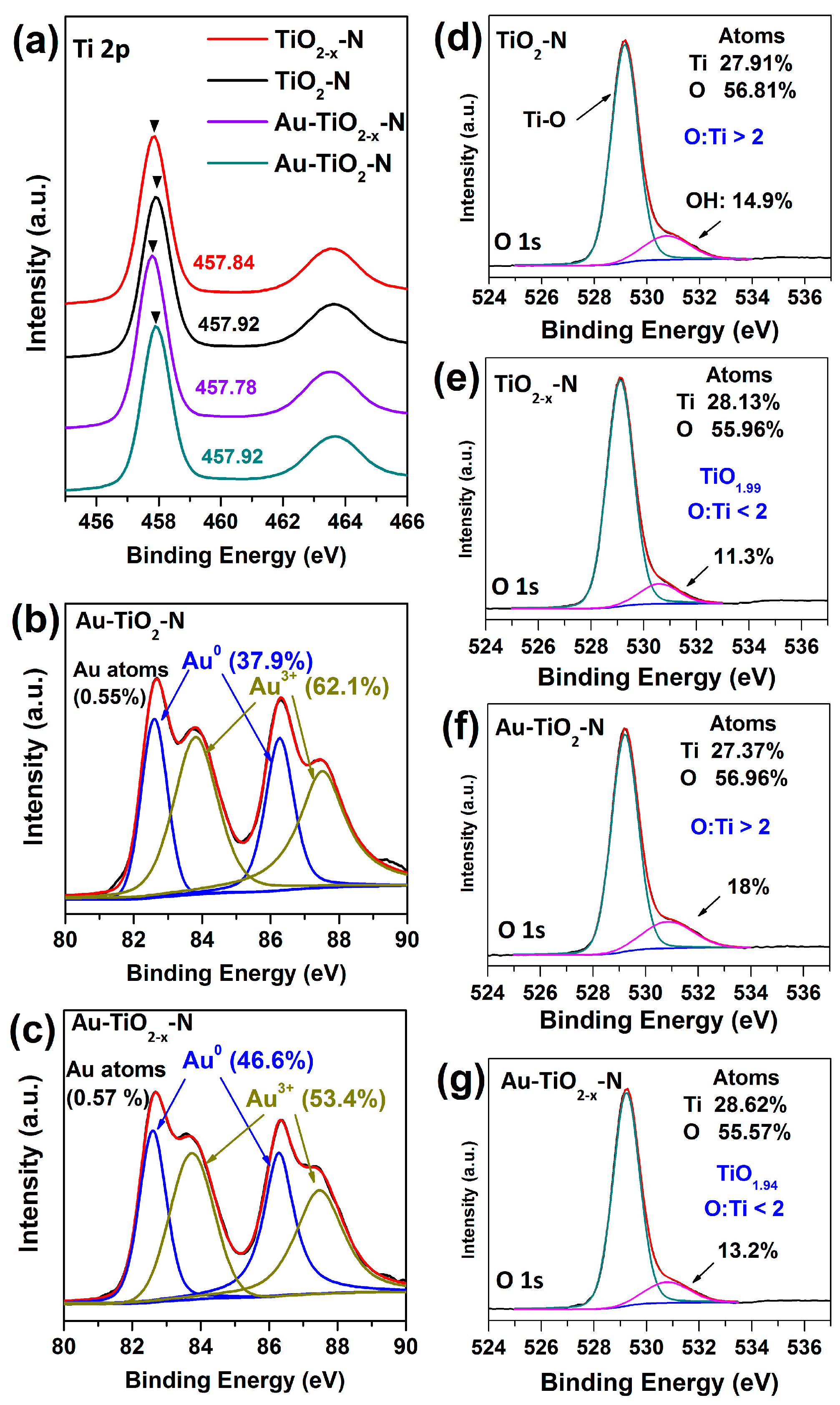
As presented in Figure S1a,b, XPS spectra of all samples show identical elements, indicating that hydrogen-etching technology does not introduce other impurities. For Ti 2p spectra (Figure 7a), compared with the TiO2-N and Au-TiO2-N calcined in air, the binding energy of TiO2−x-N and Au-TiO2−x-N shift to a low energy range, which indicates the increase in electron density around Ti atoms [64]. Thus, a certain amount of Ti3+ appears in TiO2−x-N and Au-TiO2−x-N, in accordance with EPR results.
The O 1s peaks can be fitted into two peaks at about 529.2 and 531 eV, which are due to Ti-O and surface OH species, respectively. As shown in Figure 7d–g, the O 1s spectra show that TiO2−x-N and Au-TiO2−x-N has much less surface OH species than does TiO2-N and Au-TiO2-N, respectively. Meanwhile, for the ratio of O to Ti atoms, the ratios of TiO2−x-N and Au-TiO2−x-N are all less than two, whereas the ratios of TiO2-N and Au-TiO2-N are all greater than two, indicating that TiO2−x-N and Au-TiO2−x-N present a nonstoichiometric titanium dioxide with the shortage of oxygen atoms. In short, the results indicate that the decrease of surface OH species should be due to H2 reduction during the hydrogen-etching technology [52], with the forming of surface oxygen vacancies, consistent with the results of TEM, H2-TPR, XRD, Raman and EPR.
For Au 4f spectra, Au-TiO2-N and Au-TiO2−x-N present almost equal amount of Au (Figure 7b,c). Both Au-TiO2-N and Au-TiO2-x-N present the different ratios of metallic Au0 and Au3+ species. Au-TiO2-x-N presents more metallic Au0 species (46.6%) than Au-TiO2-N (37.9%). Considering that HAuCl4 (Au3+) was used as gold source, we inferred that the obtained metallic Au0 species should be reduced by surface oxygen vacancies of support with the charge transfer from oxygen vacancies to Au particles [65]. Because the surface oxygen vacancies of TiO2−x-N are far richer than those of TiO2-N, during the deposition of gold species (i.e., Au(OH)3), the amount of metallic Au0 species supported on the TiO2−x-N is far larger than on TiO2-N. The metallic Au0 species have been regarded as a requisite for a high CO oxidation and WGS activity of Au/TiO2 catalysts [11,12,66,67]; thus, Au-TiO2−x-N present higher catalytic activities. Therefore, the difference in their catalytic activities mainly depends on the difference in Au valence, which is influenced by the amount of support surface oxygen vacancies.
2.3. Optoelectronic Properties
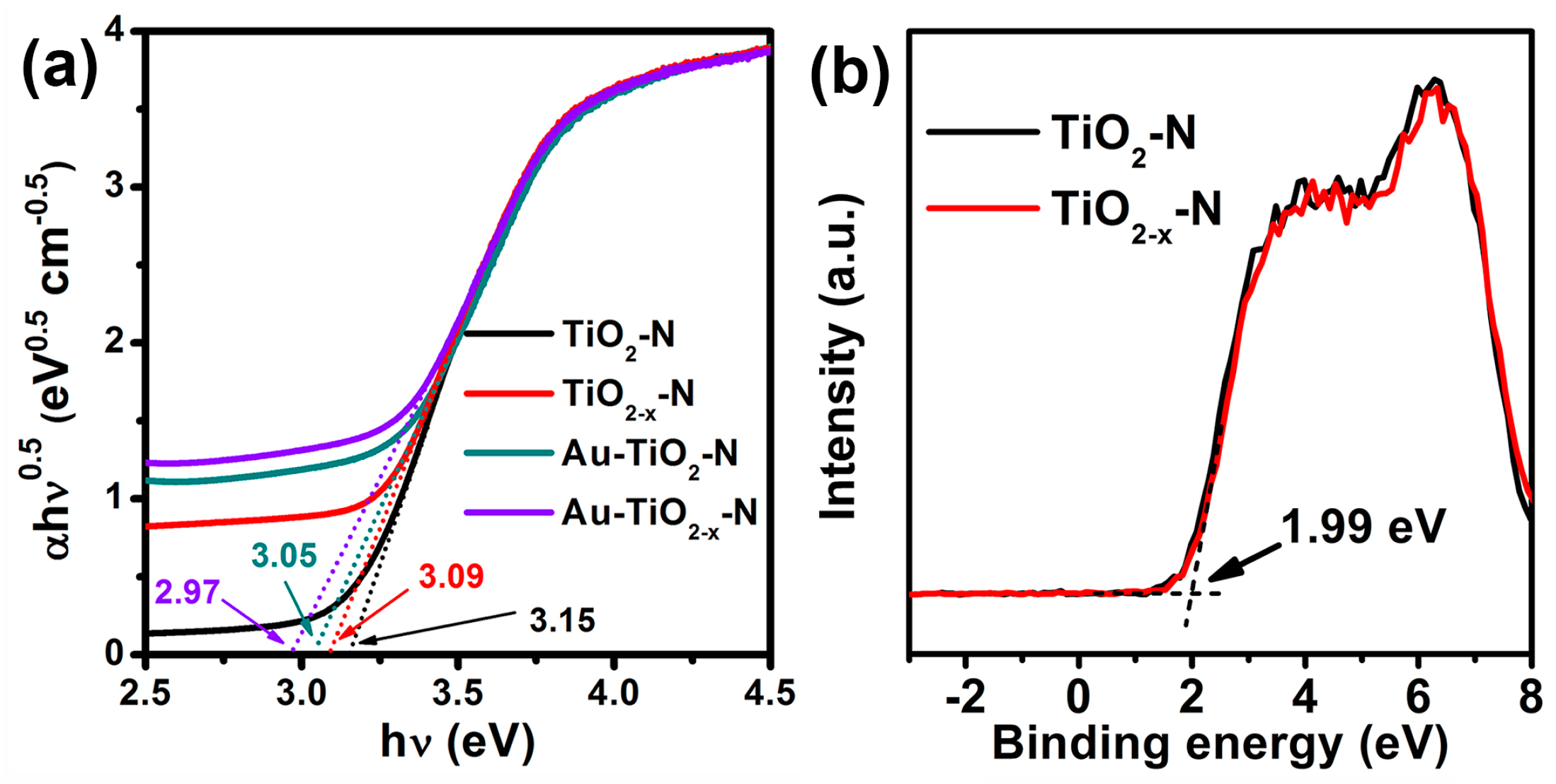
As shown in Figure 8a, the TiO2−x-N and Au-TiO2−x-N exhibit decreased Eg value (3.09 and 2.97 eV) compared with TiO2-N and Au-TiO2-N (3.15 and 3.05 eV). Then, valence band spectra of TiO2-N and TiO2−x-N were measured, and the valence band maximum (VBM) almost locates a similar value (1.99 eV below the Fermi energy, Figure 8b), consistent with the reported results [53]. Due to the same VBM, the above-mentioned decrease of Eg value should be ascribed to the decrease of the conduction band minimum (CBM). The CBM of TiO2−x-N (−1.1 eV above the Fermi energy) locates lower than that of TiO2-N (−1.16 eV above the Fermi energy).
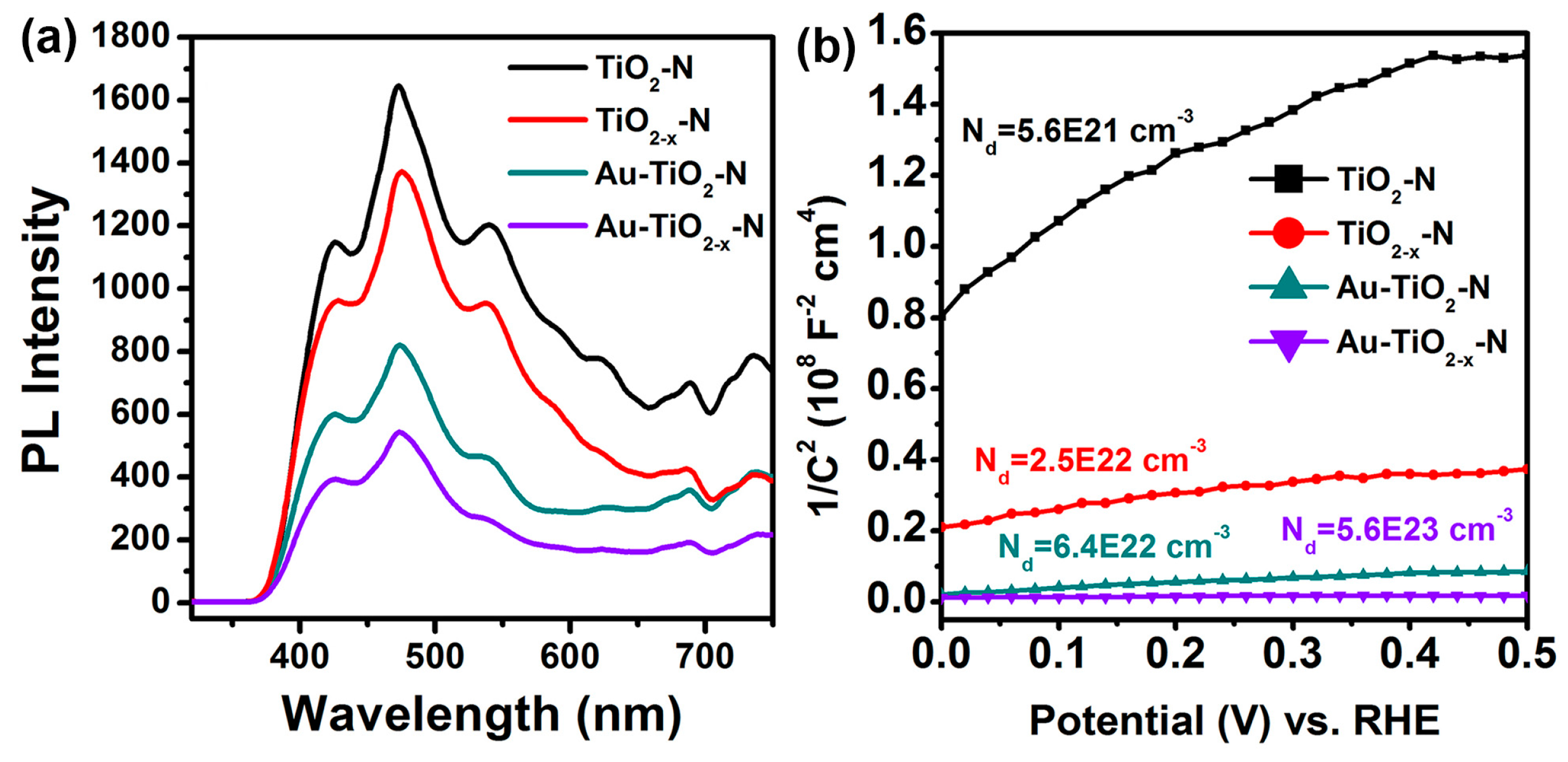
Photoluminescence (PL) spectra were also measured to understand the behaviour of light-generated electrons and holes since PL emission is a result of the recombination of the free carriers. As shown in Figure 9a, the shapes of the emission spectra of different samples are similar. Importantly, the PL peak intensities of TiO2−x-N and Au-TiO2−x-N are much lower than those of TiO2-N and Au-TiO2-N, respectively. This observation indicates that the TiO2−x-N and Au-TiO2−x-N have relatively low recombination rates of electrons and holes.
Additionally, electrochemical impedance measurements on the various TiO2 supports and Au-TiO2 catalysts were performed to investigate the effect of hydrogen-etching on their electronic properties. All samples show a positive slope in the Mott–Schottky plots (Figure 9b), as expected for n-type semiconductor. Carrier densities of these samples were calculated from the slopes of Mott–Schottky plots using the equation
where e0 is the electron charge, ε is the dielectric constant of TiO2 (ε = 170) [68], ε0 is the permittivity of vacuum (8.85 × 10−12 F/m), Nd is the electron density, and V is the applied bias at the electrode. The calculated electron densities of the TiO2-N, TiO2−x-N, Au-TiO2-N and Au-TiO2−x-N were 5.6 × 1021 cm−3, 2.5 × 1022 cm−3, 6.4 × 1022 cm−3 and 5.6 × 1023 cm−3, respectively. In other words, TiO2−x-N and Au-TiO2−x-N show substantially smaller slopes of Mott–Schottky plots compared to TiO2-N and Au-TiO2-N samples, suggesting an increase of electron densities [54,68]. The enhanced electron density is due to the increased oxygen vacancies, which are known as an electron donor for TiO2 [68]. The increased electron density improves the charge transport in TiO2−x-N and Au-TiO2−x-N. Moreover, the increased electron density is expected to shift the Fermi level of TiO2 toward the conduction band, which facilitates the charge separation at the semiconductor/metal interface. The enhanced charge separation and transportation are in very good agreement with the results of PL (Figure 9a).
Nd = (2/e0εε0)[d(1/C2)/dV]−1
3. Discussion
Proposed Electron Flow Process
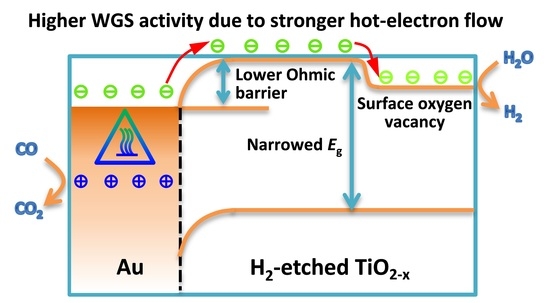
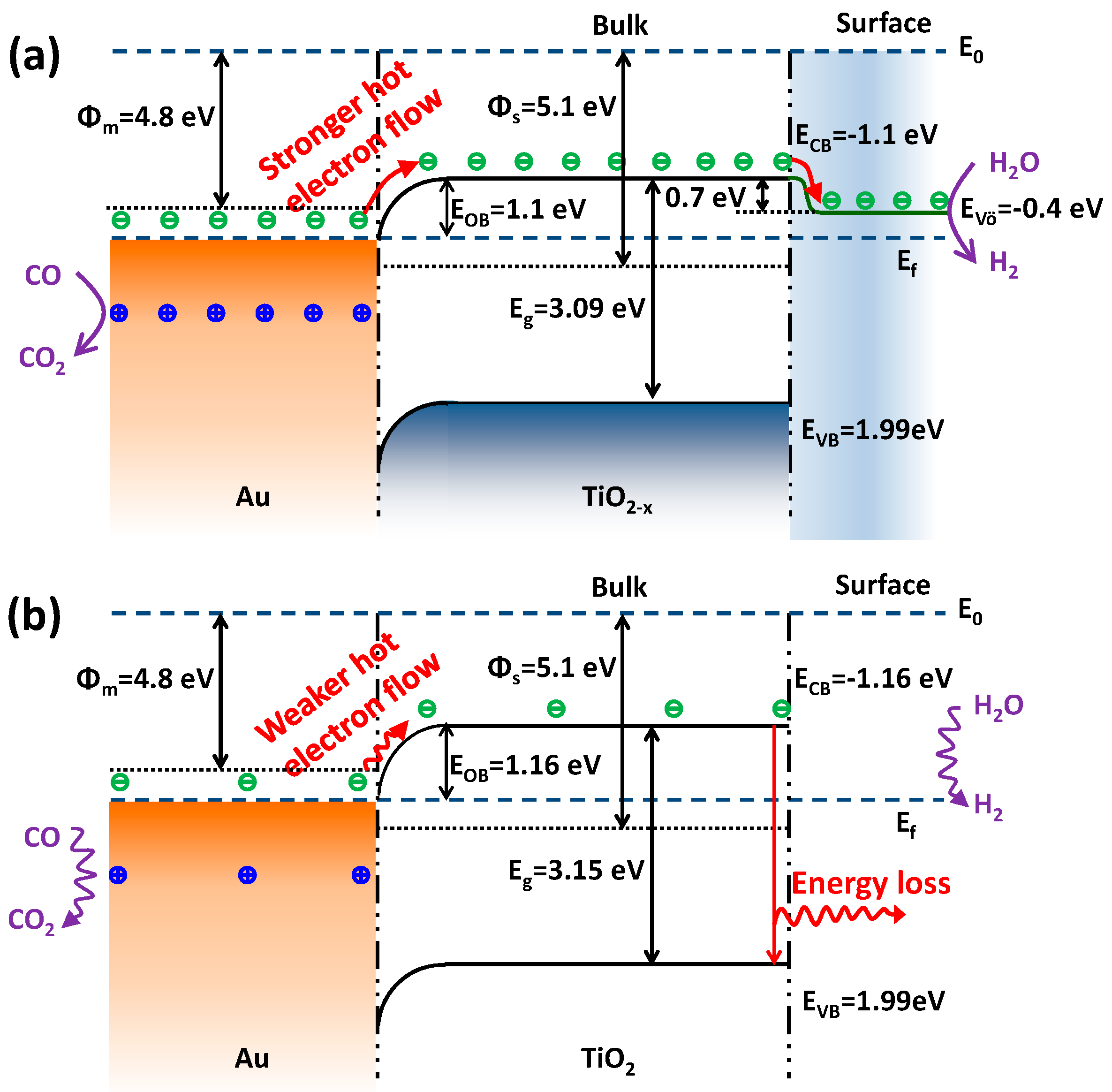
Finally, a question arises: what causes the differences in WGS catalytic activities over Au-TiO2−x-N and Au-TiO2-N? To answer this question, it is essential to discuss the reaction mechanism. From the view of the traditional WGS reaction mechanism, Au-TiO2 has been considered as a bifunctional catalyst [39,40,41]: CO adsorbs on the gold nanoparticles and the adsorption and dissociation of water takes place on the oxide or oxide-metal interfaces. Recently, Sastre et al. [69] reported the photocatalytic version of WGS reaction over Au-TiO2 catalysts. They proposed a possible photocatalytic mechanism: under visible light irradiation the surface plasmon band of Au nanoparticles will be excited and will inject electrons (−) to the conduction band of the TiO2, which facilitates water reduction to hydrogen, and the holes (+) remaining on Au nanoparticles will be responsible for the oxidation of adsorbed CO. Furthermore, “hot-electron flow” can be formed between metal-oxide interfaces during exothermic chemical processes, and the “hot-electron flow” plays a key role in determining the catalytic activity of CO oxidation [46,56,57,58]. Accordingly, correlating traditional and photocatalytic WGS reaction mechanisms with the optoelectronic properties, we try to understand the WGS pathway from the view of electron flow processes, as shown in Figure 10.
At first, more surface oxygen vacancies of TiO2−x-N lead to the fact that gold of Au-TiO2−x-N mainly exists as metallic Au0 species (XPS results). As shown in Figure 10a, since the work function of metal Au (Φm = 4.8 eV) is smaller than that of semiconductor TiO2 (Φs = 5.1 eV) [70], the electrons can diffuse from the metal into the semiconductor when the two phases are in contact [71,72]. This electron transfer was called “hot-electron flow” [56,57,73,74,75] or “chemicurrent” [76,77,78], which usually happened in exothermic catalytic reactions [56,57,73,74,75] and low-energy reactions [74] or even nonthermal directions [75]. For example, it has been reported that electron excitation was also involved in atomic/molecular adsorption processes [73,77,78]. The chemicurrent concept was first applied by Nienhaus et al. to observe the generation of hot electrons and holes during chemisorption of monatomic gas particles on the Ag/n-Si nanodiodes [78]. In this case, CO can chemisorb on coordinately unsaturated Au sites, and the heat of adsorption was reported to be in the range of 52.3–76.6 kJ/mol [79,80]. Meanwhile, the electron transfer can be promoted by the energies from the WGS reaction heat (molar enthalpy of reaction at 200 °C: −41.7 kJ/mol) and/or external heat (QAu = 4.4 kJ/mol at 200 °C). Thus, energetic electrons can be powered by the energy of 98.4–122.7 kJ/mol (1.02–1.27 eV), which is enough for electron transfer from metal Au to TiO2−x support. Hence, the gold carries positive charge and TiO2−x carries negative charge. Gold species with a positive charge (+) are similar to the Au-(OH)x species with a mildly oxidized state (Auδ+), which were identified as active gold sites [10,81]. For example, Hammer et al. investigated the valence shifts in Au of AuTiO2 catalysts in the WGS reaction using in situ X-ray absorption spectroscopy (XAS) [81]. When the feed is changed from inert to WGS reaction composition, there are some extent valence shifts in gold from Au0 to Au+. It means that Au0 can give the electrons to the near TiO2 supports under the WGS reaction conditions. Meanwhile, after the WGS reaction, the positive Au+ can go back to metallic Au0. Therefore, the formation of the mildly oxidized gold species with positive charge (+) actually happen under the WGS reaction conditions because of the electron transmission from gold to TiO2−x support. In subsequent steps, the mildly oxidized gold species with positive charge (+) can easily react with electronic donors (adsorbed CO reactant) to further form the CO2 product.
In addition, the energy bands (ECB and EVB) of the semiconductor are bent downwards and form their new Fermi levels [56,71,72], which forms the Ohmic barrier (EOB, i.e., energy difference between metal Au and the bent ECB of the semiconductor). As mentioned in the paragraph above, energetic electrons with energy of 98.4–122.7 kJ/mol (1.02–1.27 eV) are enough to move across the Ohmic barrier (EOB = 1.1 eV, Figure 10), suggesting that the electrons can transfer to the conduction band of the semiconductor TiO2 to form hot-electron flow. According to the results of Figure 8, the Ohmic barrier (EOB = 1.1 eV) of Au-TiO2−x-N is smaller than that of Au-TiO2-N (EOB = 1.16 eV). Thus, compared with the excited electrons in Au-TiO2-N, the excited electrons in Au-TiO2−x-N can more easily move across a lower Ohmic barrier and form a stronger “hot-electron flow”. Because oxygen vacancies are considered as electron traps [68], the “hot-electrons” of conduction band further transfer to the oxygen vacancies, which locate at about 0.7 eV below the conduction band minimum [51]. Thus, the surface oxygen vacancies of TiO2−x-N and Au-TiO2−x-N suppress the recombination of electron from conduction band to valence band. It is reflected in lower intensities of PL of TiO2−x-N and Au-TiO2−x-N (Figure 9a). The abundant “hot-electrons” located in oxygen vacancies can be easily trapped by electronic acceptors (H2O reactant) to form the H2 product. In other words, due to the lower Ohmic barrier and more surface oxygen vacancies, Au-TiO2−x-N presents a higher transmission efficiency of “hot-electron flow”, which are correlated with higher electron density based on the results of Mott–Schottky plots (Figure 9b), so as to enhance the WGS catalytic activity. On the contrary, for Au-TiO2-N catalyst (Figure 10b), a larger forbidden band gap leads to a bigger Ohmic barrier, which makes a weaker “hot-electron flow”. Meanwhile, due to few surface oxygen vacancies, few formed “hot-electrons” easily jump from conduction band to valence band and make energy loss. Therefore, Au-TiO2-N catalyst presents a lower WGS catalytic activity.
From the above discussion, a stronger “hot-electron flow” effect of Au-TiO2−x-N should be attributed to the lower Ohmic barrier (i.e., decrease of the forbidden band gap) and abundant surface oxygen vacancies (i.e., higher transmission efficiency of “hot-electron flow”). The stronger hot-electron flow can easily boost the reaction rate [73,74]. Considering that Au-TiO2−x-N catalyst showed a bigger TOF (Figure 1b), we concluded that the bigger TOF mainly stems from a stronger “hot-electron flow”, in very good agreement with the conclusions of some studies [73,74]. In addition, Au-TiO2−x-N presented better stability than Au-TiO2-N (Figure 1c). It has been reported that partially reduced support (with oxygen vacancies) is the best substrate to anchor Au particles [82]. Therefore, the results further prove that oxygen vacancies can anchor Au particles to avoid aggregation of Au particles and the deactivation of WGS catalysts.
Certainly, the present results do not provide direct mechanistic information on the effect of hydrogen-etched TiO2−x support on the WGS activity of Au-TiO2−x catalysts. A deeper understanding on the reaction mechanism of the WGS reaction over the Au-TiO2−x catalyst needs detailed studies of elementary steps and intermediate productions. Nevertheless, it is also interesting to understand WGS reaction pathways from the new standpoint of electron flow processes. In this sense, the involved hydrogen-etching method resulting in abundant stable surface oxygen vacancies can be extended to other supported catalysts (e.g., CeO2 and ZrO2 based catalysts) for WGS reaction or other thermocatalytic reactions.
4. Materials and Methods
4.1. Preparation of White TiO2-N and Blue-Black TiO2−x-N Supports
TiO2 nanoribbons were prepared by alkaline hydrothermal route as reported in our previous works [83]. 1.6 g of Ti(OC4H9)4, 17 g of NaOH, and 40 mL of distilled water were added into a 50 mL of Teflon-lined autoclave and mixed homogeneously. Then, they were reacted at 200 °C for 48 h in an electrical oven. The obtained sodium titanate (Na2Ti3O7) nanoribbon was rinsed with distilled water, acidified in 0.1 M HNO3 overnight, and dried at 100 °C. After that, the protonated titanate (H2Ti3O7) was calcined at 550 °C for 4 h in air and H2 atmospheres, the white TiO2 nanoribbon (TiO2-N) and the corresponding blue-black TiO2 nanoribbon (TiO2−x-N) were obtained, respectively.
4.2. Preparation of Au-TiO2-N and Au-TiO2−x-N Catalysts
Gold supported on the white TiO2-N and blue-black TiO2−x-N catalysts with Au content of 4 wt % were fabricated by deposition-precipitation. The synthetic route was described as follows: 0.7 g of the above as-prepared supports was dispersed in 200 mL of deionized water under sonication for 30 min. Then, 60 mL of 0.0025 mol/L HAuCl4 solution and an appropriate amount of 0.05 mol/L NH3·H2O solution were simultaneously added into the above suspension. During this process, the pH value and the temperature of the solution were held at 10 ± 1 and 80 °C, respectively. The precipitate was aged at this temperature for another 5 h, washed with deionized water until pH value of supernatant is neutral, dried at 105 °C for 12 h in air. The obtained samples were directly used as catalyst and denoted as Au-TiO2-N or Au-TiO2−x-N.
4.3. Evaluation of Catalytic Activity and Stability
The catalytic activities and stabilities of the catalysts for WGS reaction were tested in a fixed bed reactor at atmospheric pressure. 0.1 g of catalysts (60–80 mesh) were placed on quartz wool layer in a quartz glass tube (inner diameter = 7 mm). The reaction temperature was monitored by two thermocouples, which are inserted into the catalyst bed and the reactor wall, respectively. The experiment was directly performed without any pre-reduction. The flow of dry feed gas (12.5% CO and 87.5% N2) was at 50 mL/min. The ratio of vapor to dry feed gas was maintained at 1:1. The residual water of the outlet was removed by a condenser before entering a gas chromatography (GC 2060) equipped with a thermal conductivity detector (TCD). The by-product (e.g., methane) was not checked in the temperature range investigated. The catalytic activity was measured from 200 to 600 °C, staying for approximately 1 h at each temperature. Then, an isothermal test (600 °C) was run for about 32 h to investigate the deactivation.
The conversion of CO was defined as: XCO (%) = (1 − V’CO/VCO) × 100%/(1 + V’CO) [60,61], where VCO and V’CO are the inlet and outlet CO content of dry gas due to condensation, respectively.
To make a more realistic comparison of catalytic activities, turnover frequencies (TOFs) were measured in the kinetic region with negligible heat and mass-transfer effects. Differential conditions were maintained by keeping CO conversion below 10%. This was accomplished by decreasing the catalyst weight and adjusting the flow rate of feed gas (12.5% CO and 87.5% N2). Meanwhile, the catalyst was diluted with low surface area quartz sands in order to maintain a constant bed height and volume. The measurements were conducted at 10 °C intervals for temperatures of 200–240 °C. The TOF (s−1) was calculated shown as [84]:
where F is the total flow rate (μmol s−1), W the mass of catalyst (g), Au loading (wt %) was measured by inductively coupled plasma optical emission spectrometer (ICP-OES), gold dispersion (D) was estimated from TEM results (Supplementary Materials), MAu is the molar mass of Au (197 g/mol).
4.4. Characterization
Powder XRD patterns of the as-synthesized samples were recorded by a PANalytical X’ pert Pro diffractometer (PANalytical, Almelo, The Netherlands) equipped with Cu-Kα (λ = 0.1541 nm) radiation (X’Celerator detector) operating at 40 kV and 40 mA with a scanning rate of 0.12 °/min. For Rietveld analysis, the XRD pattern of standard reference material (NIST 640A silicon), which is a material with no microstrain nor size broadening, was measured from 10° to 140° on the same instrument with the same experimental parameters. The patterns of the standard and experimental samples were fitted with a pseudo-Voigt function (PVF), which were carried out with X’pert highscore plus software. The actual Au loading in each catalyst was measured by ICP-OES using a Varian 710-ES analyzer (Varian, Englewood, CO, USA). The morphology was examined by using field emission scanning electron microscopy (SEM, Hitachi S-4800, Hitachi, Ibaraki, Japan) and transmission electron microscopy (TEM, JEM-2100, JEM Ltd., Tokyo, Japan). Gold cluster size distributions were measured using >200 particles for each sample to determine surface-averaged cluster diameters (dTEM) [84]:
where ni is the number of clusters with diameter di. Metal dispersions (D), defined as the fraction of Au atoms exposed at cluster surfaces, and were estimated from dTEM:
where νm is the bulk atomic density of Au (16.49 × 10−3 nm3) and am is the area occupied by an Au atom (8.75 × 10−2 nm2) on a polycrystalline surface [84].
Raman spectra were collected at room temperature on a Renishaw Invia Plus instrument (Renishaw, Gloucetershire, UK) using a semiconductor laser as an illumination source (532 nm). The electron paramagnetic resonance (EPR) spectra were collected using a Bruker EMX-8 spectrometer (Bruker Instruments Inc., Rheinstetten, Germany) at room temperature. The settings were center field, 3510 G; microwave frequency, 9.859 GHz; and power, 10.02 mW. The X-ray photoelectron spectroscopy (XPS) measurements were performed on a VG ESCALAB 250 spectrophotometer (Thermo Electron Co., Eastern Grinstead, UK) with Al Ka radiation (1486.6 eV), operating at 15 kV × 10 mA, in FAT mode (Fixed Analyser Transmission), with a pass energy of 30 eV for regions ROI and 100 eV for survey. The base pressure of the main chamber was kept at about 1 × 10−9 mbar. Each sample was first placed in a copper holder mounted on a sample-rod in the pretreatment chamber of the spectrometer, and it was then outgassed at 100 °C for 1 h before being transferred to the analysis chamber. A flood gun was always used for charge compensation. The spot size is 500 μm and each high-resolution spectrum was scanned for ten times with an energy step size of 0.05 eV. All binding energies (BE) were calibrated by using that of C 1s (284.5 eV). The peaks obtained after a Shirley background subtraction were fitted to Lorentzian-Gaussian curves using a public software XPSPEAK (Version 4.1, programmed by R. Kwok, Chinese University of Hong Kong, China).
Temperature-programmed reduction (TPR) measurements were carried out on an AutoChem 2910 apparatus equipped with a thermal conductivity detector (TCD) (Micromeritics, Norcross, GA, USA) for analysis. 50 mg of each sample was purged with high purity argon gas at 110 °C for 1 h, and cooled under the same gas flow to room temperature, finally reduced by H2/Ar (H2: 10 vol. %) with a flow of 30 mL/min in the temperature range of 50–600 °C at a heating rate of 10 °C/min. Diffuse reflectance spectroscopy (DRS) was measured on Agilent Cary 5000 UV-vis spectrophotometer (Agilent, Santa Clara, CA, USA) in the range of 200–800 nm. The band gaps were estimated by extrapolating a linear part of the plots to (αhν)0.5 = 0. The photoluminescence (PL) spectra were obtained using Hitachi F-4500 fluorescence spectrophotometer (Hitachi, Ibaraki, Japan) with optical filter and the excitation wavelength at 300 nm. Mott–Schottky plots were derived from impedance-potential at a frequency of 10 KHz by a CHI 660D electrochemical station (Chenhua Ltd., Shanghai, China) in the dark. 1 M NaOH aqueous solution (pH = 13.6) was used as an electrolyte. Saturated calomel electrode (SCE) and Pt wire were used as reference and counter electrode, respectively.
5. Conclusions
TiO2−x nanoribbons with abundant stable surface oxygen vacancies was prepared by hydrogen-etching technology. The WGS catalytic activities and stabilities of gold supported on hydrogen-etched TiO2−x catalyst are obviously higher than those of gold supported on traditional white TiO2 catalysts. Firstly, the reduction of the surface oxygen species led to disorder of layers and abundant stable surface oxygen vacancies, which resulted in higher microstrain and more metallic Au0 species, respectively. These are beneficial to higher WGS activities. Secondly, disorder layers and stable surface oxygen vacancies led to the narrowed forbidden band gap, enhanced electron density and efficient charge separation and transportation. Finally, an electron flow process for understanding the difference in WGS activity has been proposed. Hereinto, the narrowed forbidden band gap facilitates the reduction of the Ohmic barrier; thus, the transmission efficiency of “hot-electron flow” is improved. Meanwhile, the abundant surface oxygen vacancies are functional as electron traps; thus, promoting the flow of “hot-electron” and reduction reaction of H2O, so as to enhance the WGS catalytic activity.
Supplementary Materials
The following are available online at www.mdpi.com/2073-4344/8/1/26/s1, Figure S1: XPS spectra of various TiO2 supports and Au-TiO2 catalysts, Figure S2: Schematic structure of white and blue black TiO2, Table S1: Physical properties of Au-TiO2 and Au-TiO2−x catalysts, Table S2: Comparison of water-gas shift rates of Au-TiO2−x-N catalysts with literature data, Table S3: Microstructure parameters of monoclinic TiO2 in various TiO2 supports and Au-TiO2 catalysts.
Acknowledgments
This research was financially supported by the National Natural Science Foundation of China (21503092), the Zhejiang Provincial Natural Science Foundation of China (LQ18B030006) and Zhejiang Provincial Analysis and Measurement Foundation of China (2018C37077).
Author Contributions
Li Song and Lei Li conceived and designed the experiments; Li Song, Zhufeng Lu, Yuting Zhang, Qi Su and Lei Li performed the experiments; Li Song and Lei Li analyzed the data; Li Song and Lei Li wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Flytzani-Stephanopoulos, M. Gold atoms stabilized on various supports catalyze the water-gas shift reaction. Acc. Chem. Res. 2013, 47, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Bunluesin, T.; Gorte, R.J.; Graham, G.W. Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: Implications for oxygen-storage properties. Appl. Catal. B Environ. 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Hakeem, A.A.; Vásquez, R.S.; Rajendran, J.; Li, M.; Berger, R.J.; Delgado, J.J.; Kapteijn, F.; Makkee, M. The role of rhodium in the mechanism of the water–gas shift over zirconia supported iron oxide. J. Catal. 2014, 313, 34–45. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Q.; Flytzani-Stephanopoulos, M. Low-temperature water-gas shift reaction over Cu- and Ni-loaded cerium oxide catalysts. Appl. Catal. B Environ. 2000, 27, 179–191. [Google Scholar] [CrossRef]
- Ren, Z.; Peng, F.; Li, J.; Liang, X.; Chen, B. Morphology-Dependent Properties of Cu/CeO2 Catalysts for the Water-Gas Shift Reaction. Catalysts 2017, 7, 48. [Google Scholar] [CrossRef]
- Sakurai, H.; Ueda, A.; Kobayashi, T.; Haruta, M. Low-temperature water-gas shift reaction over gold deposited on TiO2. Chem. Commun. 1997, 0, 271–272. [Google Scholar] [CrossRef]
- Ma, Z.; Yin, H.F.; Dai, S. Performance of Au/MxOy/TiO2 Catalysts in water-gas shift reaction. Catal. Lett. 2010, 136, 83–91. [Google Scholar] [CrossRef]
- Shi, J.; Mahr, C.; Mangir Murshed, M.; Zielasek, V.; Rosenauer, A.; Gesing, T.M.; Bäumer, M.; Wittstock, A. A versatile sol–gel coating for mixed oxides on nanoporous gold and their application in the water gas shift reaction. Catal. Sci. Technol. 2016, 6, 5311–5319. [Google Scholar] [CrossRef]
- Faust, M.; Dinkel, M.; Bruns, M.; Bräse, S.; Seipenbusch, M. Support Effect on the Water Gas Shift Activity of Chemical Vapor Deposition-Tailored-Pt/TiO2 Catalysts. Ind. Eng. Chem. Res. 2017, 56, 3194–3203. [Google Scholar] [CrossRef]
- Yang, M.; Allard, L.F.; Flytzani-Stephanopoulos, M. Atomically Dispersed Au−(OH)x Species Bound on Titania Catalyze the Low-Temperature Water-Gas Shift Reaction. J. Am. Chem. Soc. 2013, 135, 3768–3771. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kispersky, V.F.; Nicholas Delgass, W.; Ribeiro, F.H. Determination of the Au active site and surface active species via operando transmission FTIR and isotopic transient experiments on 2.3 wt. % Au/TiO2 for the WGS reaction. J. Catal. 2012, 289, 171–178. [Google Scholar] [CrossRef]
- Shekhar, M.; Wang, J.; Lee, W.S.; Williams, W.D.; Kim, S.M.; Stach, E.A.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Size and support effects for the water-gas shift catalysis over gold nanoparticles supported on model Al2O3 and TiO2. J. Am. Chem. Soc. 2012, 134, 4700–4708. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, P.; Kondarides, D.I. Effects of alkali promotion of TiO2 on the chemisorptive properties and water-gas shift activity of supported noble metal catalysts. J. Catal. 2009, 267, 57–66. [Google Scholar] [CrossRef]
- Pérez, P.; Soria, M.A.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Mendes, A.; Madeira, L.M. Application of Au/TiO2 catalysts in the low-temperature water–gas shift reaction. Int. J. Hydrogen Energy 2016, 41, 4670–4681. [Google Scholar] [CrossRef]
- Hinojosa-Reyes, M.; Rodríguez-González, V.; Zanella, R. Gold nanoparticles supported on TiO2–Ni as catalysts for hydrogen purification via water–gas shift reaction. RSC Adv. 2014, 4, 4308–4316. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, M.; Lobban, L.L.; Mallinson, R.G. Structural effects of Na promotion for high water gas shift activity on Pt-Na/TiO2. J. Catal. 2011, 278, 123–132. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ramírez, P.J.; Asara, G.G.; Viñes, F.; Evans, J.; Liu, P.; Ricart, J.M.; Illas, F. Charge Polarization at a Au–TiC Interface and the Generation of Highly Active and Selective Catalysts for the Low-Temperature Water-Gas Shift Reaction. Angew. Chem. Int. Ed. 2014, 53, 11270–11274. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhan, Y.; Zheng, Q.; Zheng, Y.; Chen, C.; She, Y.; Lin, X.; Wei, K. Water-Gas Shift Reaction over CuO/CeO2 Catalysts: Effect of the Thermal Stability and Oxygen Vacancies of CeO2 Supports previously prepared by different methods. Catal. Lett. 2009, 130, 532–540. [Google Scholar] [CrossRef]
- Duarte de Farias, A.M.; Nguyen-Thanh, D.; Fraga, M.A. Discussing the use of modified ceria as support for Pt catalysts on water–gas shift reaction. Appl. Catal. B Environ. 2010, 93, 250–258. [Google Scholar] [CrossRef]
- Hwang, K.R.; Park, J.S.; Ihm, S.K. Si-modified Pt/CeO2 catalyst for a single-stage water-gas shift reaction. Int. J. Hydrogen Energy 2011, 36, 9685–9693. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef] [PubMed]
- González-Castaño, M.; Ivanova, S.; Ioannides, T.; Centeno, M.A.; Odriozola, J.A. Deep insight into Zr/Fe combination for successful Pt/CeO2/Al2O3 WGS catalyst doping. Catal. Sci. Technol. 2017, 7, 1556–1564. [Google Scholar] [CrossRef]
- Bobrova, L.; Andreev, D.; Ivanov, E.; Mezentseva, N.; Simonov, M.; Makarshin, L.; Cribovskii, A.; Sadykov, V. Water-gas shift reaction over Ni/CeO2 catalysts. Catalysts 2017, 7, 310. [Google Scholar] [CrossRef]
- Wyvratt, B.M.; Gaudet, J.R.; Thompson, L.T. Effects of passivation on synthesis, structure and composition of molybdenum carbide supported platinum water–gas shift catalysts. J. Catal. 2015, 330, 280–287. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Gutiérez, R.A.; Zuo, Z.; Ramírez, P.J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. Highly active Au/δ-MoC and Au/β-Mo2C catalysts for the low-temperature water gas shift reaction: Effects of the carbide metal/carbon ratio on the catalyst performance. Catal. Sci. Technol. 2017, 7, 5332–5342. [Google Scholar] [CrossRef]
- Schweitzer, N.M.; Schaidle, J.A.; Ezekoye, O.K.; Pan, X.; Linic, S.; Thompson, L.T. High Activity Carbide Supported Catalysts for Water Gas Shift. J. Am. Chem. Soc. 2011, 133, 2378–2381. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wang, A.; Qiao, B.; Liu, X.; Yang, X.; Wang, X.; Liang, J.; Li, J.; Liu, J.; Zhang, T. Remarkable performance of Ir1/FeOx single-atom catalyst in water-gas shift reaction. J. Am. Chem. Soc. 2013, 135, 15314–15317. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.; Martos, C.; Ruiz, A.; Ayuela, F.J. Effect of the precursor on the activity of high temperature water gas shift catalysts. Int. J. Hydrogen Energy 2013, 38, 7647–7653. [Google Scholar] [CrossRef]
- Soria, M.A.; Pérez, P.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Mendes, A.; Madeira, L.M. Effect of the preparation method on the catalytic activity and stability of Au/Fe2O3 catalysts in the low-temperature water–gas shift reaction. Appl. Catal. A Gen. 2014, 470, 45–55. [Google Scholar] [CrossRef]
- Kono, E.; Tamura, S.; Yamamuro, K.; Ogo, S.; Sekine, Y. Pd/K/Co-oxide catalyst for water gas shift. Appl. Catal. A Gen. 2015, 489, 247–254. [Google Scholar] [CrossRef]
- Cybulskis, V.J.; Wang, J.; Pazmiño, J.H.; Ribeiro, F.H.; Delgass, W.N. Isotopic transient studies of sodium promotion of Pt/Al2O3 for the water–gas shift reaction. J. Catal. 2016, 339, 163–172. [Google Scholar] [CrossRef]
- Lenite, B.A.; Galletti, C.; Specchia, S. Studies on Au catalysts for water gas shift reaction. Int. J. Hydrogen Energy 2011, 36, 7750–7758. [Google Scholar] [CrossRef]
- Tibiletti, D.; Meunier, F.C.; Goguet, A.; Reid, D.; Burch, R.; Boaro, M.; Vicario, M.; Trovarelli, A. An investigation of possible mechanisms for the water–gas shift reaction over a ZrO2-supported Pt catalyst. J. Catal. 2006, 244, 183–191. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhan, Y.; Chen, C.; Cao, Y.; Lin, X.; Zheng, Q. Highly efficient Au/ZrO2 catalysts for low-temperature water-gas shift reaction: Effect of pre-calcination temperature of ZrO2. Int. J. Hydrogen Energ. 2012, 37, 12292–12300. [Google Scholar] [CrossRef]
- Luo, S.; Barrio, L.; Nguyen-Phan, T.D.; Vovchok, D.; Johnston-Peck, A.C.; Xu, W.; Stach, E.A.; Rodriguez, J.A.; Senanayake, S.D. Importance of Low Dimensional CeOx Nanostructures in Pt/CeOx-TiO2 Catalysts for the Water-Gas Shift Reaction. J. Phys. Chem. C 2017, 121, 6635–6642. [Google Scholar] [CrossRef]
- Silva, L.P.C.; Terra, L.E.; Coutinho, A.C.; Passos, F.B. Sour water-gas shift reaction over Pt/CeZrO2 catalysts. J. Catal. 2016, 341, 1–12. [Google Scholar] [CrossRef]
- Daly, H.; Goguet, A.; Hardacre, C.; Meunier, F.C.; Pilasombat, R.; Thompsett, D. The effect of reaction conditions on the stability of Au/CeZrO4 catalysts in the low-temperature water–gas shift reaction. J. Catal. 2010, 273, 257–265. [Google Scholar] [CrossRef]
- Sun, Y.; Hla, S.S.; Duffy, G.J.; Cousins, A.J.; French, D.; Morpeth, L.D.; Edwards, J.H.; Roberts, D.G. A comparative study of CeO2-La2O3-based Cu catalysts for the production of hydrogen from simulated coal-derived syngas. Appl. Catal. A Gen. 2010, 390, 201–209. [Google Scholar] [CrossRef]
- Rodriguez, J.A. Gold-based catalysts for the water–gas shift reaction: Active sites and reaction mechanism. Catal. Today 2011, 160, 3–10. [Google Scholar] [CrossRef]
- Williams, W.D.; Greeley, J.P.; Delgass, W.N.; Ribeiro, F.H. Water activation and carbon monoxide coverage effects on maximum rates for low temperature water-gas shift catalysis. J. Catal. 2017, 347, 197–204. [Google Scholar] [CrossRef]
- Ammal, S.C.; Heyden, A. Water-Gas Shift Catalysis at Corner Atoms of Pt Clusters in Contact with a TiO2 (110) Support Surface. ACS Catal. 2014, 4, 3654–3662. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Z.; Wen, J.; Ding, K.; Yang, X.; Poeppelmeier, K.R.; Marks, L.D. Adhesion and Atomic Structures of Gold on Ceria Nanostructures: The Role of Surface Structure and Oxidation State of Ceria Supports. Nano Lett. 2015, 15, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Graciani, J.; Evans, J.; Park, J.B.; Yang, F.; Stacchiola, D.; Senanayake, S.D.; Ma, S.; Pérez, M.; Liu, P.; et al. Water-Gas Shift Reaction on a Highly Active Inverse CeOx/Cu(111) Catalyst: Unique Role of Ceria Nanoparticles. Angew. Chem. 2009, 121, 8191–8194. [Google Scholar] [CrossRef]
- Graciani, J.; Sanz, J.F. Designing a new generation of catalysts: Water gas shift reaction example. Catal. Today 2015, 240, 214–219. [Google Scholar] [CrossRef]
- Rodríguez, J.A.; Evans, J.; Graciani, J.; Park, J.B.; Liu, P.; Hrbek, J.; Sanz, J.F. High Water−Gas Shift Activity in TiO2(110) Supported Cu and Au Nanoparticles: Role of the Oxide and Metal Particle Size. J. Phys. Chem. C 2009, 113, 7364–7370. [Google Scholar] [CrossRef]
- Goddeti, K.C.; Kim, S.M.; Lee, Y.K.; Kim, S.H.; Park, J.Y. Chemical Doping of TiO2 with Nitrogen and Fluorine and Its Support Effect on Catalytic Activity of CO Oxidation. Catal. Lett. 2014, 144, 1411–1417. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Peter, Y.Y.; Mao, S.S. Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals. Science 2011, 331, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, P.; Christodoulakis, A.; Kondarides, D.I.; Boghosian, S. Particle size effects on the reducibility of titanium dioxide and its relation to the water–gas shift activity of Pt/TiO2 catalysts. J. Catal. 2006, 240, 114–125. [Google Scholar] [CrossRef]
- Yang, C.; Wang, Z.; Lin, T.; Yin, H.; Lü, X.; Wan, D.; Xu, T.; Zheng, C.; Lin, J.; Huang, F.; et al. Core-Shell Nanostructured “Black” Rutile Titania as Excellent Catalyst for Hydrogen Production Enhanced by Sulfur Doping. J. Am. Chem. Soc. 2013, 135, 17831–17838. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Li, W.; Wang, J.; Qu, Y.; Yang, Y.; Xie, Y.; Zhang, K.; Wang, L.; Fu, H.; Zhao, D. Ordered Mesoporous Black TiO2 as Highly Efficient Hydrogen Evolution Photocatalyst. J. Am. Chem. Soc. 2014, 136, 9280–9283. [Google Scholar] [CrossRef] [PubMed]
- Naldoni, A.; Allieta, M.; Santangelo, S.; Marelli, M.; Fabbri, F.; Cappelli, S.; Bianchi, C.L.; Psaro, R.; Santo, V.D. Effect of Nature and Location of Defects on Bandgap Narrowing in Black TiO2 Nanoparticles. J. Am. Chem. Soc. 2012, 134, 7600–7603. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Huang, B.; Lu, J.; Wang, Z.; Qin, X.; Zhang, X.; Dai, Y.; Whangbo, M.H. Hydrogenated titania: Synergy of surface modification and morphology improvement for enhanced photocatalytic activity. Chem. Commun. 2012, 48, 5733–5735. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, C.; Lin, T.; Yin, H.; Chen, P.; Wan, D.; Xu, F.; Huang, F.; Lin, J.; Xie, X.; et al. Visible-light Photocatalytic, Solar Thermal and Photoelectrochemical Properties of Aluminium-reduced Black Titania. Energy Environ. Sci. 2013, 6, 3007–3014. [Google Scholar] [CrossRef]
- Lu, X.; Wang, G.; Zhai, T.; Yu, M.; Gan, J.; Tong, Y.; Li, Y. Hydrogenated TiO2 Nanotube Arrays for Supercapacitors. Nano Lett. 2012, 12, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Peng, J.F.; Chu, W.; Li, Y.Z.; Tong, D.G. Black mesoporous anatase TiO2 nanoleaves: A high capacity and high rate anode for aqueous Al-ion batteries. J. Mater. Chem. A 2014, 2, 1721–1731. [Google Scholar] [CrossRef]
- Kim, S.M.; Lee, H.; Park, J.Y. Charge Transport in Metal–Oxide Interfaces: Genesis and Detection of Hot Electron Flow and Its Role in Heterogeneous Catalysis. Catal. Lett. 2015, 145, 299–308. [Google Scholar] [CrossRef]
- Park, J.Y.; Lee, H.; Renzas, J.R.; Zhang, Y.; Somorjai, G.A. Probing Hot Electron Flow Generated on Pt Nanoparticles with Au/TiO2 Schottky Diodes during Catalytic CO Oxidation. Nano Lett. 2008, 8, 2388–2392. [Google Scholar] [CrossRef] [PubMed]
- Furube, A.; Du, L.; Hara, K.; Katoh, R.; Tachiya, M. Ultrafast Plasmon-Induced Electron Transfer from Gold Nanodots into TiO2 Nanoparticles. J. Am. Chem. Soc. 2007, 129, 14852–14853. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lu, G.; Zhou, R.; Mao, J.; Chen, Y.; Zheng, X. Studies of pore structure, temperature-programmed reduction performance, and micro-structure of CuO/CeO2 catalysts. Appl. Surf. Sci. 2001, 173, 208–220. [Google Scholar] [CrossRef]
- Li, L.; Song, L.; Chen, C.; Zhang, Y.; Zhan, Y.; Lin, X.; Zheng, Q.; Wang, H.; Ma, H.; Ding, L.; et al. Modified precipitation processes and optimized copper content of CuO-CeO2 catalysts for water-gas shift reaction. Int. J. Hydrogen Energy 2014, 39, 19570–19582. [Google Scholar] [CrossRef]
- Li, L.; Song, L.; Wang, H.; Chen, C.; She, Y.; Zhan, Y.; Lin, X.; Zheng, Q. Water-Gas Shift Reaction over CuO/CeO2 Catalysts: Effect of CeO2 Supports previously prepared by precipitation with different precipitants. Int. J. Hydrogen Energy 2011, 36, 8839–8849. [Google Scholar] [CrossRef]
- Beuvier, T.; Richard-Plouet, M.; Brohan, L. Accurate Methods for Quantifying the Relative Ratio of Anatase and TiO2(B) Nanoparticles. J. Phys. Chem. C 2009, 113, 13703–13706. [Google Scholar] [CrossRef]
- Hemmingson, S.L.; James, T.E.; Feeley, G.M.; Tilson, A.M.; Campbell, C.T. Adsorption and Adhesion of Au on Reduced CeO2(111) Surfaces at 300 and 100 K. J. Phys. Chem. C 2016, 120, 12113–12124. [Google Scholar] [CrossRef]
- Li, B.; Zhao, Z.; Zhou, Q.; Meng, B.; Meng, X.; Qiu, J. Highly Efficient Low-Temperature Plasma-Assisted Modification of TiO2 Nanosheets with Exposed {001} Facets for Enhanced Visible-Light Photocatalytic Activity. Chem. Eur. J. 2014, 20, 14763–14770. [Google Scholar] [CrossRef] [PubMed]
- Kruse, N.; Chenakin, S. XPS characterization of Au/TiO2 catalysts: Binding energy assessment and irradiation effects. Appl. Catal. A Gen. 2011, 391, 367–376. [Google Scholar] [CrossRef]
- Kast, P.; Kučerová, G.; Behm, R.J. On the nature of the active Au species: CO oxidation on cyanide leached Au/TiO2 catalysts. Catal. Today 2015, 244, 146–160. [Google Scholar] [CrossRef]
- Magadzu, T.; Yang, J.H.; Henao, J.D.; Kung, M.C.; Kung, H.H.; Scurrell, M.S. Low-Temperature Water-Gas Shift Reaction over Au Supported on Anatase in the Presence of Copper: EXAFS/XANES Analysis of Gold-Copper Ion Mixtures on TiO2. J. Phys. Chem. C 2017, 121, 8812–8823. [Google Scholar] [CrossRef]
- Wang, G.; Wang, H.; Ling, Y.; Tang, Y.; Yang, X.; Fitzmorris, R.C.; Wang, C.; Zhang, J.Z.; Li, Y. Hydrogen-Treated TiO2 Nanowire Arrays for Photoelectrochemical Water Splitting. Nano Lett. 2011, 11, 3026–3033. [Google Scholar] [CrossRef] [PubMed]
- Sastre, F.; Oteri, M.; Corma, A.; García, H. Photocatalytic water gas shift using visible or simulated solar light for the efficient, room-temperature hydrogen generation. Energy Environ. Sci. 2013, 6, 2211–2215. [Google Scholar] [CrossRef]
- Deáka, P.; Kullgren, J.; Aradi, B.; Frauenheim, T.; Kavan, L. Water splitting and the band edge positions of TiO2. Electrochim. Acta 2016, 199, 27–34. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, L.; Zhan, Y.; Lin, X.; Zheng, Q.; Wei, K. Ag/ZnO Heterostructure Nanocrystals: Synthesis, Characterization, and Photocatalysis. Inorg. Chem. 2007, 46, 6980–6986. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kong, X.; Yu, Y.; Zhang, H. Synthesis and Characterization of Water-Soluble and Bifunctional ZnO−Au Nanocomposites. J. Phys. Chem. C 2007, 111, 3836–3841. [Google Scholar] [CrossRef]
- Park, J.Y.; Baker, L.R.; Somorjai, G.A. Role of Hot Electrons and Metal–Oxide Interfaces in Surface Chemistry and Catalytic Reactions. Chem. Rev. 2015, 115, 2781–2817. [Google Scholar] [CrossRef] [PubMed]
- Nedrygailov, I.I.; Lee, C.; Moon, S.Y.; Lee, H.; Park, J.Y. Hot Electrons at Solid-Liquid Interfaces: A Large Chemoelectric Effect during the Catalytic Decomposition of Hydrogen Peroxide. Angew. Chem. Int. Ed. 2016, 55, 10859–10862. [Google Scholar] [CrossRef] [PubMed]
- Renzas, J.R.; Somorjai, G.A. Rh Thin-Film Nanocatalysts as Chemical Sensors—The Hot Electron Effect. J. Phys. Chem. C 2010, 114, 17660–17664. [Google Scholar] [CrossRef]
- Maximoff, S.N.; Head-Gordon, M.P. Chemistry of fast electrons. Proc. Natl. Acad. Sci. USA 2009, 106, 11460–11465. [Google Scholar] [CrossRef] [PubMed]
- Gergen, B.; Nienhaus, H.; Weinberg, W.H.; McFarland, E.W. Chemically Induced Electronic Excitations at Metal Surfaces. Science 2001, 294, 2521–2523. [Google Scholar] [CrossRef] [PubMed]
- Nienhaus, H.; Bergh, H.S.; Gergen, B.; Majumdar, A.; Weinberg, W.H.; McFarland, E.W. Electron-Hole Pair Creation at Ag and Cu Surfaces by Adsorption of Atomic Hydrogen and Deuterium. Phys. Rev. Lett. 1999, 82, 446–449. [Google Scholar] [CrossRef] [Green Version]
- Meier, D.C.; Goodman, D.W. The Influence of Metal Cluster Size on Adsorption Energies: CO Adsorbed on Au Clusters Supported on TiO2. J. Am. Chem. Soc. 2004, 126, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T., Jr. Spectroscopic Observation of Dual Catalytic Sites during Oxidation of CO on a Au/TiO2 Catalyst. Science 2011, 333, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Hammer, N.; Kvande, I.; Van Beek, W.; Chen, D.; Rønning, M. Identification of valence shifts in Au during the water-gas shift reaction. Top. Catal. 2007, 45, 25–29. [Google Scholar] [CrossRef]
- Liu, Z.; Jenkins, S.J.; King, D.A. Origin and Activity of Oxidized Gold in Water-Gas-Shift Catalysis. Phys. Rev. Lett. 2005, 94, 196102–196104. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Zhou, J.; Wu, Y.; Tang, L.; Zhu, L.; Gu, L. Self-assembled, robust titanate nanoribbon membranes for highly efficient nanosolid capture and molecule discrimination. Nanoscale 2013, 5, 3486–3495. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Zones, S.I.; Iglesia, E. Challenges and strategies in the encapsulation and stabilization of monodisperse Au clusters within zeolites. J. Catal. 2016, 339, 195–208. [Google Scholar] [CrossRef]
Figure 1.
(a) The CO conversions of Au-TiO2 and Au-TiO2−x catalysts as a function of temperature; (b) Arrhenius plots of TOF over Au-TiO2 and Au-TiO2−x catalysts in the reaction temperature range of 200–240 °C; (c) The CO conversions at 600 °C as a function of time on stream. Dried feed gas: 12.5% CO and 87.5% N2.
Figure 1.
(a) The CO conversions of Au-TiO2 and Au-TiO2−x catalysts as a function of temperature; (b) Arrhenius plots of TOF over Au-TiO2 and Au-TiO2−x catalysts in the reaction temperature range of 200–240 °C; (c) The CO conversions at 600 °C as a function of time on stream. Dried feed gas: 12.5% CO and 87.5% N2.

Figure 2.
(a,b) The photographs of white TiO2 support calcined in air and blue black TiO2−x support calcined in H2; (c,d) Scanning electron microscopy (SEM) images of TiO2−x-N and TiO2-N.
Figure 2.
(a,b) The photographs of white TiO2 support calcined in air and blue black TiO2−x support calcined in H2; (c,d) Scanning electron microscopy (SEM) images of TiO2−x-N and TiO2-N.

Figure 3.
TEM images of Au-TiO2-N (a) and Au-TiO2−x-N (d). HRTEM images of TiO2−x-N (b), Au-TiO2−x-N (c), TiO2-N (e) and Au-TiO2-N (f). The dash line indicates the interface between the disordered layer and crystalline core. Surface-averaged Au cluster diameters (dTEM) and cluster diameter distributions of Au-TiO2-N (g) and Au-TiO2−x-N (h). H2-TPR profiles of TiO2−x-N and TiO2-N supports (i).
Figure 3.
TEM images of Au-TiO2-N (a) and Au-TiO2−x-N (d). HRTEM images of TiO2−x-N (b), Au-TiO2−x-N (c), TiO2-N (e) and Au-TiO2-N (f). The dash line indicates the interface between the disordered layer and crystalline core. Surface-averaged Au cluster diameters (dTEM) and cluster diameter distributions of Au-TiO2-N (g) and Au-TiO2−x-N (h). H2-TPR profiles of TiO2−x-N and TiO2-N supports (i).

Figure 4.
X-ray Diffraction (XRD) patterns of various TiO2 supports and Au-TiO2 catalysts.

Figure 5.
Raman spectra of various TiO2 supports and Au-TiO2 catalysts.

Figure 6.
The electron paramagnetic resonance (EPR) spectra of various TiO2 supports and Au-TiO2 catalysts.
Figure 6.
The electron paramagnetic resonance (EPR) spectra of various TiO2 supports and Au-TiO2 catalysts.

Figure 7.
XPS spectra of various TiO2 supports and Au-TiO2 catalysts (a) Ti 2p; (b,c) Au 4f; (d–g) O 1s.
Figure 7.
XPS spectra of various TiO2 supports and Au-TiO2 catalysts (a) Ti 2p; (b,c) Au 4f; (d–g) O 1s.

Figure 8.
(a) The optical absorption edges and (b) XPS valence band spectra of various TiO2 supports and Au-TiO2 catalysts.
Figure 8.
(a) The optical absorption edges and (b) XPS valence band spectra of various TiO2 supports and Au-TiO2 catalysts.

Figure 9.
(a) Photoluminescence spectra and (b) Mott-Schottky plots collected at a frequency of 10 kHz in the dark of various TiO2 supports and Au-TiO2 catalysts.
Figure 9.
(a) Photoluminescence spectra and (b) Mott-Schottky plots collected at a frequency of 10 kHz in the dark of various TiO2 supports and Au-TiO2 catalysts.

Figure 10.
Proposed electron flow process of Au-TiO2−x (a) and Au-TiO2 (b) for the WGS reaction based on hot-electron flow. Φm: work function of metal; Φs: work function of semiconductor; Ef: Fermi level; E0: vacuum level; EOB: Ohmic barrier, VÖ: oxygen vacancy; ECB: conduction band; EVB: valence band.
Figure 10.
Proposed electron flow process of Au-TiO2−x (a) and Au-TiO2 (b) for the WGS reaction based on hot-electron flow. Φm: work function of metal; Φs: work function of semiconductor; Ef: Fermi level; E0: vacuum level; EOB: Ohmic barrier, VÖ: oxygen vacancy; ECB: conduction band; EVB: valence band.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Microstructure parameters of various TiO2 supports and Au-TiO2 catalysts.
| Sample | Anatase | |||||||
|---|---|---|---|---|---|---|---|---|
| 2theta (°) | d-Spacing (Å) | FWHM (°) | Crystal Size (nm) | Microstrain (Δd/d) (%) | a (Å) | C (Å) | V (Å3) | |
| TiO2-N | 25.287 | 3.519 | 0.564 | 14.52 | 0.600 | 3.7847 | 9.5181 | 136.33 |
| TiO2−x-N | 25.365 | 3.508 | 0.597 | 14.63 | 0.617 | 3.7845 | 9.5146 | 136.27 |
| Au-TiO2-N | 25.244 | 3.525 | 0.602 | 14.85 | 0.615 | 3.7838 | 9.5058 | 136.09 |
| Au-TiO2−x-N | 25.274 | 3.521 | 0.609 | 14.89 | 0.625 | 3.7818 | 9.5087 | 135.99 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Song, L.; Lu, Z.; Zhang, Y.; Su, Q.; Li, L. Hydrogen-Etched TiO2−x as Efficient Support of Gold Catalysts for Water–Gas Shift Reaction. Catalysts 2018, 8, 26. https://doi.org/10.3390/catal8010026
AMA Style
Song L, Lu Z, Zhang Y, Su Q, Li L. Hydrogen-Etched TiO2−x as Efficient Support of Gold Catalysts for Water–Gas Shift Reaction. Catalysts. 2018; 8(1):26. https://doi.org/10.3390/catal8010026
Chicago/Turabian StyleSong, Li, Zhufeng Lu, Yuting Zhang, Qi Su, and Lei Li. 2018. "Hydrogen-Etched TiO2−x as Efficient Support of Gold Catalysts for Water–Gas Shift Reaction" Catalysts 8, no. 1: 26. https://doi.org/10.3390/catal8010026
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.