Ruthenium(II)-Arene Complexes of the Water-Soluble Ligand CAP as Catalysts for Homogeneous Transfer Hydrogenations in Aqueous Phase †



1
Consiglio Nazionale delle Ricerche (CNR), Istituto di Chimica dei Composti OrganoMetallici (ICCOM), Via Madonna del Piano 10, 50019 Sesto Fiorentino (Florence), Italy
2
Consiglio Nazionale delle Ricerche (CNR), Dipartimento di Scienze Chimiche e Tecnologia dei Materiali (DSCTM), Via dei Taurini 19, 00185 Rome, Italy
*
Authors to whom correspondence should be addressed.
†
Dedicated to Prof. Peter M. Maitlis on occasion of his 85th birthday.
Catalysts 2018, 8(2), 88; https://doi.org/10.3390/catal8020088
Submission received: 29 December 2017
/
Revised: 14 February 2018
/
Accepted: 15 February 2018
/
Published: 22 February 2018
(This article belongs to the Special Issue Homogeneous Catalysis and Mechanisms in Water and Biphasic Media)
Abstract
:The neutral Ru(II) complex κP-[RuCl2(η6-p-cymene)(CAP)] (1), and the two ionic complexes κP-[RuCl(η6-p-cymene)(MeCN)(CAP)]PF6 (2) and κP-[RuCl(η6-p-cymene)(CAP)2]PF6 (3), containing the water-soluble phosphine 1,4,7-triaza-9-phosphatricyclo[5.3.2.1]tridecane (CAP), were tested as catalysts for homogeneous hydrogenation of benzylidene acetone, selectively producing the saturated ketone as product. The catalytic tests were carried out in aqueous phase under transfer hydrogenation conditions, at mild temperatures using sodium formate as hydrogen source. Complex 3, which showed the highest stability under the reaction conditions applied, was also tested for C=N bond reduction from selected cyclic imines. Preliminary NMR studies run under pseudo-catalytic conditions starting from 3 showed the formation of κP-[RuH(η6-p-cymene)(CAP)2]PF6 (4) as the pivotal species in catalysis.
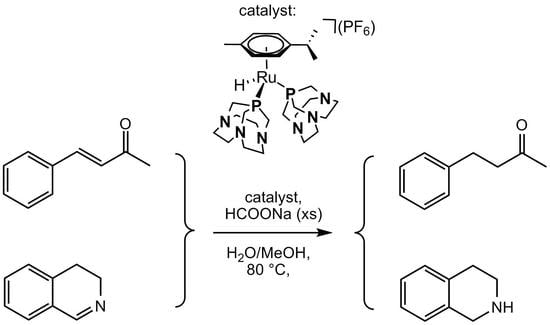
1. Introduction
The importance of ruthenium compounds as homogeneous catalysts for the synthesis of fine chemicals and pharmaceutical intermediates has been widely demonstrated [1,2,3,4,5]. Some of them display high reactivity and selectivity in many catalytic transformations and, in particular, in the hydrogenation of different unsaturated compounds, such as α,β-unsaturated ketones and imines, using molecular H2 and transfer hydrogenation (TH) protocols as reducing agents [6,7,8,9]. The employment of these last methodologies offers attractive advantages including the use of inexpensive and easy to handle hydrogen donors instead of explosive hydrogen gas, mild reaction conditions, and the possibility of using environmentally friendly solvents such as water [10,11,12]. The development of metal complexes containing water soluble ligands is the most common strategy to perform catalytic hydrogenations in water or under aqueous biphasic conditions and many organophosphines have been prepared for this purpose [13,14].
The water soluble phosphine 1,3,5-triaza-7-phospaadamantane (PTA, Figure 1) and its derivatives found large use as ligands for transition metal complexes active in homogeneous aqueous phase and biphasic catalysis and in the course of last decade many reports concerning Ru-PTA catalysts appeared in the literature [15,16,17]. For example, cyclopentadienyl (Cp) and pentamethylcyclopentadienyl (Cp*) Ru(II) complexes such as [RuCpCl(PTA)2], [RuCp(MeCN)(PTA)2](PF6), [RuCp*Cl(PTA)2], and [RuCp*(MeCN)(PTA)2](PF6) were proven to be active catalysts for the hydrogenation of benzylidene acetone (BZA) under H2 pressure in a biphasic water/octane solvent mixture showing a high chemoselectivity to C=C double bond reduction [18,19]. Another class of catalytically active compounds is represented by [RuCl2(η6-p-cymene)(PTA)] (RAPTA-C) and [RuCl(η6-p-cymene)(PTA)2](BF4), which were shown to be active catalysts for the full hydrogenation of various substituted arenes into the corresponding cyclohexanes under biphasic conditions [20]. Some Ru(II) complexes bearing ‘upper rim’ PTA derivatives—i.e., pending arms on the C atom adjacent to the P donor (Figure 1)—were also tested in catalytic hydrogenation of acetophenone giving good conversions even at room temperature using protocols based on the presence of both H2 gas and tBuOK/iPrOH [21].
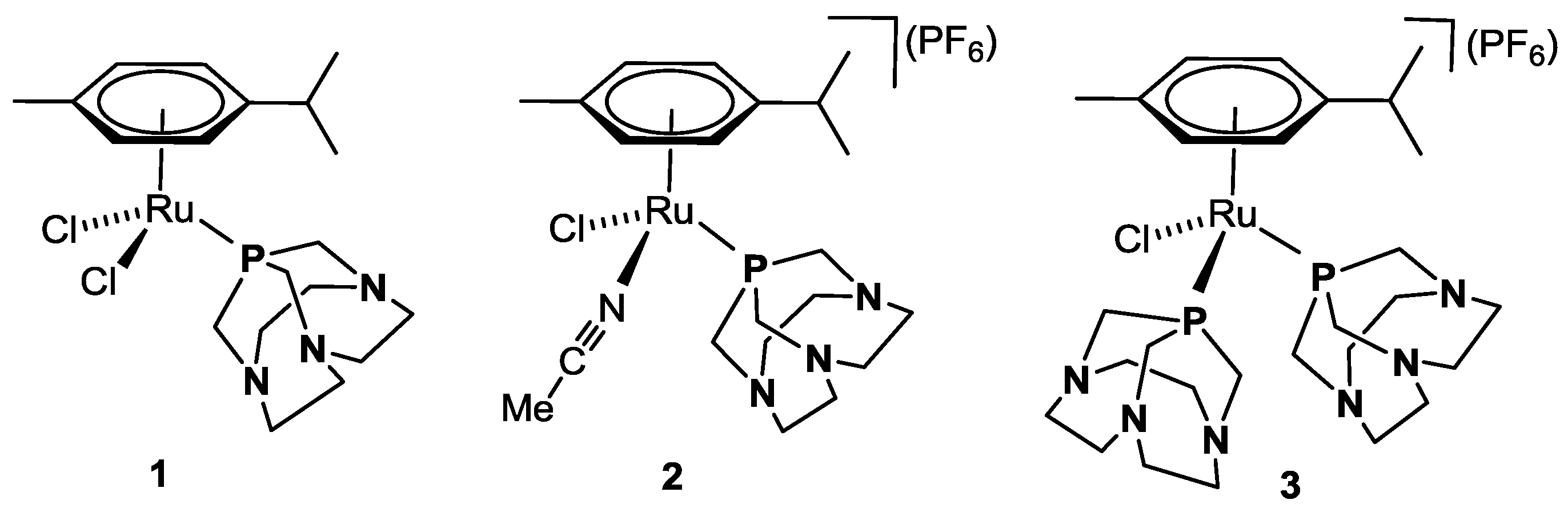
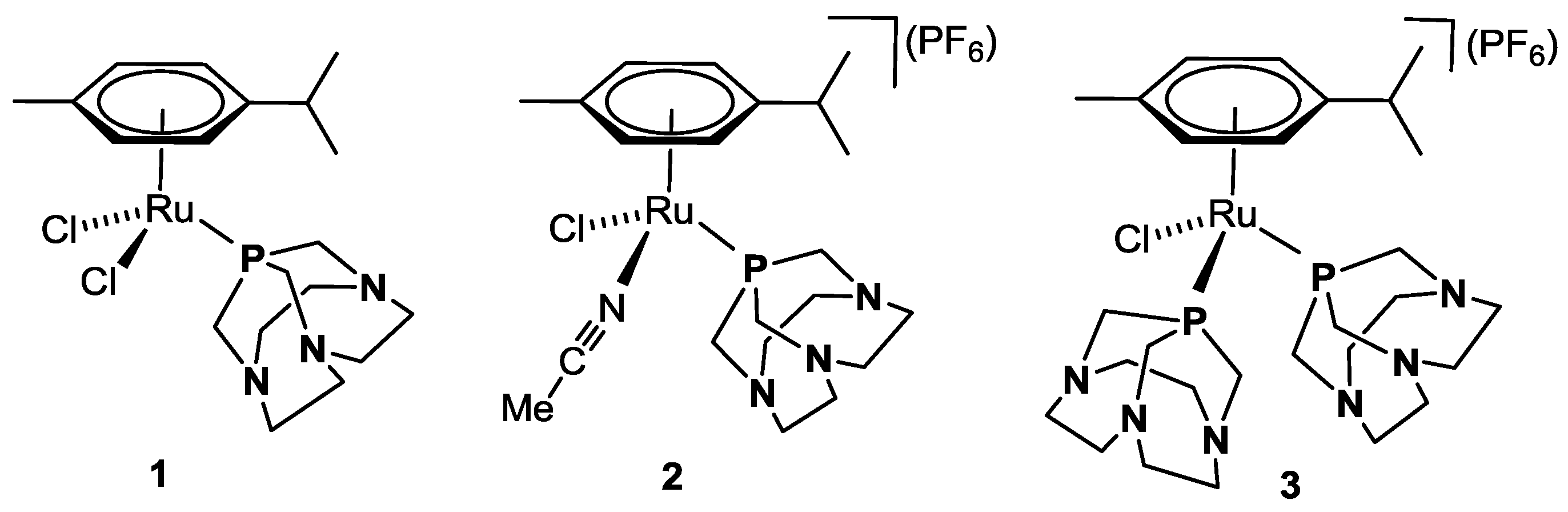
Recently, we reported on the synthesis of a new class of half-sandwich ruthenium arene complexes bearing a higher homologue of PTA, namely 1,4,7-triaza-9-phosphatricyclo[5.3.2.1]tridecane, CAP [22]. As PTA, this compound is a white, air-stable solid under standard conditions, and it is moderately water soluble with S(H2O)25 °C = 20 g L−1, about one order of magnitude lower than for PTA. Although structurally very similar to PTA, CAP has a higher cage flexibility due to the presence of two CH2 spacers between the N atoms instead of one [23,24]. The Ru(II) complex κP-[RuCl2(η6-p-cymene)(CAP)] (1, Figure 2) was obtained by reacting [RuCl2(η6-p-cymene)]2 with 2 equiv. of CAP in CH2Cl2 at room temperature. The two cationic complexes κP-[RuCl(η6-p-cymene)(MeCN)(CAP)](PF6) (2) and κP-[RuCl(η6-p-cymene)(CAP)2](PF6) (3, Figure 2) were synthesized by chloride abstraction from 1 with TlPF6 either in the presence of MeCN or a second equivalent of CAP, respectively. These compounds were tested in vitro for their cytotoxic activity against selected cancer cell lines with good results [22].
Based on the good catalytic properties shown by Ru(II) arene PTA complexes, we tested complexes 1–3 as catalysts for the reduction of α,β-unsaturated compounds by a mild TH protocol (HCOONa/H2O/MeOH), choosing BZA as model substrate. Complex 3 was also tested under the same conditions for the C=N bond hydrogenation of selected cyclic imines to produce the corresponding amines.
Finally, in order to clarify the nature of catalytically active species involved in the hydrogenation process, some preliminary mechanistic NMR experiments under pseudo-catalytic conditions were run starting from 3 and the results are presented below.
2. Results
2.1. Catalytic Transfer Hydrogenation of BZA
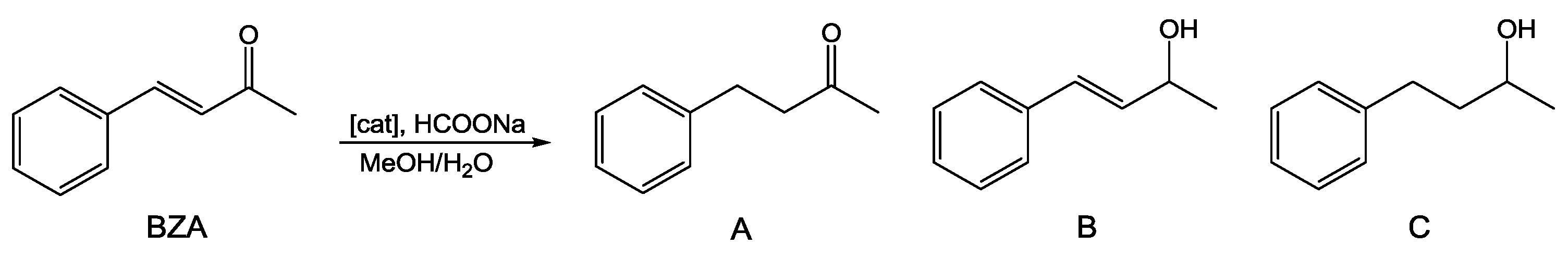
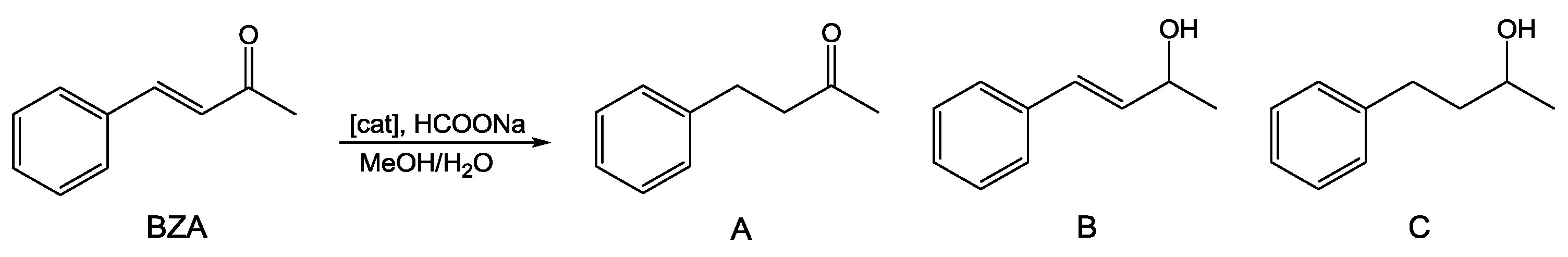
As discussed above, several Ru(II) complexes including PTA and derivatives have been used as homogeneous catalysts for hydrogenation of a wide variety of unsaturated substrates [15,16,17]. We selected benzylidene acetone (BZA) as model substrate for α,β-unsaturated ketones hydrogenation (Scheme 1) and tested compounds 1–3 as catalysts using a TH protocol involving HCOONa/H2O with a standard catalyst/substrate/formate ratio of 1:100:1000.
Sodium formate was chosen as it is one of the mildest reducing agents with large compatibility with various functional groups, also having good water solubility. To facilitate the formation of a homogeneous liquid phase and to ensure complete solubility of all reagents, the tests were carried out in a water/methanol 1:1 mixture. The results are summarized in Table 1.
At first, the catalytic run was carried out at 60 °C in the presence of complex 1 (entry 1) showing a conversion of 60% after 24 h and a ca. 3.5:1 ratio between the saturated ketone (A) and the unsaturated alcohol (B). When the temperature was raised to 80 °C (entry 3), the conversion reached 99.4% after 4 h only. A high conversion of 82.6% was also observed with 2 after 4 h reaction under the same conditions (entry 5), also in this case with high selectivity (83.4%) towards the saturated ketone. However, darkening of the reaction mixture and the appearance of a black precipitate during tests run either at 60 °C or 80 °C with both catalysts 1 and 2 suggested that these complexes were at least partially decomposing under these conditions. This was confirmed by repeating the runs using the standard Hg(0) poisoning test which resulted in the reduction of the catalytic activity. This was particularly evident for complex 2, with an important decrease of conversion after 4 h at 80 °C (51.9%, entry 6, vs. 82.6%, entry 5).
For comparison, we tested complex RAPTA-C, i.e., the PTA analogue of 1, under the same reaction conditions as in Table 1, entry 3. Also in this case a substantial drop of conversion was observed in the presence of Hg (30%, entry 10, vs. 99.5% without Hg, entry 9), indicating that a significant degree of decomposition leading to undefined catalytically active phosphine-free species was probably occurring also in the case of RAPTA-C.
Complex 3 was then tested and showed to be more stable than 1 and 2 under the catalytic conditions applied as no black precipitate was observed during the catalytic runs even after 48 h at 80 °C. The catalytic activity was only slightly reduced in the presence of Hg(0), giving 92.5% BZA conversion after 48 h at 80 °C (entry 8) compared to the 96.0% achieved in the test run without addition of mercury (entry 7). The higher stability was accompanied by a lower reaction rate to have almost complete conversion that was reached only after 48 h at 80 °C. On the other hand, the choice of this higher temperature resulted to be essential to achieve good activity with 3, as only 7.7% BZA conversion was obtained after 24 h in tests run at 60 °C.
As with 1 and 2, complex 3 also proved to be a rather chemoselective catalyst for C=C bond hydrogenation, converting BZA mainly to the saturated ketone 4-phenyl-butan-2-one (A) with a selectivity of 71.5% at 92.5% conversion after 48 h.
As an example of cyclic α,β-unsaturated ketone, 2-cyclohexen-1-one was used as substrate. In the case of complex 1, after 4 h at 80 °C using the standard conditions for TH described above, the almost complete conversion was observed. However, mainly cyclohexanol (62.9%) was obtained, demonstrating poor chemoselectivity to either C=C or C=O bond reduction. Moreover, a non-negligible amount of cyclohexanone dimethyl ketal (ca. 36.6%) was identified in solution by GC-MS at the end of the run, suggesting that C=O bond protection was competing with hydrogenation under these conditions. When the tests were performed with catalyst 3, low yields (ca. 1%) of hydrogenation products were observed after 24 h at 80 °C. The formation of the corresponding ketal was again observed (ca. 27%).
2.2. Transfer Hydrogenation of Cyclic Imines
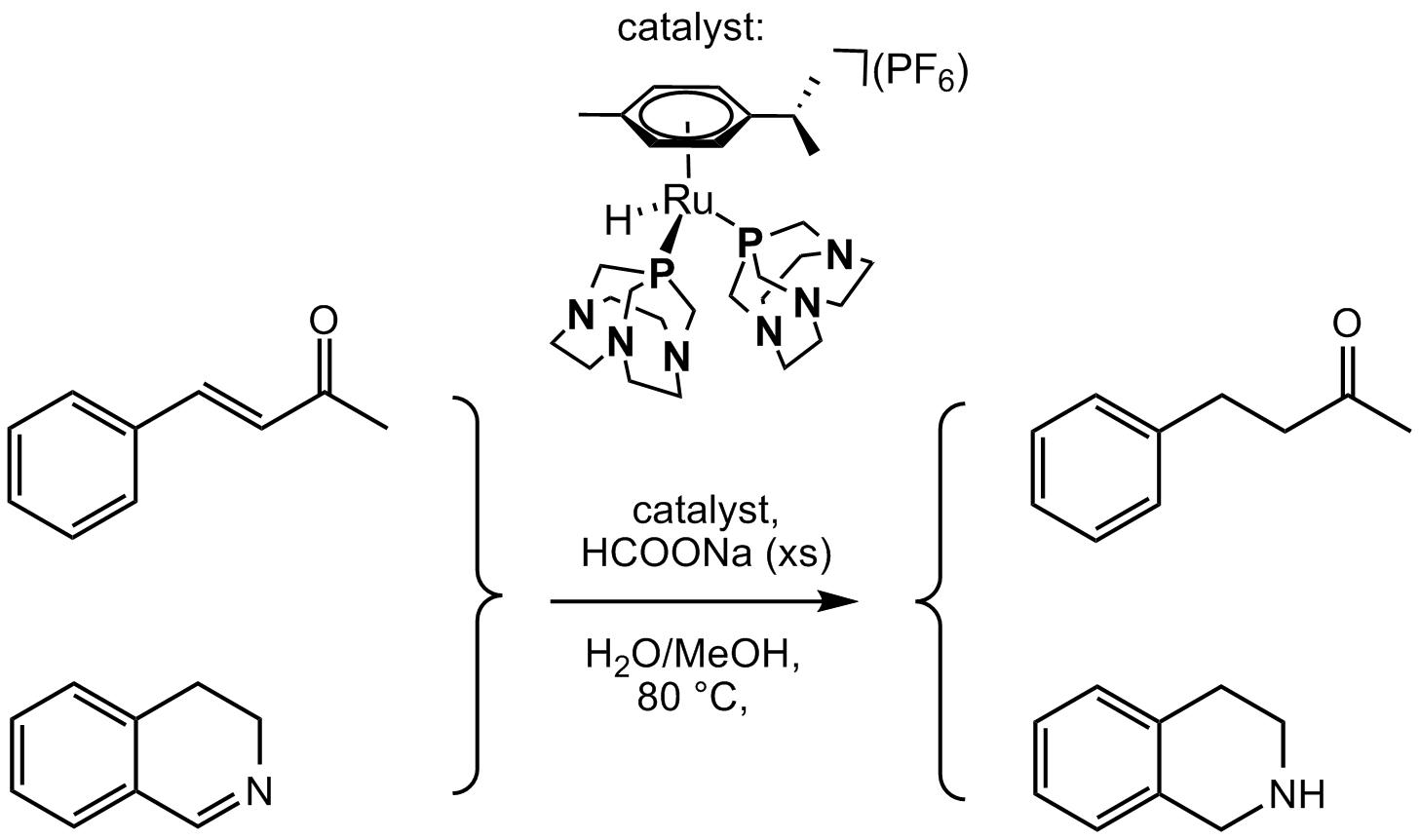
Hydrogenation of imines and, in particular, cyclic imines is a worthy synthetic tool to obtain amines, that in turn are synthetic intermediates of great importance especially in pharmaceutical industry [25]. Among Ru catalysts described in the literature for this class of homogeneously catalyzed reaction, outstanding performances were obtained for example with Noyori’s complex [RuCl(η6-p-cymene)(TsDPEN)] (TsDPEN=N-(p-toluene-sulfonyl)-1,2-diphenylethylenediamine), using a formic acid-triethylamine azeotropic mixture as reducing agent [26,27]. Examples of water soluble ruthenium–arene catalysts for TH of imines in aqueous phase with HCOONa are also known [28,29]. For cyclic imines hydrogenation, we also described an efficient system based on a heterogenized Ir(I) complex bearing PTA supported into an ion-exchange resin, using water and H2 pressure [30].
On the basis of the higher stability demonstrated by complex 3 compared to 1 and 2 under the catalytic conditions previously discussed, this compound was selected as a catalyst for tests in TH of the cyclic imines shown in Table 2.
The catalytic tests were at first performed with 3,4-dihydroisoquinoline to obtain selectively 1,2,3,4-tetrahydroisoquinoline, a moiety often present in pharmaceutical drugs [31]. As for TH of BZA, a catalyst/substrate/HCOONa ratios of 1:100:1000 were used. The first experiment was carried out in neat water, but as in the case of BZA a MeOH/H2O (1:1) mixture was required to guarantee the formation of a homogeneous phase. In fact, a conversion of only ca. 40% was obtained after 24 h at 80 °C in neat water (Table 2, entry 1). On the other hand, when the reaction was carried out in MeOH/H2O (1:1) mixture, the conversion to 1,2,3,4-tetrahydroisoquinoline reached 74.1% after 24 h and 92.5% after 48 h at 80 °C (Table 2, entry 3). As already noted for BZA, a temperature of 80 °C was required to achieve high conversions, as demonstrated by the test performed at 60 °C (Table 2, entry 2) that gave only ca. 11% conversion after 24 h.
The optimized test conditions were used with 3 and three cyclic imines, namely 2-methylquinoxaline, 5-methylquinoxaline, and quinoline (Table 2, entries 4–6). Disappointingly, these tests showed only minor or no substrate conversion even after 48 h at 80 °C.
2.3. NMR Mechanistic Studies under Pseudo-Catalytic Conditions
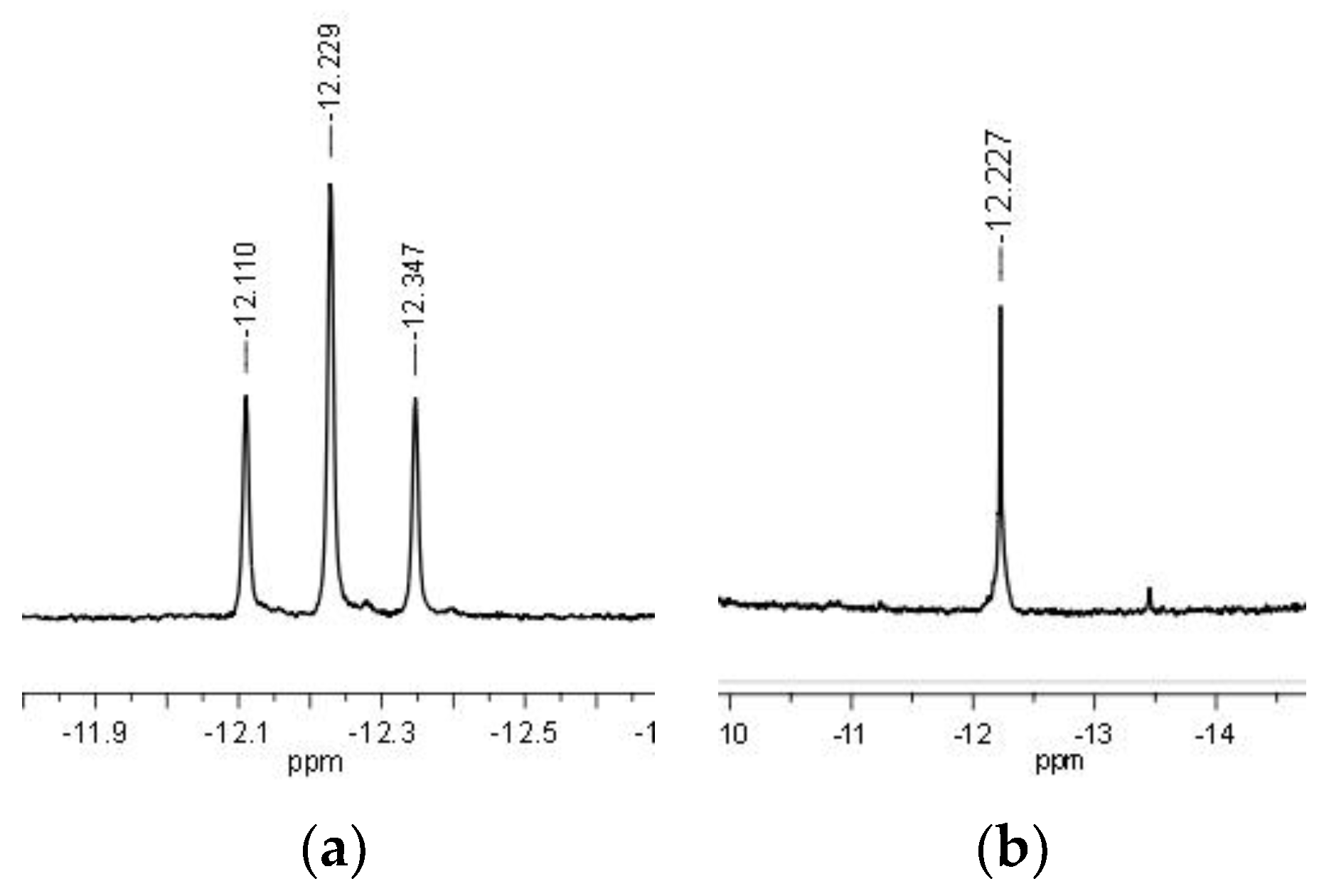
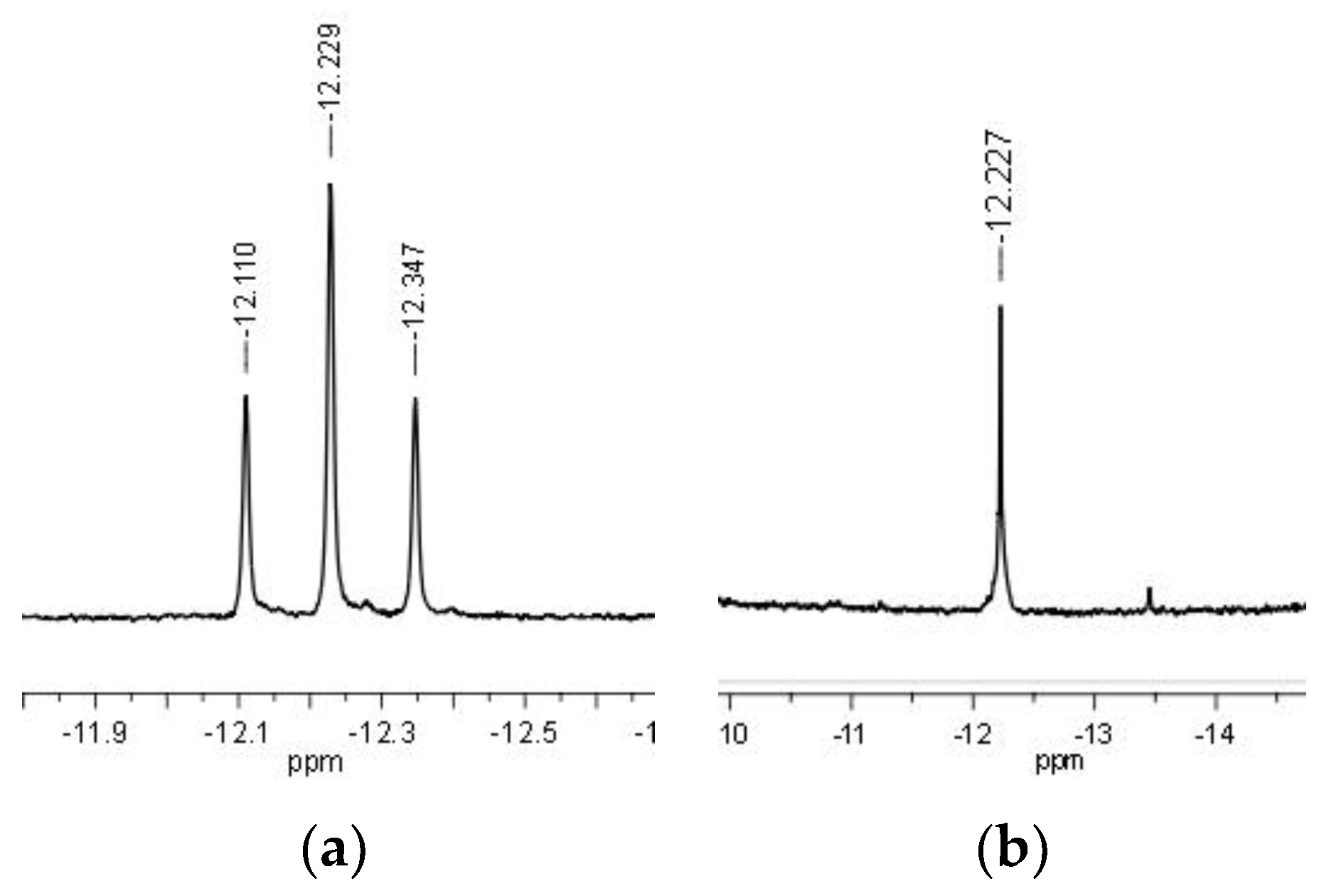
Preliminary mechanistic studies were carried out by NMR spectroscopy with complex 3 under pseudo-catalytic conditions. Firstly, 20 equiv. of HCOONa were added to compound 3 dissolved in MeOH and the resulting orange solution was immediately analyzed by 31P{1H} NMR. The spectrum showed a singlet at 52.56 ppm, corresponding to unreacted 3, and an accompanying septet at −143.76 ppm due to the PF6 anion. No changes were observed after heating the solution to 60 °C for 2 h; thus, an additional 30 equiv. of HCOONa were added to the reaction mixture and the NMR tube was left standing at 60 °C for 24 h. After this time, the 31P{1H} NMR spectrum showed complete conversion of 3 to a new species (4) characterized by a singlet at 74.34 ppm and a septet at −143.79 ppm (PF6). The 1H NMR spectrum gave a triplet in the negative region at −11.43 ppm (2JHP = 35.3 Hz), as expected for the formation of a Ru-H bond. In another test, 3 was reacted directly with 50 equiv. of HCOONa in MeOH/H2O (1:1) at 80 °C. After 3 h, in addition to the singlet at 55.77 ppm and the septet at −144.45 ppm due to 3 (slight differences in the chemical shift values derive from the use of MeOH/H2O mixture instead of pure MeOH), the singlet due to 4 was detected in the 31P{1H} NMR spectrum at 78.26 ppm, in 4:1 ratio with 3. Complete conversion of 3 to 4 was reached after leaving the tube standing at 80 °C for 17 h. Upon solvent removal in vacuo, and dissolving the obtained solid in CD2Cl2, the 31P{1H} NMR spectrum of 4 showed a singlet at 67.66 ppm and a septet at −149.50 (1JPF = 711 Hz) ppm due to PF6. The corresponding 1H NMR spectrum showed in the negative region a triplet at −12.23 ppm (2JHP = 35.5 Hz) which became a singlet in the corresponding P-decoupled 1H {31P} NMR spectrum (Figure 3).
Based on this evidence and by comparison with data reported for [RuCp(H)(PTA)2], whose hydride signal was identified by 1H NMR as a triplet at −14.36 ppm (2JHP = 36.6 Hz) in CD2Cl2 [32], we attribute the new pattern to the formation of the cationic monohydrido complex κP-[RuH(η6-p-cymene)(CAP)2](PF6) (4, Scheme 2). A confirmation of this attribution came from the independent synthesis of 4 (see Experimental Section).
A final NMR scale experiment was carried out with 3 and HCOONa (1:50 ratio) under the same conditions of solvents (MeOH/H2O 1:1) and temperature (80 °C) described above, adding 3,4-dihydroisoquinoline (5 equiv. to 3) to the mixture before heating. The pattern due to 4 was observed initially after 4 h, to become the only P-containing species after 24 h. The conversion of the imine to the corresponding amine 1,2,3,4-tetrahydroisoquinoline was confirmed by GC-MS analysis of the crude mixture after 24 h.
3. Experimental Section
3.1. Materials and Methods
All manipulations were carried out under a purified N2 atmosphere using standard Schlenk techniques unless otherwise noted. Deuterated solvents and other reagents were bought from commercial suppliers and used without further purification. Doubly distilled water was used. All solvents were distilled, dried, and degassed prior to use. Compounds κP-[RuCl2(η6-p-cymene)(CAP)] (1), κP-[RuCl(η6-p-cymene)(MeCN)(CAP)](PF6) (2), κP-[RuCl(η6-p-cymene)(CAP)2](PF6) (3) [22], RAPTA-C [33] and [RuCl2(η6-p-cymene)]2 [34] were prepared as described in the literature. 1H and 31P{1H} NMR spectra were recorded on a Bruker DRX300 spectrometer (operating at 300.13 and 121.50 MHz, respectively). The 31P spectra were normally run with proton decoupling and are reported in ppm relative to an external H3PO4 standard, with downfield positive shifts. For catalytic tests, all substrates were bought from Aldrich and used without further purification, with the exception of benzylidene acetone which was recrystallized from hot toluene. 3,4-dihydroisoquinoline and BZA together with their reduction products—namely 1,2,3,4-tetrahydroisoquinoline (95%, Sigma-Aldrich S.r.l., Milan, Italy), 4-phenyl-2-butanone (98%, Sigma-Aldrich S.r.l., Milan, Italy), and 4-phenyl-2-butanol (97%, Sigma-Aldrich S.r.l., Milan, Italy,)—were used as standards for GC analyses; on the contrary, compound 4-phenyl-3-buten-2-ol has been identified by GC-MS: m/z (%) 148 [M]+ (50), 129 (100) [35]. All GC analyses were performed on a Shimadzu GC 2010 Plus (Shimadzu _Italia S.r.l., Milan, Italy) gas chromatograph (carrier gas: He; injection mode: split at 250 °C,) equipped with flame ionization detector and a Supelco (part of Sigma-Aldrich Inc., St. Louis, MO, USA) SPBTM-1 capillary column (30 m, 0.25 mm ID, 0.25 μm film thickness). In the case of hydrogenation of BZA, the GC method started from a column temperature of 110 °C (hold time: 12 min) to increase 12 °C/min up to 240 °C (hold time: 5 min); The initial pressure was 111.8 kPa and the split ratio 80.0; the linear velocity was set at 30.0 cm/s. All products were identified at different retention times (rt) as indicated here in detail: 4-phenyl-2-butanone, rt = 8.38 min; 4-phenyl-2-butanol, rt = 9.21 min; 4-phenyl-3-buten-2-ol, rt = 12.26 min; BZA, rt = 13.54 min. In the case of 3,4-dihydroisoquinoline, the GC method started from a column temperature of 115 °C (hold time: 12 min) to increase 5 °C/min up to 250 °C (hold time: 5 min); the initial pressure was 120.8 kPa and the split ratio 80.0; the linear velocity was set at 32.0 cm/sec. Compound 3,4-dihydroisoquinoline and 1,2,3,4-tetrahydroisoquinoline showed retention times of 6.72 min and 7.52 min, respectively.
GC-MS analyses were performed on a Shimadzu GCMS-QP2010S apparatus (Shimadzu _Italia S.r.l., Milan, Italy) equipped with a flame ionization detector and a Supelco (part of Sigma-Aldrich Inc., St. Louis, MO, USA) SPBTM-1 fused silica capillary column (30 m, 0.25 mm ID, 0.25 μm film thickness), mass analyzer metal quadrupole mass filter with pre-rod, ionisation mode EI, split 250 °C, ion source temperature 200 °C, and interface temperature 280 °C. The specific analysis parameters where chosen to match those used for the corresponding GC method.
3.2. Transfer Hydrogenation Tests
All reactions were carried out in a Schlenk tube under an inert atmosphere of nitrogen using degassed solvents. In a typical experiment, HCOONa was dissolved in 2.0 mL of water and added by syringe to the solution of the catalyst in MeOH/H2O (1:1, 2.0 mL). The temperature was set and when reached, the solution of the substrate in MeOH (2.0 mL) was added to the reaction mixture. Both during the sampling and at the end of the catalytic runs, an aliquot of the reaction mixture (0.1 mL) was taken by syringe, diluted with methanol (0.4 mL), and analyzed by GC. The products of the tests were confirmed by GC-MS analyses. Each catalytic test was repeated at least twice to check for reproducibility.
3.3. NMR Scale Experiments
In a first experiment, a Schlenk tube was charged under an inert atmosphere of nitrogen with complex 3 (4.0 mg, 0.005 mmol) and HCOONa (17.0 mg, 0.25 mmol), which were dissolved in 1.0 mL of degassed solvent (MeOH or MeOH/H2O 1:1 mixture). Then, 0.7 mL of the resulting clear solution was trasferred by syringe into a NMR tube containing a C6D6 capillary for deuterium lock. In a second experiment, the same procedure was repeated, also adding solid 3,4-dihydroisoquinoline (3.3 mg, 0.025 mmol) to the solution before heating. In both cases, the solutions were first analyzed at room temperature, then the NMR tubes were placed in a oil bath set at the desired temperature, taking 1H and 31P{1H} NMR spectra at room temperature and different intervals of time to monitor the course of the reactions.
3.4. Synthesis of κP-[RuH(η6-p-cymene)(CAP)2](PF6) 4
A Schlenk tube was charged under an inert atmosphere of nitrogen with complex 3 (30 mg, 0.037 mmol) and HCOONa (125.3 mg, 1.84 mmol), adding 8 mL of degassed MeOH/H2O (1:1 mixture). The resulting orange mixture was left stirring at 80 °C for 28 h. After this time, the solvent was removed in vacuo and the solid residue dissolved in CH2Cl2. After filtration, to the resulting solution, diethylether was added to precipitate the product as a light orange solid, that was finally dried under vacuum (isolated yield ca. 50%).
Analysis: 1H NMR, negative region: δ(ppm, CD2Cl2) −12.23 ppm (2JHP = 35.5 Hz); 31P{1H} NMR: δ(ppm, CD2Cl2) 67.51 (s); −149.53 (sept, 1JPF = 711.1 Hz, PF6).
4. Conclusions
The half-sandwich ruthenium(II) arene complexes 1–3 bearing the water soluble ligand CAP were tested in homogeneous catalytic transfer hydrogenations of some selected unsaturated substrates. Whereas the monophosphine complexes 1 and 2 showed poor overall stability under the reaction conditions applied for the tests, the bisphosphine complex 3 proved to be more stable and active in the hydrogenation of benzylidene acetone and 3,4-dihydroisoquinoline using a very mild reduction protocol such as transfer hydrogenation with sodium formate. NMR studies in solution performed with 3 under pseudo-catalytic conditions clearly showed the formation of the monohydride derivative 4, which is likely the active form of the catalyst during the reaction.
Acknowledgments
The Italian Ministry for Education and Research (MIUR) is kindly acknowledged for financial support through Project PRIN 2015 (grant number 20154X9ATP).
Author Contributions
A.G. and L.G. conceived and designed the experiments; A.G. performed the experiments and analyzed the data; M.P. contributed with all the authors to write and approve the final version of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Morris, R.H. Ruthenium and Osmium. In The Handbook of Homogeneous Hydrogenation; de Vries, J.G., Elsevier, C.J., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; Chapter 3; pp. 45–70. ISBN 978-3-527-31161-3. [Google Scholar]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Evans, D.; Osborn, J.A.; Jardine, F.H.; Wilkinson, G. Homogeneous Hydrogenation and Hydroformylation using Ruthenium Complexes. Nature 1965, 208, 1203–1204. [Google Scholar] [CrossRef]
- Blaser, H.U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Selective Hydrogenation for Fine Chemicals: Recent Trends and New Developments. Adv. Synth. Catal. 2003, 345, 103–151. [Google Scholar] [CrossRef]
- Zanotti-Gerosa, A.; Hems, W.; Groarke, M.; Hancock, F. Ruthenium-Catalysed Asymmetric Reduction of Ketones. Platin. Met. Rev. 2005, 49, 158–165. [Google Scholar] [CrossRef]
- Clarke, M.L.; Dìaz-Valenzuela, M.B.; Slawin, A.M.Z. Hydrogenation of Aldehydes, Esters, Imines, and Ketones Catalyzed by a Ruthenium Complex of a Chiral Tridentate Ligand. Organometallics 2007, 26, 16–19. [Google Scholar] [CrossRef]
- James, B.R. Synthesis of chiral amines catalyzed homogeneously by metal complexes. Catal. Today 1997, 37, 209–221. [Google Scholar] [CrossRef]
- Samec, J.S.M.; Bäckvall, J.E. Ruthenium-Catalyzed Transfer Hydrogenation of Imines by Propan-2-ol in Benzene. Chem. Eur. J. 2002, 8, 2955–2961. [Google Scholar] [CrossRef]
- Foubelo, F.; Nájera, C.; Yus, M. Catalytic asymmetric transfer hydrogenation of ketones: Recent advances. Tetrahedron Asymmetry 2015, 26, 769–790. [Google Scholar] [CrossRef]
- Ito, J.-I.; Nishiyama, H. Recent topics of transfer hydrogenation. Tetrahedron Lett. 2014, 55, 3133–3146. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, K.H. Hydrophilic ligands and their application in aqueous-phase metal-catalyzed reactions. Chem. Rev. 2009, 109, 643–710. [Google Scholar] [CrossRef] [PubMed]
- James, B.R.; Lorenzini, F. Developments in the chemistry of tris(hydroxymethyl)phosphine. Coord. Chem. Rev. 2010, 254, 420–430. [Google Scholar] [CrossRef]
- Phillips, A.D.; Gonsalvi, L.; Romerosa, A.; Vizza, F.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phospaadamantane (PTA). Transition metal complexes and related catalytic, medicinal and photoluminescent applications. Coord. Chem. Rev. 2004, 248, 955–993. [Google Scholar] [CrossRef]
- Bravo, J.; Bolaño, S.; Gonsalvi, L.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phospaadamantane (PTA) and derivatives. Part II. The quest for tailored ligands, complexes and related applications. Coord. Chem. Rev. 2010, 254, 555–607. [Google Scholar] [CrossRef]
- Guerriero, A.; Peruzzini, M.; Gonsalvi, L. Coordination chemistry of 1,3,5-triaza-7-phospaadamantane (PTA) and derivatives. Part III. Variations on a theme: Novel architectures, materials and applications. Coord. Chem. Rev. 2018, 355, 328–361. [Google Scholar] [CrossRef]
- Akbayeva, D.N.; Gonsalvi, L.; Oberhauser, W.; Peruzzini, M.; Vizza, F.; Brüggeller, P.; Romerosa, A.; Sava, G.; Bergamo, A. Synthesis, catalytic properties and biological activity of new water soluble ruthenium cyclopentadienyl PTA complexes [(C5R5)RuCl(PTA)2] (R= H, Me; PTA= 1,3,5-triaza-7-phosphaadamantane). Chem. Commun. 2003, 264–265. [Google Scholar] [CrossRef]
- Bolaño, S.; Gonsalvi, L.; Zanobini, F.; Vizza, F.; Bertolasi, V.; Romerosa, A.; Peruzzini, M. Water soluble ruthenium cyclopentadienyl and aminocyclopentadienyl PTA complexes as catalysts for selective hydrogenation of α,β-unsaturated substrates (PTA = 1,3,5-triaza-7-phosphaadamantane). J. Mol. Catal. A Chem. 2004, 224, 61–70. [Google Scholar] [CrossRef]
- Dyson, P.J.; Ellis, D.J.; Laurenczy, G. Minor Modifications to the Ligands Surrounding a Ruthenium Complex Lead to Major Differences in the Way in which they Catalyse the Hydrogenation of Arenes. Adv. Synth. Catal. 2003, 345, 211–215. [Google Scholar] [CrossRef]
- Krogstad, D.A.; Guerriero, A.; Ienco, A.; Manca, G.; Peruzzini, M.; Reginato, G.; Gonsalvi, L. Imidazolyl-PTA Derivatives as Water-Soluble (P,N) Ligands for Ruthenium-Catalyzed Hydrogenations. Organometallics 2011, 30, 6292–6302. [Google Scholar] [CrossRef]
- Guerriero, A.; Oberhauser, W.; Riedel, T.; Peruzzini, M.; Dyson, P.J.; Gonsalvi, L. New Class of Half-Sandwich Ruthenium(II) Arene Complexes Bearing the Water-Soluble CAP Ligand as an in Vitro Anticancer Agent. Inorg. Chem. 2017, 56, 5514–5518. [Google Scholar] [CrossRef] [PubMed]
- Britvin, S.N.; Lotnyk, A. Water-Soluble Phosphine Capable of Dissolving Elemental Gold: The Missing Link between 1,3,5-triaza-7-phosphaadamantane (PTA) and Verkade’s Ephemeral Ligand. J. Am. Chem. Soc. 2015, 137, 5526–5535. [Google Scholar] [CrossRef] [PubMed]
- Britvin, S.N.; Rumyantsev, A.M.; Zobnina, A.E.; Padkina, M.V. Between Adamantane and Atrane: Intrabridgehead Interactions in the Cage-Like Phosphane Related to a Novel Tris(homoadamantane) Ring System. Chem. Eur. J. 2016, 22, 14227–14235. [Google Scholar] [CrossRef] [PubMed]
- Fleury-Brégeot, N.; de la Fuente, V.; Castillón, S.; Claver, C. Highlights of Transition Metal-Catalyzed Asymmetric Hydrogenation of Imines. ChemCatChem 2010, 2, 1346–1371. [Google Scholar] [CrossRef]
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric Transfer Hydrogenation of Aromatic Ketones Catalyzed by Chiral Ruthenium(II) Complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Asymmetric Transfer Hydrogenation of Imines. J. Am. Chem. Soc. 1996, 118, 4916–4917. [Google Scholar] [CrossRef]
- Wu, J.; Wang, F.; Ma, Y.; Cui, X.; Cun, L.; Zhu, J.; Deng, J.; Yu, B. Asymmetric transfer hydrogenation of imines and iminiums catalyzed by a water-soluble catalyst in water. Chem. Commun. 2006, 1766–1768. [Google Scholar] [CrossRef] [PubMed]
- Canivet, J.; Süss-Fink, G. Water-soluble arene ruthenium catalysts containing sulfonated diamine ligands for asymmetric transfer hydrogenation of α-aryl ketones and imines in aqueous solution. Green Chem. 2007, 9, 391–397. [Google Scholar] [CrossRef]
- Barbaro, P.; Gonsalvi, L.; Guerriero, A.; Liguori, F. Facile heterogeneous catalytic hydrogenation of C=N and C=O bonds in neat water: Anchoring of water-soluble metal complexes onto ion-exchange resins. Green Chem. 2012, 14, 3211–3219. [Google Scholar] [CrossRef]
- Antkiewicz-Michaluk, L.; Wąsik, A.; Michaluk, J. 1-Methyl-1,2,3,4-Tetrahydroisoquinoline, an Endogenous Amine with Unexpected Mechanism of Action: New Vistas of Therapeutic Application. Neurotox. Res. 2014, 25, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.J.; Mebi, C.A. Aqueous organometallic chemistry: Synthesis, structure and reactivity of the water-soluble metal hydride CpRu(PTA)2H. Organometallics 2004, 23, 5317–5323. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.L. [Ru(η6-p-cymene)Cl2(pta)] (pta = 1,3,5-triaza-7-phosphatricyclo[3.3.1.1]-decane): A Water Soluble Compound That Exhibits pH Dependent DNA Binding Providing Selectivity for Diseased Cells. Chem. Commun. 2001, 2, 1396–1397. [Google Scholar] [CrossRef]
- Bennett, M.A.; Huang, T.N.; Matheson, T.W.; Smith, A.K. (η6-Hexamethylbenzene)Ruthenium Complexes. Inorg. Synth. 1982, 21, 74–78. [Google Scholar] [CrossRef]
- Xu, W.; Zhou, Y.; Wang, R.; Wu, G.; Chen, P. Lithium amidoborane, a highly chemoselective reagent for the reduction of α,β-unsaturated ketones to allylic alcohols. Org. Biomol. Chem. 2012, 10, 367–371. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
PTA ligand and Ru(II)-arene complexes bearing ‘upper rim’ PTA derivatives.
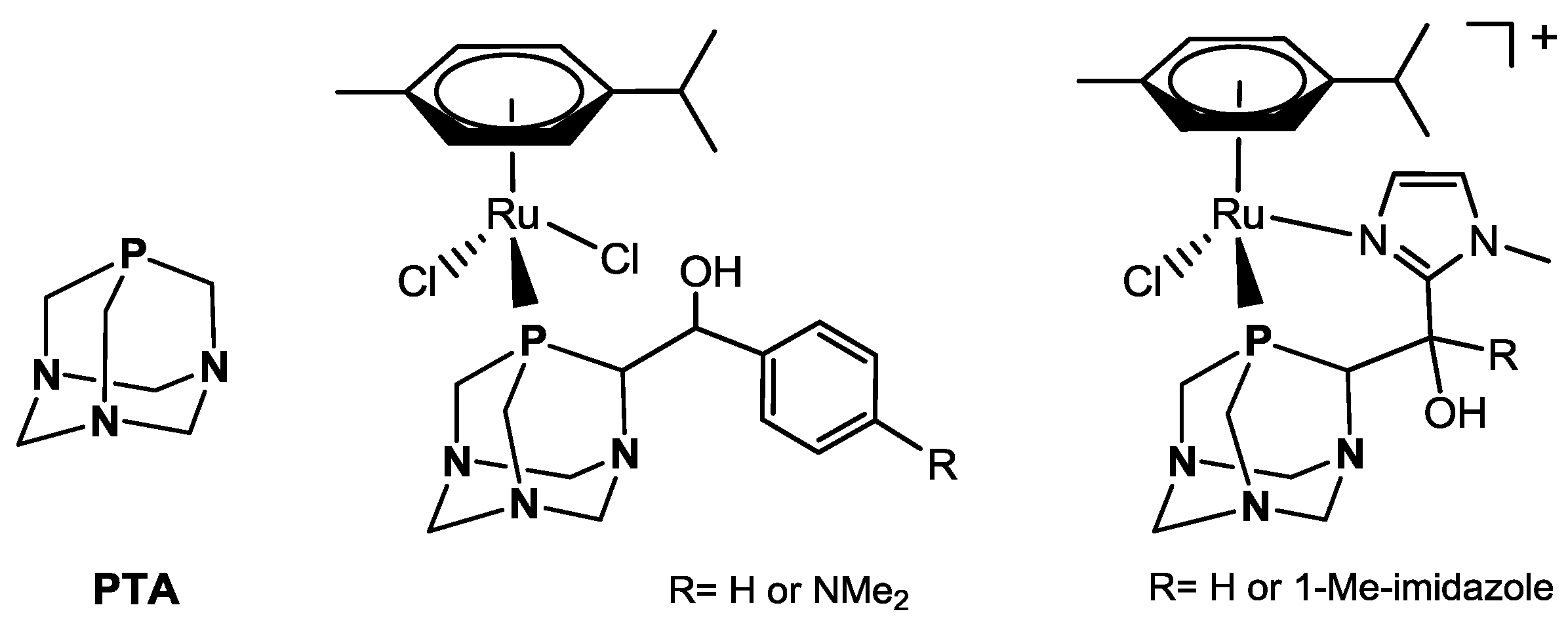
Figure 2.
Ru(II)-arene complexes 1–3 bearing ligand CAP [22].
Figure 2.
Ru(II)-arene complexes 1–3 bearing ligand CAP [22].
Scheme 1.
Hydrogenation of benzylidene acetone (BZA).
Scheme 2.
Conversion of 3 to 4 by reaction with HCOONa in H2O/MeOH.
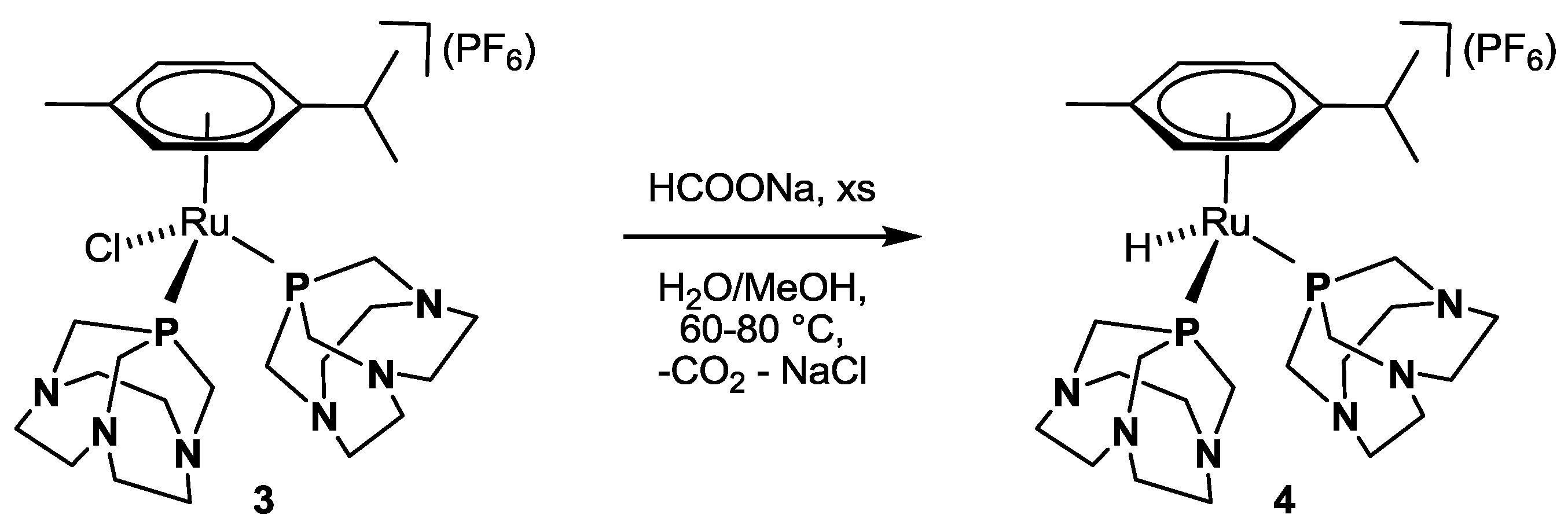
Figure 3.
1H (a) and 1H {31P} NMR (b) spectra (negative region only, CD2Cl2) showing the change from triplet to singlet for the Ru-H signal in 4.
Figure 3.
1H (a) and 1H {31P} NMR (b) spectra (negative region only, CD2Cl2) showing the change from triplet to singlet for the Ru-H signal in 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
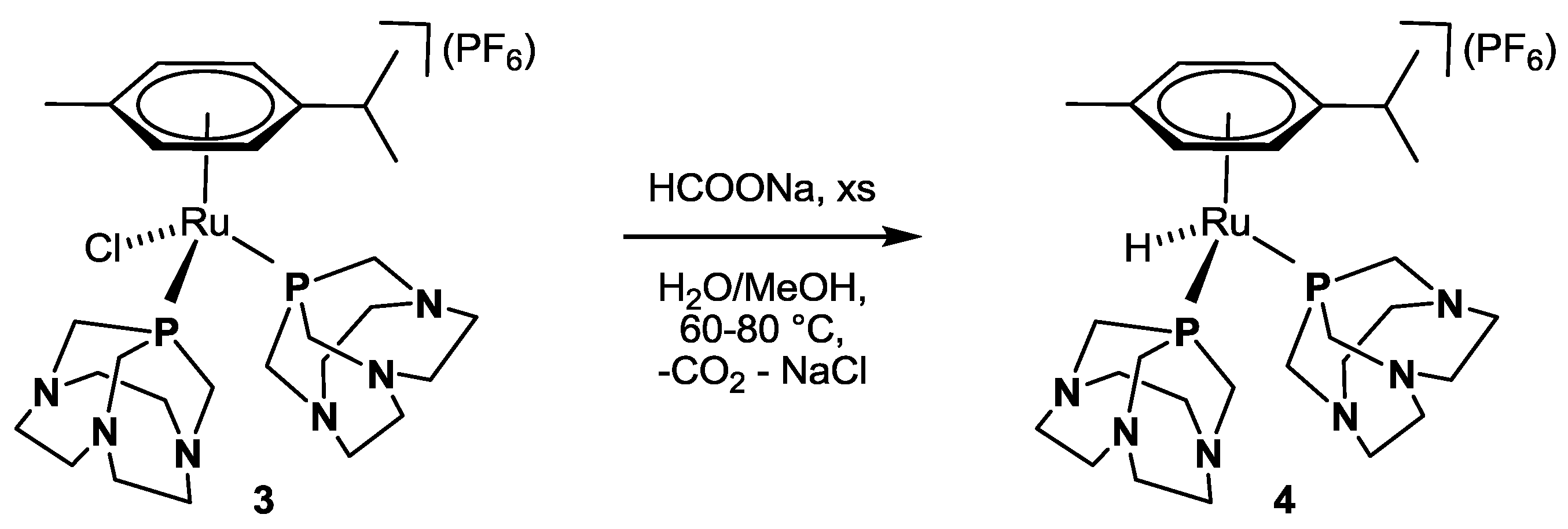{kind=link}
Table 1.
Catalytic transfer hydrogenation of benzylidene acetone (BZA) with complexes 1–3 a.
| Entry | Catalyst | T (°C) | % Conv. | Time (h) | Yield A (%) b | Yield B (%) b | Yield C (%) b |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 60 | 60.0 | 24 | 45.3 | 13.0 | 1.7 |
| 2 c | 1 | 60 | 18.7 | 24 | 14.2 | 4.4 | 0.1 |
| 3 | 1 | 80 | 99.4 | 4 | 82.0 | 5.2 | 12.2 |
| 4 c | 1 | 80 | 76.3 | 4 | 61.7 | 12.8 | 1.8 |
| 5 | 2 | 80 | 82.6 | 4 | 68.9 | 11.5 | 2.2 |
| 6 c | 2 | 80 | 51.9 | 4 | 40.4 | 10.8 | 0.7 |
| 7 | 3 | 80 | 71.8 | 24 | 52.3 | 15.8 | 3.7 |
| 96.0 | 48 | 68.7 | 13.5 | 13.8 | |||
| 8 c | 3 | 80 | 66.5 | 24 | 46.6 | 17.8 | 2.1 |
| 92.5 | 48 | 73.5 | 13.0 | 6.0 | |||
| 9 | RAPTA-C | 80 | 99.5 | 24 | 93.2 | 3.2 | 3.1 |
| 10 c | RAPTA-C | 80 | 30.0 | 24 | 23.8 | 6.0 | 0.2 |
a General conditions: catalyst, 9.8 × 10−3 mmol; BZA, 0.98 mmol; HCOONa, 9.8 mmol; MeOH:H2O (1:/1), 6 mL. b GC values based on pure samples, except B (see Experimental Section): A = 4-phenyl-2-butanone; B = 4-phenyl-3-buten-2-ol; C = 4-phenyl-2-butanol. c Hg(0) added (one drop).
Table 2.
Catalytic transfer hydrogenation of cyclic imines with 3 a.
| Entry | T (°C) | Solvent | Substrate | Product b | Yield c | Time (h) |
|---|---|---|---|---|---|---|
| 1 | 80 | H2O |  |  | 39.9 | 24 |
| 3,4-dihydroisoquinoline | 1,2,3,4-tetrahydroisoquinoline | |||||
| 2 | 60 | MeOH/H2O 1:1 |  |  | 10.9 | 24 |
| 3 | 80 | MeOH/H2O 1:1 |  |  | 74.1 92.5 | 24 48 |
| 4 | 80 | MeOH/H2O 1:1 |  |  | 0.4 | 24 |
| 2-methylquinoxaline | 2-methyl-1,2,3,4-tetrahydroquinoxaline | |||||
| 5 | 80 | MeOH/H2O 1:1 |  |  | 0.0 | 48 |
| 5-methylquinoxaline | 5-methyl-1,2,3,4-tetrahydroquinoxaline | |||||
| 6 | 80 | MeOH/H2O 1:1 |  |  | 1.5 3.0 | 24 48 |
| quinoline | 1,2,3,4-tetrahydroquinoline |
a General conditions: catalyst, 1.0 × 10−2 mmol; substrate, 1.0 mmol; HCOONa, 10.0 mmol; solvent, 6 mL. b Products confirmed by GC and GC-MS analyses. c GC values based on pure samples where available.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Guerriero, A.; Peruzzini, M.; Gonsalvi, L. Ruthenium(II)-Arene Complexes of the Water-Soluble Ligand CAP as Catalysts for Homogeneous Transfer Hydrogenations in Aqueous Phase. Catalysts 2018, 8, 88. https://doi.org/10.3390/catal8020088
AMA Style
Guerriero A, Peruzzini M, Gonsalvi L. Ruthenium(II)-Arene Complexes of the Water-Soluble Ligand CAP as Catalysts for Homogeneous Transfer Hydrogenations in Aqueous Phase. Catalysts. 2018; 8(2):88. https://doi.org/10.3390/catal8020088
Chicago/Turabian StyleGuerriero, Antonella, Maurizio Peruzzini, and Luca Gonsalvi. 2018. "Ruthenium(II)-Arene Complexes of the Water-Soluble Ligand CAP as Catalysts for Homogeneous Transfer Hydrogenations in Aqueous Phase" Catalysts 8, no. 2: 88. https://doi.org/10.3390/catal8020088
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.