A Biorefinery Cascade Conversion of Hemicellulose-Free Eucalyptus Globulus Wood: Production of Concentrated Levulinic Acid Solutions for γ-Valerolactone Sustainable Preparation

 , , and
, , and
Abstract
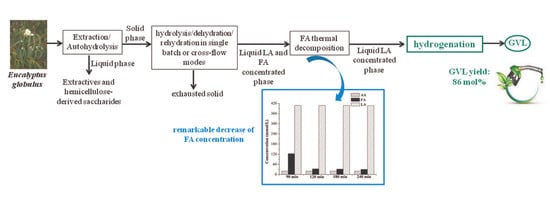
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Autohydrolysis of Eucalyptus Globulus Wood
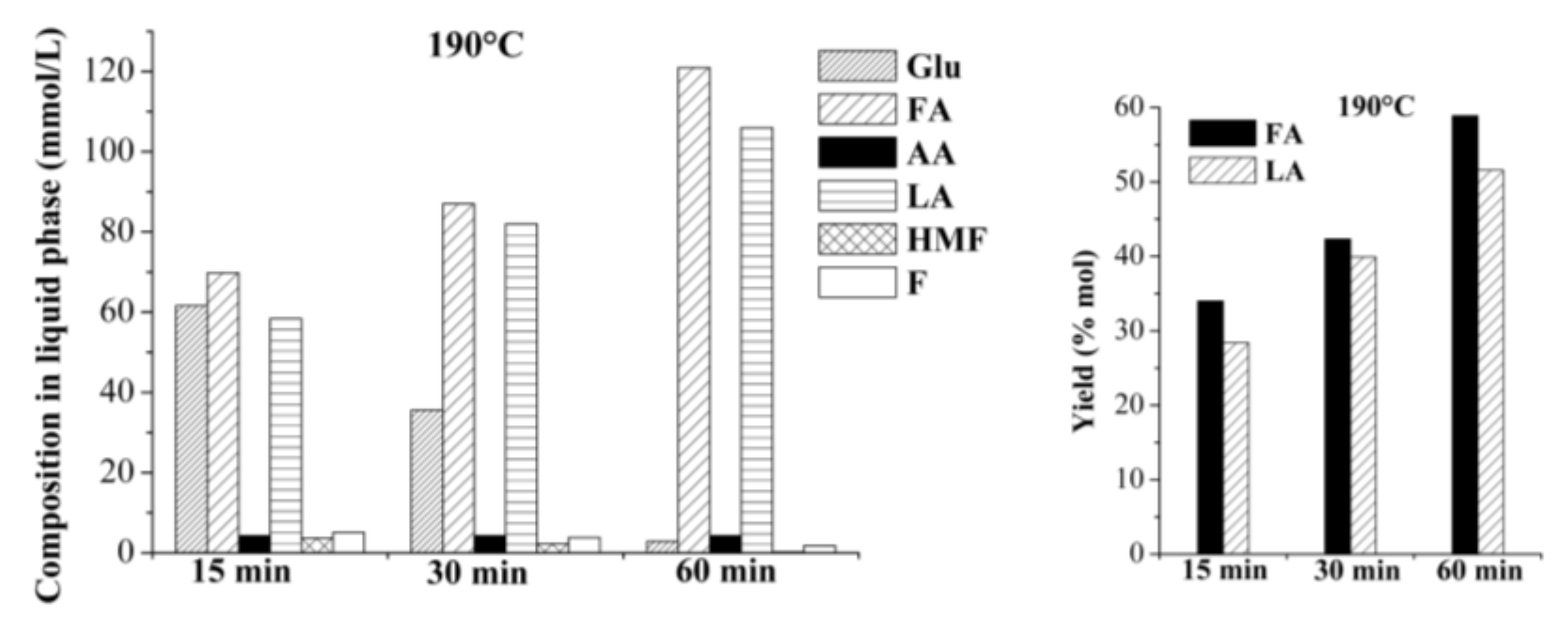
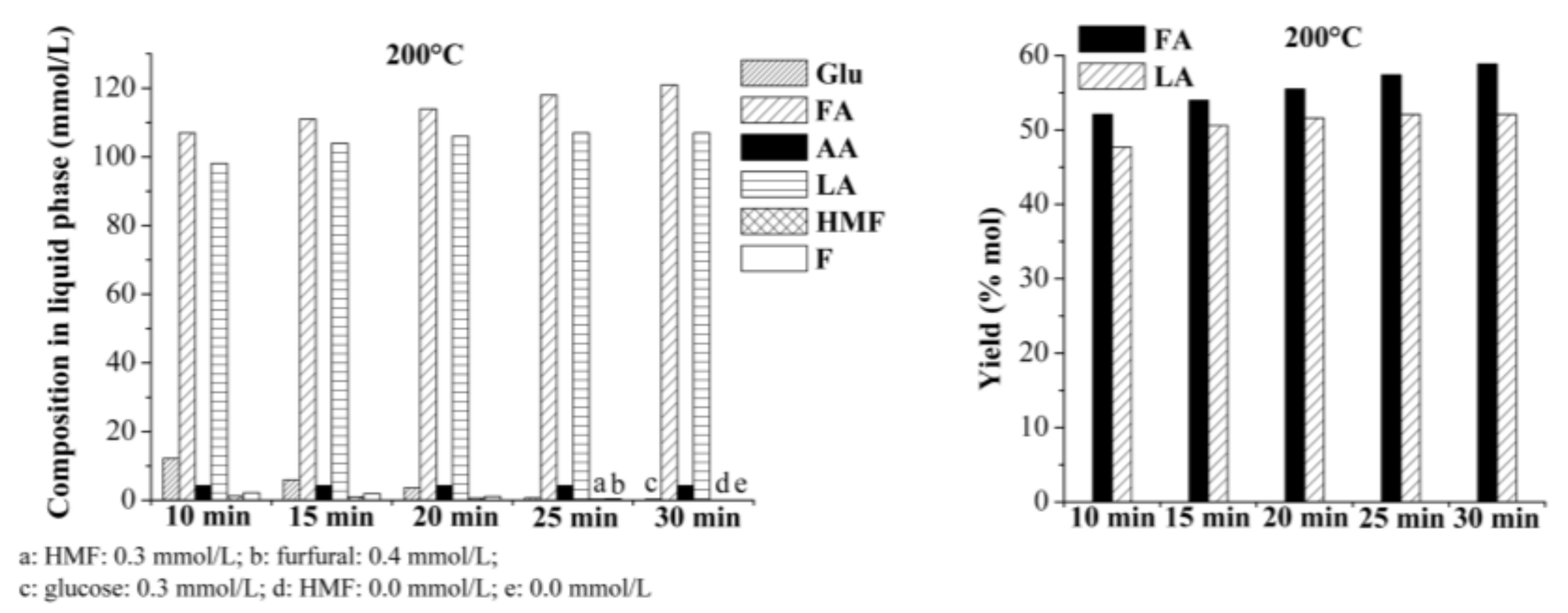
2.2. Production of Levulinic Acid (LA) and Formic Acid (FA) from Auto-Hydrolyzed Wood
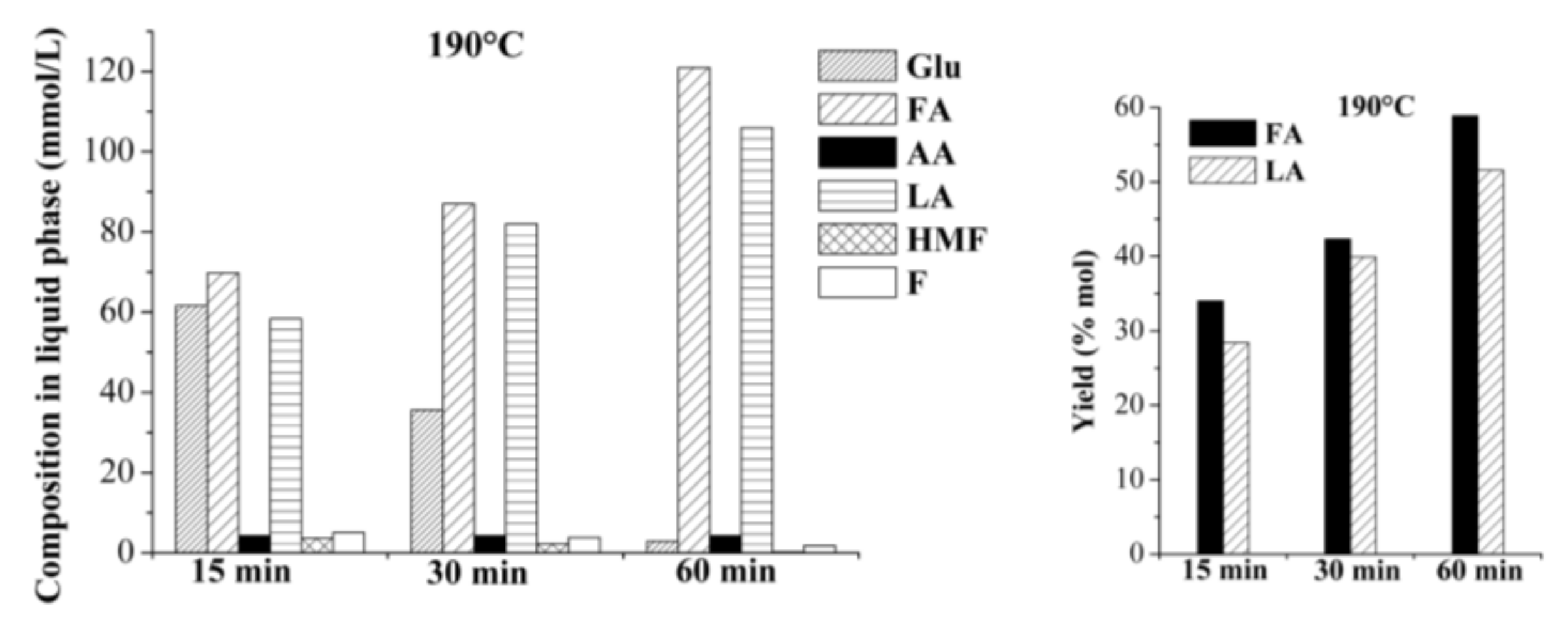
2.2.1. Single-Batch Experiments
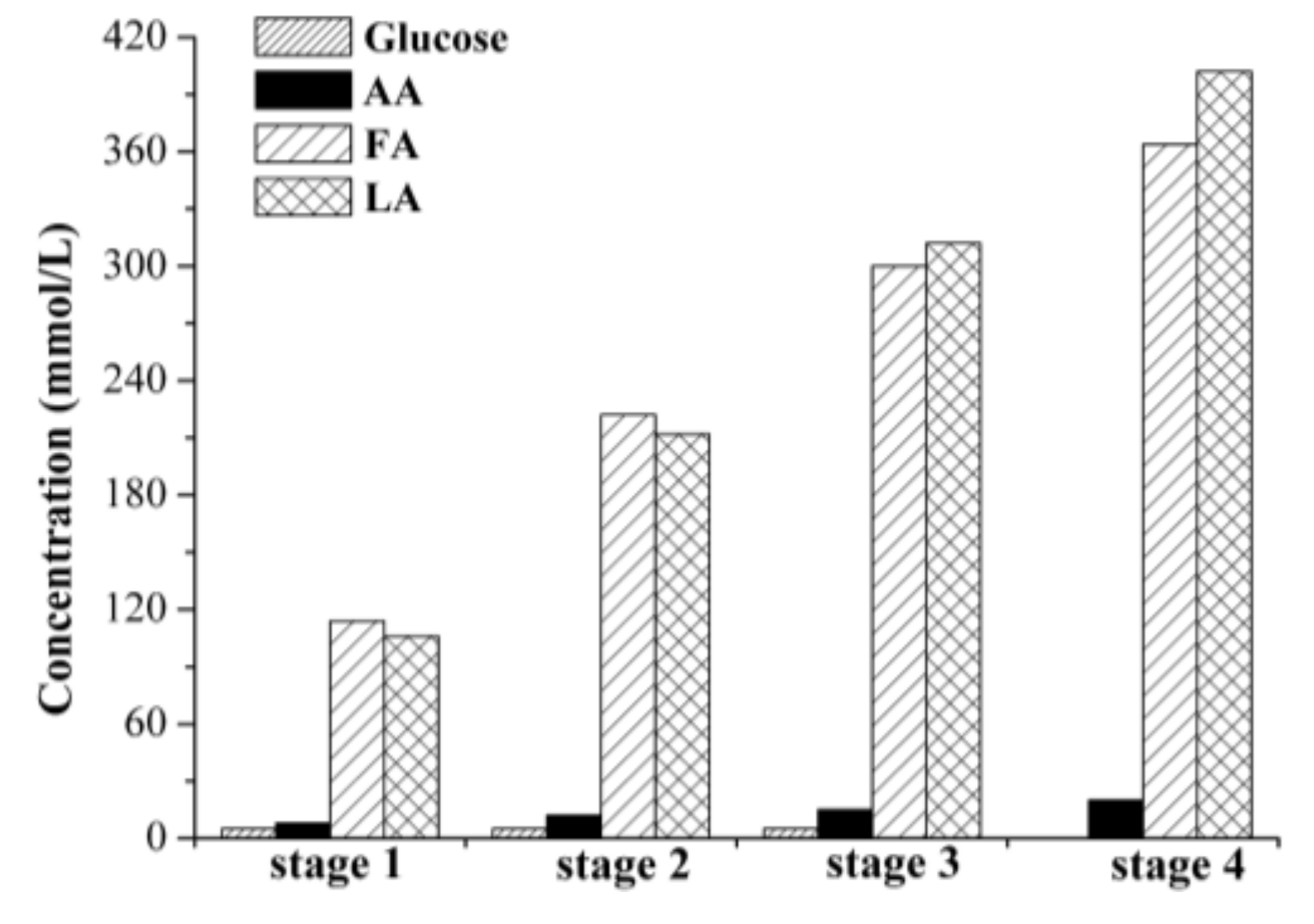
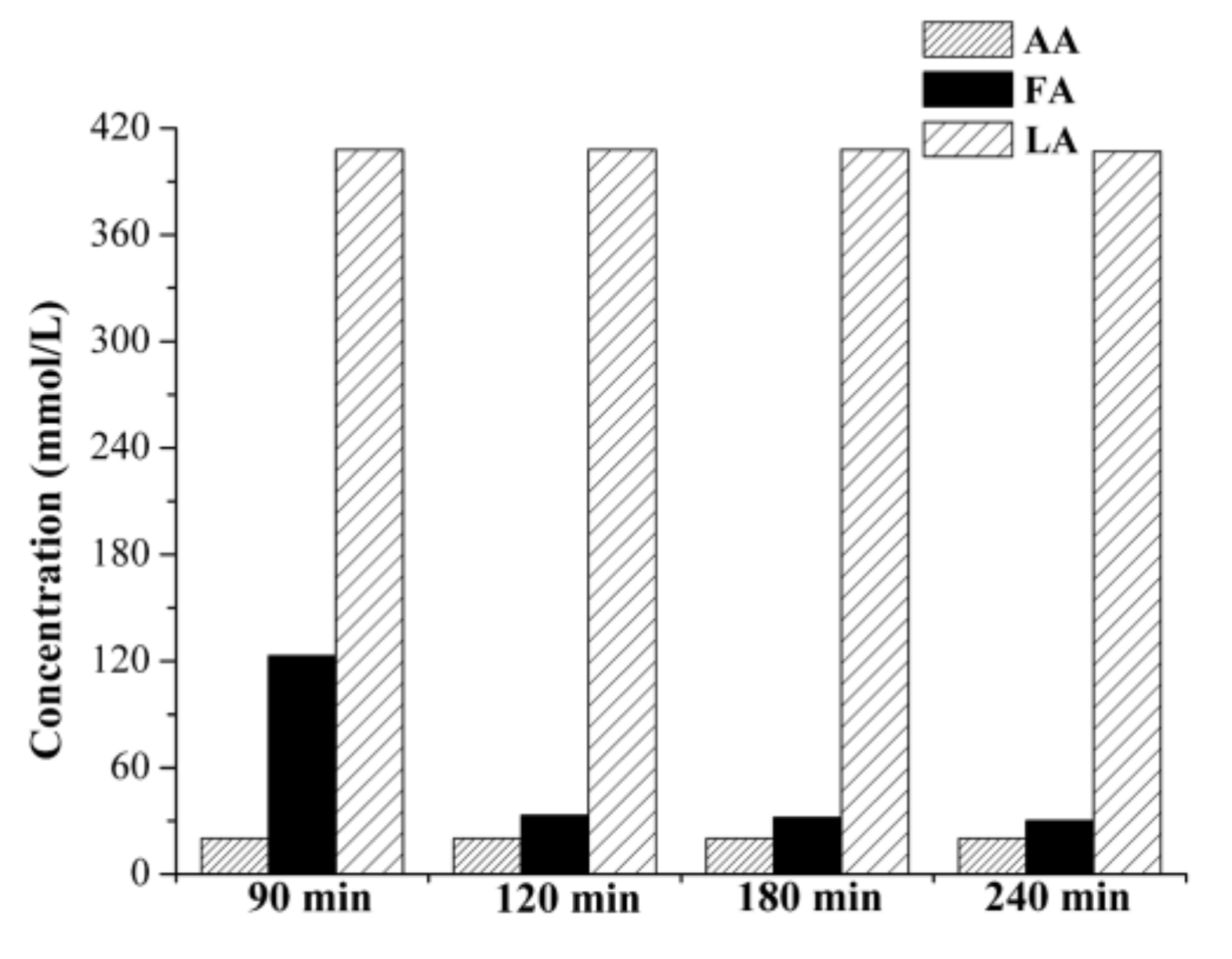
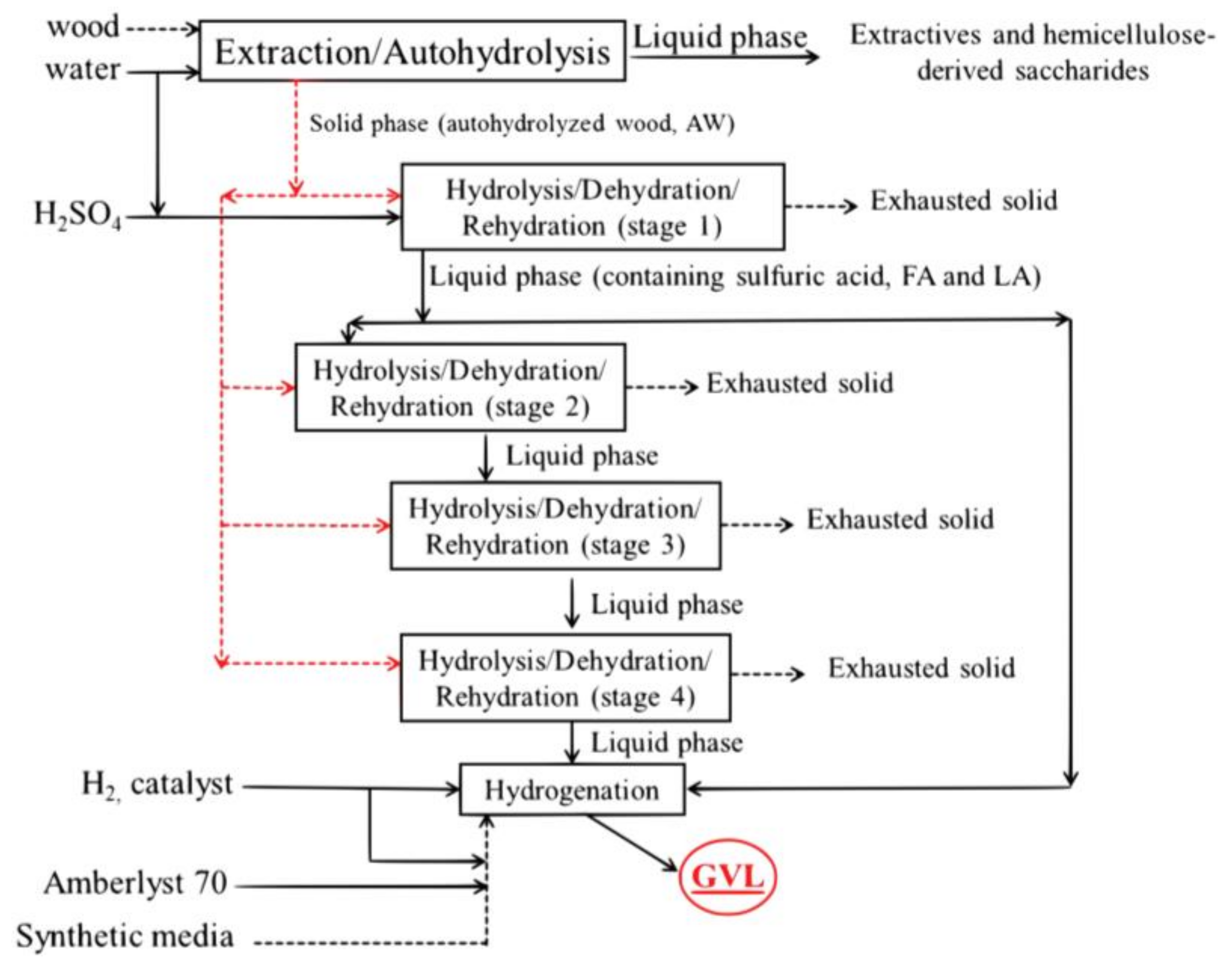
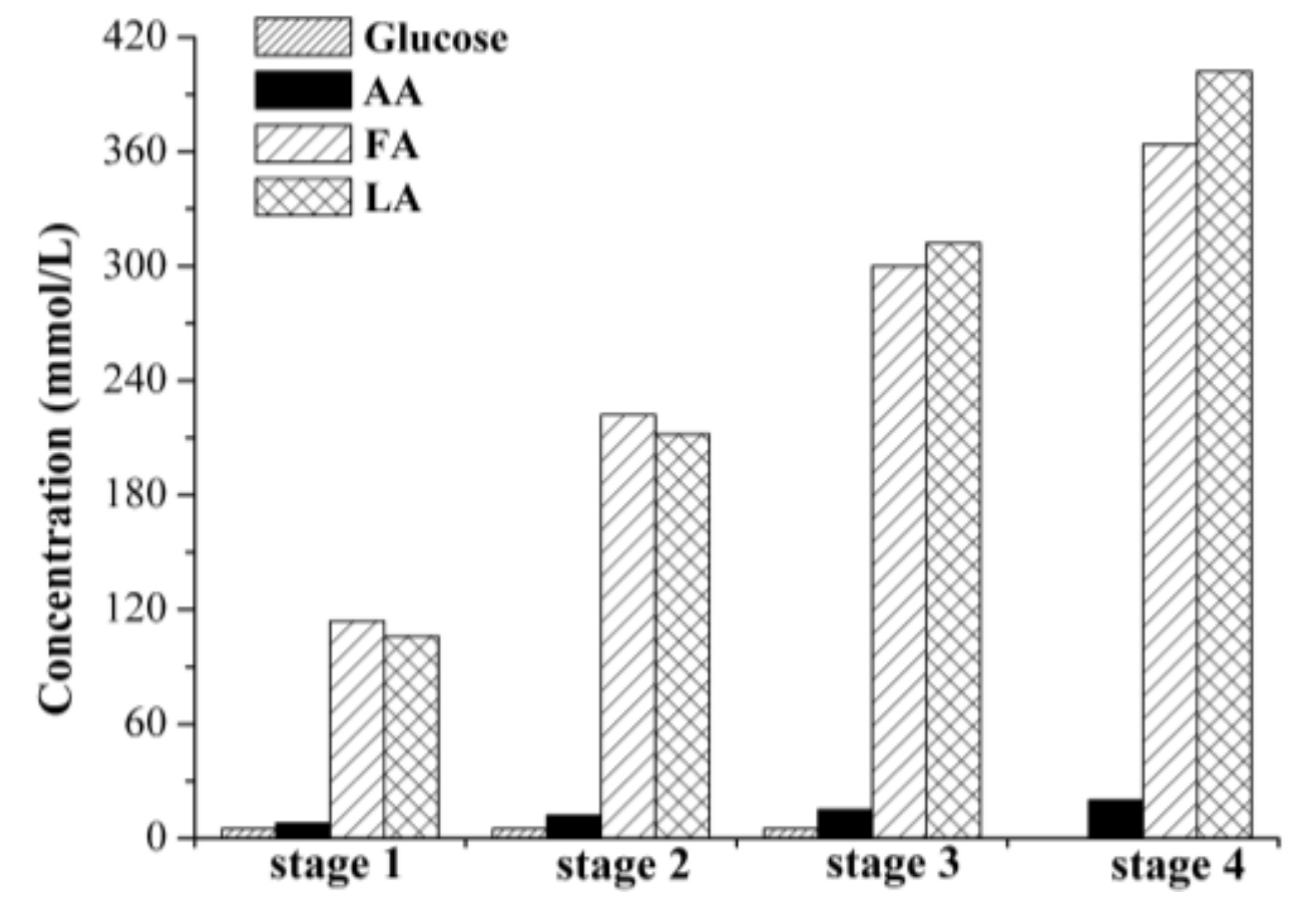
2.2.2. Cross-Flow Batch Experiments
2.3. γ-Valerolactone (GVL) Production
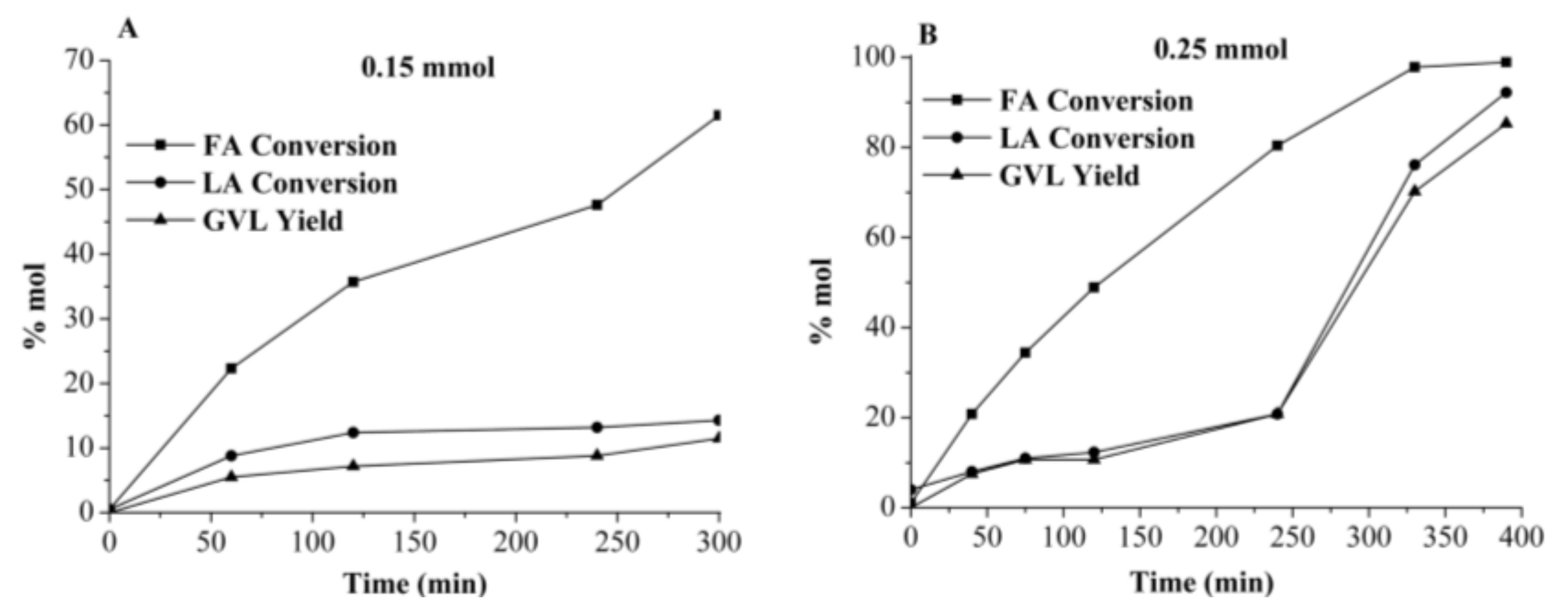
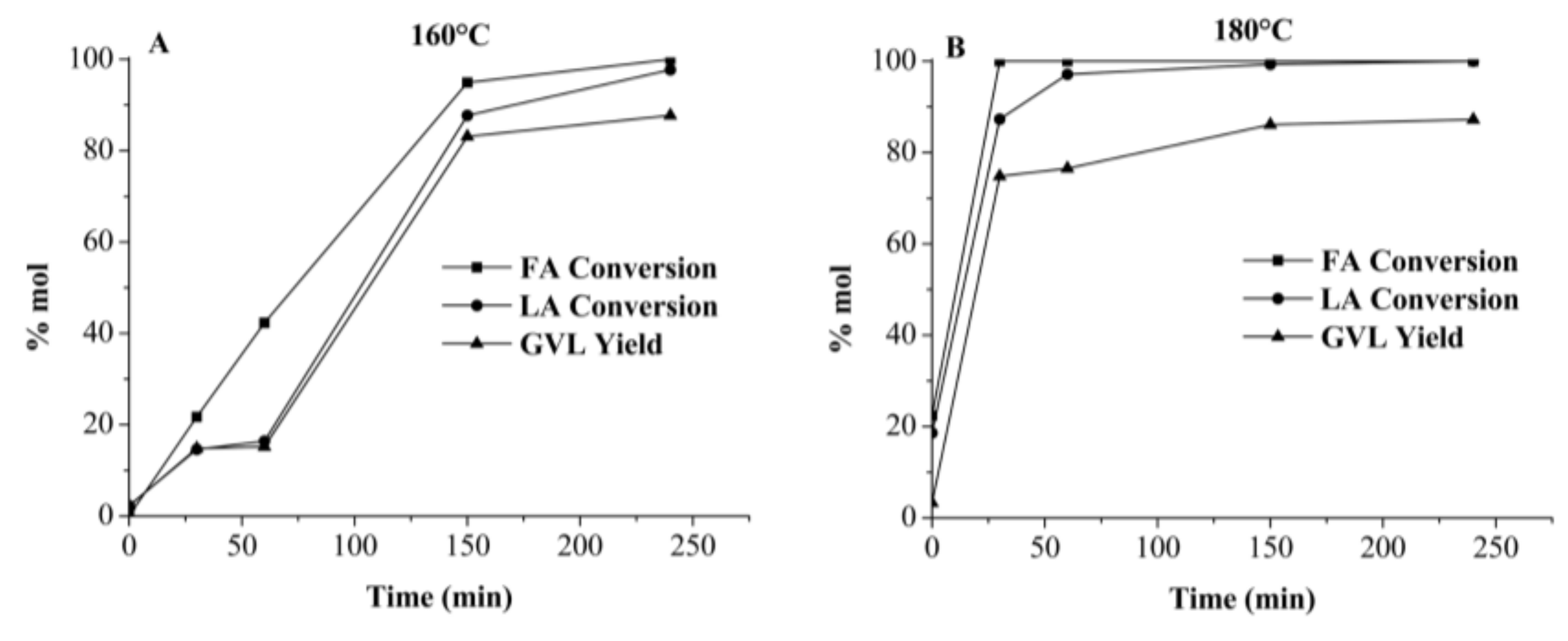
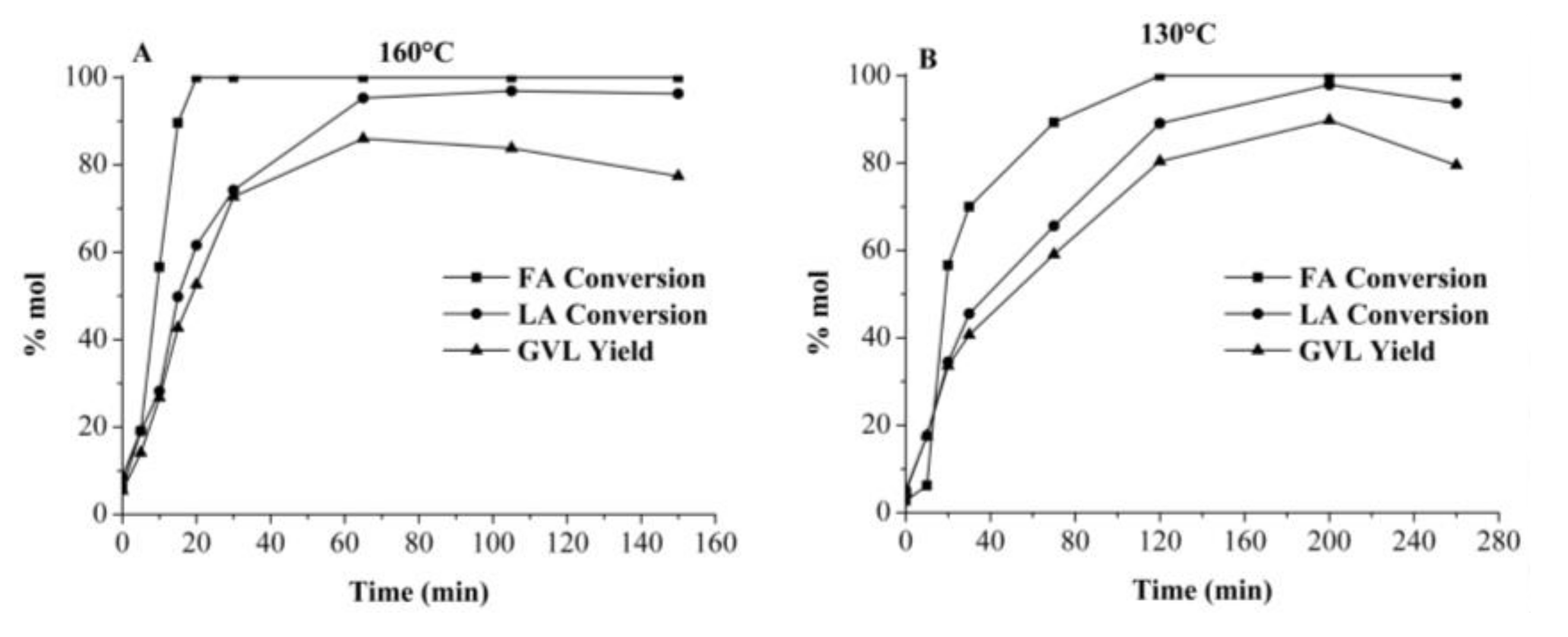
2.3.1. γ-Valerolactone GVL Production Using Crude Media from Autohydrolyzed Wood (AW) Processing in Single-Batch Mode
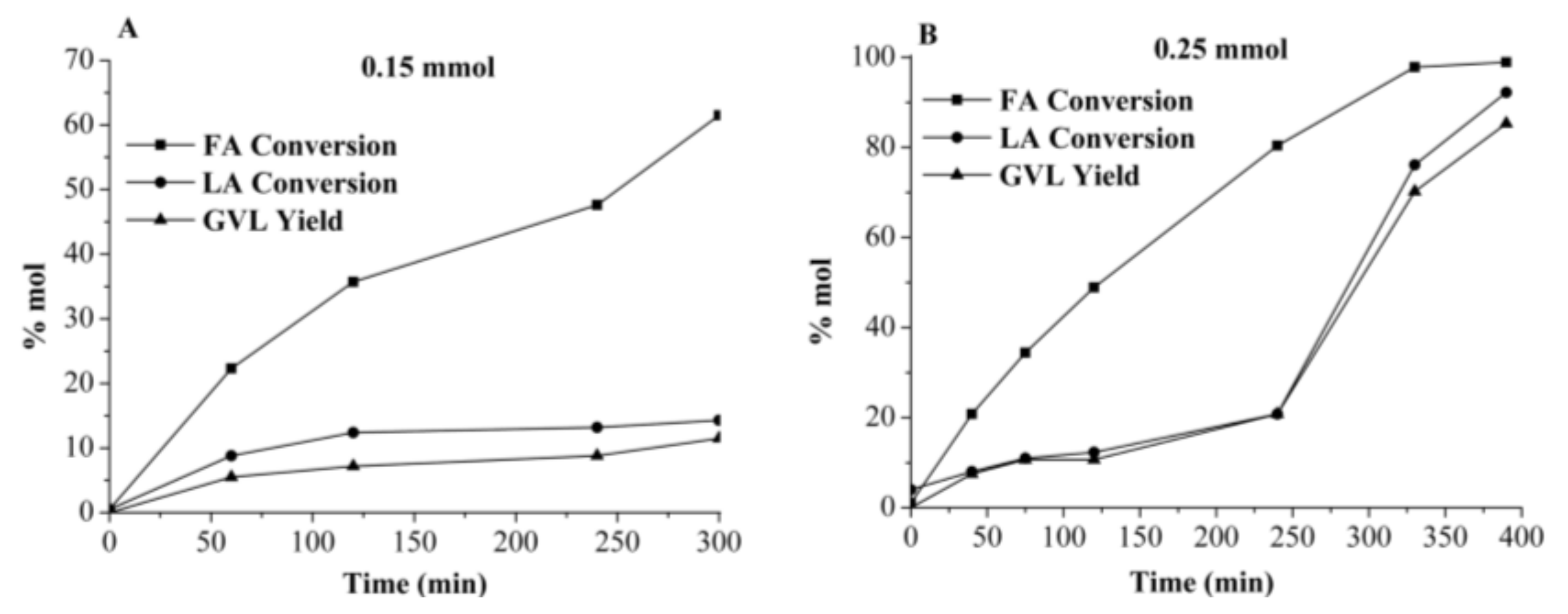
2.3.2. GVL Production Using Crude Media from AW Processing Cross-Flow Operation
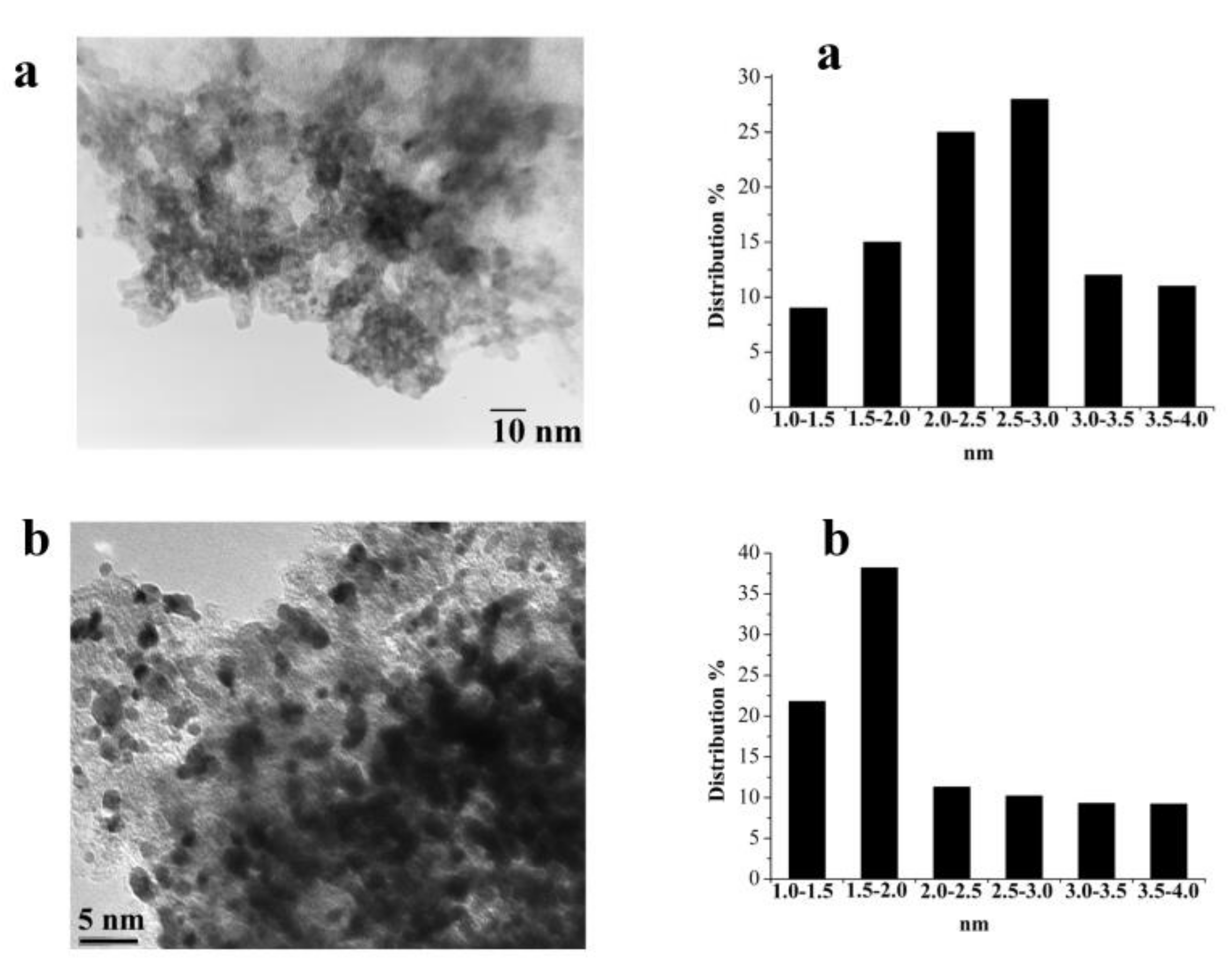
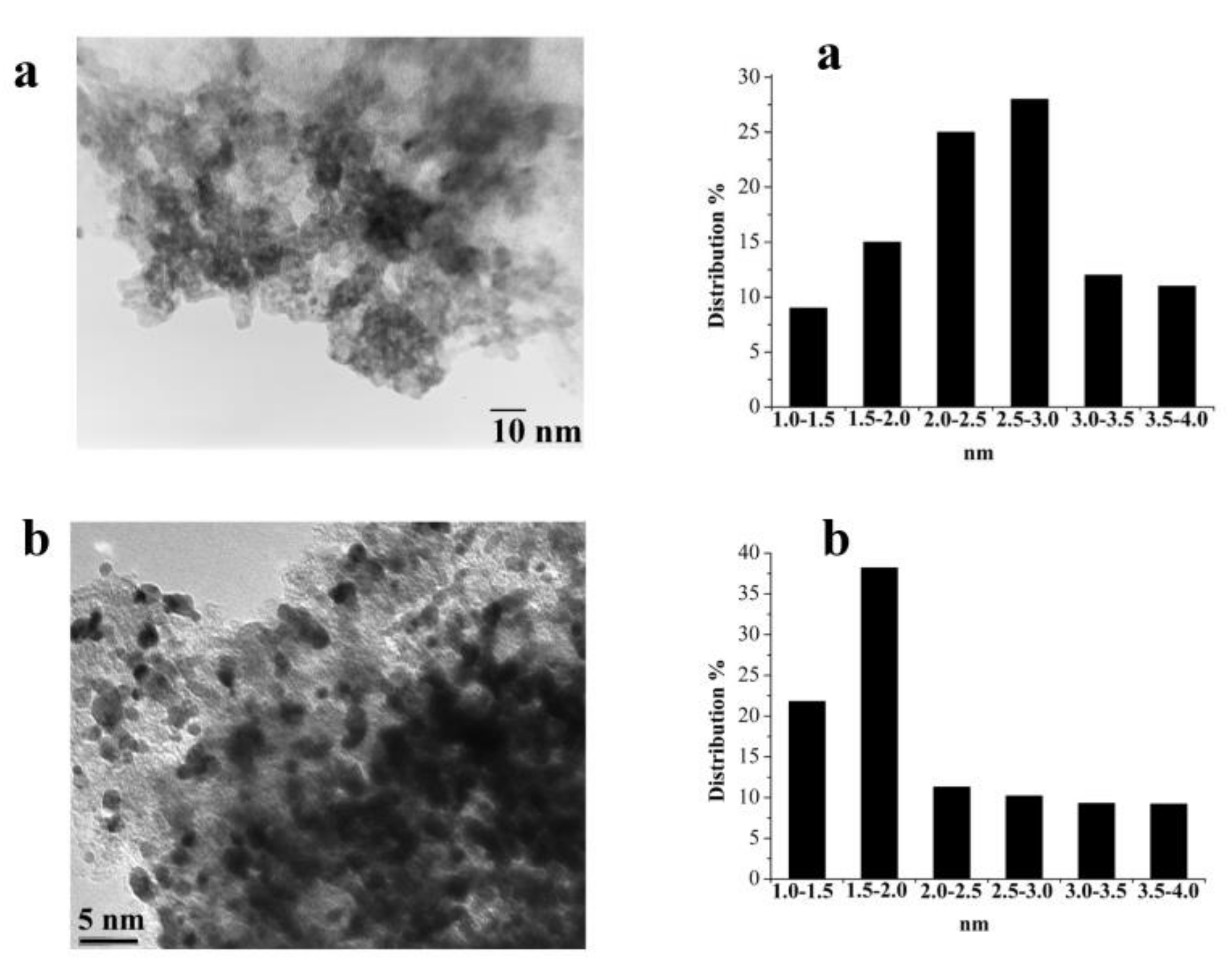
2.3.3. Catalyst Characterization before and after Reaction
3. Materials and Methods
3.1. Materials
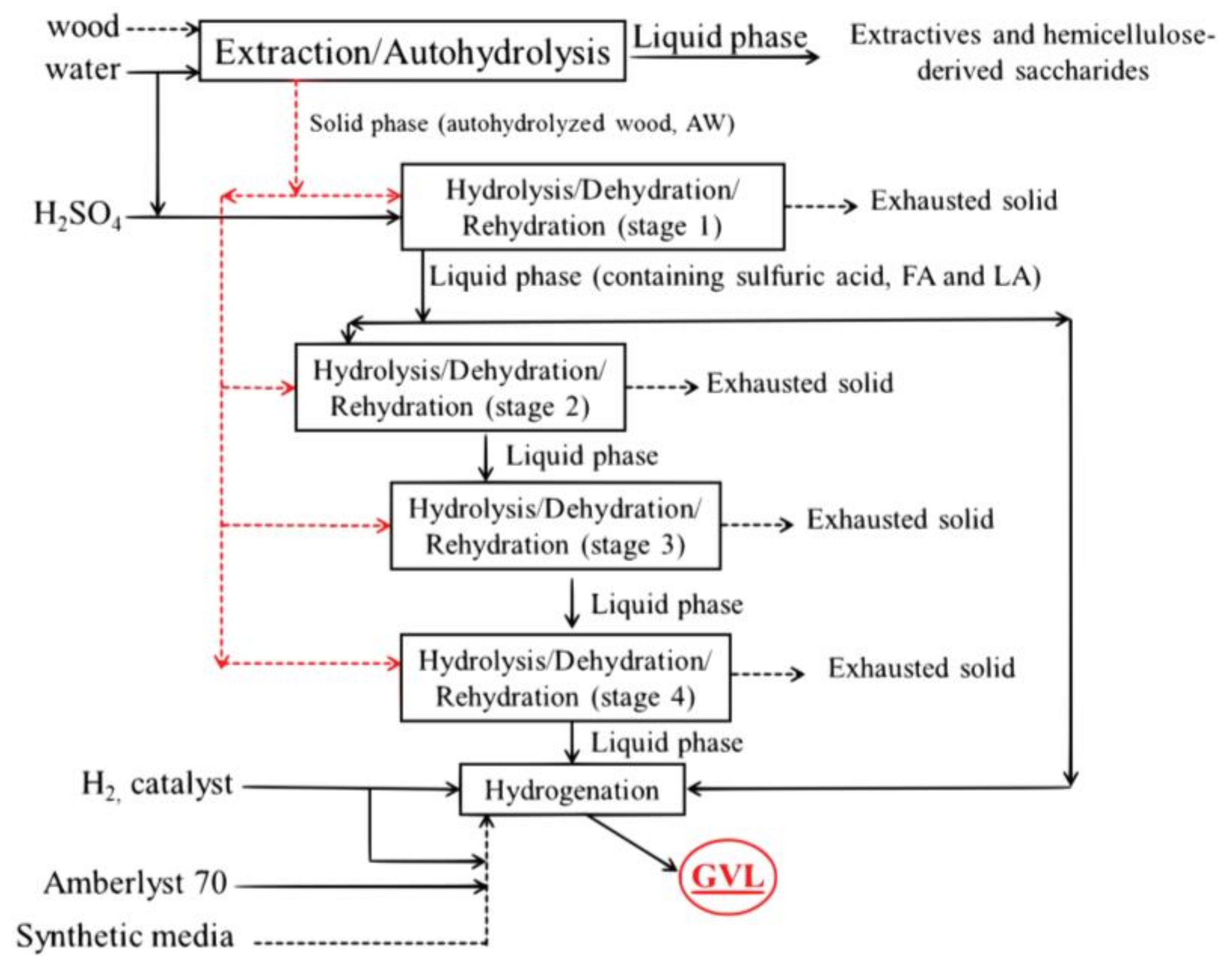
3.2. Aqueous Processing of Wood
3.3. Wood and Solid Analysis
3.4. LA Production from AW in Acidic Media
3.5. LA Hydrogenation to GVL
3.6. Analytical Methods and Structural Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Jong, E.; Higson, A.; Walsh, P.; Wellisch, M. Value Added Products from Biorefineries. IEA Bioenergy. Task 42: Biorefinery. 2012. Available online: http://www.ieabioenergy.com/publications/bio-based-chemicals-value-added-products-from-biorefineries (accessed on 1 February 2018).
- Sheldon, R.A. Green and sustainable manufacture of chemicals from biomass: State of the art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- FitzPatrick, M.; Champagne, P.; Cunningham, M.F.; Whitney, R.A. A biorefinery processing perspective: Treatment of lignocellulosic materials for the production of value-added products. Bioresour. Technol. 2010, 101, 8915–8922. [Google Scholar] [CrossRef] [PubMed]
- Gullón, P.; Romaní, A.; Vila, C.; Garrote, G.; Parajó, J.C. Potential of hydrothermal treatments in lignocellulose biorefineries. Biofuels Bioprod. Biorefin. 2012, 6, 219–232. [Google Scholar] [CrossRef]
- Garrote, G.; Domínguez, H.; Parajó, J.C. Generation of xylose solutions from Eucalyptus Globulus wood by autohydrolysis-posthydrolysis processes: Posthydrolysis kinetics. Bioresour. Technol. 2001, 79, 155–164. [Google Scholar] [CrossRef]
- Gullón, P.; González-Muñoz, M.J.; van Gool, M.P.; Schols, H.A.; Hirsch, J.; Ebringerová, A.; Parajó, J.C. Structural features and properties of soluble products derived from Eucalyptus globulus hemicelluloses. Food Chem. 2011, 127, 1798–1807. [Google Scholar] [CrossRef]
- Girisuta, B.; Dussan, K.; Haverty, D.; Leahy, J.J.; Hayes, M.H.B. A kinetic study of acid catalysed hydrolysis of sugar cane bagasse to levulinic acid. Chem. Eng. J. 2013, 217, 61–70. [Google Scholar] [CrossRef]
- Galletti, A.M.; Antonetti, C.; De Luise, V.; Licursi, D.; Nassi, N. Levulinic acid production from waste biomass. Bioresources 2012, 7, 1824–1835. [Google Scholar] [CrossRef]
- Galletti, A.M.; Antonetti, C.; Ribechini, E.; Colombini, M.P.; Nassi o Di Nasso, N.; Bonari, E. From giant reed to levulinic acid and gamma-valerolactone: A high yield catalytic route to valeric biofuels. Appl. Energy 2013, 102, 157–162. [Google Scholar] [CrossRef]
- Ribechini, E.; Zanaboni, M.; Galletti, A.M.; Antonetti, C.; Nassi o Di Nasso, N.; Bonari, E.; Colombini, M.P. Py-GC/MS characterization of a wild and a selected clone of Arundo Donax, and of its residues after catalytic hydrothermal conversion to high added-value products. J. Anal. Appl. Pyrolysis 2012, 94, 223–229. [Google Scholar] [CrossRef]
- Rivas, S.; Raspolli Galletti, A.M.; Antonetti, C.; Santos, V.; Parajó, J.C. Sustainable production of levulinic acid from the cellulosic fraction of Pinus Pinaster wood: Operation in aqueous media under microwave irradiation. J. Wood Chem. 2015, 35, 315–324. [Google Scholar] [CrossRef]
- Rivas, S.; Raspolli Galletti, A.M.; Antonetti, C.; Santos, V.; Parajó, J.C. Sustainable conversion of Pinus Pinaster wood into biofuel precursors: A biorefinery approach. Fuel 2016, 164, 51–58. [Google Scholar] [CrossRef]
- Bensah, E.C.; Mensah, M. Chemical pretreatment methods for the production of cellulosic ethanol: Technologies and innovations. Int. J. Chem. Eng. 2013, 2013, 719607. [Google Scholar] [CrossRef]
- National Research Council; Division on Engineering and Physical Sciences; National Materials Advisory Board; Commission on Engineering and Technical Systems; Committee on Microwave Processing of Materials: An Emerging Industrial Technology. Microwave Processing of Materials; The National Academic Press: Washington, DC, USA, 1994; ISBN 978-0-309-05027-2. [Google Scholar]
- Galia, A.; Schiavo, B.; Antonetti, C.; Raspolli Galletti, A.M.; Interrante, L.; Lessi, M.; Scialdone, O.; Valenti, M.G. Autohydrolysis pretreatment of Arundo donax: A comparison between microwave-assisted batch and fast heating rate flow-through reaction systems. Biotechnol. Biofuels 2015, 8, 218–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteil-Rivera, F.; Huang, G.H.; Paquet, L.; Deschamps, S.; Beaulieu, C.; Hawari, J. Microwave-assisted extraction of lignin from triticale straw: Optimization and microwave effects. Bioresour. Technol. 2012, 104, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, C.; Bonari, E.; Licursi, D.; Nassi o Di Nasso, N.; Raspolli Galletti, A.M. Hydrothermal conversion of giant reed to furfural and levulinic acid: Optimization of the Process under microwave irradiation and investigation of distinctive agronomic parameters. Molecules 2015, 20, 21232–21253. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Yan, Y.; Zhang, Y.; Tang, Y. Direct production of levulinic acid in high yield from cellulose: Joint effect of high ion strength and microwave field. RSC Adv. 2016, 6, 39131–39136. [Google Scholar] [CrossRef]
- Antonetti, C.; Licursi, D.; Fulignati, S.; Valentini, G.; Raspolli Galletti, A.M. New frontiers in the catalytic synthesis of levulinic acid: From sugars to raw and waste biomass as starting feedstock. Catalysts 2016, 6, 196–224. [Google Scholar] [CrossRef]
- Pileidis, F.D.; Titirici, M.-M. Levulinic acid biorefineries: New challenges for efficient utilization of biomass. ChemSusChem 2016, 9, 562–582. [Google Scholar] [CrossRef] [PubMed]
- Climent, M.J.; Corma, A.; Iborra, S. Converting carbohydrates to bulk chemicals and fine chemicals over heterogeneous catalysts. Green Chem. 2011, 13, 520–540. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem. 2014, 16, 516–547. [Google Scholar] [CrossRef]
- Raspolli Galletti, A.M.; Antonetti, C.; De Luise, V.; Martinelli, M. A sustainable process for the production of gamma-valerolactone by hydrogenation of biomass-derived levulinic acid. Green Chem. 2012, 14, 688–694. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Jędrzejczyk, M.; Sneka-Płatek, O.; Keller, N.; Dumon, A.S.; Michel, C.; Sautet, P.; Grams, J. Ru Catalysts for levulinic acid hydrogenation with formic acid as a hydrogen source. Green Chem. 2016, 18, 2014–2028. [Google Scholar] [CrossRef]
- Jain, A.B.; Vaidya, P.D. Kinetics of the ruthenium-catalyzed hydrogenation of levulinic acid to γ-valerolactone in aqueous solutions. Can. J. Chem. Eng. 2016, 94, 2364–2372. [Google Scholar] [CrossRef]
- Piskun, A.; Winkelman, J.G.M.; Tang, Z.; Heeres, H.J. Support screening studies on the hydrogenation of levulinic acid to γ-valerolactone in water using Ru catalysts. Catalysts 2016, 6, 131–150. [Google Scholar] [CrossRef]
- Piskun, A.S.; van de Bovenkamp, H.H.; Rasrendra, C.B.; Winkelman, J.G.M.; Heeres, H.J. Kinetic modeling of levulinic acid hydrogenation to γ-valerolactone in water using a carbon supported Ru catalyst. Appl. Catal. A Gen. 2016, 525, 158–167. [Google Scholar] [CrossRef]
- Molinari, V.; Antonietti, M.; Esposito, D. An integrated strategy for the conversion of cellulosic biomass into γ-valerolactone. Catal. Sci. Technol. 2014, 4, 3626–3630. [Google Scholar] [CrossRef]
- Al-Naji, M.; Yepez, A.; Balu, A.M.; Romero, A.A.; Chen, Z.; Wilde, N.; Li, H.; Shih, K.; Gläser, R.; Luqueb, R. Insights into the selective hydrogenation of levulinic acid to γ-valerolactone using supported mono- and bimetallic catalysts. J. Mol. Catal. A Chem. 2016, 417, 145–152. [Google Scholar] [CrossRef]
- Putro, J.N.; Kurniawan, A.; Soetaredjo, F.E.; Lin, S.-Y.; Ju, Y.-H.; Ismadji, S. Production of gamma valerolactone from sugarcane bagasse over TiO2-supported platinum and acid-activated bentonite as co-catalyst. RSC Adv. 2015, 5, 41285–41299. [Google Scholar] [CrossRef] [Green Version]
- Wachala, M.; Grams, J.; Kwapiński, W.; Ruppert, A.M. Influence of ZrO2 on catalytic performance of Ru catalyst in hydrolytic hydrogenation of cellulose towards γ-valerolactone. Int. J. Hydrogen Energy 2016, 41, 8688–8695. [Google Scholar] [CrossRef]
- Flannelly, T.; Lopes, M.; Kupiainen, L.; Dooley, S.; Leahy, J.J. Non-stoichiometric formation of formic and levulinic acids from the hydrolysis of biomass derived hexose carbohydrates. RSC Adv. 2016, 6, 5797–5804. [Google Scholar] [CrossRef]
- Szabolcs, A.; Molnár, M.; Dibó, G.; Mika, L.T. Microwave-assisted conversion of carbohydrates to levulinic acid: An essential step in biomass conversion. Green Chem. 2013, 15, 439–445. [Google Scholar] [CrossRef]
- Kang, S.; Yu, J. An intensified reaction technology for high levulinic acid concentration from lignocellulosic biomass. Biomass Bioenergy 2016, 95, 214–220. [Google Scholar] [CrossRef]
- Matubayasi, N.; Nakahara, M. Hydrothermal reactions of formic acid: Free-energy analysis of equilibrium. J. Chem. Phys. 2005, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Savage, P.E. Decomposition of formic acid under hydrothermal conditions. Ind. Eng. Chem. Res. 1998, 37, 2–10. [Google Scholar] [CrossRef]
- Akiya, N.; Savage, P.E. Role of water in formic acid decomposition. AIChe J. 1998, 44, 405–415. [Google Scholar] [CrossRef]
- Yan, K.; Jarvis, C.; Gu, J.; Yan, Y. Production and catalytic transformation of levulinic acid: A platform for speciality chemicals and fuels. Renew. Sustain. Energy Rev. 2015, 51, 986–997. [Google Scholar] [CrossRef]
- Liguori, F.; Moreno-Marrodan, C.; Barbaro, P. Environmentally friendly synthesis of γ-valerolactone by direct catalytic conversion of renewable sources. ACS Catal. 2015, 5, 1882–1894. [Google Scholar] [CrossRef]
- Abdelrahman, O.A.; Heyden, A.; Bond, J.Q. Analysis of kinetics and reaction pathways in the aqueous-phase hydrogenation of levulinic acid to form γ-valerolactone over Ru/C. ACS Catal. 2014, 4, 1171–1181. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gamma-Valerolactone, a sustainable platform molecule derived from lignocellulosic biomass. Green Chem. 2013, 15, 584–595. [Google Scholar] [CrossRef]
- Ftouni, J.; Genuino, H.C.; MuÇoz-Murillo, A.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Influence of sulfuric acid on the performance of ruthenium-based catalysts in the liquid-phase hydrogenation of levulinic acid to γ-valerolactone. ChemSusChem 2017, 10, 2891–2896. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.Y.; Mullen, G.M.; Flaherty, D.W.; Mullins, C.B. Selective hydrogen production from formic acid decomposition on Pd-Au bimetallic surfaces. J. Am. Chem. Soc. 2014, 136, 11070–11078. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Pulleri, J.K.; Ting, S.-W.; Chan, K.-Y. Activity of Pd/C for hydrogen generation in aqueous formic acid solution. Int. J. Hydrogen Energy 2014, 39, 381–390. [Google Scholar] [CrossRef]
- Gilkey, M.J.; Xu, B. Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal. 2016, 6, 1420–1436. [Google Scholar] [CrossRef]
- Wettstein, S.G.; Bond, J.Q.; Alonso, D.M.; Pham, H.N.; Datye, A.K.; Dumesic, J.A. RuSn bimetallic catalysts for selective hydrogenation of levulinic acid to γ-valerolactone. Appl. Catal. B Environ. 2012, 117–118, 321–329. [Google Scholar] [CrossRef]
- Deng, L.; Zhao, Y.; Li, J.; Fu, Y.; Liao, B.; Guo, Q.-X. Conversion of levulinic acid and formic acid into γ-valerolactone over heterogeneous catalysts. ChemSusChem 2010, 3, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Testa, M.L.; Corbel-Demailly, L.; La Parola, V.; Venezia, A.M.; Pinel, C. Effect of Au on Pd supported over HMS and Ti doped HMS as catalysts for the hydrogenation of levulinic acid to γ-valerolactone. Catal. Today 2015, 257, 291–296. [Google Scholar] [CrossRef]
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics; Wiley-VCH Verlag GmbH & Co KGaA: Weinheim, Germany, 2003; ISBN 3-527-30574-2. [Google Scholar]
- Iqbal, S.; Kondrat, S.A.; Jones, D.R.; Schoenmakers, D.C.; Edwards, J.K.; Lu, L.; Yeo, B.R.; Wells, P.P.; Gibson, E.K.; Morgan, D.J.; et al. Ruthenium nanoparticles supported on carbon: an active catalyst for the hydrogenation of lactic acid to 1,2-propanediol. ACS Catal. 2015, 5, 5047–5059. [Google Scholar] [CrossRef]
- Romanì, A.; Garrote, G.; Alonso, J.L.; Parajò, J.C. Bioethanol production from hydrothermally pretreated Eucalyptus Globulus wood. Bioresour. Technol. 2010, 101, 8706–8712. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivas, S.; Raspolli Galletti, A.M.; Antonetti, C.; Licursi, D.; Santos, V.; Parajó, J.C. A Biorefinery Cascade Conversion of Hemicellulose-Free Eucalyptus Globulus Wood: Production of Concentrated Levulinic Acid Solutions for γ-Valerolactone Sustainable Preparation. Catalysts 2018, 8, 169. https://doi.org/10.3390/catal8040169
Rivas S, Raspolli Galletti AM, Antonetti C, Licursi D, Santos V, Parajó JC. A Biorefinery Cascade Conversion of Hemicellulose-Free Eucalyptus Globulus Wood: Production of Concentrated Levulinic Acid Solutions for γ-Valerolactone Sustainable Preparation. Catalysts. 2018; 8(4):169. https://doi.org/10.3390/catal8040169
Chicago/Turabian StyleRivas, Sandra, Anna Maria Raspolli Galletti, Claudia Antonetti, Domenico Licursi, Valentín Santos, and Juan Carlos Parajó. 2018. "A Biorefinery Cascade Conversion of Hemicellulose-Free Eucalyptus Globulus Wood: Production of Concentrated Levulinic Acid Solutions for γ-Valerolactone Sustainable Preparation" Catalysts 8, no. 4: 169. https://doi.org/10.3390/catal8040169