Molecular Rh(III) and Ir(III) Catalysts Immobilized on Bipyridine-Based Covalent Triazine Frameworks for the Hydrogenation of CO2 to Formate

 , ,
, ,
Abstract
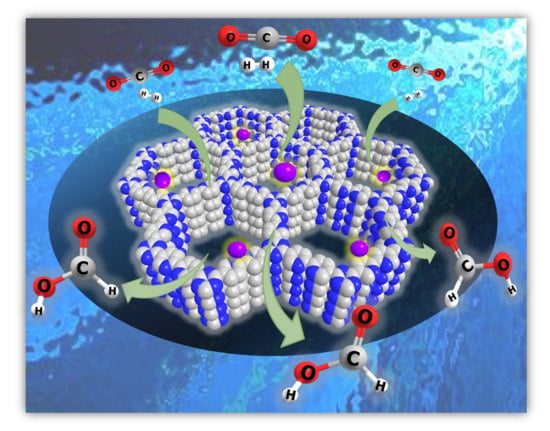
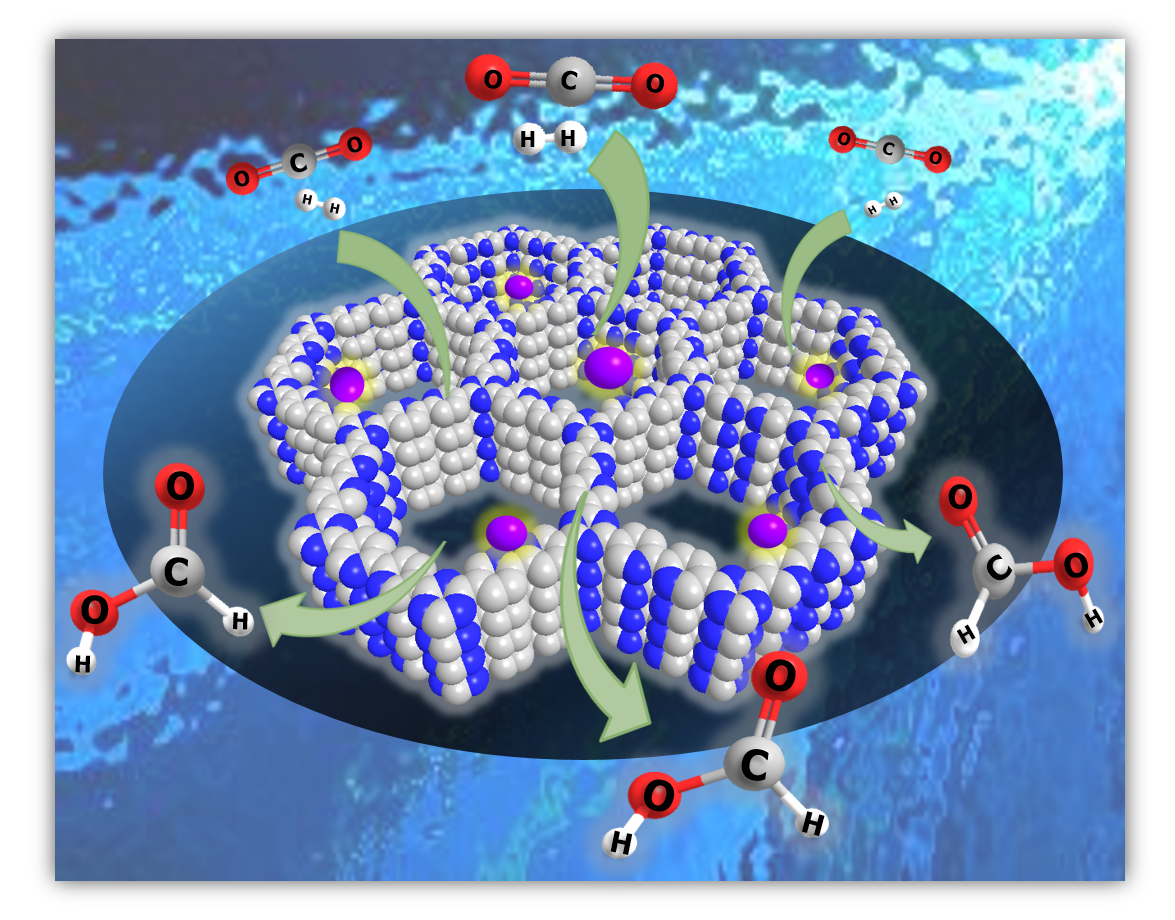
:
1. Introduction
2. Results and Discussion
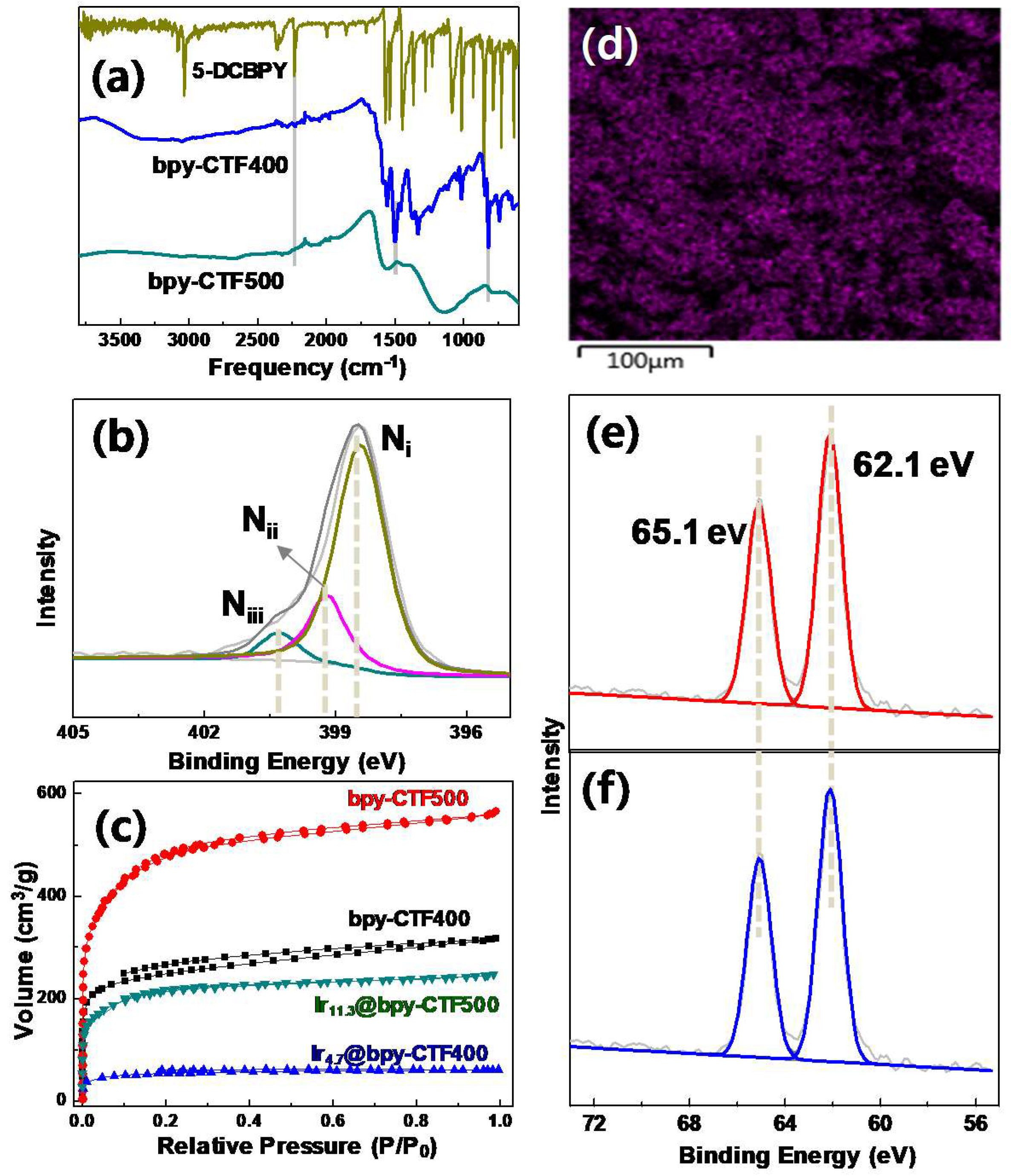
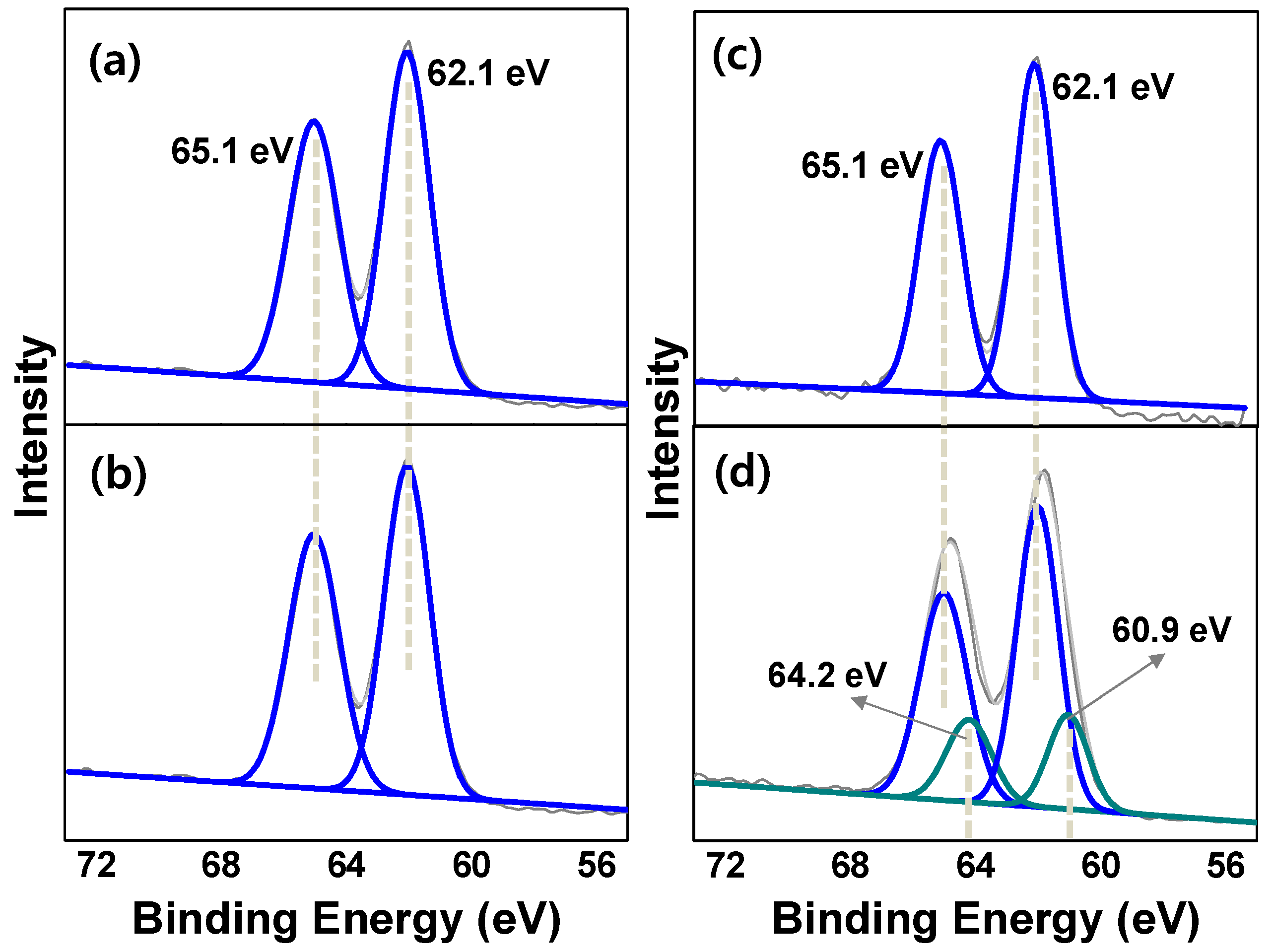
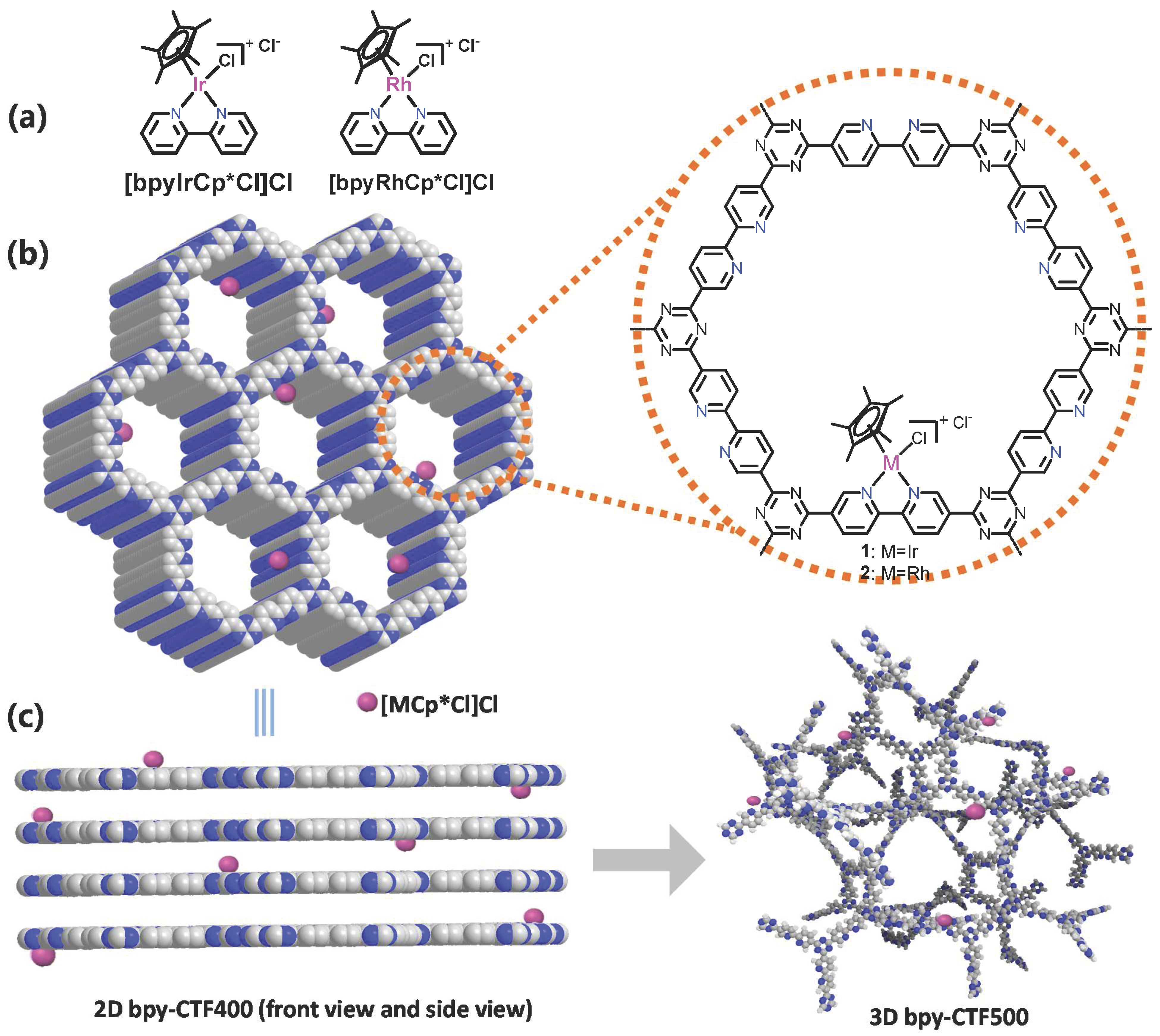
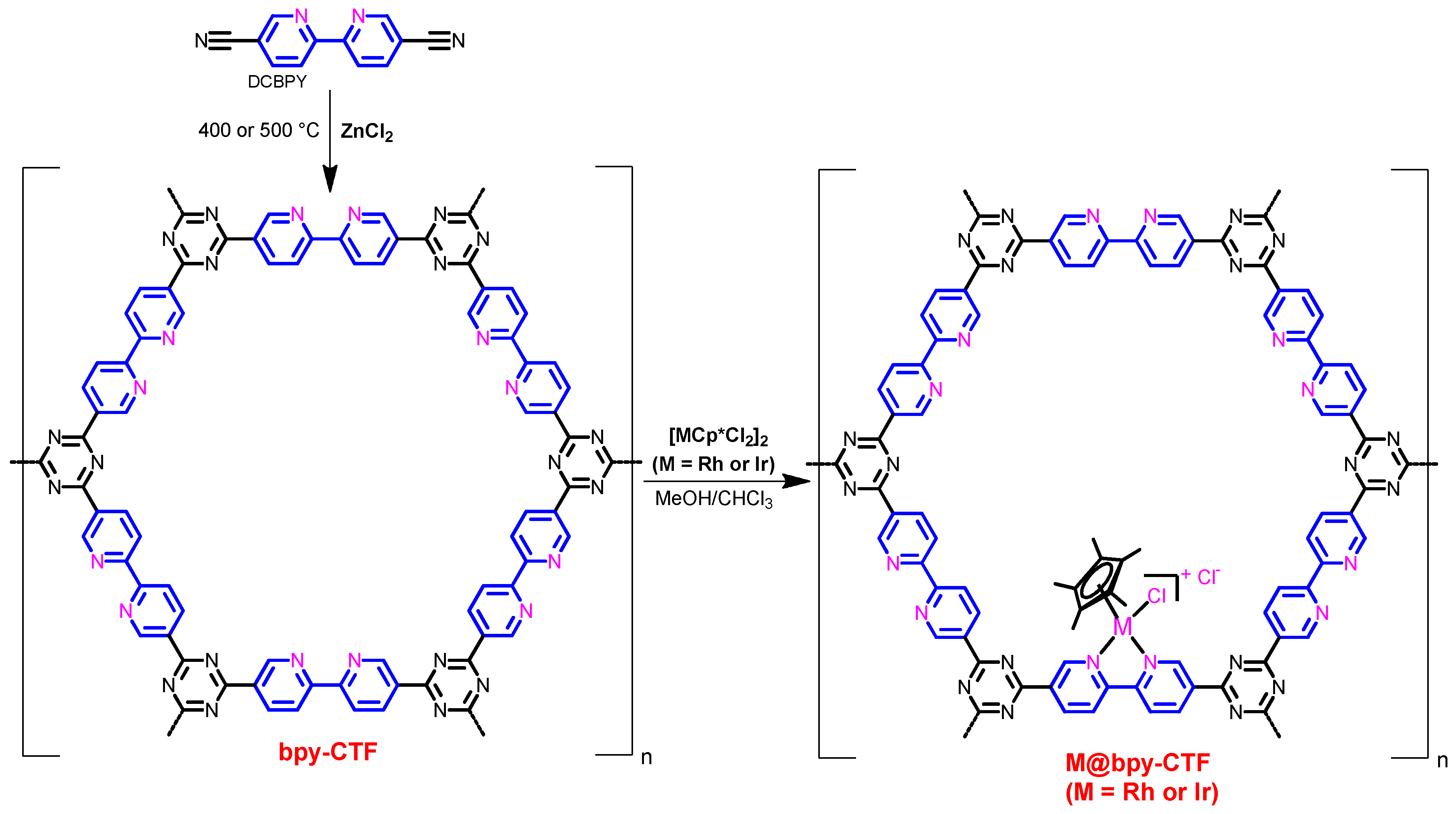
2.1. Synthesis and Characterization
2.1.1. Rh@bpy-CTF400
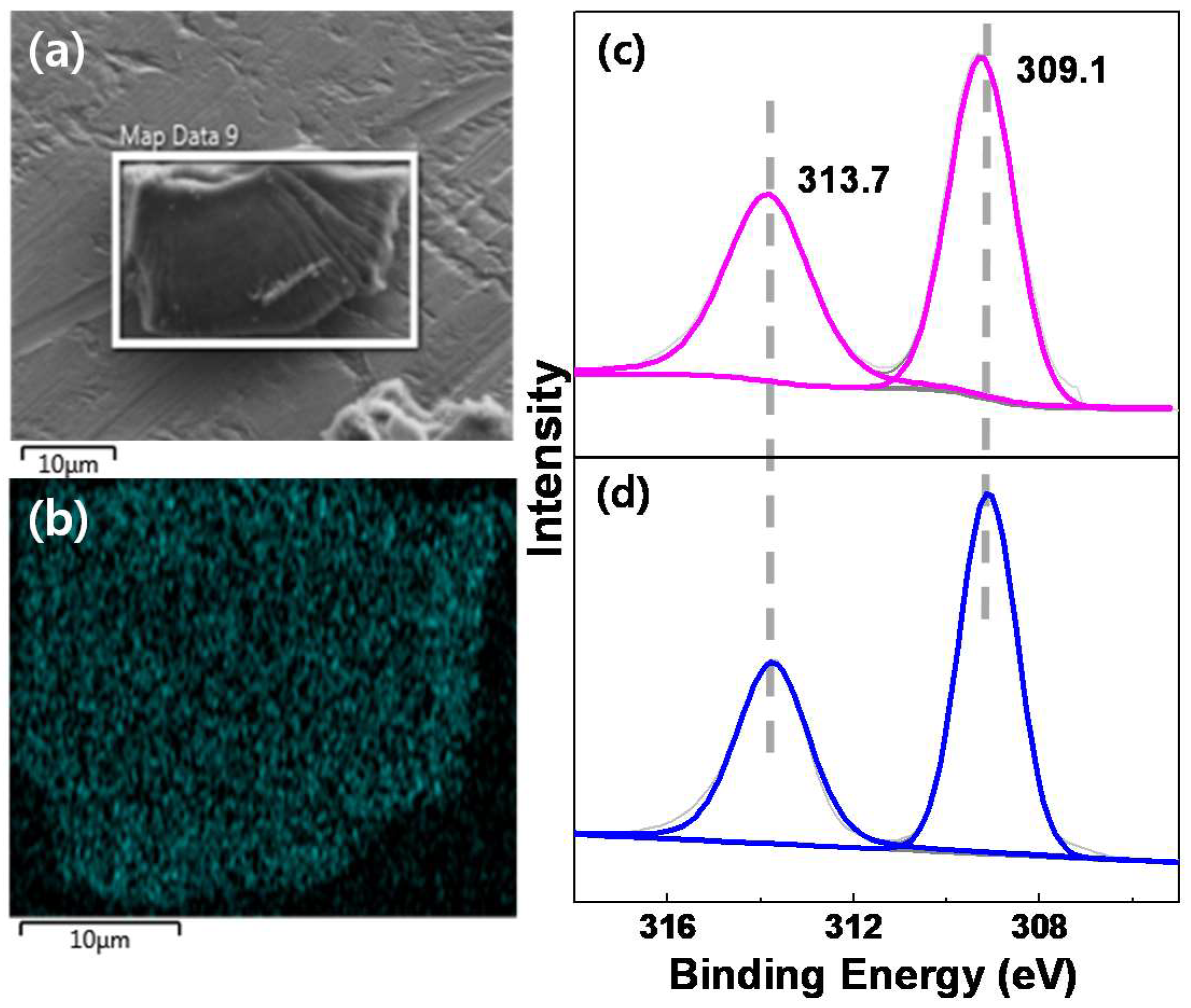
2.1.2. Three-Dimensional Architecture of Ir@bpy-CTF500
2.1.3. Ir@bpy-CTF400 with Various Ir Loadings
2.2. Hydrogenation of CO2 into Formate
2.2.1. Effect of Rh and Ir Metal Centers
2.2.2. Influence of Three-Dimensional Architecture of Ir@bpy-CTF500
2.2.3. Effect of Ir Loadings on Ir@bpy-CTF400
2.2.4. Effect of Base Concentrations on the Production of Formate
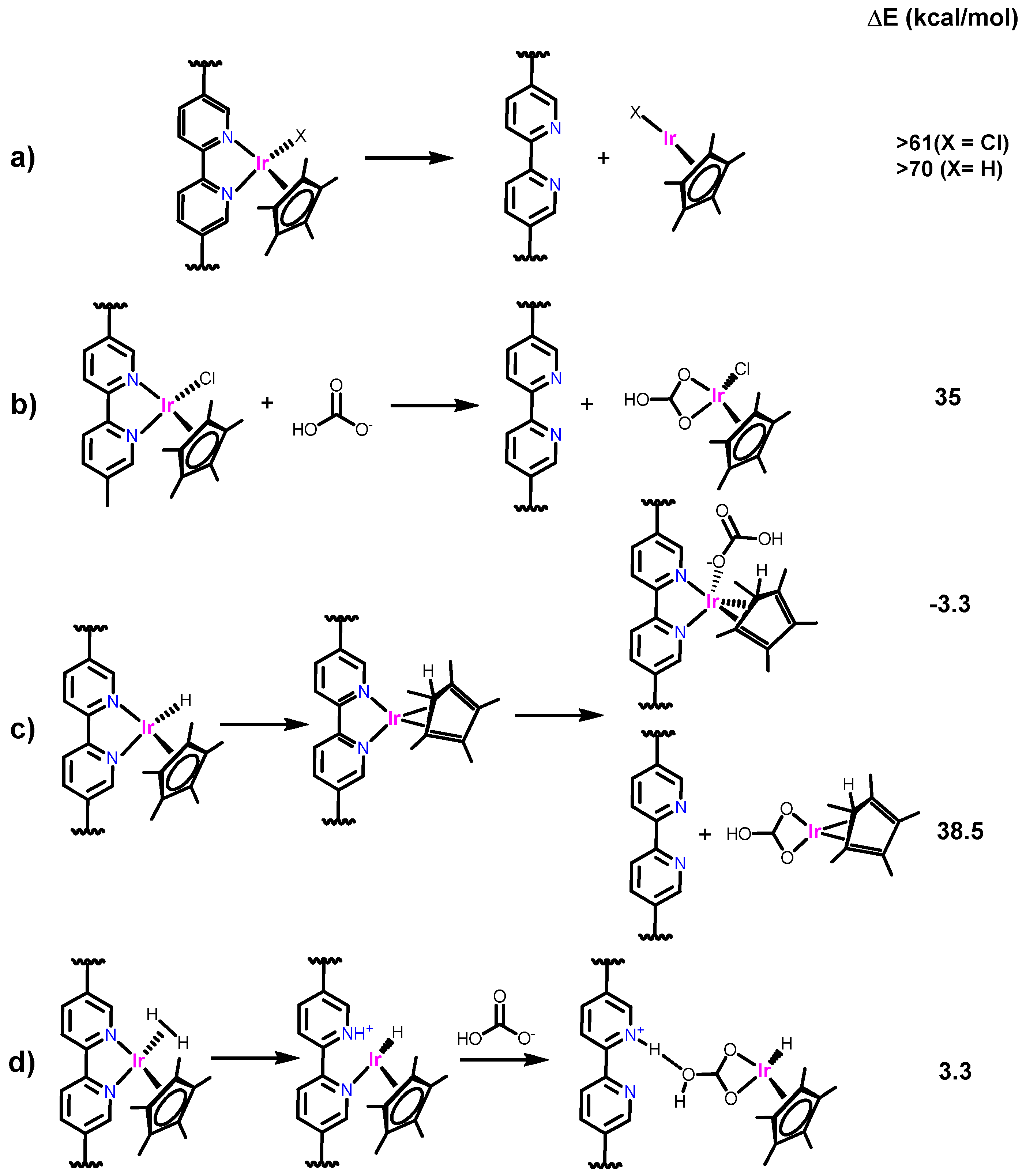
2.2.5. Leaching and Computational Studies
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Centi, G. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry. Angew. Chem. Int. Ed. 2016, 55, 7296–7343. [Google Scholar] [CrossRef] [PubMed]
- Enthaler, S.; von Langermann, J.; Schmidt, T. Carbon dioxide and formic acid—The couple for environmental-friendly hydrogen storage? Energy Environ. Sci. 2010, 3, 1207–1217. [Google Scholar] [CrossRef]
- Hull, J.F.; Himeda, Y.; Wang, W.-H.; Hashiguchi, B.; Periana, R.; Szalda, D.J.; Muckerman, J.T.; Fujita, E. Reversible hydrogen storage using CO2 and a proton-switchable iridium catalyst in aqueous media under mild temperatures and pressures. Nat. Chem. 2012, 4, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Sudakar, P.; Sivanesan, D.; Yoon, S. Copolymerization of Epichlorohydrin and CO2 Using Zinc Glutarate: An Additional Application of ZnGA in Polycarbonate Synthesis. Macromol. Rapid Commun. 2016, 37, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Sivanesan, D.; Kim, Y.E.; Youn, M.H.; Park, K.T.; Kim, H.-J.; Grace, A.N.; Jeong, S.K. The salt-based catalytic enhancement of CO2 absorption by a tertiary amine medium. RSC Adv. 2016, 6, 64575–64580. [Google Scholar] [CrossRef]
- Sivanesan, D.; Choi, Y.; Lee, J.; Youn, M.H.; Park, K.T.; Grace, A.N.; Kim, H.J.; Jeong, S.K. Carbon Dioxide Sequestration by Using a Model Carbonic Anhydrase Complex in Tertiary Amine Medium. ChemSusChem 2015, 8, 3977–3982. [Google Scholar] [CrossRef] [PubMed]
- Sivanesan, D.; Youn, M.H.; Murnandari, A.; Kang, J.M.; Park, K.T.; Kim, H.J.; Jeong, S.K. Enhanced CO2 absorption and desorption in a tertiary amine medium with a carbonic anhydrase mimic. J. Ind. Eng. Chem. 2017, 52, 287–294. [Google Scholar] [CrossRef]
- Kieczska, H.; Reutemann, W. Formic acid. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2002. [Google Scholar]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source—Recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Lau, C.P.; Chen, Y.Z. Hydrogenation of carbon dioxide to formic acid using a 6,6′-dichloro-2,2′-bipyridine complex of ruthenium,cis-[Ru(6,6′-Cl2bpy)2(H2O)2](CF3SO3)2. J. Mol. Catal. A Chem. 1995, 101, 33–36. [Google Scholar] [CrossRef]
- Jessop, P.G.; Hsiao, Y.; Ikariya, T.; Noyori, R. Homogeneous catalysis in supercritical fluids: Hydrogenation of supercritical carbon dioxide to formic acid, alkyl formates, and formamides. J. Am. Chem. Soc. 1996, 118, 344–355. [Google Scholar] [CrossRef]
- Gunasekar, G.H.; Yoon, Y.; Baek, I.; Yoon, S. Catalytic reactivity of an iridium complex with a proton responsive N-donor ligand in CO2 hydrogenation to formate. RSC Adv. 2018, 8, 1346–1350. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed]
- Schmeier, T.J.; Dobereiner, G.E.; Crabtree, R.H.; Hazari, N. Secondary coordination sphere interactions facilitate the insertion step in an iridium(III) CO2 reduction catalyst. J. Am. Chem. Soc. 2011, 133, 9274–9277. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Nowakowski, K. Catalytic Hydrogenation of Carbon Dioxide to Formic Acid, 1st ed.; Elsevier Inc.: New York, NY, USA, 2014; Volume 66, ISBN 0898-8838. [Google Scholar]
- Himeda, Y.; Onozawa-komatsuzaki, N.; Sugihara, H.; Kasuga, K. Simultaneous Tuning of Activity and Water Solubility of Complex Catalysts by Acid—Base Equilibrium of Ligands for Conversion of Carbon Dioxide. Organometallics 2007, 26, 702–712. [Google Scholar] [CrossRef]
- Wang, W.H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Hull, J.F.; Muckerman, J.T.; Fujita, E.; Himeda, Y. Second-coordination-sphere and electronic effects enhance iridium(iii)-catalyzed homogeneous hydrogenation of carbon dioxide in water near ambient temperature and pressure. Energy Environ. Sci. 2012, 5, 7923–7926. [Google Scholar] [CrossRef]
- Maenaka, Y.; Suenobu, T.; Fukuzumi, S. Catalytic interconversion between hydrogen and formic acid at ambient temperature and pressure. Energy Environ. Sci. 2012, 5, 7360–7367. [Google Scholar] [CrossRef]
- Gunasekar, G.H.; Park, K.; Jung, K.-D.; Yoon, S. Recent developments in the catalytic hydrogenation of CO2 to formic acid/formate using heterogeneous catalysts. Inorg. Chem. Front. 2016, 3, 882–895. [Google Scholar] [CrossRef]
- Stalder, C.J.; Chao, S.; Summers, D.P.; Wrighton, M.S. Supported Palladium Catalysts for the Reduction of Sodium Bicarbonate to Sodium Formate in Aqueous Solution at Room Temperature and One Atmosphere of Hydrogen. J. Am. Chem. Soc. 1983, 105, 6318–6320. [Google Scholar] [CrossRef]
- Srivastava, V. Functionalized Hydrotalcite Tethered Ruthenium Catalyst for Carbon Sequestration Reaction. Catal. Lett. 2018, 148, 1879–1892. [Google Scholar] [CrossRef]
- Zhang, Y.; Fei, J.; Yu, Y.; Zheng, X. Silica immobilized ruthenium catalyst used for carbon dioxide hydrogenation to formic acid (I): The effect of functionalizing group and additive on the catalyst performance. Catal. Commun. 2004, 5, 643–646. [Google Scholar] [CrossRef]
- Wesselbaum, S.; Hintermair, U.; Leitner, W. Continuous-flow hydrogenation of carbon dioxide to pure formic acid using an integrated scCO2 process with immobilized catalyst and base. Angew. Chem. Int. Ed. 2012, 51, 8585–8588. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Mcnamara, N.D.; Neumann, G.T.; Schneider, W.F.; Hicks, J.C. Catalytic hydrogenation of CO2 to formic acid with silica-tethered iridium catalysts. ChemCatChem 2013, 5, 1769–1771. [Google Scholar] [CrossRef]
- McNamara, N.D.; Hicks, J.C. CO2 capture and conversion with a multifunctional polyethyleneimine-tethered iminophosphine iridium catalyst/adsorbent. ChemSusChem 2014, 7, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Gunasekar, G.H.; Shin, J.; Jung, K.-D.; Park, K.; Yoon, S. Design Strategy Toward Recyclable and Highly Efficient Heterogeneous Catalysts for the Hydrogenation of CO2 to Formate. ACS Catal. 2018, 8, 4346–4353. [Google Scholar] [CrossRef]
- Park, K.; Gunasekar, G.H.; Prakash, N.; Jung, K.-D.; Yoon, S. A Highly Efficient Heterogenized Iridium Complex for the Catalytic Hydrogenation of Carbon Dioxide to Formate. ChemSusChem 2015, 8, 3410–3413. [Google Scholar] [CrossRef] [PubMed]
- Bavykina, A.V.; Rozhko, E.; Goesten, M.G.; Wezendonk, T.; Seoane, B.; Kapteijn, F.; Makkee, M.; Gascon, J. Shaping Covalent Triazine Frameworks for the Hydrogenation of Carbon Dioxide to Formic Acid. ChemCatChem 2016, 8, 2217–2221. [Google Scholar] [CrossRef]
- Gunniya Hariyanandam, G.; Hyun, D.; Natarajan, P.; Jung, K.D.; Yoon, S. An effective heterogeneous Ir(III) catalyst, immobilized on a heptazine-based organic framework, for the hydrogenation of CO2 to formate. Catal. Today 2016, 265, 52–55. [Google Scholar] [CrossRef]
- Gunasekar, G.H.; Park, K.; Ganesan, V.; Lee, K.; Kim, N.K.; Jung, K.D.; Yoon, S. A Covalent Triazine Framework, Functionalized with Ir/N-Heterocyclic Carbene Sites, for the Efficient Hydrogenation of CO2 to Formate. Chem. Mater. 2017, 29, 6740–6748. [Google Scholar] [CrossRef]
- Preti, D.; Resta, C.; Squarcialupi, S.; Fachinetti, G. Carbon dioxide hydrogenation to formic acid by using a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2011, 50, 12551–12554. [Google Scholar] [CrossRef] [PubMed]
- Bi, Q.Y.; Lin, J.D.; Liu, Y.M.; Du, X.L.; Wang, J.Q.; He, H.Y.; Cao, Y. An aqueous rechargeable formate-based hydrogen battery driven by heterogeneous Pd catalysis. Angew. Chem. Int. Ed. 2014, 53, 13583–13587. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Lu, M.; Lin, H. High yield production of formate by hydrogenating CO2 derived ammonium carbamate/carbonate at room temperature. Green Chem. 2015, 17, 2769–2773. [Google Scholar] [CrossRef]
- Nguyen, L.T.M.; Park, H.; Banu, M.; Kim, J.Y.; Youn, D.H.; Magesh, G.; Kim, W.Y.; Lee, J.S. Catalytic CO2 hydrogenation to formic acid over carbon nanotube-graphene supported PdNi alloy catalysts. RSC Adv. 2015, 5, 105560–105566. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Vrijburg, W.L.; Hensen, E.J.M.; Pidko, E.A. On the activity of supported Au catalysts in the liquid phase hydrogenation of CO2 to formates. J. Catal. 2016, 343, 97–105. [Google Scholar] [CrossRef]
- Umegaki, T.; Enomoto, Y.; Kojima, Y. Metallic ruthenium nanoparticles for hydrogenation of supercritical carbon dioxide. Catal. Sci. Technol. 2016, 6, 409–412. [Google Scholar] [CrossRef]
- Wang, T.; Ren, D.; Huo, Z.; Song, Z.; Jin, F.; Chen, M.; Chen, L. A nanoporous nickel catalyst for selective hydrogenation of carbonates into formic acid in water. Green Chem. 2017, 19, 716–721. [Google Scholar] [CrossRef]
- Patel, P.; Nandi, S.; Maru, M.S.; Kureshy, R.I.; Khan, N.U.H. Nitrogen-rich graphitic carbon stabilized cobalt nanoparticles as an effective heterogeneous catalyst for hydrogenation of CO2 to formate. J. CO2 Util. 2018, 25, 310–314. [Google Scholar] [CrossRef]
- Jiang, J.; Gunasekar, G.H.; Park, S.; Kim, S.-H.; Yoon, S.; Piao, L. Hierarchical Cu nanoparticle-aggregated cages with high catalytic activity for reduction of 4-nitrophenol and carbon dioxide. Mater. Res. Bull. 2018, 100, 184–190. [Google Scholar] [CrossRef]
- Maru, M.S.; Ram, S.; Adwani, J.H.; Shukla, R.S. Selective and Direct Hydrogenation of CO2 for the Synthesis of Formic Acid over a Rhodium Hydrotalcite (Rh-HT) Catalyst. ChemistrySelect 2017, 2, 3823–3830. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Zhang, H.; Yu, B.; Zhao, Y.; Ji, G.; Liu, Z. A Tröger’s base-derived microporous organic polymer: Design and applications in CO2/H2 capture and hydrogenation of CO2 to formic acid. Chem. Commun. 2015, 51, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Kann, A.; Hartmann, H.; Besmehn, A.; Hausoul, P.; Palkovits, R. Hydrogenation of CO2 to Formate over Ru Immobilized on Solid Molecular Phosphines. ChemSusChem 2018, 11, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Bavykina, A.V.; Olivos-Suarez, A.I.; Osadchii, D.; Valecha, R.; Franz, R.; Makkee, M.; Kapteijn, F.; Gascon, J. Facile Method for the Preparation of Covalent Triazine Framework coated Monoliths as Catalyst Support: Applications in C1 Catalysis. ACS Appl. Mater. Interfaces 2017, 9, 26060–26065. [Google Scholar] [CrossRef] [PubMed]
- Rajendiran, S.; Gunasekar, G.H.; Yoon, S. A heterogenized cobaltate catalyst on a bis-imidazolium-based covalent triazine framework for hydroesterification of epoxides. New J. Chem. 2018, 42, 12256–12262. [Google Scholar] [CrossRef]
- Hao, L.; Ning, J.; Luo, B.; Wang, B.; Zhang, Y.; Tang, Z.; Yang, J.; Thomas, A.; Zhi, L. Structural evolution of 2D microporous covalent triazine-based framework toward the study of high-performance supercapacitors. J. Am. Chem. Soc. 2015, 137, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Puthiaraj, P.; Lee, Y.-R.; Zhang, S.; Ahn, W.-S. Triazine-based covalent organic polymers: Design, synthesis and applications in heterogeneous catalysis. J. Mater. Chem. A 2016, 4, 16288–16311. [Google Scholar] [CrossRef]
- Rajendiran, S.; Natarajan, P.; Yoon, S. A covalent triazine framework-based heterogenized Al-Co bimetallic catalyst for the ring-expansion carbonylation of epoxide to beta-lactone. RSC Adv. 2017, 7, 4635–4638. [Google Scholar] [CrossRef]
- Park, K.; Lee, K.; Kim, H.; Ganesan, V.; Cho, K.; Jeong, S.K.; Yoon, S. Preparation of covalent triazine frameworks with imidazolium cations embedded in basic sites and their application for CO2 capture. J. Mater. Chem. A 2017, 5, 8576–8582. [Google Scholar] [CrossRef]
- Puthiaraj, P.; Kim, S.S.; Ahn, W.S. Covalent triazine polymers using a cyanuric chloride precursor via Friedel-Crafts reaction for CO2 adsorption/separation. Chem. Eng. J. 2016, 283, 184–192. [Google Scholar] [CrossRef]
- Kuhn, P.; Forget, A.; Su, D.; Thomas, A.; Antonietti, M. From microporous regular frameworks to mesoporous materials with ultrahigh surface area: Dynamic reorganization of porous polymer networks. J. Am. Chem. Soc. 2008, 130, 13333–13337. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Antonietti, M.; Thomas, A. Porous, covalent triazine-based frameworks prepared by ionothermal synthesis. Angew. Chem. Int. Ed. 2008, 47, 3450–3453. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Thomas, A.; Antonietti, M. Toward tailorable porous organic polymer networks: A high-temperature dynamic polymerization scheme based on aromatic nitriles. Macromolecules 2009, 42, 319–326. [Google Scholar] [CrossRef]
- Palkovits, R.; Antonietti, M.; Kuhn, P.; Thomas, A.; Schüth, F. Solid catalysts for the selective low-temperature oxidation of methane to methanol. Angew. Chem. Int. Ed. 2009, 48, 6909–6912. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Forget, A.; Hartmann, J.; Thomas, A.; Antonietti, M. Template-free tuning of nanopores in carbonaceous polymers through lonothermal synthesis. Adv. Mater. 2009, 21, 897–901. [Google Scholar] [CrossRef]
- Palkovits, R.; von Malotki, C.; Baumgarten, M.; Müllen, K.; Baltes, C.; Antonietti, M.; Kuhn, P.; Weber, J.; Thomas, A.; Schüth, F. Development of molecular and solid catalysts for the direct low-temperature oxidation of methane to methanol. ChemSusChem 2010, 3, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Hug, S.; Tauchert, M.E.; Li, S.; Pachmayr, U.E.; Lotsch, B.V. A functional triazine framework based on N-heterocyclic building blocks. J. Mater. Chem. 2012, 22, 13956–13964. [Google Scholar] [CrossRef]
- Hug, S.; Stegbauer, L.; Oh, H.; Hirscher, M.; Lotsch, B.V. Nitrogen-Rich Covalent Triazine Frameworks as High-Performance Platforms for Selective Carbon Capture and Storage. Chem. Mater. 2015, 27, 8001–8010. [Google Scholar] [CrossRef]
- Rajendiran, S.; Park, K.; Lee, K.; Yoon, S. Ionic-Liquid-Based Heterogeneous Covalent Triazine Framework Cobalt Catalyst for the Direct Synthesis of Methyl 3-Hydroxybutyrate from Propylene Oxide. Inorg. Chem. 2017, 56, 7270–7277. [Google Scholar] [CrossRef] [PubMed]
- Sudakar, P.; Gunasekar, G.H.; Baek, I.H.; Yoon, S. Recyclable and efficient heterogenized Rh and Ir catalysts for the transfer hydrogenation of carbonyl compounds in aqueous medium. Green Chem. 2016, 18, 6456–6461. [Google Scholar] [CrossRef]
- Bavykina, A.V.; Goesten, M.G.; Kapteijn, F.; Makkee, M.; Gascon, J. Efficient production of hydrogen from formic acid using a Covalent Triazine Framework supported molecular catalyst. ChemSusChem 2015, 8, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lim, S.; Baik, J.H.; Kim, H.; Jung, K.-D.; Yoon, S. Exceptionally Stable Rh-based Molecular Catalyst Heterogenized on a Cationically charged Covalent Triazine Framework Support for Efficient Methanol Carbonylation. Catal. Sci. Technol. 2018, 8, 2894–2900. [Google Scholar] [CrossRef]
- Jena, H.S.; Krishnaraj, C.; Wang, G.; Leus, K.; Schmidt, J.; Chaoui, N.; Van Der Voort, P. Acetylacetone Covalent Triazine Framework: An Efficient Carbon Capture and Storage Material and a Highly Stable Heterogeneous Catalyst. Chem. Mater. 2018, 30, 4102–4111. [Google Scholar] [CrossRef]
- Sharifi, T.; Gracia-Espino, E.; Reza Barzegar, H.; Jia, X.; Nitze, F.; Hu, G.; Nordblad, P.; Tai, C.W.; Wågberg, T. Formation of nitrogen-doped graphene nanoscrolls by adsorption of magnetic γ-Fe2O3 nanoparticles. Nat. Commun. 2013, 4, 2319. [Google Scholar] [CrossRef] [PubMed]
- Chan-Thaw, C.E.; Villa, A.; Katekomol, P.; Su, D.; Thomas, A.; Prati, L. Covalent triazine framework as catalytic support for liquid phase reaction. Nano Lett. 2010, 10, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Himeda, Y. Conversion of CO2 into formate by homogeneously catalyzed hydrogenation in water: Tuning catalytic activity and water solubility through the acid-base equilibrium of the ligand. Eur. J. Inorg. Chem. 2007, 2007, 3927–3941. [Google Scholar] [CrossRef]
- Balcells, D.; Nova, A.; Clot, E.; Gnanamgari, D.; Crabtree, R.H.; Eisenstein, O. Mechanism of homogeneous iridium-catalyzed alkylation of amines with alcohols from a DFT study. Organometallics 2008, 27, 2529–2535. [Google Scholar] [CrossRef]
- Bartoszewicz, A.; González Miera, G.; Marcos, R.; Norrby, P.O.; Martĺn-Matute, B. Mechanistic studies on the alkylation of amines with alcohols catalyzed by a bifunctional iridium complex. ACS Catal. 2015, 5, 3704–3716. [Google Scholar] [CrossRef]
- Ertem, M.Z.; Himeda, Y.; Fujita, E.; Muckerman, J.T. Interconversion of Formic Acid and Carbon Dioxide by Proton-Responsive, Half-Sandwich CpIrIII Complexes: A Computational Mechanistic Investigation. ACS Catal. 2016, 6, 600–609. [Google Scholar] [CrossRef]
- Suna, Y.; Ertem, M.Z.; Wang, W.; Kambayashi, H.; Manaka, Y.; Muckerman, J.T.; Fujita, E.; Himeda, Y. Positional Effects of Hydroxy Groups on Catalytic Activity of Proton- Responsive Half-Sandwich Cp*Iridium(III) Complexes. Organometallics 2014, 33, 6519–6530. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | T (°C) | Final Formate Concentration (M) | TON b | TOF c (h−1) |
|---|---|---|---|---|---|
| 1 | Rh1.7@bpy-CTF400 | 80 | 0.01 | 168 | 84 |
| 2 | Rh1.7@bpy-CTF400 | 100 | 0.03 | 440 | 220 |
| 3 | Rh1.7@bpy-CTF400 | 120 | 0.11 | 1410 | 705 |
| 4 | Rh1.7@bpy-CTF400 | 120 | 0.02 | 240 | 960 |
| 5 | Ir1.4@bpy-CTF400 | 120 | 0.60 | 5000 | 2500 |
| 6 d | Rh1.7@bpy-CTF400 | 80 | 0.01 | 78 | 39 |
| 7 e | Rh1.7@bpy-CTF400 | 80 | <0.01 | 57 | 29 |
| Entry | Catalyst | Final Formate Concentration (M) | TON b | TOF (h−1) c |
|---|---|---|---|---|
| 1 d | Ir4.7@bpy-CTF400 | 0.60 | 5000 | 2500 |
| 2 | Ir11.3@bpy-CTF500 | 0.26 | 2720 | 1360 |
| 3 e | Ir11.3@bpy-CTF500 | 0.60 | 5000 | 2480 f |
| 4 | Ir1.4@bpy-CTF400 | 0.60 | 5000 | 2500 |
| 5 | Ir4.1@bpy-CTF400 | 0.60 | 5000 | 2500 |
| 6 | Ir10.6@bpy-CTF400 | 0.37 | 3032 | 1516 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunasekar, G.H.; Park, K.; Jeong, H.; Jung, K.-D.; Park, K.; Yoon, S. Molecular Rh(III) and Ir(III) Catalysts Immobilized on Bipyridine-Based Covalent Triazine Frameworks for the Hydrogenation of CO2 to Formate. Catalysts 2018, 8, 295. https://doi.org/10.3390/catal8070295
Gunasekar GH, Park K, Jeong H, Jung K-D, Park K, Yoon S. Molecular Rh(III) and Ir(III) Catalysts Immobilized on Bipyridine-Based Covalent Triazine Frameworks for the Hydrogenation of CO2 to Formate. Catalysts. 2018; 8(7):295. https://doi.org/10.3390/catal8070295
Chicago/Turabian StyleGunasekar, Gunniya Hariyanandam, Kwangho Park, Hyeonseok Jeong, Kwang-Deog Jung, Kiyoung Park, and Sungho Yoon. 2018. "Molecular Rh(III) and Ir(III) Catalysts Immobilized on Bipyridine-Based Covalent Triazine Frameworks for the Hydrogenation of CO2 to Formate" Catalysts 8, no. 7: 295. https://doi.org/10.3390/catal8070295