1. Introduction
In recent decades, the impact of chemistry on society has largely been influenced by its capacity to make new molecules in an environmentally considerate and sustainable way. This has led to the development of catalysts with increasing efficiency and selectivity, which, when combined with the improvements in reaction engineering and purification methods, leads to greater atom economies and energy efficiencies [
1]. The industrial production of adipic acid historically relies on the aerobic oxidation of cyclohexane at 125–165 °C and 8–15 atm, usually in the presence of a homogenous cobalt-based catalyst. The resulting product mixture of cyclohexanol and cyclohexanone, known as KA oil, is then oxidised by nitric acid to adipic acid [
2,
3]. Alternative technology based on cyclohexene as a raw material for cyclohexanol synthesis is also now practiced commercially, with the product still being converted to adipic acid using nitric acid oxidation. The process for producing adipic acid from KA oil via nitric acid oxidation historically produced 400,000 metric tons of N
2O globally. N
2O has a global warming potential approximately 300 times that of CO
2 [
4]. However, recent catalytic and thermal abatement of these emissions within both adipic acid and nitric acid processes has resulted in very significant reductions in attendant N
2O emissions, and therefore of their environmental significance. However, in principle, processes for adipic acid synthesis that avoid the use of nitric acid as an oxidant eliminate concerns with N
2O emissions [
5,
6,
7] and offer a higher atom economy. Routes to adipic acid based on aerobic oxidation with air as the terminal oxidant are therefore of continuing research interest. The possibility of using cyclohexene as a feedstock has lately become an option because of recent improvements in the partial hydrogenation of benzene to cyclohexene in the Asahi process [
8]. The higher reactivity of cyclohexene allows it to be oxidised at lower temperatures by oxidants such as O
2 and hydrogen peroxide, and therefore avoids N
2O formation when using nitric acid. The first step in forming adipic acid from cyclohexene is epoxidation of the alkene, which can subsequently undergo hydrolysis to form cyclohexane diol. The diol can then be converted into adipic acid through the oxidative cleavage of the C–C bond, as demonstrated by Obara et al. [
9] and Rozhko et al. [
10] However, the prospect of achieving the one-step conversion of cyclohexene to adipic acid is hindered by the low miscibility of cyclohexane diol in cyclohexene, therefore, a second step would be required to form adipic acid.
Current epoxidations of cyclohexene require the use of stoichiometric oxygen donors such as chromates, permanganates, and periodates, which could result in complex heat management needs and by-products that are harmful to the environment [
11]. Peroxides are also commonly used to act as radical initiators [
12] or stoichiometric oxidants, however, these can increase the cost of the process and produce unwanted waste [
13]. High yields of cyclohexene oxide are also mostly achieved using specific solvents, which can become uneconomical and contribute to the high levels of waste produced [
14]. Therefore, the most sustainable alternative would be to run the reaction under solventless conditions using air [
15] or oxygen [
16] as the oxidant. However, this leads to lower selectivity where mainly products of allylic oxidation are observed [
17,
18].
In previous work, Sato et al. achieved the one-pot solventless oxidation of cyclohexene to adipic acid in the presence of an Na
2WO
4∙2H
2O catalyst, a phase transfer catalyst, and 30% hydrogen peroxide [
19]. This produced a 90% yield of adipic acid after 8 h at 90 °C in air. However, this reaction required 4.0 to 4.4 molar equivalents of H
2O
2 to cyclohexene. This ratio renders the process uneconomical on an industrial scale because of the expense of using hydrogen peroxide in these relative amounts. The presence of a toxic phase transfer catalyst is also a major drawback of this approach.
Gold nanoparticles have been shown to be active in a number of reactions, despite being originally considered chemically inactive. Since the pioneering work on gold nanoparticles by Haruta and Hutchings [
20,
21], gold has been shown to be active in several reactions, namely, low-temperature CO oxidation [
22,
23], C–C bond coupling [
24,
25], selective hydrogenation [
26,
27,
28], and oxidations [
29,
30]. Ovoshchnikov et al. assessed the reactivity of Au-based catalysts for cyclohexene oxidations using solvent-free and initiator-free conditions [
31]. It was found that the selectivity of the oxidations could be altered from allylic products to the epoxide via the choice of co-catalyst. A WO
3 co-catalyst was found to shift the selectivity towards cyclohexene oxide, while MIL-101 catalysed conversion of cyclohexenyl hydroperoxide to the allylic products. Using a WO
3 and Au/SiO
2 system, they could achieve 50% conversion and 26% selectivity towards the epoxide as the main product at 65 °C and 1 atm O
2. WO
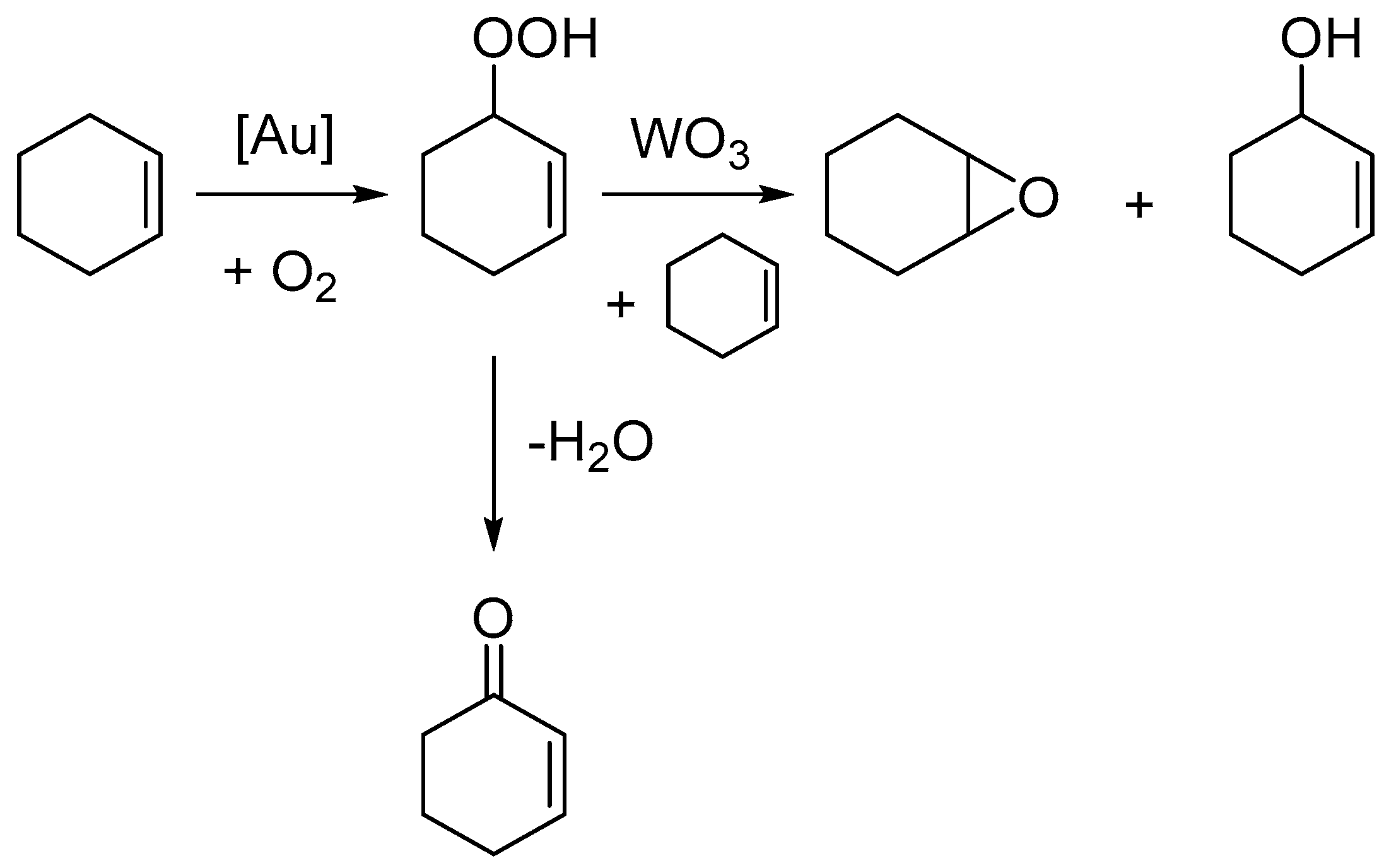
3 achieves this by promoting the reaction between cyclohexenyl hydroperoxide and cyclohexene to create cyclohexene oxide and 2-cyclohexen-1-ol, as shown in
Scheme 1. As a result of these findings, herein we present an investigation into the selective oxidation of cyclohexene under solventless and radical initiator free conditions to target cyclohexanediol with Au-based catalysts.
2. Results
The aim of this work was to modify conditions used by Ovoshchnikov et al. in order to achieve further oxidation of the epoxide to yield cyclohexane diol. For all catalysts, the Au content was maintained at 1 wt % with either a graphite or graphene support. The catalysts were prepared via an impregnation method or a sol immobilisation method and then analysed by TEM (transmission electron microscopy) and STEM (scanning transmission electron microscopy) techniques to quantify nanoparticle size and establish a relationship between nanoparticle size and selectivity to the desired products. Unless otherwise stated, all reactions were run at 3 bar O
2 at 60 °C for 24 h, with 0.1 g of catalyst (5.08 × 10
−6 Au mol %). The results are presented in
Table 1.
It was found in this study that the highest activity for a catalyst without any co-catalyst was observed for 1% Au/graphene (73.2%) prepared via sol immobilisation, however, this catalyst gave high selectivity to the allylic products. Similarly, the 1% Au/graphite catalyst prepared via sol immobilisation also gave high conversion, but also suffered from the same selectivity problem. The catalysts prepared via impregnation are significantly less active, with only 8.5% and 25.9% conversion for the 1% Au/graphite and 1% Au/graphene catalysts, respectively. Another observation was that the impregnation catalysts showed high batch to batch variability in the observed catalytic activity. Presumably, this is due to a large variation in the Au particle size distribution obtained between samples. Contrastingly, the sol immobilisation catalysts consistently gave high conversions between batches, with 61.4% over the graphite supported catalyst and 73.2% over the graphene supported analogue. The type of support appears to have a slight impact on the total conversion observed. This is evidenced by the result utilising graphene in the absence of gold, which gave a conversion of 4.3%, higher than the graphite, which showed only trace amounts of conversion. Furthermore, the impregnation and sol immobilisation catalysts supported on graphene both appear to give higher conversions than the graphite supported analogues. This may be due to some stabilising effect of the graphene sheets on the cyclohexenyl radical as observed by Yang et al. in the aerobic oxidation of cyclohexane, thereby increasing the rate of the autocatalytic reaction [
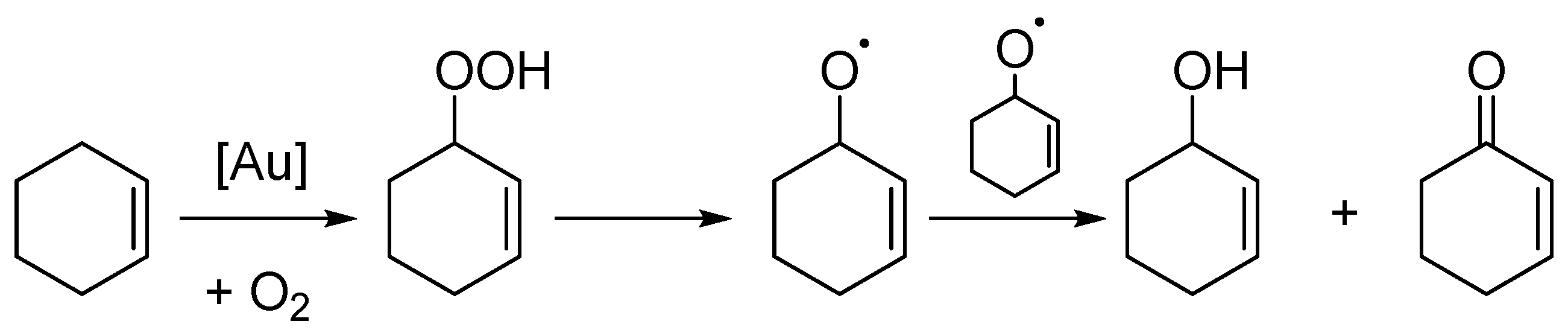
32]. In addition, the Au/graphene catalysts consistently provided higher selectivities for the allylic oxidation products in comparison with the Au/graphite catalysts. This adds further weight to the hypothesis that graphene promotes the allylic oxidation route, which, as suggested before, may be a result of a stabilising effect on the cyclohexenyl radical, which leads to allylic oxidation, as shown in
Scheme 2.
The effect of adding WO
3 to the reaction is seen to significantly increase the selectivity towards epoxide from 13.7% to 29.8% for the 1% Au/graphite (impregnation) catalyst, with the conversion also increasing from 8.5% to 17.7%. This increase in conversion occurs due to the WO
3 co-catalyst showing slight activity in converting cyclohexene to cyclohexene oxide in the absence of Au. The effect of adding WO
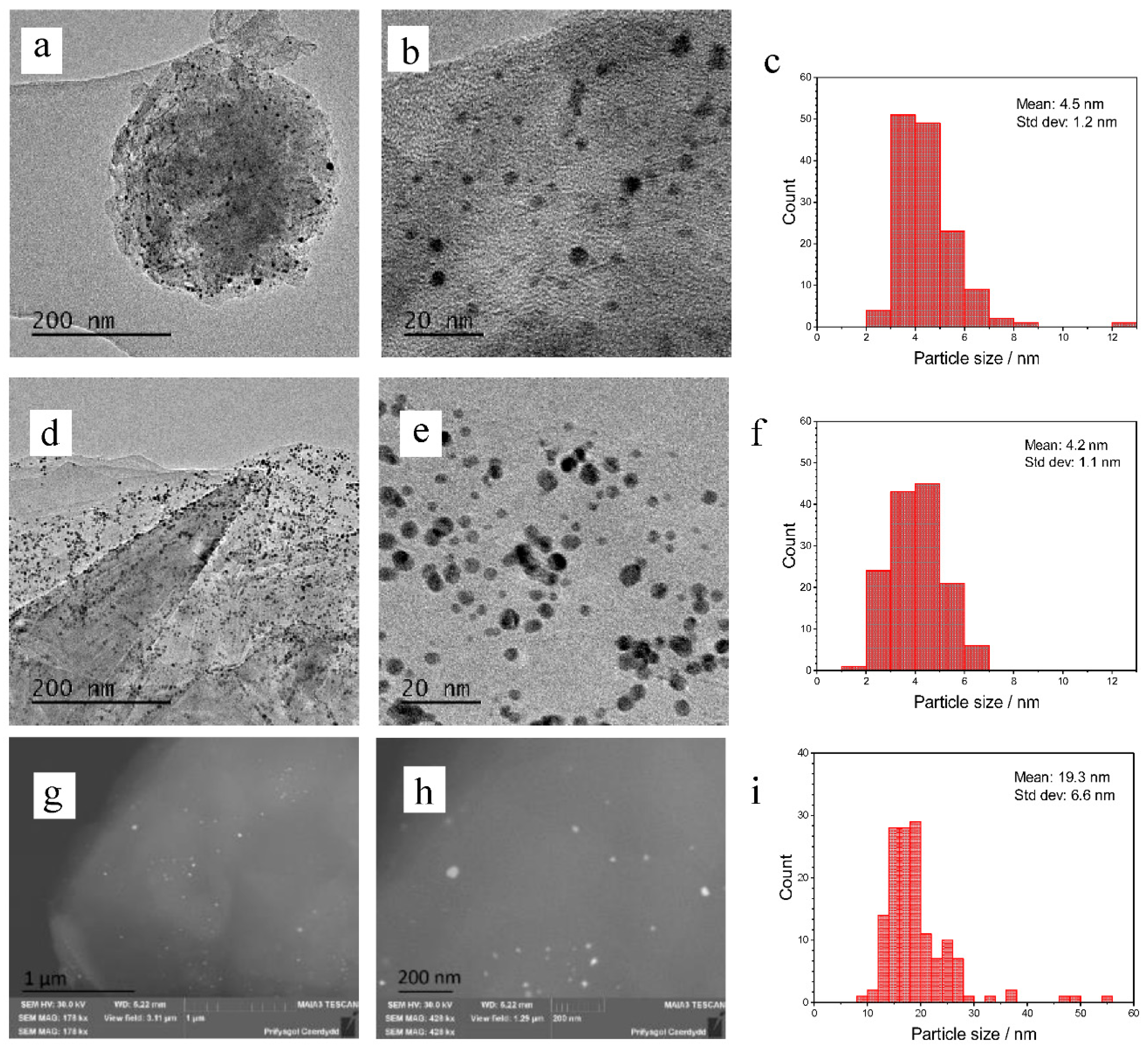
3 to the Au/graphene catalysts has a similar effect by increasing conversion and selectivity to the diol, however, this is observed to a lesser extent. Bright field TEM and STEM images of the 1% Au catalysts on graphene and graphite are shown in
Figure 1. The mean diameters of Au nanoparticles were determined by TEM for the sol-immobilised catalysts and BSE (back-scattered electrons) for the catalysts prepared via impregnation. We were unable to determine the Au nanoparticle sizes of Au/graphite as synthesised by impregnation using a TEM approach. This was predicted to be the result of a wide dispersion of nanoparticles across the catalyst surface and the possibility of an agglomeration of large, highly dispersed particles that could be more difficult to find. However, the particles could then be located after switching to BSE. The TEM images of the sol-immobilised catalyst show an equal dispersion of nanoparticles across the graphite and graphene surface, as would be expected of this technique. The Au graphite catalyst has a mean particle size diameter of 4.2 nm and the Au graphene has a similar mean particle size of 4.5 nm. The standard deviations are also similar at 1.1 and 1.2 nm for the graphite and graphene supported catalysts, respectively, suggesting that the supports themselves have no impact on the particle sizes or dispersion. The particle size distribution is also much narrower, as would be expected of the technique, which can allow good control over nanoparticle size. The BSE images of the impregnation catalyst show a much wider dispersion and distribution of particles, as is common in this technique. These particles had a mean diameter of 19.3 nm and a standard deviation of 6.6 nm. The particles also ranged from 8.7 nm to 55.3 nm. This large range in particle size could account for its lower activity relative to the sol-immobilised catalysts due to the general inactivity of large particles towards catalytic epoxidation [
33]. However, this particle size range may be inaccurate due to the difficulty in measuring particles, which may be too small to resolve. The BSE images suggest that the large particles mainly reside in the centre of the graphitic sheets, rather than at the sheet edges as with the sol catalysts. This explains why they were not observed during initial TEM studies.
Despite their high activity, the sol immobilisation catalysts do not display promising selectivity to the epoxide even with the addition of a co-catalyst. In addition to this, the graphene supported materials seemed to favour allylic products. Therefore, further studies were conducted on the 1% Au/graphite, prepared via impregnation, to further oxidise the epoxide to the diol due to its high selectivity to the epoxide (29.8%) in the presence of WO
3. Small amounts of diol were observed in all the previous reactions, through hydrolysis of the epoxide. However, these may have been limited by the amount of available water in the reaction. Water is formed as a by-product in the dehydration of the hydroperoxyl intermediate to the allylic ketone. We thus decided to add additional water at the start of the reaction to assess this theory and attempt to hydrolyse all epoxide through to the diol. On addition of water, the epoxide was almost entirely converted to the diol, achieving a total selectivity of 17.0% (
Table 2). However, there was a noticeable drop in conversion to 11.8%, which suggests that water has a detrimental effect on overall reaction and the amount may need to be further tuned or added in a stepwise manner.
3. Discussion
These results demonstrate the challenges in utilising cyclohexene as an alternative substrate for the formation of adipic acid through solventless and green conditions. The use of diatomic oxygen as the oxidant is highly desirable both from an economic and environmental viewpoint. However, as described here, and in previous work by Ovoshchnikov et al., the formation of the allylic hydroperoxyl intermediate ultimately limits the possible yield of epoxide to 50% because of the formation of equimolar amounts of the allylic alcohol. This then limits possible yield to cyclohexane diol, a key intermediate in the formation of adipic acid. An adapted mechanism from the paper by Ovoshchnikov et al. is shown in
Scheme 3 for the formation of cyclohexane diol with the addition of water [
31]. The results in
Table 2 demonstrate the importance of water in the formation of cyclohexane diol, and therefore any process that achieves high selectivity to the epoxide would be limited because of the lack of in situ water formed via dehydration of the hydroperoxyl intermediate. Furthermore, any large improvements made to yield cyclohexane diol would face additional problems due to its insolubility in cyclohexene. Synthesis of adipic acid would thus require separation of the produced diol, solvation in water, and subsequent oxidation, as shown by the work of Obara et al. and Rozhko et al.
In this study, we have also demonstrated the variation in activities and selectivies attainable through simple modification of gold catalysts. The previous study by Ovoshchnikov focused on the synthesis of PPh
3 stabilised gold nanoclusters, which were then deposited on various oxide supports. This work shows high activity for cyclohexene oxidation is achievable through the use of simple sol immobilisation catalysts. However, despite promising activation of diatomic oxygen and subsequent cleavage of the resulting peroxyl intermediates, considerable further improvements are required in order to obtain high yields of cyclohexene epoxide or diol. It is possible that there is a strong dependence on nanoparticle size of the gold for epoxidation, considering the large differences observed here between sol immobilised and impregnation catalysts. The high activity of the sol immobilisation catalysts was such that only trace amounts of epoxide were observed even in the presence of WO
3, which is able to direct the breakdown of the hydroperoxy intermediate (
Table 1). However, with these catalysts, there were appreciable amounts of diol formation, which suggests that epoxide was formed at some stage in the reaction and further oxidised to the diol as a consequence of in situ generated water. The highest selectivity to the epoxide was achieved by the standard impregnation catalyst in combination with WO
3. This large discrepancy between sol immobilisation and impregnation selectivity is likely linked to their activity, as the small nanoparticles observed in the sol immobilisation catalyst rapidly catalyse the breakdown of the intermediate species to allylic products. This also suggests that optimisation of reaction temperature and the stoichiometry of catalyst and co-catalyst used could yield higher amounts of either epoxide or diol. The latter may be limited at lower temperatures because of hydrolysis of the epoxide occurring at around 60 °C [
34]. Temperature ramping studies could allow for the higher formation of epoxide followed by ring opening to the diol. However, as per the previously mentioned limitations with respect to the reaction mechanism, the diol yield would likely peak at 50%.
Taking into consideration the above points, the use of this system for production of cyclohexane diol would not be a suitable replacement of the current industrial process. Formation of adipic acid by further oxidation of this diol in a one-pot approach is further limited because of the formation of cyclohexane dione rather than the hydroxyketone, as seen in aqueous systems with either oxygen or hydrogen peroxide as oxidant. The lack of water also provides significant barriers to the formation of diol and hydration of adipic anhydride to adipic acid.
We suggest that these catalysts may be more interesting when utilised in a biphasic system for activation of oxygen and subsequent oxidation of a co-catalyst for the highly selective oxidation of cyclohexene. For this system, an emulsion may need to be stabilised in work similar to that described by Zhou et al., who designed amphiphilic silica particles for the solvent-free acetalisation of long-chain fatty aldehydes with ethylene glycol [
35]. In addition to this work, He et al. has suggested that graphene oxide particles can also be used to stabilise emulsions of the oil-in-water type [
36]. In this case, gold would need to be in a separate phase to the cyclohexene because of its high activity in cyclohexene oxidation, which could be achieved by the use of hydrophilic supports. Alternatively, the equimolar use of hydrogen peroxide per mole of epoxide produced would be a more economically viable process. Formation of the diol from this would be achieved by hydration and subsequent aqueous phase oxidation to adipic acid would be achieved by the use of diatomic oxygen as previously discussed. The majority of current epoxidations either use excess hydrogen peroxide or fail to achieve economic use to the epoxide, as a result of the breakdown of peroxide under reaction conditions or over oxidation of the initial product. Our recent work has suggested that highly efficient use of hydrogen peroxide can be achieved by the use of colloidal gold and palladium bimetallic catalysts partnered with the stepwise addition of peroxide for oxidation of methane [
37]. A similar approach in this system could provide an economically viable approach to epoxidation of cyclohexene and subsequent formation of cyclohexane diol.
4. Materials and Methods
4.1. Materials
Gold precursor (99.8%) was purchased from Strem, Sodium borohydride (>97%), hydrochloric acid (37%), nitric acid (65%), poly vinyl alcohol (80% hydrolysed), sulfuric acid (95–98%), n-decane (99%), cyclohexene (99%, inhibitor-free), 2-cyclohexen-1-ol (95%), 2-cyclohexen-1-one (98%), and cyclohexene oxide (98%) were purchased from Sigma-Aldrich. The graphite and tungsten (VI) oxide (nanopowder, <100 nm particle size) were purchased from Sigma-Aldrich (Gillingham, Dorset, UK). Graphene nanoplatelets aggregates (<1 μm) were purchased from Alfa Aesar. All materials were used without further purification.
4.2. Catalyst Preparation
4.2.1. Impregnation
HAuCl4 (0.816 mL, 12.25 mg/mL) was added to graphite or graphene (0.99 g) in a vial with enough water to create a thick slurry. This mixture was then heated to 80 °C and stirred for 30 min or until the stirrer bar became immobilised. The resulting paste was then dried for 17 h at 110 °C, yielding a black powder.
4.2.2. Sol Immobilisation
HAuCl4 (0.816 mL, 12.25 mg/mL) was diluted in de-ionised water (400 mL) and stirred for 15 min. PVA (0.65 mL, 1 mM) was added to the mixture and stirring was continued for 15 min. NaBH4 (1.269 mL, 0.2 M) was then added to the solution, causing a colour change from pale yellow to red. This mixture was then stirred for 30 min. Graphite or graphene (0.99 g) was then added to the solution with five drops of H2SO4 to turn the pH to 2. This mixture was then stirred for 1 h and filtered and washed with de-ionised water (1 L). The resulting powder was dried for 17 h at 110 °C, yielding a black powder.
4.3. Catalyst Characterization
Scanning transmission electron microscopy (STEM) was performed on a Tescan MAIA 3 field emission gun scanning electron microscope (FEG-SEM) fitted with secondary and backscattered electron detection. Transmission electron microscopy (TEM) was performed on a Jeol JEM 2100 LaB6 microscope operating at 200 kV. Samples were suspended in ethanol and then deposited on holey carbon-coated copper grids. At least 150 particles were measured to plot size distributions.
4.4. Catalyst Testing
Cyclohexene oxidation was performed in a Colaver reactor heated using an oil bath. The system was flushed with oxygen and remained connected to an O2 line throughout the reaction. Typically, a mixture of cyclohexene (10 mL), catalyst (0.1 g), and n-decane (1 mL, as an internal standard) was magnetically stirred (900 rpm) at 60 °C. If required, WO3 (0.1 g) and deionised water (1 mL) were also added to the mixture. After 24 h, the reactor was cooled down to room temperature and the reaction mixture was separated from the solid catalyst by centrifugation. Where two phases were formed, ethanol (1 mL) was added to solubilise both layers. The liquid samples were analysed via gas chromatography (GC). Quantification of conversion and selectivity was conducted using n-decane as an internal standard with calibrations against bought analytical samples. Selectivity was found in most cases to be largely to the allylic ketone and alcohol, cyclohexane oxide, and cyclohexane diol. However, because of the high activity of the catalysts, there was a large number of unknowns. Gaseous products were not analysed because of the use of a Colaver reactor, however, this contribution is likely to be negligible. The presence of cyclohexenyl hydroperoxide was not confirmed as mentioned in the literature, despite the use of GC-MS and H-NMR.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}