Constructing A Rational Kinetic Model of the Selective Propane Oxidation Over A Mixed Metal Oxide Catalyst


1
Stirnerstr. 12, 12169 Berlin, Germany
2
Department of Energy, Environmental and Chemical Engineering, Washington University, St. Louis, MO 63130, USA
3
Fritz-Haber-Institute of the Max-Planck-Society, Department of Inorganic Chemistry, Faradayweg 4–6, 14195 Berlin, Germany
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(8), 330; https://doi.org/10.3390/catal8080330
Submission received: 14 July 2018
/
Revised: 6 August 2018
/
Accepted: 9 August 2018
/
Published: 13 August 2018
(This article belongs to the Special Issue New Developments in Heterogeneous Partial and Total Oxidation Catalysis)
Abstract
:This research presents a kinetic investigation of the selective oxidation of propane to acrylic acid over a MoVTeNb oxide (M1 phase) catalyst. The paper contains both an overview of the related literature, and original results with a focus on kinetic aspects. Two types of kinetic experiments were performed in a plug flow reactor, observing (i) steady-state conditions (partial pressure variations) and (ii) the catalyst evolution as a function of time-on-stream. For this, the catalyst was treated in reducing atmosphere, before re-oxidising it. These observations in long term behaviour were used to distinguish different catalytic routes, namely for the formation of propene, acetic acid, acrylic acid, carbon monoxide and carbon dioxide. A partial carbon balance was introduced, which is a ‘kinetic fingerprint’, that distinguishes one type of active site from another. Furthermore, an ‘active site’ was found to consist of one or more ‘active centres’. A rational mechanism was developed based on the theory of graphs and includes two time scales belonging to (i) the catalytic cycle and (ii) the catalyst evolution. Several different types of active sites exist, at least as many, as kinetically independent product molecules are formed over a catalyst surface.
1. Introduction
Light alkenes are largely produced from crude oil feedstock by steam-cracking. These hydrocarbons are transformed into valuable intermediates, such as ethylene oxide, acrylic acid, and maleic anhydride. Current research efforts are undertaken to establish alkanes instead of alkenes as feedstock for the latter processes, hence, facilitating the change from a crude oil to natural gas-based economy in this respect. The synthesis of maleic anhydride from butane has already been realised in larger scale, whereas the analogous selective oxidation of propane and ethane still faces challenges in their performance to compete with the established alkene-based processes [1,2].
For the complex reaction of propane oxidation on oxide catalysts, particularly the MoVTeNbO in the M1 structure, has been studied by many researchers under different conditions, including atmospheric conditions and high vacuum (cf. Table 1).
In this article, we will analyse this process as an example of the selective oxidation process over an MoVTeNb oxide catalyst.
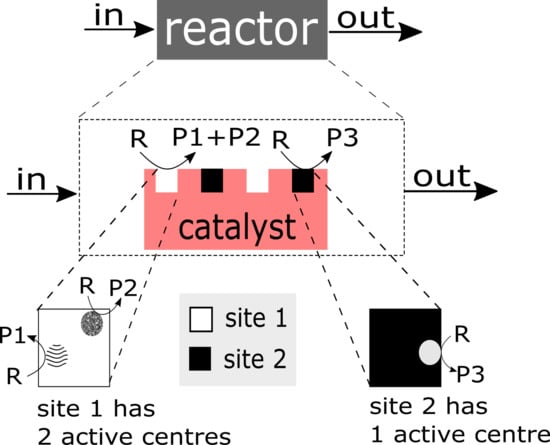
As many catalytic heterogeneous reactions, the selective oxidation of propane over MoVTeNbO phase, is a complex physico-chemical process. Three interwoven aspects have to be stressed: (a) it is a multi step process, (b) it occurs over the active catalytic site (centre) of complex structure, and (c) it is a process with multi-scale temporal behaviour. A categorisation is presented in Figure 1 to visualise different levels of complexity.
- (a)
- Multi step process: The main steps of this process are: (i) interactions between components of the reaction mixture and the catalytic surface, i.e., the impact reaction between the molecule of gaseous propane and catalyst oxygen, adsorption of water etc; (ii) mass transfer processes, i.e., oxygen exchange between the catalyst surface and bulk; (iii) transport of charge (electrons and/or ions) within the catalytic unit.
- (b)
- Complex structure of the active site/centre: The ‘structure of the catalyst’ tackles the nature of the catalyst surface; i.e., the architecture of catalytically active site/centre, how many active centres are considered per site, and furthermore, how many sites are distinguishable etc.An empirical concept of site isolation proposed by Grasselli [19,20,21] can be used as one of basic structure pre-assumptions for the rational kinetic model. This concept is presented as follows: “The catalytic reaction sequence should occur on an ensemble of atoms (‘the active site’) that holds together with the educt molecule all other reactants and exchanges electrons between the reactants. For these operations neither the influx of active species during the unit conversion time (as long as the educt is adsorbed) nor the participation of electrons from outside of the active site are required ... The catalytic cycle is only finished when the site is regenerated into its initial active form, being here fully oxidised and holding all oxygen atoms belonging to its initial structure” [22].In this concept, many additional processes are not considered, such as processes of the formation of active sites, processes of spill-over, rather to say, all processes external regarding the isolated sites. From the kinetic point of view, the process over the isolated site will be characterised by the specific relationship between the rate of reactants and products which participate in this process. However, these relationships are still not recognised.In the literature, there are many considerations on the complex functional structure of the active sites which is responsible for generation of different products. It makes sense to distinguish: a ‘whole active site’ and ‘active centre’ (or ‘centres’) within this site, which generates (or generate) the specific product(s). One ‘active site’ consists of at least one ‘active centre’. There are speculations about dimer- and trimer structures within the whole active site. These different centres can be considered as ingredients over which different catalytic routes are performed and different products are generated. In particular, it was assumed that an active site for oxidation of propane may consist of dimer- and trimer-ingredients. Dimers and more complex structures (trimers and even ‘dimers of dimers’) can store redox-equivalents as a part of the process of oxygen activation.It is clear that structures of active sites and/or active centres mean surface structures. The surface of a catalyst material (e.g., a metal or metal oxide) may significantly differ from its bulk structure and composition, and may additionally be affected strongly by the gas phase surrounding it [12,23,24,25,26,27].
- (c)
- Multi-scale temporal behaviour: As for multi-scale temporal behaviour of the catalytic process, at least three characteristic times have to be distinguished; ‘fast’ and ‘moderate’ intrinsic times of the catalytic cycle, and the ‘slow’ time, which is external. The latter is caused by the modification and evolution of the catalytic centre/site under the influence of reactive medium. In the catalytic literature, this phenomenon was described and conceptualised by Boreskov [28], see also Yablonskii et al. [29]. A variety of physical methods for the investigation of the surface and bulk of catalysts and different methods of kinetic analysis, mostly steady state methods, have been used to investigate this process.
1.1. Investigations of the Selective Oxidation of Propane. Methods and Results
1.1.1. Physical Methods
Several methods were applied to characterise the catalyst before and after reaction to report their change. Micro calorimetric experiments showed a lower adsorption enthalpy of propane after reaction, suggesting a significant change in the nature of the catalyst surface [25]. Furthermore, ESR (electron spin resonance) and IR (infra-red) investigations found different sites for propane activation and nucleophilic oxygen addition on MoO catalysts, as well as different types of lattice oxygen and their role during the reaction are discussed [30]. A combined LEIS (low-energy ion scattering), SEM (scanning electron microscopy) and TEM (transmission electron microscopy) investigation of M1 catalyst found that the (001) plane of the M1 crystals being not solely responsible for the selectivity during propane (amm)oxidation [31].
In situ experiments are very valuable for the characterisation of catalysts under working conditions. Most important, and still missing, is their link to kinetic interpretations. In situ XPS (X-ray photoelectron spectroscopy) investigation of MoVTeNbO M1 catalysts at 623 K and dry and wet CH/O feed found the production of acrylic acid correlated with the Te/V ratio and inversely correlated with the abundance of Mo [25,32].
Bulk and surface V content correlated with the selectivity to acrylic acid, determined by in situ XPS [5].
Experiments were performed in propane/oxygen feed with and without steam. The addition of steam was beneficial for the selectivity to acrylic acid, as it was found before, which was observed with an enrichment in V and Te, and depletion in Mo on the surface of the catalyst. Simultaneously, the conductivity decreased together with the work function and a modification of the valence band structure. A sub-surface space charge region was formed, which was depleted in electrons under reaction conditions. The extension of band bending was not affected by the addition of steam, hence no affect on the number of charge carriers in this space charge region. Near-edge X-ray absorption fine structure (NEXAFS) investigations point to a significantly modified electronic structure of the surface, especially the vanadyl bond motif (suggesting an enrichment of covalently bonded V) [33].
1.1.2. Kinetic Methods. Model-Free Kinetic Data
For the investigation of the selective oxidation of propane; mostly the plug flow reactor (PFR) has been applied. Examples for these investigations are summarised in Table 1.
Typically, there was investigated an influence of the three reactants, i.e., propane, oxygen, and water.
The essential role of water in activating the catalyst is widely accepted. Without the presence of its large excess (in comparison to stoichiometric requirements), neither acetic acid nor acrylic acid would form on the catalyst. Centi et al. demonstrated the necessity of surface modification by water and/or ammonia prior to observable selective (amm)oxidation products [34].
Novakova et al. [6] determined apparent kinetic orders for propane disappearance, first order with respect to propane, 0.26 with respect to oxygen. The formation rate of propene is first order with respect to propane and 0.26 order with respect to oxygen. Formation of acrylic acid is close to first order in propane and one half with respect to oxygen. Water increased propane conversion and selectivity to oxygenated products. Space time variations show non-zero intercept for acrylic acid and acetic acid. Propene is considered a major intermediate, for the formation of acetic acid and acrylic acid.
Vitry et al. [7] determined kinetic orders for different reactants: propene (0.02/0.86), acrylic acid (0.15/0.55), acetone (−0.34/0.88), acetic acid (0.09/0.56) with respect to p(O)/p(CH).
The kinetic order was determined to be 0.88 and 0.06 with respect to propane and oxygen partial pressure, respectively [10]. The independence of oxygen partial pressure was considered for all rates, if its concentration is above 1 vol-%. Below that threshold the re-oxidation becomes very slow and the oxygen partial pressure becomes kinetically relevant [10].
A constant ratio between propene formation and consumption, being 1:25, was observed [35].
Several O/CH and HO/CH ratios have been investigated for testing the predictability of power law, Eley-Rideal (ER) and Mars-van-Krevelen (MvK) models. Mazloom et al. [11] analysed these models and found that the oxygen dependency was described most accurately by the MvK model. In addition, they concluded that under water free conditions the propane activation is rate determining, whereas under wet conditions it is the oxygen activation. Generally, most of researchers support the MvK mechanism for the selective oxidation reactions over mixed metal oxide catalysts [1,2,36,37].
Naumann d’Alnoncourt et al. investigated the propane oxidation over MoVTeNbO in the M1 structure. As found before by Novakova et al. [6] steam is beneficial for acrylic acid production. The apparent activation energy was found to depend on the feed composition [12].
Summing up, one can say, that most of the kinetic data are likely obtained in the small conversion domain. The apparent kinetic order of propane was determined close to first order with respect to propane and zero or small with respect to oxygen. As for the influence of water, its beneficial character can be considered as a reasonable consideration. However, literature data regarding the kinetic order of water are still uncertain.
1.1.3. Mechanisms
Since the 1970s, in the literature there was discussion regarding the details of selective propane oxidation focused on main questions:
- Is the mechanism parallel or consecutive, or parallel-consecutive, regarding the different gas products, in particular propene?
- Does this complex reaction occur on a single site or multiple sites?
- What is the nature of an active site?
In early isotopic labelling experiments for the reaction of propene with HO over a MoWSnTeO catalyst, the incorporation of O into the acrylic acid molecule was observed [15].
A comprehensive isotopic labelling (D and C) investigation over Fe-Sb oxide catalysts was performed. Starting from propene; no significant H/D scrambling in the propene precursor could be observed, neither a double bond isomerisation. The slow step is the allyl formation from adsorbed propene [38].
Two different active sites were proposed, i.e., an acidic and an oxidizing site (cf. Figure 2), in investigation on BiMo-oxide catalysts [39,40]. Either site may contribute to scrambling of the deuterium labelled carbon atom being oxidised. The rate determining step is the abstraction of the allyl hydrogen to form the -allyl complex.
Ammoxidation and oxidation of propene over molybdate and antimonate catalysts were investigated. For molybdates and oxidation, the allyl formation was considered as slow compared to other reaction steps, and the first oxygen insertion reversible [41].
Investigation of the role of water was performed by Saleh-Alhamed et al. on the partial oxidation of propene [42,43]. Water suppresses the CO formation, by blocking the most active sites and also keeps the catalyst at a high oxidation state. Water competes with acrylic acid for adsorption sites.
According to Barrault et al. [3] the addition of water changed selectivity, not the activity of the catalyst. Mo species are responsible for the formation of acetic acid, where as Mo species support the formation of acrylic acid, via acrolein. Both reaction pathways branch off from propene as a central intermediate product.
Furthermore, Barrault et al. [3] found a parallel reaction pathway for acetic and acrylic acid. Allylic oxidation is considered to occur over coordinatively unsaturated Mo sites in combination with lattice oxygen. If a site is highly reduced and slowly reoxidised with molecular oxygen, the lattice may not be able to provide enough lattice oxygen to achieve high selectivity for the allylic oxidation.
For the similar ODH of propane process, Chen et al. [13] proposed a first order reaction network (propane → propene, propane → CO, propene → CO), and detailed mechanism for ODH over MoO/ZrO and VO/ZrO catalysts, and these mechanisms were similar. The initial C–H bond cleavage was to be rate determining. It was suggested, that the relative adsorption enthalpies of propene and propane influence the relative reaction rates of these molecules, as well as the Lewis acidity of the cations involved in the adsorption. Propane ODH and propane combustion show similar activation energies, hence, the fate of reaction is decided after the kinetically relevant step. Activation energies are higher for propene combustion. Additionally, a large number of isotopic labelling experiments was performed.
Lin et al. [44] investigated the selective oxidation of propane on several catalyst systems, mainly the mixed metal oxide MoVTeNbO in the M1 structure, and proposed a first order reaction network [45]. Also Lin et al. [44] proposed propene as the central intermediate from which the major pathway to acrylic acid via acrolein branches off, together with the minor pathway to acetic acid via acetone.
Luo et al. [46] studied the partial oxidation of propane, emphasising the differences in reaction pathways over zeolite VO-H-beta and MoVTeNb-oxide. The schemes in Figure 3 compare both catalysts suggesting the reaction in zeolite H-beta being driven by its acidity, whereas in MoVTeNb-oxide the oxidation of a C-chain rather than the cracking and total oxidation was favoured. Furthermore, two sites for MoVTeNb-oxide are suggested; the first site catalyses the reaction propane-propene-acrylic acid, whereas the second supports the pathway propene-acetic acid.
Centi et al. [34] proposed a reaction mechanisms for the selective (amm)oxidation reaction. It was pointed out the role of surface dynamics, i.e., the modification of the surface reactivity due to chemisorbed species (reactants, intermediates, and products), hence, the dynamics of catalyst reconstruction. Multiple pathways exist even for the same reaction product.
Vitry et al. [7] assumed at least three catalytic oxidation sites to be present on the catalyst surface (i.e., for propane activation, allylic oxidation, and acrolein oxidation) and supported this assumption by data on different kinetic orders.
A more comprehensive mechanism including acetic acid and propionic acid was presented by Novakova et al. [6]. Their investigations were performed over a Mo-V-Sb-Nb oxide catalyst. The scheme is presented in Figure 4a. The order of CO and CO formation with respect to oxygen is 1.1 and 0.8, respectively. Acrylic acid was found to be first order in propane and close to one half in oxygen [6].
Balcells et al. [47] employed a first-order power rate law network, where propene is considered to be the only primary intermediate, form which acrolein, acrylic acid and by-products are formed in parallel pathways.
Grabowski et al. compared two kinetic models, namely related to the Langmuir-Hinshelwood (LH) mechanism and Eley-Ridel (ER) mechanism. The latter one achieved a better description of the experimental data [48]. Kinetics of the oxidative dehydrogenation of C–C alkanes was summarised in a comprehensive Grabowski article [49].
Based on the network by Lin et al. [45], Naraschewski et al. [10] modified the scheme of the reaction by introducing two additional reaction pathways (cf. Figure 4b). The rate equations were fitted to experimental data [10].
It is assumed that only one active site associated with vanadium exists, and furthermore, that not all vanadium present is part of the catalytically active cycle. Hence, there are active and inactive sites containing vanadium. The initial C–H bond cleavage is considered to be rate determining, however, the subtle change in V concentration, hence, substitution for Mo in the M1 structure, affects the activity and selectivity of the catalyst. Not all V sites are catalytically active. Sites are distinguished for propane adsorption and oxygen insertion, and C–C bond cleavage. The formation of acetic acid via acetone can be lowered by decreasing the Brønsted acid sites [10].
Temporal analysis of products (TAP) pulse-response investigations were performed over Mo-V-Te-Nb oxide catalysts [9]. The proposed reaction scheme is presented in Figure 5. The main features are the competition of water, propene and oxygen for the same vacancy. Acrylic acid may originate from propane directly (no re-adsorption of propene). Acetic acid, however, originates from re-adsorbed propene.
Selective oxidation of propane was investigated by Naumann et al. [12,50] leading to acrylic acid. An interesting peculiarity was found, i.e., a maximum in the selectivity of acrylic acid as a function of propane conversion. Furthermore, the dependence of apparent activation energies was determined as a function of reaction conditions.
Ammoxidation of propane provides insight into the reducibility of the MoVNbTeO catalyst. The catalyst could be reduced with CH/NH pulses (analogy to the results of reduction in CH/HO feed of this work) up to a depth of 70 layers. The catalyst could be re-oxidised without structural collapse using O pulses [14].
The measurement of kinetic isotope effects (KIE) in propane oxidation revealed essential differences in the activation of the propane molecule over MoVTeNb oxide with M1 structure and silica-supported vanadium oxide. Whereas the abstraction of the first hydrogen atom at the methylene group of the CH molecule is rate limiting over silica-supported vanadium oxide, activation of propane over M1 clearly involves both methyl and methylene groups simultaneously in rate-limiting process(es) [51]. The formation pathways of carbon oxides during propane oxidation were studied by means of pulse experiments with C-labeled/unlabeled propane/oxygen mixtures. Over M1, oxidation of acrolein to acrylic acid via oxygen insertion into the C–H bond of the CHO group is faster than oxidation of the vinyl group followed by decarbonylation of the resulting surface species and acetaldehyde formation. In contrast, decarbonylation of acrolein is preferred over silica-supported vanadium oxide showing that surface structure and electronic properties of the catalyst have an impact on the reaction mechanism.
1.1.4. Catalyst Structure and Nature of the Active Site
Generally it is agreed, that vanadium sites are required for the activation of alkanes. However in their investigation, Holmberg et al. [52] found the importance of the chemical surrounding of such V-sites, thus, only the orthorhombic (not the hexagonal) phase of a MoVNbTeO catalyst was active for paraffin activation. Furthermore, they found that both orthorhombic and hexagonal phases are selective for the ammoxidation of propene.
Vitry et al. [53] found that the addition of niobium to a MoVTeO catalyst increased the selectivity to acrylic acid.
Grasselli et al. [14] investigated MoVNbTeO catalysts in M1 and M2 structure, namely their physical mixture as particles and powders. The latter exhibit a much higher selectivity for the selective oxidation of propane. Furthermore, the redox behaviour was investigated. The catalyst changed its selectivity depending on its oxidation-reduction state. This reduction-oxidation process was found to be reversible.
Oxygen exchange between the catalyst bulk and surface is traditionally taken into account for the explanation of kinetic behaviour of selective oxidation of propane. As a development of this concept there was proposed, that a reactive site acts as a collective electron storage unit. [18] The first consideration is: this ‘exchange’ idea contradicts to the ‘site isolation’ concept. However, it maybe considered as an apparent contradiction: it is a subject of the further analysis.
The parameter for this exchange, which can be found by conductivity analysis, may be used as a coefficient in our kinetic model for oxygen exchange [12].
1.2. Aim
In the present work, we are aiming to develop a methodology for designing rational kinetic models employing the selective oxidation of propane over MoVTeNbO catalysts as an example. Also we attempt to link features of temporal kinetic behaviour to mechanistic and structural aspects, proposing a kinetic fingerprint for the justification for the isolated site concept.
2. Results
2.1. Characterisation
The catalyst has been characterised by several methods. One of the most important information is presented in Figure 6. Both the ‘fresh’ and ‘used’ catalyst (i.e., after at least the here presented kinetic investigations) show typical reflexes for the M1 structure. The reflex positions and width are, in comparison to the ‘fresh’ sample, preserved for the ‘used’ sample. The structural features of M1 phase remain unchanged, despite repeated reducing/oxidising treatment to the catalyst.
The BET surface area was determined to be 7.31 m g. X-ray fluorescence (XRF) investigations gave an element distribution of Mo/V/Te/Nb/O = 46.27%/6.74%/6.82%/7.01%/33.2%. The catalyst appeared in highly crystalline rods of several 100 nm in length with an homogeneously distributed elemental composition, revealed by TEM and SEM characterisation, respectively.
2.2. Reaction Equations
In our reaction system with eight components (, , , , , , and , i.e., propane, oxygen, water, carbon monoxide, carbon dioxide, acetic acid and acrylic acid, respectively), which consist of three elements (i.e., C, H, and O), we assume the following set of five () independent overall reactions (Equations (1)–(5)). Reaction (1), (3)–(5) have only one carbon containing product, , , , and , respectively. Reaction (2) has two carbon containing products, i.e., and .
2.3. Justification of Kinetic Data
The first step of kinetic analysis is testing the validity of obtained data using elemental balances. In this work it was done via checking the elemental balances for carbon and oxygen with an accuracy of ±3%.
2.4. Kinetic Experiments under Catalyst Steady-State Conditions
Partial pressure variation experiments were performed under steady-state conditions (CH/O/HO/N = 3/12/10/75, 573 K and a weight hourly space velocity (WHSV) of 10 mL g h). The feed of one reactant (i.e., CH, O, and HO, respectively) was changed, maintaining the feed of the respective other reactants and adjusting the flow of nitrogen in order to keep the WHSV constant. The observations are shown in Figure 7 and Figure 8. All product formation rates are proportional to propane partial pressure, most pronounced for the production of acrylic acid and propene. CO, acetic acid and CO form a group of similar dependence (cf. Figure 7a). Only acrylic acid showed a slight positive dependence on the partial pressure of oxygen, whereas all other products were observed to be independent (cf. Figure 7b). The dependence on water partial pressure is presented in Figure 8. Acrylic acid, acetic acid and CO are clearly proportional with respect to water partial pressure. From the dependence of the latter two, it was immediately concluded, that the acetic acid production was accompanied by the production of CO, which is also shown in Equation (2).
We distinguish , which originates solely from non-selective oxidation (cf. Equation (4)) and related to the formation of acetic acid, as presented in Equation (2). To evaluate the production separately from the production of acetic acid, the latter was subtracted from , determined according to Equation (6).
Thus, the turquoise bullets in Figure 7 and Figure 8 represent the previously described formation rate of , independent of the production due to acetic acid formation.
Table 2 summarises the apparent kinetic orders of product formation rates with respect to all three reactants, i.e., propane, oxygen, and water. The apparent kinetic order of a product formation rate was determined according to Equation (7).
The apparent kinetic order of CO, propene, and acrylic acid with respect to propane are similar, in the range from 0.6–0.8, however, their kinetic coefficients vary within one order of magnitude. The trends of CO and CO-aca are rather similar, certainly with respect to oxygen and water, in which both kinetic coefficients and apparent kinetic orders are much alike. Propene and both carboxylic acids have similar apparent orders with respect to propane and oxygen, also their kinetic coefficients with respect to propane are rather similar. A significant difference is observed in their kinetics with respect to water. Propene clearly does not depend, whereas both acetic and acrylic acid show an apparent order of one half. Water is, thus, necessary for the formation of acetic and acrylic acid, whereas it is at the same time a reaction product, hence, to some extend we can call it auto-catalysis regarding water.
Overall, the sum of apparent kinetic orders is smaller or about one for CO, CO, and propene. Acetic and acrylic acid apparent kinetic orders sum up to a value of 1.5.
2.5. Kinetic Experiments during Catalyst Evolution (‘Slow Dynamics’ Scale of Min and Hours)
The experimental data in Figure 9 and Figure 10a was recorded after a treatment of the catalyst in reducing conditions (i.e., 12 h in CH/O/HO/N = 3/0/10/87 at 573 K and 10 mL g h). Subsequently, the oxygen feed was added (i.e., CH/O/HO/N = 3/12/10/75 at 573 K and 10 mL g h). The gas analysis was started in this moment, representing time zero. The chemical composition of gases from the PFR exit was measured every 20 min to observe the ‘slow’ process of the kinetic steady-state (rather say pseudo-steady-state) of the catalyst. Before the observed different trends are described, the non-destructive treatment in reducing conditions needs to be explained.
Reducing conditions were applied several times according to the same procedure described before. The reproducibility of sequential treatment in reducing and oxidising conditions is presented in Figure 9. The same catalyst loading was treated for 2 h in reducing (i.e., propane/water/inert) feed for 2 h, before applying an oxidising feed (i.e., propane/oxygen/water/inert). This procedure was repeated after the catalyst has reached stable performance in terms of product evolution (i.e., not changing with time). Figure 9 confirms the reproducibility of such sequential treatment in reducing and oxidising conditions. Both experiments show essentially the same product evolution as a function of time-on-stream. Certainly, the initial state of the catalyst (at time zero) was affected by the length of pre-treatment time (compare Figure 9 and Figure 10a).
Clearly, the catalyst was affected by the pretreatment, since the catalyst regains, thus, recovers its original performance to produce acrylic acid with a high selectivity. In Figure A1 the course of propane conversion and product evolution during oxidising and reducing feed is shown. In reducing feed, the propane conversion drops rapidly (cf. Figure A1a). After 10 min the conversion approaches practically zero. The concentration of all products except acetone decreases, the concentration of the acids decreases faster than the concentration of CO and CO (cf. Figure A1b).
Figure 10a shows the evolution of all product species as a function of time, after a treatment in reducing conditions for 12 h. Significant trends are observed. CO and CO decreased in a similar fashion, with an simultaneous increase of acrylic acid. Both acetic acid and propene respond much faster to the suddenly added oxygen in the feed, in comparison to the formerly mentioned products. A rapid increase was observed for acetic acid from the first to the second data point and decreasing subsequently. The concentration of propene increases slightly with time-on-stream. Furthermore, the ratio of (CO-aca)/CO is 0.8 and constant as a function of time-on-stream. All concentrations achieved a plateau, eventually.
2.6. Data Interpretation and Rational Mechanism
2.6.1. Data Interpretation
In our interpretation and discussion, we are going to combine data of both types of experiments with ‘slow’ and ‘fast’ times. First, we would like to stress the obvious fact, that we do have this ‘slow’ trend in kinetic dependences, and this tendency is exhibited for special conditions of the catalyst, i.e., for a reduced catalyst. As experiments show, this trend is very reproducible, and the catalyst, analysed after all experimental treatments, is not damaged, i.e., reduced vs. oxidised. The fact whether this ‘slow’ trend is observed or not, will only depend on reduction and oxidation degree of the catalyst. The existence of this trend, reveals the fact of some ‘slow’ process, which exist inside the whole catalytic process. Regarding the whole catalytic cycle (cycles), it is likely external, e.g., it may combine different catalytic cycles.
The plateau of all observed dependencies can be identified with the stage when the catalyst is already re-oxidised. Obviously all the data presented in Figure 10a can be attributed to the stage of catalyst evolution from its reduced state to the oxidised one.
Figure 10a contains information about a significant difference in the temporal behaviour of all products, regarding the TOS-scale. Three cases can be distinguished:
- no temporal trend for propene concentration;
- a strongly decreasing trend in CO and CO concentration; and a strongly increasing trend in acrylic acid concentration;
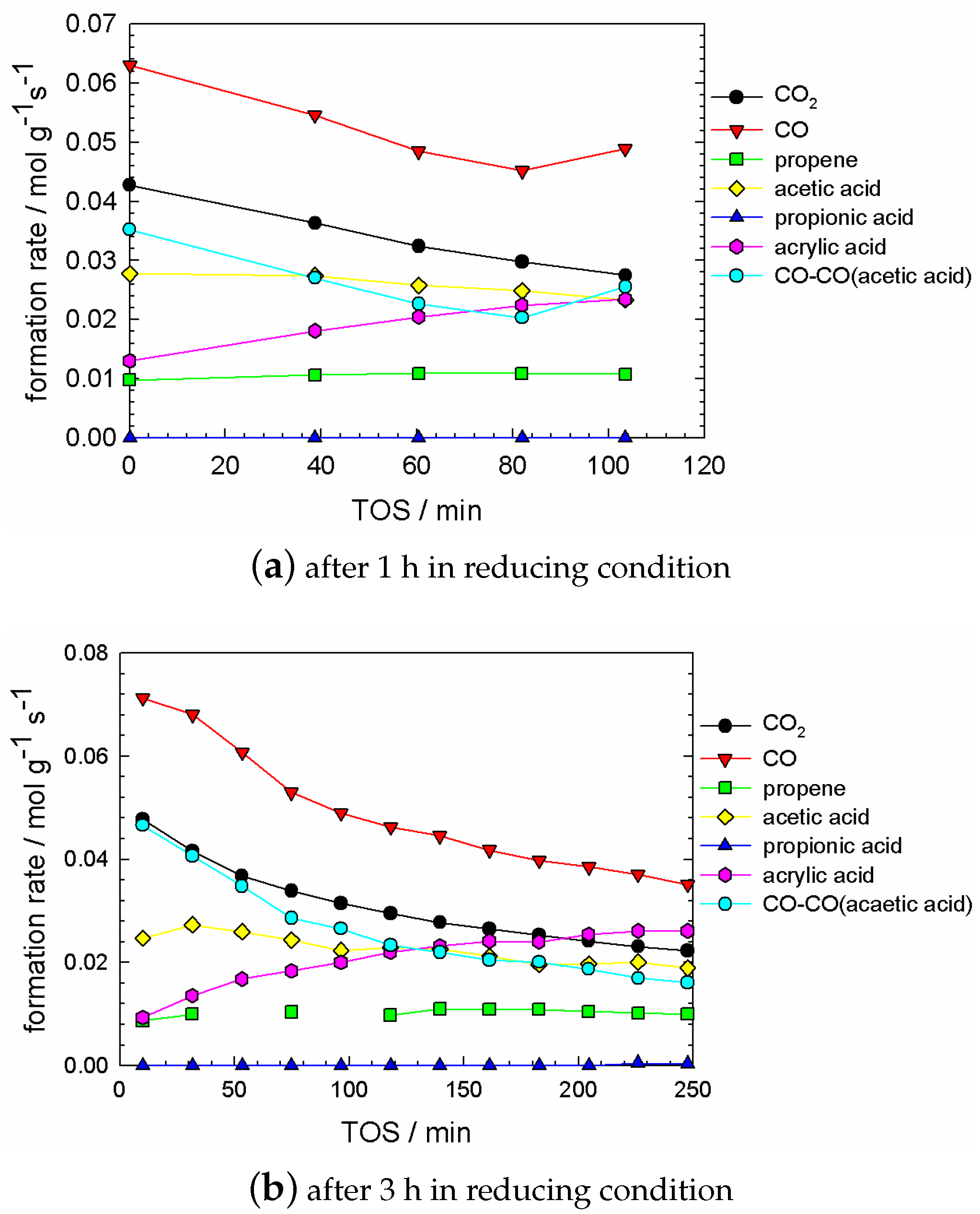
- some trend in acetic acid concentration, which can be considered between ‘small’ and ‘significant’, depending on the degree of reduction, which is governed by the pretreatment time. This trend was small if the pretreatment time was up to 2–3 h (cf. Figure A2).
Because of this difference in trends, we can hypothesise that in our process at least three different active cites are present.
The cite for , , and acrylic acid formation was affected by oxidation, whereas the oxidation to propene was not affected at all.
In Figure 10b the mass balances are plotted as a function of time-on-stream. The propane conversion (named as all) is the sum of all components, and it is slightly decreasing.
The interesting feature observed during TOS-investigations was the following: at any time the relationship
see Figure 10b. It can be termed as a ‘partial carbon balance’. This partial carbon balance may be employed as a fingerprint for the existence of the special ‘isolated site’. (Generally speaking, this site can have a complex structure, combining different centres.)
How to interpret this balance? It is not a total carbon balance, since this equation does not include concentrations of other carbon-containing substances, i.e., propane, propene and acetic acid.
It is possible that Equation (8) relates to the corresponding overall reaction in which acrylic acid is oxidised into , , and water.
The partial balance in Equation (8) consists of the products of Equations (3)–(5). Let’s present Equation (3) with a factor of two,
and Equations (4) and (5) together as follows.
In accordance with this stoichiometric equation, the following relationships can be written:
Hence,
and
or
This linear relationship in Equation (16) is a consequence of the stoichiometric Equation (11). However, a hypothesis about the oxidation of acrylic acid into and via reaction (11), or even the formation of acrylic acid from and (the reverse of reaction (11)), can be eliminated easily. It makes sense to say, that Reaction (11) has no physical meaning. Equation (11) reflects a hidden process of surface evolution, a bridge mechanism, as it will be explained later.
From our point of view, this intriguing invariant [] + [] + []–balance (in Figure 10b) (the CO employed in this balances is only produced other than from the reaction forming acetic acid), which does not depend on the propane conversion, gives a hint on a ‘closed’ structure, within which the complete transformation of admitted substances (propane) occurs, and substances acrylic acid, CO, and CO are produced. This structure can be identified with an isolated site. Generally, this conclusion relates to Grasselli’s concept, that a reaction may occur on an isolated site, and phase cooperation plays a significant role in the process as well [19,20,39]. Several catalysts are composed of different phases, which are proven to outperform as a mixture compared to the single phase catalysts. Site isolation is the distribution and isolation of active sites/components/centres from each other. However, since these products (acrylic acid from one side and CO/CO from another ones) are characterised by different kinetic dependences, we hypothesize that this site has two centres, which are responsible for two different routes.
One of these centres is responsible for the production of CO and CO, and the second one performs production of acrylic acid. Our hypothesis about these two processes (i.e., ‘two processes‘–’two centres’) is justified by the significant difference in long term kinetic dependencies for acrylic acid, from one site, and CO and CO, from another one.
The mentioned centres, say X and , may be linked by a slow oxidation reaction. Therefore, such reaction will be responsible for slow kinetic behaviour presented in Figure 10a. The detailed transformation over the ‘isolated site’ is depicted schematically in Figure 11. The transformation is fed by [O], which represents oxygen from either the bulk catalyst phase or the gas phase. The possible sources of oxygen shall not be limited to one or the other phase here.
Since the slow kinetic evolution results in some steady-state with the ‘non-zeroth’ concentration, process of site transformation has to be considered reversible, and the schematic drawing will be modified. The transformation is ‘slow’ in comparison with the ‘fast’ cycles of product generation. Therefore, cycle A (CO and CO production) and cycle B (acrylic acid production) can be presented and analysed separately. Slow dynamics of the separate ‘−’ transformation is qualitatively presented in Equation (17):
where and are dimensionless concentrations of the two centres, respectively, and the balance equation
Has to be fulfilled, with = and = and t is in the scale of time-on-stream. and are surface/bulk intermediates/phases which belong to the total oxidation (TO) cycle and and are intermediates/phases which belong to the selective oxidation (SO) cycle, respectively.
2.6.2. Rational Mechanism
Comparing data on both types of experiments, it is evident that long term-measurements (TOS experiments) exhibit dramatic differences between temporal behaviour of products, in comparison with short time experiments (partial pressure variation experiments).
In fact, partial pressure experiments do not show significant differences in the kinetic trends of the reaction products with respect to oxygen, i.e., all kinetic orders are zero. All product formation rates depend positively on propane partial pressure. The only exception is found for the influence of water partial pressure, both acetic and acrylic acids are positively depending on water partial pressure (auto-catalysis), whereas all other products (i.e., propene, CO, and CO) are independent.
Oppositely, in long term experiments, as mentioned before, the big difference in time-dependences for all product formation rates was observed, shown in Figure 9 and Figure 10a the preliminary analysis above. We did not find a stoichiometric relationships, which relate these dependences to each other. There was clearly a strong coupling observed between the formation rate of acrylic acid and the formation rates of CO and CO. At the same time, there is no kinetic correlation between other product formation rates: i.e., between (i) propene and acetic acid, (ii) propene and CO, CO, (iii) propene and acrylic acid (iv) acetic acid and CO.
Difference in long term behaviour for some products reflects a difference in specific catalytic routes of these product formation. Using this hypothesis, we are proposing the following mechanism. It has such routes: (a) route of propene formation, (b) route of acetic acid formation, and (c) and (d) routes of CO and CO formation bridged by a slow oxidation step to the route of acrylic acid formation.
Route of CO and CO formation have one joined step, in which propane is consumed and some specific steps, in which CO and CO are produced, respectively. CO produced in route (c) and also in route (b), together with acetic acid.
Therefore, every specific product is characterised by a specific route over a specific centre (principle ‘specific product–specific centre’). There is an obvious advantage of such a principle, i.e., simplicity in writing the steady-state (or pseudo-steady-state) rate equation.
From Figure 7, Figure 8, Figure 9 and Figure 10a it can be easily seen, that the ratio of (CO-aca)/CO is roughly around one, at least not strictly bound to each other. For general purpose, CO and CO are produced by different centres, but our experimental data is not enough to distinguish these. It can, however, be concluded, that CO and CO do not originate from the same process. Otherwise, a constant 2:1 or 1:2 ratio of (CO-aca)/CO would be expected, since propane has three carbon atoms. Thus, propane forms solely CO via one route, and solely CO via another route.
Characterising our mechanisms, we would like to stress, that all mechanisms are rational. Mechanisms for propene and CO and CO formation, are two step mechanisms, i.e., they are minimal catalytic cycles. As for mechanisms for the production of acetic and acrylic acid, they are four step cycles, containing one step of interaction with propane, one step for the reaction with water and two steps for the reaction with oxygen. In our opinion, it is a reasonable level of simplification. Also, these two mechanisms for the formation of acrylic and acetic acid, have a buffer step (step out of the cycle), in which water is a participant. As indicated in the introduction, the presence of water is necessary to activate the cycles for the production of selective oxidation products. Additionally, based on theoretical kinetic considerations [54] we need to have a reversible buffer step of the interaction with water to obtain a sum of kinetic orders greater than one, which is in accord with experimental findings (cf. Table 2).
All these mechanisms relate to ‘fast’ processes/reactions, and their rates were monitored on the level of seconds (Figure 7 and Figure 8). The ‘slow’ process of oxidation/reduction (), is presented in Figure 10a. The slow dynamics of this process controls the evolution of pseudo-steady-state kinetic dependences for the formation of CO and CO, and acrylic acid.
The mechanism presented in Figure 12 can be modified to take into account the peculiarities of acetic acid kinetic behaviour, i.e., the concentration goes through a maximum and then a slow decrease occurred. Presently, we do not have enough data to secure such interpretation.
2.6.3. Kinetic Equations
In this section, the kinetic equations related to our rational mechanism together with their corresponding apparent kinetic orders are presented route-by-route. As mentioned above, many routes are uncoupled, and can be easily derived based on theory of graphs [54]. For all cases R is the rate of product formation and is the kinetic coefficient of corresponding reaction. and are the partial pressures of propane and water, respectively, and is the concentration of oxygen in the bulk catalyst phase, which participates in the reaction. Thus, the foundation for our mechanism and the resulting kinetics is the well accepted Mars-van-Krevelen mechanism. is the apparent kinetic order of rate of substance A with respect to component B.
Rate of propene formation:
if
Equation (19) becomes:
The apparent kinetic orders for this cycle are:
where and are dimensionless reduced and oxidised surface intermediates in active site Z in the route of propene formation.
Rate of acetic acid formation:
with
If terms containing will dominate over the other terms, and the rate expression reduces to
where
The apparent kinetic orders for this cycle are:
The physical meaning of these characteristics is understandable. Typically, the apparent order regarding some substance, is equal to the dimensionless concentration (or sum of them), which react with this substance [54]. As found for this cycle, the apparent order regarding oxygen, is equal to the sum of concentration of intermediates + , the apparent order regarding water is equal to + , as for the apparent order regarding propane, it is equal to show, that it is equal to the sum of concentrations of + .
Rate of acrylic acid formation:
with
If terms containing will dominate over the other terms, and the rate expression reduces to
The apparent kinetic orders for this cycle are:
It can be noted, that the kinetic expressions and apparent kinetic orders for both acids are similar, reflecting the similarity of both mechanisms on different centres.
If terms containing will dominate over the other terms, the rate expressions reduce to
and
respectively.
The apparent kinetic orders for this cycle are:
Table 3 summarises all apparent kinetic orders of the proposed mechanism in Figure 12. The obtained apparent kinetic orders allow to make a comparison with experimental values, presented previously. The non-trivial experimental fact is, that the sum of experimental kinetic orders is larger than one, 1.64 for acetic acid and 1.32 for acrylic acid. It is confirmed by our theoretical analysis. The theoretical values of these sums are for acetic and acrylic acid are 1 + and 1 + , respectively. The introduction of a buffer step, increased the sum of kinetic orders on the value of concentration of buffer intermediate, which reacts with water.
Rate of ‘slow’ dynamics: Slow dynamics is represented by the differential equation:
where
The starting point is = 1, when all the catalyst is reduced. At the final equilibrated state
The solution of the differential equation is:
where t is in the scale of time-on-stream. Or
and ,
Hypothetically, starting from the completely oxidised catalyst (), we will have
and ,
It is easy to show, that dividing Equation (51) by Equation (54)
which is constant for a given temperature and partial pressure of oxygen. This apparent equilibrium constant for the oxidation-reduction process can be estimated as follows:
For a fixed temperature and gas composition is constant. For a completely reduced catalyst at (t = 0; = 1)
Similarly, for the catalyst state at which we assume an oxidation-reduction equilibrium
Roughly considering proportionality between measured concentration and the rate of []-change (inlet concentration of = 0), we have
where and are the concentrations at t = 0 and at equilibrium. Therefore,
and
Thus, = 1 − = 0.71, and
3. Discussion
3.1. Kinetics and Mechanism
The catalyst has been treated in reducing and oxidising reactant medium. Figure 9 and Figure 10a report the catalyst evolution under oxidising atmosphere after a reductive treatment. The data suggest, that such reducing treatment of the M1 catalyst was reversible. These observations are in line with experimental findings on reduction and oxidation behaviour of MoVO from Ueda et al. [55,56]. In their contribution, two different types of lattice oxygen were distinguished. In early stages of treatments in reducing conditions type-1 oxygen evolves from the structure irreversibly, whereas type-2 oxygen leaves the structure continuously and returns reversibly in oxidising atmosphere [55]. The structure of the M1 catalyst, however, is stable, even under extreme conditions [12].
We correspond to Ueda et al.’s [55] concept, reflecting it kinetically by our ‘slow dynamics’ in Figure 11. This mechanism visualises this reversible reduction-oxidation behaviour of the catalyst from a kinetic point of view, expressed through the kinetic equation for this slow dynamics (cf. Equation (17)). The apparent equilibrium constant in Equations (55) and (56) may therefore be correlated with the degree of reduction (parameter in [55]). The alkane conversion was significantly affected by the degree of reduction [55], which confirms the importance of oxygen (and water) in providing a certain chemical potential in the gas phase to stabilise the catalyst surface structure [2,12]. According to Barrault et al. [3], and supporting the statements above, a high selectivity towards selective oxidation is only provided by an oxidised site (not a reduced site). This is expressed in our proposed bridge mechanism in Figure 11.
The Mars-van-Krevelen (MvK) mechanism is widely accepted for selective oxidation reactions over mixed metal oxide catalysts [1,2,36,37]. Further significant inspiration to our mechanistic development was provided by different concepts: (i) the site isolation concept by Grasselli [19,20,21]; (ii) the concept of an ensemble of atoms forming an active site by Schlögl [22]; (iii) the proposed multi-route parallel reaction pathways by Schlögl et al. [8] and Barrault et al. [3]; and (iv) the change in product distribution upon surface acidity by Barrault et al. [3] and Luo et al. [46].
Based on these concepts, the experimental observations in Figure 7, Figure 8, and Figure 10a were interpreted. Our mechanism was presented in Figure 12 and consists of four independent catalytic cycles for the formation of propene, acetic acid, acrylic acid, and total oxidation, respectively. All products (except total oxidation) originate from a specific catalytic cycle, a specific active centre, which is in line with Schlögl [22] and Grasselli [19,20,21]. Acetic acid and acrylic acid are formed via different reaction pathways [3]. The total oxidation products CO and CO are proposed to as well originate each from an individual specific active centre. This was also earlier proposed by Naumann d’Alnoncourt et al. [12]. The data presented in this work is, however, not sufficient to support this hypothesis. Therefore both products are summarised in one cycle. Such a separate reaction pathway from propane to total oxidation products was earlier proposed [8,46], together with the stability of other reaction products against further oxidation under the given reaction condition [8]. In most mechanisms, however, the total oxidation is considered to be the final product of all species involved (e.g., [6,9,10]).
Water has a special role in many mechanisms presented in the past. In our mechanism, water is both a reactant in the selective oxidation of propane, and at the same time an activator for both catalytic cycles forming acetic acid and acrylic acid. For example Centi et al. [34] and Novakova et al. [6] confirm the necessity of water for the activation of the surface and to increase the selectivity to selective oxidation products, respectively. Schlögl et al. confirm these observations by in situ XPS investigation [25,32].
Propene has been chosen as a product during the selective oxidation of propane, which re-adsorbs for further oxidation, e.g., [3,6,9,10,12,46]. Certainly, if the data in Figure 8 would be plotted as selectivity against conversion or feed concentration, the propene-selectivity would drop (since the selectivity is affected by the concentration of all products). This would easily lead to the conclusion that propene re-adsorbs to be further converted to acrylic acid. Due to the observations in Figure 8 and Figure 10a, i.e., a constant propene concentration during increasing acrylic acid concentration (and, thus, increasing propane conversion), propene is in our mechanism (cf. Figure 12) kinetically independent. The tools for the mechanistic development were provided by the theory of graphs [54].
Our apparent kinetic orders were presented in Table 2 are in good agreement with other investigations. Apparent kinetic orders are high with respect to propane for propene and intermediate high for acetic and acrylic acid. All products show an apparent kinetic order close to zero with respect to oxygen (which could be expected for a MvK-type reaction) [6,7,8]. Kinetic orders with respect to water are hardly reported.
3.2. Structure-Kinetic Interrelationship
Based on the results, interpretations and discussions in this work, an overall model of our kinetic understanding for catalytic surface reactions is provided in Figure 13. It incorporates the concepts of isolated active sites presented by Grasselli [19,20,21] and the concept of an ensemble of atoms forming an active site presented by Schlögl [22].
Figure 13 depicts our understanding of the combination of different ‘active sites’ on a catalyst surface. An active site can be characterised by a complex structural architecture. An ‘active site’ consists of one or more ‘active centres’. There are possibly as many active centres as there are kinetically independent products of a reaction. Thus, if a reactant splits on one active site into two product molecules, they are not kinetically independent.
Therefore, an active site is in our understanding characterised by a ‘kinetic fingerprint’ of the product(s) evolving from it.
Furthermore, in Figure 13 are two levels of isolation expressed: (i) Two active sites are separated on a slow and on a fast time scale. (ii) Two active centres (on the same site) are separated on a fast time scale, and joint together by a slow process.
The final hypothesis in our understanding is: Each kinetically independent product originates from a specific active centre. The chemical nature of an active centre determines uniquely how and to which product the reactant molecule(s) are converted.
3.3. Presenting Logics in Developing a Rational Mechanism
Development of a rational mechanism can be presented as an attempt to answer a series of questions:
- 1.
- Do we have an isolated site or not?
- 2.
- Does this site consist of one centre or more?
Remember we distinguished the terms ‘site’ and ‘centre’: A ‘site’ is a complex construction, which may consist of one or more active ‘centres’.
- 3.
- Do we have a single- or a multi-route process for a centre?
- 4.
- Is there hierarchy in times of reaction or not?
- 5.
- What kind of limiting steps are there for single- or multi-route reaction?
- 6.
- What are the kinetic equations and kinetic orders?
A series of steps in kinetic strategy is depicted in Figure 14.
Manifestation of the isolated sites showed in the previous analysis, using the partial balance of carbon atoms [CO] + [CO] + [acrylic acid], which was found experimentally. The complex nature of the isolated active site, was shown based on the difference long-term time-on-stream dependences for CO and CO from one side and acrylic acid from another side, respectively. We assumed, that the ‘fast’ processes of product formation are linked by the ‘slow’ process of centre oxidation-reduction. For acetic acid and propene formation, we also assume different centres, considering the differences in long-term kinetic trends.
As for distinguishing multi-route and single-route scenarios, there is a potential to answer this question, based on selectivity analysis.
An analysis of the selectivity, rather to say, the differential selectivity, in particular the ratio of rates of different products or the ratio of product rate over the reactant rate can be very fruitful. As known, the typical kinetic rate expression has the form of Langmuir-Hinshelwood-Hougen-Watson (LHHW) expression, and the form of this expression is ‘a numerator over denominator’-equation. For the multi-route reaction on the same centre, kinetic equations for all routes have the same denominator. In this case, the differential selectivity will be presented by the ratio ‘of numerators’ which can be a quite simple function. For the multi-route reactions on different centres, denominators of kinetic expressions (LHHW-equations) will be not the same, and selectivities will be functionally not so simple.
Therefore, the observed partial balances can be considered as a potential fingerprint of the multi-route process over the isolated site.
Comparing rate of CO and CO formation, we conclude: the corresponding route rates (according to Temkin-Horiuti terminology [29]) are equal, because the routes include the same step of interaction between the centre X and propane. Rate of oxygen consumption and water production for these routes will be different, according to the stoichiometric equations.
Analysing kinetic equations for different products, and assuming oxygen excess, we can consider that steps, which do not contain oxygen are limiting in the steady-state process. These steps may be assumed slow in comparison with oxygen-containing steps of the same route. However, they are certainly ‘fast’ in comparison with the ‘slow’ oxidation-reduction step.
Generally, if the reaction is considered to occur on different sites via a single route, the apparent kinetic orders and apparent energy of activation can be used as a traditional tool for estimating surface intermediates and energetic parameters of elementary steps. We presented analytic expressions for the apparent orders of all five products with respect to all three reactants, i.e., for propane, oxygen, and water (cf. Table 3).
4. Materials and Methods
4.1. Synthesis and Characterisation
A phase pure M1 MoVTeNbO catalyst (internal identification number 17817) was synthesised according to Kolen’ko et al. [57], following a slurry-spray-drying protocol. The as-prepared catalyst was characterised by powder X-ray diffractometry (XRD), nitrogen adsorption (Brunauer–Emmett–Teller method–BET), Raman, and electron microscopy (i.e., SEM and TEM) methods. A second series of TEM and XRD analysis was carried out for the used catalyst, after (at least) all experiments described in the present work.
4.2. Reactor Set-Up and Reaction Conditions
The kinetic experiments were carried out in one reactor of a 10-fold parallel reactor unit, which has been presented in more detail before [50]. A gas chromatograph (7890A, Agilent, Santa Clara, California, United States) was used to perform online gas analysis. It was equipped with a combination of PoraplotQ and Plot Molesieve columns connected to a thermal conductivity detector, and a combination of HP-FFAP and PoraplotQ columns connected to a flame ionisation detector. All reactant and product gases were analysed (including water). 100 mg of catalyst were placed over 50 mg of SiC, both in a sieve fraction of 250–355 m. The catalyst was pressed (2.5 t) and sieved to receive the desired sieve fraction.
All experiments were carried out at 573 K, 1 bar pressure and a weight hourly space velocity (WHSV) of 10 mL g h. A typical reactant composition was CH/O/HO/N = 3/12/10/75. At partial pressure variation experiments, one reactant partial pressure was changed. The flow of nitrogen was adjusted accordingly to keep the WHSV constant. The propane conversion was kept below 10%.
The formation rate of products is determined as follows:
where is the formation rate of product j in mol g s, the molar total flow rate in mol s, m the catalyst mass in g, and the determined outlet concentration of product j by the gas chromatograph divided by 100%.
4.3. Types of Kinetic Experiments
In our work, two types of kinetic experiments have been performed:
- Kinetic experiments under catalyst steady-state conditions: Partial pressure variations of propane (1.5–4.5%), oxygen (2–12%), and water (5–20%) at 573 K and a constant WHSV of 10 mL g h.
- Kinetic experiments during catalyst evolution (’slow dynamics’ scale of min and hours): Observation of catalyst evolution with time-on-stream (TOS) under CH/O/HO/N = 3/12/10/75 feed, after treatment in reducing conditions for 12 h in CH/O/HO/N = 3/0/10/87. Both parts of the experiment were carried out at 573 K and a constant WHSV of 10 mL g h.
Both experiments were performed in a tube reactor (the plug flow reactor, PFR). In both experiments, the steady-state kinetic data have been produced. Obviously, there is a big difference between the two experiments.
The first one corresponds to the ‘fast’ and ‘moderate’ processes of gas transformation over the solid catalyst at the given catalyst state (catalyst composition). The second one relates to the ‘slow’ process within the ‘gas-solid’ catalytic system. Such ‘slow’ kinetic behaviour was observed in studies over the preliminary reduced catalyst. In experiments over oxide catalysts, which have been described in the literature previously, such ‘slow’ trends were not observed. Kinetic evolution demonstrated in our experiments can be explained by gradual oxidation of the catalyst under the influence of the reactive medium.
All experiments, both in with a scale of seconds and with a scale of minute/hours, were reproducible and undermine its validity.
5. Conclusions
The selective oxidation of propane to acrylic acid was kinetically investigated over a phase pure M1 MoVTeNbO catalyst at 573 K and 1 bar pressure in a plug flow reactor.
The investigation was performed on two time scales, i.e., ‘slow’ (minutes and hours) and ‘fast’ (seconds). Characterisation by XRD showed no structural changes of the catalyst, despite repeated oxidation and reduction treatments. Observed differences in long term behaviour, have been used for distinguishing catalytic routes for the formation of propene, acetic acid, acrylic acid, CO, and CO. Based on the partial carbon balance ([CO] + [CO] + [aa] = const.), the existence of an isolated site was assumed for the production of these substances. Two different centres are assumed within this site for the production of CO and CO and for the production of acrylic acid, respectively. The relationship between these routes is linked by a slow step of catalyst oxidation.
The partial carbon balance, established here, can be considered as a ‘kinetic fingerprint’, which is a special combination of products originating from one active site, for the consideration of the known concept of isolated sites. A ‘kinetic fingerprint’ distinguishes types of active sites. An active site consists of one or more active centres. Therefore, each kinetically independent product originates from a specific active centre. The chemical nature of an active centre determines uniquely how and to which product the reactant molecule(s) are converted.
In the development of the rational mechanism, the principle was used, ‘every specific route is distinguished by a specific centre’. Kinetic rate expressions have been found for all routes and for a slow oxidation-reduction process.
The main message of our article is: the kinetic investigations of ‘slow dynamics’ in the scale of min and hours reveal long term trends in catalyst evolution. In combination with investigations over a catalyst at steady-state, such experiments give an insight in mechanistic aspects. The performed research can be considered as a development of methodology for revealing a rational mechanism of complex catalytic reactions.
It is interesting to mention, at fixed external parameters (i.e., flow rate, concentration, and temperature) the selectivity of highly molecular substances (i.e., acrylic acid) is growing during the slow re-oxidation of the catalyst, exhibiting a simple example of chemical evolution.
Author Contributions
Conceptualization, C.S.; Methodology, C.S. and G.S.Y.; Validation, C.S., G.S.Y. and A.T.; Formal Analysis, C.S. and G.S.Y.; Investigation, C.S.; Resources, R.S.; Data Curation, C.S.; Writing—Original Draft Preparation, C.S. and G.S.Y.; Writing—Review & Editing, C.S., G.S.Y. and A.T.; Visualization, C.S.; Supervision, G.S.Y. and A.T.; Project Administration, R.S. and A.T.; Funding Acquisition, R.S. Please turn to the CRediT taxonomy for the term explanation.
Funding
This work was conducted in the framework of the BasCat collaboration between BASF SE, TU Berlin, FHI, and the cluster of excellence “Unified Concepts in Catalysis” (UniCat www.unicat.tu-berlin.de).
Acknowledgments
The authors gratefully acknowledge M. Jastak for the synthesis of the catalyst and P. Kube for additional catalytic testing results. Furthermore, the authors acknowledge F. Girgsdies, M. Hashagen, E. Ider, M. Jastak, A. Klein-Hoffmann, T. Lunkenbein, and G. Weinberg for the characterisation of the catalyst.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MDPI | Multidisciplinary Digital Publishing Institute |
| PFR | Plug Flow Reactor |
| TAP | Temporal Analysis of Products |
| ER | Eley-Rideal |
| MvK | Mars-van-Krevelen |
| LH | Langmuir-Hinshelwood |
| LHHW | Langmuir-Hinshelwood-Hougen-Watson |
| WHSV | Weight Hourly Space Velocity |
| aca | acetic acid |
| aa | acrylic acid, CHO |
| TOS | Time-On-Stream |
| a.u. | arbitrary units |
| apparent kinetic order | |
| product formation rate | |
| kinetic coefficient | |
| apparent equilibrium constant | |
| is the concentration of component A | |
| the concentration of oxygen in the bulk catalyst phase | |
| dimensionless concentration of surface intermediate j | |
| M1 | phase (ICSD No. 55097) |
| XRF | X-ray Fluorescence |
| BET | nitrogen adsorption (Brunauer–Emmett–Teller method) |
| XRD | X-ray powder Diffractometry |
| LEIS | Low-Energy Ion Scattering |
| SEM | Scanning Electron Microscopy |
| TEM | Transmission Electron Microscopy |
| XPS | X-ray Photoelectron Spectroscopy |
| NEXAFS | Near-Edge X-ray Absorption Fine Structure |
Appendix A. Supporting Information–Additional Experimental Data
Figure A1.
Catalyst behaviour under reducing and oxidising conditions: (a) Propane conversion; (b) Concentration of formed products: “Steady State 1” feed (CH/O/He/HO = 3/12/75/10), “Reduction” feed (CH/O/He/HO = 3/0/87/10), and “Steady State 2” feed (CH/O/He/HO = 3/12/75/10); reaction temperature 573 K.
Figure A1.
Catalyst behaviour under reducing and oxidising conditions: (a) Propane conversion; (b) Concentration of formed products: “Steady State 1” feed (CH/O/He/HO = 3/12/75/10), “Reduction” feed (CH/O/He/HO = 3/0/87/10), and “Steady State 2” feed (CH/O/He/HO = 3/12/75/10); reaction temperature 573 K.
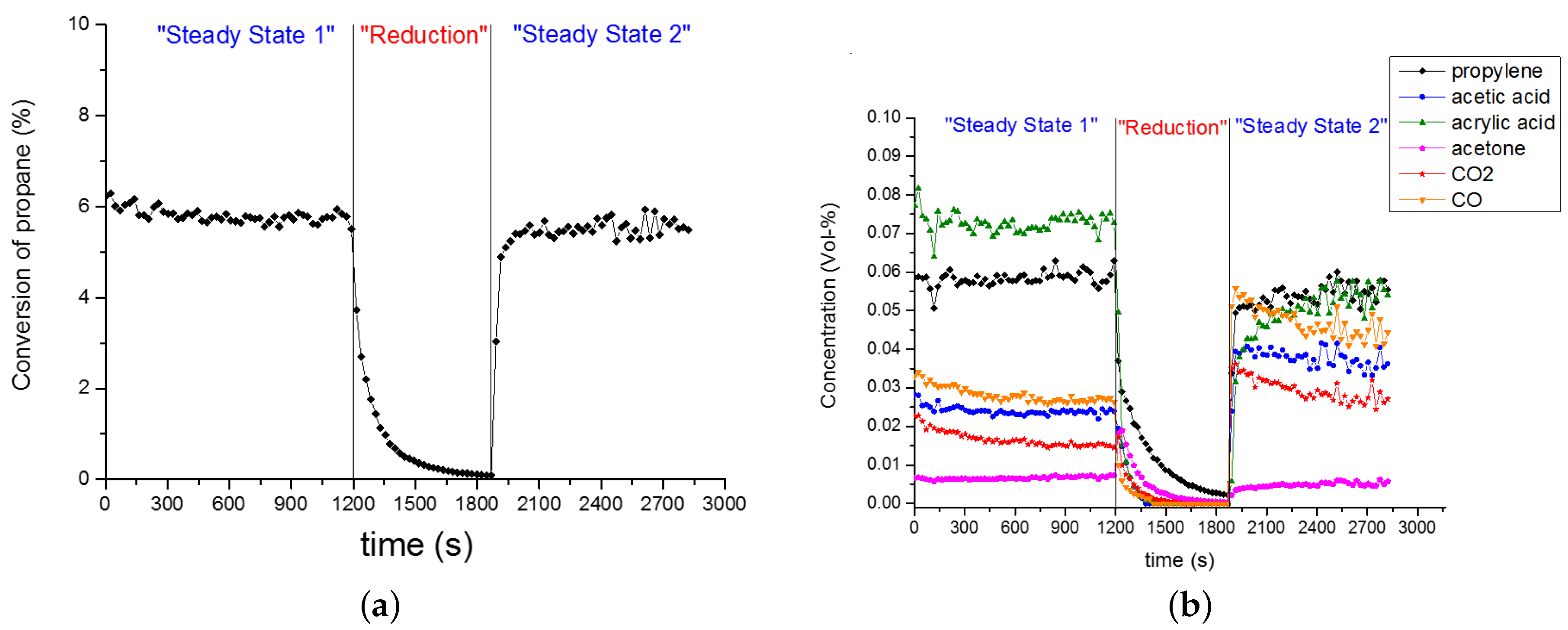
Figure A2.
C3H8/O2/H2O/N2 = 3/12/10/75 at 573 K and 104 mL g−1 h−1 over M1 catalysts MoVTeNbOx: course of outlet concentrations as a function of time-on-stream after a treatment in reducing condition (C3H8/O2/H2O/N2 = 3/0/10/87 at 573 K and 104 mL g−1 h−1): (a) 1 h and (b) 3 h.
Figure A2.
C3H8/O2/H2O/N2 = 3/12/10/75 at 573 K and 104 mL g−1 h−1 over M1 catalysts MoVTeNbOx: course of outlet concentrations as a function of time-on-stream after a treatment in reducing condition (C3H8/O2/H2O/N2 = 3/0/10/87 at 573 K and 104 mL g−1 h−1): (a) 1 h and (b) 3 h.
References
- López Nieto, J.M.; Solsona, B. 5–Gas phase heterogeneous partial oxidation reactions. In Metal Oxides in Heterogeneous Catalysis: 5—Gas Phase Heterogeneous Partial Oxidation Reactions; Védrine, J.C., Korotcenkov, G., Eds.; Elsevier Inc.: New York, NY, USA, 2018; pp. 211–286. [Google Scholar]
- Védrine, J.C. Heterogeneous catalytic partial oxidation of lower alkanes (C1–C6) on mixed metal oxides. J. Energy Chem. 2016, 25, 936–946. [Google Scholar] [CrossRef]
- Barrault, J.; Batiot, C.; Magaud, L.; Ganne, M. Selective oxidation of propane into oxygenated compounds over promoted nickel-molybdenum catalysts. Stud. Surf. Sci. Catal. 1997, 110, 375–382. [Google Scholar] [CrossRef]
- Li, W.; Ueda, W. Catalytic selective oxidation of C2–C4 alkanes over reduced heteropolymolybdates. Stud. Surf. Sci. Catal. 1997, 110, 433–442. [Google Scholar] [CrossRef]
- Botella, P.; Lopez Nieto, J.M.; Solsona, B.; Mifsud, A.; Marquez, F. The Preparation, Characterization, and Catalytic Behavior of MoVTeNbO Catalysts Prepared by Hydrothermal Synthesis. J. Catal. 2002, 209, 445–455. [Google Scholar] [CrossRef]
- Novakova, E.K.; Vedrine, J.C.; Derouane, E.G. Propane Oxidation on Mo–V–Sb–Nb Mixed-Oxide Catalysts. J. Catal. 2002, 211, 226–234. [Google Scholar] [CrossRef]
- Vitry, D.; Morikawa, Y.; Dubois, J.L.; Ueda, W. Propane selective oxidation over monophasic Mo–V–Te–O catalysts prepared by hydrothermal synthesis. Top. Catal. 2003, 23, 47–53. [Google Scholar] [CrossRef]
- Widi, R.K.; Hamid, S.B.A.; Schlögl, R. Kinetic investigation of propane oxidation on diluted Mo1V0.3Te0.23Nb0.12Ox mixed-oxide catalyst. React. Kinet. Catal. Lett. 2009, 98, 273–286. [Google Scholar] [CrossRef]
- Fushimi, R.; Shekhtman, S.O.; Gaffney, A.; Han, S.; Yablonsky, G.S.; Gleaves, J.T. TAP Vacuum Pulse-Response and Normal-Pressure Studies of Propane Oxidation over MoVTeNb Oxide Catalysts. Ind. Eng. Chem. Res. 2005, 44, 6310–6319. [Google Scholar] [CrossRef]
- Naraschewski, F.N.; Jentys, A.; Lercher, J.A. On the Role of the Vanadium Distribution in MoVTeNbO x Mixed Oxides for the Selective Catalytic Oxidation of Propane. Top. Catal. 2011, 54, 639. [Google Scholar] [CrossRef]
- Mazloom, G.; Alavi, S.M. Kinetic study of selective propane oxidation to acrylic acid over Mo1V0.3Te0.23Nb0.12Ox using the genetic algorithm. React. Kinet. Mech. Catal. 2013, 110, 387–403. [Google Scholar] [CrossRef]
- Naumann d’Alnoncourt, R.; Csepei, L.I.; Hävecker, M.; Girgsdies, F.; Schuster, M.E.; Schlögl, R.; Trunschke, A. The reaction network in propane oxidation over phase-pure MoVTeNb M1 oxide catalysts. J. Catal. 2014, 311, 369–385. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Bell, A.T.; Iglesia, E. Kinetics and Mechanism of Oxidative Dehydrogenation of Propane on Vanadium, Molybdenum, and Tungsten Oxides. J. Phys. Chem. B 2000, 104, 1292–1299. [Google Scholar] [CrossRef] [Green Version]
- Grasselli, R.K.; Buttery, D.J.; Burrington, J.D.; Andersson, A.; Holmberg, J.; Ueda, W.; Kubo, J.; Lugmair, C.G.; Volpe, A.F., Jr. Active centers, catalytic behavior, symbiosis and redox properties of MoV(Nb,Ta)TeO ammoxidation catalysts. Top. Catal. 2006, 38, 7–16. [Google Scholar] [CrossRef]
- Novakova, J.; Dolejsek, Z.; Habersberger, K. A Note on the Influence of Water Vapour in the Oxidation of Propene to Acrylic Acid on a Mixed Oxide Catalyst. React. Kinet. Catal. Lett. 1976, 4, 389–395. [Google Scholar] [CrossRef]
- Chen, K.; Iglesia, E.; Bell, A.T. Kinetic Isotopic Effects in Oxidative Dehydrogenation of Propane on Vanadium Oxide Catalysts. J. Catal. 2000, 192, 197–203. [Google Scholar] [CrossRef]
- Chen, K.; Bell, A.T.; Iglesia, E. Isotopic Tracer Studies of Reaction Pathways for Propane Oxidative Dehydrogenation on Molybdenum Oxide Catalysts. J. Phys. Chem. B 2001, 105, 646–653. [Google Scholar] [CrossRef] [Green Version]
- Mestl, G. MoVW mixed metal oxides catalysts for acrylic acid production: from industrial catalysts to model studies. Top. Catal. 2006, 38, 69–82. [Google Scholar] [CrossRef]
- Grasselli, R.K. Genesis of site isolation and phase cooperation in selective oxidation catalysis. Top. Catal. 2001, 15, 93–101. [Google Scholar] [CrossRef]
- Grasselli, R.K.; Andersson, A.; Buttery, D.J.; Burrington, J.D.; Lugmair, C.G.; Volpe, A.F., Jr. Proceedings of the Abstract of papers 228th ACS National Meeting, Philadelphia, PA, USA, 22–26 August 2004.
- Grasselli, R.K. Site isolation and phase cooperation: Two important concepts in selective oxidation catalysis: A retrospective. Catal. Today 2014, 238, 10–27. [Google Scholar] [CrossRef]
- Schlögl, R. Active Sites for Propane Oxidation: Some Generic Considerations. Top. Catal. 2011, 54, 627. [Google Scholar] [CrossRef]
- Haber, J. Catalysis by Transition Metal Oxides. In Solid State Chemistry in Catalysis: Catalysis by Transition Metal Oxides; Grasselli, R.K., Brazdil, J.F., Eds.; American Chemical Society: Washington, DC, USA, 1985; pp. 3–21. [Google Scholar]
- Somorjai, G.A. The 13th International Symposium on Relations Between Homogeneous and Heterogeneous Catalysis—An Introduction. Top. Catal. 2008, 48, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hävecker, M.; Wrabetz, S.; Kröhnert, J.; Csepei, L.I.; Naumann d’Alnoncourt, R.; Kolen’ko, Y.V.; Girgsdies, F.; Schlögl, R.; Trunschke, A. Surface chemistry of phase-pure M1 MoVTeNb oxide during operation in selective oxidation of propane to acrylic acid. J. Catal. 2012, 285, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Wachs, I.E.; Routray, K. Catalysis Science of Bulk Mixed Oxides. ACS Catal. 2012, 2, 1235–1246. [Google Scholar] [CrossRef]
- Védrine, J.C. Revisiting active sites in heterogeneous catalysis: Their structure and their dynamic behaviour. Appl. Catal. A Gen. 2014, 474, 40–50. [Google Scholar] [CrossRef]
- Boreskov, G.K. Heterogeneous Catalysis; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2003. [Google Scholar]
- Yablonskii, G.S.; Bykov, V.I.; Elokhin, V.I.; Gorban, A.N. Comprehensive Chemical Kinetics. In Kinetic Models of Catalytic Reactions: Comprehensive Chemical Kinetics; Elsevier Inc.: New York, NY, USA, 1991; Volume 32, p. 492. [Google Scholar]
- Haber, J.; Serwicka, E.M. The Role of Oxygen in Catalysis. React. Kinet. Catal. Lett. 1987, 35, 369–379. [Google Scholar] [CrossRef]
- Sanfiz, A.C.; Hansen, T.W.; Sakthivel, A.; Trunschke, A.; Schlögl, R.; Knoester, A.; Brongersma, H.H.; Looi, M.H.; Hamid, S.B.A. How important is the (001) plane of M 1 for selective oxidation of propane to acrylic acid? J. Catal. 2008, 258, 35–43. [Google Scholar] [CrossRef]
- Sanfiz, A.C.; Hanen, T.W.; Teschner, D.; Schnörch, P.; Girgsdies, F.; Trunschke, A.; Schlögl, R.; Looi, M.H.; Hamid, S.B.A. Dynamics of the MoVTeNb Oxide M1 Phase in Propane Oxidation. J. Phys. Chem. C 2010, 114, 1912–1921. [Google Scholar] [CrossRef]
- Heine, C.; Hävecker, M.; Trunschke, A.; Schlögl, R.; Eichelbaum, M. The impact of steam on the electronic structure of the selective propane oxidation catalyst MoVTeNb oxide (orthorhombic M1 phase). Phys. Chem. Chem. Phys. 2015, 17, 8983–8993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centi, G.; Perathoner, S. Reaction Mechanism and Control of Selectivity in Catalysis by Oxides: Some Challenges and Open Questions. Int. J. Mol. Sci. 2001, 2, 183–196. [Google Scholar] [CrossRef]
- Naraschewski, F.N.; Kumar, C.P.; Jentys, A.; Lercher, J.A. Phase formation and selective oxidation of propane over MoVTeNbOx catalysts with varying compositions. Appl. Catal. A Gen. 2011, 391, 63–69. [Google Scholar] [CrossRef]
- Védrine, J.C. 9—Concluding remarks and 9 challenges of heterogeneous catalysis on metal oxides. In Metal Oxides in Heterogeneous Catalysis: 9—Concluding Remarks and Challenges of Heterogeneous Catalysis on Metal Oxides; Védrine, J.C., Korotcenkov, G., Eds.; Elsevier Inc.: New York, NY, USA, 2018; p. 551. [Google Scholar]
- Andrushkevich, T.V.; Chesalov, Y.A. Mechanism of heterogeneous catalytic oxidation of organic compounds to carboxylic acids. Russ. Chem. Rev. 2018, 87, 586. [Google Scholar] [CrossRef]
- Keulks, G.W.; Lo, M.Y. Catalytic Oxidation of Propylene. 11. An Investigation of the Kinetics and Mechanism over Iron-Antimony Oxide. J. Phys. Chem. 1986, 90, 4768–4775. [Google Scholar] [CrossRef]
- Grasselli, R.K.; Burrington, J.D.; Brazdil, J.F. Mechanistic Features of Selective Oxidation and Ammoxidation Catalysis. Faraday Discuss. Chem. Soc. 1981, 72, 203–223. [Google Scholar] [CrossRef]
- Burrington, J.D.; Kartisek, C.T.; Grasselli, R.K. Aspects of Selective Oxidation and Ammoxidation Mechanisms over Bismuth Molybdate Catalysts. J. Catal. 1980, 63, 235. [Google Scholar] [CrossRef]
- Burrington, J.D.; Kartisek, C.T.; Grasselli, R.K. Surface Intermediates in Selective Propylene Oxidation and Ammoxidation over Heterogeneous Molybdate and Antimonate Catalysts. J. Catal. 1984, 87, 363–380. [Google Scholar] [CrossRef]
- Saleh-Alhamed, Y.A.; Hudgins, R.R.; Silveston, P.L. Investigations of catalytic mechanisms for selective propene oxidation in the presence of steam. Appl. Catal. A Gen. 1995, 127, 177–199. [Google Scholar] [CrossRef]
- Saleh-Alhamed, Y.A.; Hudgins, R.R.; Silveston, P.L. Role of Water Vapor in the Partial Oxidation of Propene. J. Catal. 1996, 161, 430–440. [Google Scholar] [CrossRef]
- Lin, M.M. Selective oxidation of propane to acrylic acid with molecular oxygen. Appl. Catal. A Gen. 2001, 207, 1–16. [Google Scholar] [CrossRef]
- Lin, M.H.; Desai, T.B.; Kaiser, f.W.; Klugherz, P.D. Reaction pathways in the selective oxidation of propane over a mixed metal oxide catalyst. Catal. Today 2000, 61, 223–229. [Google Scholar] [CrossRef]
- Luo, L.; Labinger, J.A.; Davis, M.E. Comparison of Reaction Pathways for the Partial Oxidation of Propane over Vanadyl Ion-Exchanged Zeolite Beta and Mo1V0.3Te0.23Nb0.12Ox. J. Catal. 2001, 200, 222–231. [Google Scholar] [CrossRef]
- Balcells, E.; Borgmeier, F.; Griesstede, I.; Lintz, H.G.; Rosowski, F. Partial oxidation of propane to acrylic acid at a Mo–V–Te–Nb-oxide catalyst. Appl. Catal. A Gen. 2004, 266, 211–221. [Google Scholar] [CrossRef]
- Grabowski, R. Kinetics of the oxidative dehydrogenation of propane on vanadia/titania catalysts, pure and doped with rubidium. Appl. Catal. A Gen. 2004, 270, 37–47. [Google Scholar] [CrossRef]
- Grabowski, R. Kinetics of Oxidative Dehydrogenation of C2 -C3 Alkanes on Oxide Catalysts. Catal. Rev. 2006, 48, 199–268. [Google Scholar] [CrossRef]
- Naumann d’Alnoncourt, R.; Kolenko, Y.V.; Schlögl, R.; Trunschke, A. A New Way of Probing Reaction Networks: Analyzing Multidimensional Parameter Space. Comb. Chem. High Throughput Screen. 2012, 15, 161. [Google Scholar] [CrossRef]
- Kube, P.; Frank, B.; Schlögl, R.; Trunschke, A. Synthesis of MoVTeNb Oxide Catalysts with Tunable Particle Dimens ions. ChemCatChem 2017, 9, 3446. [Google Scholar] [CrossRef]
- Holmberg, J.; Grasselli, R.K.; Andersson, A. A study of the functionalities of the phases in Mo–V–Nb–Te oxides for propane ammoxidation. Top. Catal. 2003, 23, 55–63. [Google Scholar] [CrossRef]
- Vitry, D.; Morikawa, Y.; Dubois, J.L.; Ueda, W. Mo-V-Te-(Nb)-O mixed metal oxides prepared by hydrothermal synthesis for catalytic selective oxidations of propane and propene to acrylic acid. Appl. Catal. A Gen. 2003, 251, 411–424. [Google Scholar] [CrossRef]
- Marin, G.; Yablonsky, G.S. Kinetics of Chemical Reactions—Decoding Complexity; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Ishikawa, S.; Kobayashi, D.; Konya, T.; Ohmura, S.; Murayama, T.; Yasuda, N.; Sadakane, M.; Ueda, W. Redox Treatment of Orthorhombic Mo 29V11O112 and Relationships between Crystal Structure, Microporosity and Catalytic Performance for Selective Oxidation of Ethane. J. Phys. Chem. C 2015, 119, 7195–7206. [Google Scholar] [CrossRef]
- Ueda, W. Personal communication at NGSC 11. Tromsø, Norway, 8 June 2016. [Google Scholar]
- Kolen’ko, Y.V.; Zhang, W.; Naumann d’Alnoncourt, R.; Girgsdies, F.; Hansen, T.W.; Wolfram, T.; Schlögl, R.; Trunschke, A. Synthesis of MoVTeNb Oxide Catalysts with Tunable Particle Dimens ions. ChemCatChem 2011, 3, 1597–1606. [Google Scholar] [CrossRef]
Figure 1.
Scheme of three levels of kinetic complexity.

Figure 2.
Reaction scheme: “Allyl alcohol-1,1- isomerization, ether formation, and oxidation over MoO” Ref. [40] (Figure 2).
Figure 2.
Reaction scheme: “Allyl alcohol-1,1- isomerization, ether formation, and oxidation over MoO” Ref. [40] (Figure 2).

Figure 3.
Reaction schemes taken from Ref. [46]. Scheme 4 (left): “Reaction network for propane oxidation over MoVTeNbO. Shaded arrow refers to main reaction pathway; compounds with underlines refer to the main products involved in propane and propene oxidation reaction.” and Scheme 5 (right): “Reaction network for propane oxidation over VO–H beta. Shaded arrow refers to main reaction pathway; compounds with underlines refer to the main products involved in propane and propene oxidation reaction”.
Figure 3.
Reaction schemes taken from Ref. [46]. Scheme 4 (left): “Reaction network for propane oxidation over MoVTeNbO. Shaded arrow refers to main reaction pathway; compounds with underlines refer to the main products involved in propane and propene oxidation reaction.” and Scheme 5 (right): “Reaction network for propane oxidation over VO–H beta. Shaded arrow refers to main reaction pathway; compounds with underlines refer to the main products involved in propane and propene oxidation reaction”.

Figure 4.
Reaction scheme: (a) “Proposed reaction pathways for propane oxidation over Mo–V–Sb–Nb mixed oxide catalysts (*, not detected).“ Ref. [6] (Scheme 2) and (b) “Reaction network and rate constants for MoV0.27Te0.17Nb0.12Ox at 653 K derived from experiments with single reactants. (rate constants in mL g−1 s−1)” Ref. [10] (Figure 4).
Figure 4.
Reaction scheme: (a) “Proposed reaction pathways for propane oxidation over Mo–V–Sb–Nb mixed oxide catalysts (*, not detected).“ Ref. [6] (Scheme 2) and (b) “Reaction network and rate constants for MoV0.27Te0.17Nb0.12Ox at 653 K derived from experiments with single reactants. (rate constants in mL g−1 s−1)” Ref. [10] (Figure 4).

Figure 5.
Reaction scheme: “The simplified graph of the presented detailed mechanism.” Ref. [9] (Figure 19).
Figure 5.
Reaction scheme: “The simplified graph of the presented detailed mechanism.” Ref. [9] (Figure 19).

Figure 6.
XRD diffractogram of the fresh and used sample - typical reflexes for M1 structure appear, the bulk structure of the catalyst is preserved. Each diffractogram was normalised with respect to its corresponding maximum value.
Figure 6.
XRD diffractogram of the fresh and used sample - typical reflexes for M1 structure appear, the bulk structure of the catalyst is preserved. Each diffractogram was normalised with respect to its corresponding maximum value.
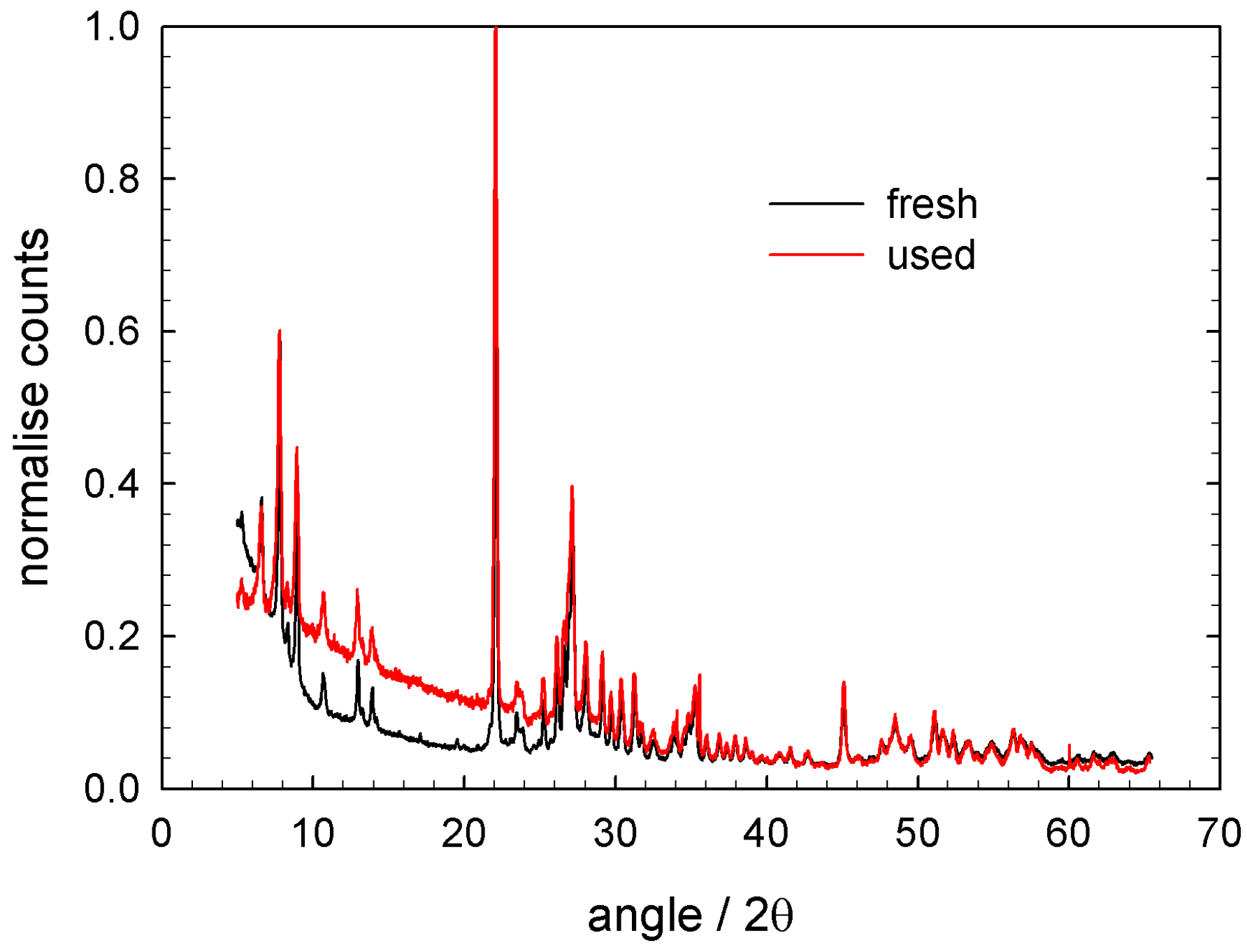
Figure 7.
Dependence of product formation rates during partial pressure variation experiments at 573 K and WHSV = 104 mL g−1 h−1: (a) Propane variation (C3H8/O2/H2O/N2 = x/12/10/78 − x); (b) Oxygen variation (C3H8/O2/H2O/N2 = 3/x/10/87 − x). Legend: ‘black circle’ –CO2; ‘red triangle down’ –CO; ‘green square’–propene; ‘yellow diamond’—acetic acid; ‘blue triangle up’—propionic acid; ‘magenta hexagon’—acrylic acid; ‘cyan cirlce’ –CO–CO(acetic acid).
Figure 7.
Dependence of product formation rates during partial pressure variation experiments at 573 K and WHSV = 104 mL g−1 h−1: (a) Propane variation (C3H8/O2/H2O/N2 = x/12/10/78 − x); (b) Oxygen variation (C3H8/O2/H2O/N2 = 3/x/10/87 − x). Legend: ‘black circle’ –CO2; ‘red triangle down’ –CO; ‘green square’–propene; ‘yellow diamond’—acetic acid; ‘blue triangle up’—propionic acid; ‘magenta hexagon’—acrylic acid; ‘cyan cirlce’ –CO–CO(acetic acid).
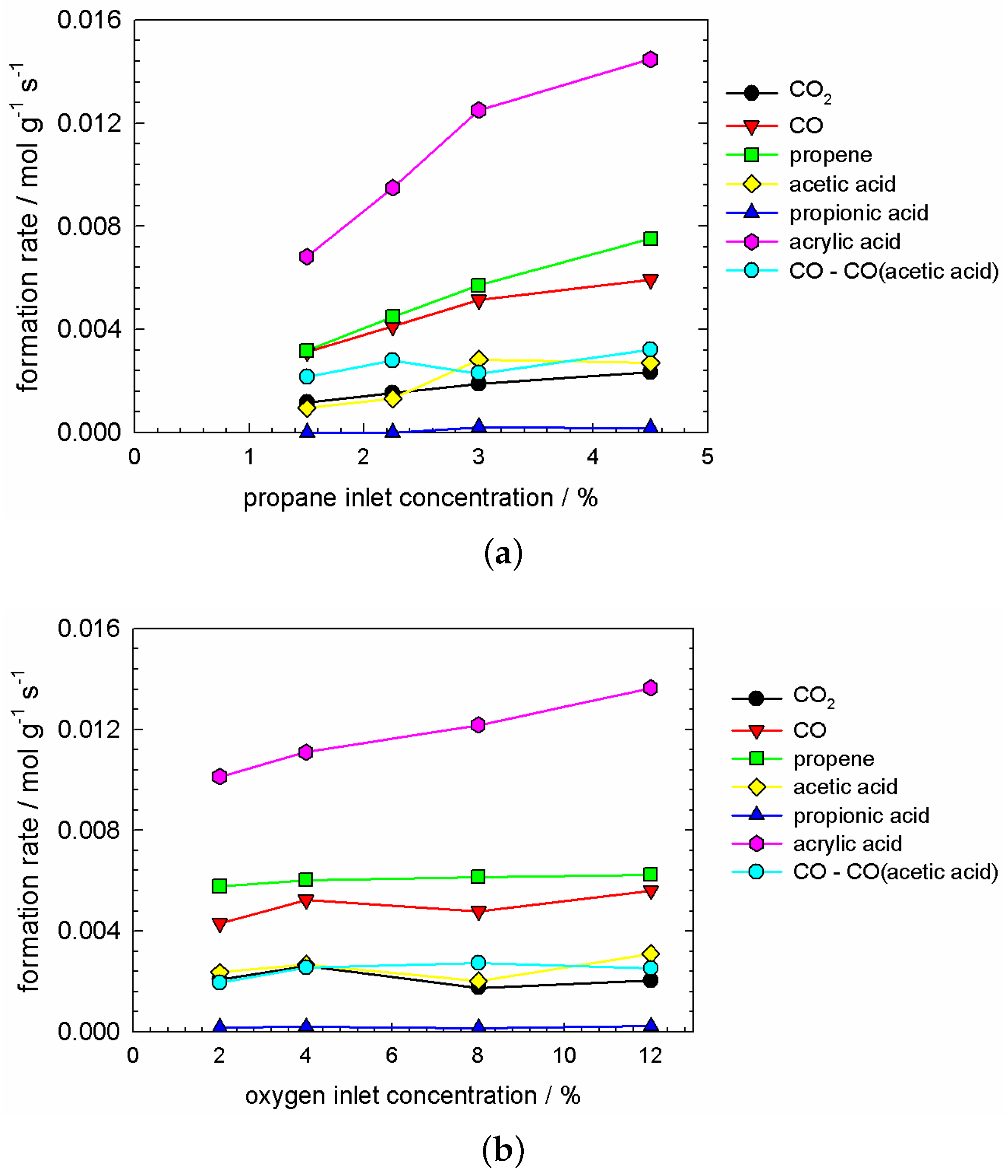
Figure 8.
Dependence of product formation on water partial pressure in the reactor feed at 573 K and WHSV = 10 mL g h (CH/O/HO/N = 3/12/x/). Legend: ‘black circle’ –CO; ‘red triangle down’ –CO; ‘green square’—propene; ‘yellow diamond’—acetic acid; ‘blue triangle up’—propionic acid; ‘magenta hexagon’—acrylic acid; ‘cyan cirlce’ –CO–CO(acetic acid).
Figure 8.
Dependence of product formation on water partial pressure in the reactor feed at 573 K and WHSV = 10 mL g h (CH/O/HO/N = 3/12/x/). Legend: ‘black circle’ –CO; ‘red triangle down’ –CO; ‘green square’—propene; ‘yellow diamond’—acetic acid; ‘blue triangle up’—propionic acid; ‘magenta hexagon’—acrylic acid; ‘cyan cirlce’ –CO–CO(acetic acid).

Figure 9.
CH/O/HO/N = 3/12/10/75 at 573 K and 10 mL g h over M1 catalysts MoVTeNbO: course of outlet concentrations as a function of time-on-stream after a treatment for 2 h in CH/O/HO/N = 3/0/10/87 at 573 K and 10 mL g h. Parts (a,b) show the experiment performed under the exact same conditions on two consecutive days, expressing the reproducibility of the experiment and reversibility of the catalyst performance. Legend: ‘black circle’ –CO; ‘red triangle down’ –CO; ‘green square’—propene; ‘yellow diamond’–acetic acid; ‘blue triangle up’—propionic acid; ‘magenta hexagon’—acrylic acid; ‘cyan cirlce’ –CO–CO(acetic acid).
Figure 9.
CH/O/HO/N = 3/12/10/75 at 573 K and 10 mL g h over M1 catalysts MoVTeNbO: course of outlet concentrations as a function of time-on-stream after a treatment for 2 h in CH/O/HO/N = 3/0/10/87 at 573 K and 10 mL g h. Parts (a,b) show the experiment performed under the exact same conditions on two consecutive days, expressing the reproducibility of the experiment and reversibility of the catalyst performance. Legend: ‘black circle’ –CO; ‘red triangle down’ –CO; ‘green square’—propene; ‘yellow diamond’–acetic acid; ‘blue triangle up’—propionic acid; ‘magenta hexagon’—acrylic acid; ‘cyan cirlce’ –CO–CO(acetic acid).

Figure 10.
(a) C3H8/O2/H2O/N2 = 3/12/10/75 at 573 K and 104 mL g−1 h−1 over M1 catalysts MoVTeNbOx: course of outlet concentrations as a function of time-on-stream after a treatment for 12 h in C3H8/O2/H2O/N2 = 3/0/10/87 at 573 K and 104 mL g−1 h−1. Legend: ‘black circle’ –CO2; ‘red triangle down’ –CO, ‘green square’–propene, ‘yellow diamond’–acetic acid, ‘blue triangle up’—propionic acid, ‘magenta hexagon’—acrylic acid, ‘cyan cirlce ’–CO–CO(acetic acid). (b) Data treatment of the data presented in (a): partial mass balances, including the stoichiometry of carbon atoms. Legend: ‘black circle’– CO2 − CO − CO (acetic acid); ‘red triangle down’ –CO + CO2 + 3*acrylic acid; ‘green square’ –CO + CO2 + 3*acrylic acid + 2*acetic acid; ‘yellow diamond’—3*propene + 2*acetic acid; ‘blue triangle up’—all; ‘magenta hexagon’ –CO + CO2 + 3*acrylic acid –CO(acetic acid).
Figure 10.
(a) C3H8/O2/H2O/N2 = 3/12/10/75 at 573 K and 104 mL g−1 h−1 over M1 catalysts MoVTeNbOx: course of outlet concentrations as a function of time-on-stream after a treatment for 12 h in C3H8/O2/H2O/N2 = 3/0/10/87 at 573 K and 104 mL g−1 h−1. Legend: ‘black circle’ –CO2; ‘red triangle down’ –CO, ‘green square’–propene, ‘yellow diamond’–acetic acid, ‘blue triangle up’—propionic acid, ‘magenta hexagon’—acrylic acid, ‘cyan cirlce ’–CO–CO(acetic acid). (b) Data treatment of the data presented in (a): partial mass balances, including the stoichiometry of carbon atoms. Legend: ‘black circle’– CO2 − CO − CO (acetic acid); ‘red triangle down’ –CO + CO2 + 3*acrylic acid; ‘green square’ –CO + CO2 + 3*acrylic acid + 2*acetic acid; ‘yellow diamond’—3*propene + 2*acetic acid; ‘blue triangle up’—all; ‘magenta hexagon’ –CO + CO2 + 3*acrylic acid –CO(acetic acid).

Figure 11.
Scheme of a bridge mechanism. The ‘active site’ consists of two ‘active centres’ X and X’. On centre X the total oxidation (cycle A) takes place, and the selective oxidation (cycle B) takes place on centre X’. X(TO) corresponds to the sum of all intermediates, which are part of cycle A (for total oxidation). X’(SO) corresponds to the sum of all intermediates, which are part of cycle B (for selective oxidation). The bridge mechanism is fed by [O], i.e., oxygen from the catalyst bulk or the gas phase.
Figure 11.
Scheme of a bridge mechanism. The ‘active site’ consists of two ‘active centres’ X and X’. On centre X the total oxidation (cycle A) takes place, and the selective oxidation (cycle B) takes place on centre X’. X(TO) corresponds to the sum of all intermediates, which are part of cycle A (for total oxidation). X’(SO) corresponds to the sum of all intermediates, which are part of cycle B (for selective oxidation). The bridge mechanism is fed by [O], i.e., oxygen from the catalyst bulk or the gas phase.

Figure 12.
Summary of the proposed reaction mechanism of selective oxidation of propane.

Figure 13.
The data interpretation lead to the following model: (a) A catalyst has different types of ‘active sites’ (here, 1 and 2). A specific product (mixture) is formed over each active site. Active sites have one or more ‘active centres’. (b) The evolution of the active centres on site 1 is linked together (one appears, the other disappears), thus, the sum of product evolution as a function of time over site 1 is constant. This phenomenon is called a ‘kinetic fingerprint’.
Figure 13.
The data interpretation lead to the following model: (a) A catalyst has different types of ‘active sites’ (here, 1 and 2). A specific product (mixture) is formed over each active site. Active sites have one or more ‘active centres’. (b) The evolution of the active centres on site 1 is linked together (one appears, the other disappears), thus, the sum of product evolution as a function of time over site 1 is constant. This phenomenon is called a ‘kinetic fingerprint’.

Figure 14.
Decision tree–Strategy of kinetic analysis.


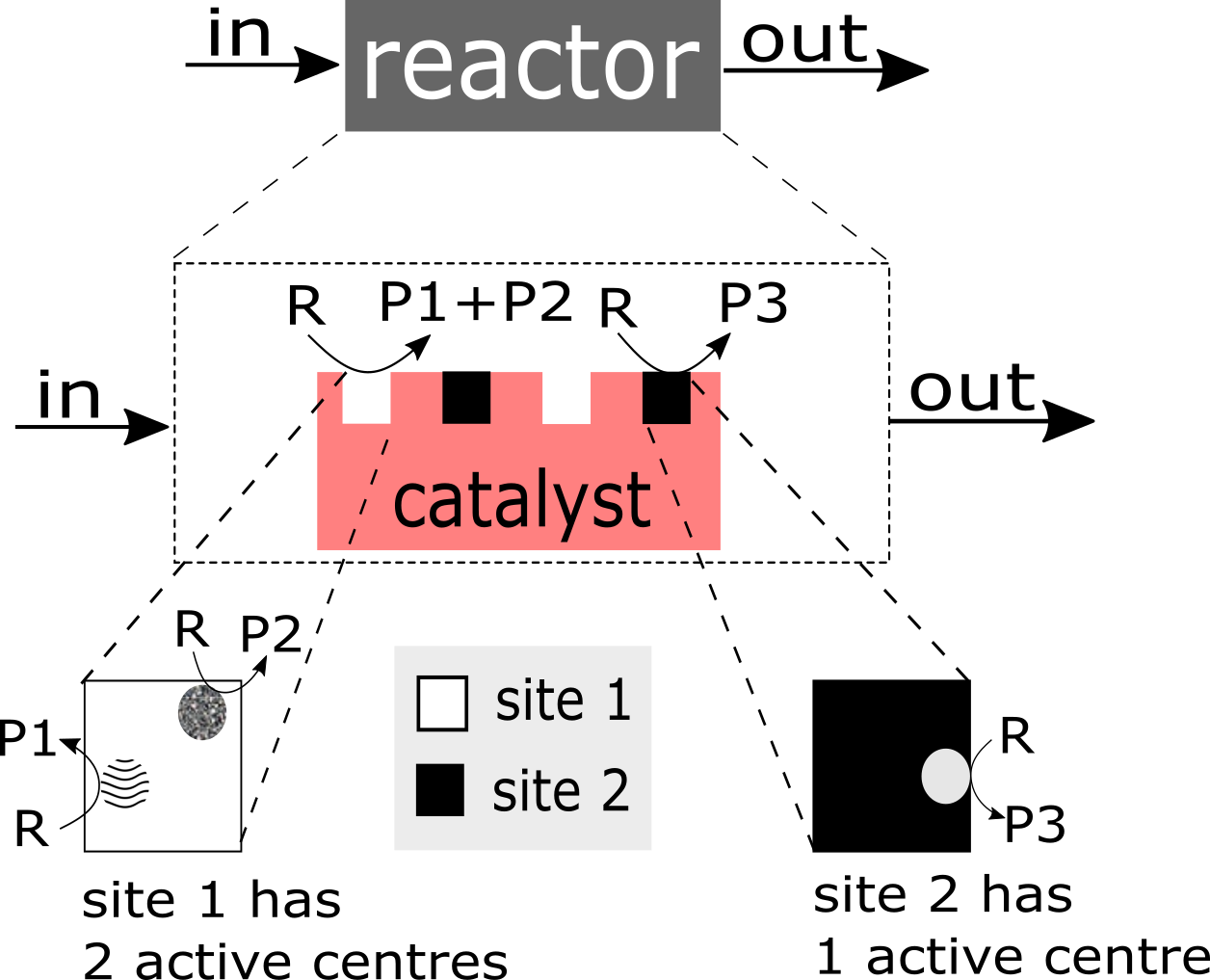{kind=link}
{kind=link}
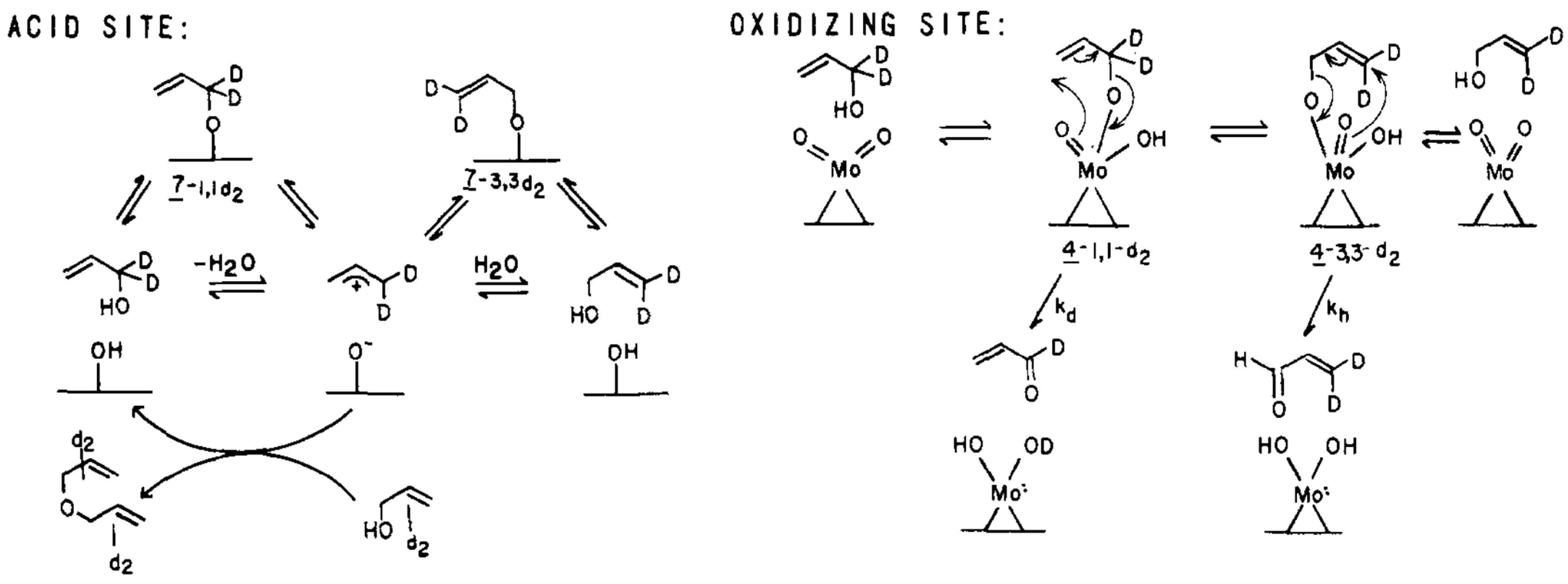{kind=link}
{kind=link}
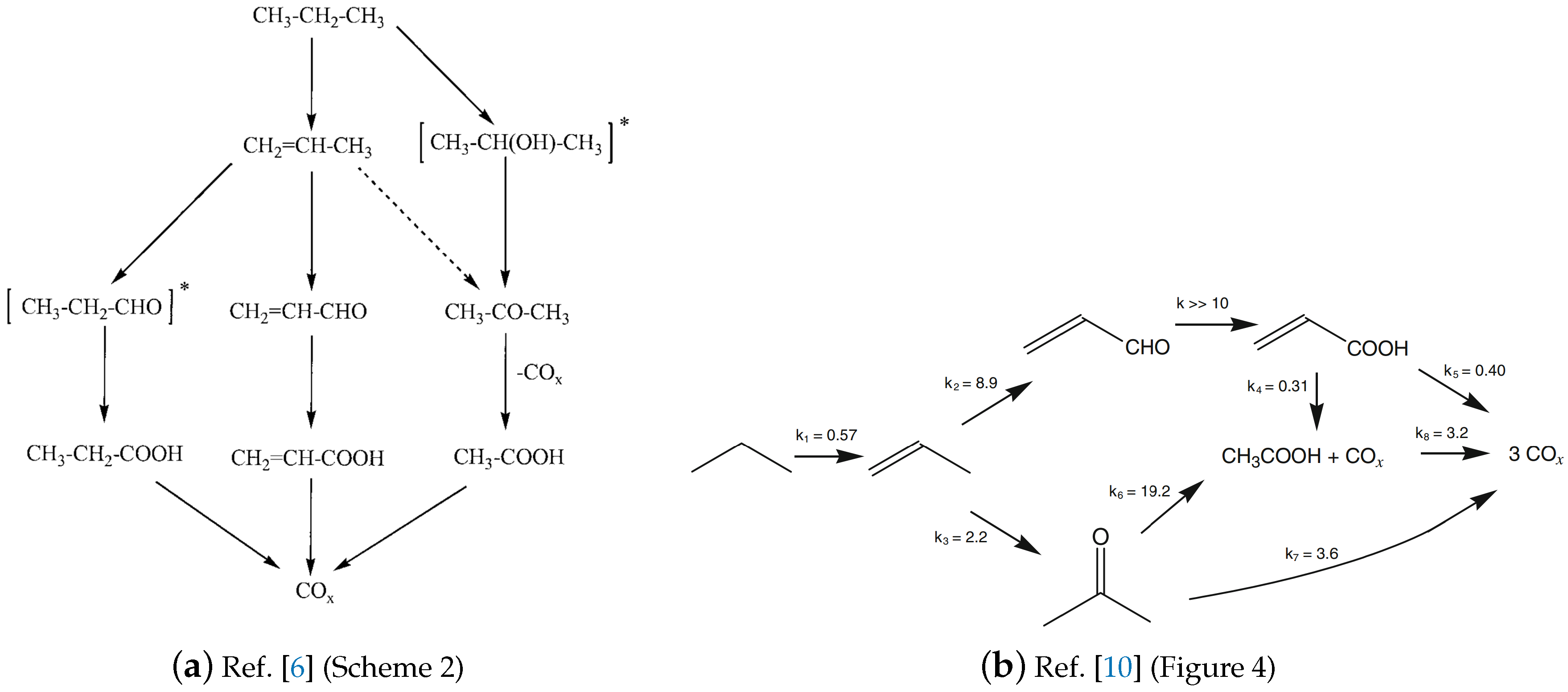{kind=link}
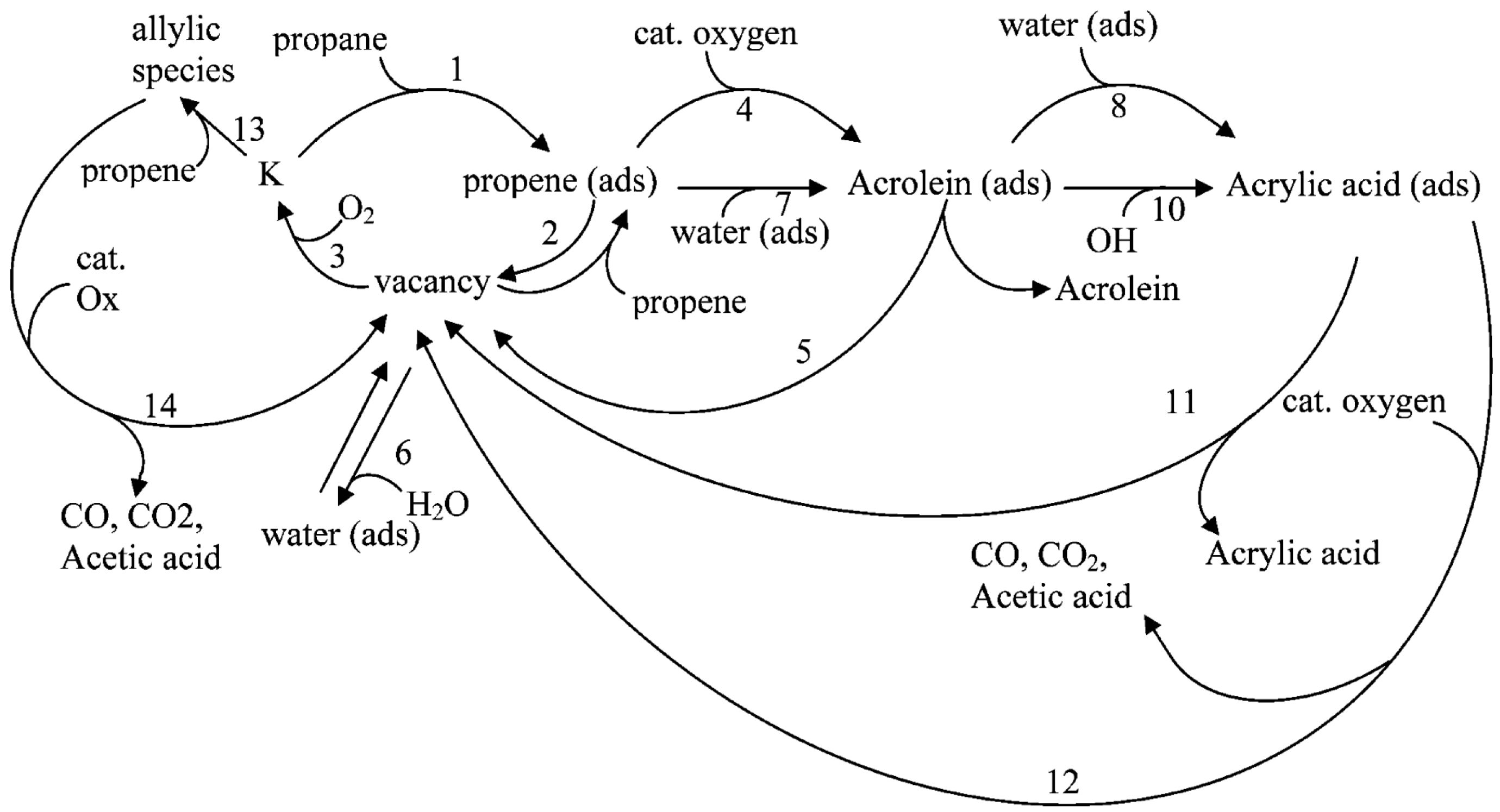{kind=link}
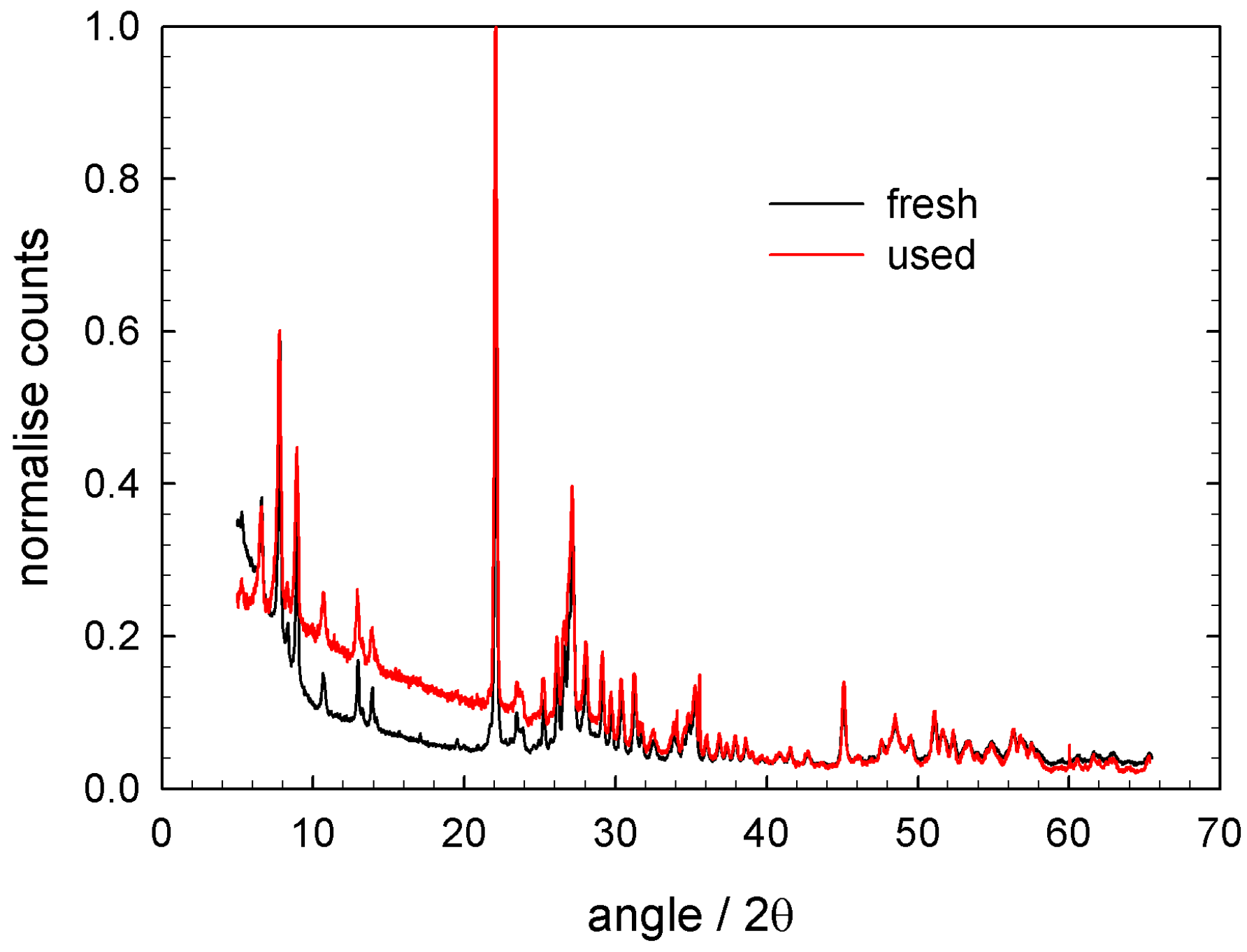{kind=link}
{kind=link}
{kind=link}
{kind=link}
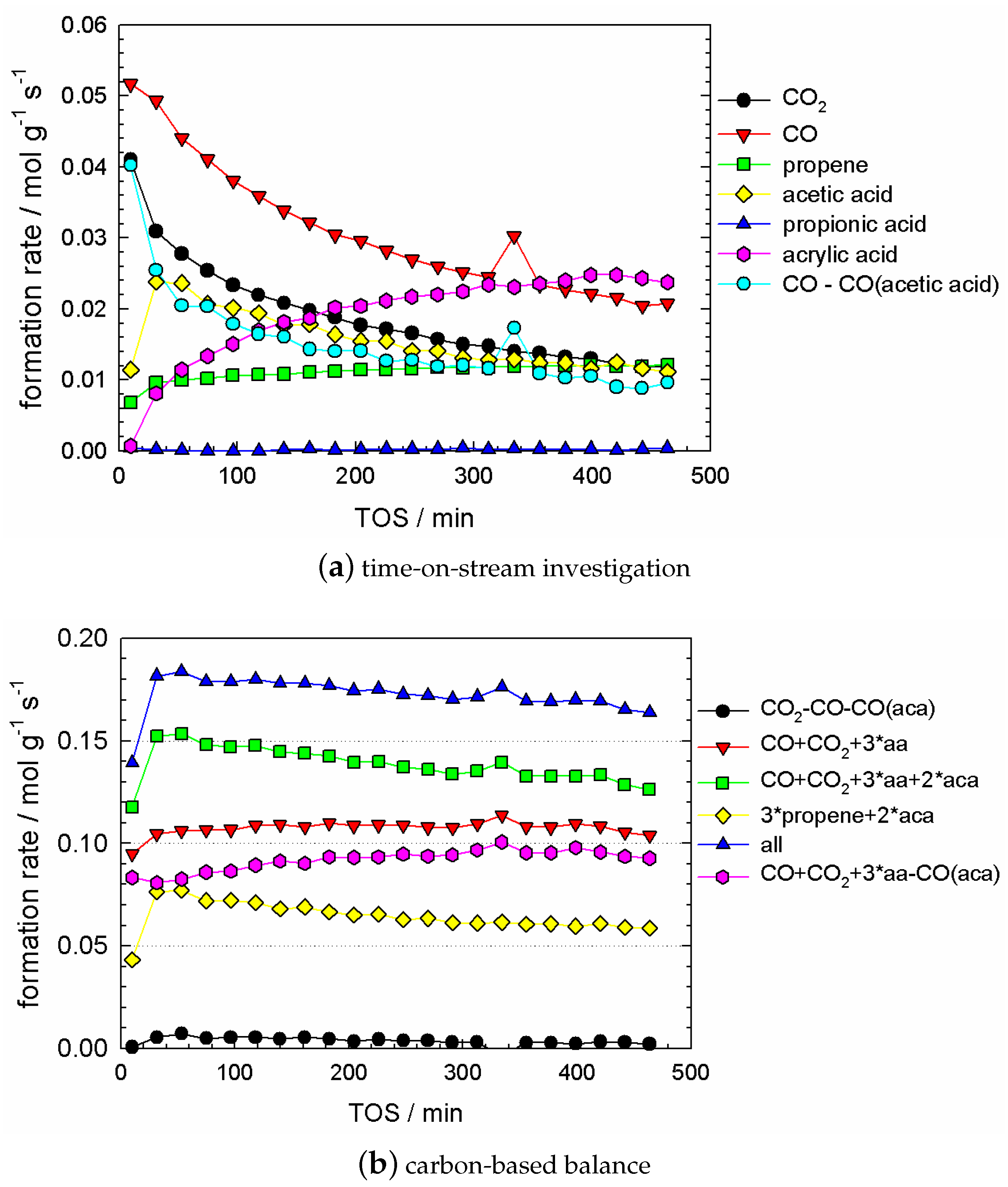{kind=link}
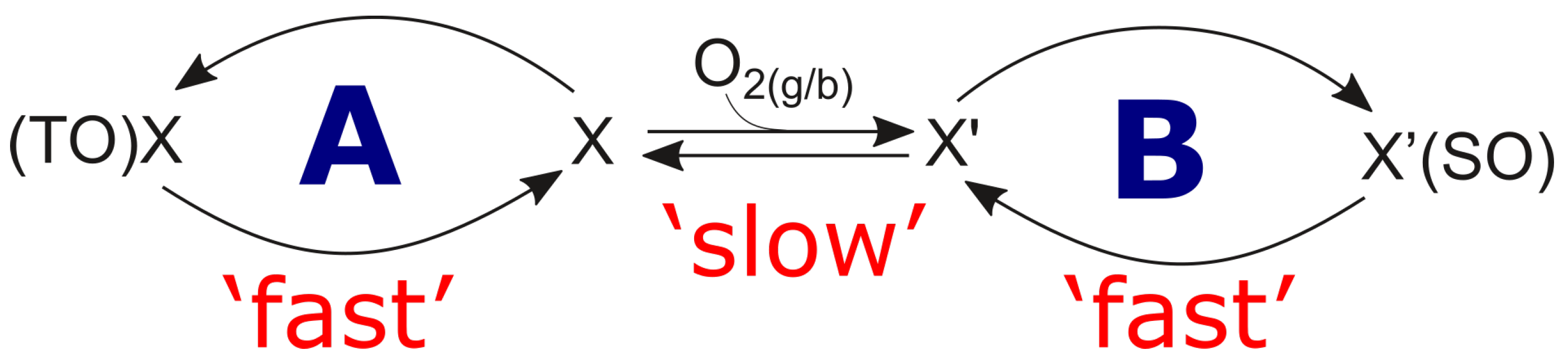{kind=link}
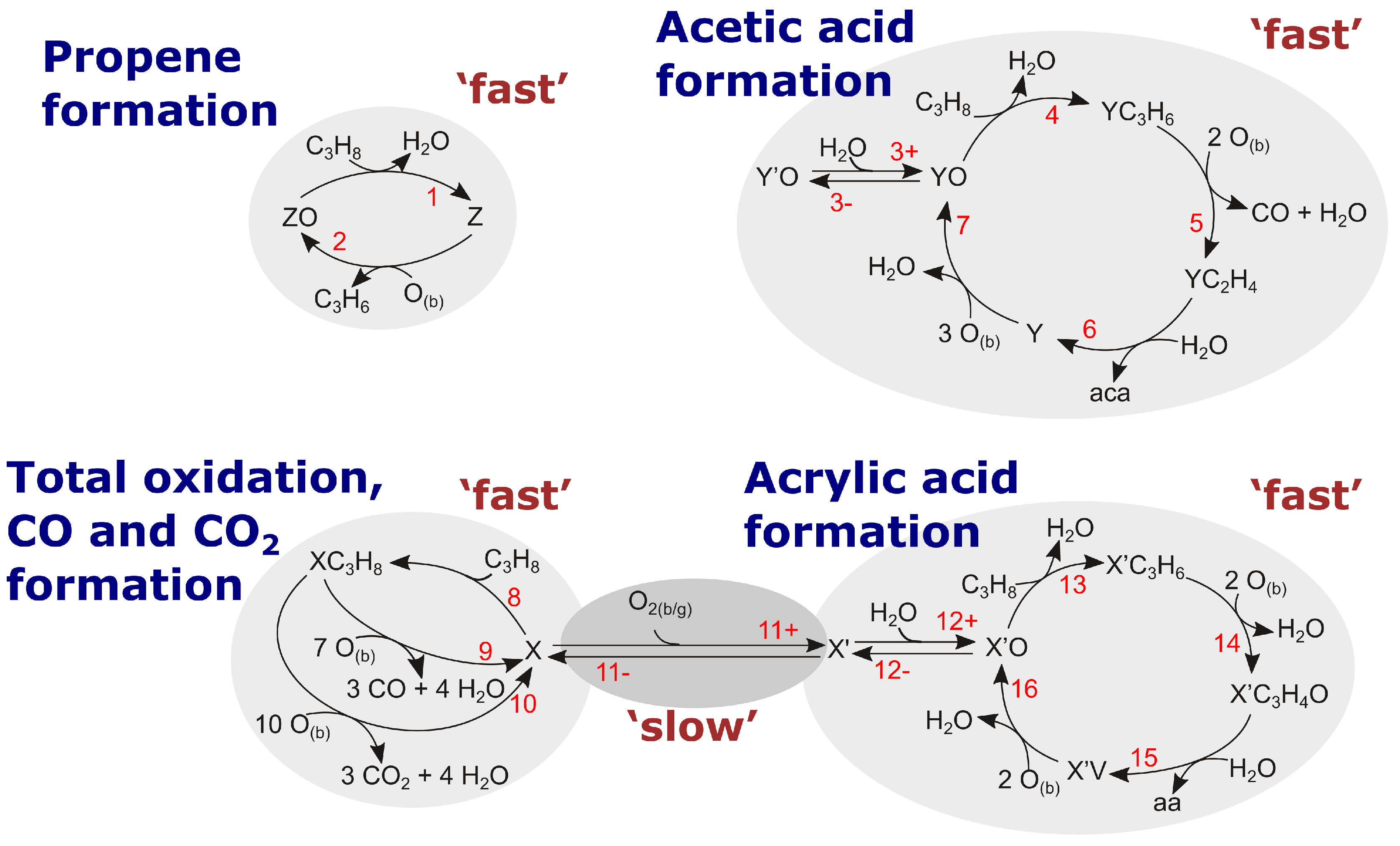{kind=link}
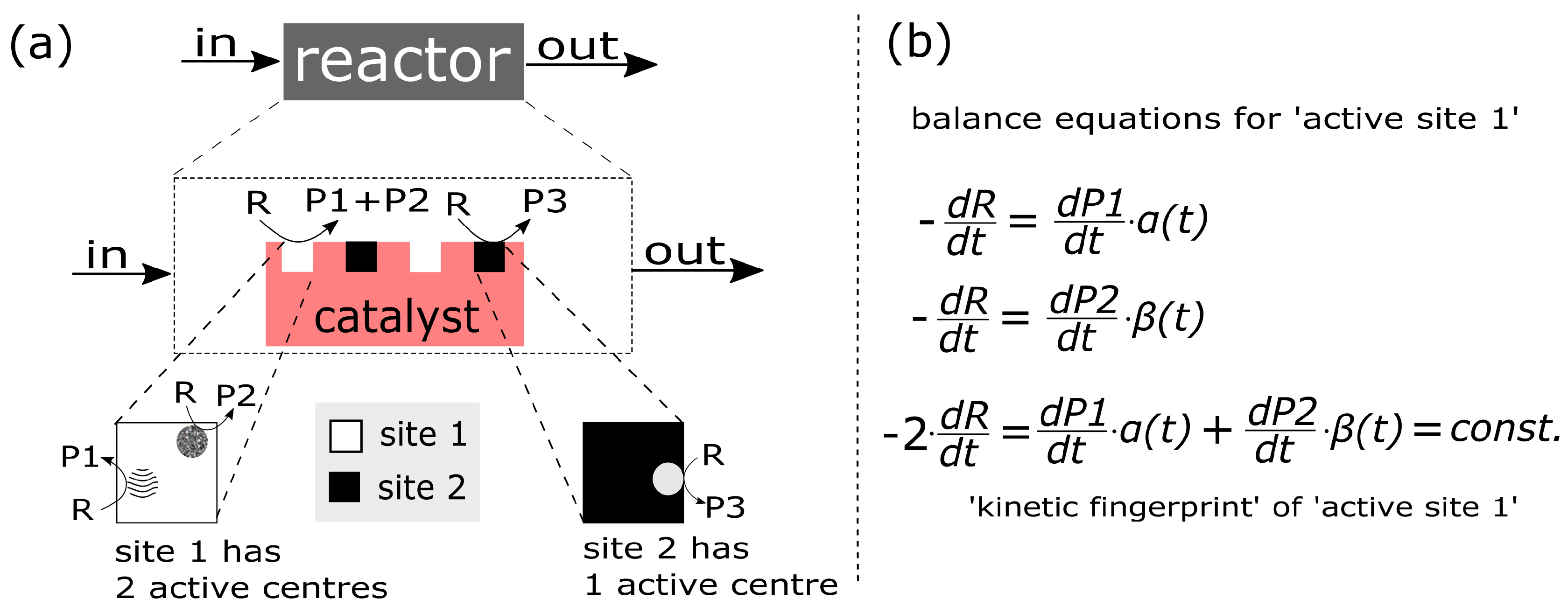{kind=link}
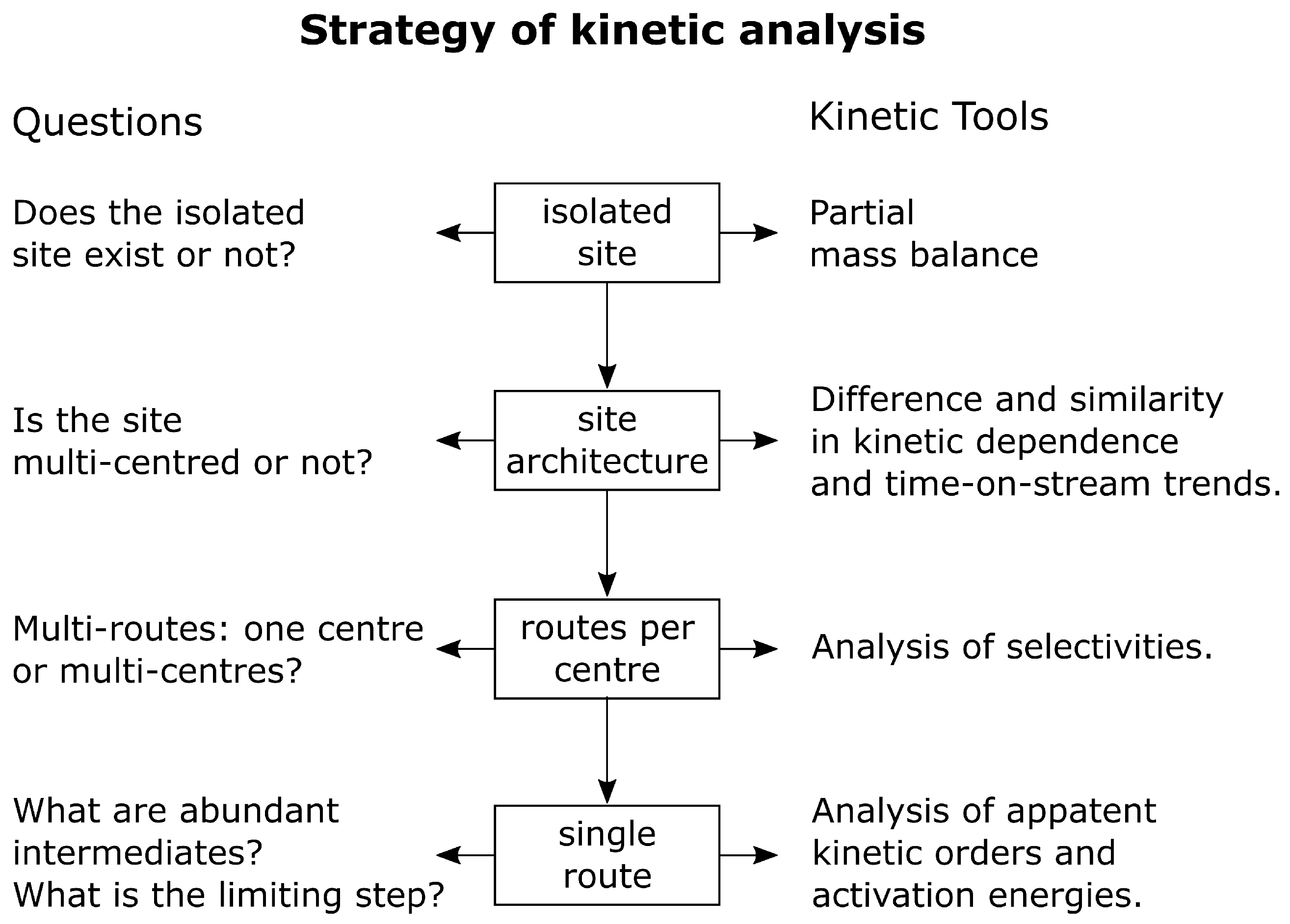{kind=link}
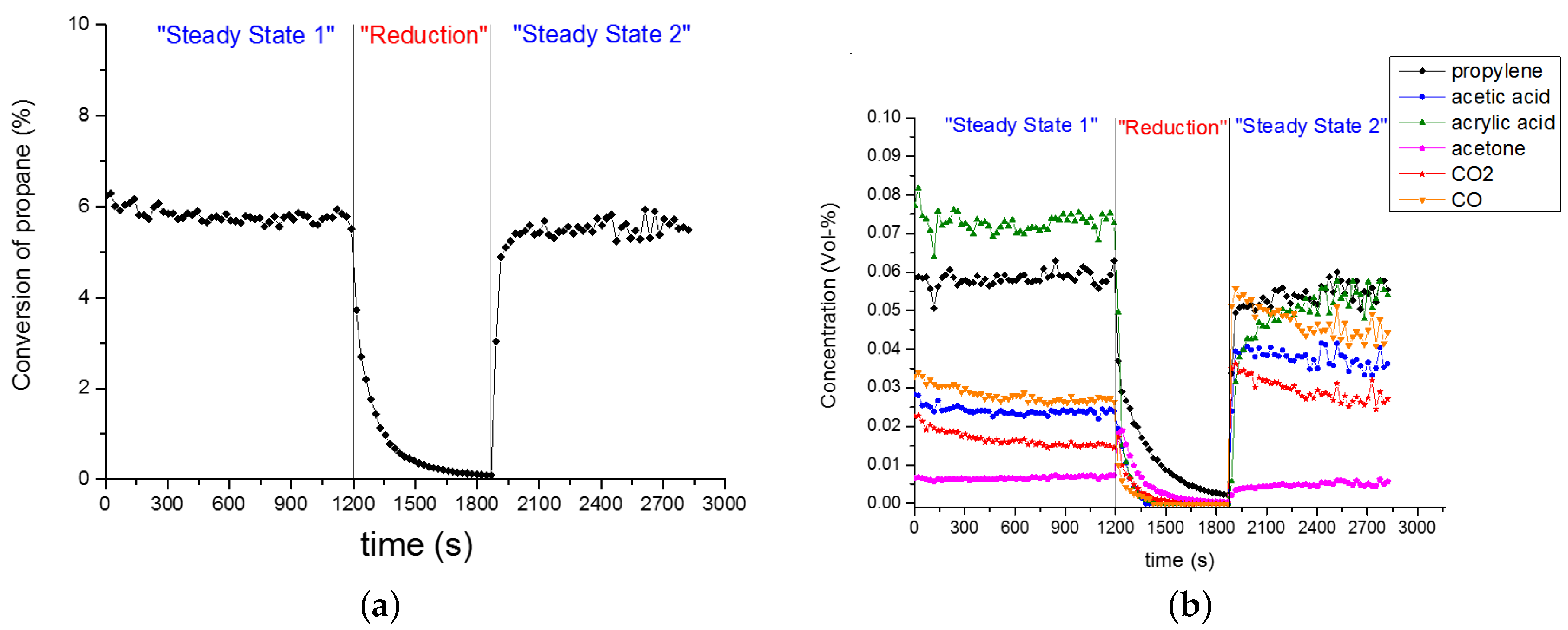{kind=link}
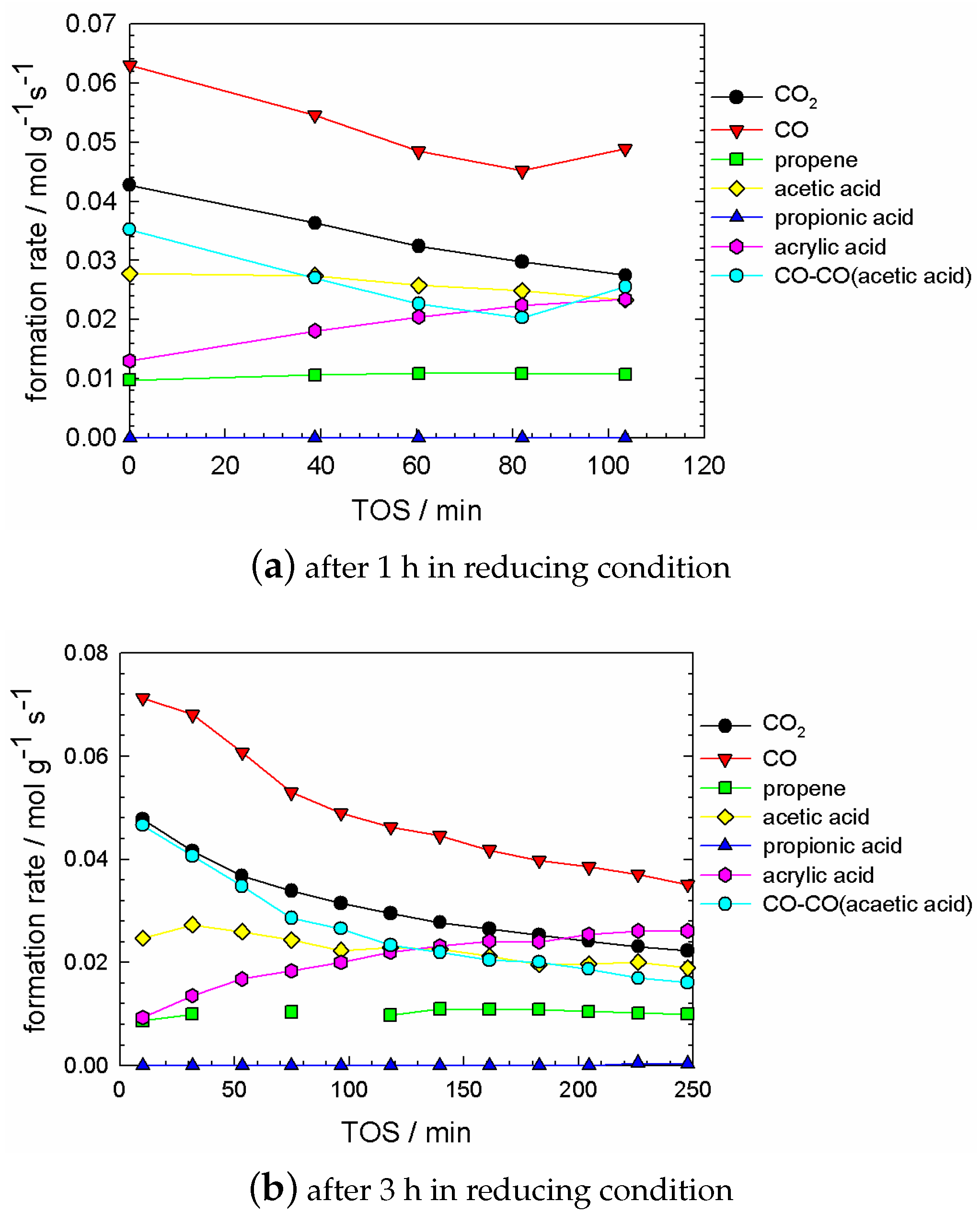{kind=link}
Table 1.
Experiments: catalysts and working conditions.
| Kinetic Tool | Experimental Details | Temperature/K | Gas Phase Composition | Reference | ||
|---|---|---|---|---|---|---|
| CH | O | HO | ||||
| PFR | reduced and oxidised Mo-species | 648–698 | 60 | 20 | 20 | [3] |
| MoO/NiMoO | ||||||
| PFR | importance of reduced states | 613 | 3.3 | 3.3 | 33.3 | [4] |
| HPMoO(Py) | ||||||
| PFR | catalyst composition change | 623–693 | 4 | 8 | 30 | [5] |
| MoVTeNbO | ||||||
| PFR | propane/propene oxidation, three route network | 653–713 | 2–6 | 5–60 | 0–60 | [6] |
| MoVSbNbO | ||||||
| PFR | discrimination of active bulk structure | 613–653 | 8 (2.5–20) | 10 (2.5–20) | 44 | [7] |
| MoVTeO | ||||||
| PFR | multi-route mechanism, partial pressure variation | 653–683 | ≤10% | ≤10% | ≤10% | [8] |
| MoVTeNbO | ||||||
| TAP | influence of reactants and products tested | 293–623 | 6.2–40 | 15–50 | 25 | [9] |
| MoVTeNbO | ||||||
| PFR | first order network, structural analysis | 653 | 5 | 10 | 20 | [10] |
| MoVTeNbO M1 | ||||||
| PFR | O/CH and HO/CH change | 653–763 | 1 | 1, 2, 3 | 5, 7.5 | [11] |
| MoVTeNbO | ||||||
| PFR | extended characterisation and testing | 623–663 | 3 | 4.5–12 | 0–40 | [12] |
| MoVTeNbO M1 | ||||||
| PFR | steady-state and non-steady-state kinetics | 573 | 1.5–4.5 | 2–12 | 5–20 | |
| MoVTeNbO M1 | ||||||
| PFR | ODH | 703 | 5–15 | 1–4 | 0–5 | [13] |
| MoO, VO, WO/ZrO | ||||||
| PFR | ammoxidation of propane | 693 | 1 | 1.2 | (NH) 3 | [14] |
| MoVNbTeO & MoVTaTeO | ||||||
| PFR | isotopic labelling HO | 703 | CH 1 | 6.1 | 7 | [15] |
| MoWSnTeO | ||||||
| PFR | ODH; isotopic labelling | 593 | 14.2 | 1.7 | – | [16] |
| 10 wt% VO/ZrO | ||||||
| PFR | ODH; isotopic labelling C, O and H/D | 688–703 | 5–15 | 1–4 | – | [17] |
| MoO/ZrO | ||||||
| PFR | acrolein oxidation | 533–573 | CHO 4–8 | 6–20 | 20 | [18] |
| (MoVW)O | ||||||
PFR: Plug Flow Reactor; TAP: Temporal Analysis of Products; balance to 100% is inert gas; relative to total composition, balance nitrogen; CO 0–50%; relative to CH; this work.
Table 2.
Apparent kinetic orders of product formation rates presented in Figure 7 and Figure 8 as a function of the three reactants.
| Product | p(CH) | p(O) | p(HO) | ∑ Kin. Order | |||
|---|---|---|---|---|---|---|---|
| log k | log k | log k | |||||
| CO | 0.59 | 0.11 | 0.36 | 1.06 | |||
| CO-aca | 0.29 | 0.15 | 0.21 | 0.65 | |||
| CO | 0.63 | 0.2 | 0.77 | ||||
| CH | 0.78 | 0.04 | 0.04 | 0.86 | |||
| acetic acid | 1.06 | 0.07 | 0.51 | 1.64 | |||
| acrylic acid | 0.7 | 0.16 | 0.46 | 1.32 | |||
Table 3.
Apparent kinetic orders with respect to the three reactants ([], [], and [], respectively) of the mechanism proposed in Figure 12.
Table 3.
Apparent kinetic orders with respect to the three reactants ([], [], and [], respectively) of the mechanism proposed in Figure 12.
| Product | [] | [] | [] |
|---|---|---|---|
| CO | + | 0 | + + |
| CO-aca | 0 | ||
| CO | 0 | ||
| CH | 0 | ||
| acetic acid | + | + | + |
| acrylic acid | + | + | + |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sprung, C.; Yablonsky, G.S.; Schlögl, R.; Trunschke, A. Constructing A Rational Kinetic Model of the Selective Propane Oxidation Over A Mixed Metal Oxide Catalyst. Catalysts 2018, 8, 330. https://doi.org/10.3390/catal8080330
AMA Style
Sprung C, Yablonsky GS, Schlögl R, Trunschke A. Constructing A Rational Kinetic Model of the Selective Propane Oxidation Over A Mixed Metal Oxide Catalyst. Catalysts. 2018; 8(8):330. https://doi.org/10.3390/catal8080330
Chicago/Turabian StyleSprung, Christoph, Gregory S. Yablonsky, Robert Schlögl, and Annette Trunschke. 2018. "Constructing A Rational Kinetic Model of the Selective Propane Oxidation Over A Mixed Metal Oxide Catalyst" Catalysts 8, no. 8: 330. https://doi.org/10.3390/catal8080330
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.