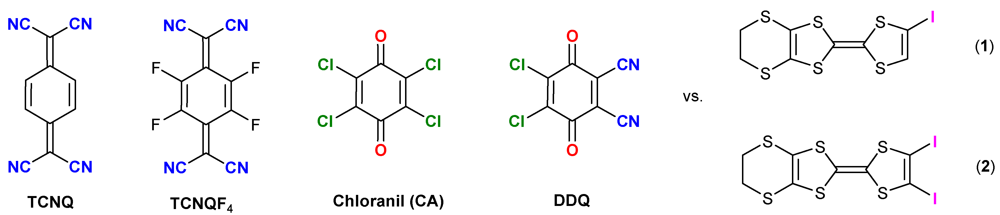
Halogen Bonding Interactions in DDQ Charge Transfer Salts with Iodinated TTFs

Abstract
:1. Introduction

2. Results and Discussion
2.1. Structural and Magnetic Properties of (1)(DDQ)
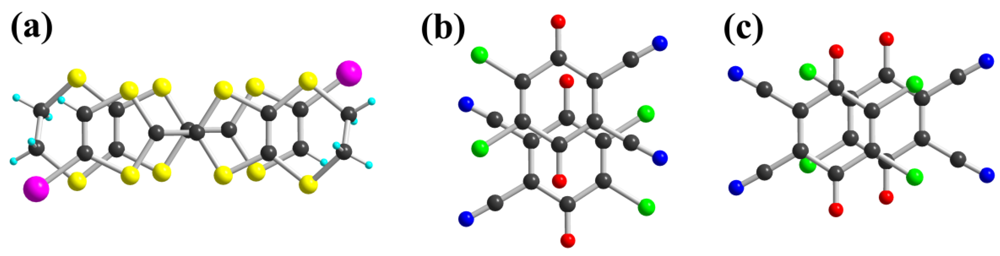
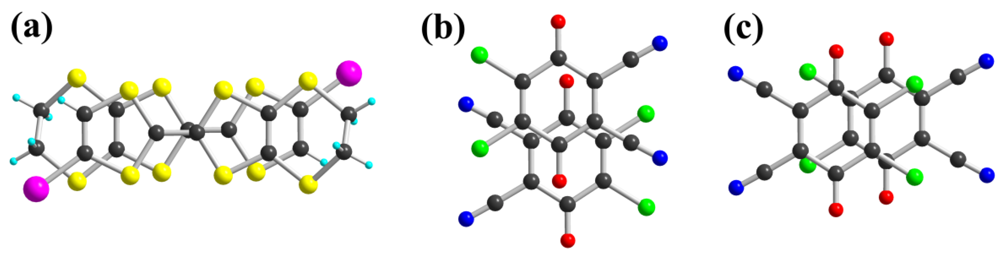
 with both donor 1 and DDQ in general position in the unit cell (Figure 1). Intramolecular bond lengths within the TTF core in 1 and the DDQ molecule are highly sensitive to their oxidation state. As shown in Table 1, Table 2, by comparison with reference compounds, a full charge transfer between 1 and DDQ has occurred, leading unambiguously to a (1+·)(DDQ−·) formulation. In the solid state, the 1+· cations are associated into face-to-face homo-dyads, which interact laterally along a-axis. A projection view of these dyads (Figure 2a) shows a typical bond-over-ring overlap, with a short plane-to-plane distance of 3.27 Å and a calculated βHOMO−HOMO interaction energy of 0.71 eV. On the other hand, the DDQ−· radical anions are stacked to form alternated chains running along a-axis with two different overlap modes shown in Figure 2b,c. Plane-to-plane distances amount to 2.79 and 3.42 Å respectively, demonstrating the strong dimerization of the DDQ chains. It is further confirmed by the calculated βLUMO−LUMO interaction energies for the two overlap modes, which amount to 0.65 and 0.04 eV respectively. It should be stressed here that these strong overlap between respectively 1+· and DDQ−· species let us infer that the radical species are strongly associated into the bonding combination of respectively the TTF’s HOMO and the DDQ LUMO. This is confirmed by the temperature dependence of the magnetic susceptibility of the salt, which exhibits an essentially diamagnetic behavior, with a Curie tail associated to 2.5% magnetic defaults.
with both donor 1 and DDQ in general position in the unit cell (Figure 1). Intramolecular bond lengths within the TTF core in 1 and the DDQ molecule are highly sensitive to their oxidation state. As shown in Table 1, Table 2, by comparison with reference compounds, a full charge transfer between 1 and DDQ has occurred, leading unambiguously to a (1+·)(DDQ−·) formulation. In the solid state, the 1+· cations are associated into face-to-face homo-dyads, which interact laterally along a-axis. A projection view of these dyads (Figure 2a) shows a typical bond-over-ring overlap, with a short plane-to-plane distance of 3.27 Å and a calculated βHOMO−HOMO interaction energy of 0.71 eV. On the other hand, the DDQ−· radical anions are stacked to form alternated chains running along a-axis with two different overlap modes shown in Figure 2b,c. Plane-to-plane distances amount to 2.79 and 3.42 Å respectively, demonstrating the strong dimerization of the DDQ chains. It is further confirmed by the calculated βLUMO−LUMO interaction energies for the two overlap modes, which amount to 0.65 and 0.04 eV respectively. It should be stressed here that these strong overlap between respectively 1+· and DDQ−· species let us infer that the radical species are strongly associated into the bonding combination of respectively the TTF’s HOMO and the DDQ LUMO. This is confirmed by the temperature dependence of the magnetic susceptibility of the salt, which exhibits an essentially diamagnetic behavior, with a Curie tail associated to 2.5% magnetic defaults. 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ρ | Ci=Ci | Ci−S | Co−S | Co−Co | Ref | |
|---|---|---|---|---|---|---|---|
| Neutral 1 | mol A | 0 | 1.332 (13) | 1.763 (18) | 1.759 (19) | 1.331 (14) | [18] |
| mol B | 1.323 (14) | 1.760 (17) | 1.747 (16) | 1.331 (13) | |||
| (1)2[Ag(CN)2] | +0.5 | 1.373 | 1.739 | 1.746 | 1.347 | [9] | |
| (1)2[Ni(mnt)2] | +1 | 1.382 (19) | 1.723 (25) | 1.731 (3) | 1.348 (19) | [18] | |
| (1)(DDQ) | 1 | 1.390 (7) | 1.721 (6) | 1.736 (6) | 1.350 (8) | this work | |
| Compound | ρ | C=O | CO−CCl | CO−CCN | CCl=CCl | CCN=CCN | νCN (cm−1) | Ref |
|---|---|---|---|---|---|---|---|---|
| DDQ | 0 | 1.202 | 1.483 | 1.501 | 1.340 | 1.343 | 2234 | [19] |
| (1)(DDQ) | –1 | 1.239 (5) | 1.475 (7) | 1.453 (8) | 1.360 (6) | 1.384 (6) | 2202 | – |
| (2)2(DDQ) | –1 | 1.265 (10) | 1.479 (15) | 1.422 (17) | 1.379 (12) | 1.402 (12) | 2212 | – |
| (Et4N)DDQ | –1 | 1.246 | 1.463 | 1.444 | 1.363 | 1.385 | 2217 | [20] |

2.2. Structural Properties of (2)2(DDQ)·(CH3CN)
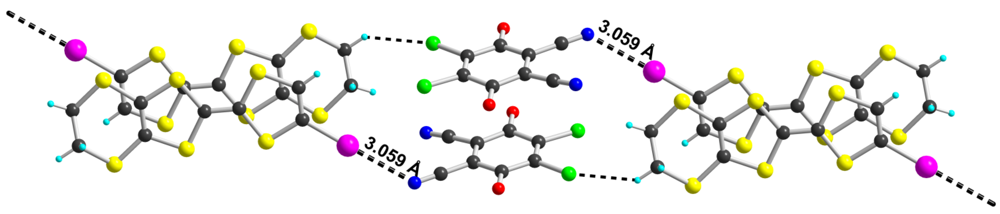
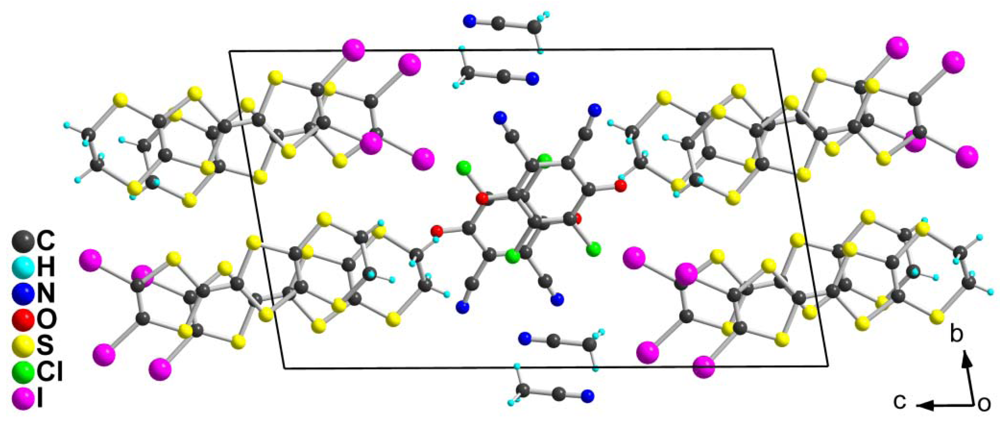
with two crystallographically independent TTF molecules 2, one DDQ in general position and one acetonitrile solvent molecule. A projection of the unit cell along a-axis (Figure 4), shows that the donor molecules form layers, separated from each other by the acetonitrile and DDQ molecules. As shown in Table 2, the DDQ molecule appears as fully reduced radical anion form. Bond lengths within the TTF core (Table 3) in 2 are also comparable to those described in other mixed-valence (ρ = 0.5) salts of 2 [11,12,13,14,15,16,17]. Note that between the two crystallographically independent donor molecules 2, the one bearing I3/I4 iodine atoms appears slightly more oxidized than molecule bearing I1/I2 iodine atoms. As shown in Figure 5, halogen bonding interactions develop at the interface between the mixed-valence slabs and the DDQ/CH3CN layer. Both crystallographically independent molecules 2 show short contacts with DDQ−· anion. Very short and linear interactions are found with I(3) and I(4) atoms, with the carbonyl oxygen atoms, the nitrile nitrogen atoms of both the DDQ and the CH3CN molecules. This donor molecule is also the one which appeared as the most oxidized one from comparison of intramolecular bond lengths (Table 4). Comparison of the two structures demonstrates that the reduced DDQ acts as a powerful halogen bond acceptor, involving the oxygen and nitrogen atoms. Earlier theoretical calculations of the charge and spin density in DDQ−· have shown a negative charge on the N, O and Cl atoms in the order −0.24, −0.20 and −0.18 [22], demonstrating a halogen bonding preference for the most negatively charged atoms. This behavior is in accordance with recent ab initio calculations showing that the electrostatic interaction between the positive σ-hole [23] located on the halogen atom along the C−Hal bond and Lewis bases such as pyridine is indeed the main component of the halogen bonding interaction [24].
| Compound | ρ | Ci=Ci | Ci−S | Co−S | Co−Co | Ref | |
|---|---|---|---|---|---|---|---|
| (2)2(I3) | +0.5 | 1.365 (29) | 1.740 (20) | 1.749 (20) | 1.348 (16) | [11] | |
| (2)2(DDQ) | mol (I1,I2) | ≈0.5 | 1.354 (9) | 1.757 (8) | 1.762 (8) | 1.336 (9) | this work |
| mol (I3,I4) | ≈0.5 | 1.365 (9) | 1.736 (8) | 1.745 (8) | 1.348 (9) | ||
| (2)(I3) | +1 | 1.40 (2) | 1.718 (9) | – | – | [18] | |

| Interaction | ∑vdw (Å) | Danis (Å) | I···(O,N) (Å) | C−I···(O,N) (°) | I···(O,N)−C (°) |
|---|---|---|---|---|---|
| I1···N2 | 3.53 | 3.36 | 3.163 (12) | 178.2 (3) | 147.8 (9) |
| I3···N3 | 3.53 | 3.36 | 2.895 (12) | 171.0 (3) | 139.2 (9) |
| I4···O1 | 3.50 | 3.30 | 2.761 (8) | 178.2 (2) | 138.7 (5) |


3. Experimental Section
3.1. Syntheses
3.2. Crystallography
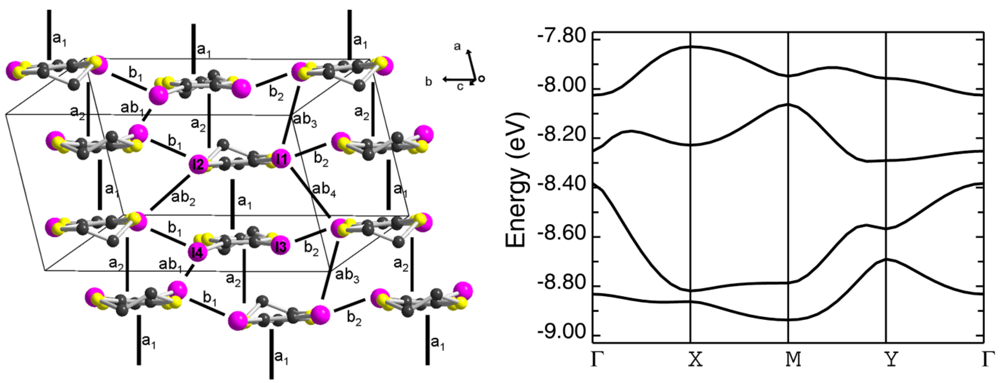
, a = 7.0227(2), b = 12.6760(3), c = 13.1732(3) Å, α = 62.169(1), β = 76.282(1), γ = 78.615(1)°, V = 1002.31(4) Å3, Z = 2, Dc = 2.145 g cm−3, T = 150(2) K, 14723 reflections collected, 4566 unique (Rint = 0.0303) with among them 3871 with I > 2σ(I), R[I > 2σ(I)] = 0.0341, wR2 (F2, all data) = 0.0967, GoF = 1.083., a = 7.4173(9), b = 12.9354(15), c = 21.258(3) Å, σ = 79.600(4), β = 85.564(4), γ = 75.854(4)°, V = 1944.1(4) Å3, Z = 2, Dc = 2.324 g cm−3, T = 150(2) K, 23021 reflections collected, 8631 unique (Rint = 0.0373) with among them 6389 with I > 2σ(I), R[I > 2σ(I)] = 0.0426, wR2 (F2, all data) = 0.1444, GoF = 1.051.3.3. Band Structure Calculations
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Fourmigué, M.; Batail, P. Activation of Hydrogen- and Halogen-Bonding Interactions in Tetrathiafulvalene-Based Crystalline Molecular Conductors. Chem. Rev. 2004, 104, 5379–5418. [Google Scholar] [CrossRef]
- Dolbecq, A.; Fourmigué, M.; Batail, P.; Coulon, C. From Racemic Mixtures of Chiral π-Donor molecules to Mixed Stacks of H-Bonded Centrosymmetrical Dimers of Cation and Anion Radicals with Singlet-Triplet Excitations: The Example of [(±)Me3TTF-C*HMe(OH)·+]2[TCNQ·–]2. Chem. Mater. 1994, 6, 1413–1418. [Google Scholar] [CrossRef]
- Baudron, S.A.; Mézière, C.; Heuzé, K.; Fourmigué, M.; Batail, P.; Auban-Senzier, P. Interplay of charge transfer, dimensionality and amide hydrogen bond network adaptability in TCNQF4 complexes of EDO-TTF-CONH2 and EDT-TTF-CONH2. J. Solid State Chem. 2002, 168, 668–674. [Google Scholar] [CrossRef]
- Ono, G.; Terao, H.; Higuchi, S.; Sugawara, T.; Izuoka, A.; Mochida, T. One-dimensional double chain composed of carbamoylmethylthio-substituted TTF-based donor in ion radical salt. J. Mater. Chem. 2000, 10, 2277–2282. [Google Scholar] [CrossRef]
- Iyoda, M.; Kuwatani, Y.; Ogura, E.; Hara, K.; Suzuki, H.; Takano, T.; Takeda, K.; Takano, J.-I.; Ugawa, K.; Yoshida, M.; et al. Synthesis, structure and conducting properties of halogenated ethylenedioxytetrathiafulvalenes. Heterocycles 2001, 54, 833–848. [Google Scholar] [CrossRef]
- Batsanov, A.S.; Bryce, M.R.; Chesney, A.; Howard, J.A.K.; John, D.E.; Moore, A.J.; Wood, C.L.; Gershtenman, H.; Becker, J.Y.; Khodorkovsky, V.Y.; et al. Synthesis and crystal engineering of new halogenated tetrathiafulvalene (TTF) derivatives and their charge transfer complexes and radical ion salts. J. Mater. Chem. 2001, 11, 2181–2191. [Google Scholar]
- Iyoda, M.; Suzuki, H.; Sasaki, S.; Yoshino, H.; Kikuchi, K.; Saito, K.; Ikemoto, I.; Matsuyama, H.; Mori, T. Charge-transfer complex and radical cation salt of a new donor EDT-TTFCl2 unique conductivities and crystal structures. J. Mater. Chem. 1996, 6, 501–503. [Google Scholar]
- Iyoda, M.; Kuwatani, Y.; Hara, K.; Ogura, E.; Suzuki, H.; Ito, H.; Mori, T. Halogenated Bis(methylthio)tetrathiafulvalenes as a Unique Donor System. Chem. Lett. 1997, 26, 599–600. [Google Scholar]
- Imakubo, T.; Sawa, H.; Kato, R. Novel radical cation salts of organic π-donors containing iodine atom(s): the first application of strong intermolecular-I···X− (X = CN, halogen atom) interaction to molecular conductors. Synth. Metals 1995, 73, 117–122. [Google Scholar] [CrossRef]
- Imakubo, T.; Shirahata, T.; Hervé, K.; Ouahab, L. Supramolecular organic conductors based on diiodo-TTFs and spherical halide ion X- (X = Cl, Br). J. Mater. Chem. 2006, 16, 162–173. [Google Scholar] [CrossRef]
- Domercq, B.; Devic, T.; Fourmigué, M.; Auban-Senzier, P.; Canadell, E. Hal···Hal interactions in a series of three isostructural salts of halogenated tetrathiafulvalenes. Contribution of the halogen atoms to the HOMO–HOMO overlap interactions. J. Mater. Chem. 2001, 11, 1570–1575. [Google Scholar] [CrossRef]
- Alberola, A.; Fourmigué, M.; Gómez-García, C.J.; Llusar, R.; Triguero, S. Halogen halogen interactions with the [Mo3S7Cl6]2− cluster anion in the mixed valence salt [EDT-TTFI2]4[Mo3S7Cl6]·CH3CN. New J. Chem. 2008, 32, 1103–1109. [Google Scholar] [CrossRef]
- Devic, T.; Evain, M.; Moëlo, Y.; Canadell, E.; Senzier, P.; Fourmigué, M.; Batail, P. Single Crystalline Commensurate Metallic Assemblages of π-Slabs and CdI2-type Layers: Synthesis and Properties of β-[EDT-TTF-I2]2[Pb5/6 square 1/6I2]3 and β-[EDT-TTF-I2]2[Pb2/3 + xAg1/3 - 2x square xI2]3, X = 0.05. 05. J. Am. Chem. Soc. 2003, 125, 3295–3301. [Google Scholar] [CrossRef]
- Imakubo, T.; Sawa, H.; Kato, R. Crystal engineering in molecular conductors based on iodine-bonded π-donors. Synth. Metals 1997, 86, 1847–1848. [Google Scholar] [CrossRef]
- Ranganathan, A.; El-Ghayoury, A.; Mézière, C.; Harté, E.; Clérac, R.; Batail, P. Balancing framework densification with charged, halogen-bonded-π-conjugated linkages: [PPh4]2{[E-TTF-I2][Re6Se8(CN)6]} vs. [PPh4]2[EDT-TTF-I]2{[EDT-TTF-I][Re6Se8(CN)6]}. Chem. Commun. 2006, 21, 2878–2880. [Google Scholar]
- Hervé, K.; Cador, O.; Golhen, S.; Costuas, K.; Halet, J.-F.; Shirahata, T.; Muto, T.; Imakubo, T.; Miyazaki, A.; Ouahab, L. Substituted tetrathiafulvalene radical cation salts with [M(isoq)2(NCS)4]− anions where M = CrIII, GaIII: role of I···S and S···S contacts on structural and magnetic properties. Chem. Mater. 2006, 18, 790–797. [Google Scholar]
- Fourmigué, M.; Auban-Senzier, P. Anionic layered networks reconstructed from [Cd(SCN)3]∞- chains in pseudo one dimensional conducting salts of halogenated tetrathiafulvalenes. Inorg. Chem. 2008, 47, 9979–9986. [Google Scholar] [CrossRef]
- Devic, T.; Domercq, B.; Auban-Senzier, P.; Molinié, P.; Fourmigué, M. Cyano-Halogen Interactions in [EDT-TTF-I]2[Ni(mnt)2] and [EDT-TTF-I2]2[Ni(mnt)2] and Geometrical Evolutions within Mixed-Valence or Fully Oxidized Dyads. Eur. J. Inorg. Chem. 2002, 2002, 2844–2849. [Google Scholar]
- Zanotti, G.; Bardi, R.; Del Pra, A. Structure of 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ). Acta Crystallogr. 1980, B36, 168–171. [Google Scholar]
- Zanotti, G.; Del Pra, A.; Bozio, R. Structure of tetraethylammonium-2,3-dichloro-5,6-dicyano-p-benzoquinone. Acta Crystallogr. 1982, B38, 1225–1229. [Google Scholar]
- Nyburg, S.C.; Faerman, C.H. A Revision of van der Waals Atomic Radii for Molecular Crystals: N, O, F, S, Cl, Se, Br and I Bonded to Carbon. Acta Cryst 1985, B41, 274–279. [Google Scholar]
- Miller, J.S.; Krusic, P.J.; Dixon, D.A.; Reiff, W.M.; Zhang, J.H.; Anderson, E.C.; Epstein, A.J. Radical Ion Salts of 2,3-Dichloro-5,6-dicyanobenzoquinone and Metallocenes. A Reexamination of their Magnetic and Spectroscopic Properties. J. Am. Chem. Soc. 1986, 108, 4459–4466. [Google Scholar]
- Politzer, P.; Murray, J.S.; Lane, P. Hole bonding and hydrogen bonding: Competitive interactions. Int. J. Quantum Chem. 2007, 107, 3046–3052. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Wakisaka, A.; Ono, T.; Sonoda, T. Magnitude and Origin of the Attraction and Directionality of the Halogen Bonds of the Complexes of C6F5X and C6H5X (X = I, Br, Cl and F) with Pyridine. Chem. Eur. J. 2012, 18, 951–960. [Google Scholar] [CrossRef]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: a new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELX97—Programs for Crystal Structure Analysis (Release 97-2); University of Göttingen: Göttingen, Germany, 1998. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Whangbo, M.-H.; Hoffmann, R. The band structure of the tetracyanoplatinate chain. J. Am. Chem. Soc. 1978, 100, 6093–6098. [Google Scholar]
- Ren, J.; Liang, W.; Whangbo, M.-H. Crystal and Electronic Structure Analysis Using CAESAR; PrimeColor Software, Inc.: Cary, NC, USA, 1998. [Google Scholar]
- Ammeter, J.; Bürgi, H.-B.; Thibeault, J.; Hoffmann, R. Counterintuitive orbital mixing in semiempirical and ab initio molecular orbital calculations. J. Am. Chem. Soc. 1978, 100, 3686–3692. [Google Scholar]
- Clementi, E.; Roetti, C. Roothaan-Hartree-Fock atomic wavefunctions: Basis functions and their coefficients for ground and certain excited states of neutral and ionized atoms, Z ≤ 54. At. Data Nucl. Data Tables 1974, 14, 177–478. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lieffrig, J.; Jeannin, O.; Shin, K.-S.; Auban-Senzier, P.; Fourmigué, M. Halogen Bonding Interactions in DDQ Charge Transfer Salts with Iodinated TTFs. Crystals 2012, 2, 327-337. https://doi.org/10.3390/cryst2020327
Lieffrig J, Jeannin O, Shin K-S, Auban-Senzier P, Fourmigué M. Halogen Bonding Interactions in DDQ Charge Transfer Salts with Iodinated TTFs. Crystals. 2012; 2(2):327-337. https://doi.org/10.3390/cryst2020327
Chicago/Turabian StyleLieffrig, Julien, Olivier Jeannin, Kyoung-Soon Shin, Pascale Auban-Senzier, and Marc Fourmigué. 2012. "Halogen Bonding Interactions in DDQ Charge Transfer Salts with Iodinated TTFs" Crystals 2, no. 2: 327-337. https://doi.org/10.3390/cryst2020327