3.2. Synthesis of Complexes
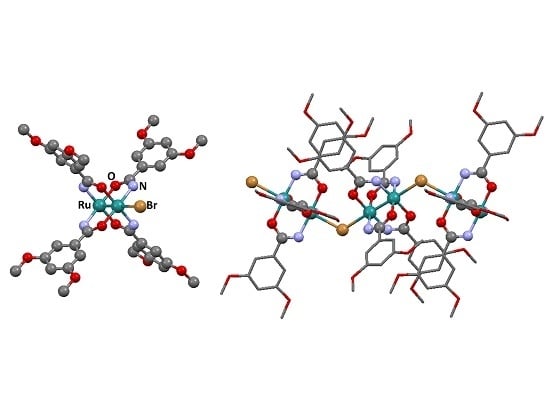
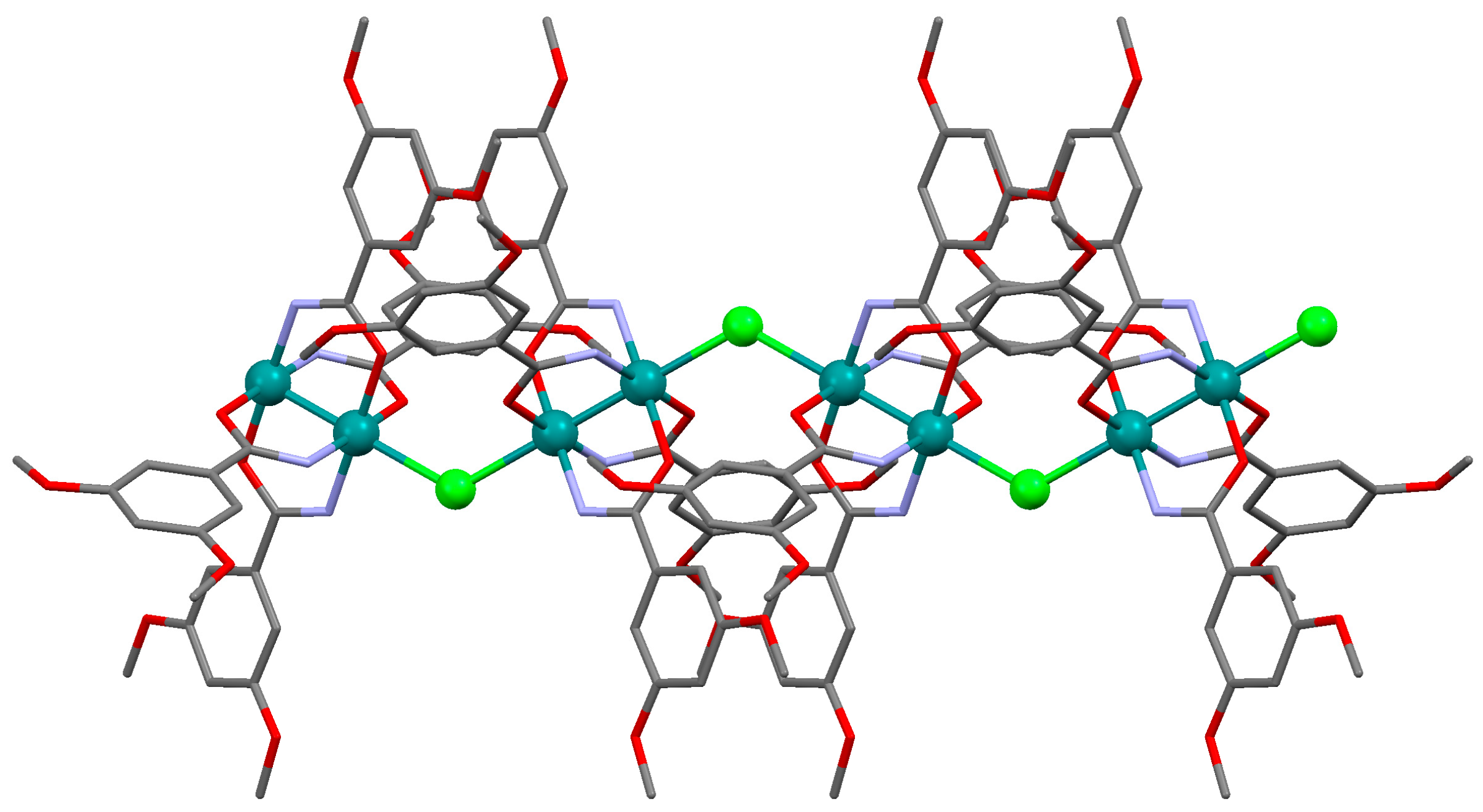
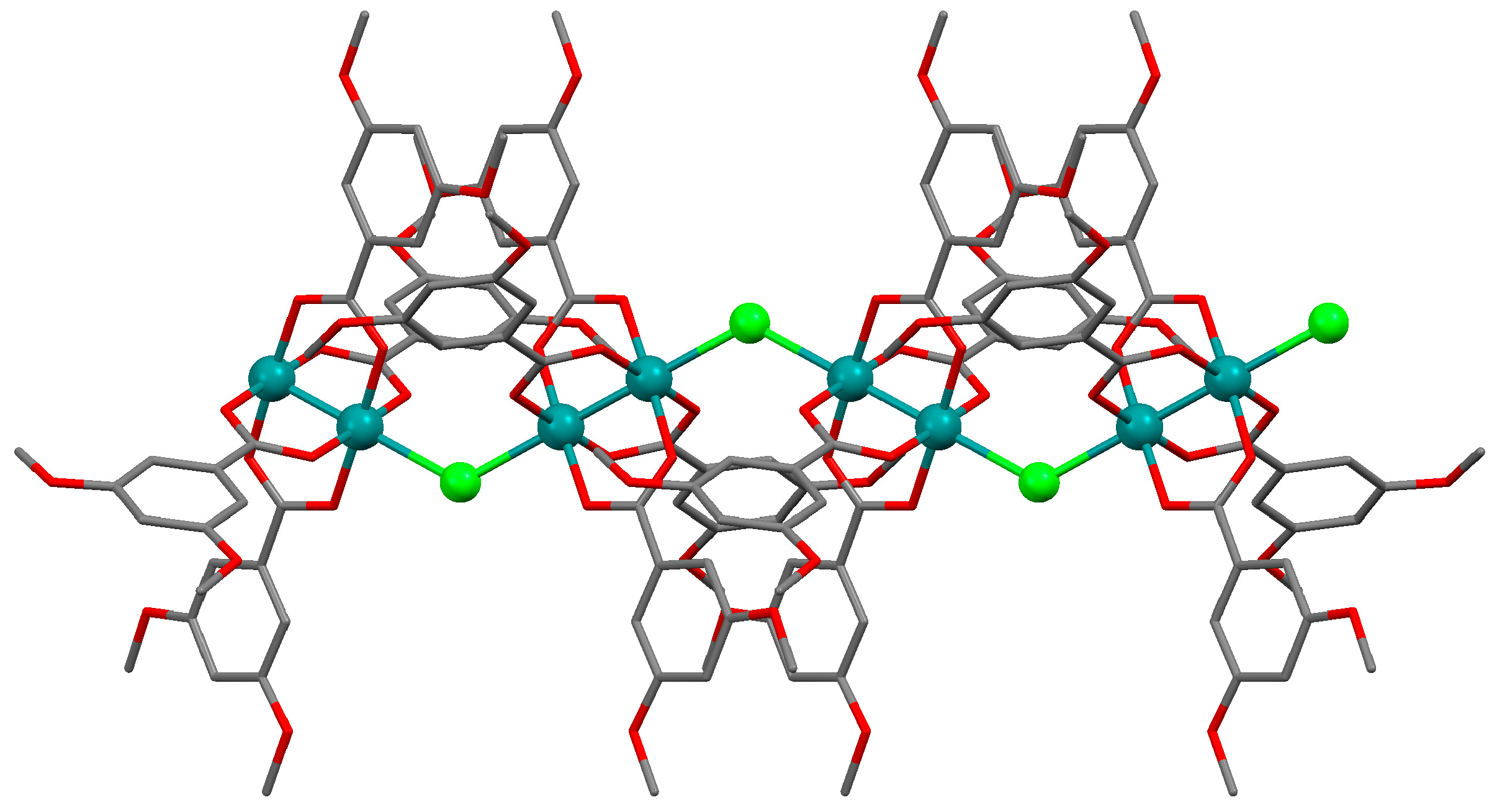
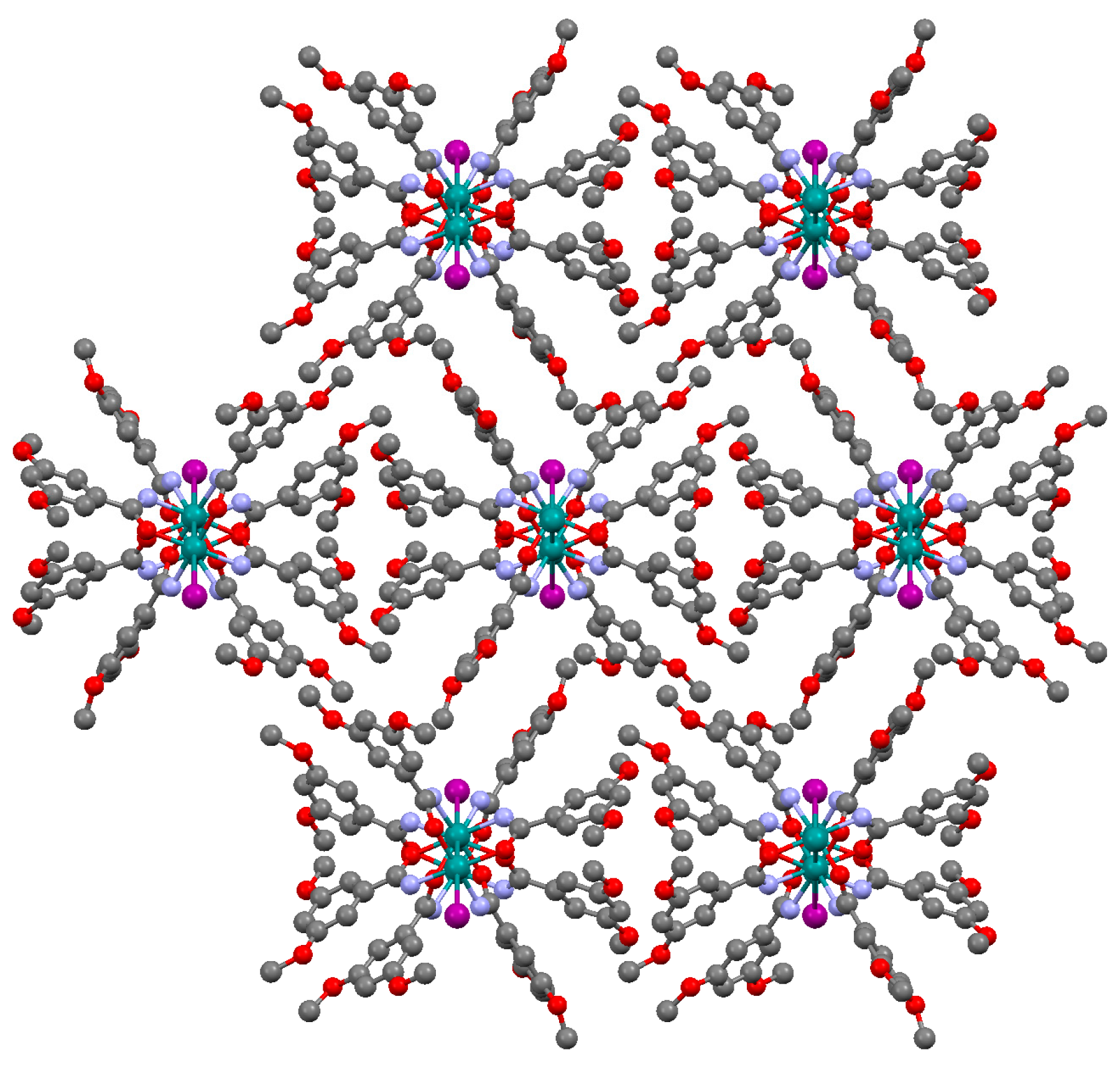
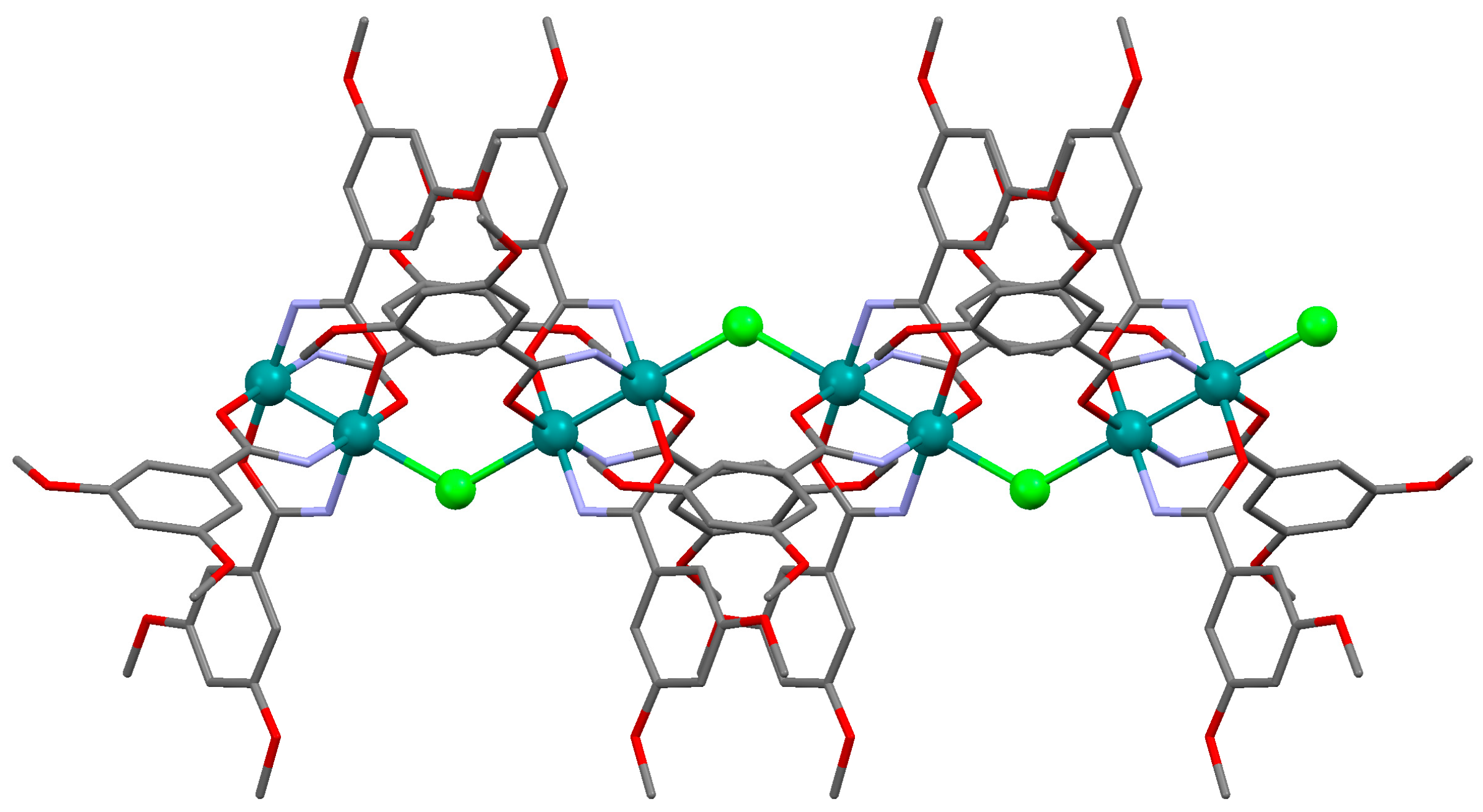
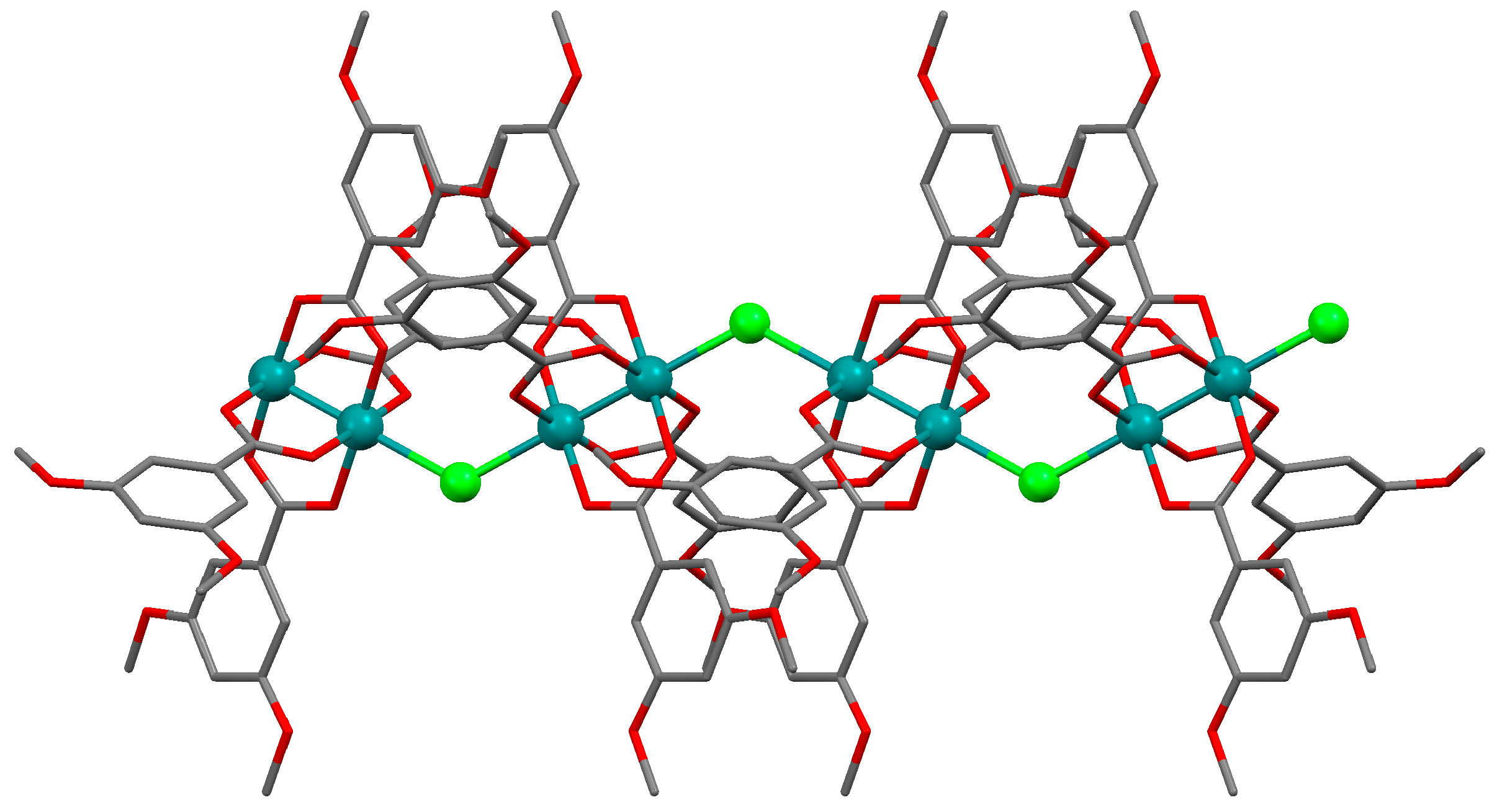
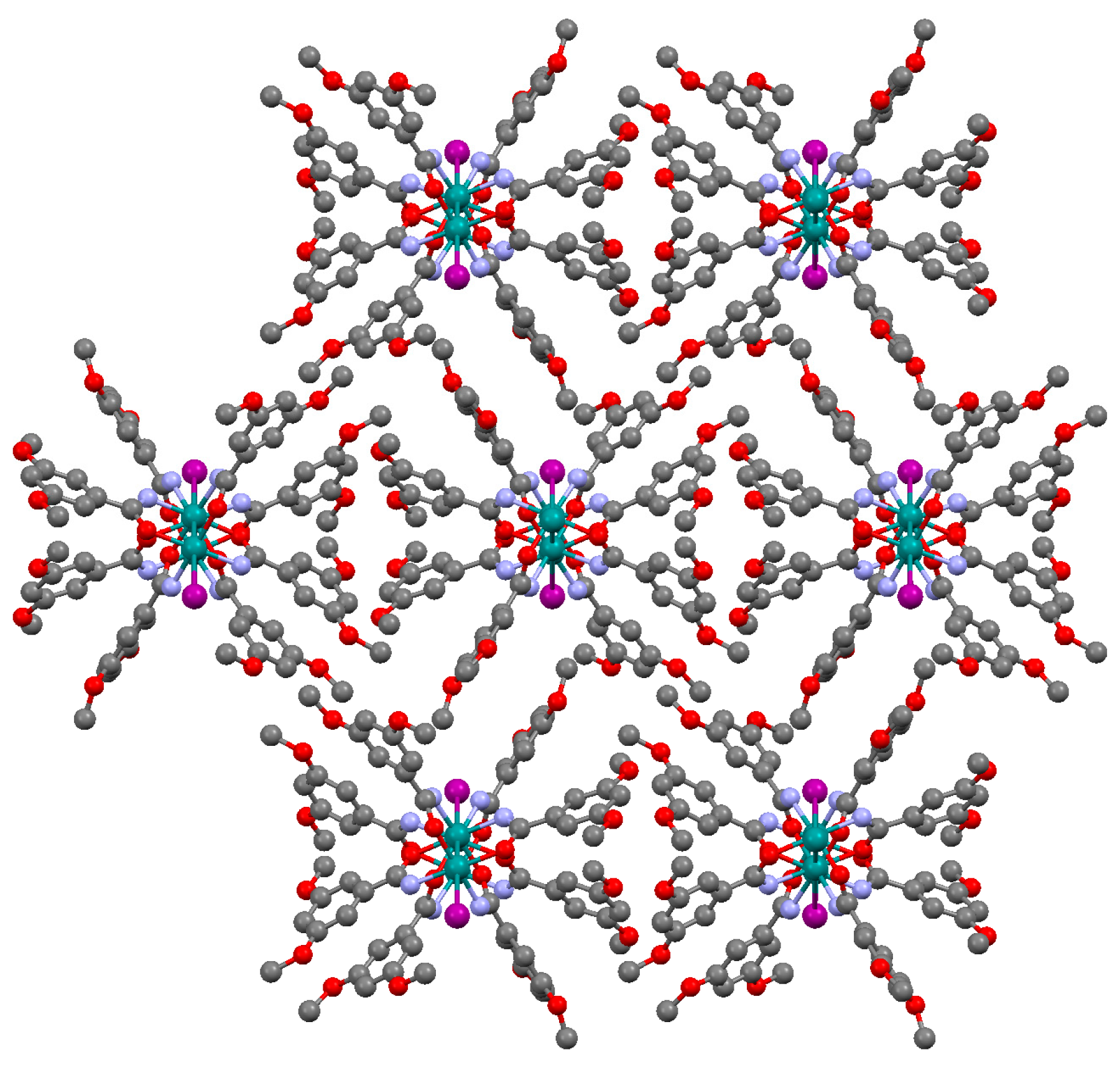
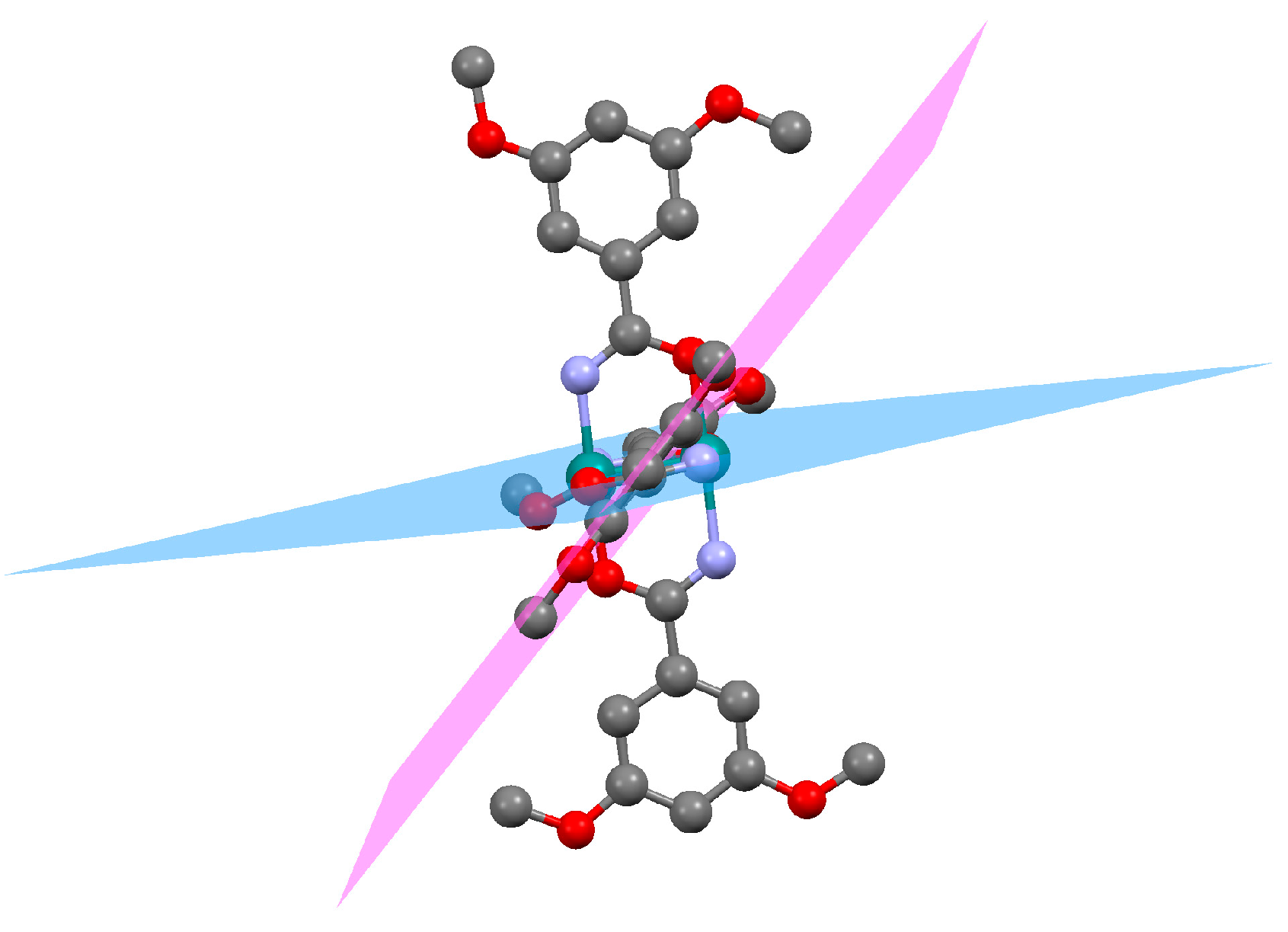
3.2.1. Synthesis of [Ru2Cl(µ-NHOCC6H3-3,5-(OMe)2)4]n (1)
Microwave assisted synthesis (method a). Chloridotetrakis(acetato)diruthenium(II,III) (0.12 g, 0.25 mmol), 3,5-dimethoxybenzamide (0.27 g, 1.50 mmol), lithium chloride (0.08 g, 2 mmol), triethylamine (0.25 mL) and absolute ethanol (8 mL) were added into a 85 mL TFM Teflon vessel with magnetic stirrer bar. The vessel was sealed with a lid equipped with temperature and pressure sensors and placed in the microwave oven. Reaction mixture was then treated by a three-step program consisting of (i) 15 min heating ramp up to 130 °C; (ii) 16 h isotherm at 130 °C; and (iii) 20 min cooling ramp up to room temperature. A brown suspension was obtained. The solid was obtained by filtration and washed twice with 10 mL of cold ethanol. Yield: 52%. Anal. Calcd. for Ru2ClC36H43N4O13.5 (1·1.5H2O): %C 43.88; %H 4.40; %N 5.69. Found: %C 43.57; %H 4.09; %N 5.61.
Solvothermal synthesis (method b). MeOH was used as solvent. Same reagents and quantities used in method (a) were added to a 23 mL Teflon-lined autoclave and stirred several minutes to become homogenised. The reactor was closed and heated under a three-step program consisting of (i) 2 h heating ramp up to 130 °C; (ii) 24 h isotherm at 130 °C; and (iii) 24 h cooling ramp up to room temperature. The microcrystalline brown solid obtained was filtered and washed with cold ethanol (2 × 10 mL). Yield: 62%. Anal. Calcd. for Ru2ClC36H46N4O15 (1·3H2O): %C 42.71; %H 4.58; %N 5.53. Found: %C 42.52; %H 4.14; %N 5.48.
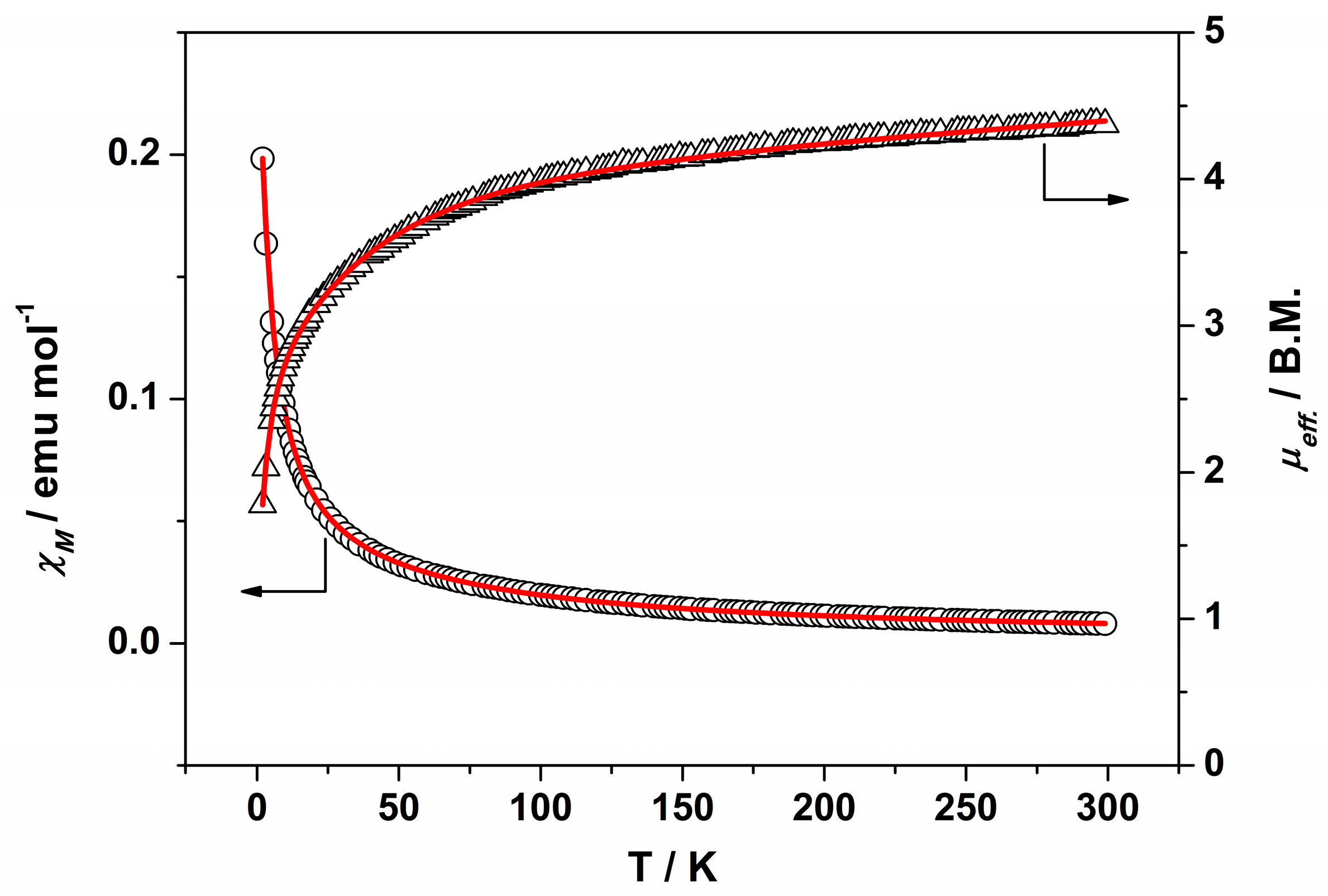
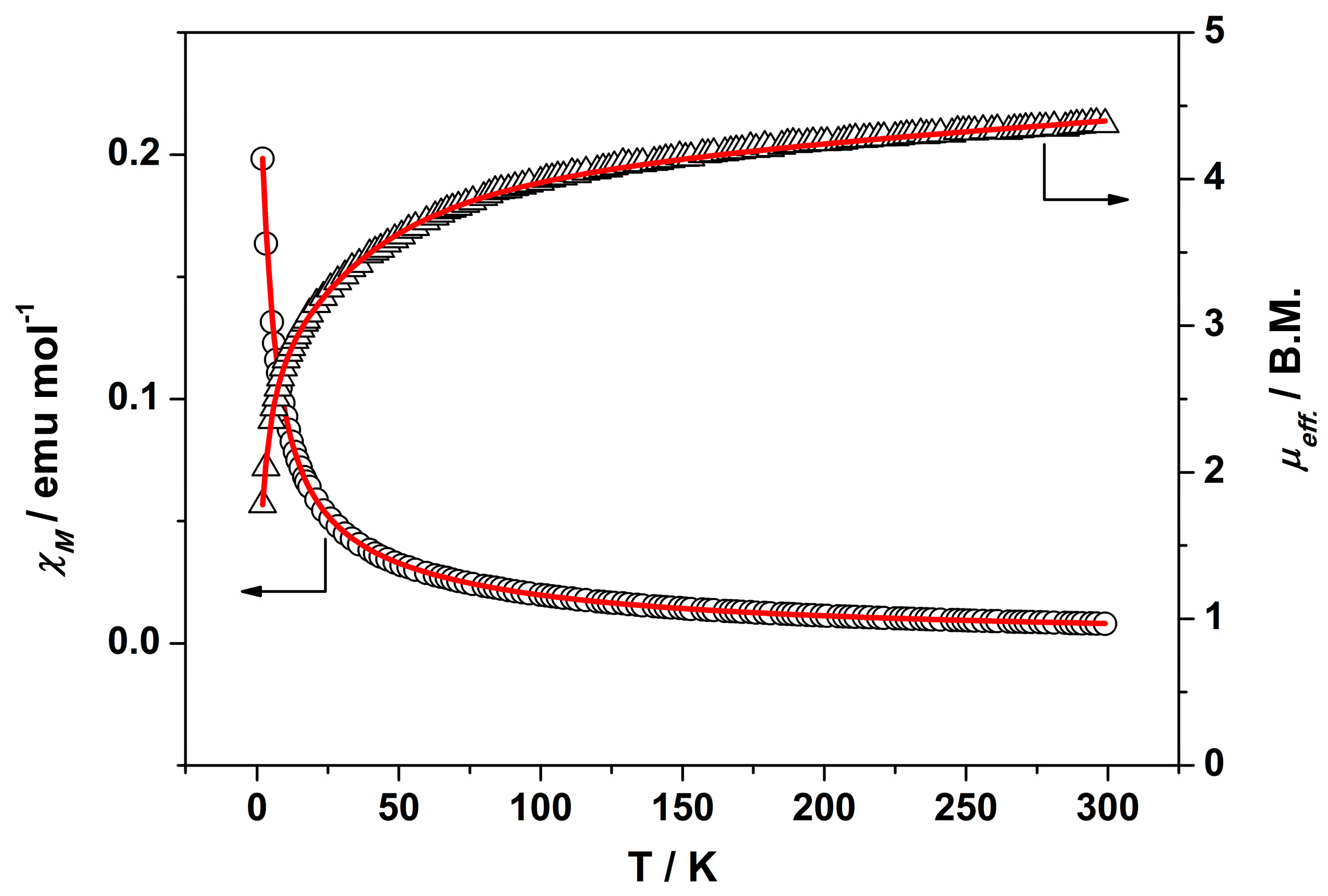
IR (cm−1): 3325w, 3017w, 2937w, 2833w, 1595m, 1509m, 1483w, 1452m, 1420m, 1343w, 1315m, 1300m, 1202m, 1180s, 1102w, 1046s, 942w, 921w, 851w, 919m, 756w, 744s, 686m. UV-Vis-NIR (diffuse reflection): [λ, nm] 338, 379, 473, 995. μeff (rt): 4.37 μB.
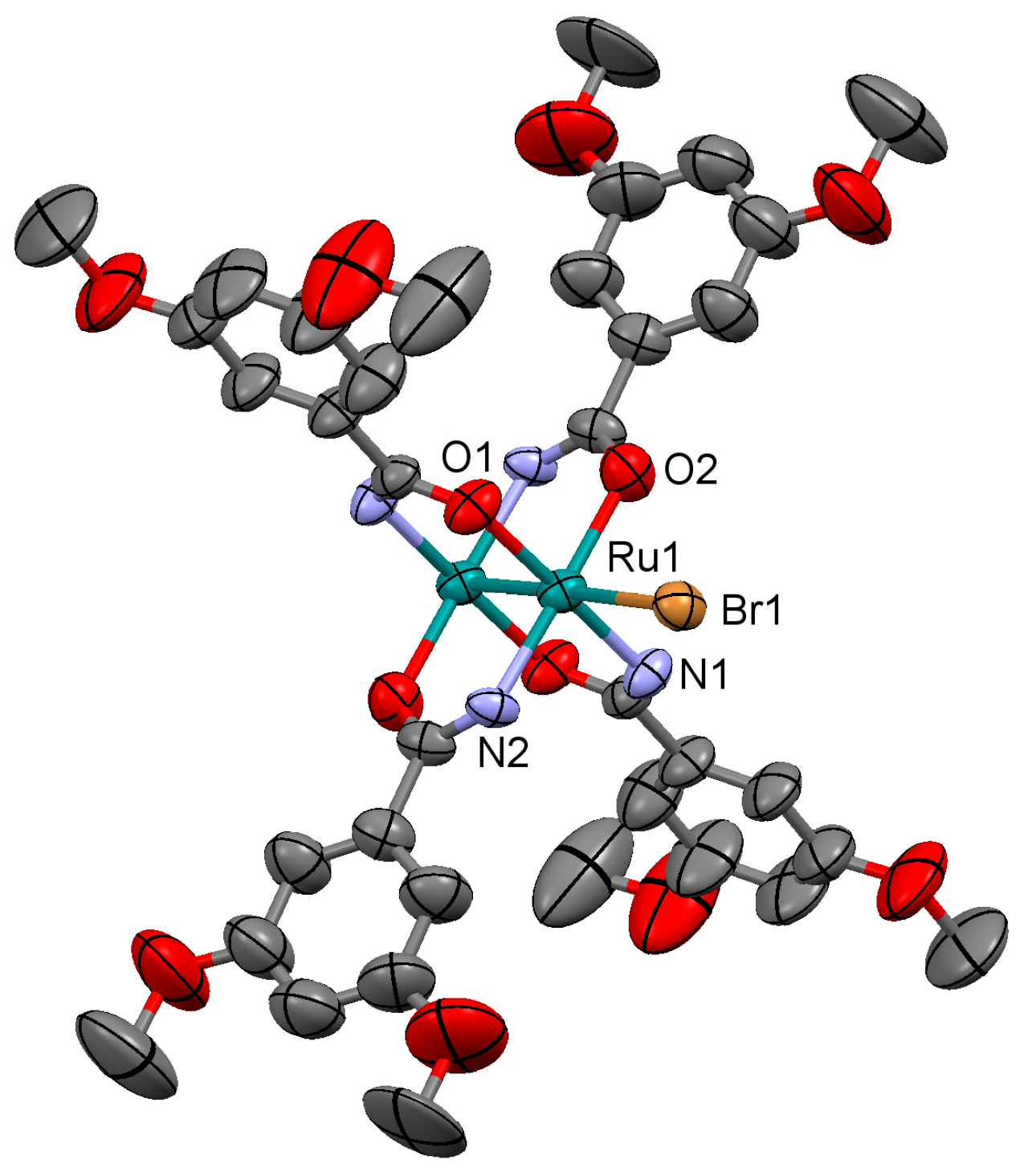
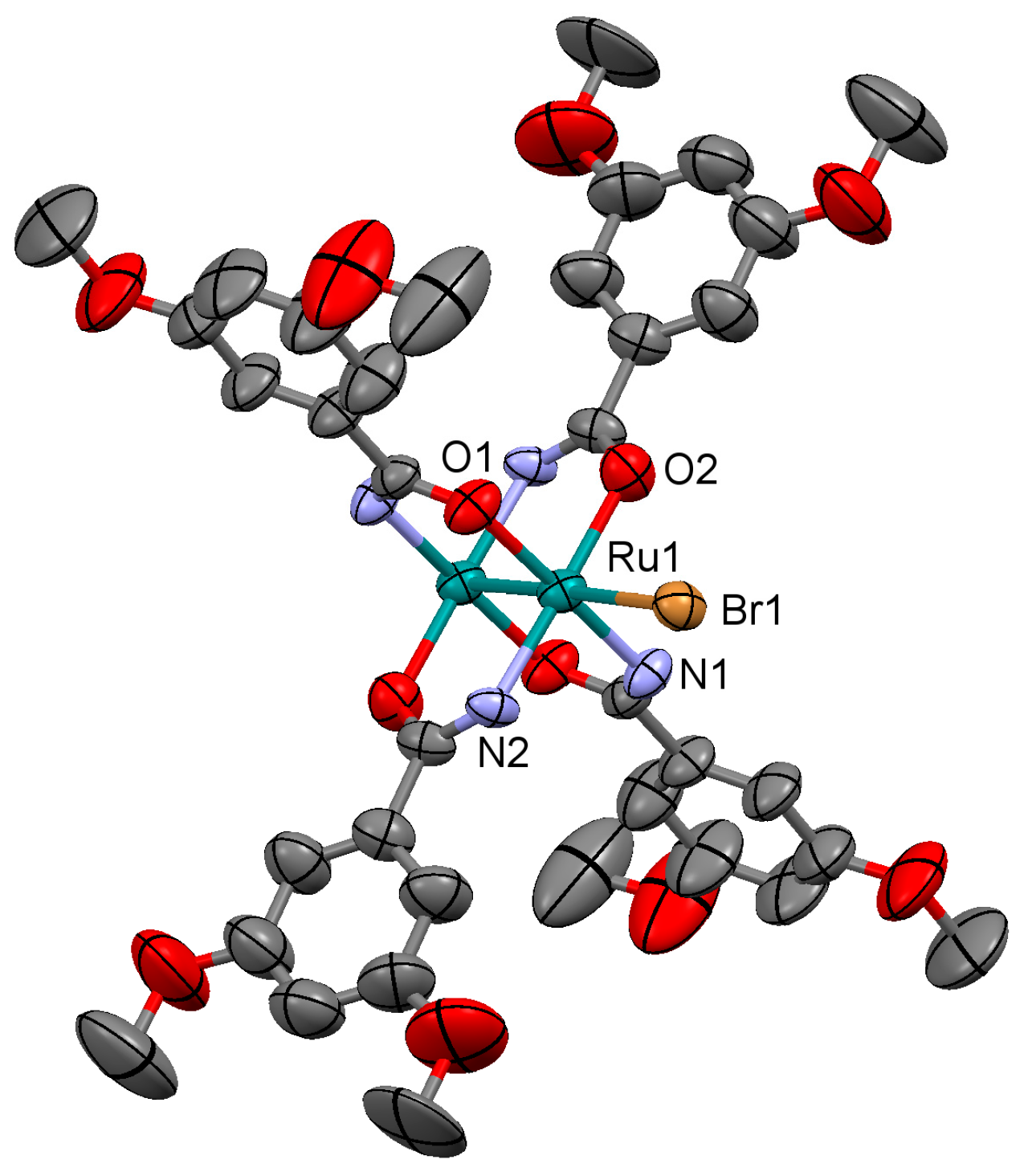
3.2.2. Synthesis of [Ru2Br(µ-NHOCC6H3-3,5-(OMe)2)4]n (2)
Microwave assisted synthesis (method a). Bromidotetrakis(acetato)diruthenium(II,III) (0.13 g, 0.25 mmol), 3,5-dimethoxybenzamide (0.27 g, 1.50 mmol), potassium bromide (0.24 g, 2 mmol), triethylamine (0.25 mL) and absolute ethanol (8 mL) were added into a 85 mL TFM Teflon vessel with magnetic stirrer bar. The vessel was sealed with a lid equipped with temperature and pressure sensors, and placed in the microwave oven. Reaction mixture was then treated by a three-step program consisting of (i) 15 min heating ramp up to 120 °C; (ii) 16 h isotherm at 120 °C; and (iii) 20 min cooling ramp up to room temperature. A brown suspension was obtained. Solid is obtained by filtration and washed twice with 10 mL of cold ethanol and with 10 mL of water. Yield: 2%.
Solvothermal synthesis (method b). Same reagents and quantities used in method (a) were added to a 23 mL Teflon-lined autoclave and stirred several minutes to become homogenised. The reactor was closed and heated under a three-step program consisting of (i) 2 h heating ramp up to 100 °C; (ii) 24 h isotherm at 100 °C; and (iii) 48 h cooling ramp up to room temperature. The microcrystalline brown solid obtained was filtered and washed with cold ethanol (2 × 10 mL) and water (2 × 10 mL). Yield: 40%.
Anal. Calcd. for Ru2BrC36H44N4O14 (2·2H2O): %C 41.62; %H 4.27; %N 5.39. Found: %C 40.37; %H 3.59; %N 5.61. IR (cm−1): 3321w, 3003w, 2930w, 2832w, 1596s, 1509s, 1483m, 1451s, 1420s, 1342m, 1316s, 1300s, 1285w, 1202s, 1172m, 1152s, 1101m, 1052s, 1047s, 944m, 922w, 862w, 851m, 819w, 755m, 744s, 686m. UV-Vis-NIR (diffuse reflection): [λ, nm] 343, 364, 467, 999. μeff (rt): 4.21 μB.
3.2.3. Synthesis of [Ru2I(µ-NHOCC6H3-3,5-(OMe)2)4]n (3)
Microwave assisted synthesis (method a). Iodotetrakis(acetato)diruthenium(II,III) (0.14 g, 0.25 mmol), 3,5-dimethoxybenzamide (0.27 g, 1.50 mmol), potassium iodide (0.34 g, 2 mmol), triethylamine (0.25 mL) and absolute ethanol (8 mL) were added into a 85 mL TFM Teflon vessel with magnetic stirrer bar. The vessel was sealed with a lid equipped with temperature and pressure sensors, and placed in the microwave oven. Reaction mixture was then treated by a three-step program consisting of (i) 15 min heating ramp up to 100 °C; (ii) 16 h isotherm at 100 °C; and (iii) 20 min cooling ramp up to room temperature. A brown suspension was obtained. Solid is obtained by filtration and washed twice with 10 mL of cold ethanol and with 10 mL of water. Yield: 48%. Anal. Calcd. for Ru2IC36H44N4O14 (3·2H2O): %C 39.82; %H 4.08; %N 5.16. Found: %C 39.60; %H 3.81; %N 5.05.
Solvothermal synthesis (method b). Same reagents and quantities used in method (a) were added to a 23 mL Teflon-lined autoclave and stirred several minutes to become homogenised. The reactor was closed and heated under a three-step program consisting of (i) 2 h heating ramp up to 100 °C; (ii) 24 h isotherm at 100 °C; and (iii) 48 h cooling ramp up to room temperature. The microcrystalline brown solid obtained was filtered and washed with cold ethanol (2 × 10 mL) and water (2 × 10 mL). Yield: 35%. Anal. Calcd. for Ru2IC36H44N4O14 (3·2H2O): %C 39.82; %H 4.08; %N 5.16. Found: %C 39.65; %H 3.94; %N 5.20.
IR (cm−1): 3299w, 2997w, 2935w, 2835w, 1707w, 1595s, 1508s, 1482m, 1453s, 1417s, 1343m, 1315m 1297m, 1257w, 1202s, 1155s, 1112m, 1062s, 1049s, 992w, 940w, 923w, 841m, 756s, 743s, 689w. UV-Vis-NIR (diffuse reflection): [λ, nm] 324, 1002. μeff (rt): 4.04 μB.
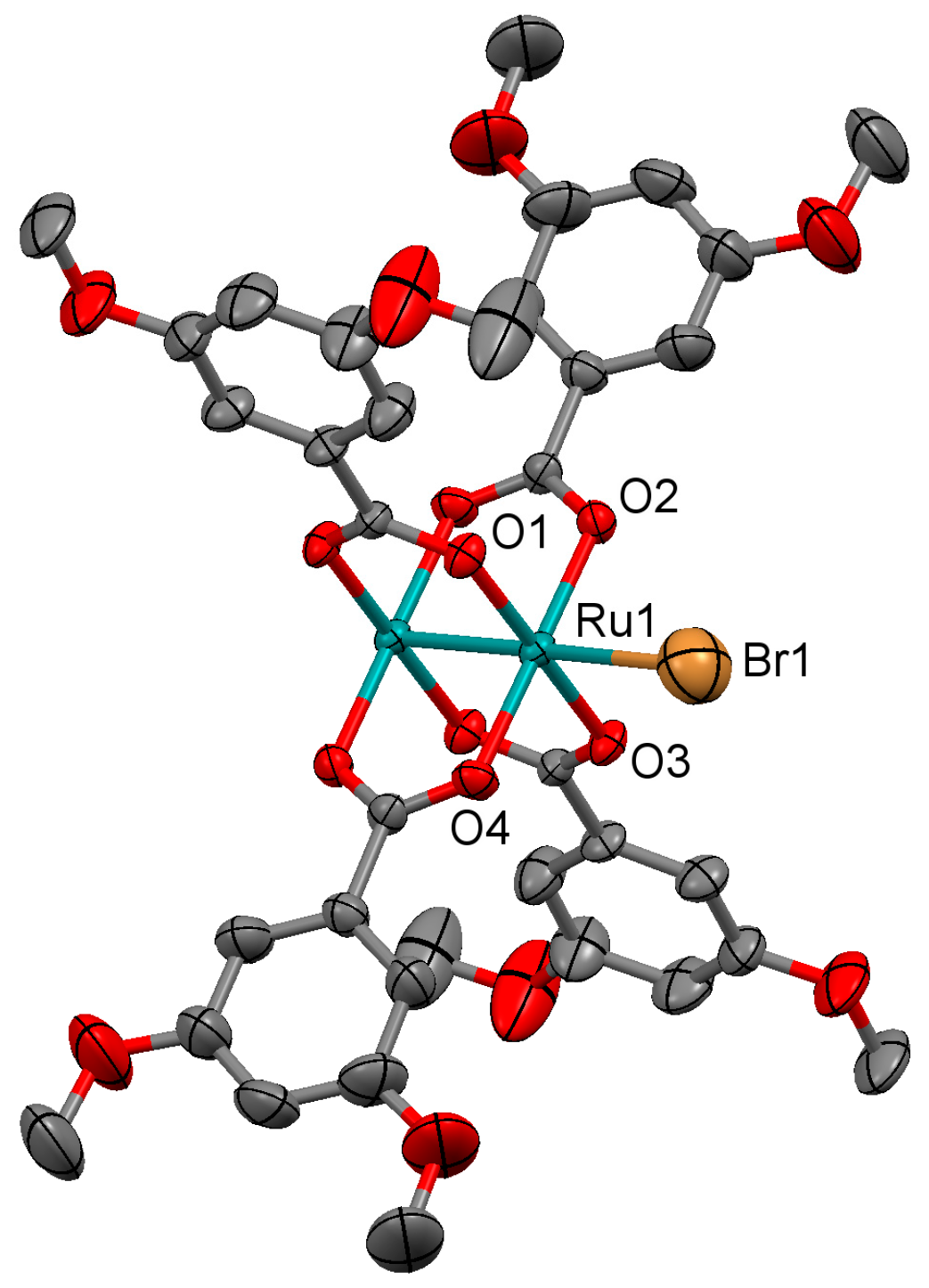
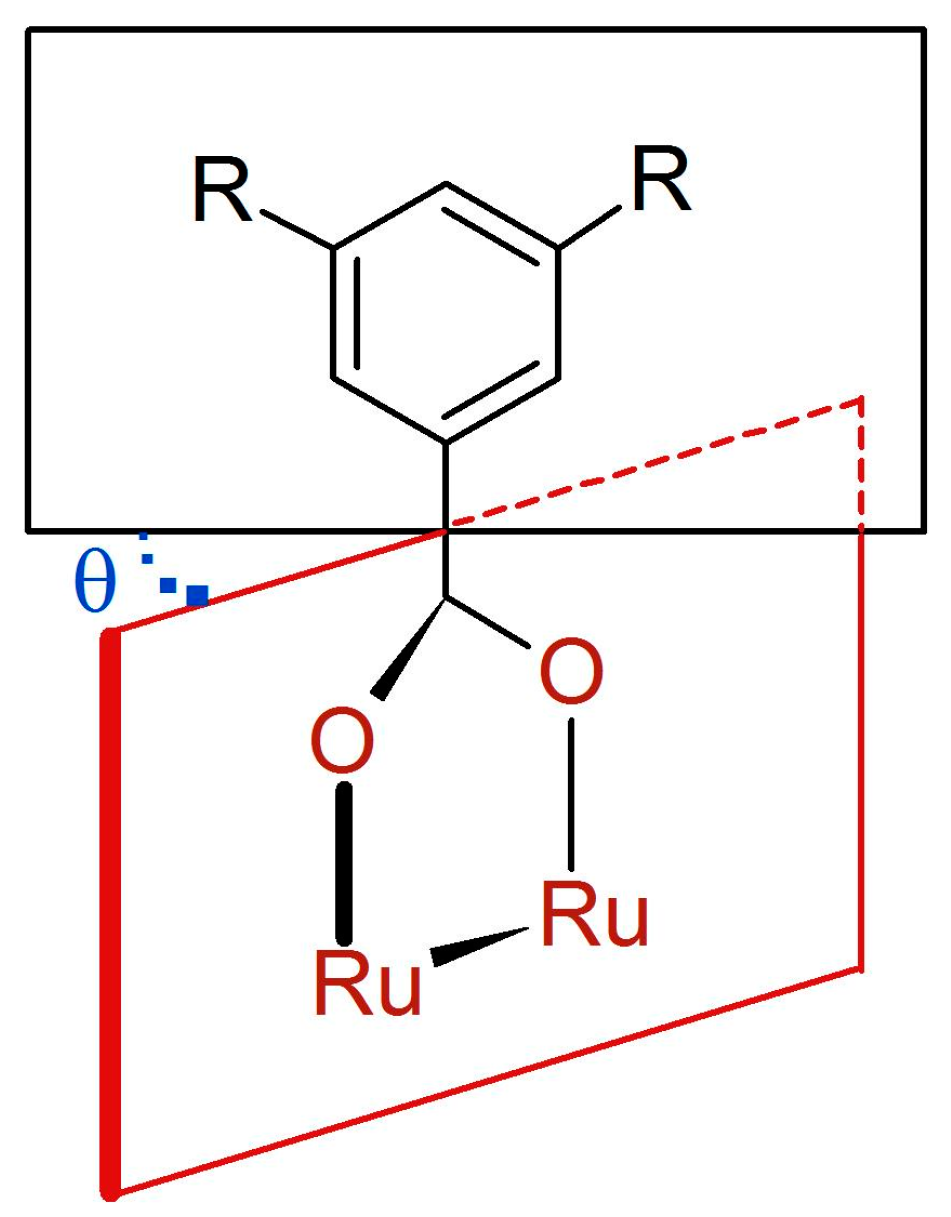
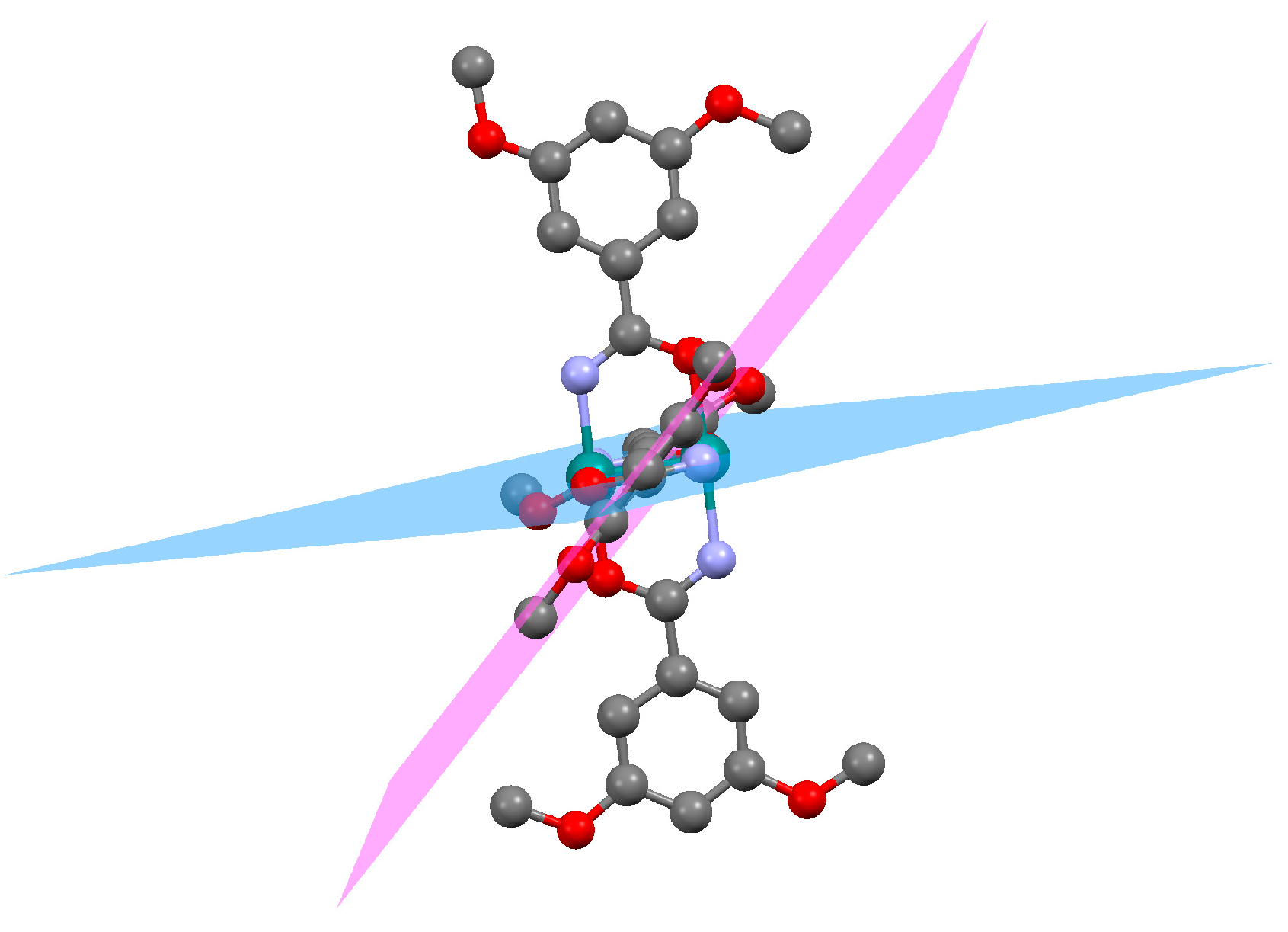
3.2.4. Synthesis of [Ru2Cl(µ-O2CC6H3-3,5-(OMe)2)4]n (4)
Microwave assisted synthesis (method a). Chloridotetrakis(acetato)diruthenium(II,III) (0.12 g, 0.25 mmol), 3,5-dimethoxybenzoic acid (0.27 g, 1.50 mmol) and methanol (8 mL) were added into a 85 mL TFM Teflon vessel with magnetic stirrer bar. The vessel was sealed with a lid equipped with temperature and pressure sensors, and placed in the microwave oven. Reaction mixture was then treated by a three-step program consisting of (i) 15 min heating ramp up to 130 °C; (ii) 16 h isotherm at 130 °C; and (iii) 20 min cooling ramp up to room temperature. A brown suspension was obtained. Solid is obtained by filtration and washed twice with 10 mL of cold methanol. Yield: 83%. Anal. Calcd. for Ru2ClC36H40O18 (4·2H2O): %C 43.31; %H 4.04. Found: %C 43.34; %H 3.69.
Solvothermal synthesis (method b). Same reagents and quantities used in method (a) were added to a 23 mL Teflon-lined autoclave and stirred several minutes to become homogenised. The reactor was closed and heated under a three-step program consisting of (i) 2 h heating ramp up to 85 °C; (ii) 24 h isotherm at 85 °C; and (iii) 72 h cooling ramp up to room temperature. The microcrystalline brown solid obtained was filtered and washed with cold methanol (2 × 10 mL). Yield: 90%. Anal. Calcd. for Ru2ClC36H42O19 (4·3H2O): %C 42.55; %H 4.17. Found: %C 42.33; %H 3.64.
Conventional synthesis (method c). 0.27 g of 3,5-dimethoxybenzoic acid (1.50 mmol) were added to a suspension of chloridotetrakis(acetato)diruthenium(II,III) (0.12 g, 0.25 mmol) in 24 mL of MeOH/H2O (1:1). The reaction mixture was refluxed for 4 h, yielding a brown precipitate. The solvent was eliminated by filtration and the brown solid was washed twice with 10 mL of cold methanol. Yield: 82%. Anal. Calcd. for Ru2ClC36H40O18 (4·2H2O): %C 43.31; %H 4.04. Found: %C 43.07; %H 3.70.
IR (cm−1): 3094w, 3003w, 2957w, 2937w, 2840w, 1593m, 1520w, 1481m, 1448m, 1433m, 1381s, 1312w, 1296w, 1251w, 1201m, 1157m, 1107w, 1058m, 1045m, 991w, 943m, 926w, 873w, 853w, 827w, 773m, 759s, 696w, 675w. UV-Vis-NIR (diffuse reflection): [λ, nm] 321, 482, 1153. μeff (rt): 4.04 μB.
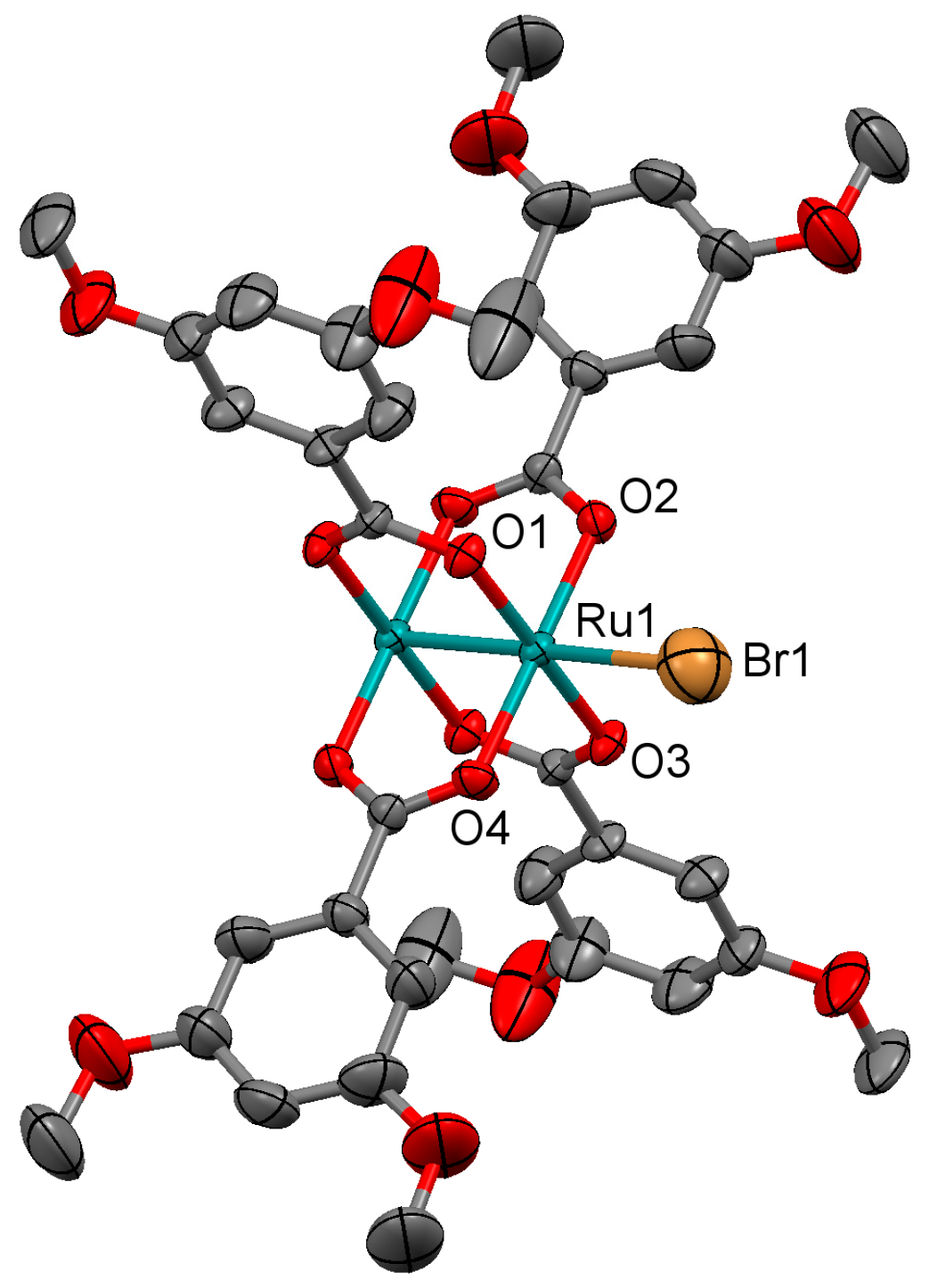
3.2.5. Synthesis of [Ru2Br(µ-O2CC6H3-3,5-(OMe)2)4]n (5)
Microwave assisted synthesis (method a). Bromidotetrakis(acetato)diruthenium(II,III) (0.13 g, 0.25 mmol), 3,5-dimethoxybenzoic acid (0.27 g, 1.50 mmol) and methanol (8 mL) were added into a 85 mL TFM Teflon vessel with magnetic stirrer bar. The vessel was sealed with a lid equipped with temperature and pressure sensors, and placed in the microwave oven. Reaction mixture was then treated by a three-step program consisting of (i) 15 min heating ramp up to 130 °C; (ii) 16 h isotherm at 130 °C; and (iii) 20 min cooling ramp up to room temperature. A brown suspension was obtained. Solid is obtained by filtration and washed twice with 10 mL of cold methanol. Yield: 84%. Anal. Calcd. for Ru2BrC36H38O17 (5·H2O): %C 42.20; %H 3.74. Found: %C 42.19; %H 3.54.
Solvothermal synthesis (method b). Same reagents and quantities used in method (a) were added to a 23 mL Teflon-lined autoclave and stirred several minutes to become homogenised. The reactor was closed and heated under a three-step program consisting of (i) 2 h heating ramp up to 85 °C; (ii) 24 h isotherm at 85 °C; and (iii) 72 h cooling ramp up to room temperature. The microcrystalline brown solid obtained was filtered and washed with cold methanol (2 × 10 mL). Yield: 76%. Anal. Calcd. for Ru2BrC36H36O16 (5): %C 42.95; %H 3.60. Found: %C 43.30; %H 3.69.
Conventional synthesis (method c). 0.27 g of 3,5-dimethoxybenzoic acid (1.50 mmol) were added to a suspension of bromidotetrakis(acetato)diruthenium(II,III) (0.13 g, 0.25 mmol) in 30 mL of MeOH/H2O (1:1). The reaction mixture was refluxed for 4 h, yielding a brown precipitate. The solvent was eliminated by filtration and the brown solid was washed twice with 10 mL of cold methanol. Yield: 80%. Anal. Calcd. for Ru2BrC36H39O17.5 (5·1.5H2O): %C 41.83; %H 3.80. Found: %C 41.49; %H 3.49.
IR (cm−1): 3003w, 2957w, 2935w, 2840w, 1593m, 1481w, 1449m, 1434w, 1383s, 1312w, 1296w, 1249w, 1202m, 1158m, 1107m, 1059w, 1046w, 991w, 943m, 926w, 873w, 853m, 828w, 773w, 760m, 697w, 675w. UV-Vis-NIR (diffuse reflection): [λ, nm] 322, 494, 1137. μeff (rt): 4.48 μB.
3.2.6. Synthesis of [Ru2I(µ-O2CC6H3-3,5-(OMe)2)4]n (6)
Microwave assisted synthesis (method a). Iodotetrakis(acetato)diruthenium(II,III) (0.14 g, 0.25 mmol), 3,5-dimethoxybenzoic acid (0.27 g, 1.50 mmol) and methanol (8 mL) were added into a 85 mL TFM Teflon vessel with magnetic stirrer bar. The vessel was sealed with a lid equipped with temperature and pressure sensors, and placed in the microwave oven. Reaction mixture was then treated by a three-step program consisting of (i) 15 min heating ramp up to 130 °C; (ii) 16 h isotherm at 130 °C; and (iii) 20 min cooling ramp up to room temperature. A brown suspension was obtained. Solid is obtained by filtration and washed twice with 10 mL of cold ethanol. Yield: 71%. Anal. Calcd. for Ru2IC36H42O19 (6·3H2O): %C 39.03; %H 3.82. Found: %C 38.74; %H 3.64.
Solvothermal synthesis (method b). Same reagents and quantities used in method (a) were added to a 23 mL Teflon-lined autoclave and stirred several minutes to become homogenised. The reactor was closed and heated under a three-step program consisting of (i) 2 h heating ramp up to 85 °C; (ii) 24 h isotherm at 85 °C; and (iii) 72 h cooling ramp up to room temperature. The microcrystalline brown solid obtained was filtered and washed with cold ethanol (2 × 10 mL). Yield: 59%. Anal. Calcd. for Ru2IC36H36O16 (6): %C 41.04; %H 3.44. Found: %C 40.93; %H 3.59.
Conventional synthesis (method c). 0.27 g of 3,5-dimethoxybenzoic acid (1.50 mmol) were added to a suspension of iodotetrakis(acetato)diruthenium(II,III) (0.14 g, 0.25 mmol) in 30 mL of MeOH/H2O (1:1). The reaction mixture was refluxed for 4 h, yielding a brown precipitate. The solvent was eliminated by filtration and the brown solid was washed twice with 10 mL of cold ethanol. Yield: 43%. Anal. Calcd. for Ru2IC36H38O17 (6·H2O): %C 40.35; %H 3.57. Found: %C 39.01; %H 3.58.
IR (cm−1): 3003w, 2938w, 2839w, 1596m, 1519w, 1481w, 1449m, 1380s, 1310w, 1290w, 1252w, 1204m, 1155s, 1110w, 1062m, 1048m, 1017m, 1002m, 945w, 925w, 873w, 845w, 827w, 792w, 760m, 698m, 673w. UV-Vis-NIR (diffuse reflection): [λ, nm] 346, 500, 1100. μeff (rt): 4.68 μB.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}