
The Interplay between Various σ- and π-Hole Interactions of Trigonal Boron and Trigonal Pyramidal Arsenic Triiodides


Abstract
:1. Introduction
2. Methods
2.1. Experimental
2.2. Computations
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Cavall, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Primagi, A.; Ransani, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bundhun, A.; Ramasami, P.; Murray, J.S.; Politzer, P. Trends in σ-hole strengths and interactions of F3MX molecules (M = C, Si, Ge and X = F, Cl, Br, I). J. Mol. Model. 2013, 19, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Kolář, M.; Hostaš, J.; Hobza, P. The strength and directionality of a halogen bond are co-determined by the magnitude and size of the σ-hole. Phys. Chem. Chem. Phys. 2014, 16, 23279–23280. [Google Scholar]
- Riley, K.E.; Murray, J.S.; Fanfrlik, J.; Rezac, J.; Sola, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen Bond Tunability I: The Effects of Aromatic Fluorine Substitution on the Strengths of Halogen-Bonding Interactions Involving Chlorine, Bromine, and Iodine. J. Mol. Model. 2011, 17, 3309–3318. [Google Scholar] [CrossRef] [PubMed]
- Herdegger, L.A.; Kuhn, B.; Spinnler, B.; Anselm, L.; Ecabert, R.; Stihle, M.; Gsell, B.; Thoma, R.; Diez, J.; Benz, J.; et al. Systematic Investigation of Halogen Bonding in Protein−Ligand Interactions. Angew. Chem. Int. Ed. 2011, 50, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Kolář, M.; Kamlar, M.; Hurny, D.; Ruiz, F.X.; Cousido-Siah, A.; Mitschler, A.; Řezáč, J.; Munusamy, E.; Lepšík, M.; et al. The Modulation of Aldose Reductase Inhibition by Halogen Bond Tuning. ACS Chem. Biol. 2013, 8, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Ring, M.A.; Donnay, J.D.H.; Koski, W.S. The Crystal Structure of Boron Triiodide. Inorg. Chem. 1962, 1, 109–111. [Google Scholar] [CrossRef]
- Trotter, J. The crystal structure of arsenic triiodide, AsI3. Z. Kristallogr. Cryst. Mater. 1965, 121, 81–86. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bergner, M.; Dolg, M.; Küchle, W.; Stoll, H.; Preuss, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Flűkiger, P.; Lűthi, H.P.; Portmann, S.; Weber, J. MOLEKEL 4.3; Swiss Center for Scientific Computing: Manno, Switzerland, 2000. [Google Scholar]
- Portmann, S.; Luthi, H.P. MOLEKEL: An Interactive Molecular Graphic Tool. CHIMIA Int. J. Chem. 2000, 54, 766–770. [Google Scholar]
- Riley, K.E.; Tran, K.-A.; Lane, P.; Murray, J.S.; Politzer, P. Comparative analysis of electrostatic potential maxima and minima on molecular surfaces, as determined by three methods and a variety of basis sets. J. Comput. Sci. 2016, 17, 273–284. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Řezáč, J. Cuby: An integrative framework for computational chemistry. J. Comput. Chem. 2016, 37, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Koch, H.; Olsen, J.; Wilson, A.K. Basis-Set Convergence in Correlated Calculations on Ne, N2, and H2O. Chem. Phys. Lett. 1998, 286, 243–252. [Google Scholar] [CrossRef]
- Foord, A.; Beagley, B.; Reader, W.; Steer, I.A. A gas-phase electron-diffraction study of trvinylborane. J. Mol. Struct. 1975, 24, 131–137. [Google Scholar] [CrossRef]
- Cornu, D.; Miele, P.; Faure, R.; Bonnetot, B.; Mongeot, H.; Bouix, J. Conversion of B (NHCH3)3 into boron nitride and polyborazine fibres and tubular BN structures derived therefrom. J. Mater. Chem. 1999, 9, 757–761. [Google Scholar] [CrossRef]
- Lo, R.; Svec, P.; Ruzickova, Z.; Ruzicka, A.; Hobza, P. On the nature of the stabilisation of the E···π pnicogen bond in the SbCl3···toluene complex. Chem. Commun. 2016, 52, 3500–3503. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H.; Schier, A. π-Complexation of Post-Transition Metals by Neutral Aromatic Hydrocarbons: The Road from Observations in the 19th Century to New Aspects of Supramolecular Chemistry. Organometallics 2008, 27, 2361–2395. [Google Scholar] [CrossRef]
- Fanfrlík, J.; Sedlak, R.; Pecina, A.; Rulíšek, L.; Dostál, L.; Moncóľ, J.; Růžička, A.; Hobza, P. The non-planarity of the benzene molecule in the X-ray structure of the chelated bismuth(III) heteroboroxine complex is not supported by quantum mechanical calculations. J. Chem. Soc. Dalton Trans. 2016, 45, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Grund, S.C.; Hanusch, K.; Wolf, H.U. Arsenic and Arsenic Compounds, Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Scholes, R.S. Arsenic in Glass. Ind. Eng. Chem. 1912, 4, 16–17. [Google Scholar] [CrossRef]
- Delgado-Friedrichs, O.; Foster, M.D.; O’Keeffe, M.; Proserpio, D.M.; Treacy, M.M.J.; Yaghi, O.M. What do we know about three-periodic nets? J. Solid State Chem. 2005, 178, 2533–2554. [Google Scholar] [CrossRef]
- Haiduc, I. Inverse coordination—An emerging new chemical concept. Oxygen and other chalcogens as coordination centers. Coord. Chem. Rev. 2017, 338, 1–26. [Google Scholar] [CrossRef]
- Honda, D.; Ikegami, S.; Inoue, T.; Ozeki, T.; Yagasaki, A. Protonation and methylation of an Anderson-type polyoxoanion [IMo6O24]5−. Inorg. Chem. 2007, 46, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Long, D.L.; Song, Y.F.; Wilson, E.F.; Kögerler, P.; Guo, S.X.; Bond, A.M.; Hargreaves, J.S.; Cronin, L. Capture of Periodate in a {W18O54} Cluster Cage Yielding a Catalytically Active Polyoxometalate [H3W18O56(IO6)]6− Embedded with High-Valent Iodine. Angew. Chem. Int. Ed. 2008, 47, 4384–4387. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Chen, W.L.; Wang, E.B.; Li, Y.G.; Feng, X.J.; Liu, L. Three new polyoxometalate-based hybrids prepared from choline chloride/urea deep eutectic mixture at room temperature. Inorg. Chem. Commun. 2010, 13, 972–975. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, C.; Comotti, A.; Ward, M.D. Supramolecular Archimedean cages assembled with 72 hydrogen bonds. Science 2011, 333, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Krautscheid, H.; Lode, C.; Vielsack, F.; Vollmer, H. Synthesis and crystal structures of iodoplumbate chains, ribbons and rods with new structural types. J. Chem. Soc. Dalton Trans. 2001, 7, 1099–1104. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (dft-d) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
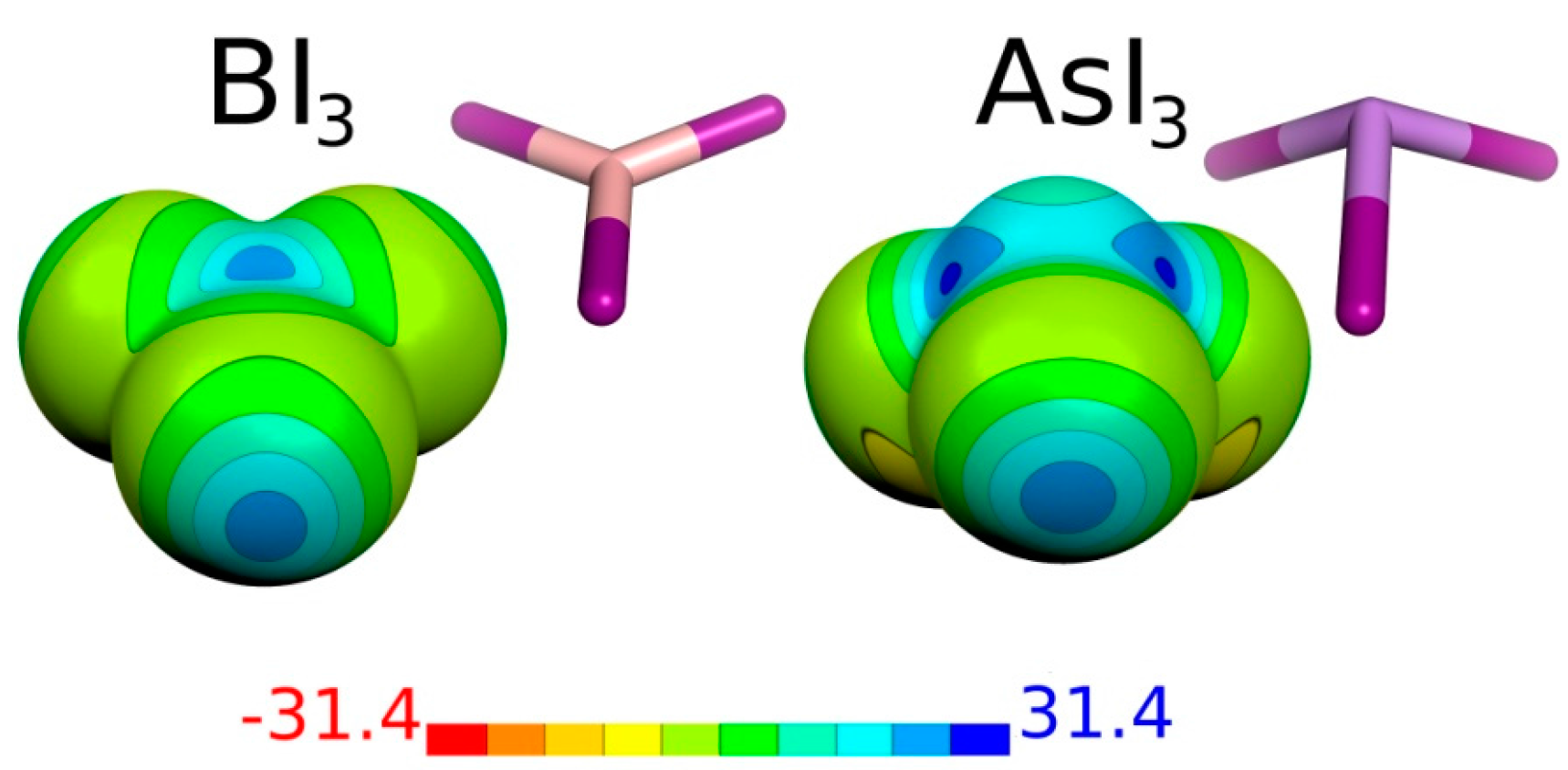
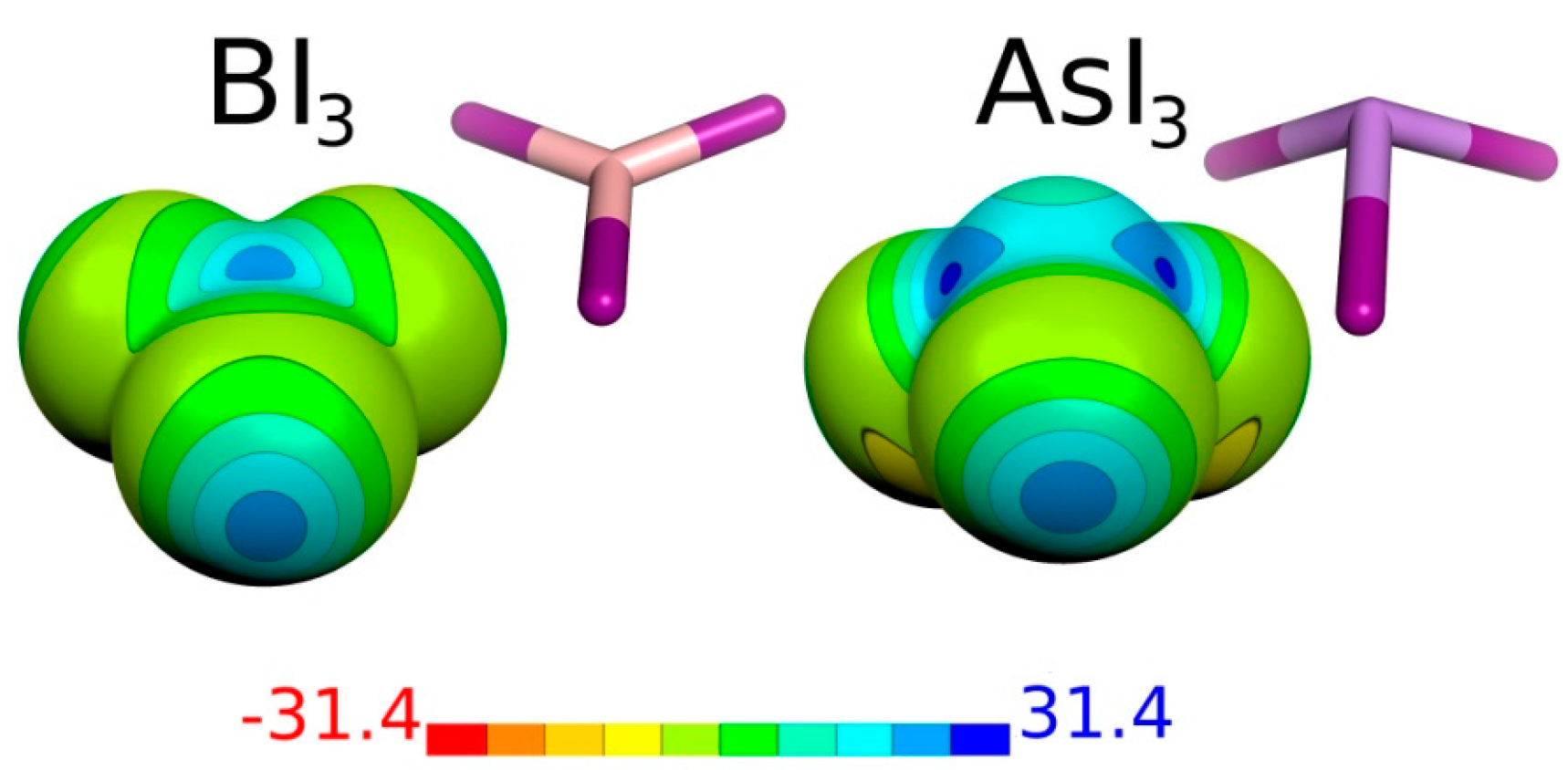
| Molecule | Atom | Vmax | μ |
|---|---|---|---|
| BI3 | B | 2 × 23.5 | 0.00 |
| I | 22.3 | ||
| AsI3 | As | 3 × 25.7 | 0.74 |
| I | 23.2 |
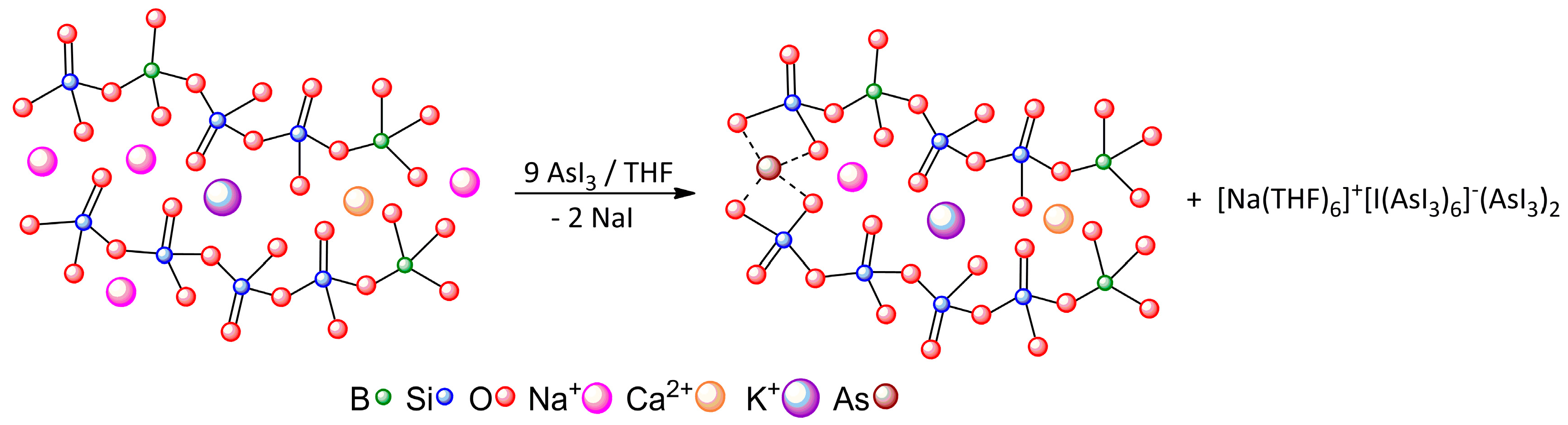
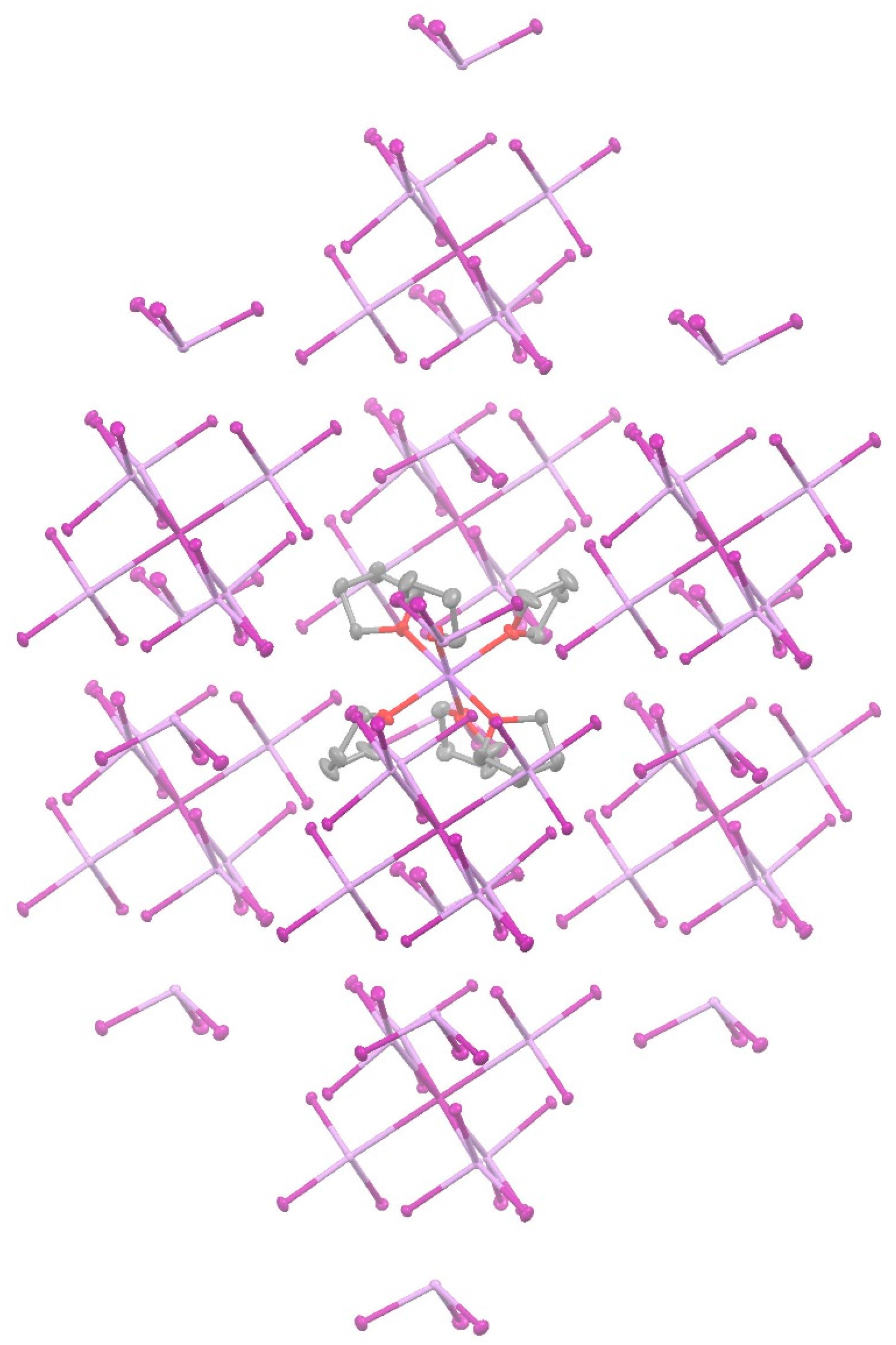
| Crystal | Motif | Interaction | CCSD(T)/CBS |
|---|---|---|---|
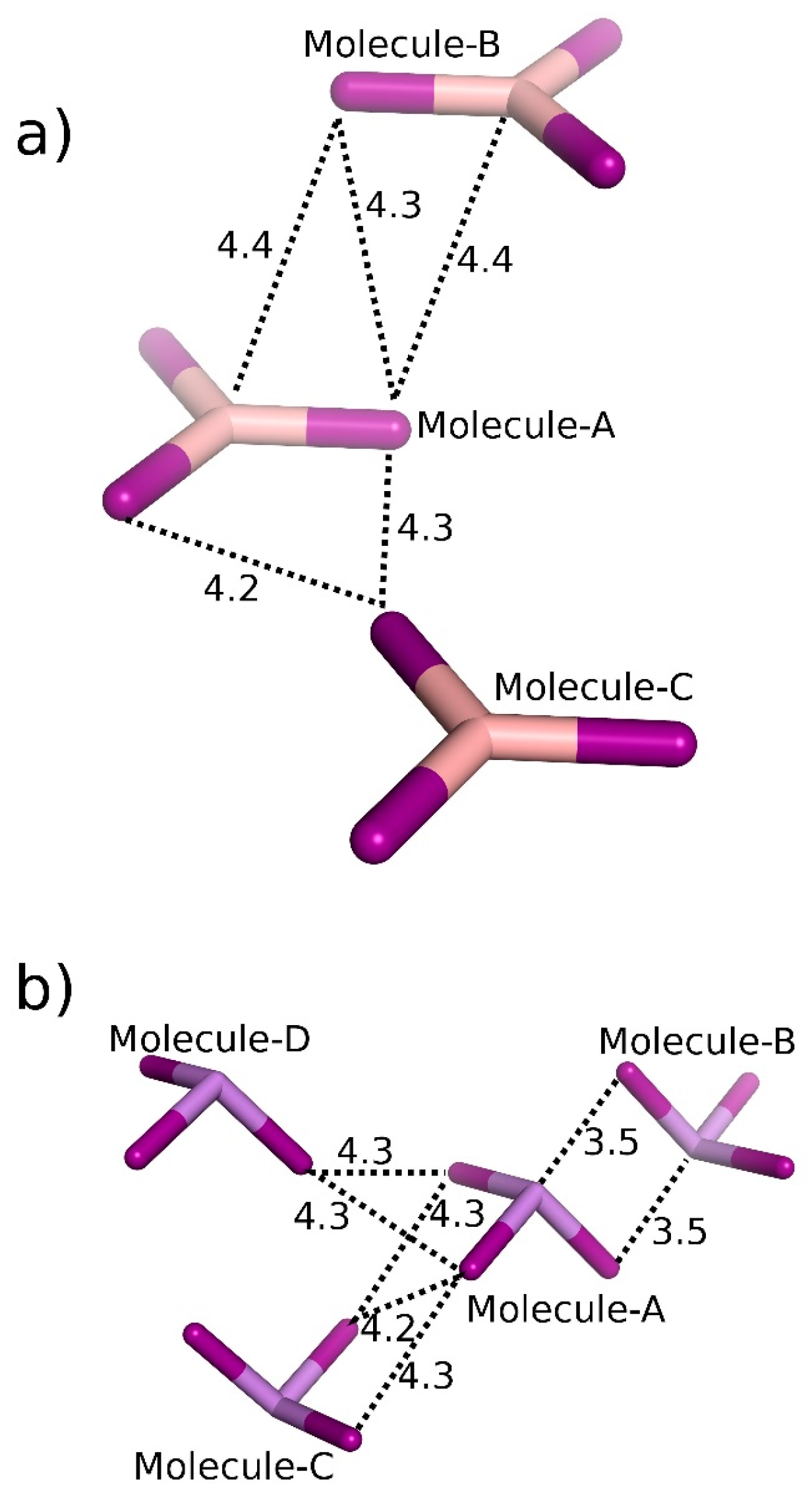
| BI3 | A···B | 2 × π-hole bonding | −6.13 |
| A···C | Bifurcated diX-bond | −2.78 | |
| AsI3 | A···B | 2 × Pn-bonding | −7.52 |
| A···C | 2 × Bifurcated diX-bonding | −4.43 | |
| A···D | Bifurcated diX-bonding | −3.26 | |
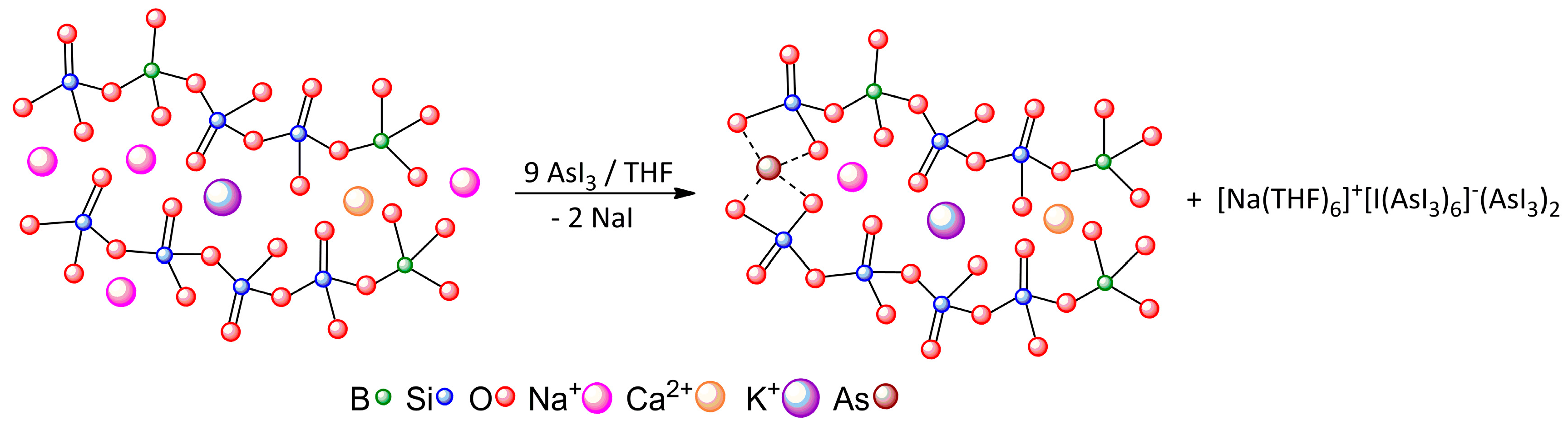
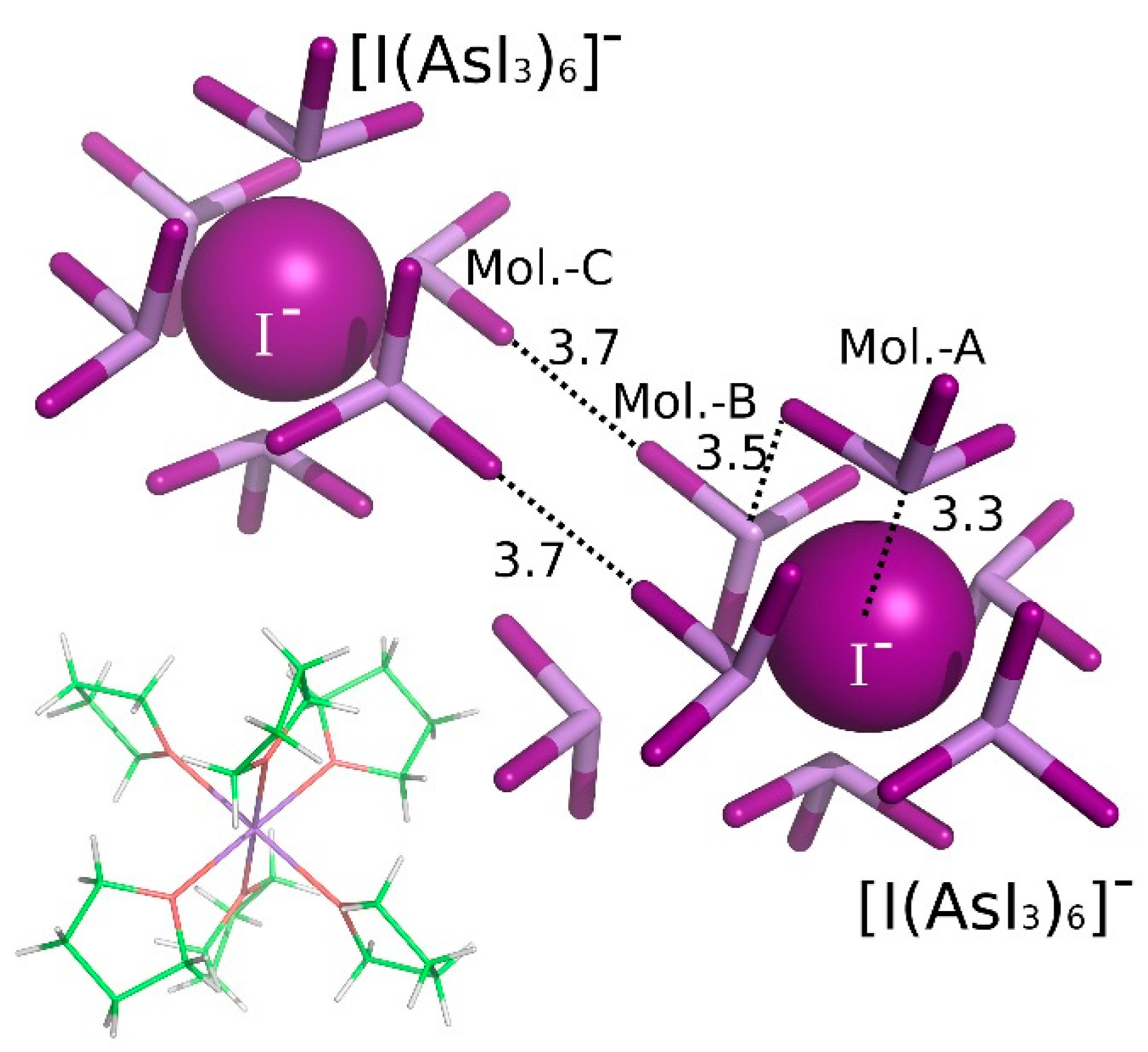
| [Na(THF)6]+[I(AsI3)6]−(AsI3)2 | A···I− | Pn-bonding | −23.59 |
| A···B | Pn-bonding | −4.64 | |
| B···C | diX-bonding | −2.24 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fanfrlík, J.; Švec, P.; Růžičková, Z.; Hnyk, D.; Růžička, A.; Hobza, P. The Interplay between Various σ- and π-Hole Interactions of Trigonal Boron and Trigonal Pyramidal Arsenic Triiodides. Crystals 2017, 7, 225. https://doi.org/10.3390/cryst7070225
Fanfrlík J, Švec P, Růžičková Z, Hnyk D, Růžička A, Hobza P. The Interplay between Various σ- and π-Hole Interactions of Trigonal Boron and Trigonal Pyramidal Arsenic Triiodides. Crystals. 2017; 7(7):225. https://doi.org/10.3390/cryst7070225
Chicago/Turabian StyleFanfrlík, Jindřich, Petr Švec, Zdeňka Růžičková, Drahomír Hnyk, Aleš Růžička, and Pavel Hobza. 2017. "The Interplay between Various σ- and π-Hole Interactions of Trigonal Boron and Trigonal Pyramidal Arsenic Triiodides" Crystals 7, no. 7: 225. https://doi.org/10.3390/cryst7070225