The Jahn-Teller Distortion at High Pressure: The Case of Copper Difluoride


1
Centre of New Technologies, University of Warsaw, 02-097 Warsaw, Poland
2
Faculty of Mathematics and Natural Sciences, Cardinal Stefan Wyszyński University, 01-038 Warsaw, Poland
Crystals 2018, 8(3), 140; https://doi.org/10.3390/cryst8030140
Submission received: 14 February 2018
/
Revised: 15 March 2018
/
Accepted: 16 March 2018
/
Published: 19 March 2018
(This article belongs to the Special Issue High-Pressure Studies of Crystalline Materials)
Abstract
:The opposing effects of high pressure (in the GPa range) and the Jahn-Teller distortion led to many intriguing phenomena which are still not well understood. Here we report a combined experimental-theoretical study on the high-pressure behavior of an archetypical Jahn-Teller system, copper difluoride (CuF2). At ambient conditions this compound adopts a distorted rutile structure of P21/c symmetry. Raman scattering measurements performed up to 29 GPa indicate that CuF2 undergoes a phase transition at 9 GPa. We assign the novel high-pressure phase to a distorted fluorite structure of Pbca symmetry, iso-structural with the ambient-pressure structure of AgF2. Density functional theory calculations indicate that the Pbca structure should transform to a non-centrosymmetric Pca21 polymorph above 30 GPa, which, in turn, should be replaced by a cotunnite phase (Pnma symmetry) at 72 GPa. The elongated octahedral coordination of the Cu2+ cation persists up to the Pca21–Pnma transition upon which it is replaced by a capped trigonal prism geometry, still bearing signs of a Jahn-Teller distortion. The high-pressure phase transitions of CuF2 resembles those found for difluorides of transition metals of similar radius (MgF2, ZnF2, CoF2), although with a much wider stability range of the fluorite-type structures, and lower dimensionality of the high-pressure polymorphs. Our calculations indicate no region of stability of a nanotubular polymorph observed for the related AgF2 system.
1. Introduction
In 1937, Jahn and Teller showed that non-linear molecules exhibiting orbital degeneracy will undergo a distortion leading to a lower-energy, and orbitally non-degenerate, structure [1]. The so-called Jahn-Teller (JT) effect is particularly strong in systems containing divalent copper (3d9 electron count). Due to operation of the JT effect the first coordination sphere of the Cu2+ cation is distorted and most often forms an elongated instead of a regular octahedron with four shorter equatorial bonds and two longer axial ones [2,3,4].
The Jahn-Teller effect, also present in compounds containing the iso-electronic Ag2+ cation (4d9) [5], has a large impact on material properties such as magnetism [6], and electronic structure [7,8,9]. Even subtle distortions in the first coordination sphere of the JT-active cation can lead to large changes in material properties, as exemplified by the case of Ag2+-bearing fluorides [10,11]. Due to the fluxional nature of the Jahn-Teller effect many studies have been devoted to tuning this distortion either by chemical substitution [12,13,14,15,16], or by high external pressure [17,18,19,20,21,22].
In the latter case pressures above 1 GPa (=10 kbar) are used to induce substantial volume reduction which in turn leads to changes in the electronic and structural properties of the studied system. Large compression generally leads to the reduction of the JT distortion; it was found, however, that in compounds containing both Cu2+ and Mn3+ cations (the latter has a 3d4 configuration and is JT-active in the high-spin state) this distortion is surprisingly robust. In LaMnO3 JT-distorted domains persist up to the insulator-to-metal transition at 34 GPa [18], while for CsMnF4 it was found that the effect is quenched only above 37 GPa when Mn3+ cations enter the low-spin state [17]. The JT distortion seems to be even more stable in the case of divalent copper [19,20]. For CuWO4 it initially decreases upon compression, but then increases abruptly during a phase transition at 9.9 GPa, and remains in place up to at least 20 GPa [19]. For Rb2CuCl4 it was found that the JT-distorted first coordination sphere of Cu2+ is stiffer than the rest of the crystal structure, which leads to tilting distortions at high pressure [20].
In order to elucidate the complex interplay between the effect of large compression and the Jahn-Teller distortions we studied the high pressure phase transitions of copper difluoride (CuF2). This compound is one of the simplest binary connections containing the Cu2+ cation. It belongs to the family of metal difluorides, which have been extensively studied at high pressure [23,24,25,26,27,28,29,30,31,32,33,34,35,36,37]. Due to the operation of the JT effect CuF2 adopts at ambient conditions a rarely-encountered crystal structure found only in one other compound (CrF2) [38].
Here we present experimental and computational evidence that up to 100 GPa CuF2 undergoes three phase transitions. The four lowest-enthalpy structures can be assigned to the rutile, fluorite, and cotunnite structure families, and the general phase transition sequence found for CuF2 (rutile → fluorite → cotunnite) resembles that observed in other difluorides. However due to the operation of the Jahn-Teller effect the coordination of Cu2+, as well as the dimensionality of the structures, differs from that found for other MF2 systems. The high-pressure phase transitions of CuF2 are reminiscent of those recently reported for its heavier analogue, AgF2 [37], with the exception that a nanotubular phase found for AgF2 is not observed [36].
2. Materials and Methods
Copper difluoride supplied by Sigma-Aldrich (Saint Louis, Missouri, United States) in a form of a powder (98% purity) was used in the study. Due to its hygroscopic nature all loadings were performed in an argon-filled glovebox with both water and oxygen content below 0.5 ppm. Measurements at ambient condition were performed on samples flame-sealed in quartz capillaries (OD 0.3 mm). The purity of the sample was confirmed by powder X-ray diffraction measurements (see Figure S1 in Supplementary Materials).
Raman spectra were acquired with the use of the Alpha300M+ system (Witec Gmbh, Ulm, Germany). We used a 532 nm laser line (35 mW power at sample) delivered to a confocal microscope by a single-mode optical fiber. The signal was collected through a 20× long working distance objective, and passed through a multi-mode optical fiber to a lens-based spectrometer (Witec UHTS 300, f/4 aperture and a focal length of 300 mm) coupled with an Andor iDUS 401 detector (Oxford Instruments, Abingdon-on-Thames, UK). The spectra were collected with the use of a 1800 mm grating resulting in a 1.5 cm–1 spectral resolution.
A total of three high-pressure runs were conducted with the use of a diamond anvil cell (DAC) supplied by D’Anvils (Hod-Hasharon, Israel). The DAC was equipped with low-fluorescence Ia diamonds (single-beveled with culet sizes of 400 µm and 500 µm) and a pre-indented stainless-steel gasket (35 µm thick). The gasket hole of 250 µm was drilled by spark-erosion. The pressure was determined from the shift of the R1 ruby fluorescence line [39]. The position of Raman bands was established with Fityk 0.9.8 software (Marcin Wojdyr, Poland) by background subtraction and fitting of the observed spectra with Lorentzian profiles [40].
Periodic DFT calculations utilized the rotationally-invariant DFT+U method [41], with the PBE exchange-correlation functional [42]. We set the U and J values of the DFT+U method to 7 eV and 0.9 eV, respectively, as suggested in a recent study on KCuF3 [43]. These value are similar to those used in other studies [44,45]. The employed method yielded lattice constants and Cu-F bond lengths overestimated by less than 2% compared to the experimental structure of CuF2 determined at low temperature [46].
The projector–augmented-wave (PAW) method was used [47], as implemented in the VASP 5.4 code. The cutoff energy of the plane waves was set to 920 eV with a self-consistent-field convergence criterion of 10−6 eV. Valence electrons (Zn, Cu: 3d, 4s; F: 2s, 2p) were treated explicitly, while standard VASP pseudopotentials (accounting for scalar relativistic effects) were used for the description of core electrons. The k-point mesh was set at 2π × 0.03 Å−1. All structures were optimized using a conjugate-gradient algorithm until the forces acting on the atoms were smaller than 5 meV/Å. For each structure the optimization was performed for the lowest-energy spin state, that is: (i) AFM ordering within [CuF4/2] sheets for P21/c, Pbca, and Pca21; (ii) FM ordering within chains of the cotunnite Pnma structure; and (iii) AFM ordering within nanotubes present in the Pbcn polymorph.
Evolutionary algorithm searches were performed at 20, 60, and 100 GPa for Z = 8 with XtalOpt software (version r9 [48]) which was coupled with the DFT+U method described above. The searches yielded the Pbca/Pca21/Pnma structures as the lowest-enthalpy polymorphs of CuF2 at 20/60/100 GPa, in accordance with results presented in this work.
Calculations of Γ-point vibration frequencies were conducted in VASP within the DFT finite-displacement method (0.007 Å displacement was used) and a tighter SCF convergence (10−8 eV). Visualization of all structures was performed with the VESTA software package [49]. For symmetry recognition we used the FINDSYM program [50]. Group theory analysis of the vibrational modes was performed with the use of the Bilbao Crystallographic Server [51].
3. Results
3.1. Ambient Pressure
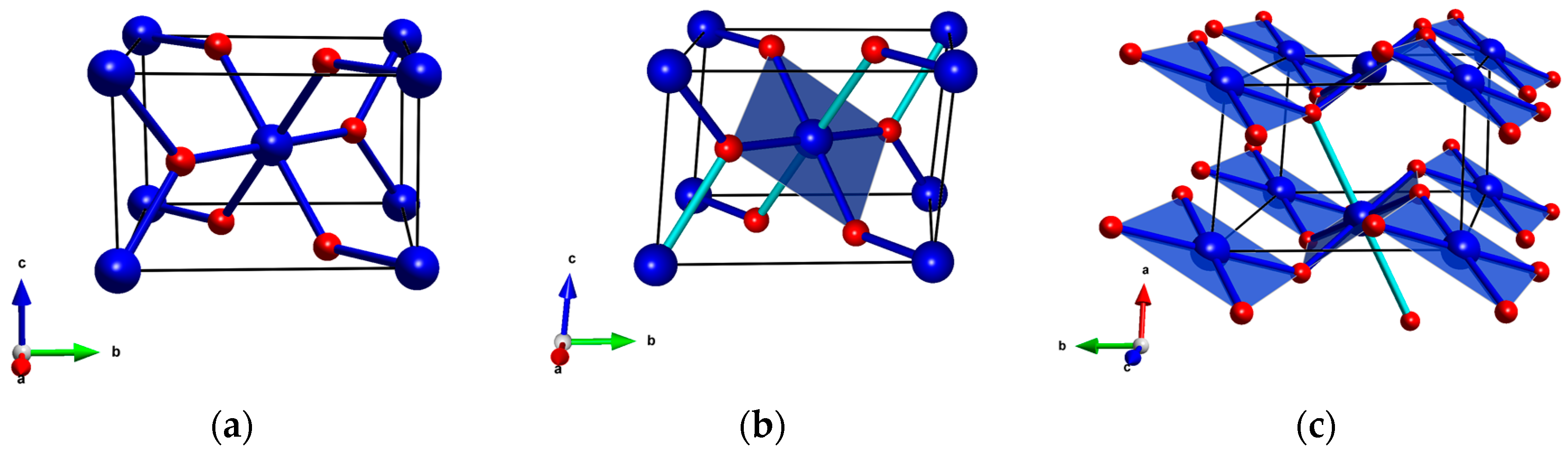
At ambient conditions CuF2 crystallizes in a structure belonging to the rutile-type family. The rutile (TiO2) aristotype, adopted by most of the first row transition metal difluorides, has tetragonal (P42/mnm) symmetry, and features a six-fold coordination of the metal center (Figure 1a). Due to operation of a collective JT distortion copper difluoride adopts a structure with lower symmetry (monoclinic, P21/n), exhibiting a 4 + 2 coordination of Cu2+ [46,52,53,54,55]. This structure, shown in Figure 1b, can be also transformed to a P21/c setting (Figure 1c) which more clearly illustrates the presence of puckered sheets of [CuF4/2] stoichiometry. These sheets host a relatively strong antiferromagnetic (AFM) interaction between the neighboring Cu2+ sites [56], which together with a weak ferromagnetic (FM) inter-layer coupling leads to a spin-canted 2D AFM state below 70 K [46,57,58]. Hereinafter when referring to the ambient pressure rutile-type structure of CuF2 we will use the P21/c setting.
Up to date various techniques have been employed in the characterization of CuF2, but to our knowledge there are no reports concerning the Raman spectrum of this material. Group theory analysis of the P21/c structure of copper difluoride (Z = 2), performed with the use of the Bilbao Crystallographic Server [51], indicates that among the 18 Γ-point vibrational modes (3Ag + 6Au + 3Bg + 6Bu) six are Raman-active (3Ag + 3Bg).
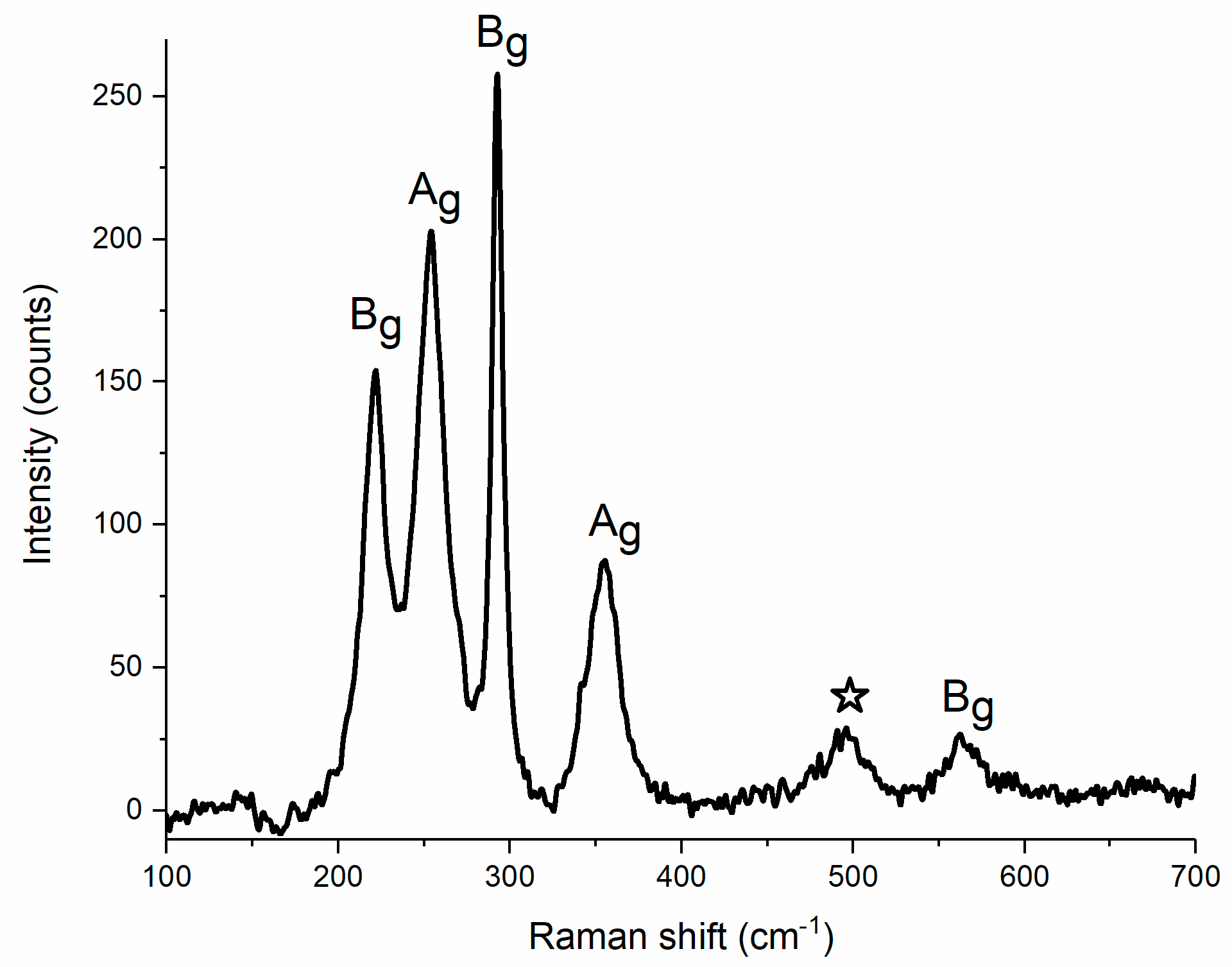
We performed calculations of the Γ-point frequencies for the P21/c structure of CuF2 with the use of the density functional theory with the inclusion of the on-site Coulomb repulsion (DFT+U method, for calculation details see Materials and Methods). The resulting values are compared in Table 1 with those obtained from ambient-pressure Raman measurements (Figure 2). The lowest-energy Ag mode is not observed experimentally as its predicted frequency of 70 cm−1 lies below the detection limit of our Raman setup. Two Bg and two Ag modes with calculated frequencies in the 200–350 cm−1 range can be assigned to the four strongest Raman bands found in experiment (Figure 2). The frequency of these four bands is on average only 4.7% higher than those predicted theoretically. Finally, the highest-frequency Bg mode is found experimentally at a Raman shift 8.0% higher than predicted from DFT+U. One additional band at 496 cm−1 is observed in the Raman spectrum of powder CuF2. This transition, marked by a star in Figure 2, can be tentatively assigned as an overtone of the strongest Ag mode at 254 cm−1, or as a combination mode of two Bg vibrations at 221 and 293 cm−1.
The good accordance between the experimental and theoretical Raman frequencies gives confidence that DFT+U method employed here can accurately simulate the pressure dependence of the frequencies of Raman active modes of CuF2. In particular the theoretical values should fall close to the experimental ones after scaling by 1.047. This is indeed the case, as will be shown in the subsequent section
3.2. Raman Scattering up to 29 GPa
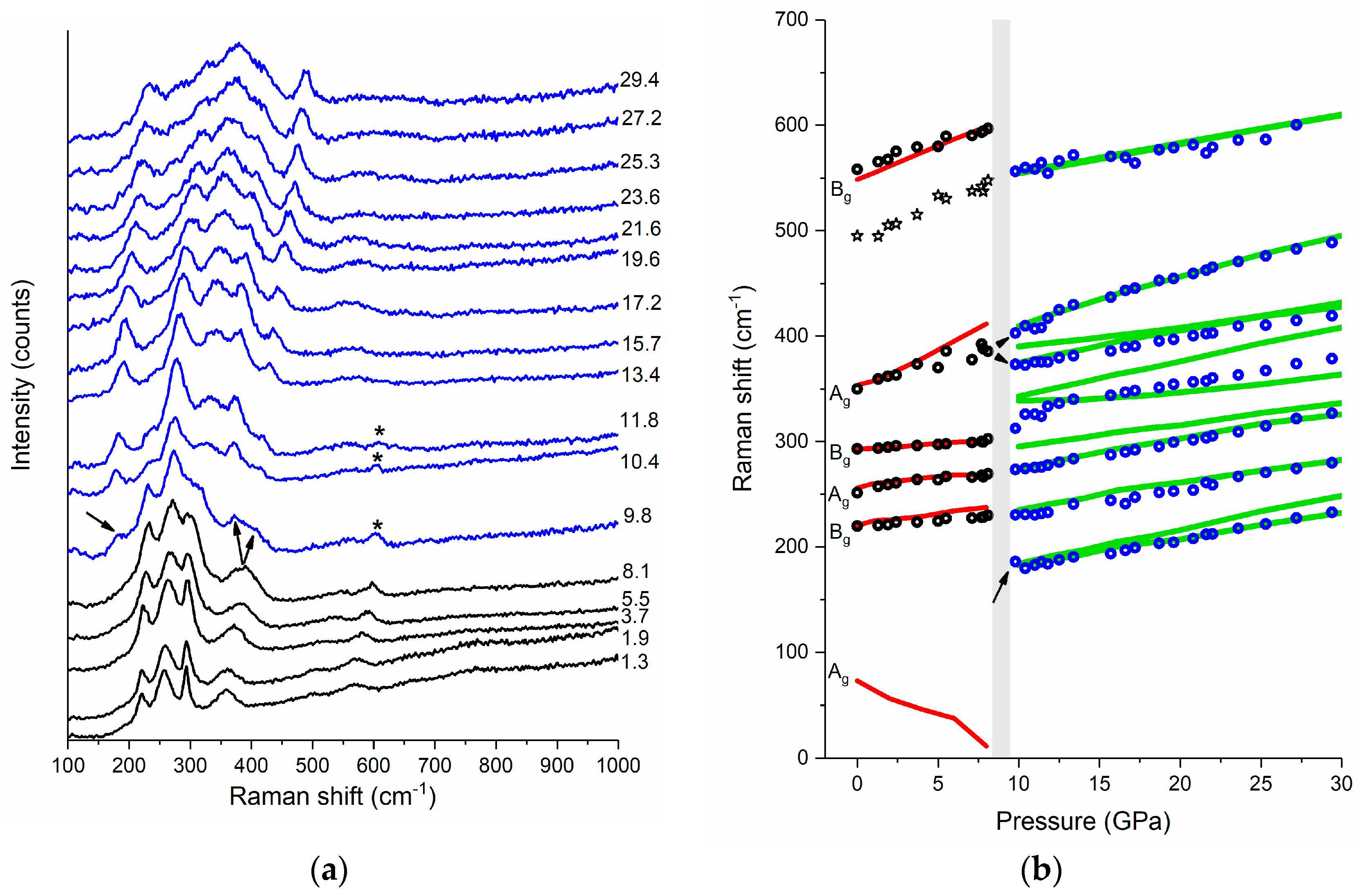
Powder samples of CuF2 were loaded into the DAC and compressed to 29.4 GPa with Raman spectra taken upon compression in ca. 2 GPa intervals (for more details see Materials and Methods section). At high pressure all of the observed Raman modes shift to higher frequencies and broaden (Figure 3a). Around 9 GPa a splitting of the highest-frequency Ag band is observed, as well as a new band appears at 185 cm−1 signaling changes in the structure of CuF2 (Figure 3b, see also Figure S2 in Supplementary Materials, for a deconvolution of the Raman spectra at 19.6 GPa). As we will argue below the changes in the Raman pattern at 9 GPa are a result of a phase transition from the ambient pressure rutile-type P21/c structure to a fluorite-like polymorph of Pbca symmetry.
Before we discuss this transition we note that the pressure dependence of the Raman frequencies below 9 GPa is in very good agreement with that predicted theoretically for the P21/c structure. Interestingly, the lowest-frequency Ag mode (not observed experimentally) is predicted to soften upon compression. This behavior resembles the one found in compounds adopting at ambient conditions the rutile aristotype, for example ZnF2 [24], CoF2 [28,31], FeF2 [30], and MnF2 [33]. In these systems the pressure-induced softening of a low-frequency B1g mode leads to a second order phase transition from the P42/mnm structure to a CaCl2-type polymorph (Pnnm symmetry, Z = 2). The latter structure can be obtained from the rutile aristotype by introducing tilts of the MF6 octahedra about the c axis (compare Figure 1a).
One might, therefore, expect that the ambient-pressure P21/c structure of CuF2 will undergo a similar transition. Indeed in our calculations we find another structure of P21/c symmetry and Z = 2 which is related to the ambient-pressure structure by rotation of the CuF6 units about the a axis (compare Figure 1c). At 9 GPa this polymorph, which we will refer to as P21/c (I), has a marginally lower enthalpy than P21/c (ΔH = −1.4 meV per f.u.). We note, however, that the frequency of its Raman modes is very similar to that of the original P21/c structure (differences not exceeding 3%), with the exception of the lowest-frequency Ag mode which is shifted from 11 cm−1 for P21/c to 74 cm−1 for P21/c (I). Hence the Raman bands predicted for P21/c (I) cannot account for the changes observed in the spectral region above 100 cm−1.
A possible candidate for the high-pressure polymorph of CuF2 is a fluorite type structure of Pbca symmetry (Z = 4, Figure 4a), which is adopted at ambient conditions by AgF2 [59,60]. Indeed, as can be seen in Figure 3b, in the whole pressure range studied there is a good match between the frequencies of the Raman-active modes predicted for this structure and those observed in experiment. Therefore the phase transition at 9 GPa can be assigned to the transformation from P21/c to Pbca. This notion is further corroborated by DFT+U calculation which predict a phase transition between these two CuF2 polymorphs at the same pressure (vide infra). Interestingly the 2D puckered sheets present in P21/c are retained in the Pbca polymorph (Figure 4a).
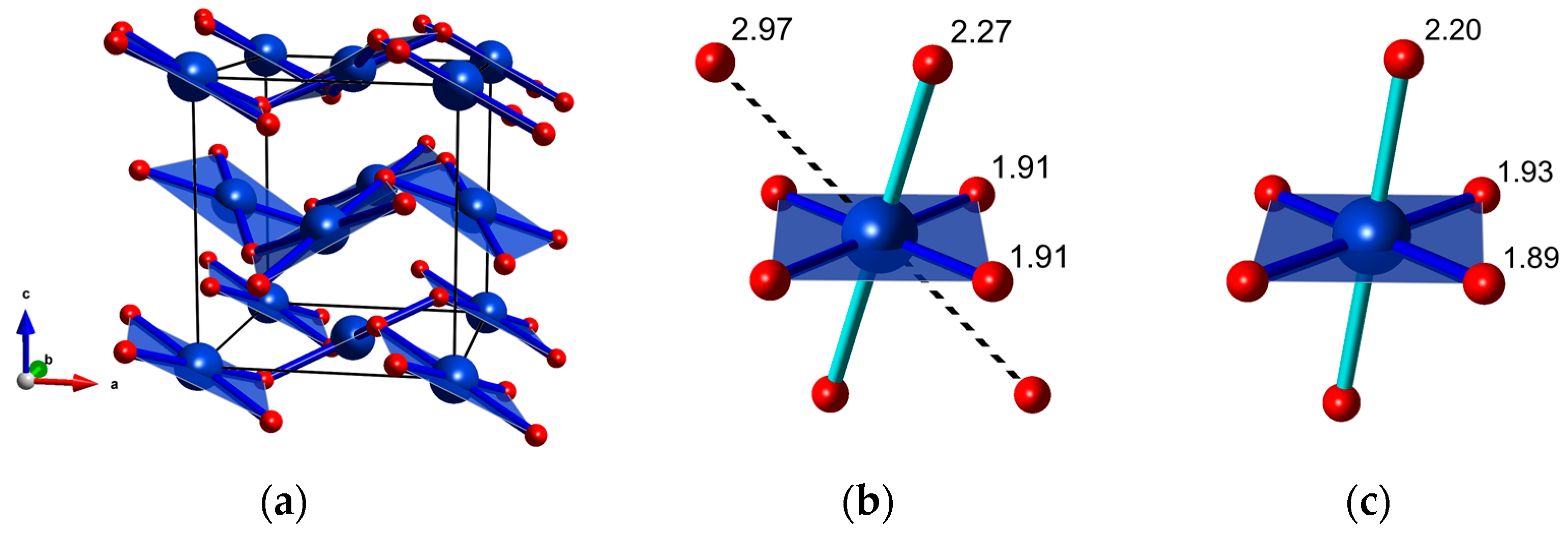
The Pbca structure can be related to the fluorite aristotype (CaF2, Fm-3m symmetry, Z = 4) [37]. Therefore the high-pressure transition from rutile-type P21/c to Pbca is analogous to the rutile-fluorite transition found in difluorides containing non-JT ions (e.g., MgF2 [23], ZnF2 [26], CoF2 [31]). The Pbca structure exhibits a 4 + 2 + 2 coordination of Cu2+ with two Cu-F contacts considerably longer (≈30%) than the remaining six. Therefore the number of neighbors in the first coordination sphere of Cu2+ (6) remains unchanged upon transition from P1/c to Pbca.
3.3. Calculations up to 100 GPa
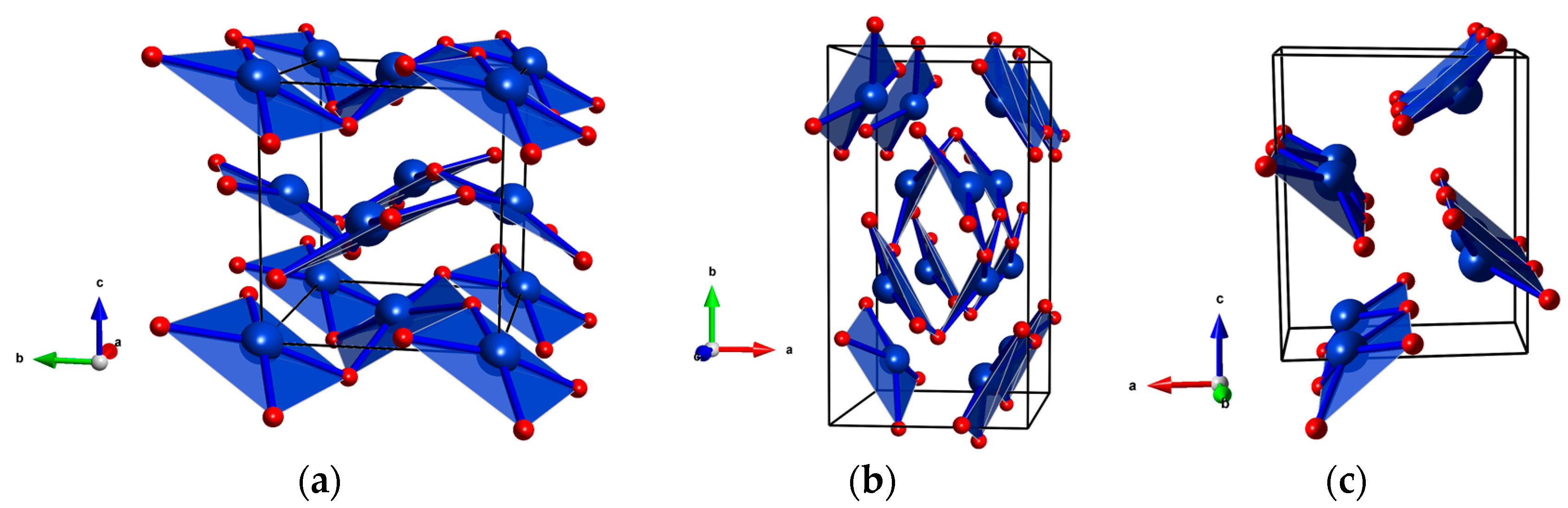
In order to further validate the interpretation of experiment, and to extend our study to higher pressures we performed DFT+U calculations for various CuF2 phases up to a pressure of 100 GPa. Apart from the P21/c and Pbca polymorphs mentioned earlier we took into account three other possible structures: Pca21 (Z = 4, Figure 5a), Pbcn (Z = 8, Figure 5b), and Pnma (Z = 4, Figure 5c). These structure were proposed as high-pressure polymorphs of AgF2 with Pca21 and Pbcn indeed observed experimentally [36,37]. We also searched for other candidate structures with the use of the XtalOpt evolutionary algorithm [48], but did not find any structure competitive in terms of enthalpy with the five mentioned above.
For AgF2, the Pca21 polymorph (HP1-AgF2) is stable between 9 and 14 GPa (Figure 5a). This structure arises from a phonon instability of the ambient-pressure Pbca polymorph stable up to 9 GPa [37]. These two fluorite-type structures are closely related and both feature 2D sheets. The main difference between Pca21 and Pbca is that, in the former structure, the metal cations are displaced out of the plane formed by the four nearest F atoms which results in a non-centrosymmetric coordination of the metal cation
The Pbcn polymorph (HP2-AgF2), observed for AgF2 from 15 GPa up to at least 36 GPa [36,37], features nanotubes built from AgF4 plaquettes distorted in the same way as in Pca21 (Figure 5b). Finally, the Pnma structure consists of chains built from analogous AgF4 units (Figure 5c). The Pnma phase is isostructural with the cotunnite (α-PbCl2) aristotype, a structure featuring nine-fold coordination of the metal center. The α-PbCl2 polytype is adopted by many metal difluorides at large compression [27]. The Pbcn phase also belongs to the cotunnite structure family [37].
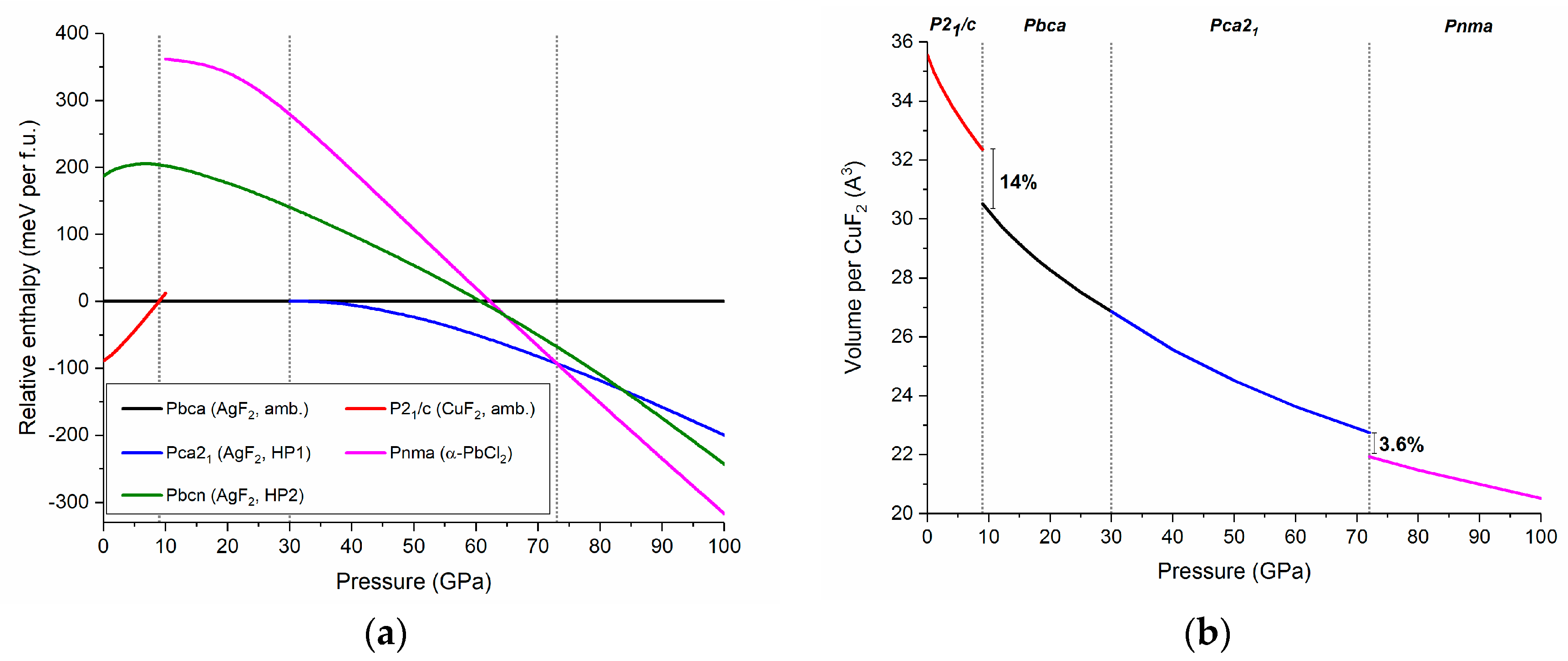
Optimization of the Pca21, Pbcn, and Pnma structures assuming a CuF2 stoichiometry does not lead to changes in the bonding topology between fluorine atoms and metal centers with respect to that found in the respective AgF2 polymorphs. By performing calculations at various pressures we were able to extract and compare the enthalpy of each of the five studied phases up to 100 GPa. In accordance with experiment we find that at ambient conditions (p ≈ 0 GPa) the P21/c rutile-type structure is the lowest energy polymorph of CuF2 (Figure 6a). Calculations indicate that at 9 GPa CuF2 should undergo a phase transition from P21/c to Pbca, in accordance with the high-pressure experimental results presented in the previous section. We predict a substantial volume decrease (14%) at this transition (Figure 6b).
Upon further compression Pbca is predicted to transform into the Pca21 polymorph at 30 GPa. The smooth enthalpy change upon the transition, as well as the lack of a volume discontinuity suggests that this is a second order transition, in analogy with what was previously reported for an analogous transition in AgF2 [37]. The last structural transition, between Pca21 and Pnma is predicted to occur at 72 GPa with a 3.6% volume reduction. We note that in contrast to the P21/c, Pbca, and Pca21 polymorphs Pnma features 1D chains. The calculations indicate no region of stability for the nanotubular Pbcn phase which is observed for AgF2.
For the rutile (P21/c) and fluorite (Pbca) phases of CuF2 we fitted the calculated volumes with the Birch-Murnaghan equation of state [61]. The obtained values of the bulk modulus (B0), given in Table 2, indicate that, surprisingly, the low-pressure P21/c structure is less compressible than the rutile-like polymorph (at the same time P21/c has a larger volume than Pbca). The B0 values calculated for the CuF2 phases are about 30% lower than those calculated for the rutile and fluorite phases of ZnF2 (Table 2). Given the fact that Zn2+ has nearly identical radius to Cu2+ (Roct(Zn2+) = 0.88 Å; Roct(Cu2+) = 0.87 Å [62]), one would expect a similar value of B0 for both CuF2 and ZnF2. The lower bulk moduli found for copper difluoride phases most likely stems from the 2D character of its structures which results in facile compression in the direction perpendicular to the sheets. This notion is corroborated by the fact that both P21/c and Pbca exhibit anisotropic compression with the inter-sheet distances more compressible than the intra-sheet ones (see Figure S3 in the Supplementary Materials).
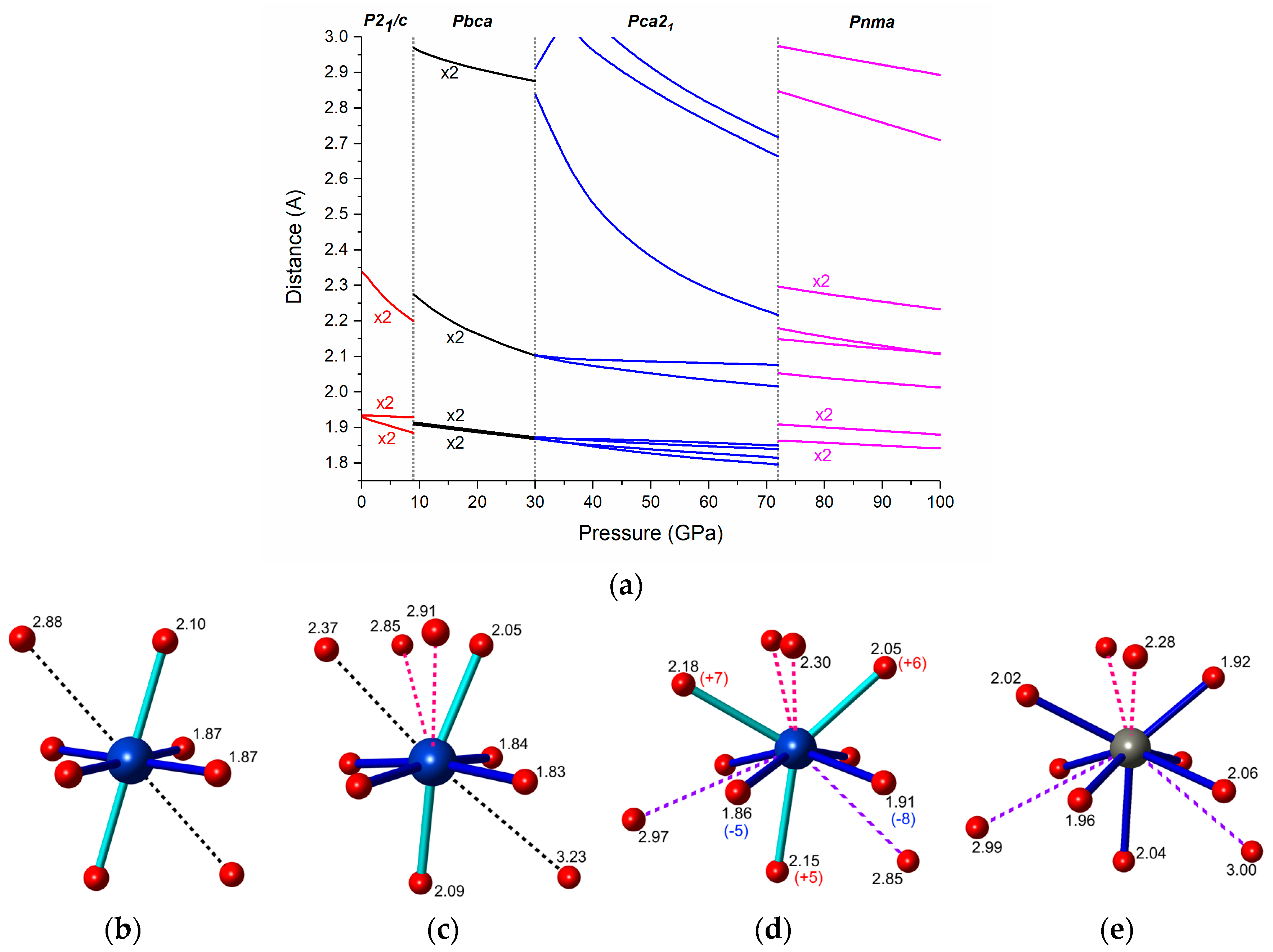
We now move to the analysis of the bonding pattern in the high-pressure polymorphs of CuF2. As can be seen in Figure 7a the Jahn-Teller distortion in P21/c is reduced upon compression. This observation is further corroborated by comparing the compressibility of M-F distances in CuF2 and ZnF2 (see Figures S4 and S5 in Supplementary Materials). As mentioned earlier the number of neighbors in the first coordination sphere of Cu2+ remains at six upon the P21/c to Pbca transition. This can be well seen in the pressure dependence of Cu-F contacts shown in Figure 7a. It is noteworthy to point out that the distortion of the CuF6 octahedron becomes larger at the transition. This signals an increase of the JT effect upon the P21/c–Pbca phase transition in analogy to what was found for CuWO4 [19].
The elongated octahedral coordination is also retained during the Pbca-Pca21 transition, although due to additional secondary contacts the CuF6 units become more distorted in Pca21 (compare Figure 7b,c). Upon compression of Pca21 one of the Cu-F contacts in the second coordination sphere of Cu2+ shortens considerably (by 22% from 30 to 72 GPa), and at 72 GPa is only 6.7% longer than the longer of the two Cu-F axial bonds.
The most dramatic changes in the coordination of Cu2+ are seen upon the Pca21-Pnma transition. The four short equatorial bonds, and the two axial ones elongate upon the transition. Additionally, the longer axial bond becomes nearly equal in length with one of the secondary Cu-F contacts (compare Figure 7c,d). As a result the first coordination sphere of Cu2+ can no longer be described as a distorted octahedron, but rather as a capped trigonal prism (coordination number equal to 7). In fact, it closely resembles that of the Zn2+ cation in the same Pnma phase of ZnF2 (Figure 7d,e). This might suggest that the Jahn-Teller effect, present in the P21/c, Pbca, and Pca21 phases, is quenched in the Pnma phase.
However, the four shortest Cu-F bonds in Pnma (dark blue cylinders in Figure 7d) are 5% to 8% shorter than the corresponding distances in ZnF2, while the three longer bonds (light blue cylinders) are longer by approximately the same amount. Those differences in the coordination spheres of Cu2+ and Zn2+ resemble the Jahn-Teller effect found for the octahedral environment. Therefore, it is highly probable that the JT effect is still operational in the Pnma phase of CuF2, although in a different coordination environment. We note that in our calculations that magnetic moments on Cu2+ atoms (mCu), as well as a substantial the band gap (Eg) are retained in the Pnma polymorph even at 100 GPa (mCu = 0.83 µB, Eg = 2.4 eV). Moreover the shape of the spin-density of Pnma at this pressure (Figure S6 in Supplementary Materials) suggests occupation of a local d(x2 − y2) orbital on each Cu2+ site, in analogy with the situation found for an elongated octahedral coordination of a d9 cation.
4. Discussion
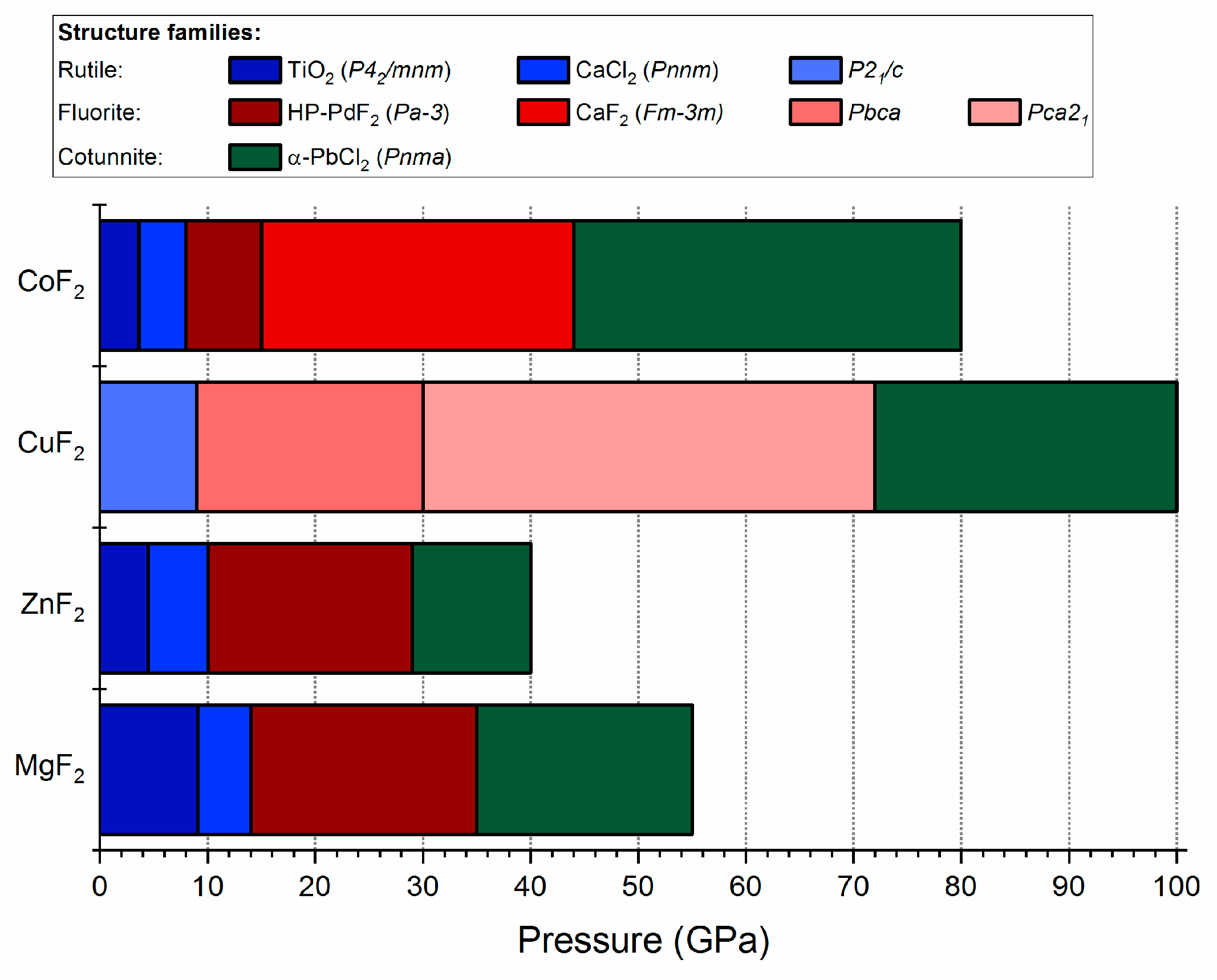
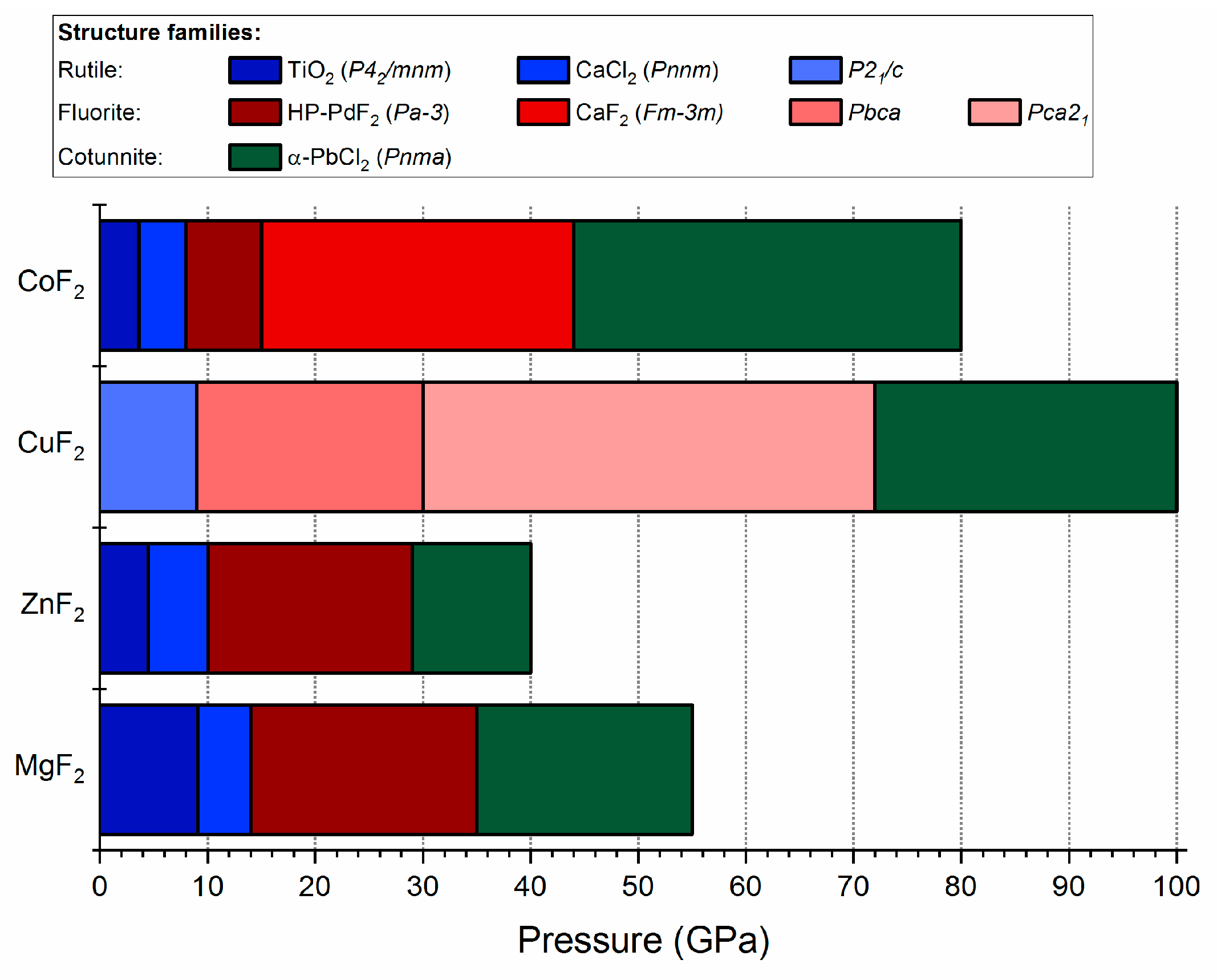
The high-pressure transformations of CuF2 can be compared to that of other metal difluorides, in particular those containing cations of similar size [62]: Mg2+ (0.86 Å), Zn2+ (0.88 Å), and Co2+ (0.89 Å in the high-spin state). The MF2 systems (M = Mg, Zn, Co), all adopting the undistorted rutile structure at ambient conditions, exhibit a similar phase transition sequence upon compression [23,26,31]: rutile (P42/mnm) → CaCl2-type (distorted rutile, Pnnm) → HP-PdF2 (distorted fluorite, Pa-3) → α-PbCl2 (cotunnite, Pnma). Only in the case of CoF2 an additional undistorted fluorite phase (Fm-3m) exhibits a region of stability between the HP-PdF2 and α-PbCl2 phases [31].
The corresponding transition pressures are summarized and compared with that of CuF2 in Figure 8. The subsequent high-pressure transitions of CuF2 from rutile P21/c to fluorite Pbca and Pca21 up to cotunnite Pnma matches that found for MF2 (M = Mg, Zn, Co). The differences between copper difluoride and other systems lies in the lower symmetry of CuF2 phases, which is a result of the JT effect. Moreover, for CuF2, the stabilization pressure of the cotunnite structure is shifted to much higher pressures compared to the MF2 systems.
The Pbca → Pca21 phase transition predicted to occur at 30 GPa for CuF2 is analogous to that found at 9 GPa for its heavier analogue, AgF2 [37]. The difference between the two compounds lies in the fact that for CuF2 the Pca21 polymorph is predicted to transform to the cotunnite Pnma phase at 72 GPa, while for AgF2 Pca21 transforms to a nanotubular cotunnite-like Pbcn structure at 14 GPa. Calculations on the AgF2 system indicate that Pnma and Pbcn polymorphs become nearly degenerate in terms of enthalpy above 50 GPa [37]. We do find for CuF2 that Pbcn is more stable than Pnma below 64 GPa (see Figure 6a), but at this pressure both are less stable than the Pca21 polymorph, and above this pressure Pnma is more stable.
In conclusion, Raman measurements indicate that CuF2 undergoes a phase transition at 9 GPa between the rutile-type P21/c structure and the fluorite-type Pbca structure. This result is corroborated by DFT+U calculations, which further indicate that, at 30 GPa, it should transform to a structurally-related Pca21 polymorph. Upon further compression copper difluoride should adopt a cotunnite Pnma structure at 72 GPa. Due to the low dimensionality of its high-pressure phases CuF2 should be more compressible than ZnF2. Surprisingly for CuF2 high pressure induces a transition from 2D structure (P21/c, Pbca, Pca21) t0 a 1D polymorph (Pnma).
The classical Jahn-Teller effect leading to an elongated octahedral coordination of Cu2+ can be observed in the P21/c, Pbca, and Pca21 phases up to 72 GPa. Upon entering the Pnma phase at that pressure the first coordination sphere of Cu2+ changes substantially, but the Jahn-Teller effect seems to be still operational. We hope that our results will motivate further studies into CuF2 subject to high pressure, in particular measurements which will enable direct probing of the local electronic structure of the Cu2+ cations.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/8/3/140/s1, Table S1: Comparison of Γ-point Raman-active modes of ZnF2 and CuF2, Figure S1: The experimental powder X-ray diffraction pattern of a sample of CuF2 together with patterns simulated for the CuF2 crystal and CuF2·2H2O, Figure S2: The experimental Raman spectrum of CuF2 together with the deconvolution into Lorentzian profiles, Figure S3: A comparison of the compressibility of inter-sheet and intra-sheet Ag-Ag distances in P21/c and Pbca, Figure S4: A omparison of the eigenvectors of the B2g mode of ZnF2, and the symmetry-related Bg mode of CuF2, Figure S5: Calculated pressure evolution of the difference between the Zn-F/Cu-F bonds together with the predicted differences in the frequencies of the highest Bg mode of CuF2 and the B2g mode of ZnF2, Figure S6: A comparison of the spin-density calculated for Pbca at 30 GPa and Pnma at 100 GPa, Figure S7: The pressure dependence of the relative enthalpy of the cotunnite phase of ZnF2 referenced to that of the HP-PdF2 phase.
Acknowledgments
The author acknowledges the support from the Polish National Science Centre (NCN) within grant no. UMO-2014/13/D/ST5/02764. This research was carried out with the support of the Interdisciplinary Centre for Mathematical and Computational Modelling (ICM) University of Warsaw under grant no. GA67-13. Comments from Jakub Gawraczyński and Adam Grzelak are greatly appreciated.
Conflicts of Interest
The author declares no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Jahn, H.A.; Teller, E. Stability of Polyatomic Molecules in Degenerate Electronic States. I. Orbital Degeneracy. Proc. R. Soc. A Math. Phys. Eng. Sci. 1937, 161, 220–235. [Google Scholar] [CrossRef]
- Reinen, D.; Friebel, C. Local and Cooperative Jahn-Teller Interactions in Model Structures Spectroscopic and Structural Evidence. In Structural Problems; Springer: Berlin/Heidelberg, Germany, 1979; Volume 37, pp. 1–60. [Google Scholar]
- Falvello, L.R. Jahn–Teller effects in solid-state co-ordination chemistry. Dalton Trans. 1997, 4463–4476. [Google Scholar] [CrossRef]
- Halcrow, M.A. Jahn-Teller distortions in transition metal compounds, and their importance in functional molecular and inorganic materials. Chem. Soc. Rev. 2013, 42, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Mazej, Z.; Kurzydłowski, D.; Grochala, W. Unique Silver(II) Fluorides: The Emerging Electronic and Magnetic Materials. In Photonic and Electronic Properties of Fluoride Materials; Tressaud, A., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 231–260. ISBN 9780128016398. [Google Scholar]
- Kugel’, K.I.; Khomskii, D.I. The Jahn-Teller effect and magnetism: Transition metal compounds. Sov. Phys. Uspekhi 1982, 25, 231–256. [Google Scholar] [CrossRef]
- Guennou, M.; Bouvier, P.; Toulemonde, P.; Darie, C.; Goujon, C.; Bordet, P.; Hanfland, M.; Kreisel, J. Jahn-Teller, Polarity, and Insulator-to-Metal Transition in BiMnO3 at High Pressure. Phys. Rev. Lett. 2014, 112, 75501. [Google Scholar] [CrossRef] [PubMed]
- Reinen, D. The Modulation of Jahn–Teller Coupling by Elastic and Binding Strain Perturbations—A Novel View on an Old Phenomenon and Examples from Solid-State Chemistry†. Inorg. Chem. 2012, 51, 4458–4472. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, I.B. The Jahn-Teller and pseudo Jahn-Teller effect in materials science. J. Phys. Conf. Ser. 2017, 833, 12001. [Google Scholar] [CrossRef]
- McLain, S.E.; Dolgos, M.R.; Tennant, D.A.; Turner, J.F.C.; Barnes, T.; Proffen, T.; Sales, B.C.; Bewley, R.I. Magnetic behaviour of layered Ag(II) fluorides. Nat. Mater. 2006, 5, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Kurzydłowski, D.; Derzsi, M.; Mazej, Z.; Grochala, W. Crystal, electronic, and magnetic structures of M2AgF4 (M = Na–Cs) phases as viewed from the DFT+U method. Dalton Trans. 2016, 45, 16255–16261. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.A.; Martínez-Lope, M.J.; Casais, M.T.; Fernández-Dáz, M.T. Evolution of the Jahn-Teller distortion of MnO6 octahedra in RMnO3 perovskites (R = Pr, Nd, Dy, Tb, Ho, Er, Y): A neutron diffraction study. Inorg. Chem. 2000, 39, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Lufaso, M.W.; Woodward, P.M. Jahn-Teller distortions, cation ordering and octahedral tilting in perovskites. Acta Crystallogr. B. 2004, 60, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, J.B. Electronic and ionic transport properties and other physical aspects of perovskites. Rep. Prog. Phys. 2004, 67, 1915–1993. [Google Scholar] [CrossRef]
- Kurzydłowski, D.; Mazej, Z.; Grochala, W. Na2AgF4: 1D antiferromagnet with unusually short Ag2+···Ag2+ separation. Dalton Trans. 2013, 42, 2167–2173. [Google Scholar] [CrossRef] [PubMed]
- Kurzydłowski, D.; Jaroń, T.; Ozarowski, A.; Hill, S.; Jagličić, Z.; Filinchuk, Y.; Mazej, Z.; Grochala, W. Local and Cooperative Jahn–Teller Effect and Resultant Magnetic Properties of M2AgF4 (M = Na–Cs) Phases. Inorg. Chem. 2016, 55, 11479–11489. [Google Scholar] [CrossRef] [PubMed]
- Aguado, F.; Rodríguez, F.; Núñez, P. Pressure-induced Jahn-Teller suppression and simultaneous high-spin to low-spin transition in the layered perovskite CsMnF4. Phys. Rev. B 2007, 76, 94417. [Google Scholar] [CrossRef]
- Baldini, M.; Struzhkin, V.V.; Goncharov, A.F.; Postorino, P.; Mao, W.L. Persistence of Jahn-Teller Distortion up to the Insulator to Metal Transition in LaMnO3. Phys. Rev. Lett. 2011, 106, 66402. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Fuertes, J.; Segura, A.; Rodríguez, F.; Errandonea, D.; Sanz-Ortiz, M.N. Anomalous High-Pressure Jahn-Teller Behavior in CuWO4. Phys. Rev. Lett. 2012, 108, 166402. [Google Scholar] [CrossRef] [PubMed]
- Aguado, F.; Rodríguez, F.; Valiente, R.; Itiè, J.-P.; Hanfland, M. Pressure effects on Jahn-Teller distortion in perovskites: The roles of local and bulk compressibilities. Phys. Rev. B 2012, 85, 100101. [Google Scholar] [CrossRef]
- Calestani, G.; Orlandi, F.; Mezzadri, F.; Righi, L.; Merlini, M.; Gilioli, E. Structural evolution under pressure of BiMnO3. Inorg. Chem. 2014, 53, 8749–8754. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A.; Winkler, B.; Morgenroth, W.; Perlov, A.; Milman, V. Pressure-induced spin collapse of octahedrally coordinated Mn3+ in the tetragonal hydrogarnet henritermierite Ca3Mn2[SiO4]2 [O4 H4]. Phys. Rev. B 2015, 92, 014117. [Google Scholar] [CrossRef]
- Haines, J.; Léger, J.M.; Gorelli, F.; Klug, D.D.; Tse, J.S.; Li, Z.Q. X-ray diffraction and theoretical studies of the high-pressure structures and phase transitions in magnesium fluoride. Phys. Rev. B 2001, 64, 134110. [Google Scholar] [CrossRef]
- Perakis, A.; Lampakis, D.; Boulmetis, Y.C.; Raptis, C. High-pressure Raman study of the ferroelastic rutile-to-CaCl2 phase transition in ZnF2. Phys. Rev. B 2005, 72, 144108. [Google Scholar] [CrossRef]
- Wu, X.; Wu, Z. Theoretical calculations of the high-pressure phases of ZnF2 and CdF2. Eur. Phys. J. B 2006, 50, 521–526. [Google Scholar] [CrossRef]
- Kusaba, K.; Kikegawa, T. In situ X-ray observation of phase transitions in under high pressure and high temperature. Solid State Commun. 2008, 145, 279–282. [Google Scholar] [CrossRef]
- Dorfman, S.M.; Jiang, F.; Mao, Z.; Kubo, A.; Meng, Y.; Prakapenka, V.B.; Duffy, T.S. Phase transitions and equations of state of alkaline earth fluorides CaF2, SrF2, and BaF2 to Mbar pressures. Phys. Rev. B 2010, 81, 174121. [Google Scholar] [CrossRef]
- Wang, H.; Liu, X.; Li, Y.; Liu, Y.; Ma, Y. First-principles study of phase transitions in antiferromagnetic XF2 (X = Fe, Co and Ni). Solid State Commun. 2011, 151, 1475–1478. [Google Scholar] [CrossRef]
- Liu, G.; Wang, H.; Ma, Y.; Ma, Y. Phase transition of cadmium fluoride under high pressure. Solid State Commun. 2011, 151, 1899–1902. [Google Scholar] [CrossRef]
- López-Moreno, S.; Romero, A.H.; Mejía-López, J.; Muñoz, A.; Roshchin, I.V. First-principles study of electronic, vibrational, elastic, and magnetic properties of FeF2 as a function of pressure. Phys. Rev. B 2012, 85, 134110. [Google Scholar] [CrossRef]
- Barreda-Argüeso, J.A.; López-Moreno, S.; Sanz-Ortiz, M.N.; Aguado, F.; Valiente, R.; González, J.; Rodríguez, F.; Romero, A.H.; Muñoz, A.; Nataf, L.; et al. Pressure-induced phase-transition sequence in CoF2: An experimental and first-principles study on the crystal, vibrational, and electronic properties. Phys. Rev. B 2013, 88, 214108. [Google Scholar] [CrossRef]
- Torabi, S.; Hammerschmidt, L.; Voloshina, E.; Paulus, B. Ab initio investigation of ground-state properties of group-12 fluorides. Int. J. Quantum Chem. 2014, 114, 943–951. [Google Scholar] [CrossRef]
- López-Moreno, S.; Romero, A.H.; Mejía-López, J.; Muñoz, A. First-principles study of pressure-induced structural phase transitions in MnF2. Phys. Chem. Chem. Phys. 2016, 18, 33250–33263. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, E.; Yao, Y.; Goncharov, A.F.; Konôpková, Z.; Raptis, C. High-pressure structural study of MnF2. Phys. Rev. B 2016, 93, 54101. [Google Scholar] [CrossRef]
- Barreda-Argüeso, J.A.; Aguado, F.; González, J.; Valiente, R.; Nataf, L.; Sanz-Ortiz, M.N.; Rodríguez, F. Crystal-Field Theory Validity Through Local (and Bulk) Compressibilities in CoF2 and KCoF3. J. Phys. Chem. C 2016, 120, 18788–18793. [Google Scholar] [CrossRef]
- Grzelak, A.; Gawraczyński, J.; Jaroń, T.; Kurzydłowski, D.; Mazej, Z.; Leszczyński, P.J.; Prakapenka, V.B.; Derzsi, M.; Struzhkin, V.V.; Grochala, W. Metal fluoride nanotubes featuring square-planar building blocks in a high-pressure polymorph of AgF2. Dalton Trans. 2017, 46, 14742–14745. [Google Scholar] [CrossRef] [PubMed]
- Grzelak, A.; Gawraczyński, J.; Jaroń, T.; Kurzydłowski, D.; Budzianowski, A.; Mazej, Z.; Leszczyński, P.J.; Prakapenka, V.B.; Derzsi, M.; Struzhkin, V.V.; et al. High-Pressure Behavior of Silver Fluorides up to 40 GPa. Inorg. Chem. 2017, 56, 14651–14661. [Google Scholar] [CrossRef] [PubMed]
- Maitland, R.; Jack, K.H. The Crystal Structure and Interatomic Bonding of Chromous and Chromic Fluorides. Proc. Chem. Soc. 1957, 1957, 232–233. [Google Scholar]
- Dewaele, A.; Torrent, M.; Loubeyre, P.; Mezouar, M. Compression curves of transition metals in the Mbar range: Experiments and projector augmented-wave calculations. Phys. Rev. B 2008, 78, 104102. [Google Scholar] [CrossRef]
- Wojdyr, M. Fityk : A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Liechtenstein, A.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Legut, D.; Wdowik, U.D. Vibrational properties and the stability of the KCuF3 phases. J. Phys. Condens. Matter 2013, 25, 115404. [Google Scholar] [CrossRef] [PubMed]
- Pavarini, E.; Koch, E.; Lichtenstein, A.I. Mechanism for Orbital Ordering in KCuF3. Phys. Rev. Lett. 2008, 101, 266405. [Google Scholar] [CrossRef] [PubMed]
- Binggeli, N.; Altarelli, M. Orbital ordering, Jahn-Teller distortion, and resonant x-ray scattering in KCuF3. Phys. Rev. B 2004, 70, 85117. [Google Scholar] [CrossRef]
- Fischer, P.; Hälg, W.; Schwarzenbach, D.; Gamsjäger, H. Magnetic and crystal structure of copper(II) fluoride. J. Phys. Chem. Solids 1974, 35, 1683–1689. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Falls, Z.; Lonie, D.C.; Avery, P.; Shamp, A.; Zurek, E. XtalOpt version r9: An open-source evolutionary algorithm for crystal structure prediction. Comput. Phys. Commun. 2016, 199, 178–179. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Stokes, H.T.; Hatch, D.M. FINDSYM : Program for identifying the space-group symmetry of a crystal. J. Appl. Crystallogr. 2005, 38, 237–238. [Google Scholar] [CrossRef]
- Kroumova, E.; Aroyo, M.I.; Perez-Mato, J.M.; Kirov, A.; Capillas, C.; Ivantchev, S.; Wondratschek, H. Bilbao Crystallographic Server: Useful Databases and Tools for Phase-Transition Studies. Phase Transit. 2003, 76, 155–170. [Google Scholar] [CrossRef]
- Billy, C.; Haendler, H.M. The Crystal Structure of Copper(II) Fluoride. J. Am. Chem. Soc. 1957, 79, 1049–1051. [Google Scholar] [CrossRef]
- Taylor, J.C.; Wilson, P.W. The structures of fluorides VI. Precise structural parameters in copper difluoride by neutron diffraction. J. Less Common Met. 1974, 34, 257–259. [Google Scholar] [CrossRef]
- Chatterji, T.; Hansen, T.C. Magnetoelastic effects in Jahn–Teller distorted CrF2 and CuF2 studied by neutron powder diffraction. J. Phys. Condens. Matter 2011, 23, 276007. [Google Scholar] [CrossRef] [PubMed]
- Burns, P.C.; Hawthorne, F.C. Rietveld Refinement of the Crystal Structure of CuF2. Powder Diffr. 2013, 6, 156–158. [Google Scholar] [CrossRef]
- Reinhardt, P.; Moreira, I.D.P.R.; de Graaf, C.; Dovesi, R.; Illas, F. Detailed ab-initio analysis of the magnetic coupling in CuF2. Chem. Phys. Lett. 2000, 319, 625–630. [Google Scholar] [CrossRef]
- Joenk, R.J.; Bozorth, R.M. Magnetic Properties of CuF2. J. Appl. Phys. 1965, 36, 1167–1168. [Google Scholar] [CrossRef]
- Boo, W.O.J.; Stout, J.W. Heat capacity and entropy of CuF2 and CrF2 from 10 to 300 °K. Anomalies associated with magnetic ordering and evaluation of magnetic contributions to the heat capacity. J. Chem. Phys. 1979, 71, 9–16. [Google Scholar] [CrossRef]
- Fischer, P.; Roult, G.; Schwarzenbach, D. Crystal and magnetic structure of silver difluoride-II. Weak 4d-ferromagnetism of AgF2. J. Phys. Chem. Solids 1971, 32, 1641–1647. [Google Scholar] [CrossRef]
- Jesih, A.; Lutar, K.; Žemva, B.; Bachmann, B.; Becker, S.; Müller, B.G.; Hoppe, R. Einkristalluntersuchungen an AgF2. Z. Anorg. Allg. Chem. 1990, 588, 77–83. [Google Scholar] [CrossRef]
- Birch, F. Finite Elastic Strain of Cubic Crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
Figure 1.
(a) The rutile aristotype (P42/mnm); (b) The CuF2 structure in the P21/n setting; and (c) the same structure in the P21/c setting. Blue/red balls mark metal/ligand atoms (Cu/F in case of CuF2); for CuF2 dark blue cylinders depict short Cu-F bonds (1.9 Å); light blue cylinders depict long bonds (2.3 Å).
Figure 1.
(a) The rutile aristotype (P42/mnm); (b) The CuF2 structure in the P21/n setting; and (c) the same structure in the P21/c setting. Blue/red balls mark metal/ligand atoms (Cu/F in case of CuF2); for CuF2 dark blue cylinders depict short Cu-F bonds (1.9 Å); light blue cylinders depict long bonds (2.3 Å).
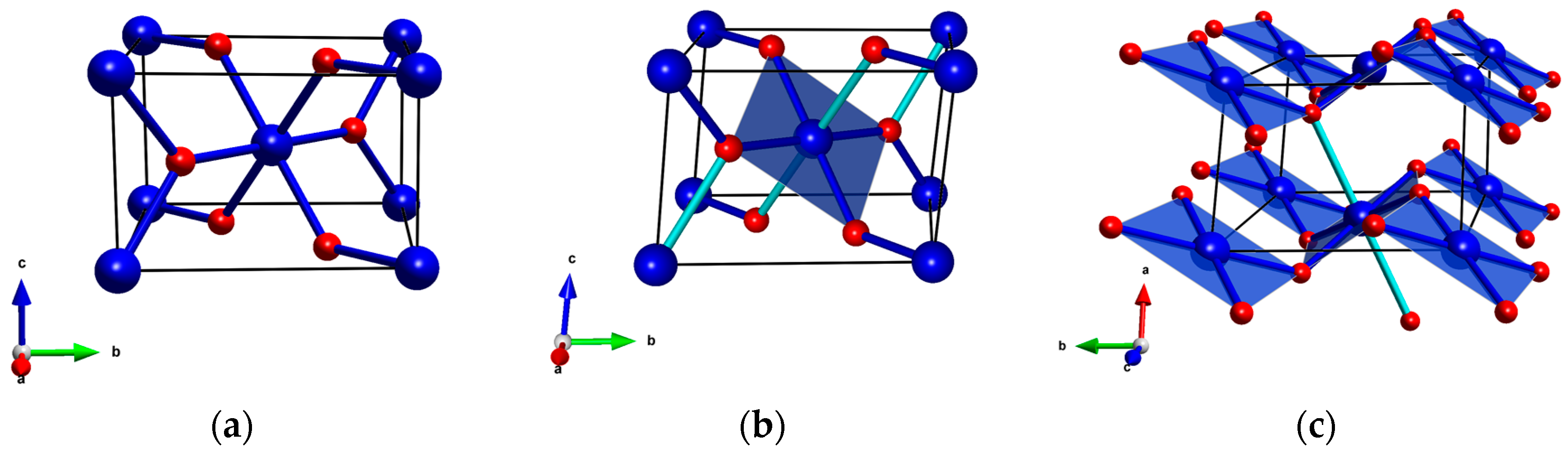
Figure 2.
Raman spectrum of powder CuF2 at ambient condition.
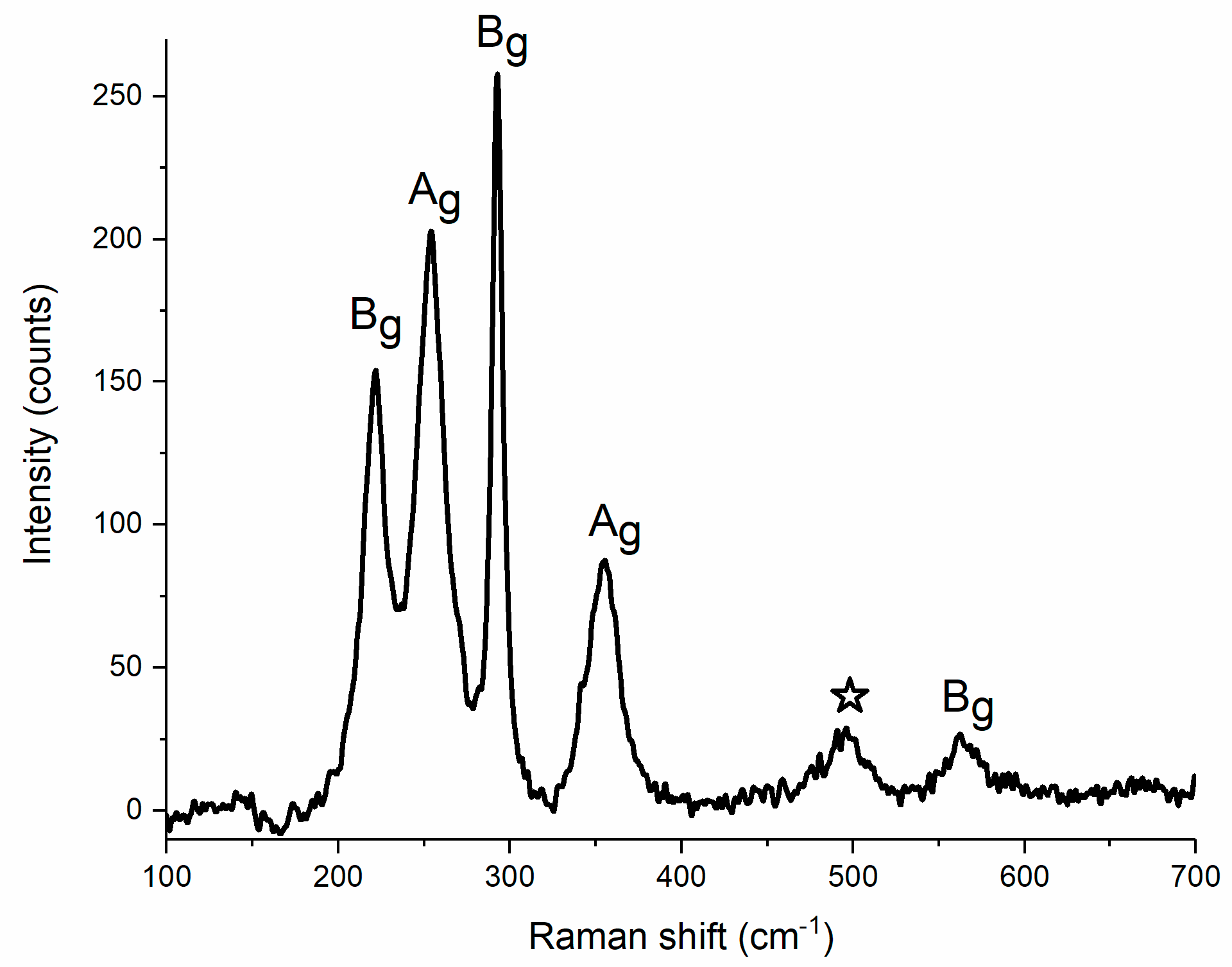
Figure 3.
(a) Raman spectrum of powder CuF2 at selected pressures (their values are given in GPa), the spectra corresponding to the rutile P21/c phase are shown in black while those assigned to the fluorite-type Pbca phase are shown in blue; and (b) pressure dependence of the frequency of the Raman bands (circles for experiment: black—P21/c; blue—Pbca; lines for DFT+U calculations: red—P21/c; green—Pbca). Arrows mark the appearance of a new low-frequency band and splitting of the Ag band. Asterisks in (a) mark the Bg band originating from traces of the P21/c structure still present above the phase transition, while stars in (b) indicate the pressure dependence of the Ag overtone or Bg combination mode of rutile CuF2 (see text). The calculated frequencies were scaled by 1.047.
Figure 3.
(a) Raman spectrum of powder CuF2 at selected pressures (their values are given in GPa), the spectra corresponding to the rutile P21/c phase are shown in black while those assigned to the fluorite-type Pbca phase are shown in blue; and (b) pressure dependence of the frequency of the Raman bands (circles for experiment: black—P21/c; blue—Pbca; lines for DFT+U calculations: red—P21/c; green—Pbca). Arrows mark the appearance of a new low-frequency band and splitting of the Ag band. Asterisks in (a) mark the Bg band originating from traces of the P21/c structure still present above the phase transition, while stars in (b) indicate the pressure dependence of the Ag overtone or Bg combination mode of rutile CuF2 (see text). The calculated frequencies were scaled by 1.047.
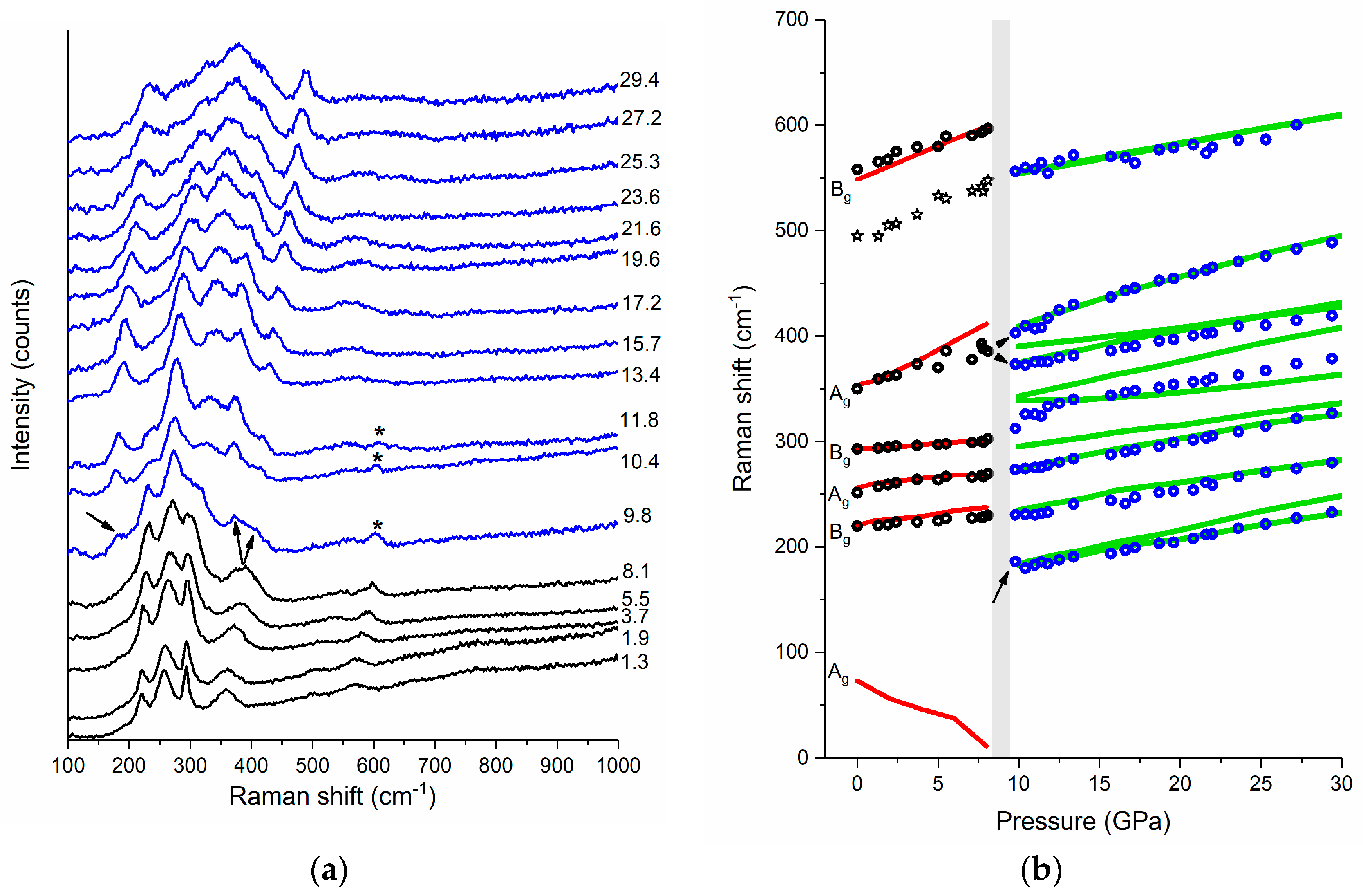
Figure 4.
(a) The fluorite-type Pbca structure CuF2 (for clarity only the four shortest Cu-F bonds are shown); (b) the coordination of Cu2+ in Pbca calculated at 9 GPa; and (c) the coordination of Cu2+ in P21/c calculated at 9 GPa; Cu-F distances are given in Å.
Figure 4.
(a) The fluorite-type Pbca structure CuF2 (for clarity only the four shortest Cu-F bonds are shown); (b) the coordination of Cu2+ in Pbca calculated at 9 GPa; and (c) the coordination of Cu2+ in P21/c calculated at 9 GPa; Cu-F distances are given in Å.
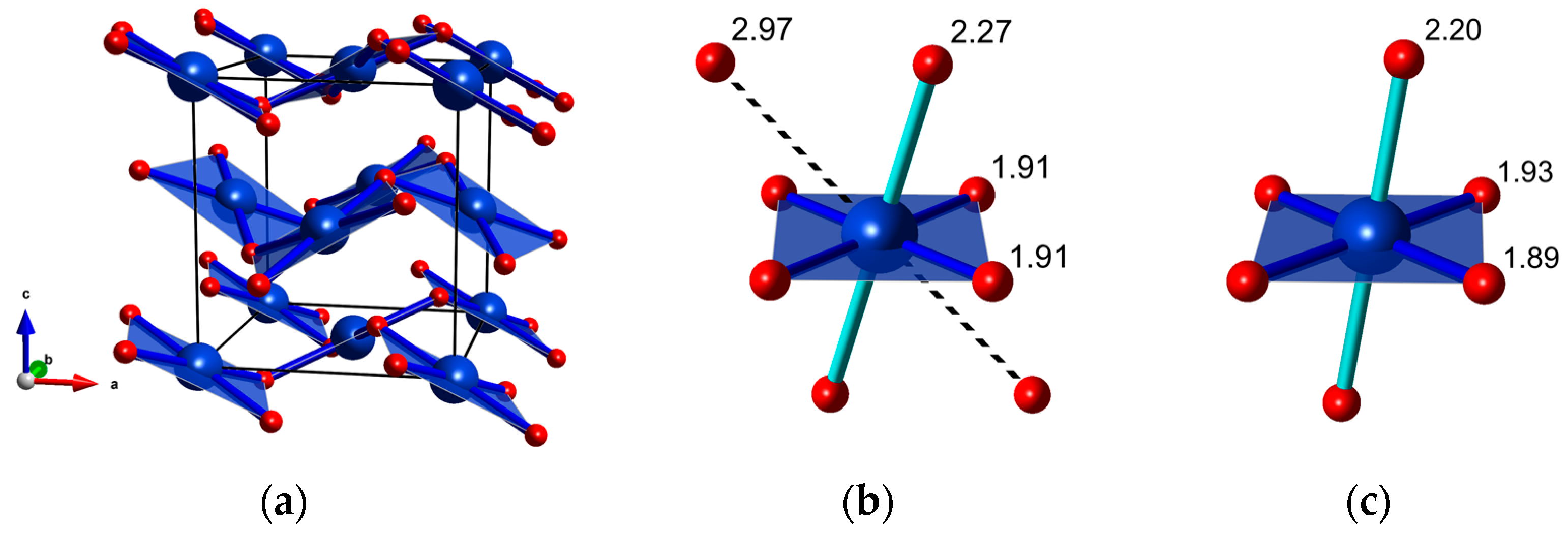
Figure 5.
Possible high-pressure structures of CuF2 (a) Pca21 (Z = 4); (b) Pbcn (Z = 8); and (c) Pnma (Z = 4). For clarity only the four shortest Cu-F bonds are shown.
Figure 5.
Possible high-pressure structures of CuF2 (a) Pca21 (Z = 4); (b) Pbcn (Z = 8); and (c) Pnma (Z = 4). For clarity only the four shortest Cu-F bonds are shown.

Figure 6.
(a) The pressure dependence of the relative enthalpies (referenced to that of Pbca) of various CuF2 high-pressure polymorphs; and (b) the pressure dependence of the volume per one CuF2 unit. Dotted lines mark P21/c → Pbca, Pbca → Pca21, and Pca21 → Pnma phase transition predicted at 9, 30, and 72 GPa, respectively.
Figure 6.
(a) The pressure dependence of the relative enthalpies (referenced to that of Pbca) of various CuF2 high-pressure polymorphs; and (b) the pressure dependence of the volume per one CuF2 unit. Dotted lines mark P21/c → Pbca, Pbca → Pca21, and Pca21 → Pnma phase transition predicted at 9, 30, and 72 GPa, respectively.
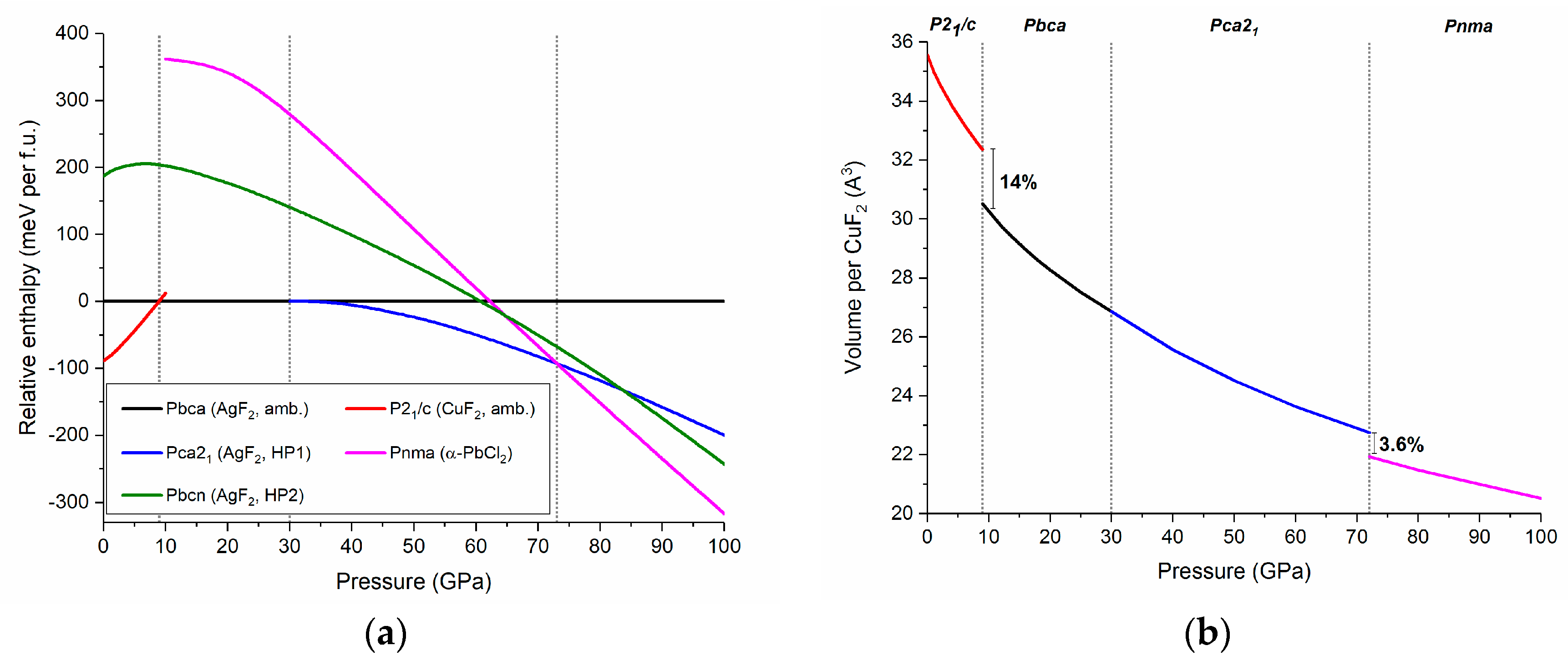
Figure 7.
(a) Calculated pressure dependence of the Cu-F distances in the high-pressure polymorphs of CuF2. The coordination of the Cu2+ cation in (b) Pbca at 30 GPa; (c) Pca21 at 50 GPa; and (d) Pnma at 72 GPa; together with (e) the Zn2+ coordination in the Pnma phase of ZnF2 optimized at 72 GPa. Distances are given in Å; numbers in parentheses indicate the percentage difference between the Cu-F and Zn-F distances in the Pnma polymorphs.
Figure 7.
(a) Calculated pressure dependence of the Cu-F distances in the high-pressure polymorphs of CuF2. The coordination of the Cu2+ cation in (b) Pbca at 30 GPa; (c) Pca21 at 50 GPa; and (d) Pnma at 72 GPa; together with (e) the Zn2+ coordination in the Pnma phase of ZnF2 optimized at 72 GPa. Distances are given in Å; numbers in parentheses indicate the percentage difference between the Cu-F and Zn-F distances in the Pnma polymorphs.
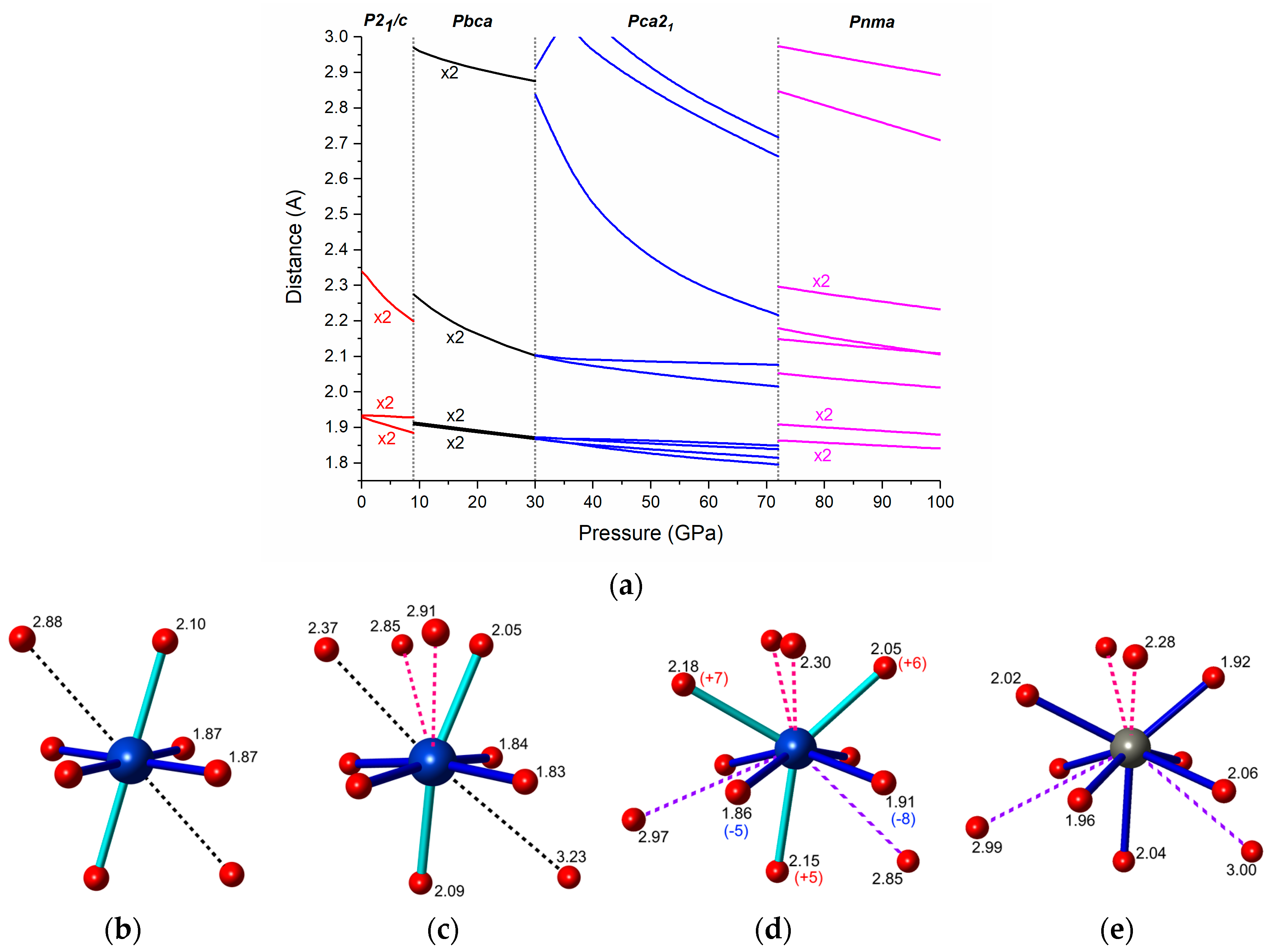
Figure 8.
Bar diagram showing the pressure stability intervals of the different structural modifications of MF2 fluorides. Experimental results for CoF2, ZnF2, and MgF2 are taken from [23,26,31], respectively. The HP-PdF2 to cotunnite phase transition for ZnF2 at 29 GPa is taken from our calculations (see Figure S7 in Supplementary Materials).
Figure 8.
Bar diagram showing the pressure stability intervals of the different structural modifications of MF2 fluorides. Experimental results for CoF2, ZnF2, and MgF2 are taken from [23,26,31], respectively. The HP-PdF2 to cotunnite phase transition for ZnF2 at 29 GPa is taken from our calculations (see Figure S7 in Supplementary Materials).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of calculated (ωth) and experimental (ωexp) Γ-point Raman frequencies (in cm−1) of the rutile-type P21/c structure of CuF2 at ambient pressure. No scaling was applied to the calculated frequencies.
Table 1.
Comparison of calculated (ωth) and experimental (ωexp) Γ-point Raman frequencies (in cm−1) of the rutile-type P21/c structure of CuF2 at ambient pressure. No scaling was applied to the calculated frequencies.
| Symmetry | ωth | ωexp |
|---|---|---|
| Bg | 524 | 566 |
| Ag | 338 | 355 |
| Bg | 280 | 293 |
| Ag | 245 | 254 |
| Bg | 210 | 221 |
| Ag | 70 | n.d. |
Table 2.
The bulk modulus in GPa (B0), and its derivative (B0’) calculated for CuF2 phases. Results obtained for the rutile (P42/mnm) and fluorite (Fm-3m) structures of ZnF2 are shown for comparison.
Table 2.
The bulk modulus in GPa (B0), and its derivative (B0’) calculated for CuF2 phases. Results obtained for the rutile (P42/mnm) and fluorite (Fm-3m) structures of ZnF2 are shown for comparison.
| Phase | B0 | B0’ |
|---|---|---|
| P21/c | 75 | 6.1 |
| Pbca | 71 | 5.4 |
| ZnF2 (P42/mnm) | 101 (105) 1 | 4.3 |
| ZnF2 (Fm-3m) | 116 (120) 1 | 4.7 |
1 DFT calculations with the PBE functional from ref. [32].
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kurzydłowski, D. The Jahn-Teller Distortion at High Pressure: The Case of Copper Difluoride. Crystals 2018, 8, 140. https://doi.org/10.3390/cryst8030140
AMA Style
Kurzydłowski D. The Jahn-Teller Distortion at High Pressure: The Case of Copper Difluoride. Crystals. 2018; 8(3):140. https://doi.org/10.3390/cryst8030140
Chicago/Turabian StyleKurzydłowski, Dominik. 2018. "The Jahn-Teller Distortion at High Pressure: The Case of Copper Difluoride" Crystals 8, no. 3: 140. https://doi.org/10.3390/cryst8030140
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.