The Rietveld Refinement in the EXPO Software: A Powerful Tool at the End of the Elaborate Crystal Structure Solution Pathway



Abstract
:1. Introduction
- determine the cell parameters;
- identify the space group;
- solve the structure ab initio in the reciprocal space via direct methods (extract the integrated intensities from the experimental profile for estimating the observed structure factor modulus associated to each reflection, probabilistically evaluate the phases of reflections, calculate the electron density map for deriving the structure model, optimize the model);
- solve the structure in the direct space (search the best model, compatible with the expected molecular geometry, by global optimization methods;
- refine the structure model by the Rietveld method.
2. The Crystal Structure Solution Process in EXPO
2.1. Solution in Reciprocal Space
- (i)
- Data resolution. Experimental diffraction data far away from the atomic resolution can prevent the success of the structure solution process or lead to a poor electron density map, and can be difficult to interpret;
- (ii)
- Structure complexity. RS methods are usually able to solve successfully crystal structures with number of atoms in the asymmetric unit (Nau) up to about 40 [13]. Nau >> 40 is a challenging task for RS methods.
- Direct methods basic principles
- (1)
- the positivity of (i.e., > 0 in the unit cell);
- (2)
- the atomicity of (i.e., corresponds to discrete atoms).
- (1)
- Normalization: the integrated intensities are normalized by the Wilson method and the values are calculated. Statistical analysis on is performed in order to detect: (a) the presence or absence of an inversion center; (b) the possible presence and type of pseudotranslational symmetry [18]; (c) the preferred orientation effects [19]. The largest values (i.e., ≥ 1) reflections, the so called strong reflections, whose number is Nlarg, are considered because they strongly contribute to the DM phasing process (see next point 2). The default number of strong reflections to be phased (Nphas) is automatically calculated by EXPO by taking into account the number of atoms in the asymmetric unit and the type of symmetry. Nphas should be at most Nlarg; if Nphas > Nlarg, EXPO sets Nphas = Nlarg;
- (2)
- structure invariants (s.i.) calculation: s.i. are magnitudes that are independent of change of the origin and depend only on the structure. They are fundamental in the phasing process. For example, a s.i. of order n is the product of n normalized structure factors with h1 + h2 + … + hn = 0. In the phasing process, a special role is occupied by triplet invariants (n = 3). EXPO estimates the triplet phase via the probabilistic formula [20] that depends on ten values. The reliability parameter of the estimate of the triplet phase is proportional to the product (it is large for strong reflections) and, in the case of a crystal structure with N non-H equal atoms in the unit cell, is inversely proportional to N: with the increasing of N, becomes negligible and, consequently, the probability of DM failure increases. can be positive or negative; if positive, attains its maximum at (i.e., positive triplets), if negative, is maximum at (i.e., negative triplets). The positive triplets with ≥ 0.6 are stored by EXPO and are strongly involved in the phasing process;
- (3)
- Phases estimate: a milestone for DM is the tangent formula, proposed by Karle & Hautpman [21], that derives the phases of the Nphas reflections starting from a subset of selected reflections (the so called starting set) and actively uses the triplets in which the reflections to be phased are embedded. The phases of the starting set can be set via a multisolution method based on magic integers [22] (this is the default choice of EXPO), or, alternatively, by a random approach starting with random phase values. DM provide several possible sets of phases that are ranked according to a suitable mathematical tool, the combined figure of merit (CFOM) [23,24], mainly based on the values. The largest CFOM value phase set should correspond to the correct solution. When it does not provide a plausible and chemically interpretable structure solution, EXPO offers the graphic option to conveniently explore all the generated and stored phasing sets (their number is usually 20);
- (4)
- Electron density map calculation: the calculation of (3) is carried out on the largest CFOM value phase set or on some or all the DM generated and stored sets. The interpretation of in terms of positions and intensities of its peaks supplies the fractional coordinates and the chemical labels of the atoms in the structure. Because of uncertainties on the experimental structure factors moduli extracted from the powder profile, the entire DM process can be unsatisfactory and the final structure model approximate. Consequently, the completion and/or optimization of the DM structure model are a fundamental request for a successful and meaningful application of the Rietveld method.
- Model optimization
- (1)
- WLSQFR (weighted least-squares Fourier recycling) [6] consists of a two-step approach alternating suitably weighted least-squares refinement (aiming to minimize the weighted squared difference between the observed and calculated intensities) and Fourier map calculations, which add missing atom positions to the refined model. The weights take into account the low accuracy of the integrated intensities estimates of the overlapping reflections and tend to prevent the domination of the refinement process by the largest intensity reflections. This optimization tool is automatically applied in a default run of EXPO in case of inorganic compounds solution.
- (2)
- RBM (resolution bias modification). The procedure can work in direct or reciprocal space or in both spaces [25,26,27,28,29] and is a powerful approach able to reduce the errors on the electron density map mainly due to the limited experimental resolution. RBM is able to discard false peaks and recover the missing ones. It has revealed itself to be particularly effective for organic and metal-organic compounds (for them it is the default choice of EXPO).
- (3)
- COVMAP [30], a procedure aiming to correct the electron density map by exploiting the principle of covariance between two points of the map, nevertheless its quality. It can locate the missing atoms by modifying the electron density map taking into account some basic crystal chemistry rules, in particular, the expected bond distances between couples of atoms.
- (4)
- Shift_and_Fix [31], the last developed approach and effectively introduced in EXPO based on the optimization of the models derived from all the stored DM phasing sets that are automatically and sequentially processed and analyzed. Shift_and_Fix consists of two main steps, cyclically combined:
- (a)
- The Shift step, which randomly shifts a suitably chosen part of the DM structure model;
- (b)
- The Fix step, which carries out a weighted least-squares refinement of the shifted model, followed by Fourier map calculations whose coefficients are functions of the chemical content of the compound under study.
2.2. Solution in Direct Space
- (1)
- The Rwp weighted profile reliability parameter (see Equation (2)) which represents the default choice;
- (2)
- The RI agreement factor, which compares the experimental integrated intensities Ih(obs) and the intensities Ih(calc) calculated by the model:
- Simulated Annealing
- GHBB-BC Method
- (1)
- The Big Bang-Big Crunch global optimization method (BB-BC) [43]. It is inspired by one of the cosmological theories of the universe and involves two phases: (i) the Big Bang, corresponding to the disorder caused by the energy dissipation in which a completely random population is generated; (ii) the Big Crunch, corresponding to the order due to gravitational attraction where the population shrinks to a single good quality element represented by the centre of mass, for converging to a global optimum point.
- (2)
- The metaheuristic Greedy Randomized Adaptive Search Procedure (GRASP) [44]. It is an iterative approach, particularly effective in finding empirically good quality solutions in a reasonable computational time for most of the real-world combinatorial optimization problems that are computationally difficult and have enormous solution spaces. Each GRASP iteration is made up of two phases: construction and local search. The construction phase progressively builds a set of feasible solutions from scratch; the local search phase investigates their neighbourhood until a local minimum is found. The best overall solution is kept as the final result.
- (3)
- The traditional Simulated Annealing.
2.3. The Rietveld Refinement
- (1)
- Parameters for correcting the systematic line-shift errors due to sample displacement, sample transparency, and zero-shift.
- (2)
- Background parameters. The background is automatically described by empirical functions: the classical polynomial function, the Chebyshev polynomial function, and the cosine Fourier series. It can be also modeled by a mouse-click selection of points interpolated by the best fitting background curve.
- (3)
- Parameters related to integrated intensities: scale factor and preferred orientation. Correction for the preferred orientation can be achieved by the March–Dollase function [55].
- (4)
- Profile parameters: full width at half maximum of the peak shape and peak asymmetry. The available peak shape functions are: pseudo-Voigt, Pearson-VII, and modified Thompson–Cox–Hastings pseudo-Voigt. The correction for the peak asymmetry is applied by using the semiempirical function given in [56].Kα1 and Kα2 peak doublet, if present, can be modelled.
- (5)
- Crystal structure parameters: lattice parameters, atomic fractional coordinates, occupation factors, and isotropic displacement parameters. Atomic displacement parameters can be refined individually or in a group of atoms with the same atomic type or the same environment.
3. Application
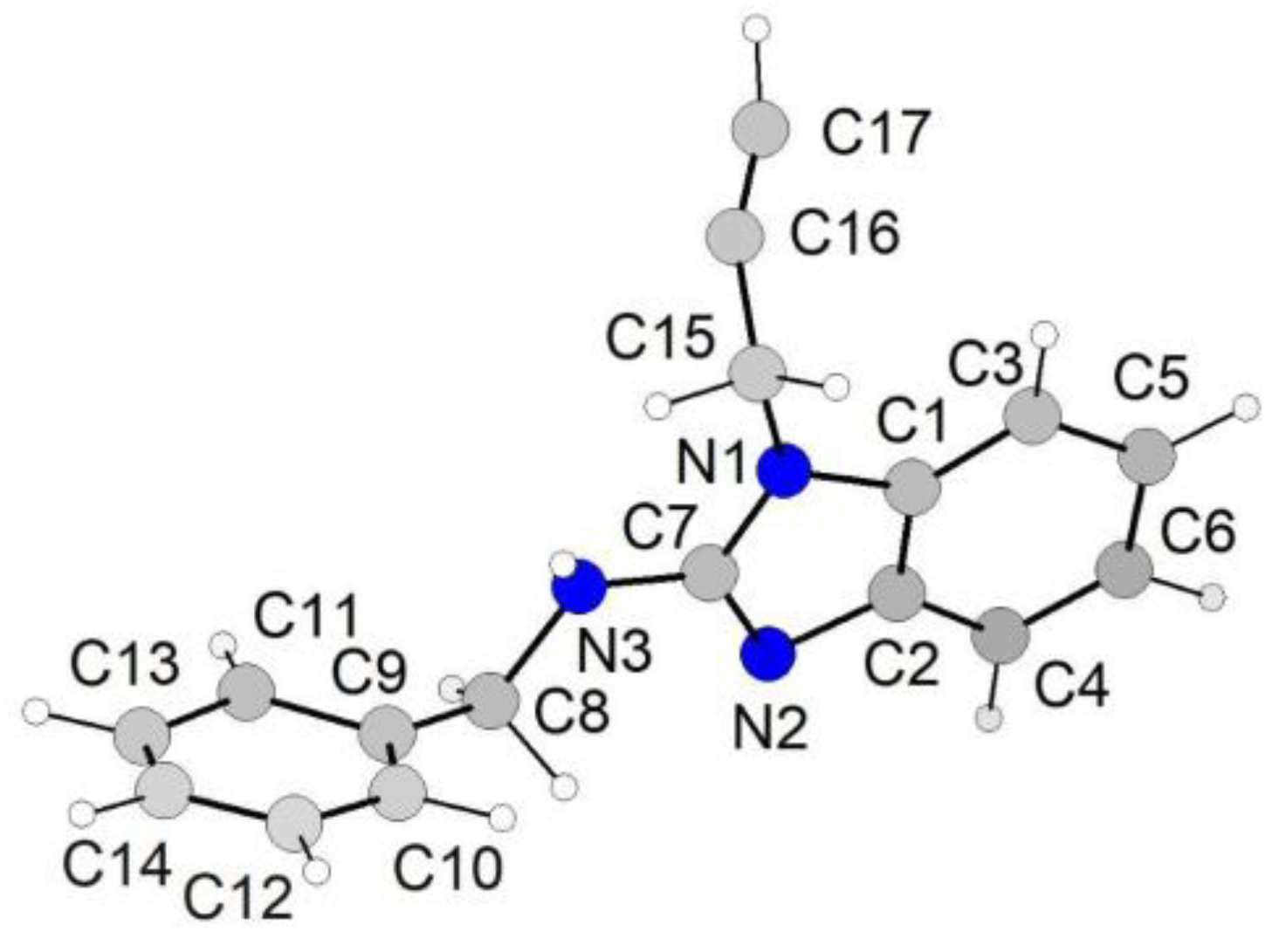
3.1. Rietveld Refinement of N-Benzyl-1-(prop-2-yn-1-yl)-1H-benzo[d]imidazol-2-amine
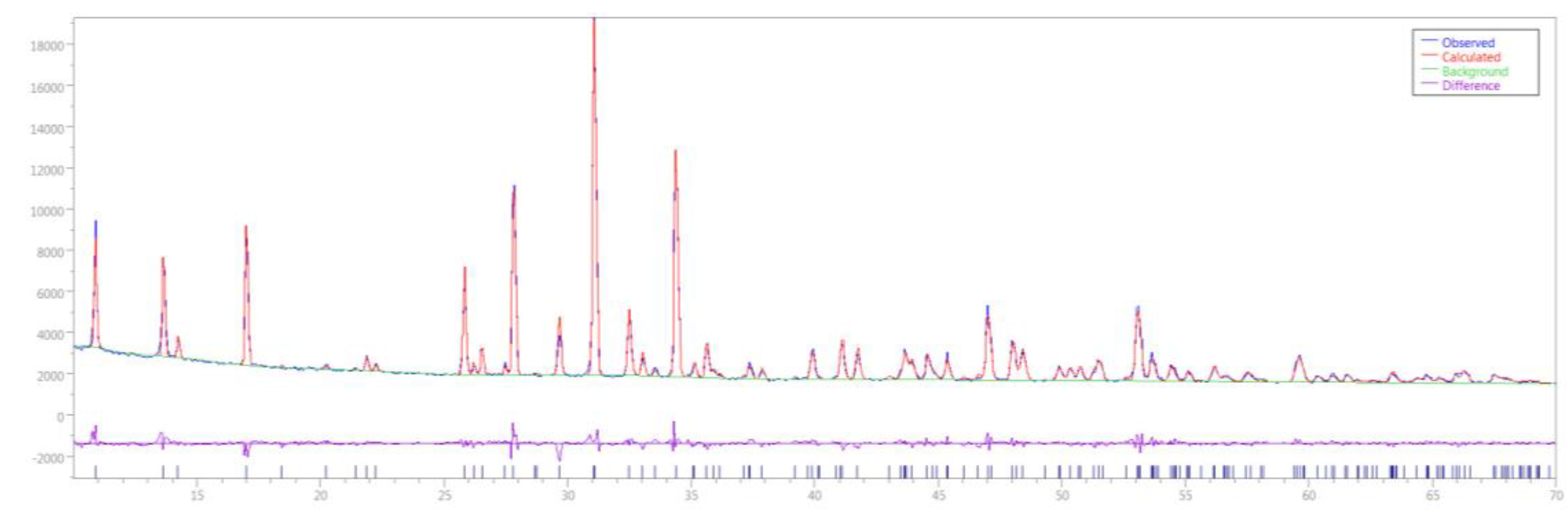
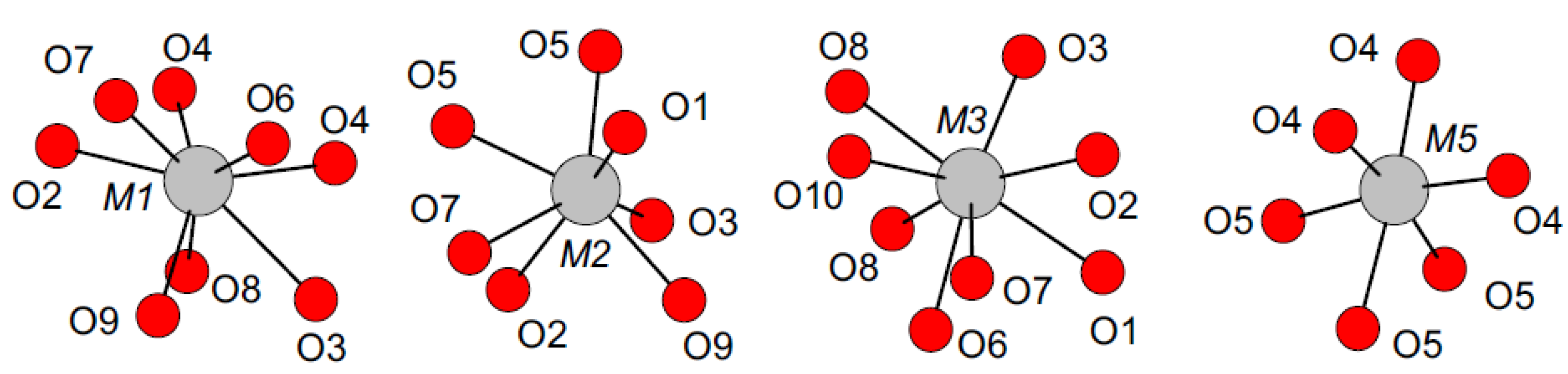
3.2. Rietveld Refinement of Ca9RE(PO4)7 (RE = La, Pr, Nd, Eu, Gd, Dy, Tm, Yb, Lu)
4. Conclusions
5. Availability of EXPO
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Cryst. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Pecharsky, V.K.; Zavalij, P.Y. Fundamentals of Powder Diffraction and Structural Characterization of Materials, 2nd ed.; Springer Science+Business Media: New York, NY, USA, 2009. [Google Scholar]
- Clearfield, A.; Reibenspies, J.; Bhuvanesh, N. Principles and Applications of Powder Diffraction; Wiley: New York, NY, USA, 2008. [Google Scholar]
- Will, G. Powder Diffraction: The Rietveld Method and the Two Stage Method to Determine and Refine Crystal Structures from Powder Diffraction Data; Springer: Berlin, Germany, 2010. [Google Scholar]
- Le Bail, A. The Profile of a Bragg Reflection for Extracting Intensities. In Powder Diffraction Theory and Practice; Dinnebier, R.E., Billinge, S.J.L., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2008; pp. 134–165. [Google Scholar]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.G.G.; Rizzi, R. Powder diffraction: The new automatic least-squares Fourier recycling procedure in EXPO2005. J. Appl. Cryst. 2006, 39, 558–562. [Google Scholar] [CrossRef]
- Cranswick, L.M.D. Computer Software for Powder Diffraction. In Powder Diffraction Theory and Practice; Dinnebier, R.E., Billinge, S.J.L., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2008; pp. 494–570. [Google Scholar]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Cryst. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Caliandro, R.; Giacovazzo, C.; Rizzi, R. Crystal Structure Determination. In Powder Diffraction Theory and Practice; Dinnebier, R.E., Billinge, S.J.L., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2008; pp. 227–265. [Google Scholar]
- Giacovazzo, C. Phasing in Crystallography: A Modern Perspective; IUCr/Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Cryst. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mat. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Moliterni, A.; Rizzi, R. Single Crystal and powder XRD techniques: An overview. In Inorganic Micro-and Nanomaterials. Synthesis and Characterization; Dibenedetto, A., Aresta, M., Eds.; De Gruyter: Berlin, Germany, 2013; pp. 57–91. [Google Scholar]
- Harker, D.; Kasper, J.S. Phases of Fourier coefficients directly from crystal diffraction data. Acta Cryst. 1948, 1, 70–75. [Google Scholar] [CrossRef]
- Sayre, D. The squaring method: A new method for phase determination. Acta Cryst. 1952, 5, 60–65. [Google Scholar] [CrossRef]
- Wilson, A.J.C. Determination of absolute from relative X-ray intensity data. Nature 1942, 150, 152. [Google Scholar] [CrossRef]
- Wilson, A.J.C. The probability distribution of X-ray intensities. Acta Cryst. 1949, 2, 318–321. [Google Scholar] [CrossRef]
- Cascarano, G.; Giacovazzo, C.; Luic, M. Direct methods and structures showing superstructures effects. III. A general mathematical model. Acta Cryst. 1988, A44, 176–183. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Early finding of preferred orientation: A new method. J. Appl. Cryst. 1994, 27, 1045–1050. [Google Scholar] [CrossRef]
- Cascarano, G.; Giacovazzo, C.; Camalli, M.; Spagna, R.; Burla, M.C.; Nunzi, A.; Polidori, G. The method of representations of structure seminvariants. The strengthening of triplet relationships. Acta Cryst. 1984, A40, 278–283. [Google Scholar] [CrossRef]
- Karle, J.; Hauptman, H. A theory of phase determination for the four types of non–centrosymmetric space groups 1P222, 2P22, 3P12, 3P22. Acta. Cryst. 1956, 9, 635–651. [Google Scholar] [CrossRef]
- White, P.S.; Woolfson, M.M. The application of phase relationships to complex structures. VII. Magic integers. Acta Cryst. 1975, A31, 53–56. [Google Scholar] [CrossRef]
- Cascarano, G.; Giacovazzo, C.; Viterbo, D. Figures of merit in direct methods: A new point of view. Acta Cryst. 1987, A43, 22–29. [Google Scholar] [CrossRef]
- Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Improved figures of merit for direct methods. Acta Cryst. 1992, A48, 859–865. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Kamel, G.S.; Moliterni, A.; Rizzi, R. Minimally resolution biased electron-density maps. Acta Cryst. 2008, A64, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R. Correcting resolution bias in electron density maps of organic molecules derived by direct methods from powder data. J. Appl. Cryst. 2008, 41, 592–599. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Maggi, S.; Moliterni, A.; Rizzi, R. Correcting electron-density resolution bias in reciprocal space. Acta Cryst. 2009, A65, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R. The dual-space resolution bias correction algorithm: Application to powder data. J. Appl. Cryst. 2010, 43, 798–804. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R. The dual-space resolution bias correction in EXPO2010. Z. Kristallogr. 2010, 225, 548–551. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R. COVMAP: A new algorithm for structure model optimization in the EXPO package. J. Appl. Cryst. 2012, 45, 789–797. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. The Shift_and_Fix procedure in EXPO: Advances for solving ab initio crystal structures by powder diffraction data. J. Appl. Cryst. 2017, 50, 1812–1820. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Symmetry in the generation of trial structures. Acta Cryst. 1968, A24, 301–311. [Google Scholar] [CrossRef]
- Andreev, Y.G.; Lightfoot, P.; Bruce, P.G. A General Monte Carlo Approach to Structure Solution from Powder-Diffraction Data: Application to Poly(ethylene oxide)3:LiN(SO2CF3)2. J. Appl. Cryst. 1997, 30, 294–305. [Google Scholar] [CrossRef]
- Kariuki, B.M.; Serrano-González, H.; Johnston, R.L.; Harris, K.D.M. The application of a genetic algorithm for solving crystal structures from powder diffraction data. Chem. Phys. Lett. 1997, 280, 189–195. [Google Scholar] [CrossRef]
- Kirkpatrick, S. Optimization by simulated annealing: Quantitative studies. J. Stat. Phys. 1984, 34, 975–986. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Cryst. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Engel, G.E.; Wilke, S.; König, O.; Harris, K.D.M.; Leusen, F.J.J. PowderSolve—A complete package for crystal structure solution from powder diffraction patterns. J. Appl. Cryst. 1999, 32, 1169–1179. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Cryst. 2018, 51, 210–218. [Google Scholar] [CrossRef]
- Favre-Nicolin, V.; Černỳ, R. FOX, ‘free objects for crystallography’: A modular approach to ab initio structure determination from powder diffraction. J. Appl. Cryst. 2002, 35, 734–743. [Google Scholar] [CrossRef]
- Altomare, A.; Corriero, N.; Cuocci, C.; Falcicchio, A.; Moliterni, A.; Rizzi, R. Direct space solution in the EXPO package: The combination of the HBB-BC algorithm with GRASP. J. Appl. Cryst. 2018, 51, 505–513. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Tremayne, M.; Lightfoot, P.; Bruce, P.G. Crystal Structure Determination from Powder diffraction data by Monte Carlo Methods. J. Am. Chem. Soc. 1994, 116, 3543–3547. [Google Scholar] [CrossRef]
- Tremayne, M.; Kariuki, B.M.; Harris, K.D.M.; Shankland, K.; Knigh, K.S. Crystal Structure Solution from Neutron Powder Diffraction Data by a New Monte Carlo Approach Incorporating Restrained Relaxation of the Molecular Geometry. J. Appl. Cryst. 1997, 30, 968–974. [Google Scholar] [CrossRef]
- Erol, O.K.; Eksin, I. A new optimization method: Big Bang-Big Crunch. Adv. Eng. Softw. 2006, 37, 106–111. [Google Scholar] [CrossRef]
- Feo, T.A.; Resende, M.G.C. Greedy randomized adaptive search procedures. J. Glob. Optim. 1995, 6, 109–113. [Google Scholar] [CrossRef]
- Johnston, J.C.; David, W.I.F.; Markvardsen, A.J.; Shankland, K. A hybrid Monte Carlo method for crystal structure determination from powder diffraction data. Acta Cryst. 2002, A58, 441–447. [Google Scholar] [CrossRef]
- Brenner, S.; McCusker, L.B.; Baerlocher, C. Using a structure envelope to facilitate structure solution from powder diffraction data. J. Appl. Cryst. 1997, 30, 1167–1172. [Google Scholar] [CrossRef]
- Wu, J.; Leinenweber, K.; Spence, J.C.H.; O’Keeffe, M. Ab initio phasing of X-ray powder diffraction patterns by charge flipping. Nature Mater. 2006, 5, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Baerlocher, C.; McCusker, L.B.; Palatinus, L. Charge flipping combined with histogram matching to solve complex crystal structures from powder diffraction data. Z. Krystallogr. 2007, 222, 47–53. [Google Scholar] [CrossRef]
- Altomare, A.; Caliandro, R.; Giacovazzo, C.; Moliterni, A.G.G.; Rizzi, R. Solution of organic crystal structures from powder diffraction by combining simulated annealing and direct methods. J. Appl. Cryst. 2003, 36, 230–238. [Google Scholar] [CrossRef]
- Altomare, A.; Caliandro, R.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.G.G.; Rizzi, R.; Platteau, C. Direct methods and simulated annealing: A hybrid approach for powder diffraction data. J. Appl. Cryst. 2008, 41, 56–61. [Google Scholar] [CrossRef]
- Giacovazzo, C.; Altomare, A.; Cuocci, C.; Moliterni, A.G.G.; Rizzi, R. Completion of crystal structure by powder diffraction data: A new method for locating atoms with polyhedral coordination. J. Appl. Cryst. 2002, 35, 422–429. [Google Scholar] [CrossRef]
- Altomare, A.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Rizzi, R. Completion of crystal structures from powder data: The use of the coordination polyhedra. J. Appl. Cryst. 2000, 33, 1305–1310. [Google Scholar] [CrossRef]
- Shankland, K. Global Rietveld refinement. J. Res. Natl. Inst. Stand. Technol. 2004, 109, 143–154. [Google Scholar] [CrossRef] [PubMed]
- McCusker, L.B.; Von Dreele, R.B.; Cox, D.E.; Louër, D.; Scardi, P. Rietveld refinement guidelines. J. Appl. Cryst. 1999, 32, 36–50. [Google Scholar] [CrossRef]
- Dollase, W.A. Correction of intensities for preferred orientation in powder diffractometry: Application of the March model. J. Appl. Cryst. 1986, 19, 267–272. [Google Scholar] [CrossRef]
- Bérar, J.-F.; Baldinozzi, G. Modeling of line-shape asymmetry in powder diffraction. J. Appl. Cryst. 1993, 26, 128–129. [Google Scholar] [CrossRef]
- Dennis, J.E., Jr.; Schnabel, R.B. Nonlinear Least Squares. In Numerical Methods for Unconstrained Optimization and Nonlinear Equations; Society for Industrial and Applied Mathematics (SIAM): Philadelphia, PA, USA, 1996; pp. 218–238. [Google Scholar]
- Young, R.A. Introduction to the Rietveld method. In The Rietveld Method; Young, R.A., Ed.; Oxford University Press: New York, NY, USA, 1996; pp. 32–36. [Google Scholar]
- Müller, P.; Herbst-Irmer, R.; Schneider, T.R.; Sawaya, M.R. Hydrogen atoms. In Crystal Structure Refinement: A Crystallographer’s Guide to SHELXL; Müller, P., Ed.; Oxford University Press: New York, NY, USA, 2006; pp. 29–31. [Google Scholar]
- Mancuso, R.; Veltri, L.; Russo, P.; Grasso, G.; Cuocci, C.; Romeo, R.; Gabriele, B. Palladium-Catalyzed Carbonylative Synthesis of Functionalized Benzimidazopyrimidinones. Synthesis 2018, 50, 267–277. [Google Scholar] [CrossRef]
- ACD/ChemSketch. Available online: http://www.acdlabs.com/resources/freeware/chemsketch/ (accessed on 20 July 2017).
- Stewart, J.J.P. MOPAC2016; Stewart Computational Chemistry: Springs, CO, USA, 2016. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- El Khouri, A.; Elaatmani, M.; Della Ventura, G.; Sodo, A.; Rizzi, R.; Rossi, M.; Capitelli, F. Synthesis, structure refinement and vibrational spectroscopy of new rare-earth tricalcium phosphates Ca9RE(PO4)7 (RE = La, Pr, Nd, Eu, Gd, Dy, Tm, Yb). Ceram. Int. 2017, 43, 15645–15653. [Google Scholar] [CrossRef]
- Capitelli, F.; Rossi, M.; El Khouri, A.; Elaatmani, M.; Corriero, N.; Sodo, A.; Della Ventura, G. Synthesis, structural model and vibrational spectroscopy of lutetium tricalcium phosphate Ca9Lu(PO4)7. J. Rare Earths 2018, in press. [Google Scholar]
- Madhukumar, K.; Varma, H.K.; Komath, M.; Elias, T.S.; Padmanabhan, V.; Nair, M.K. Photoluminescence and thermoluminescence properties of tricalcium phosphate phosphors doped with dysprosium and europium. Bull. Mater. Sci. 2007, 30, 527–534. [Google Scholar] [CrossRef]
- Dorozkhin, S.V. Calcium Orthophosphates bioceramics. Ceram. Int. 2015, 41, 13913–13966. [Google Scholar] [CrossRef]
- Lazoryak, B.I.; Kotov, R.N.; Khasanov, S.S. Crystal structure of Ca19Ce(PO4)14. Russ. J. Inorg. Chem. 1996, 41, 1225–1228. [Google Scholar]
- Ait Benhamou, R.; Bessière, A.; Wallez, G.; Viana, B.; Elaatmani, M.; Daoud, M.; Zegzouti, A. New insight in the structure–luminescence relationships of Ca9Eu(PO4)7. J. Sol. State Chem. 2009, 182, 2319–2325. [Google Scholar] [CrossRef]
- Inorganic Crystal Structure Database (ICSD); Version 2017-2; Fachinformationszentrum: Karlsruhe, Germany, 2017.
- Yashima, M.; Sakai, A.; Kamiyama, T.; Hoshikawa, A. Crystal structure analysis of β-tricalcium phosphate Ca3(PO4)2 by neutron powder diffraction. J. Sol. State Chem. 2003, 175, 272–277. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Formula, formula weight | C17H15N3, 261.32 |
| Temperature (K), λ (Å) | 293, 1.54056 |
| System, space group | Monoclinic, P21/c |
| a, b, c (Å); β (°) | 10.8944(2), 14.5650(2), 9.27713(15), 99.1021(10) |
| V (Å3), Z | 1453.53(2), 4 |
| 2θ range, step (°) | 6–80, 0.02 |
| Nr. of data points | 3701 |
| Nr. of Bragg refl., parameters | 884, 112 |
| Rp (%), Rwp (%), χ2 | 2.015, 2.787, 1.846 |
| Rexp (%), RBragg (%) | 2.051, 3.298 |
| CCDC depository nr. | CCDC1564263 |
| Formula, Formula weight | Ca9Dy(PO4)7, 1187.99 |
| Temperature (K), λ (Å) | 273, 1.54056 |
| System, space group | Rhombohedral, R3c |
| a = b, c (Å) | 10.4250(3), 37.301(2) |
| V (Å3), Z | 3510.8(3), 6 |
| 2θ range, step(°) | 10–70, 0.06 |
| Nr. of data points | 1001 |
| Nr. of Bragg refl., parameters | 174, 86 |
| Rp (%), Rwp (%), χ2 | 2.88, 4.15, 2.06 |
| Rexp (%), RBragg (%) | 2.06, 7.24 |
| ICSD depository nr. | CSD432791 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altomare, A.; Capitelli, F.; Corriero, N.; Cuocci, C.; Falcicchio, A.; Moliterni, A.; Rizzi, R. The Rietveld Refinement in the EXPO Software: A Powerful Tool at the End of the Elaborate Crystal Structure Solution Pathway. Crystals 2018, 8, 203. https://doi.org/10.3390/cryst8050203
Altomare A, Capitelli F, Corriero N, Cuocci C, Falcicchio A, Moliterni A, Rizzi R. The Rietveld Refinement in the EXPO Software: A Powerful Tool at the End of the Elaborate Crystal Structure Solution Pathway. Crystals. 2018; 8(5):203. https://doi.org/10.3390/cryst8050203
Chicago/Turabian StyleAltomare, Angela, Francesco Capitelli, Nicola Corriero, Corrado Cuocci, Aurelia Falcicchio, Anna Moliterni, and Rosanna Rizzi. 2018. "The Rietveld Refinement in the EXPO Software: A Powerful Tool at the End of the Elaborate Crystal Structure Solution Pathway" Crystals 8, no. 5: 203. https://doi.org/10.3390/cryst8050203