The Crystal Orbital Hamilton Population (COHP) Method as a Tool to Visualize and Analyze Chemical Bonding in Intermetallic Compounds



1
Institute of Inorganic Chemistry, RWTH Aachen University, D-52056 Aachen, Germany
2
Jülich-Aachen Research Alliance (JARA-FIT and -HPC), RWTH Aachen University, D-52056 Aachen, Germany
*
Author to whom correspondence should be addressed.
Crystals 2018, 8(5), 225; https://doi.org/10.3390/cryst8050225
Submission received: 25 April 2018
/
Revised: 13 May 2018
/
Accepted: 14 May 2018
/
Published: 18 May 2018
(This article belongs to the Special Issue Compounds with Polar Metallic Bonding)
Abstract
:Recognizing the bonding situations in chemical compounds is of fundamental interest for materials design because this very knowledge allows us to understand the sheer existence of a material and the structural arrangement of its constituting atoms. Since its definition 25 years ago, the Crystal Orbital Hamilton Population (COHP) method has been established as an efficient and reliable tool to extract the chemical-bonding information based on electronic-structure calculations of various quantum-chemical types. In this review, we present a brief introduction into the theoretical background of the COHP method and illustrate the latter by diverse applications, in particular by looking at representatives of the class of (polar) intermetallic compounds, usually considered as “black sheep” in the light of valence-electron counting schemes.

1. Introduction
The search for the inmost force which binds the world [1] and its constituents has generated enormous attention among scientists, and early theoretical research on the origin of this force identified a chemical power making atoms combine by so-called “valence bonds” [2]. The introduction of this very idea of valence bonds which arise from the atoms’ valence electrons facilitated the developments of certain concepts, e.g., the octet rule [3], as fundamental relationships needed to rationalize the structural arrangements and the electronic structures of diverse molecules. For the case of solid-state materials, in particular ionic salts, first explorations to establish relationships between the structural arrangements and the electron counts of such solids employed empirical data and resulted in various solid-state rules which, for instance, were based on the ratios of ionic radii, the “strengths” of the electrostatic bonds, and the connectivities between diverse coordination polyhedra [4]. Further research on the distributions of the valence electrons in intermetallic compounds revealed additional notions, e.g., those first proposed by Zintl [5,6,7,8,9,10] and Hume-Rothery [11,12,13], respectively, to somehow correlate structural arrangements and the atoms’ electronic nature. Even today, however, the existence of intermetallic compounds for which the electronic structures and, furthermore, the nature of bonding cannot be trivially categorized by applying one of the aforementioned concepts [14,15] underlines the need for different means in order to reveal the bonding nature in such materials.
To determine an unbiased picture of the nature of bonding in a given solid-state material, it seems necessary to extract the chemical bonding information from the electronic band structure that is computed for the respective material by means of a quantum-mechanical technique. For this purpose, the total energy of a many-particle system, i.e., the sum of the potential and kinetic energy, needs to be calculated solving the time-independent Schrödinger equation [16]
with H as the Hamiltonian operator, at least in principle. The wave function Ψ itself is then given by a properly symmetrized product of one-electron functions, so-called orbitals ψ. In the case of solid-state materials, the one-electron functions ψ are then constructed following Bloch´s theorem to incorporate the most important crystallographic symmetry: translation [17]. During the past nine decades, diverse techniques have been developed to approach the energies in order to construct the electronic band structure and densities-of-states of a given many-particle system [18]. The most successful trick to avoid calculating the many-electron wave function Ψ, however, is given by the framework of density-functional theory [19,20,21], the workhorse of numerous of today´s quantum-chemical computer programs. Here, the electron density, ρ(r), plays a major role in generating an effective potential Veff(r) for non-existent pseudo-electrons plus some exchange-correlation correction term. In the end, one needs to solve the single-particle Kohn–Sham equations
and deals with one-electron pseudo-functions to exactly cover the kinetic energy which is the largest contributor. One approach to eventually obtain the bonding information from the results of the electronic band structure calculations (regardless of wave-function or density-based) is the crystal orbital Hamilton population (COHP) method [22,23]. In the framework of the COHP technique, bonding, non-bonding, and antibonding interactions are identified for pairs of atoms (or orbitals) in a given solid-state material. In this review, we will present prototypical applications of the COHP procedure to solid-state materials. The outcome of this survey will demonstrate the efficiency of the method to reveal the actual nature of bonding and, furthermore, the causes for structural preferences in solids.
HΨ = EΨ
2. The COHP Method—An Introduction
Before demonstrating the applications of the COHP method for intermetallic compounds, we will provide a brief introduction into that technique. To obtain the energy of a many-particle system by solving the aforementioned Schrödinger or Kohn–Sham equations, one first needs to establish proper one-electron wave functions (orbitals). In the case of molecules consisting of a couple of atoms, these molecular orbitals are given by a linear combination of atomic orbitals (LCAO)
composed of basis functions ϕμ(r), i.e., the atomic orbitals of the atoms A, and the mixing coefficients cμi. To obtain the energies of molecular systems, the Hamiltonian acts on the respective wave functions—but what is the best wave function among all possible functions? It is the one corresponding to the lowest total energy attainable by the variational principle [24]. Hence, the energy is differentiated with respect to the coefficients cμi ( leading to the secular determinant
with and representing the on-site and off-site entries of the Hamiltonian and overlap matrices. In particular, Hμμ (μ = ν) contain the on-site or Coloumb integrals, Hμν (μ ≠ ν) are the off-site, hopping, interaction or resonance integrals, and Sμν represents the overlap integrals. Note that we use the Dirac bracket notation, in which ϕμ represent the conjugate complex wave functions, because ϕ can (and will be) a complex function including an imaginary part.
While the aforementioned procedure allows determining the energies of a given molecular system, at this point, one may wonder how the assignments of electrons to particular orbitals thereby providing information about the electron distribution can be accomplished. The numbers of electrons residing in the entire molecule and its molecular orbitals is given by
with fi as the occupation numbers, which can be 0, 1, or 2. The first term of the Equation (5) represents the net populations, while the second one contains the overlap populations. Furthermore, the sum of both terms shown in the Equation (6) comprises the gross populations, following an early suggestion of Mulliken.
While the LCAO ansatz is used to solve Schrödinger’s equation for molecules, the wave functions employed to solve Schrödinger’s or the Kohn–Sham equations for solid-state materials are constructed based on Bloch´s theorem [17]
with T as some lattice vector commutating with the Hamiltonian, ψ(k, r) as the crystal orbital at a specific site r, and k as a new quantum number from reciprocal space. Without doubt, this is the most important theorem of theoretical solid-state science. The requirement of translational symmetry for constructing the wave functions fulfilling Bloch´s theorem also demands that the crystal structure of a given material does not comprise any atomic sites showing positional of occupational disorders. Because the crystal structures of certain intermetallic compounds do comprise positionally or occupationally disordered sites (see Section 3.2 and Section 3.3), it is necessary to examine the electronic structures of models approximating the actual crystal structures of such materials. To obtain numbers of electrons occupying the crystal orbitals in a given solid-state material, the k-dependence of mixing coefficients cμi and cνi needs to be taken into consideration, leading to the k-dependent density matrix
Inserting Equation (8) in the Equations (5) and (6), respectively, and taking into account results in
with Re[PμνSμν] as the real parts of the (possibly) complex off-diagonal entries. To get hold of the numbers of electrons N being dependent of the (band) energy, it is convenient to introduce the density-of-states matrix
which is obtained from a differentiation of Pμν with respect to E
Combining the Equations (9) and (10) with Equation (11) yields
The (second) off-diagonal contribution of the gross population in Equation (14), i.e., Re[Pμν(E)Sμν], comprises the overlap-population-weighted densities-of-states and, hence, provides the essential information regarding the distributions of the electrons between the atoms in any given solid-state material. This very technique to extract the bonding information based on the overlap-population-weighted densities-of-states has been dubbed as the crystal orbital overlap population (COOP) method [25]. In the framework of the COOP method, bonding and antibonding interactions are represented by positive and negative COOP values, respectively, while nonbonding interactions are indicated by zero COOPs. The COOP method has been largely employed for electronic-structure computations based on the semiempirical extended Hückel theory (EHT), which is principally equivalent to the empirical tight-binding approach including overlap [26]. In the EHT case, only the valence orbital are taken into account. In addition, certain entries of the secular determinant Equation (4), in particular, those of the interaction (Hμν) integrals, are parameterized; to do so, the interaction matrix elements are determined solving the Wolfsberg–Helmholz formula [27] that employs Coulomb matrix elements (Hμμ) evolved from experimentally determined ionization potentials, while the overlap matrix elements are calculated utilizing Slater-type orbitals.
As denoted in the previous paragraph, theoretically determining the ground state of a given solid-state system can be carried out by different approaches, and the computational challenges arising from the notorious electron–electron interactions can be so tremendous that they easily overcome today´s (and also tomorrow’s) available computational resources [18]. To nonetheless come to numerically powerful solutions of this problem, it has turned out extraordinarily convenient to concentrate on the electron density which arises from effective one-electron wave functions (orbitals) ψi(r) in the framework of density-functional-theory [19,20,21] (DFT), leading to
Using DFT jargon, the total energy E is a functional of the electron density because the latter depends on the spatial coordinates r (i.e., a functional is a function which also depends on a function). To make DFT succeed, one sets up a pseudo-system of non-interacting electrons of the same density and adds a correction term for exchange and correlation. So, the effective potential Veff(r) employed in DFT computations (see Equation (2)) is the sum of the external potential Vext(r) (= the Coulomb potential from the nuclei), the so-called Hartree potential of the electrons VHartree(r), and the exchange-correlation potential VXC(r), a correction term:
Trivially, the accuracy of any DFT calculation mostly depends on the quality of VXC(r). As basis sets, solid-state people typically use plane waves and related functions fulfilling Bloch´s theorem Equation (7); traditionally, methods based on cellular (augmentation) techniques to separate outer and inner parts of the individual atoms were used early on [18]. Particularly, these techniques are based on the concept to first solve Schrödinger´s equation for a single atom within one cell and, subsequently, to glue the energy-dependent atomic functions (that is, the numerically derived atomic orbitals) together with those located on neighboring atoms. In this connection, it is convenient to define muffin-tin spheres being boundaries between the potentials in the regions close to the nuclei and the (zero) potentials in the interstitial regions. Accordingly, two-region potentials are obtained, and the wave functions which are constructed following the augmented-plane wave method [28] can be depicted as hybrid functions composed of atomic functions inside the muffin-tin spheres and single plane waves outside these spheres; however, the computations are slow because of the energy-dependence of the augmented plane waves, hence no simple diagonalization. To drastically enhance the speed of the computations, Taylor series of the energy for the radial parts of the functions within the spheres were developed and then truncated after the second term [29]. This approach (and its modifications) to compute the electronic structures in solid-state materials has been implemented in diverse quantum-mechanical techniques dubbed as linear methods including the (Full-potential) Linearized Augmented Plane-Wave, (F)LAPW, and Linearized Muffin-Tin Orbital, LMTO, methods. More recently, a related approach named Projector-Augmented Wave (PAW) method [30] has been introduced. In this (all-electron) technique, pseudopotentials (also dubbed effective core potentials) replace the ion-electron potentials and are optimized during the computations utilizing full wave functions. At this stage, one may wonder how the nature of covalent (hence, localized) bonding in solid-state materials may be interpreted based on the results of density-functional-theory-based computations, in particular for the reason that the use of essentially delocalized plane waves hinders the extractions of information regarding the bonding from the calculations [18]. We will come back to this point shortly.
Because extracting the bonding information from the electronic-structure computations strongly depends on the types of employed orbitals in the case of the aforementioned extended-Hückel based COOP method, applying this procedure to the results of various DFT computations with whatever kind of bases sets is rather problematic [22]. To nonetheless gain the bonding information from DFT, it is more appropriate to partition the band-structure energy leading to the Hamilton-matrix-weighted densities-of-states as shown in the following [22].
Under consideration of the secular determinant Equation (4), which can be transformed to
it is plausible that the overlap integrals used to define the overlap-population-weighted densities-of-states in the Equation (14) can be replaced by the Hamilton matrix elements; however, it should be noted that a straightforward transformation which might be implied by the relation in the Equation (18) cannot be made. Nonetheless, using an analogy to the aforementioned overlap-population-weighted densities-of-states, the densities-of-states matrix Equation (11) is weighted by the Hamilton matrix elements resulting in the Crystal Orbital Hamilton Populations (COHP), according to
Because the off-site COHP terms (A ≠ B) originate from the interactions between the pairs of atoms in a given solid-state structure, these contributions are generally taken into consideration for the bonding analyses. More recently, a generalized variant of the COHP dubbed Density-of-Energy (DOE) function has been introduced [31]. In the DOE function, the (interatomic) off-site as well as the (atomic) on-site contributions are included such that the entire band energy can be obtained from the energy integral of the DOE function
In contrast to the COOP method, bonding and antibonding interactions are represented by negative and positive values of the COHP, respectively, while non-bonding interactions are indicated by zero values of the COHP. Somewhat simplified, a positive COOP denotes bonding, just like a negative COHP; in case one plots −COHP data, the shape of the two functions look similar, with bonding spikes to the right. Furthermore, a direct comparison between the COHP values of compounds with dissimilar compositions cannot be made, because the average electrostatic potential in each density-functional-theory-based computation is set to an arbitrary ‘zero’ energy, whose relative position can vary from system to system [18]. To identify the differences between the bonding situations in compounds with dissimilar compositions, the cumulative ICOHP/cell, i.e., the sum of the ICOHP/bond values of the nearest neighboring interactions weighted by the respective bond frequencies, are typically projected as the percentage contributions to the net bonding capacities (examples of this procedure have been provided in the Section 3).
The COHP technique has been first implemented in programs employing LMTO-based methods (see above) to compute the electronic structures of solid-state materials [22]. In a sense, LMTO theory introduced first-principles techniques to solid-state chemistry. This is because the COHP approach requires the use of crystal orbitals derived from local basis sets, the LMTO success ingredient. The more popular and essentially delocalized plane-wave-based computations of today, however, are blind for chemical-bonding analysis, so one first needs to reconstruct both Hamilton and overlap matrix elements using auxiliary atomic orbitals. In other words, such information may be projected from plane waves by means of the projected Crystal Orbital Hamilton Population (pCOHP) technique, a modern descendant of the COHP method [23]. The pCOHP technique has been implemented in the Local Orbital Basis Suite Towards Electronic-Structure Reconstruction (LOBSTER) program being available at www.cohp.de free of charge and compatible with a growing list of quantum-mechanical programs (VASP [32,33,34,35,36], ABINIT [37,38,39,40], Quantum ESPRESSO [41,42]) to extract the information regarding the nature of bonding from the electronic-structure computations [22,23,43,44].
After this brief introduction into the theoretical background of the COHP method, we will now present some applications for the examples of diverse intermetallic compounds to show the strengths of this technique in revealing the bonding situations in solid-state materials (Figure 1).
3. Applications of the COHP Method to Intermetallic Compounds
3.1. Rare-Earth Transition-Metal Halides as Anti-Werner-Fashioned Complexes
Explorative research on the phase diagrams for the oxides and halides of the vanadium- to manganese-group elements revealed the existence of so-called “reduced” transition-metal halides and oxides, whose crystal structures comprise transition-metal building units of polyhedral forms dubbed as metal atom clusters [46,47,48]. In particular, the excess valence electrons of the transition metals, which do not participate in transition-metal−halide bonding, are distributed in transition-metal–transition-metal bonds formed by the d-orbitals of the metal atoms within the clusters [49]. Additional research on the phase diagrams for the halides of the scandium- to titanium-group elements being exceptionally poor in electron density identified halides composed of transition-metal clusters enclosing endohedral atoms, so-called ‘interstitials’ [48,50,51,52,53,54,55]. Furthermore, these rare-earth metal clusters encapsulating the interstitials have been observed to be surrounded solely by halide ligands (such types of clusters are typically classified as ‘isolated’), or to share common vertices, edges and faces to assemble oligomers, chains, and sheets (Figure 1) [48,50,51,52,53,54,55]. As an outcome, these compounds comprise oxidized networks of metals surrounded by monoatomic ligands such that this group of materials has been assigned to the class of the polar intermetallic compounds [15].
To date, a broad variety of elements including transition-metal as well as main-group elements has been observed to be incorporated in the centers of the rare-earth clusters [54,55]. At that point, one may wonder how the electronic structure and the bonding situation typically expected for a transition-metal atom cluster is affected by the incorporation of an endohedral atom. In fact, the orbitals of the interstitials combine with the transition-metal cluster-based orbitals such that interstitial–transition-metal bonds evolve [49]. While the electronic structures and bonding motifs of isolated transition-metal clusters enclosing endohedral atoms can be described based on a molecular orbital ansatz [49], yet, the bonding situations of interstitially centered transition-metal cluster oligomers, chains, and sheets showing more metal–metal condensation and bonding need to be examined based on the electronic band structures of these materials. In the following, we will provide a survey of analyses employing the COHP method to determine the bonding motifs for the examples of the most prolific representatives of the transition-metal (T) centered rare-earth (R) cluster halides (X).
A large number of halides which are composed of isolated rare-earth clusters encapsulating transition-metal atoms has been identified to adopt the [TR6]X10 and [TR6]X12R types of structure (note that the brackets define the components of the clusters) [56,57,58]. In particular, the endohedral transition-metal atoms which can be an element from the manganese- to zinc-groups for the [TR6]X12R-type and from the iron- to nickel-groups for the [TR6]X10-type are coordinated by octahedra constituted by the rare-earth atoms, while all edges of the octahedral [TR6] clusters are capped by halide ligands. The electronic structures and bonding situations in the isolated [TR6] clusters may be depicted by the aforementioned molecular orbital (MO) theory-based approach, for which the interstitial-based valence orbitals, i.e., the d-orbitals in the forms of t2g and eg sets, are combined with the fragment orbitals constituted by the rare-earth metal d-orbitals in the skeletons of the clusters (Figure 2) [56,57,59,60]. The results of the MO-theory-based computations on isolated empty and filled transition-metal clusters helped to substantiate electron-counting schemes which address the numbers of cluster-based electrons (CBEs) being available for the metal−metal bonds within the clusters. More specifically, the totals of the CBEs are usually obtained by subtracting the numbers of halide ligands from the sums of transition-metal valence electrons. While closed-shell configurations are achieved typically for counts of 16 CBEs in empty octahedral transition-metal clusters whose edges are capped by halide ligands [61], the outcome of the MO-theory-based examinations for the transition-metal-centered rare-earth clusters revealed that closed-shell configurations are accomplished for 18 CBEs (Figure 2) [56,57]. For instance, a closed-shell configuration is expected for the previously identified [CoY6]I12Y, for which an application of the electron-counting scheme yields a total of 18 CBEs (=9 + 7∙3 − 12); however, the observed ranges of transition-metals being incorporated in the rare-earth clusters indicate certain electronic flexibilities. Chemical-bonding analysis based on the COHP curves of a number of [TR6]X10-type compounds demonstrated that the bond energy is optimized for a halide with a nickel-group interstitial in agreement with the outcome of the previous MO-theory-based calculations [58]. Furthermore, the bonding within the octahedral [TR6] clusters was shown to be dominated by the heteroatomic T−R interactions besides much lesser R−R bonding [58].
Among the group of rare-earth transition-metal halides composed of cluster oligomers, the crystal structures of a large number of compounds have been identified to contain one particular type of tetramer, [T4R16] (Figure 1). In particular, this sort of tetramer has been observed for the crystal structures of four different types of rare-earth transition-metal halides, that are, the [T4R16]X28[R4] [62], [T4R16]X20 [63], [T4R16]X24[RX3]4 [64], and [T4R16]X24 [64] types of structure. The tetramers consist of pairs of dimers, i.e., two transition-metal centered octahedral rare-earth clusters sharing one common edge, which are positioned perpendicular to each other and condensed via four joint edges. The [T4R16] units are enclosed by 36 halide ligands, and their R16 fragments can also be depicted as all-vertices-truncated supertetrahedra or Friauf-polyhedra. An examination [65] of the hitherto identified rare-earth transition-metal halides whose crystal structures comprise such tetramers suggested that the maximum numbers of CBEs for intracluster bonding are attained for totals of 15 electrons per transition-metal centered rare-earth octahedron. Examinations [66] of the electronic structures for diverse halides composed of these tetramers bared that the Fermi levels fall into gaps corresponding to closed-shell configurations in halides for which CBE counts of 15 electrons per [TR6] cluster are achieved. An additional bonding analysis [66] based on the COHP curves and their integrated values (Table 1) indicates that the majority of the bonding interactions reside between the heteroatomic R−T as well as R−X separations, whereas the homoatomic R−R and T−T interactions play minor, but evident roles.
The group of rare-earth transition-metal halides with the net formula [TR3]X3 has emerged as a prolific class of compounds comprising cluster chains. To date, five different structure types have been identified for this group: the cubic [PtPr3]I3-type [67], the tetragonal [NiLa3]Br3-type [68], the orthorhombic [RuPr3]Cl3-type [69], and two independent monoclinic types of structure, i.e., the [RuPr3]I3 and [IrY3]I3 types of structure [70] (Figure 1). In particular, the monoclinic [RuPr3]I3-type has been observed solely for bromides and iodides which comprise early lanthanide clusters (R = La−Pr) containing iron-group elements, while the cubic [PtPr3]I3-type has been identified for early lanthanide (R = La−Pr) cluster bromides and iodides containing elements from the iron- to copper-groups as endohedral atoms [71]. In the crystal structure of the monoclinic [RuPr3]I3-type, the octahedral [TR6] clusters share four common edges with neighboring clusters forming bioctahedral chains, whereas the [TR6] octahedra in the cubic [PtPr3]I3-type structure condense via three common edges with other [TR6] clusters to helical chains. The existence of the cubic [PtPr3]I3 and monoclinic [RuPr3]I3 types of structures for systems with iron-group elements as endohedral atoms suggests that there is a competition to adopt the respective type of structure. An examination [72] of the electronic band structures for two isocompositional bromides of both [TR3]X3-types reveals that the Fermi level of the cubic compound falls in a maximum of the densities-of-states (DOS), while the Fermi level in the monoclinic representative resides in a gap. Because the location of the Fermi level at a peak of the DOS curves typically indicates an electronically unfavorable situation, the monoclinic [RuPr3]I3-type structure should be preferred. Why, then, do we observe a competition between the monoclinic [RuPr3]I3-type and the cubic [PtPr3]I3-type for early lanthanide cluster bromides and iodides comprising iron-group elements as interstitials?
A chemical bonding analysis based on the COHP curves (Figure 2) and their integrated values (ICOHP; Table 1) for both compounds reveals that the heteroatomic R−T and R−X interactions show the largest percentages to the net bonding capabilities of the respective halides. A topological inspection of the respective local atomic environments indicates that the monoclinic representative contains less ‘polar’ heteroatomic contributions, which are maximized in the cubic structure. Accordingly, the interplay between the attempts to accomplish an electronically favorable situation and to optimize overall bonding appears to regulate the structural preferences between the cubic and monoclinic structures. Further research on the electronic structure of a cubic [PtPr3]I3-type bromide containing a cobalt-group element as endohedral atom bared that the Fermi level falls in a pseudogap, indicating an electronically favorable situation. Notably, the structures of the [RuPr3]Cl3 and [IrY3]I3 types, which are observed for the chlorides of the light lanthanides and the bromides as well as iodides of the heavier lanthanides, respectively [71,74,75], can be derived from that of the monoclinic [RuPr3]I3-type through displacements of the metal chains such that double chains of transition-metal centered rare-earth unicapped trigonal prims evolve. An examination [73] of the COHP curves and ICOHP values for representatives of the [RuPr3]Cl3 and [IrY3]I3 types of structure (Table 1) brings to light that significant bonding interactions are evident for the T−T contacts within the cluster chains showing noticeable contributions to the net bonding capabilities; yet such percentages to net bonding capabilities are not identified for the large T−T separations in the cluster chains of the cubic [PtPr3]I3 and monoclinic [RuPr3]I3 types.
In summary, the outcome of the bonding analyses employing the COHP method for the examples of the most prolific representatives of the transition-metal centered rare-earth cluster halides indicates the dominant role of the heteroatomic R−T and R−X interactions in these materials. Under consideration of the topologies and polarities in Werner-type complexes, the results of the bonding analyses imply that the transition-metal centered rare-earth clusters can also be described as anti-Werner complexes, in which the central atoms have higher electronegativities than the surrounding ligands [54].
3.2. The Bonding Situations in Electron-Poorer Polar Intermetallics Containing Gold
As signaled in the previous section for the examples of the isolated transition-metal cluster halides, there have been different approaches to develop effective electron counting rules, which were considered to help recognizing bonds in solid state materials [76]. For instance, Wade´s rules [77,78] or the Zintl concept [5,6,7,8,9,10] are prominent electron-counting schemes being typically applied to recognize the valence bonds in solid-state materials with polyanionic clusters and monoatomic counterions. More recent research on the components of the active-metal (main-groups I, II, and the scandium-group elements)−gold−post-transition-metal systems identified several materials composed of polyanionic clusters possessing fewer valence electrons relative to those in Zintl phases [15]. In fact, the valence-electron concentrations of these materials called electron-poorer intermetallics place them close to the Hume–Rothery phases [11,12,13]. (Figure 3; a list of e/a ratios is provided in Table 2) [79] The propensity of gold to be incorporated in these polyanionic clusters and to contribute in hetero- as well as homoatomic bonding is expected to stem from the impact of relativistic effects [80,81,82,83] leading to a tighter binding of the 6s orbitals to the Au atom, a less tight binding of the 5d orbitals, and, ultimately, more 6s−5d orbital mixing. In a lack of electron counting rules, which help to recognize the bonds in these materials, the information of the locations and types of bonds in the electron-poorer intermetallics need to be gained from the computed electronic band structures. In this section, we present a number of examples, in which the locations and types of bonds were revealed with the aid of the COHP method.
Based on the topologies of the polyanionic clusters, each of the ternary compounds composed of an active-metal, gold, and a post-transition-element can be assigned to one of the three following groups: (I) compounds composed of one-dimensional polyanionic tunnels; (II) intermetallics constructed of hexagonal-diamond-fashioned polyanionic networks; and (III) compounds comprising polyanionic clusters in the forms of diverse (types of) polyhedra. Because the crystal structures of certain electron-poorer intermetallics presented in this survey possess atomic positions exhibiting occupational and/or positional disorders, the electronic band-structure calculations and bonding analyses were accomplished based on hypothetical models that approximate the actual crystal structures and usually show the lowest total energies. To develop a starting model for materials with disordered atomic sites, it is convenient to screen diverse feasible models for the scheme with the lowest total energy, because the structure model that shows the lowest total energy (and electronic as well as dynamic stability) among diverse possible models is considered to be the most preferable to approximate the experimentally determined model [85,86,87]. Detailed information regarding the structure determinations, the crystal structures, and generating the hypothetical models employed for the electronic band structure computations may be extracted from the respective references.
The crystal structures of the polar intermetallic compounds belonging to the first of the three categories are composed of cages which are assembled by the gold and post-transition-metal atoms and condensed along a particular crystallographic direction to yield one-dimensional tunnels. These cages enclose the active-metals that are stringed along, or slightly displaced from, the axes running through the one-dimensional tunnels parallel to the given crystallographic paths such that the cages and the encased active-metals may show certain shifts from perfect linear chains for some of the representatives. For instance, the crystal structure of Sr3Au8Sn3 [101] contains one-dimensional tunnels of pentagonal and hexagonal prisms that are assembled by the gold and tin atoms and surround the strontium atoms (Figure 4). A chemical-bonding analysis based on the COHP curves for this material indicates that a structural transformation from a high-temperature to a low-temperature polymorph of this compound is influenced by the trend to optimize the Au−Au and Au−Sn bonding interactions. The gold-rich K1.8Au6In4 [102] is another example of a polar intermetallic compound composed of one-dimensional tunnels which are constructed by the gold and post-transition-metal atoms (Figure 4). An examination based on the ICOHP values for this compound denoted considerable Au−In bonding interactions besides weaker, but evident, homoatomic interactions. Notably, a survey of the average ICOHP/bond values and their percentages to the net bonding capabilities for the Au/post-transition-metal−Au/post-transition-metal interactions in polar intermetallics with one-dimensional polyanionic tunnels in their crystal structures brings to light that the largest ICOHP/bond values and percentage contributions typically stem for the heteroatomic contacts (Table 2).
The second of the three aforementioned classes of polar intermetallic compounds contains those materials composed of hexagonal diamond-like gold networks, which have so far been identified to be present in the crystal structures of four different types of polar intermetallics with diverse combinations of post-transition-elements (Zn, Cd, Al, Ga, In, or Sn) and active-metals (Sr, Ba, Eu) [93,94,103,104,105,106,107]. The cavities within the hexagonal-diamond-like gold networks encompass the active-metal atoms, or triangles assembled by extra gold and post-transition-metal atoms (Figure 5). The COHP curves and their respective integrated values have been examined for diverse types of polar intermetallics containing hexagonal-diamond-type gold networks (see Table 2 for reported ICOHP/bond values and the respective percentages). For instance, an investigation [107] of the COHP curves for BaAu5Ga2 and BaAu4Ga3 bared that the broad majority of the bonding interactions resides between the Au−Au and Au−Ga interactions. In that connection, it is remarkable that the contributions of the homoatomic and heteroatomic contacts within the hexagonal-diamond-type networks and triangles are comparable for some of these compounds, while the largest percentages to the total bonding capabilities in the electron-poorer intermetallics often stem from the heteroatomic gold−post-transition-metal interactions (see Table 2).
The third class of three aforementioned groups of electron-poorer polar intermetallics contains those compounds whose crystal structures feature polyhedral clusters of the anionic components surrounding the active-metal and/or extra gold or post-transition-metal atoms. In particular, these compounds cannot be assigned to one of the other groups based on the spatial arrangements of the polyanionic clusters in the crystal structures of these polar intermetallics. Because of the complexity of the crystal structures showing defects such as disorders of vacancies, some components of this group can also be assigned to the broad family of complex metallic alloys [108,109]. For instance, the crystal structures of EuAu6.1Al5.9 and EuAu6.2Ga5.8 are both derived from the NaZn13-type and include icosahedra, tetrahedral stars, and europium-centered snub cubes formed by the gold and post-transition-metal atoms (Figure 5) [99]. An examination of the COHP curves and ICOHP values for models approximating the real crystal structures of these intermetallics demonstrates that the driving force stabilizing these materials stems from the maximization of the amounts of the heteroatomic Au−post-transition-metal contacts. More recent research on the sodium−gold−gallium system identified an icosahedral (Bergman-type) quasicrystal, i.e., Na13Au12Ga15, for which a COHP bonding analysis revealed extensive Au/post-transition-metal−Au/post-transition-metal bonding and, also, contributions from sodium in delocalized metal−metal bonding [84]. Additional investigations of the sodium−gold−gallium system resulted in the discoveries of a series of intermetallics, i.e., Na8Au10.1Ga6.9, NaAu4Ga2, and Na5Au10Ga16, composed of diverse sorts of polyhedra that are constituted by the gold and gallium atoms and enclose the sodium atoms [91,96]. The chemical bonding analyses were accomplished by means of the COHP method for these intermetallic compounds and exhibited extensive gold/post-transition-metal−gold/post-transition-metal bonding for these materials. CaAu4.1Bi0.9 is an example of a polar intermetallic compound, in which the largest percentages of the cumulative ICOHP per cell to the total bonding capabilities originate from the Au−Au interactions [98]. The crystal structure of the bismuth-containing intermetallic is derived from the MgCu2-type and constructed of networks of vertices-sharing gold tetrahedra enclosing the calcium and bismuth atoms. Notably, the structural motif of vertices-sharing gold tetrahedra has also been encountered for other types of polar intermetallics as, e.g., K12Au21Sn4 [95,110] Furthermore, gold clusters have been identified for the crystal structures of R3Au7Sn3 (R = Y, Gd) and Y3Au9Sb, in which the gold atoms assemble trigonal prisms and antiprisms enclosing extra gold and post-transition-metal atoms, respectively [97,100]. In the antimony-containing compound, the largest percentage contributions of the cumulative ICOHP per cell to the net bonding capabilities arise from the Au−Au interactions, while the largest shares of the cumulative ICOHP per cell to the total bonding capabilities in the tin-containing compounds stem from the Au−Sn separations (Table 2).
In summary, the COHP method has been demonstrated to be a beneficial means for identifying the bonding situations in electron-poorer, polar intermetallic compounds composed of gold. Under consideration of the outcome of the bonding analyses reported for these materials to date, the largest ICOHP per cell are often observed for the heteroatomic gold−post-transition-metal contacts showing the largest percentages to the net bonding capabilities in these compounds.
3.3. The Role of Vacancies and Structural Preferences in Phase-Change Materials
The group of phase-change materials comprises those “intermetallic” compounds which can reversibly transform from amorphous to crystalline phase after irradiation with laser light [111,112,113,114]. Because the crystalline and amorphous phases of phase-change materials significantly differ in both reflectivity and resistance, these compounds can be utilized as rewriteable data-storage materials [111,112,113]. In particular, an application of a long pulse of a low-intensity laser beam heats the amorphous regions of the materials leading to recrystallizations (set pulse), while an application of a short pulse of a high-intensity laser beam locally melts the crystalline material forming amorphous regions after fast quenching (reset pulse) [111]. After having identified fast recrystallization and good optical contrast for GeTe and Ge11Te60Sn4Au25 [115,116], diverse phase-change materials composed of different combinations of a tetrel element, a pnictogen, and tellurium have been discovered to date [117]. The quest for previously unknown materials suited for phase-change memory applications stimulated the impetus to identify the origins of the reversible phase transitions typically observed for phase-change materials. Because the measurements of the dielectric functions for phase-change materials revealed that the optical dielectric constants are evidently higher for crystalline than for the amorphous phases, it was inferred that the sorts of bonding significantly change between the two phases [118]. The subsequent developments of materials´ maps which were based on the ionicities and hybridizations determined for the bonds in diverse compounds indicated that the phase-change materials are evident solely for small segments in these maps [117,119]. Additional examinations on the nature of bonding for the local structural arrangements in the amorphous phase of the phase-change material GeTe revealed that the tetrahedral fragments in the crystal structure are stabilized by the homoatomic Ge−Ge bonds [120]. In this section, we will present two applications of the COHP method to disclose the structural preferences for phase-change materials.
A prototypical representative of the group of phase-change materials is the ternary Ge2Sb2Te5, which crystallizes with a rocksalt-type of structure in its metastable state [121]. Notably, an exanimation of that crystal structure for Ge2Sb2Te5 revealed that vacancies of about 20 at.-% are evident for the occupationally disordered Ge/Sb sites [122]. To understand the origin of the presence of vacancies for the mixed Ge/Sb sites in the rocksalt-like structure of Ge2Sb2Te5, the bonding situations were examined based on the COHP curves of three different compositions, i.e., Ge2Sb2Te4, Ge1.5Sb2Te4, and GeSb2Te4 [121] A comparison of the COHP curves for the three different compositions (Figure 6) reveals that significantly antibonding Ge−Te and Sb−Te interactions are evident at the Fermi level in the germanium-richest telluride, while less and no antibonding Ge−Te and Sb−Te states are present at the Fermi levels in the germanium-poorer Ge1.5Sb2Te4 and GeSb2Te4, respectively. Accordingly, it can be inferred that the presence of vacancies in the structure of the ternary phase-change materials corresponds to a reduction of the valence-electron concentration in order to optimize the overall bonding. In other words, nature gets rid of the antibonding states by expelling some of the electron-donating Ge atoms.
Recent research in the field of phase-change data storage materials has focused on the developments of materials suited for applications in storage-class memories beyond the scope of traditional phase-change materials. In that connection, chalcogenide superlattices (CSL) [123,124] and nanocrystals [125,126] were investigated for their capabilities to serve in such systems. In particular, interfacial phase-change materials (iPCMs) [127,128,129], in which the transformations do not occur between an amorphous and a crystalline phase, but between two crystalline phases, are of great interest as candidate systems. The more recent determinations of the crystal structure for the ternary Ge4Se3Te [31] (Figure 7) indicated that the type of structure observed for that chalcogenide may serve as an archetype for future PCMs. In particular, the crystal structure of Ge4Se3Te is composed of layers of germanium and the chalcogenides (Ch = Se/Te; note that the chalcogenide sites are occupationally disordered) with the stacking sequence of −Ch−Ge−Ge−Ch−. The stacking sequence of the germanium and chalcogenide layers in Ge4Se3Te is in stark contrast to that observed for α-GeTe, in which the layers of the tellurium and germanium atoms are arranged in a stacking sequence of −Ge−Te−Ge−Te−. At this point, one may wonder why there is a difference between the stacking sequences of the germanium and chalcogenide layers in Ge4Se3Te and α-GeTe.
To understand the origin of the differences between the stacking sequences of the germanium and chalcogenide layers in Ge4Se3Te and α-GeTe, the COHP curves and Density-of-Energy (DOE) functions of Ge4Se3Te, an α-GeTe-type Ge4Se3Te, and a Ge4Se3Te-type GeTe were examined. The COHP curves for Ge4Se3Te and the α-GeTe-type Ge4Se3Te (Figure 7) indicate that there are antibonding Ge−Ch interactions at the Fermi level in the α-GeTe-type chalcogenide, while the antibonding Ge−Ch interactions are weaker at the Fermi level in the Ge4Se3Te crystallizing with its own type of structure, because the electrons have been reshuffled into Ge−Ge bonding interactions. Thus, the electronically unfavorable situation corresponding to antibonding states at the Fermi level is not alleviated by introducing vacancies as demonstrated in the previous paragraph, but by transferring electrons in tetrel−tetrel bonding states in a different type of structure. From a comparison of the DOE functions and their integrated values, it is even clearer that the adoption of its own type of structure instead of the α-GeTe-type is favored for Ge4Se3Te because of a significant loss of destabilizing interactions from the α-GeTe-type to the Ge4Se3Te-type. An inspection of the DOE function for a Ge4Se3Te-type GeTe reveals a lack of stabilizing energy contributions at lower energies and strongly destabilizing contributions at the Fermi level such that the Ge4Se3Te-type is not preferred for GeTe.
3.4. Itinerant Antiferromagnetism and Ferromagnetism from the Viewpoint of Chemical Bonding in Intermetallic Compounds
One of the very first approaches to conveniently describe and account for the occurrence of ferromagnetic states in metals and their compounds was provided by the chemical theory of the collective electron ferromagnetism, which establishes a relationship between the incidence of ferromagnetism and the electronic band structures in metals and their compounds [130]. At this point, one may wonder how the presence of itinerant antiferromagnetism and ferromagnetism in metals and their compounds may be viewed from the perspective of chemical bonding. Indeed, the COHP technique can also be applied to the outcome of spin-polarized computations, thereby providing valuable information regarding the relationships between the itinerant magnetic states and the bonding situation in a given solid-state material. In this section, we will present some applications of the COHP method to spin-polarized first-principles-based computations on intermetallics.
An inspection of the non-spin-polarized COHP curves for 3d transition-metals (T; Figure 8) indicates that the Fermi levels in the ferromagnetic metals cross antibonding regions of the T−T COHP curves [131,132]; however, in the case of the spin-polarized COHP curves for the ferromagnetic metals, the states occupied by the majority α spins lower in energy, while the states comprising the minority β spins shift upward in energy. As a result, the formerly antibonding states at the Fermi levels have disappeared and the T−T bonding has been strengthened (e.g., by 5% in bcc-Fe). The strengthening of the chemical bonding upon onset of ferromagnetism is smaller for cobalt and even smaller for nickel, and their magnetic moments and exchange splitting are also smaller. On the other hand, positioning the Fermi level in the nonbonding levels (such as for bcc-Cr) is a clear signpost for antiferromagnetism. Hence, chemically fine-tuning the valence-electron concentration (and, because of that, the Fermi level) to properly “hit” antibonding and nonbonding states yields a simple but rational recipe for making ferromagnets and antiferromagnets [133], even entire series of such materials. For instance, chemical bonding analyses using the COHP technique provided fruitful insights into the origins of the magnetic ground states for several (intermetallic) borides [134,135,136,137] such that the magnetic orderings of the respective borides may be “tailored”.
Among the diverse binary compounds consisting of one of the 3d transition-metal, the nitrides are of fundamental importance for research as well as technical applications due to their roles in the production and hardening of steel [138]. In the case of the mononitrides, two different types of structure, i.e., the rock salt and zinc blende type of structures, have been proposed. To identify the structural preferences for the mononitrides, TN (T = Sc–Ni), the COHP curves of the nitrides were examined. A comparison of the COHP curves for the transition-metal mononitrides demonstrates that more antibonding T−T interactions are occupied as the valence-electron counts of the transition-metals increase. Furthermore, FeN and CoN tend to crystallize in the zinc blende-type of structure, because the antibonding T−T interactions are less pronounced in the zinc blende-type than in the rock salt-type of structure.
More recent research on the iron nitrides identified a previously unknown NiAs-type modification for FeN, which was obtained from high-temperature high-pressure syntheses [139]. An inspection of the non-spin-polarized DOS and projected COHP curves for the NiAs-type FeN (Figure 9) indicates an electronically unfavorable situation since the Fermi level falls in a maximum of the DOS and significantly antibonding Fe−N interactions. Because the Fermi level falls in minima of the spin-polarized DOS curves and the integrated values of the spin-polarized pCOHP denote net bonding characters for the Fe−N interactions, it can be inferred that the NiAs-type FeN alleviates the electronically unfavorable situation by approaching a magnetic state, once again. This conclusion is corroborated by the presence of a magnetic sextet in the Mößbauer spectrum of the NiAs-type FeN. An additional comparison of the integrated COHP values in the NiAs-type FeN to those in the zinc blende-type modification shows that the zinc blende-type FeN comprises shorter Fe−N contacts providing larger integrated pCOHP values relative to the NiAs-type compound and, hence, should be preferred.
4. Conclusions
Because the information concerning the spatial arrangements of elements in a given material is of fundamental interest for the materials design, there is critical need to recognize the bonding situation in a given material in order to understand the structural arrangements and preferences. The extraction of the information regarding the chemical bonding from electronic-structure computations requires the use of efficient and reliable procedures. Since its introduction 25 years ago, the COHP method has been employed to identify the bonding situations in numerous solid-state compounds (and also molecules). As shown in this contribution, the COHP technique does not depend on the employed basis set and can be applied to the results of electronic-structure computations obtained using diverse quantum-chemical means. Furthermore, we demonstrated the applications of this method to identify the bonding situations in diverse (polar) intermetallic compounds, which are traditionally considered as black sheep in the light of valence-electron counting rules [76]. The COHP analyses of the rare-earth transition-metal halides enabled identification of these compounds as anti-Werner-fashioned complexes, whereas the applications of the COHP procedure to the electron-poorer polar intermetallic compounds indicated that these materials tend to optimize the overall bonding by maximizing the amounts of heteroatomic bonds providing the largest ICOHP per bond values. Furthermore, chemical-bonding analyses based on the COHP technique have allowed identifying the structural preferences for nitrides and phase-change materials that are relevant for technical applications.
Author Contributions
This paper was completed trough contributions from both authors.
Acknowledgments
S.T. is grateful for a Liebig Stipend of the Verband der Chemischen Industrie e.V. (FCI), Frankfurt a. M.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goethe, J.W. Faust—Der Tragödie Erster Teil; Reclam Publishers: Stuttgart, Germany, 1957. [Google Scholar]
- Pauling, L. The Modern Theory of Valency. J. Chem. Soc. 1948, 1461–1467. [Google Scholar] [CrossRef]
- Lewis, G.N. The atom and the molecule. J. Am. Chem. Soc. 1916, 38, 762–785. [Google Scholar] [CrossRef]
- Pauling, L. The principles determining the structure of complex ionic crystals. J. Am. Chem. Soc. 1929, 51, 1010–1026. [Google Scholar] [CrossRef]
- Zintl, E. Intermetallische Verbindungen. Angew. Chem. 1939, 52, 1–6. [Google Scholar] [CrossRef]
- Klemm, W.; Busmann, E. Volumeninkremente und Radien einiger einfach negativ geladener Ionen. Z. Anorg. Allg. Chem. 1963, 319, 297–311. [Google Scholar] [CrossRef]
- Papoian, G.A.; Hoffmann, R. Hypervalent Bonding in One, Two, and Three Dimensions: Extending the Zintl-Klemm Concept to Nonclassical Electron-Rich Networks. Angew. Chem. Int. Ed. 2000, 39, 2408–2448. [Google Scholar] [CrossRef]
- Köhler, J.; Whangbo, M.-H. Late transition metal anions acting as p-metal elements. Solid State Sci. 2008, 10, 444–449. [Google Scholar] [CrossRef]
- Miller, G.J.; Schmidt, M.W.; Wang, F.; You, T.-S. Quantitative Advances in the Zintl-Klemm Formalism. In Structure and Bonding; Fässler, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 139, pp. 1–55. ISBN 978-3-642-21150-8. [Google Scholar]
- Nesper, R. The Zintl-Klemm Concept—A Historical Survey. Z. Anorg. Allg. Chem. 2014, 640, 2639–2648. [Google Scholar] [CrossRef]
- Jones, H. The phase boundaries in binary alloys, Part 2: The theory of the α, β phase boundaries. Proc. Phys. Soc. 1937, 49, 250–257. [Google Scholar] [CrossRef]
- Massalski, T.B.; Mizutani, U. Electronic Structure of Hume-Rothery Phases. Prog. Mater. Sci. 1978, 22, 151–262. [Google Scholar] [CrossRef]
- Mizutani, U.; Sato, H. The Physics of the Hume-Rothery Electron Concentration Rule. Crystals 2017, 7, 9. [Google Scholar] [CrossRef]
- Corbett, J.D. Exploratory Synthesis in the Solid State. Endless Wonders. Inorg. Chem. 2000, 39, 5178–5191. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.D. Exploratory Synthesis: The Fascinating and Diverse Chemistry of Polar Intermetallic Phases. Inorg. Chem. 2010, 49, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, E. Quantisierung als Eigenwertproblem. Ann. Phys. 1926, 384, 361–376. [Google Scholar] [CrossRef]
- Bloch, F. Über die Quantenmechanik der Elektronen in Kristallgittern. Z. Physik 1929, 52, 555–600. [Google Scholar] [CrossRef]
- Dronskowski, R. Computational Chemistry of Solid State Materials; WILEY-VCH Publishers: Weinheim, Germany, 2005; ISBN 978–3-527-31410-2. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blöchl, P.E. Crystal Orbital Hamilton Populations (COHP). Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis As Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef] [PubMed]
- Roothaan, C.C.J. New Developments in Molecular Orbital Theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Hughbanks, T.; Hoffmann, R. Chains of Trans-Edge-Sharing Molybdenum Octahedra: Metal-Metal Bonding in Extended Systems. J. Am. Chem. Soc. 1983, 105, 3528–3537. [Google Scholar] [CrossRef]
- Hoffmann, R. How Chemistry and Physics Meet in the Solid State. Angew. Chem. Int. Ed. Engl. 1987, 26, 846–878. [Google Scholar] [CrossRef]
- Wolfsberg, M.; Helmholz, L. The Spectra and Electronic Structure of the Tetrahedral Ions MnO4−, CrO4−, and ClO4−. J. Chem. Phys. 1952, 20, 837–843. [Google Scholar] [CrossRef]
- Slater, J.C. Wave Functions in a Periodic Potential. Phys. Rev. 1937, 51, 846–851. [Google Scholar] [CrossRef]
- Andersen, O.K. Linear methods in band theory. Phys. Rev. B Condens. Matter Mater. Phys. 1975, 12, 3060–3083. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Küpers, M.; Konze, P.M.; Maintz, S.; Steinberg, S.; Mio, A.M.; Cojocaru-Mirédin, O.; Zhu, M.; Müller, M.; Luysberg, M.; Mayer, J.; et al. Unexpected Ge–Ge Contacts in the Two-Dimensional Ge4Se3Te Phase and Analysis of Their Chemical Cause with the Density of Energy (DOE) Function. Angew. Chem. Int. Ed. 2017, 56, 10204–10208. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Marsman, M.; Furthmüller, J. Vienna Ab Initio Simulation Package (VASP); The Guide Computational Materials Physics, Faculty of Physics, Universität Wien: Vienna, Austria, 2014. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Gonze, X.; Jollet, F.; Abreu Araujo, F.; Adams, D.; Amadon, B.; Applencourt, T.; Audouze, C.; Beuken, J.-M.; Bieder, J.; Bokhanchuk, A.; et al. Recent developments in the ABINIT software package. Comput. Phys. Commun. 2016, 205, 106–131. [Google Scholar] [CrossRef]
- Gonze, X.; Amadon, B.; Anglade, P.-M.; Beuken, J.-M.; Bottin, F.; Boulanger, P.; Bruneval, F.; Caliste, D.; Caracas, R.; Côté, M.; et al. ABINIT: First-principles approach to material and nanosystem properties. Comput. Phys. Commun. 2009, 180, 2582–2615. [Google Scholar] [CrossRef]
- Gonze, X.; Rignanese, G.-M.; Verstraete, M.; Beuken, J.-M.; Pouillon, Y.; Caracas, R.; Jollet, F.; Torrent, M.; Zerah, G.; Mikami, M.; et al. A brief introduction to the ABINIT software package. Z. Kristallog. Cryst. Mater. 2005, 220, 558–562. [Google Scholar] [CrossRef]
- Gonze, X.; Beuken, J.-M.; Caracas, R.; Detraux, F.; Fuchs, M.; Rignanese, G.-M.; Sindic, L.; Verstraete, M.; Zerah, G.; Jollet, F.; et al. First-principles computation of material properties: The ABINIT software projec. Comput. Mater. Sci. 2002, 25, 478–492. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Analytic Projection From Plane-Wave and PAW Wavefunctions and Application to Chemical-Bonding Analysis in Solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. LOBSTER: A Tool to Extract Chemical Bonding from Plane-Wave Based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Gladisch, F.C.; Steinberg, S. Revealing Tendencies in the Electronic Structures of Polar Intermetallic Compounds. Crystals 2018, 8, 80. [Google Scholar] [CrossRef]
- Schäfer, H.; Schnering, H.G. Metall-Metall-Bindungen bei niederen Halogeniden, Oxyden und Oxydhalogeniden schwerer Übergangsmetalle. Angew. Chem. 1964, 76, 833–849. [Google Scholar] [CrossRef]
- Cotton, F.A. Strong Homonuclear Metal-Metal Bonds. Acc. Chem. Res. 1969, 2, 240–247. [Google Scholar] [CrossRef]
- Simon, A. Cluster of Valence Electron Poor Metals—Structure, Bonding, and Properties. Angew. Chem. Int. Ed. Engl. 1988, 27, 159–183. [Google Scholar] [CrossRef]
- Hughbanks, T. Bonding in clusters and condensed cluster compounds that extend in one, two and three dimension. Prog. Solid St. Chem. 1989, 19, 329–372. [Google Scholar] [CrossRef]
- Corbett, J.D. Exploratory synthesis of reduced rare-earth-metal halides, chalcogenides, intermetallics: New compounds, structures, and properties. J. Alloys Compds. 2006, 418, 1–20. [Google Scholar] [CrossRef]
- Corbett, J.D. Interstitially-stabilized cluster-based halides of the early transition metals. J. Alloys Compds. 1995, 229, 10–23. [Google Scholar] [CrossRef]
- Simon, A.; Mattausch, H.J.; Ryazanov, M.; Kremer, R.K. Lanthanides as d Metals. Z. Anorg. Allg. Chem. 2006, 632, 919–929. [Google Scholar] [CrossRef]
- Meyer, G. Reduced Halides of the Rare-Earth Elements. Chem. Rev. 1988, 88, 93–107. [Google Scholar] [CrossRef]
- Meyer, G. Cluster Complexes as anti-Werner Complexes. Z. Anorg. Allg. Chem. 2008, 634, 2729–2736. [Google Scholar] [CrossRef]
- Meyer, G. Rare Earth Metal Cluster Complexes. In The Rare Earth Elements; Atwood, D.A., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2012; ISBN 978-1-119-95097-4. [Google Scholar]
- Hughbanks, T.; Corbett, J.D. Encapsulation of Heavy Transition Metals in Iodide Clusters. Synthesis, Structure, and Bonding of the Unusual Cluster Phase Y6I10Ru. Inorg. Chem. 1989, 28, 631–635. [Google Scholar] [CrossRef]
- Hughbanks, T.; Corbett, J.D. Rare-Earth-Metal Iodide Clusters Centered by Transition Metals: Synthesis, Structure, and Bonding of R7II2M Compounds (R = Sc, Y, Pr, Gd; M = Mn, Fe, Co, Ni). Inorg. Chem. 1988, 27, 2022–2026. [Google Scholar] [CrossRef]
- Rustige, C.; Brühmann, M.; Steinberg, S.; Meyer, E.; Daub, K.; Zimmermann, S.; Wolberg, M.; Mudring, A.-V.; Meyer, G. The Prolific {ZR6}X12R and {ZR6}X10 Structure Types with Isolated Endohedrally Stabilized (Z) Rare-Earth Metal (R) Cluster Halide (X) Complexes. Z. Anorg. Allg. Chem. 2012, 638, 1922–1931. [Google Scholar] [CrossRef]
- Hughbanks, T.; Rosenthal, G.; Corbett, J.D. Alloy Clusters: The Encapsulation of Transition Metals (Mn, Fe, Co, Ni) within Cluster Halides of Zirconium and the Rare-Earth Metals. J. Am. Chem. Soc. 1986, 108, 8289–8290. [Google Scholar] [CrossRef]
- Sweet, L.E.; Roy, L.E.; Meng, F.; Hughbanks, T. Ferromagnetic Coupling in Hexanuclear Gadolinium Clusters. J. Am. Chem. Soc. 2006, 128, 10193–10201. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A.; Haas, T.E. A Molecular Orbital Treatment of the Bonding in Certain Metal Atom Clusters. Inorg. Chem. 1964, 3, 10–17. [Google Scholar] [CrossRef]
- Ebihara, M.; Martin, J.D.; Corbett, J.D. Novel Chain and Oligomeric Condensed Cluster Phases for Gadolinium Iodides with Manganese Interstitials. Inorg. Chem. 1994, 33, 2079–2084. [Google Scholar] [CrossRef]
- Payne, M.W.; Ebihara, M.; Corbett, J.D. A Novel Oligomer of Condensed Metal Atom Clusters in [Y16Ru4I20]. Angew. Chem. Int. Ed. Engl. 1991, 30, 856–858. [Google Scholar] [CrossRef]
- Steinwand, S.J.; Corbett, J.D. Oligomeric Rare-Earth-Metal Halide Clusters. Three Structures Built of (Y16Z4)Br36 Units (Z = Ru, Ir). Inorg. Chem. 1996, 35, 7056–7067. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.; Zimmermann, S.; Brühmann, M.; Meyer, E.; Rustige, C.; Wolberg, M.; Daub, K.; Bell, T.; Meyer, G. Oligomeric rare-earth metal cluster complexes with endohedral transition metal atoms. J. Solid State Chem. 2014, 219, 159–167. [Google Scholar] [CrossRef]
- Steinberg, S.; Bell, T.; Meyer, G. Electron Counting Rules and Electronic Structure in Tetrameric Transition-Metal (T)-Centered Rare-Earth (R) Cluster Complex Halides (X). Inorg. Chem. 2015, 54, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Dorhout, P.K.; Payne, M.W.; Corbett, J.D. Condensed Metal Cluster Iodides Centered by Noble Metals. Six Examples of Cubic R3I3Z Phases (R = La, Pr; Z = Os, Ir, Pt). Inorg. Chem. 1991, 30, 4960–4962. [Google Scholar] [CrossRef]
- Zheng, C.; Mattausch, H.J.; Hoch, C.; Simon, A. La3Br3Ni: Jahn–Teller Distortion in the Reduced Rare Earth Metal Halide. Z. Anorg. Allg. Chem. 2009, 635, 2429–2433. [Google Scholar] [CrossRef]
- Herzmann, N.; Mudring, A.-V.; Meyer, G. Seven-Coordinate Ruthenium Atoms Sequestered in Praseodymium Clusters in the Chloride {RuPr3}Cl3. Inorg. Chem. 2008, 47, 7954–7956. [Google Scholar] [CrossRef] [PubMed]
- Payne, M.W.; Dorhout, P.K.; Kim, S.-J.; Hughbanks, T.R.; Corbett, J.D. Chains of Centered Metal Clusters with a Novel Range of Distortions: Pr3I3Ru, Y3I3Ru, and Y3I3Ir. Inorg. Chem. 1992, 31, 1389–1394. [Google Scholar] [CrossRef]
- Steinberg, S.; Valldor, M.; Meyer, G. Change of magnetic and electronic features through subtle substitution in cubic, non-centrosymmetric extended rare-earth metal cluster complexes {TR3}X3. J. Solid State Chem. 2013, 206, 176–181. [Google Scholar] [CrossRef]
- Steinberg, S.; Brgoch, J.; Miller, G.J.; Meyer, G. Identifying a Structural Preference in Reduced Rare-Earth Metal Halides by Combining Experimental and Computational Techniques. Inorg. Chem. 2012, 51, 11356–11364. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Meyer, G.; Corbett, J.D. Contrasts in Structural and Bonding Representations among Polar Intermetallic Compounds. Strongly Differentiated Hamilton Populations for Three Related Condensed Cluster Halides of the Rare-Earth Elements. Inorg. Chem. 2010, 49, 9949–9957. [Google Scholar] [CrossRef] [PubMed]
- Köckerling, M.; Martin, J.D. Electronic Fine Tuning of the Structures of Reduced Rare-Earth Metal Halides. Inorg. Chem. 2001, 40, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Bell, T. Seltenerd-Clusterkomplexe mit endohedralen Übergangsmetallatomen und ihre strukturellen sowie elektronischen Beziehungen zu intermetallischen Phasen. Dr. rer. nat. Thesis, University of Cologne, Dr. Hut Publishers, Munich, Germany, 2014. [Google Scholar]
- Nesper, R. Bonding Patterns in Intermetallic Compounds. Angew. Chem. Int. Ed. Engl. 1991, 30, 789–817. [Google Scholar] [CrossRef]
- Wade, K. The Structural Significance of the Number of Skeletal Bonding Electron-pairs in Carboranes, the Higher Boranes and Borane Anions, and Various Transition-metal Carbonyl Cluster Compounds. J. Chem. Soc. D Chem. Commun. 1971, 792–793. [Google Scholar] [CrossRef]
- Wade, K. Skeletal electron counting in cluster species. some generalisations and predictions. Inorg. Nucl. Chem. Lett. 1972, 8, 559–562. [Google Scholar] [CrossRef]
- Lin, Q.; Corbett, J.D. Exploratory Syntheses and Structures of SrAu4.3In1.7 and CaAg3.5In1.9: Electron-Poor Intermetallics with Diversified Polyanionic Frameworks That Are Derived from the CaAu4In2 Approximant. Inorg. Chem. 2011, 50, 11091–11098. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P. Strong Closed-Shell Interactions in Inorganic Chemistry. Chem. Rev. 1997, 97, 597–636. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P.; Desclaux, J.-P. Relativity and the Periodic System of the Elements. Acc. Chem. Res. 1979, 12, 276–281. [Google Scholar] [CrossRef]
- Pyykkö, P. Relativistic Effects in Structural Chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Pyykkö, P. Theoretical Chemistry of Gold. Angew. Chem. Int. Ed. 2004, 43, 4412–4456. [Google Scholar] [CrossRef] [PubMed]
- Smetana, V.; Lin, Q.; Pratt, D.K.; Kreyssig, A.; Ramazanoglu, M.; Corbett, J.D.; Goldman, A.I.; Miller, G.J. A Sodium-Containing Quasicrystal: Using Gold To Enhance Sodium´s Covalency in Intermetallic Compounds. Angew. Chem. Int. Ed. 2012, 51, 12699–12702. [Google Scholar] [CrossRef] [PubMed]
- Gautier, R.; Zhang, X.; Hu, L.; Yu, L.; Lin, Y.; Sunde, T.O.L.; Chon, D.; Poeppelmeier, K.R.; Zunger, A. Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds. Nat. Chem. 2015, 7, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.; Stoffel, R.P.; Dronskowski, R. Search for the Mysterious SiTe—An Examination of the Binary Si-Te System Using First-Principles-Based Methods. Cryst. Growth Des. 2016, 16, 6152–6155. [Google Scholar] [CrossRef]
- Wang, F.; Pearson, K.N.; Miller, G.J. EuAgxAl11-x with the BaCd11-Type Structure: Phase Width, Coloring, and Electronic Structure. Chem. Mater. 2009, 21, 230–236. [Google Scholar] [CrossRef]
- Steinberg, S.; Card, N.; Mudring, A.-V. From the Ternary Eu(Au/In)2 and EuAu4(Au/In)2 with Remarkable Au/In Distributions to a New Structure Type: The Gold-Rich Eu5Au16(Au/In)6 Structure. Inorg. Chem. 2015, 54, 8187–8196. [Google Scholar] [CrossRef] [PubMed]
- Smetana, V.; Corbett, J.D.; Miller, G.J. Four Polyanionic Compounds in the K-Au-Ga System: A Case Study in Exploratory Synthesis and of the Art of Structural Analysis. Inorg. Chem. 2012, 51, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Smetana, V.; Miller, G.J.; Corbett, J.D. Three Alkali-Metal-Gold-Gallium Systems. Ternary Tunnel Structures and Some Problems with Poorly Ordered Cations. Inorg. Chem. 2012, 51, 7711–7721. [Google Scholar] [CrossRef] [PubMed]
- Smetana, V.; Miller, G.J.; Corbett, J.D. Polycluster and Substitution Effects in the Na-Au-Ga System: Remarkable Sodium Bonding Characteristics in Polar Intermetallics. Inorg. Chem. 2013, 52, 12502–12510. [Google Scholar] [CrossRef] [PubMed]
- Smetana, V.; Corbett, J.D.; Miller, G.J. Complex Polyanionic Nets in RbAu4.01(2)Ga8.64(5) and CsAu5Ga9: The Role of Cations in the Formation of New Polar Intermetallics. Z. Anorg. Allg. Chem. 2014, 640, 790–796. [Google Scholar] [CrossRef]
- Mishra, T.; Lin, Q.; Corbett, J.D. Gold Network Structures in Rhombohedral and Monoclinic Sr2Au6(Au,T)3 (T = Zn, Ga). A Transition via Relaxation. Inorg. Chem. 2013, 52, 13623–13630. [Google Scholar] [CrossRef] [PubMed]
- Palasyuk, A.; Grin, Y.; Miller, G.J. Turning Gold into “Diamond”: A Family of Hexagonal Diamond-Type Au-Frameworks Interconnected by Triangular Clusters in the Sr-Al-Au System. J. Am. Chem. Soc. 2014, 136, 3108–3117. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Kim, S.-J.; Miller, G.J.; Corbett, J.D. Synthesis, Structure, and Bonding in K12Au21Sn4. A Polar Intermetallic Compound with Dense Au20 and open AuSn4 Layers. Inorg. Chem. 2009, 48, 11108–11113. [Google Scholar] [CrossRef] [PubMed]
- Smetana, V.; Corbett, J.D.; Miller, G.J. Na8Au9.8(4)Ga7.2 and Na17Au5.87(2)Ga46.63: The diversity of pseudo 5-fold symmetries in the Na–Au–Ga system. J. Solid State Chem. 2013, 207, 21–28. [Google Scholar] [CrossRef]
- Celania, C.; Smetana, V.; Provino, A.; Pecharsky, V.; Manfrinetti, P.; Mudring, A.-V. R3Au9Pn (R = Y, Gd-Tm; Pn = Sb, Bi): A Link between Cu10Sn3 and Gd14Ag51. Inorg. Chem. 2017, 56, 7247–7256. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Corbett, J.D. Multiple Nonstoichiometric Phases with Discrete Composition Ranges in the CaAu5-CaAu4Bi-BiAu2 System. A Case Study of the Chemistry of Spinodal Decomposition. J. Am. Chem. Soc. 2010, 132, 5662–5671. [Google Scholar] [CrossRef] [PubMed]
- Smetana, V.; Steinberg, S.; Mudryk, Y.; Pecharsky, V.; Miller, G.J.; Mudring, A.-V. Cation-Poor Complex Metallic Alloys in Ba(Eu)-Au-Al(Ga) Systems: Identifying the Keys that Control Structural Arrangements and Atom Distributions at the Atomic Level. Inorg. Chem. 2015, 54, 10296–10308. [Google Scholar] [CrossRef] [PubMed]
- Provino, A.; Steinberg, S.; Smetana, V.; Kulkarni, R.; Dhar, S.K.; Manfrinetti, P.; Mudring, A.-V. Gold-rich R3Au7Sn3: Establishing the interdependence between electronic features and physical properties. J. Mater. Chem. C 2015, 3, 8311–8321. [Google Scholar] [CrossRef]
- Lin, Q.; Vetter, J.; Corbett, J.D. Disorder-Order Structural Transformation in Electron-Poor Sr3Au8Sn3 Driven by Chemical Bonding Optimization. Inorg. Chem. 2013, 52, 6603–6609. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Corbett, J.D. Different Cation Arrangements in Au-In Networks. Syntheses and Structures of Six Intermetallic Compounds in Alkali-Metal-Au-In Systems. Inorg. Chem. 2007, 46, 6022–6028. [Google Scholar] [CrossRef]
- Lin, Q.; Mishra, T.; Corbett, J.D. Hexagonal-Diamond-like Gold Lattices, Ba and (Au,T)3 Interstitials and Delocalized Bonding in a Family of Intermetallic Phases Ba2Au6(Au,T)3 (T = Zn, Cd, Ga, In, or Sr). J. Am. Chem. Soc. 2013, 135, 11023–11031. [Google Scholar] [CrossRef] [PubMed]
- Gerke, B.; Hoffmann, R.-D.; Pöttgen, R. Zn3 and Ga3 Triangles as Building Units in Sr2Au6Zn3 and Sr2Au6Ga3. Z. Anorg. Allg. Chem. 2013, 639, 2444–2449. [Google Scholar] [CrossRef]
- Gerke, B.; Korthaus, A.; Niehaus, O.; Haarmann, F.; Pöttgen, R. Triangular Zn3 and Ga3 units in Sr2Au6Zn3, Eu2Au6Zn3, Sr2Au6Ga3, and Eu2Au6Ga3—Structure, magnetism, 151Eu Mössbauer and 69;71Ga solid state NMR spectroscopy. Z. Naturforsch. B 2016, 71, 567–577. [Google Scholar] [CrossRef]
- Gerke, B.; Pöttgen, R. Sr2Au6Al3 and Eu2Au6Al3—First Representatives of the Sr2Au6Zn3 Type with Aluminium Triangles. Z. Naturforsch. B 2014, 69, 121–124. [Google Scholar] [CrossRef]
- Smetana, V.; Steinberg, S.; Card, N.; Mudring, A.-V.; Miller, G.J. Crystal Structure and Bonding in BaAu5Ga2 and AeAu4+xGa3‒x (Ae = Ba and Eu): Hexagonal Diamond-Type Au Frameworks and Remarkable Cation/Anion Partitioning in the Ae-Au-Ga Systems. Inorg. Chem. 2015, 54, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Dubois, J.-M.; Belin-Ferré, E. Complex. Metallic Alloys: Fundamentals and Applications; WILEY-VCH Publishers GmbH & Co. KGaA: Weinheim, Germany, 2011; ISBN 978–3-527-32523-8. [Google Scholar]
- Urban, K.; Feuerbach, M. Structurally complex alloy phases. J. Non. Cryst. Solids 2004, 334–335, 143–150. [Google Scholar] [CrossRef]
- Li, B.; Kim, S.-J.; Miller, G.J.; Corbett, J.D. Gold Tetrahedra as Building Blocks in K3Au5Tr (Tr = In, Tl) and Rb2Au3Tl and in Other Compounds: A Broad Group of Electron-Poor Intermetallic Phases. Inorg. Chem. 2009, 48, 6573–6583. [Google Scholar] [CrossRef] [PubMed]
- Wuttig, M.; Yamada, N. Phase-change materials for rewriteable data storage. Nat. Mater. 2007, 6, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Lencer, D.; Salinga, M.; Wuttig, M. Design Rules for Phase-Change Materials in Data Storage Applications. Adv. Mater. 2011, 23, 2030–2058. [Google Scholar] [CrossRef] [PubMed]
- Wuttig, M.; Raoux, S. The Science and Technology of Phase Change Materials. Z. Anorg. Allg. Chem. 2012, 638, 2455–2465. [Google Scholar] [CrossRef]
- Bensch, W. Phasenwechselverbindungen auf Chalkogenidbasis für die optische und elektrische Datenspeicherung. Z. Anorg. Allg. Chem. 2008, 634, 2009. [Google Scholar] [CrossRef]
- Chen, M.; Rubin, K.A.; Barton, R.W. Compound materials for reversible, phase-change optical data storage. Appl. Phys. Lett. 1986, 49, 502–504. [Google Scholar] [CrossRef]
- Ohno, E.; Yamada, N.; Kurumizawa, T.; Kimura, K.; Takao, M. TeGeSnAu Alloys for Phase Change Type Optical Disk Memories. Jpn. J. Appl. Phys. 1989, 28, 1235–1240. [Google Scholar] [CrossRef]
- Lencer, D.; Salinga, M.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Wuttig, M. A map for phase-change materials. Nat. Mater. 2008, 7, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Shportko, K.; Kremers, S.; Woda, M.; Lencer, D.; Robertson, J.; Wuttig, M. Resonant bonding in crystalline phase-change materials. Nat. Mater. 2008, 7, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Esser, M.; Maintz, S.; Dronskowski, R. Automated first-principles mapping for phase-change materials. J. Comput. Chem. 2017, 38, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Deringer, V.L.; Zhang, W.; Lumeij, M.; Maintz, S.; Wuttig, M.; Mazzarello, R.; Dronskowski, R. Bonding Nature of Local Structural Motifs in Amorphous GeTe. Angew. Chem. Int. Ed. 2014, 53, 10817–10820. [Google Scholar] [CrossRef] [PubMed]
- Wuttig, M.; Lüsebrink, D.; Wamwangi, D.; Welnic, W.; Gilleßen, M.; Dronskowski, R. The role of vacancies and local distortions in the desing of new phase-change materials. Nat. Mater. 2007, 6, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Yamada, N.; Kubota, Y. Structure of stable and metastable Ge2Sb2Te5, an intermetallic compound in GeTe-Sb2Te3 pseudo-binary systems. Acta Crystallogr. Sect. B 2004, 60, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Chong, T.C.; Shi, L.P.; Zhao, R.; Tan, P.K.; Li, J.M.; Lee, H.K.; Miao, X.S.; Du, A.Y.; Tung, C.H. Phase change random access memory cell with superlattice-like structure. Appl. Phys. Lett. 2006, 88, 122114. [Google Scholar] [CrossRef]
- Simpson, R.E.; Fons, P.; Kolobov, A.V.; Fukaya, T.; Krbal, M.; Yagi, T.; Tominaga, J. Interfacial phase-change memory. Nat. Nanotechnol. 2011, 6, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.R.; Sines, I.T.; Schaak, R.E. Liquid-Phase Synthesis of Uniform Cube-Shaped GeTe Microcrystals. Chem. Mater. 2010, 22, 3236–3240. [Google Scholar] [CrossRef]
- Konze, P.M.; Deringer, V.L.; Dronskowski, R. Understanding the Shape of GeTe Nanocrystals from First Principles. Chem. Mater. 2016, 28, 6682–6688. [Google Scholar] [CrossRef]
- Tominaga, J.; Kolobov, A.V.; Fons, P.; Nakano, T.; Murakami, S. Ferroelectric Order Control of the Dirac-Semimetal Phase in GeTe-Sb2Te3 Superlattices. Adv. Mater. Interfaces 2014, 1, 1300027. [Google Scholar] [CrossRef]
- Ohyanagi, T.; Kitamura, M.; Araidai, M.; Kato, S.; Takaura, N.; Shiraishi, K. GeTe sequences in superlattice phase change memories and their electrical characteristics. Appl. Phys. Lett. 2014, 104, 252106. [Google Scholar] [CrossRef]
- Momand, J.; Wang, R.; Boschker, J.E.; Verheijen, M.A.; Calarco, R.; Kooi, B.J. Interface formation of two- and three-dimensionally bonded materials in the case of GeTe-Sb2Te3 superlattices. Nanoscale 2015, 7, 19136–19143. [Google Scholar] [CrossRef] [PubMed]
- Stoner, E.C. Collective electron ferromagnetism. Proc. R. Soc. Lond. A 1938, 165, 372–414. [Google Scholar] [CrossRef]
- Landrum, G.A.; Dronskowski, R. The Orbital Origins of Magnetism: From Atoms to Molecules to Ferromagnetic Alloys. Angew. Chem. Int. Ed. 2000, 39, 1560–1585. [Google Scholar] [CrossRef]
- Dronskowski, R. Itinerant Ferromagnetism and Antiferromagnetism from the Perspective of Chemical Bonding. J. Quantum. Chem. 2004, 96, 89–94. [Google Scholar] [CrossRef]
- Dronskowski, R.; Korczak, K.; Lueken, H.; Jung, W. Chemically Tuning between Ferromagnetnism and Antiferromagnetism by Combining Theory and Synthesis in Iron/Manganese Rhodium Borides. Angew. Chem. Int. Ed. 2002, 41, 2528–2532. [Google Scholar] [CrossRef]
- Fokwa, B.P.T.; Lueken, H.; Dronskowski, R. Rational Synthetic Tuning between Itinerant Antiferromagnetism and Ferromagnetism in the Complex Boride Series Sc2FeRu5–nRhnB2 (0≤n≤5). Chem. Eur. J. 2007, 13, 6040–6046. [Google Scholar] [CrossRef] [PubMed]
- Brgoch, J.; Goerens, C.; Fokwa, B.P.T.; Miller, G.J. Scaffolding, Ladders, Chains, and Rare Ferrimagnetism in Intermetallic Borides: Electronic Structure Calculations and Magnetic Ordering. J. Am. Chem. Soc. 2011, 133, 6832–6840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Miller, G.J.; Fokwa, B.P.T. Computational Design of Rare-Earth-Free Magnets with the Ti3Co5B2-Type Structure. Chem. Mater. 2017, 29, 2535–2541. [Google Scholar] [CrossRef]
- Fokwa, B.P.T.; Samolyuk, G.D.; Miller, G.J.; Dronskowski, R. Ladders of a Magnetically Active Element in the Structure of the Novel Complex Borid Ti9Fe2Ru18B8: Synthesis, Structure, Bonding, and Magnetism. Inorg. Chem. 2008, 47, 2113–2120. [Google Scholar] [CrossRef]
- Eck, B.; Dronskowski, R.; Takahashi, M.; Kikkawa, S. Theoretical calculations on the structures, electronic and magnetic properties of binary 3d transition metal nitrides. J. Mater. Chem. 1999, 9, 1527–1537. [Google Scholar] [CrossRef]
- Clark, W.P.; Steinberg, S.; Dronskowski, R.; McCammon, C.; Kupenko, I.; Bykov, M.; Dubronvinsky, L.; Akselrud, L.G.; Schwarz, U.; Niewa, R. High-Pressure NiAs-Type Modification of FeN. Angew. Chem. Int. Ed. 2017, 56, 7302–7306. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representations of (a) isolated; (b) tetrameric; and (c−f) chains of transition-metal (T; yellow) centered rare-earth metal (R; red) cluster halides (X; blue). For instance, isolated clusters as shown in (a) have been identified for the crystal structure of the [TR6]X10 and [TR6]X12R types of structure, whereas tetramers as presented in (b) have been observed for the rare-earth transition-metal halides crystallizing with the [T4R16]X28[R4], [T4R16]X20, [T4R16]X24[RX3]4, and [T4R16]X24 types of structure. To date, diverse types of structure which are composed of rare-earth cluster chains enclosing the endohedral transition-metal atoms have been determined to crystallize with the net formula [TR3]X3 shown in (c−f), i.e., (c) the cubic [PtPr3]I3-type, (d) the orthorhombic [RuPr3]Cl3-type, (e) the monoclinic [IrY3]I3-type, and (f) the monoclinic [RuPr3]I3-type. Adapted in part from reference [45].
Figure 1.
Representations of (a) isolated; (b) tetrameric; and (c−f) chains of transition-metal (T; yellow) centered rare-earth metal (R; red) cluster halides (X; blue). For instance, isolated clusters as shown in (a) have been identified for the crystal structure of the [TR6]X10 and [TR6]X12R types of structure, whereas tetramers as presented in (b) have been observed for the rare-earth transition-metal halides crystallizing with the [T4R16]X28[R4], [T4R16]X20, [T4R16]X24[RX3]4, and [T4R16]X24 types of structure. To date, diverse types of structure which are composed of rare-earth cluster chains enclosing the endohedral transition-metal atoms have been determined to crystallize with the net formula [TR3]X3 shown in (c−f), i.e., (c) the cubic [PtPr3]I3-type, (d) the orthorhombic [RuPr3]Cl3-type, (e) the monoclinic [IrY3]I3-type, and (f) the monoclinic [RuPr3]I3-type. Adapted in part from reference [45].

Figure 2.
(a) Molecular-orbital scheme of an isolated transition-metal (T) centered rare-earth (R) cluster enclosed by halide ligands (X): the cluster-based orbitals (middle) are constructed by the valence orbitals of the endohedral transition-metal atom (right) and the fragment orbitals of the rare-earth skeleton (left). (b) COHP curves of the homoatomic and heteroatomic interactions for two structurally different representatives of the [TR3]X3-type, whose crystal structures comprises chains of transition-metal-centered rare-earth clusters (see main text): the Fermi levels, EF, are represented by the black horizontal lines. Parts of that figure are adapted from reference [45].
Figure 2.
(a) Molecular-orbital scheme of an isolated transition-metal (T) centered rare-earth (R) cluster enclosed by halide ligands (X): the cluster-based orbitals (middle) are constructed by the valence orbitals of the endohedral transition-metal atom (right) and the fragment orbitals of the rare-earth skeleton (left). (b) COHP curves of the homoatomic and heteroatomic interactions for two structurally different representatives of the [TR3]X3-type, whose crystal structures comprises chains of transition-metal-centered rare-earth clusters (see main text): the Fermi levels, EF, are represented by the black horizontal lines. Parts of that figure are adapted from reference [45].

Figure 3.
Positions of the Zintl phases, polar intermetallics, and Hume–Rothery phases with respect to their respective valence-electron concentrations (electrons per atom = e/a) [45,84].

Figure 4.
Representations of the crystal structures of (a) K1.8Au6In4 and (b) Sr3Au8Sn3, in which the gold (yellow) and post-transition-metal atoms (blue) assemble tunnels encompassing the active-metal atoms (red).
Figure 4.
Representations of the crystal structures of (a) K1.8Au6In4 and (b) Sr3Au8Sn3, in which the gold (yellow) and post-transition-metal atoms (blue) assemble tunnels encompassing the active-metal atoms (red).

Figure 5.
Representations of the crystal structures of (a) Sr2Au7.3Al1.7; (b) Sr2Au6.2Al2.8; (c) Sr2Au5.1Al1.9; (d) SrAu4.1Al2.9; (e) CaAu4.1Bi0.9; (f) K12Au21Sn4; (g) Y3Au7Sn3; (h) Y3Au9Sb; (i) Na8Au10.1Ga6.9; (j) NaAu4Ga2; (k) Na5Au10Ga16; (l) EuAu6.1Al5.9; and (m) Na26Au18.1Ga35.9. In the case of Na26Au18.1Ga35.9, the diverse cluster shells typically observed for Bergman-type quasicrystals are shown, while atoms which are located in the unit cell but do not assemble the cluster shells have been omitted for the benefit of a clear representation. Details regarding the crystal structures and their determinations may by extracted from the literature cited in the main text and in the Table 2. Parts of the figure are adapted from reference [45].
Figure 5.
Representations of the crystal structures of (a) Sr2Au7.3Al1.7; (b) Sr2Au6.2Al2.8; (c) Sr2Au5.1Al1.9; (d) SrAu4.1Al2.9; (e) CaAu4.1Bi0.9; (f) K12Au21Sn4; (g) Y3Au7Sn3; (h) Y3Au9Sb; (i) Na8Au10.1Ga6.9; (j) NaAu4Ga2; (k) Na5Au10Ga16; (l) EuAu6.1Al5.9; and (m) Na26Au18.1Ga35.9. In the case of Na26Au18.1Ga35.9, the diverse cluster shells typically observed for Bergman-type quasicrystals are shown, while atoms which are located in the unit cell but do not assemble the cluster shells have been omitted for the benefit of a clear representation. Details regarding the crystal structures and their determinations may by extracted from the literature cited in the main text and in the Table 2. Parts of the figure are adapted from reference [45].

Figure 6.
COHP curves of Ge2Sb2Te4 (left); Ge1.5Sb2Te4 (middle); and GeSb2Te4 (right) [121]: the Ge−Te and Sb−Te COHP curves are shown in blue and red, respectively, while the Fermi levels are represented by the black horizontal lines.
Figure 6.
COHP curves of Ge2Sb2Te4 (left); Ge1.5Sb2Te4 (middle); and GeSb2Te4 (right) [121]: the Ge−Te and Sb−Te COHP curves are shown in blue and red, respectively, while the Fermi levels are represented by the black horizontal lines.

Figure 7.
(Left): Representations of the crystal structures of and structural relationships between (a) α-GeTe and (b) Ge4Se3Te: the crystal structure of the two chalcogenides discern in the stacking sequences of the layers of the germanium atoms and the chalcogenide atoms. (Right): (a) pCOHP curves and (b) DOE functions of Ge4Se3Te, a α-GeTe-type Ge4Se3Te, and a Ge4Se3Te-type GeTe: the Fermi levels, EF, are represented by the black horizontal lines. Reprinted with permission from reference [31]. Copyright 2017, WILEY-VCH, Weinheim.
Figure 7.
(Left): Representations of the crystal structures of and structural relationships between (a) α-GeTe and (b) Ge4Se3Te: the crystal structure of the two chalcogenides discern in the stacking sequences of the layers of the germanium atoms and the chalcogenide atoms. (Right): (a) pCOHP curves and (b) DOE functions of Ge4Se3Te, a α-GeTe-type Ge4Se3Te, and a Ge4Se3Te-type GeTe: the Fermi levels, EF, are represented by the black horizontal lines. Reprinted with permission from reference [31]. Copyright 2017, WILEY-VCH, Weinheim.

Figure 8.
DOS and transition-metal−transition-metal COHP curves of the 3d transition-metals in a non-magnetic regime: the COHP curves shaded in red correspond to transition-metals showing paramagnetic-ferromagnetic transitions, while the COHP curve of chromium exhibiting a paramagnetic-antiferromagnetic transition is shaded in yellow. Reprinted with permission from reference [131]. Copyright 2000 WILEY-VCH, Weinheim.
Figure 8.
DOS and transition-metal−transition-metal COHP curves of the 3d transition-metals in a non-magnetic regime: the COHP curves shaded in red correspond to transition-metals showing paramagnetic-ferromagnetic transitions, while the COHP curve of chromium exhibiting a paramagnetic-antiferromagnetic transition is shaded in yellow. Reprinted with permission from reference [131]. Copyright 2000 WILEY-VCH, Weinheim.

Figure 9.
Energy-volume curves (a), and pressure-dependence of the enthalpies (b) of the NiAs-type and ZnS-type polymorphs of FeN; non-spin-polarized and spin-polarized densities-of-states (c,e), and pCOHP curves (d,f) of the NiAs-type FeN: the Fermi level, EF, is represented by the black horizontal line. Reprinted with permission from reference [139]. Copyright 2017 WILEY-VCH, Weinheim.
Figure 9.
Energy-volume curves (a), and pressure-dependence of the enthalpies (b) of the NiAs-type and ZnS-type polymorphs of FeN; non-spin-polarized and spin-polarized densities-of-states (c,e), and pCOHP curves (d,f) of the NiAs-type FeN: the Fermi level, EF, is represented by the black horizontal line. Reprinted with permission from reference [139]. Copyright 2017 WILEY-VCH, Weinheim.


{kind=link}
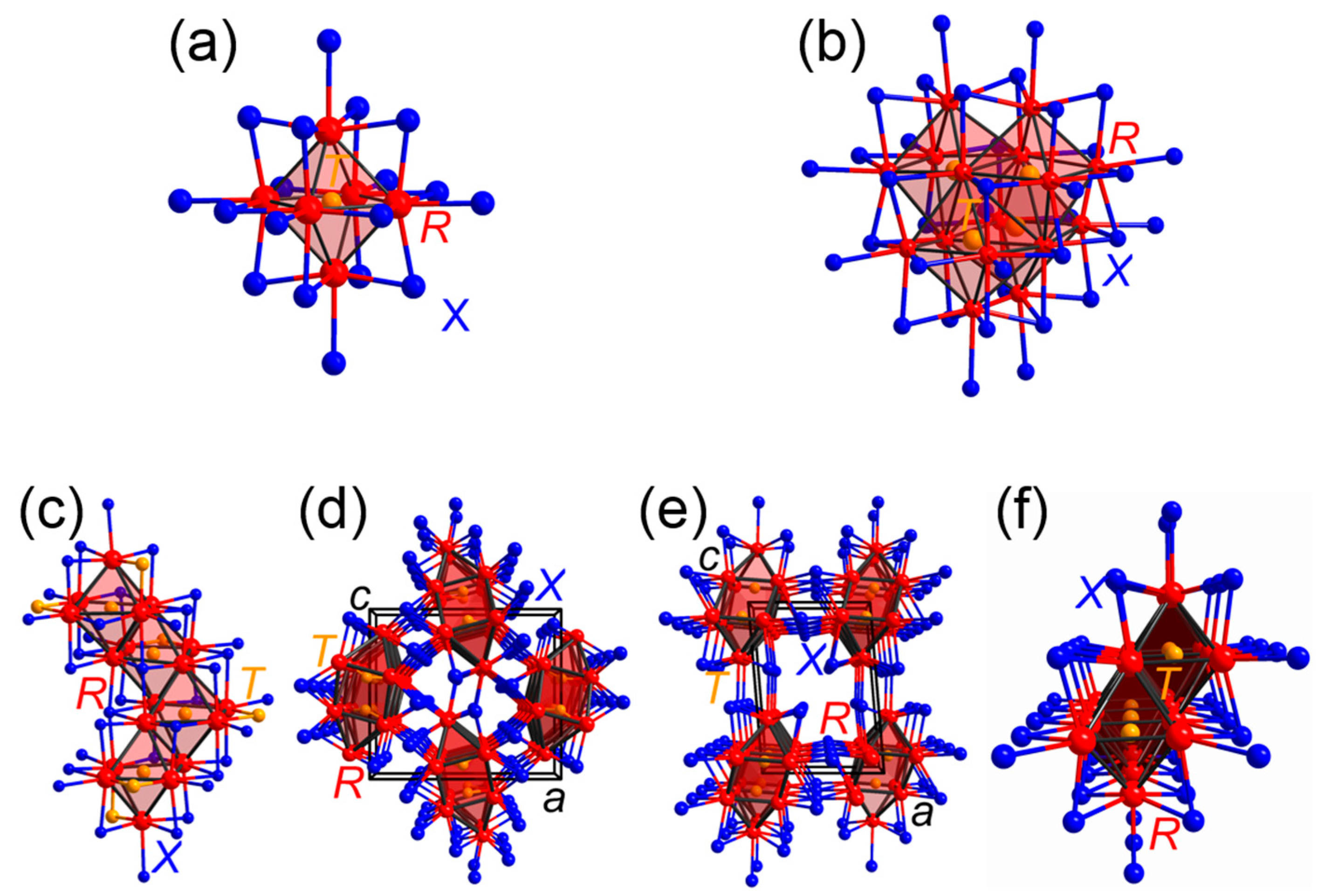{kind=link}
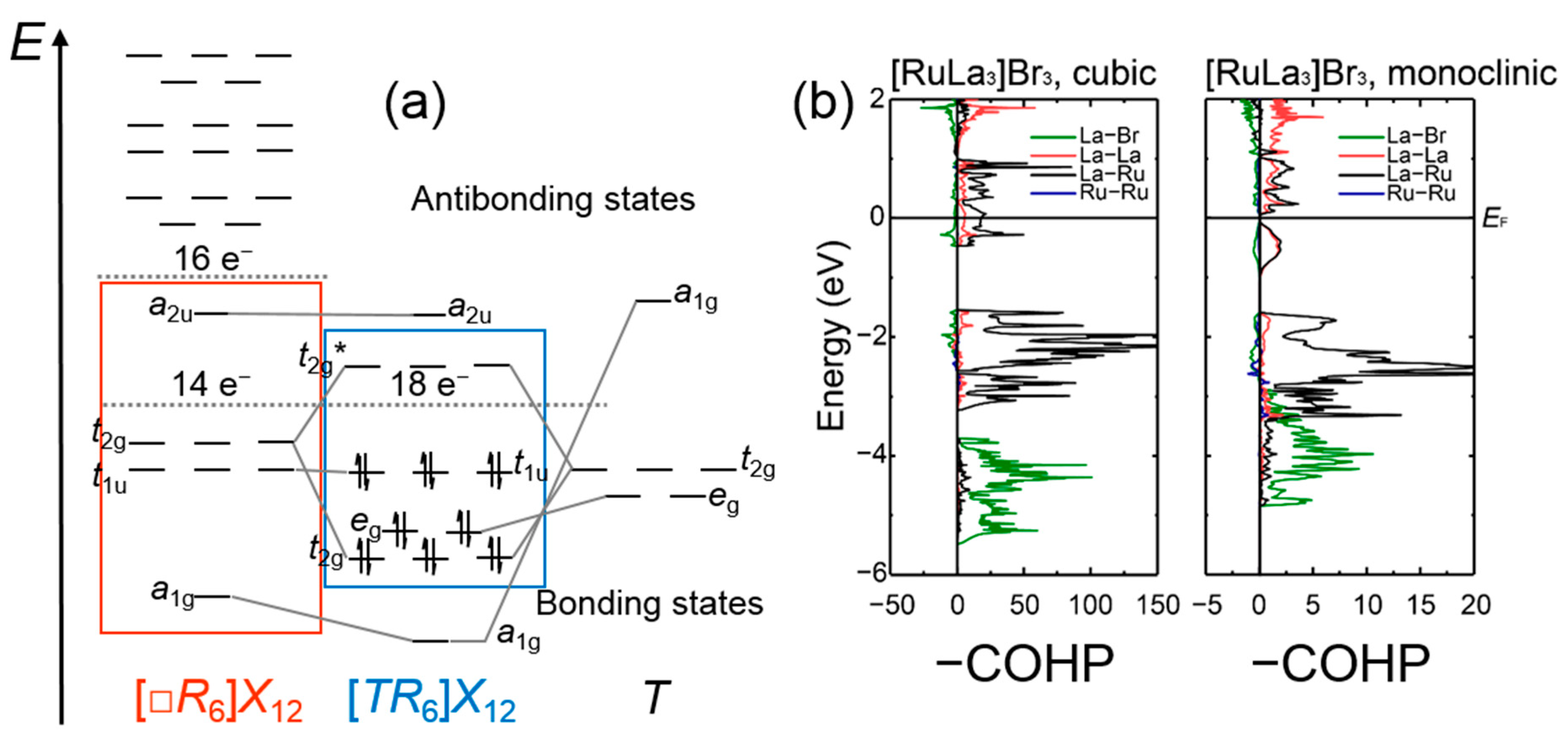{kind=link}
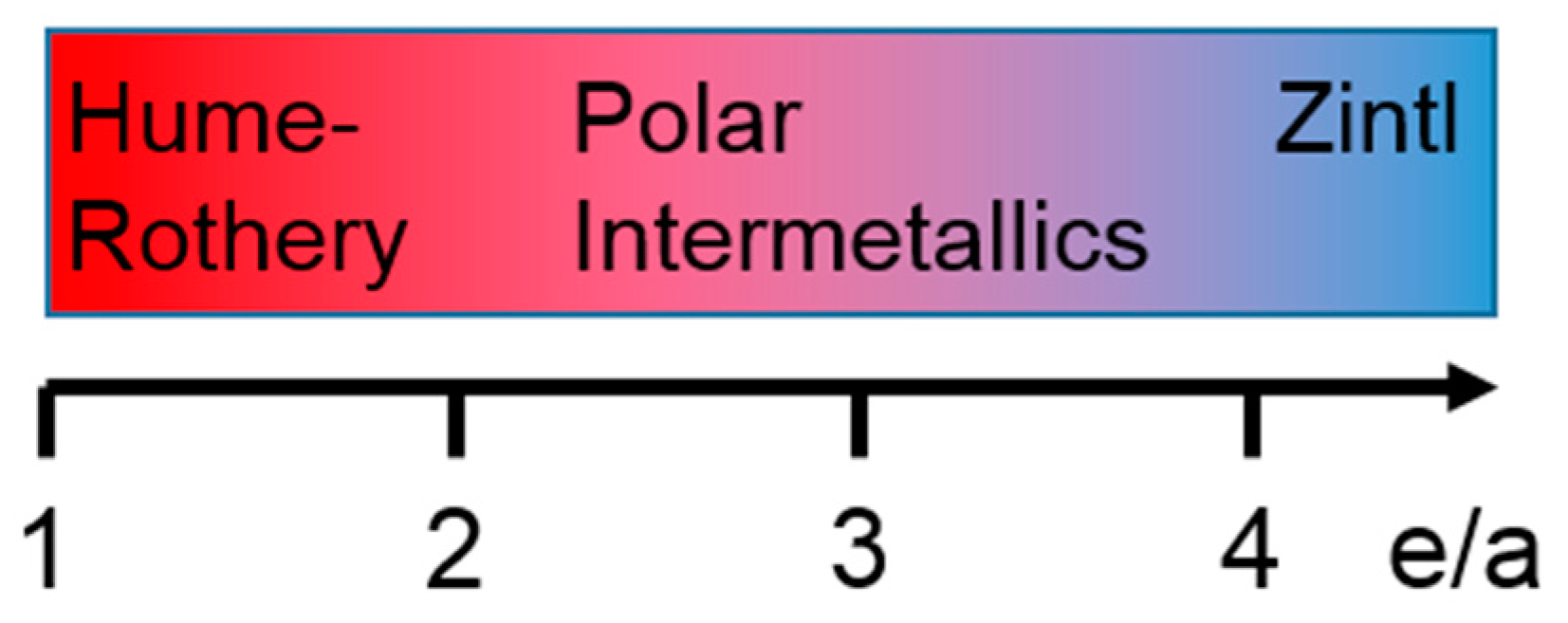{kind=link}
{kind=link}
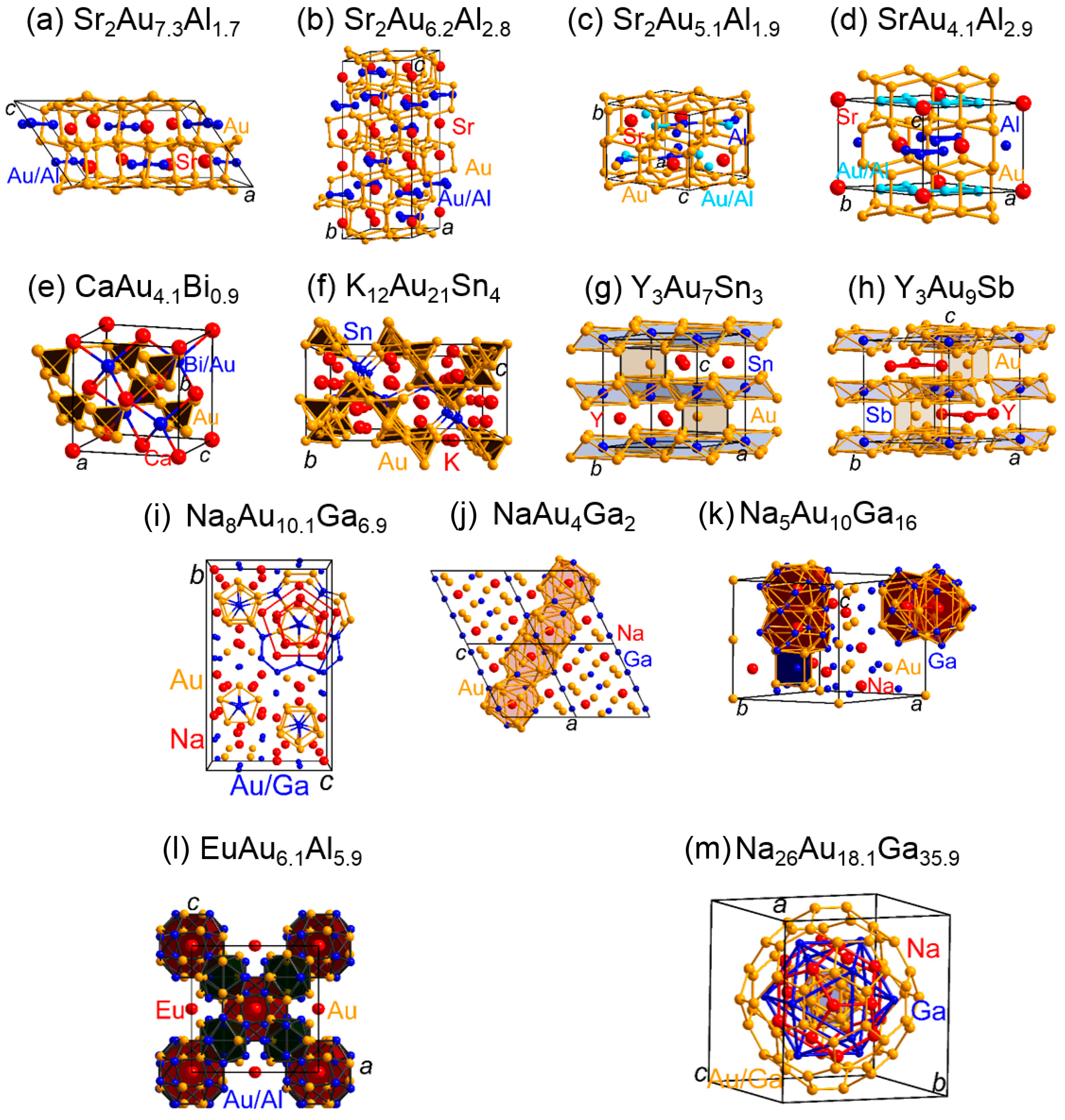{kind=link}
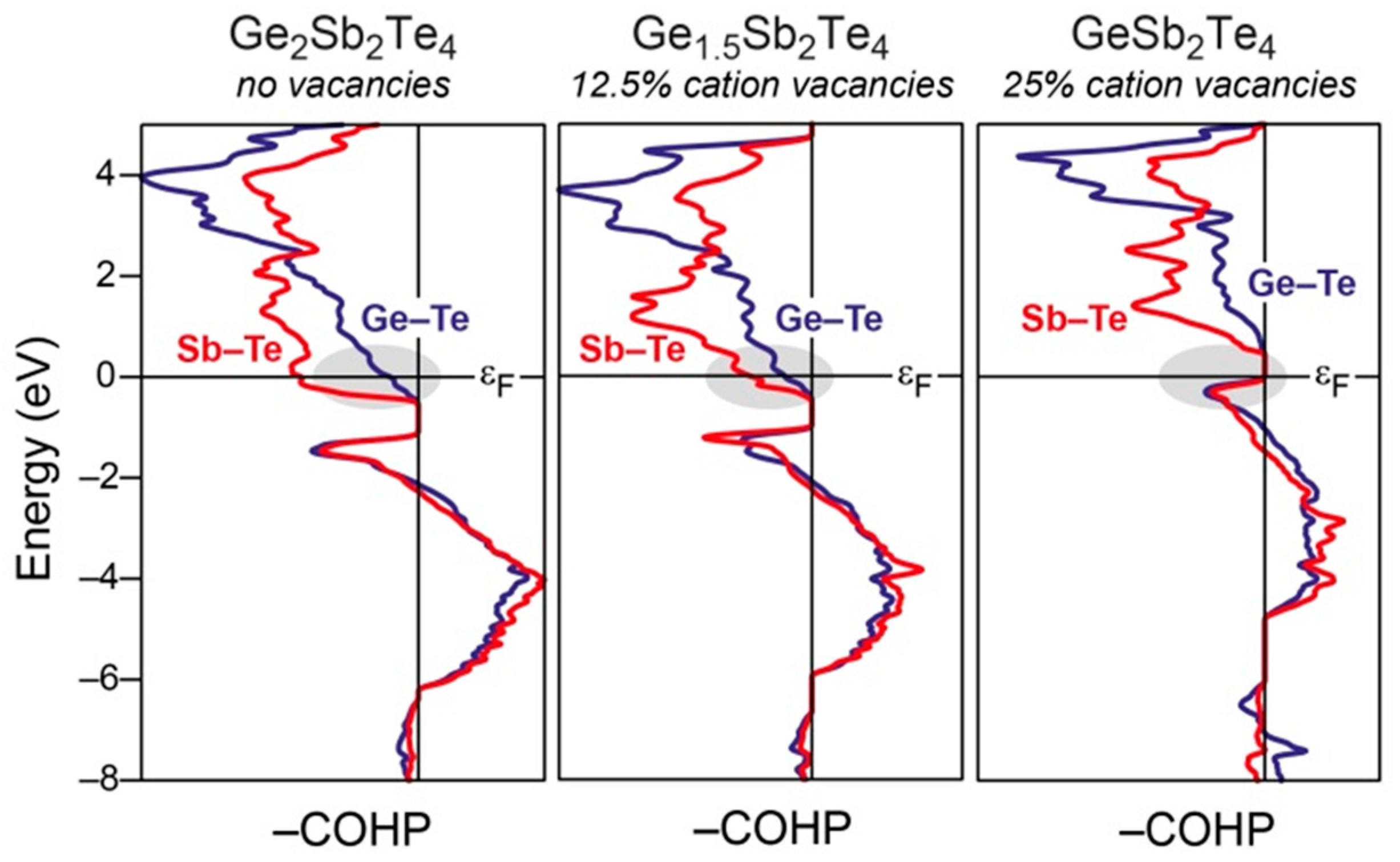{kind=link}
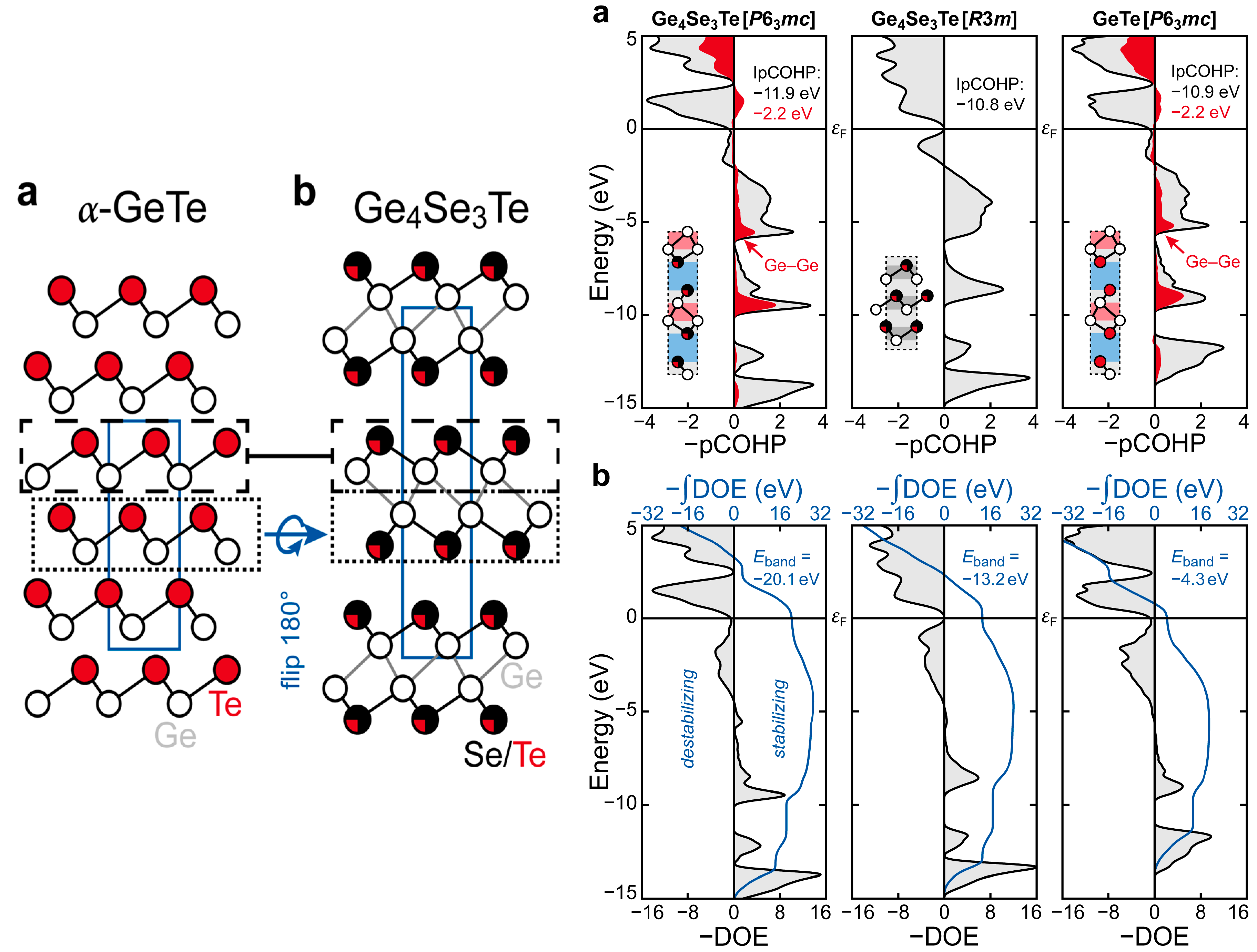{kind=link}
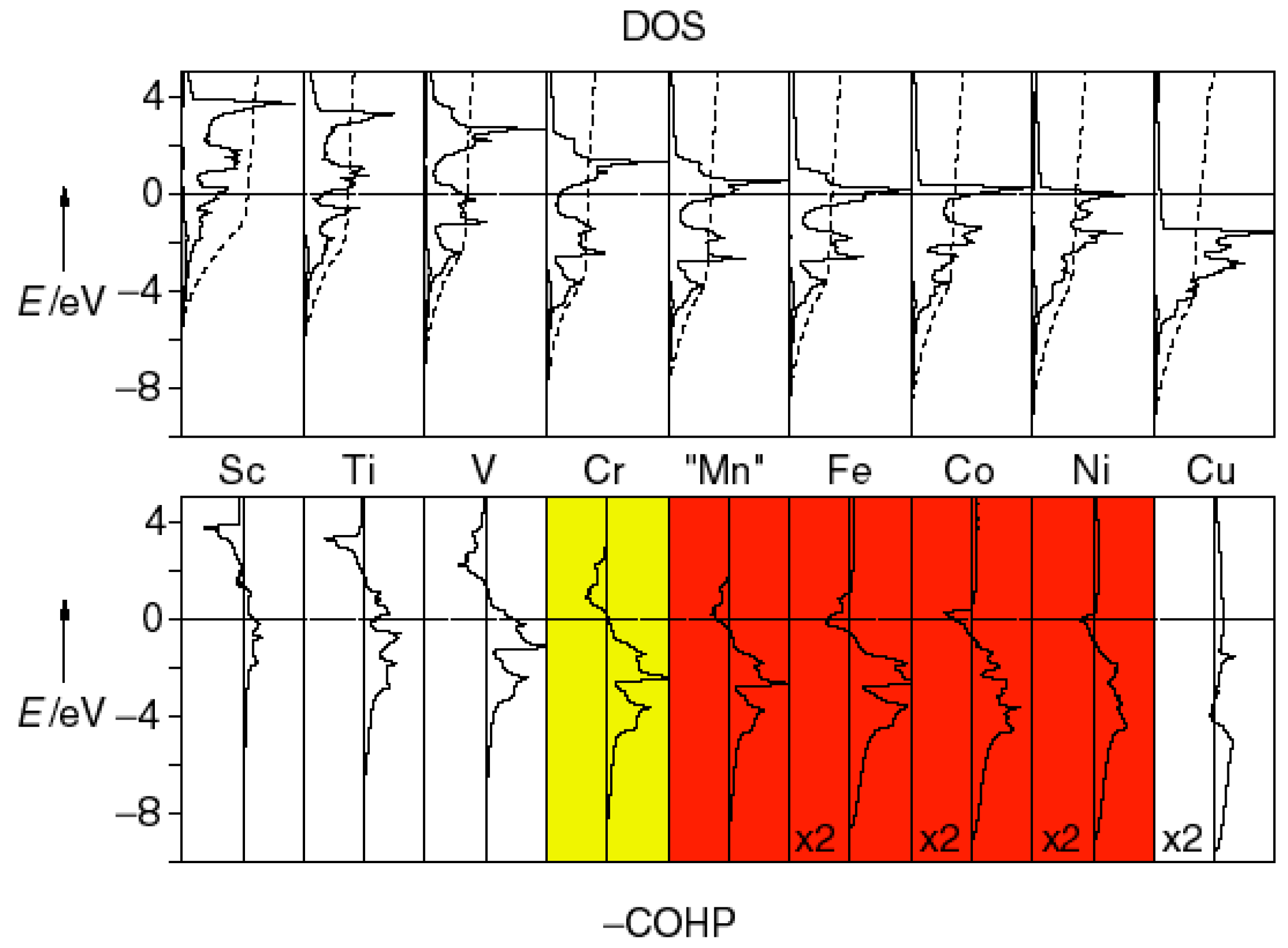{kind=link}
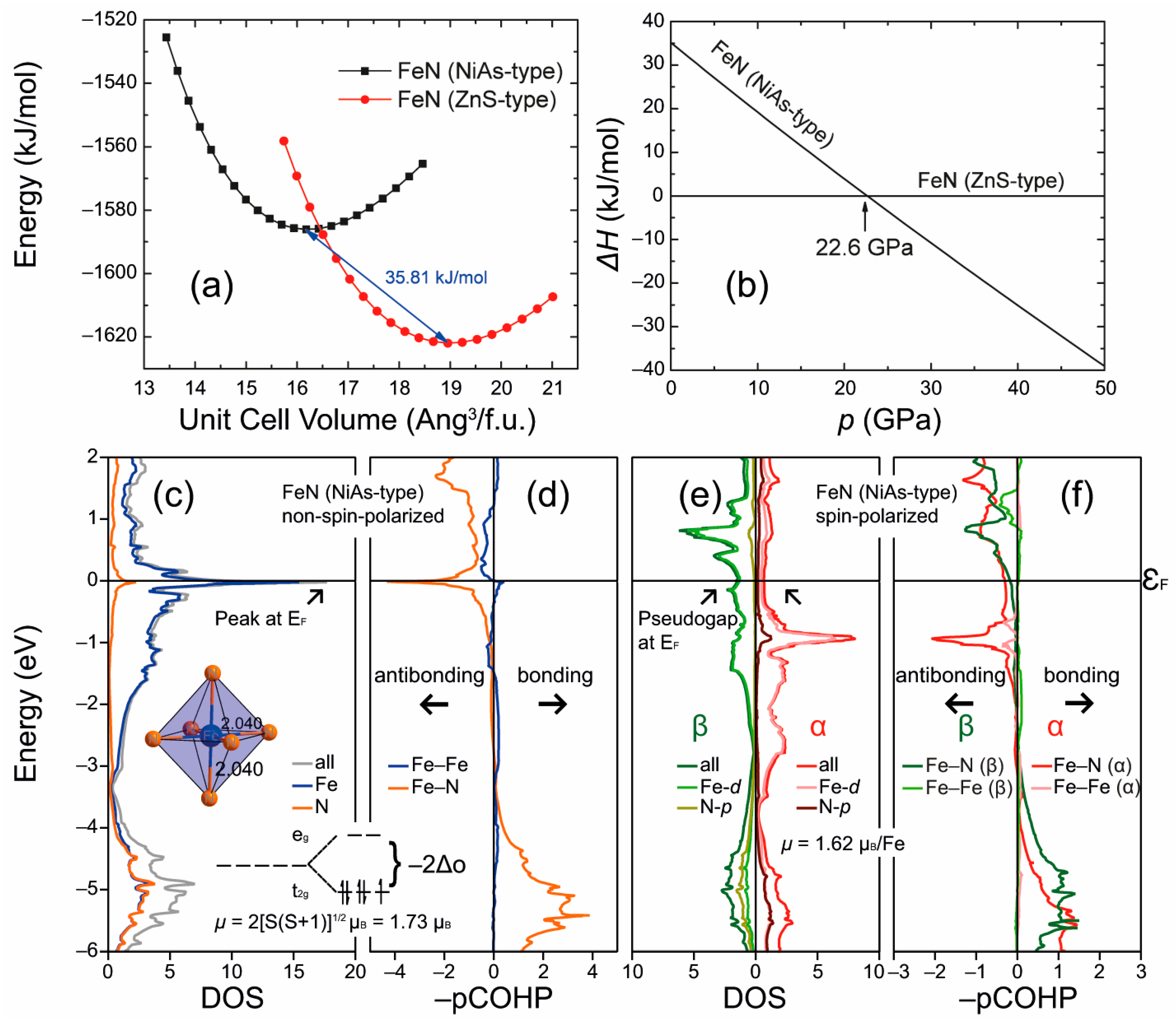{kind=link}
Table 1.
Average −ICOHP/bond values and percentage contributions of the respective interactions to the net bonding capabilities in rare-earth transition-metal halides composed of tetrameric clusters, [T4R16], and cluster chains. The details about the quantum-chemical computations, distance ranges, −ICOHP/bond ranges, and cumulative −ICOHP/cell values may be extracted from the respective literature listed in the last column. a hypothetical monoclinic [TR3]X3-type halide; details regarding the generation of the model may be obtained from the respective literature.
Table 1.
Average −ICOHP/bond values and percentage contributions of the respective interactions to the net bonding capabilities in rare-earth transition-metal halides composed of tetrameric clusters, [T4R16], and cluster chains. The details about the quantum-chemical computations, distance ranges, −ICOHP/bond ranges, and cumulative −ICOHP/cell values may be extracted from the respective literature listed in the last column. a hypothetical monoclinic [TR3]X3-type halide; details regarding the generation of the model may be obtained from the respective literature.
| Compound | R−T | R−R | R−X | T−T | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ave. −ICOHP/ Bond (eV/bond) | % | Ave. –ICOHP/ Bond (eV/bond) | % | Ave. –ICOHP/ Bond (eV/bond) | % | Ave. −ICOHP/ Bond (eV/bond) | % | ||
| Rare-earth transition-metal halides comprising tetramers | |||||||||
| [Ru4Y16]Br20 | 2.03 | 48.8 | 0.11 | 5.2 | 0.60 | 43.2 | 0.48 | 2.9 | [66] |
| [Ru4Y16]I20 | 1.89 | 50.1 | 0.08 | 4.5 | 0.56 | 44.2 | 0.18 | 1.2 | [66] |
| [Ir4Y16]Br24 | 2.03 | 41.9 | 0.08 | 3.5 | 0.80 | 51.8 | 0.60 | 2.8 | [66] |
| [Ru4Ho16]I24(Ho4I4) | 2.02 | 39.0 | 0.09 | 4.1 | 0.71 | 54.9 | 0.42 | 2.0 | [66] |
| [Ir4Tb16]Cl24(TbCl3)4 | 2.12 | 35.3 | 0.10 | 3.4 | 0.87 | 57.2 | 0.86 | 3.6 | [66] |
| [Rh4Tb16]Br24(TbBr3)4 | 2.17 | 34.0 | 0.14 | 4.5 | 0.95 | 59.6 | 0.49 | 1.9 | [66] |
| [Ir4Tb16]Br24(TbBr3)4 | 2.41 | 37.4 | 0.11 | 3.7 | 0.90 | 56.0 | 0.77 | 3.0 | [66] |
| [Ir4Sc16]Cl24(ScCl3)4 | 2.16 | 33.6 | 0.08 | 2.7 | 0.95 | 59.1 | 1.25 | 4.9 | [66] |
| [Os4Sc16]Cl24(ScCl3)4 | 2.26 | 33.5 | 0.09 | 2.7 | 0.98 | 58.0 | 1.57 | 5.8 | [66] |
| [Ru4Sc16]Cl24(ScCl3)4 | 2.08 | 31.6 | 0.10 | 3.3 | 1.01 | 61.1 | 1.07 | 4.0 | [66] |
| [Ru4Gd16]Br24(GdBr3)4 | 2.47 | 35.5 | 0.16 | 4.8 | 1.00 | 57.6 | 0.62 | 2.2 | [66] |
| Rare-earth transition-metal halides comprising cluster chains | |||||||||
| c-[RuLa3]Br3 | 1.91 | 65.0 | 0.07 | 4.2 | 0.45 | 30.7 | 0.003 | 0.1 | [72] |
| m-[RuLa3]Br3 a | 2.37 | 64.3 | 0.18 | 9.1 | 0.49 | 26.5 | −0.01 | 0.1 | [72] |
| c-[IrLa3]Br3 | 1.87 | 67.2 | 0.06 | 3.6 | 0.40 | 29.1 | 0.004 | 0.1 | [72] |
| m-[RuLa3]I3 | 2.45 | 60.8 | 0.22 | 9.9 | 0.59 | 29.2 | −0.003 | 0.1 | [72] |
| o-[RuPr3]Cl3 | 1.30 | 52.5 | 0.11 | 4.3 | 0.37 | 30.0 | 1.15 | 13.2 | [73] |
| m-[RuPr3]I3 | 2.14 | 62.7 | 0.20 | 8.7 | 0.48 | 28.4 | 0.03 | 0.26 | [73] |
| m-[MnGd3]I3 | 1.45 | 39.4 | 0.39 | 10.5 | 0.73 | 34.1 | 2.06 | 16.0 | [73] |
Table 2.
Average–ICOHP/bond values (eV/bond) and percentage contributions to the net bonding capabilities of the homoatomic Au−Au and post-transition-metal−post-transition-metal as well as heteroatomic Au−post-transition-metal interactions for diverse active-metal-poor polar intermetallics consisting of an active-metal (main-groups I, II, and scandium-group elements), gold and a post-transition-metal. The valence-electron concentrations (e/a) are given in the second column, while details regarding the quantum-chemical calculations and the crystal structures of the respective compounds may be extracted from the references listed in the last column [45].
Table 2.
Average–ICOHP/bond values (eV/bond) and percentage contributions to the net bonding capabilities of the homoatomic Au−Au and post-transition-metal−post-transition-metal as well as heteroatomic Au−post-transition-metal interactions for diverse active-metal-poor polar intermetallics consisting of an active-metal (main-groups I, II, and scandium-group elements), gold and a post-transition-metal. The valence-electron concentrations (e/a) are given in the second column, while details regarding the quantum-chemical calculations and the crystal structures of the respective compounds may be extracted from the references listed in the last column [45].
| Compound | e/a | Parent Compound Disordered? | Homoatomic Contacts | Heteroatomic Contacts | Ref. | ||
|---|---|---|---|---|---|---|---|
| Ave. −ICOHP/Bond | % | Ave. −ICOHP/Bond | % | ||||
| Compounds with anionic fragments in the forms of 1D tunnels in the crystal structures | |||||||
| EuAu5In | 1.43 | y/EuAu5.0In1.0 | Au−Au: 0.79 | 57.1 | Au−In: 0.81 | 36.5 | [88] |
| KAu3Ga2 | 1.67 | y/KAu3.1Ga1.9 | Au−Au: 0.79 Ga−Ga: 0.55 | 20.1 5.6 | Au−Ga: 1.18 | 72.2 | [89] |
| RbAu3Ga2 | 1.67 | n | Au−Au: 0.66 Ga−Ga: 0.53 | 17.7 5.7 | Au−Ga: 1.17 | 75.4 | [90] |
| Na0.5Au2Ga2 | 1.89 | y/Na0.6Au2Ga2 | Au−Au: 1.00 Ga−Ga: 0.63 | 10.2 6.5 | Au−Ga: 1.31 | 80.9 | [90] |
| K0.5Au2Ga2 | 1.89 | y/K0.6Au2Ga2 | Au−Au: 0.97 Ga−Ga: 0.51 | 8.5 4.4 | Au−Ga: 1.64 | 85.7 | [89] |
| Rb0.5Au2Ga2 | 1.89 | y/Rb0.6Au2Ga2 | Au−Au: 1.02 Ga−Ga: 0.62 | 9.9 5.9 | Au−Ga: 1.43 | 83.1 | [90] |
| NaAu2Ga4 | 2.14 | no | Ga−Ga: 1.04 | 20.0 | Au−Ga: 1.73 | 72.2 | [91] |
| KAu2Ga4 | 2.14 | y/KAu2.2Ga3.8 | Au−Au: 1.04 Ga−Ga: 1.20 | 1.6 22.7 | Au−Ga: 1.88 | 71.3 | [89] |
| CsAu5Ga9 | 2.20 | no | Au−Au: 0.59 Ga−Ga: 0.48 | 2.7 16.2 | Au−Ga: 1.42 | 78.8 | [92] |
| Compds. with hexagonal diamond-type networks as anionic fragments in the crystal structures | |||||||
| Sr2Au7Zn2 | 1.36 | y/Sr2Au6(Au,Zn)3 | Au−Au: 1.21 Zn−Zn: 0.56 | 40.5 1.0 | Au−Zn: 1.02 | 37.7 | [93] |
| Sr2Au7Al2 | 1.55 | y/Sr2Au7.3Al1.7 | Au−Au: 1.07 Al−Al: 1.26 | 32.0 2.4 | Au−Al: 1.65 | 43.0 | [94] |
| SrAu5Al2 | 1.63 | y/SrAu5.05Al1.95 | Au−Au: 1.01 Al−Al: 0.71 | 32.0 1.6 | Au−Al: 1.61 | 54.6 | [94] |
| Sr2Au6Al3 | 1.73 | y/Sr2Au6.2Al2.8 | Au−Au: 1.09 Al−Al: 1.56 | 21.5 7.7 | Au−Al: 1.68 | 50.0 | [94] |
| SrAu4Al3 | 1.88 | y/SrAu4.1Al2.9 | Au−Au: 0.93 Al−Al: 1.48 | 17.0 8.9 | Au−Al: 1.61 | 63.0 | [94] |
| Compounds with diverse (types of) polyhedrons formed by the anions in the crystal structures | |||||||
| K12Au21Sn4 | 1.32 | no | Au−Au: 1.22 | 28.0 | Au−Sn: 2.70 | 43.1 | [95] |
| Na8Au11Ga6 | 1.48 | y/Na8Au10.1Ga6.9 | Au−Au: 1.22 Ga−Ga: 1.49 | 31.9 5.2 | Au−Ga: 1.71 | 44.7 | [96] |
| NaAu4Ga2 | 1.57 | no | Au−Au: 1.20 Ga−Ga: 0.51 | 27.9 1.2 | Au−Ga: 1.61 | 65.0 | [91] |
| Y3Au9Sb | 1.77 | no | Au−Au: 1.17 | 51.7 | Au−Sb: 1.06 | 11.7 | [97] |
| CaAu4Bi | 1.83 | y/CaAu4.1Bi0.9 | Au−Au: 1.40 | 57.5 | Au−Bi: 0.54 | 22.2 | [98] |
| EuAu6Al6 | 2.00 | y/EuAu6.1Al5.9 | Au−Au: 0.88 Al−Al: 0.95 | 11.6 10.8 | Au−Al: 1.58 | 67.8 | [99] |
| EuAu6Ga6 | 2.00 | y/EuAu6.2Ga5.8 | Au−Au: 0.64 Ga−Ga: 0.91 | 11.2 11.1 | Au−Ga: 1.40 | 68.0 | [99] |
| Na5Au10Ga16 | 2.03 | no | Au−Au: 0.42 Ga−Ga: 1.10 | 0.8 22.2 | Au−Ga: 1.67 | 71.2 | [91] |
| Y3Au7Sn3 | 2.15 | no | Au−Au: 0.78 | 22.2 | Au−Sn: 1.33 | 42.1 | [100] |
| Gd3Au7Sn3 | 2.15 | no | Au−Au: 0.78 | 21.9 | Au−Sn: 1.31 | 41.1 | [100] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Steinberg, S.; Dronskowski, R. The Crystal Orbital Hamilton Population (COHP) Method as a Tool to Visualize and Analyze Chemical Bonding in Intermetallic Compounds. Crystals 2018, 8, 225. https://doi.org/10.3390/cryst8050225
AMA Style
Steinberg S, Dronskowski R. The Crystal Orbital Hamilton Population (COHP) Method as a Tool to Visualize and Analyze Chemical Bonding in Intermetallic Compounds. Crystals. 2018; 8(5):225. https://doi.org/10.3390/cryst8050225
Chicago/Turabian StyleSteinberg, Simon, and Richard Dronskowski. 2018. "The Crystal Orbital Hamilton Population (COHP) Method as a Tool to Visualize and Analyze Chemical Bonding in Intermetallic Compounds" Crystals 8, no. 5: 225. https://doi.org/10.3390/cryst8050225
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.