Ba4[Mn3N6], a Quasi-One-Dimensional Mixed-Valent Nitridomanganate (II, IV)
by
 ,
,
Alexander Ovchinnikov
1,2 ,
,
Matej Bobnar
1,
Yurii Prots
1,
Walter Schnelle
1,
Peter Höhn
1,* and
Yuri Grin
1 1
Max-Planck-Institut für Chemische Physik fester Stoffe, Nöthnitzer Straße 40, 01187 Dresden, Germany
2
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA
*
Author to whom correspondence should be addressed.
Crystals 2018, 8(6), 235; https://doi.org/10.3390/cryst8060235
Submission received: 25 April 2018
/
Revised: 17 May 2018
/
Accepted: 18 May 2018
/
Published: 25 May 2018
(This article belongs to the Special Issue Compounds with Polar Metallic Bonding)
Abstract
:The mixed-valent nitridomanganate Ba4[Mn3N6] was prepared using a gas–solid high temperature route. The crystal structure was determined employing high resolution synchrotron powder diffraction data: space group Pbcn, a = 9.9930(1) Å, b = 6.17126(8) Å, c = 14.4692(2) Å, V = 892.31(2) Å3, Z = 4. The manganese atoms in the structure of Ba4[Mn3N6] are four-fold coordinated by nitrogen forming infinite corrugated chains of edge-sharing [MnN4] tetrahedra. The chains demonstrate a complete charge order of Mn species. Magnetization measurements and first principle calculations indicate quasi-one dimensional magnetic behavior. In addition, chemical bonding analysis revealed pronounced Mn–Mn interactions along the chains.
1. Introduction
Low-dimensional magnetic systems, such as spin chains, ladders, or planes, attract much attention as perspective materials for a wide range of applications, e.g., in spintronics, quantum computing, and information storage technologies [1,2]. Such quantum magnets may display exotic physical phenomena including spin liquid behavior [3], spin-orbital Mott insulating state [4], and topological excitations [5]. Since the decrease of dimensionality implies a spatial spin confinement, the role of fluctuations becomes significant in these systems. Such fluctuations are spin-dependent and most important for S = ½ and S = 1 systems. Therefore, the electronic state of the constituting magnetic atoms, along with the magnetic topology, determines the behavior of a particular system.
Low-dimensional quantum magnets have been mainly explored in the families of halides [6], oxides [7], and higher chalcogenides [8], but little is known about the realization of such systems in nitrides. Since multicomponent nitrides often demonstrate low-dimensional crystallographic arrangements of transition-metal atoms along with low coordination numbers and oxidation states of the latter [9,10,11], they represent a natural platform to probe low-dimensional magnetism. However, the preparation of single-phase nitrides and their inherent instability make the study of this class of materials highly challenging.
In this contribution, we report on the synthesis and characterization of the first chain alkaline-earth nitridomanganate with a quasi-one-dimensional magnetic behavior.
2. Materials and Methods
Synthesis of Ba2N. All manipulations except the high-temperature treatment were done inside an Ar-filled glovebox due to the high air- and moisture-sensitivity of most of the materials. Barium nitride, Ba2N, was prepared by annealing Ba lumps (99.9%, Alfa Aesar, Thermo Fisher (Kandel) GmbH, Karlsruhe, Germany) under N2 stream (Praxair Deutschland GmbH, Dresden, Germany, 99.9999%, additionally purified by molecular sieves and a BTS-catalyst) at 973 K for 12 h, followed by cooling down to room temperature under Ar. The resulting black soft powder was single-phase according to powder X-ray diffraction (PXRD).
Synthesis of Ba4[Mn3N6]. Ba2N (Figure S1) and Mn powder (Alfa, 99.9998%) were mixed in the ratio Ba:Mn = 4.04:3 in an agate mortar and thoroughly ground. The excess of Ba2N was employed to compensate for evaporation at high temperatures. The mixture was pelletized and annealed in a Ta crucible under a constant N2 flow (7 mL/min) at T = 1023–1123 K for 108 h in total, with several intermediate re-grindings. The resulting sample was almost single-phase. The intensity of the strongest impurity peak was lower than 3% of the most intense peak of the main phase. The impurity reflections could be easily distinguished from those of the main phase by tracking the evolution of the PXRD patterns upon annealing, however, they could not be assigned to any known phases. Annealing times longer than that in the above-given protocol led to gradual decomposition of the main phase and to partial amorphisation of the sample. The composition of the sample and the absence of potential impurity elements were confirmed by chemical analysis (Table S1).
Powder X-ray diffraction (PXRD). Laboratory PXRD patterns were collected on a Huber G670 imaging plate Guinier camera (CuKα1 radiation, Huber Diffraktionstechnik GmbH & Co. KG, Rimsting, Germany). Powder samples were enclosed between two Kapton foils sealed with vacuum grease to reduce contact with air. Synchrotron PXRD data were collected at the ID22 beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). Samples were sieved to a particle size of less than 50 µm and enclosed in glass capillaries (d = 0.3 mm) sealed with Picein. Preliminary data processing was performed in the WinXPow program suite [12]. Crystal structure solution was accomplished using direct methods as implemented in EXPO2009 [13]. Rietveld refinement was performed with the Jana2006 program [14]. Further details on the crystal structure investigations can be obtained from the Fachinformationszentrum Karlsruhe, 76344 Eggenstein–Leopoldshafen, Germany (fax: (+49)7247-808-666; email: [email protected], http:///www.fiz-karlsruhe.de/request_for_deposited_data.html) on quoting the depository number CSD-434473.
Chemical analysis. Chemical analysis was performed for the constituting elements (Ba, Mn, N), as well as for expected impurities (C, H, O, Ta from the crucible). Non-metals were analyzed by a carrier-gas hot-extraction technique on LECO TCH 600 (N, H, O, LECO Corporation, Saint Joseph, MI, USA) and LECO C200 (C, LECO Corporation, Saint Joseph, MI, USA) analyzers. The metal content was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) on an Agilent Technologies 5100 spectrometer (Agilent technologies, Santa Clara, CA, USA).
Differential thermal analysis and thermogravimetry (DTA-TG). Thermal behavior was studied by means of DTA/TG measurements on a Netzsch STA 449C calorimetric setup (NETZSCH-Gerätebau GmbH, Selb, Germany) in loosely closed Ta crucibles under dynamic Ar atmosphere. To prevent sample degradation, the measurements were done inside an Ar-filled glovebox.
Electrical resistivity measurements. Electrical resistivity was measured on a cold-pressed pellet in a sapphire die cell within a cryostat using a four-contact Van-der-Pauw method. The setup was mounted inside an Ar-filled glovebox. The sample was thoroughly ground and sieved before the measurements. Only the fraction with the particle size between 20 μm and 50 μm was used in order to achieve a higher packing density and reduce the grain boundary effects.
Magnetization measurements. Temperature dependence of magnetic susceptibility was measured on a powder sample enclosed in a sealed pre-calibrated quartz tube under 400 mbar of He on a SQUID magnetometer (MPMS-XL7, Quantum Design Inc., San Diego, CA, USA) in external fields between 10 mT and 7 T within the temperature range 1.8–400 K. High-temperature magnetization measurements were performed in the temperature range 320–575 K. All data were corrected for the container diamagnetism. The Honda–Owen correction (“extrapolation to a large field”) was applied to take the possible contributions of ferro- or ferrimagnetic impurities into account [15].
Computational details. Spin-polarized electronic structure calculations were performed at the scalar relativistic level within the L(S)DA approach employing the FPLO-9 [16] or the TB-LMTO-ASA [17] code. The PW92 parametrization [18] of the LSDA functional was used in FPLO and the von Barth-Hedin parametrization [19] was employed in LMTO. Blöchl corrected linear tetrahedron method with a 8 × 12 × 6 k-mesh was employed after checking for convergence with respect to the number of k-points. For the LMTO calculations, experimentally obtained lattice parameters and atomic coordinates were used. The radial scalar-relativistic Dirac equation was solved to get the partial waves. The calculation within the atomic sphere approximation (ASA) includes corrections for the neglect of interstitial regions and partial waves of higher order [20], hence an addition of empty spheres in the case of Ba4[Mn3N6] was not necessary. The following radii of atomic spheres were applied for the calculations on: r(Ba1) = 2.116 Å; r(Ba21) = 2.187 Å, r(Mn1) = 1.415 Å, r(Mn2) = 1.217 Å, r(N1) = 1.094 Å, r(N2) = 1.003 Å and r(N3) = 1.044 Å. A basis set containing Ba(6s,5d), Mn(4s, 4p, 3d) and N(2s,2p) states was employed for the self-consistent calculations with the Ba(6p,4f) and N(3d) functions being downfolded. The electronic structures calculated with the two codes were found to be consistent. For the analysis of the Mn–Mn interactions, Crystal Orbital Hamilton Population (COHP) [21] analysis was performed using the built-in procedure in the TB-LMTO-ASA program. The topology of electron density was analyzed with the program Dgrid [22]. The calculated electron density was integrated in basins, bounded by zero-flux surfaces in the density gradient field [23]. This technique provides electron counts for each atomic basin revealing the effective charges of the QTAIM atoms.
3. Results and Discussion
Crystal structure determination. Ba4[Mn3N6] was obtained as an almost phase-pure microcrystalline product. Attempts to grow single crystals were not successful. Therefore, crystal structure determination was accomplished based on high resolution synchrotron PXRD data (Figure 1). The reflections of the major phase were indexed in the orthorhombic crystal system with the lattice parameters a = 9.9930(1) Å, b = 6.17126(8) Å, c = 14.4692(2) Å. Extinction conditions were consistent only with the space group Pbcn (#60). Crystal structure solution using direct methods provided the positions of all metal atoms and a part of the nitrogen atoms. The remaining nitrogen positions were located in a subsequent difference Fourier synthesis (Figure 2). For Ba atoms, an anisotropic refinement was possible. Mn and N atoms were refined isotropically. In addition, atomic displacement parameters for all N atoms were constrained to be the same in the final cycles of the refinement. The crystallographic data are listed in Table 1, atomic positions in Table 2, atomic displacement parameters in Table 3, and selected bond lengths/angles in Figure 3 and Table S2, respectively.
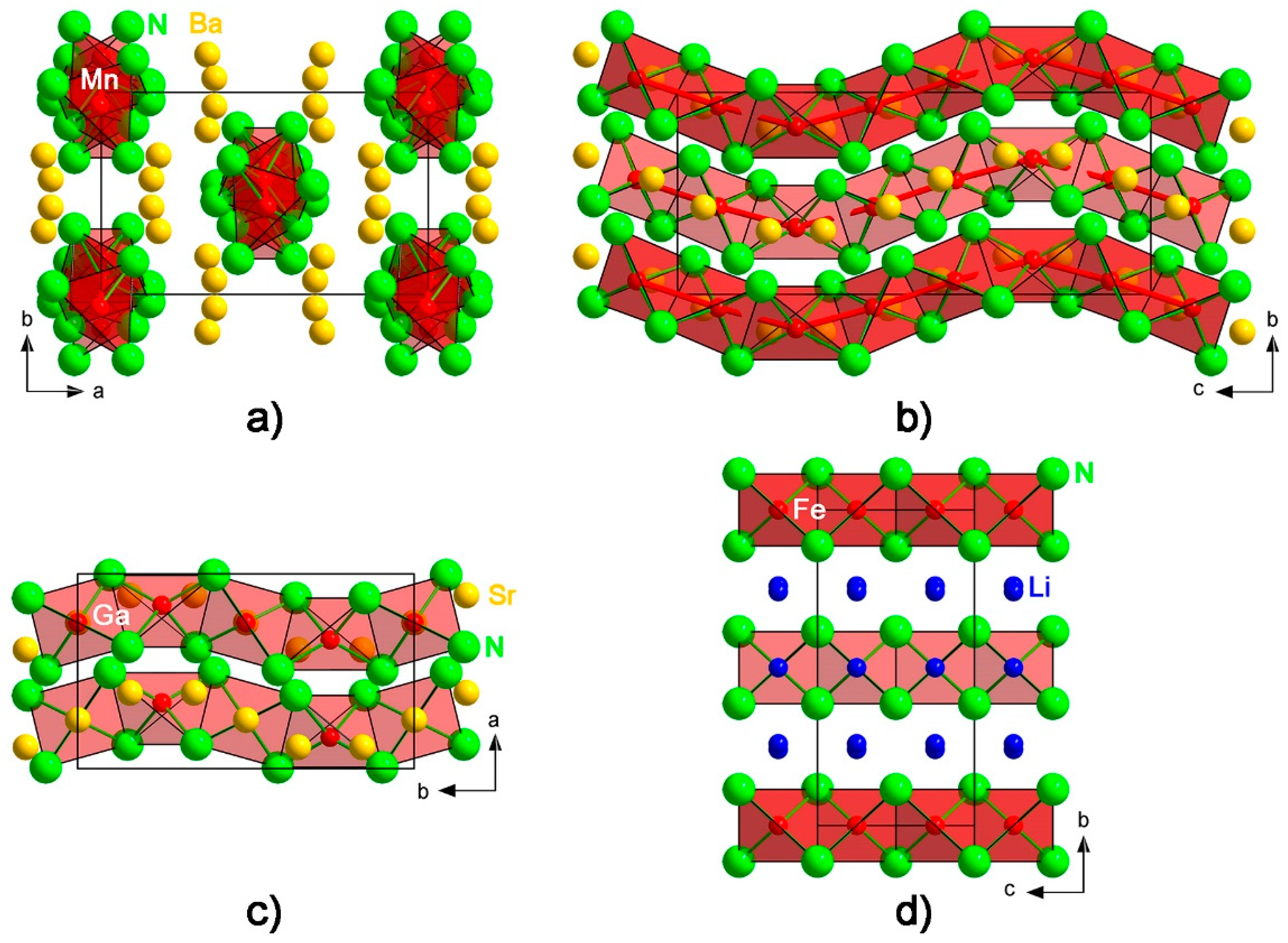
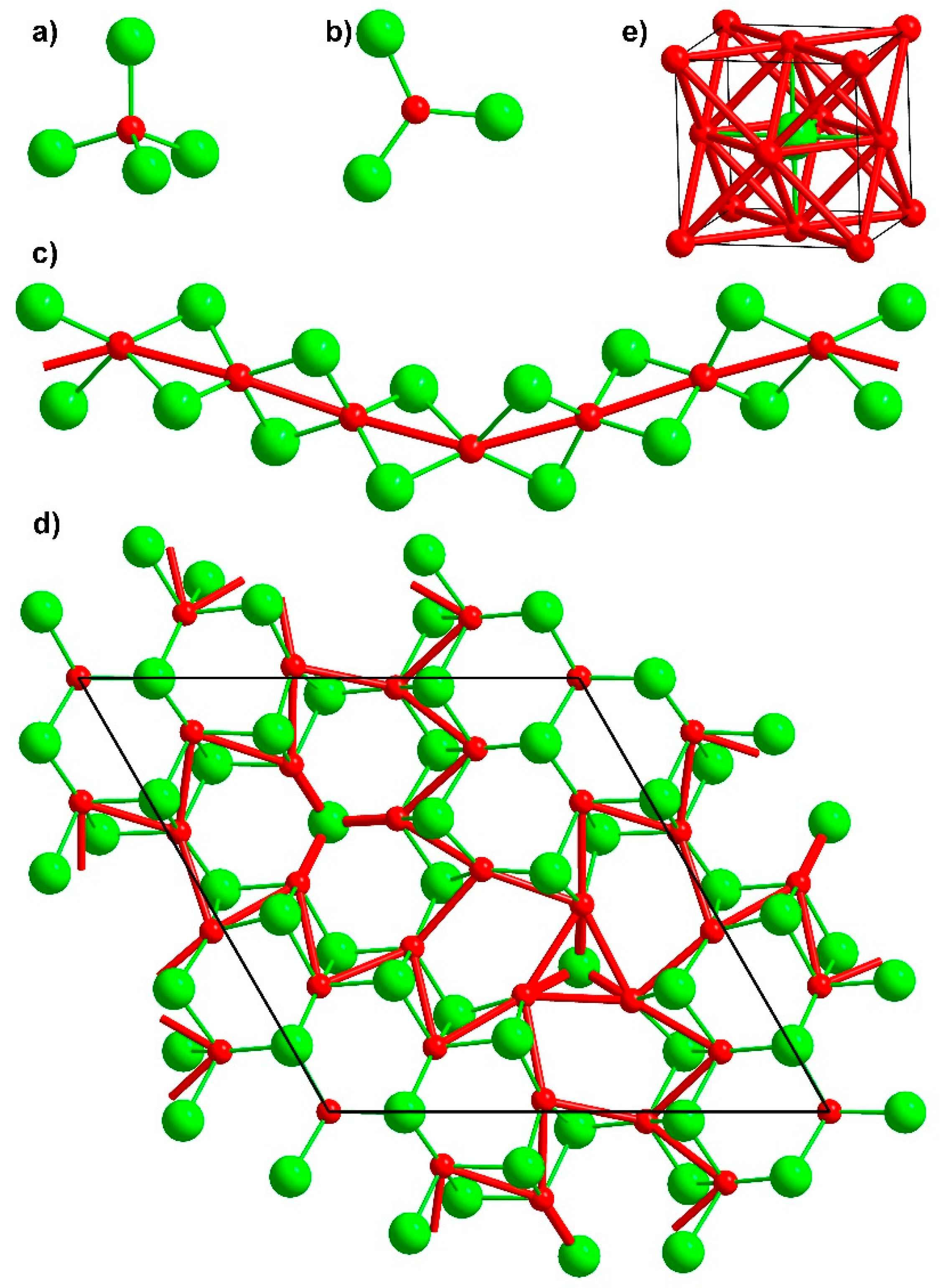
Crystal structure description. The crystal structure of Ba4[Mn3N6] represents a new structure type and can be viewed as consisting of corrugated chains of edge-sharing [MnN4] tetrahedra running along [001], and Ba atoms embedded in-between the chains (Figure 2a,b). A similar structural motif is observed for the nitridometalates Ae3[M2N4] (Ae = Sr, Ba, M = Al, Ga, Ge/Mg) [24,25,26,27,28,29] (Figure 2c), though the degree of the chain corrugation is weaker in these compounds. Perfectly linear chains of edge-sharing [Fe3+N4] tetrahedra are present in the crystal structure of the lithium nitridoferrate Li3[FeN2] [30] (Figure 2d).
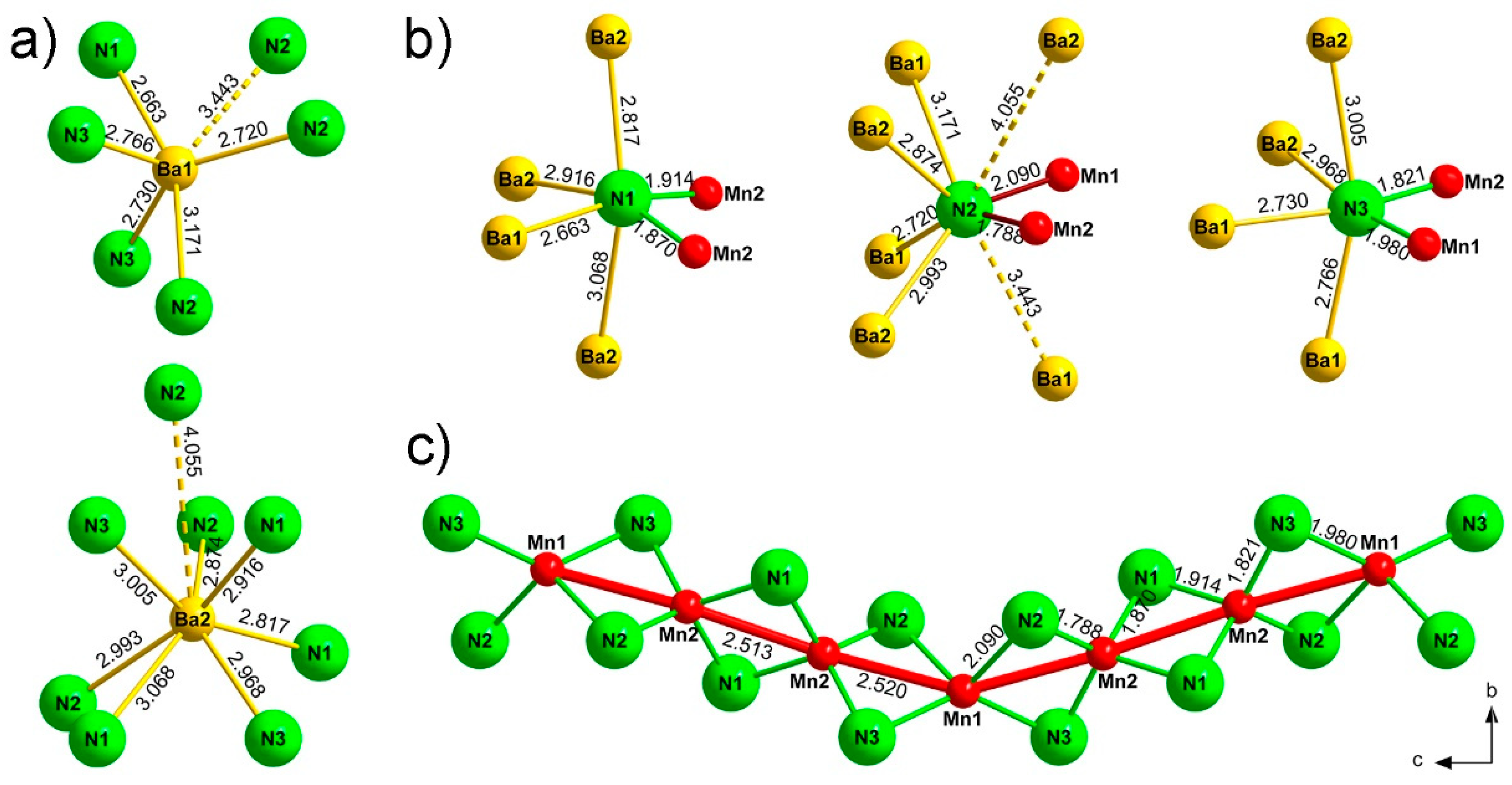
Ba1 and Ba2 atoms are (5 + 1)-fold and (7 + 1)-fold irregularly coordinated, respectively (Figure 3a). For Ba1, the five shortest Ba–N bonds range from 2.66 Å to 3.17 Å. The resulting [Ba1N5] entity resembles a distorted square pyramid, similar to the corresponding coordination polyhedron in another barium nitridomanganate, Ba3[MnN3] [32]. The sixth nitrogen atom is located at a relatively longer distance of 3.44 Å, thereby completing the (5 + 1)-fold coordination environment. Ba2 is coordinated by seven N atoms at distances of 2.82–3.07 Å and a further one at 4.06 Å. These distances are in good agreement with the typical values found in other nitridometalates containing Barium [24,27,29,32].
All nitrogen atoms in the structure have four Ba and two Mn atoms in the closest proximity (Figure 3b). The nitrogen atoms N1 and N3 show a distorted octahedral environment, whereas the coordination environment of N2 resembles a trigonal prism. An additional Ba1 atom at 3.44 Å and the Ba2 atom at the farthest corner residing at a distance of 4.06 Å complete the (6 + 2)-fold coordination of N2. A similar coordination of nitrogen is observed in the above-mentioned Ae3[Al2N4] (AE = Sr, Ba) compounds [24,25] and can be regarded as a strongly distorted square anti-prism. The corresponding longest distance in Ba3[Al2N4] is 4.04 Å [24].
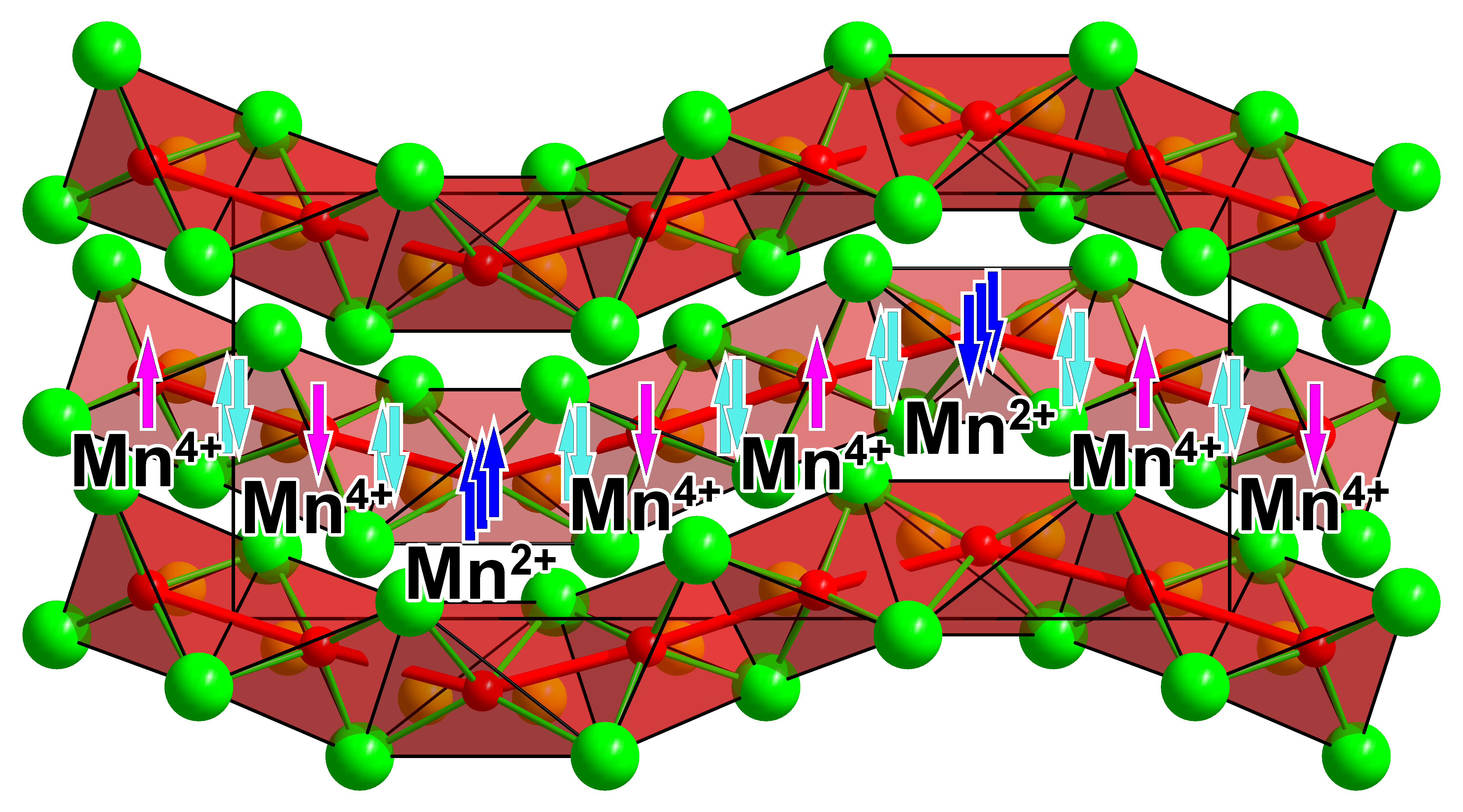
Two symmetrically independent manganese atoms, Mn1 and Mn2, alternate in the sequence [–Mn1–Mn2–Mn2–] along the chain with rather short Mn–Mn distances of 2.51 and 2.52 Å (Figure 3c). These distances fall in the range of the metal-metal contacts in metallic manganese (α-Mn: 2.26–2.93 Å [33]; β-Mn: 2.36–2.68 Å [34]; Mn2N1.08: 2.79–2.82 [35]; Mn4N: 2.74 [36]). The Mn–N contacts lie in the range 1.98–2.09 Å for Mn1 (<d> = 2.04 Å) and 1.79–1.91 Å for Mn2 (<d> = 1.85 Å), with the respective bond valence sums of 2.4 for Mn1 and 3.9 for Mn2 (based on the bond valence parameters from Brese and O’Keeffe [37]). Hence, the oxidation states balance based on the structural data can be expressed as Ba2+4[Mn2+N3−4/2][Mn4+N3−4/2]2.
Distribution of oxidation states in Ba4[Mn3N6] is further corroborated by comparing the Mn–N bond distances with those in other nitridocompounds bearing tetrahedrally coordinated Mn atoms. The range of the Mn–N distances around Mn1 is similar to that in Mn2+[GeN2] (d(Mn–N) = 2.03–2.14 Å, <d> = 2.10 Å) [38] and α-Mn2+[WN2] (d(Mn–N) = 2.01–2.19 Å, <d> = 2.10 Å) [39]. For Mn2, no proper reference compound was found, since there are no phases known with Mn4+ adopting a tetrahedral environment of nitrogen ligands. However, the Mn–N bond length distribution around Mn2 resembles that of Mn atoms in Li7[Mn5+N4] (d(Mn–N) = 1.81–1.83 Å, <d> = 1.82 Å) [40], with the bonds in the latter compound being shorter due to a higher oxidation state of Mn.
The significant difference of the Mn–N bond distances around the two independent Mn sites, which made the above-described oxidation state assignment possible, suggests a charge-ordering scenario, frequently observed for mixed-valent manganese oxides [41]. Thus, K5[Mn3O6] and Rb8[Mn5O10], possessing chains of edge-sharing [MnO4] tetrahedra, were reported to develop full charge order into di- and tri-valent manganese with the formation of the repetition units [–Mn3+–Mn2+–Mn2+–] and [–Mn3+–Mn2+–Mn2+–Mn3+–Mn2+–] for the potassium and rubidium phase, respectively [42]. In the title compound Ba4[Mn3N6], the repetition unit is [–Mn2+–Mn4+–Mn4+–]. Hence, this nitridomanganate does not conform to the known tendency of transition metals to adopt lower oxidation states in nitride compounds in comparison with the oxide analogues [9].
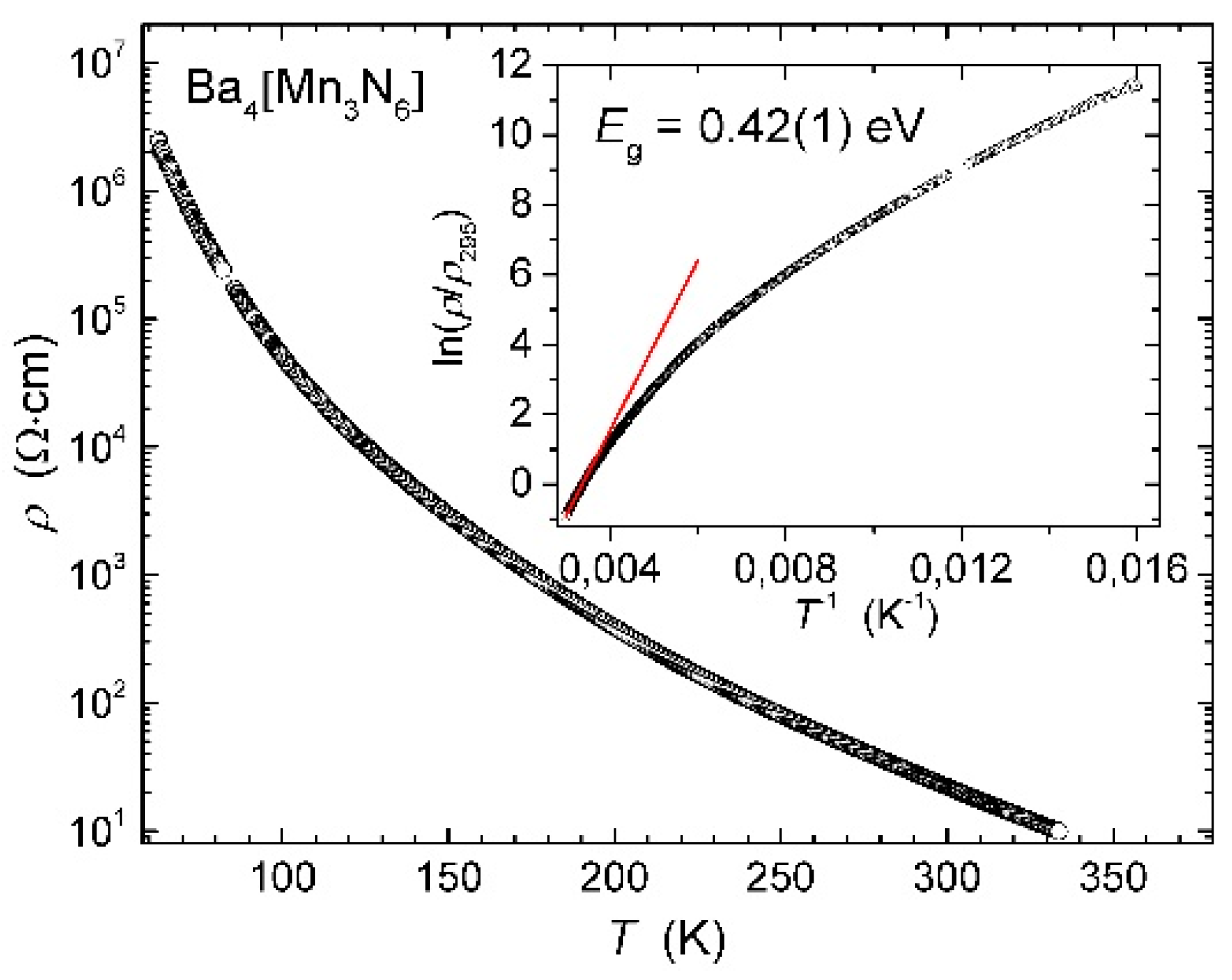
Physical properties. The temperature dependence of the electrical resistivity for Ba4[Mn3N6] is shown in Figure 4. Only data above 62 K were obtained, since at lower temperatures, the resistance exceeds the maximal measurable value achievable with the employed experimental set-up.
At room temperature, the resistivity of the sample amounts to 25 Ω∙cm. The sample displays a distinct semiconducting behavior. As it is seen from the ln(ρ/ρ295) vs. T−1 plot, the temperature behavior of resistivity does not follow a simple Arrhenius-type dependence in a wide temperature range. Linear fitting of the high-temperature region yields an estimated bandgap of 0.42(1) eV. The plot can be linearized in the ln(ρ) vs. T−1/n coordinates, which is frequently discussed as an indication of variable-range hopping conduction. However, a final decision cannot be made since relatively recent calculations showed that such kind of behavior can be observed even for a traditional band transport mechanism [43]. Additional transport measurements on not yet available single crystal samples would be necessary to get a deeper insight into the electrical conduction of Ba4[Mn3N6].
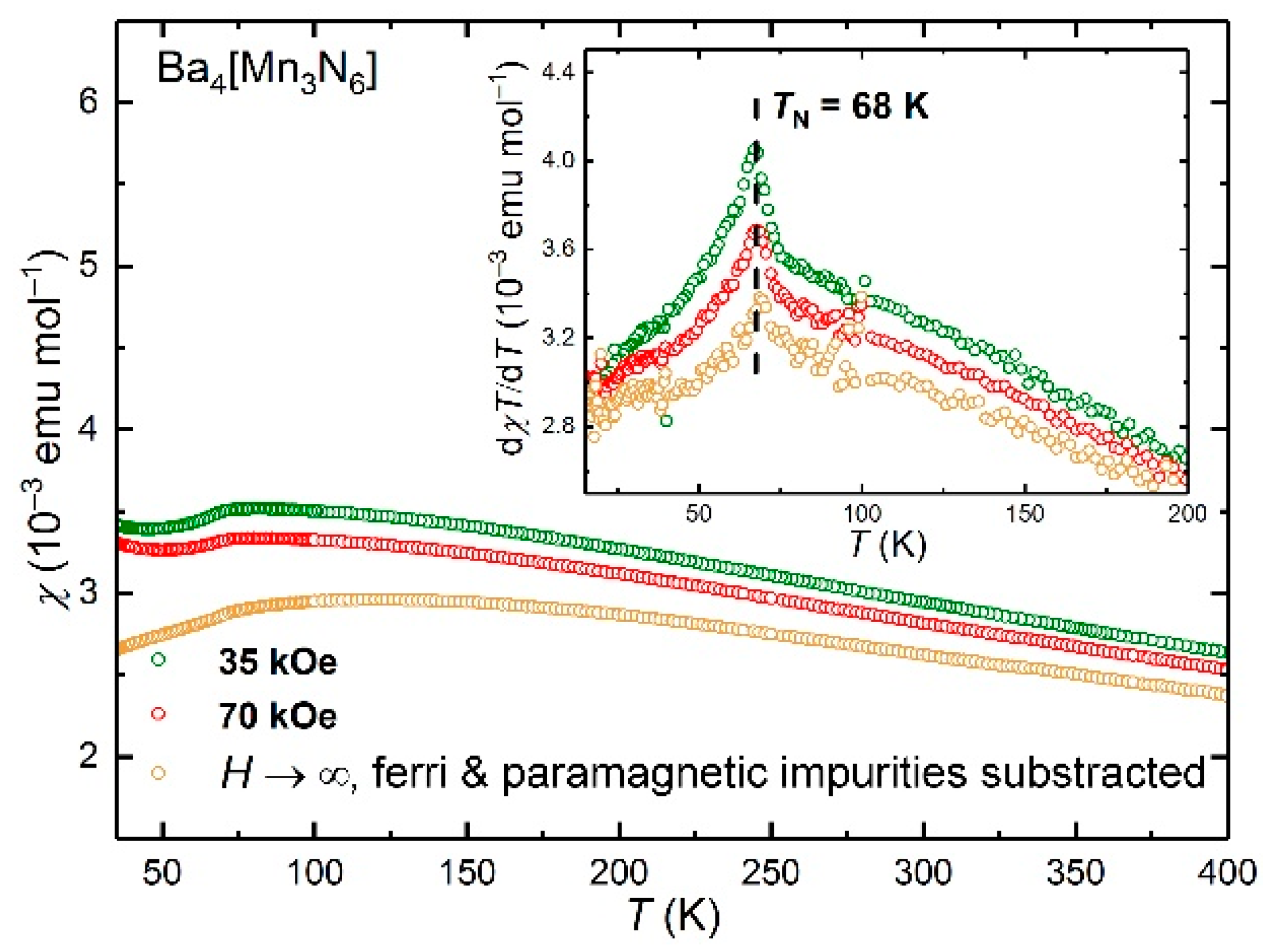
Magnetic susceptibility of Ba4[Mn3N6] versus temperature is given in Figure 5. The observed weak field dependence points to a possible ferro- or ferrimagnetic impurity, most likely ferrimagnetic Mn4N (TC = 740 K) [36]. In this case, the impurity amounts to less than 0.4 mass% as can be estimated from the magnetization data. Due to the small amount, Mn4N was not observed in the PXRD pattern of the sample under study.
The magnetic susceptibility shows an upturn at low temperatures which is probably due to paramagnetic Mn species contained in minor secondary phase(s) or due to point defects in the main phase. After the subtraction of a Curie law with C = 0.0211(2) emu mol−1 K, the susceptibility reveals a broad hump around 120 K and a clear decrease of χ(T) below TN = 68 K, where a sharp kink is observed. Such temperature dependence is typical for low-dimensional (here a quasi-1D) magnetic systems. However, we abstain from a detailed analysis of the magnetic susceptibility data. For a deeper investigation, anisotropic magnetization data on single crystals are required.
Measurements of the susceptibility to higher temperatures are hampered by the degradation of the sample. According to the DTA/TG measurements, Ba4[Mn3N6] starts to lose mass at around 673 K under inert conditions (Figure S2). For these reasons, the magnetic measurements were performed up to 575 K only to avoid possible decomposition. Between 325 and 575 K, the temperature dependence of the magnetic susceptibility was found to decrease almost linearly (Figure S3).
Electronic structure and chemical bonding. Total energy calculations were performed for eight different magnetic arrangements (Figure S4) to discern the ground state. The lowest energy was found for the AFM1 structure, which displays antiferromagnetic coupling (AFM) between manganese atoms along the chains and ferromagnetic (FM) coupling between the nearest manganese sites in adjacent chains. AFM1 with FM coupled AFM chains is only 0.7 meV f.u.−1 more stable than AFM2, possessing AFM coupled AFM chains. It is clear that the actual ground state cannot be reliably determined from LSDA calculations owing to the negligible energy difference between the two best candidates. However, the most stable solution without AFM intra-chain coupling (AFM3 in Figure S3) is by 138 meV f.u.−1 higher in energy than AFM1. These findings emphasize the strongly one-dimensional nature of exchange interactions in Ba4[Mn3N6]. The calculated magnetic moments and the bandgap are almost the same for AFM1 and AFM2 structures (Table 4).
It is worthwhile to note that FM coupled AFM chains were found to be the ground state of another quasi-one-dimensional nitridometalate, Li3[FeN2] [44]. Furthermore, the charge-ordered oxomanganates K5[Mn3O6] and Rb8[Mn5O10] also show a similar magnetic ordering with respect to the inter- and intra-chain couplings [42]. In these two oxides, the calculated magnetic moments on Mn atoms are close to the spin-only values expected for the high-spin states. Our first principle calculations confirm the charge ordering in Ba4[Mn3N6]. QTAIM charges corroborate the oxidation state assignments (Baave2+ + 1.29, Mn12+ + 0.84, Mn24+ + 1.00, Nave3− − 1.33), the low calculated values are in good accordance with other nitridometalates [45]. However, the calculated magnetic moments in the nitridomanganate are reduced by about 2 μB for each species in comparison with the anticipated spin-only values of 5.0 μB and 3.0 μB for high-spin d5 (Mn2+) and d3 (Mn4+) configurations, respectively (Table 4). LSDA calculations are known to underestimate magnetic moments in strongly correlated semiconductors. It is also well known that experimentally determined ordered magnetic moments in highly frustrated systems are often reduced in comparison with spin-only values, typically, as a consequence of quantum fluctuations [46]. Therefore, more experimental data would be necessary to probe the importance of on-site correlations in Ba4[Mn3N6].
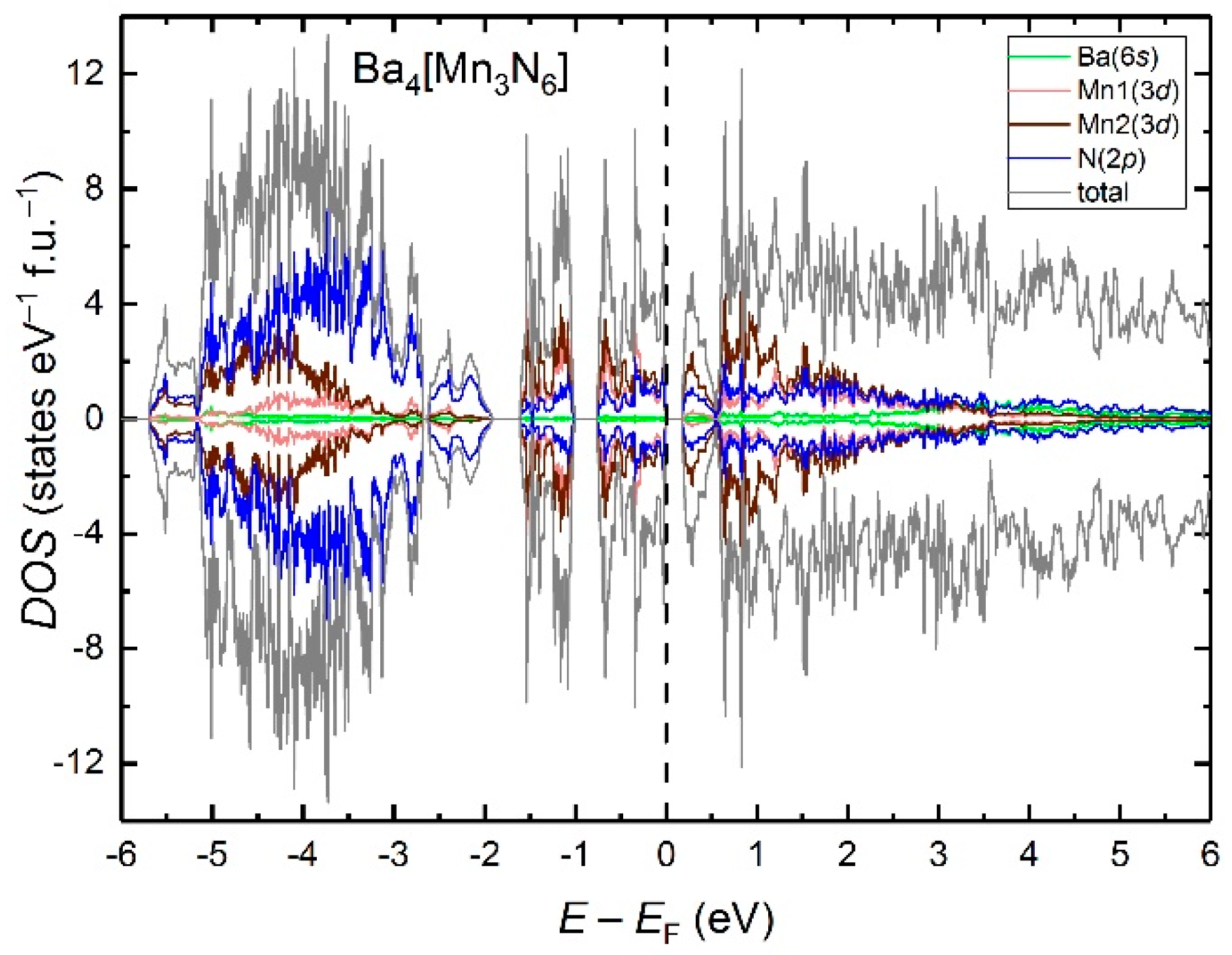
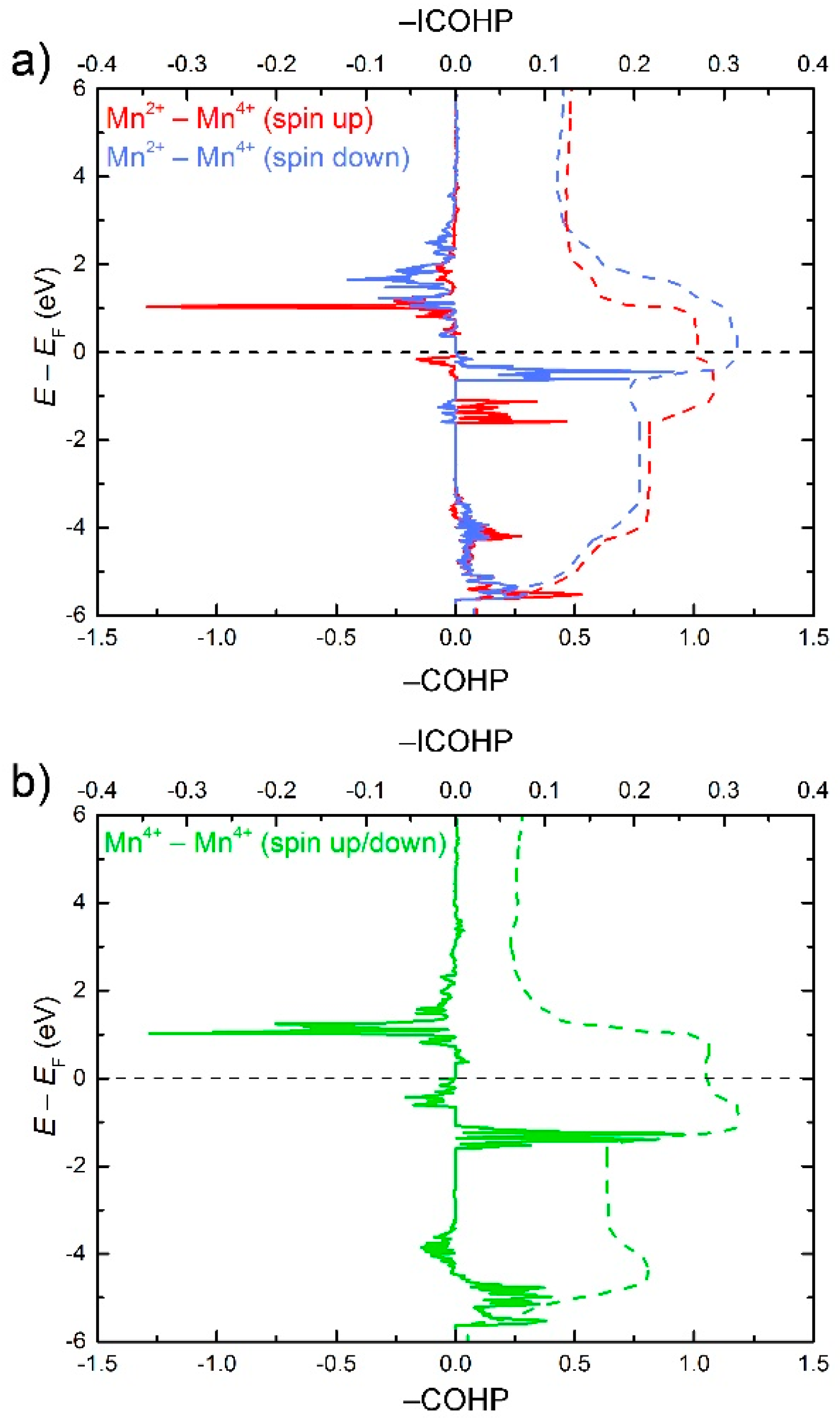
The electronic density of states for Ba4[Mn3N6] in the AFM1 structure is shown in Figure 6. Well below the Fermi level, the DOS is mainly composed of N(2p) states. A significant contribution of the Mn(3d) states is observed close to the Fermi level, where they get hybridized with the N(2p) states. Site-resolved DOS contributions from the Mn(3d) states reveal a considerable hybridization between Mn1(3d) and Mn2(3d) states in the region −1.9 eV < E < 0 eV, indicating possible Mn–Mn interactions. To study these interactions in more detail, we plotted the COHP curves for all pairs of adjacent Mn atoms in the chains (Figure 7). Below EF, all Mn–Mn contacts display a predominantly bonding character with the strongest attractive interactions falling in the energy window above −1.9 eV, in consistence with the region of the d-states hybridization. For Mn2+–Mn4+ (spin up) and Mn4+–Mn4+, the presence of some anti-bonding states just below EF reveals a slight under-optimization of bonding, whereas for Mn2+–Mn4+ (spin down), the bonding appears to be optimized at EF. For all pairs, the integrated COHP (ICOHP) values amount to 0.27–0.3 eV bond−1 spin direction−1.
Interestingly, metal-metal bonding was discussed as a reason for quenching of magnetic moments in certain nitridometalates, e.g., Ca12[Mn19N23] [47], Sc[TaN2] [48], Li6Sr2[Mn2N6] [49] and Ca6[Cr2N6]H [10]. If all adjacent Mn atoms in Ba4[Mn3N6] are linked by 2c–2e bonds, the resulting magnetic moment on every Mn atom will be lowered by 2 μB in comparison with the spin-only value. This is in line with the magnetic moments obtained from our LSDA calculations. Therefore, it can be speculated that the reduced magnetic moments for the Mn species are intrinsic to Ba4[Mn3N6] and stem from the chemical bonding between Mn atoms (Figure 8). The development of the Mn–Mn bonding in Ba4[Mn3N6] is reflected in a much shorter Mn–Mn distance (2.5 Å) in comparison with that in the structurally similar oxomanganates K5[Mn3O6] and Rb8[Mn5O10] with d(Mn-Mn) = 2.7–2.8 Å, which demonstrate the expected magnetic moments on Mn sites [41,42].
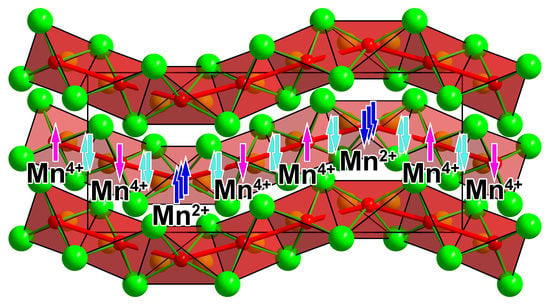
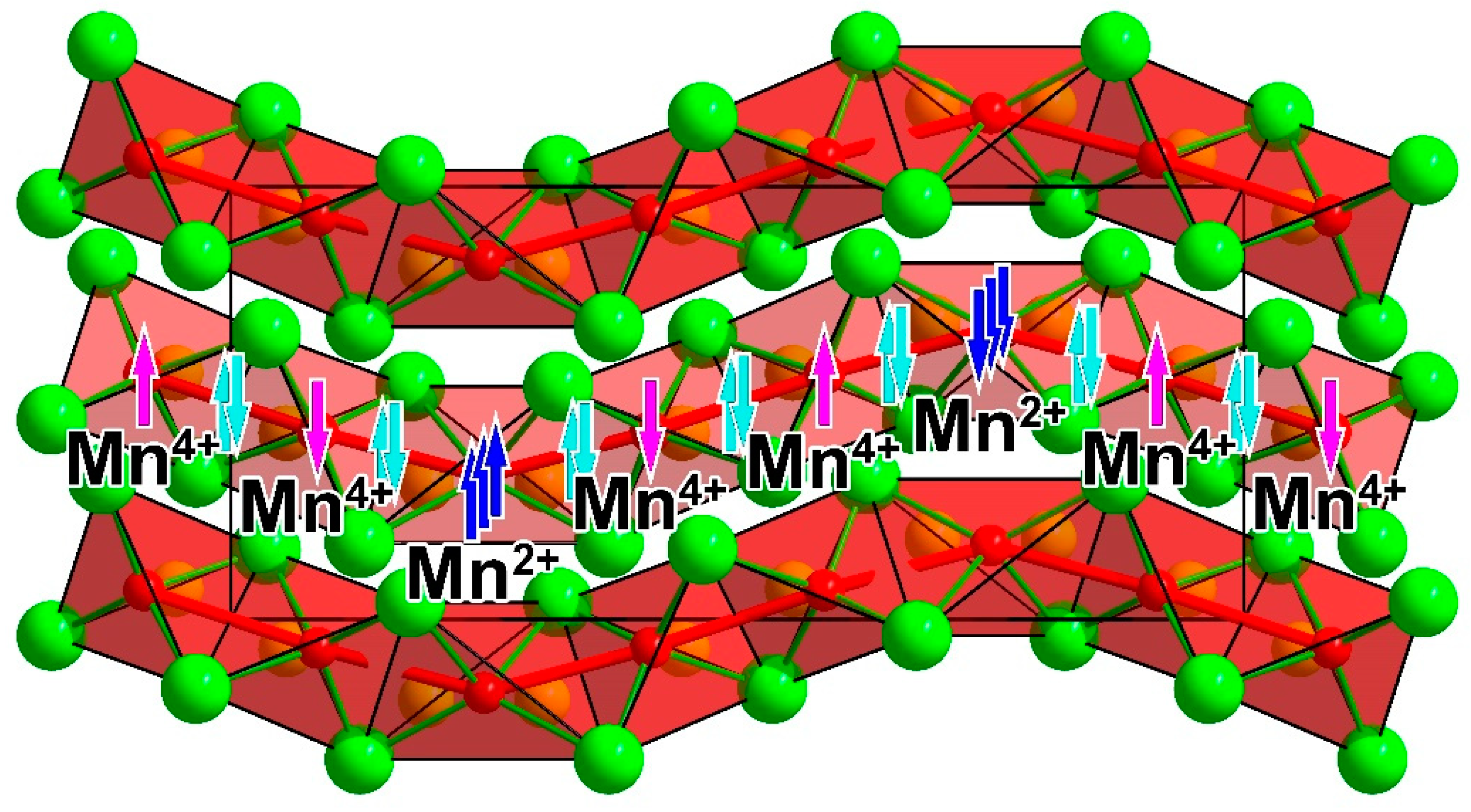
Taking into account the distinctive Mn–Mn interactions, the one-dimensional nitridomanganate anion in Ba4[Mn3N6] can be alternatively understood as a chain of chemically bound Mn atoms decorated by nitride ligands. Such a description provides a link between salt-like alkaline-earth-metal-rich nitridometalates [50], like Li7[MnN4] [40] and Ba3[MnN3] [32], and transition-metal-rich nitrides with pronounced metal-metal interactions, like Ca12[Mn19N23] [47] and Mn4N [36] (Figure 9).
4. Conclusions
The mixed-valent nitridomanganate Ba4[Mn3N6] was obtained by high-temperature nitridation of a Ba2N and Mn mixture. Its crystal structure contains infinite corrugated chains of edge-sharing [MnN4/2] tetrahedra. Analysis of the bond length distribution and first principle calculations suggest a complete charge ordering along the chains with an alternation of Mn2+ and Mn4+ species in the sequence [–Mn2+–Mn4+–Mn4+–]. Ba4[Mn3N6] shows a quasi-one-dimensional magnetic behavior stemming from strong intra-chain and weak inter-chain interactions in this compound, as shown by total energy calculations. In addition, strong bonding interactions were found between Mn atoms along the chains. The emergence of chemical bonding between transition-metal atoms in nitridometalates can provide a link to new structures with extended magnetic topologies.
Supplementary Materials
The following data are available online at https://www.mdpi.com/2073-4352/8/6/235/s1: Results of the chemical analysis, selected bond lengths/angles, DTA-TG curves, high-temperature magnetic susceptibility, and magnetic models used for LSDA calculations. Figure S1. Powder pattern of Ba2N starting material; Figure S2. DTA-TG curves for a Ba4[Mn3N6] sample (m = 41.79 mg). Heating under flowing Ar (100 mL/min) with a rate of 5 K/min. A slight mass gain in the range T = 300–575 K is probably due to sample oxidation by traces of oxygen; Figure S3. Temperature dependence of magnetic susceptibility for Ba4[Mn3N6] between 320 and 575 K; Figure S4. Model magnetic arrangements used for the total energy calculations (FM—ferromagnetic, AFM—antiferromagnetic, FiM—ferrimagnetic); Table S1. Results of chemical analysis for Ba4[Mn3N6]; Table S2. Selected interatomic distances and angles for Ba4[Mn3N6].
Author Contributions
A.O. and P.H. planned and carried out the material synthesis. A.O. and Y.P. planned and carried out the X-ray analysis. A.O., M.B., and W.S. planned and carried out resisitivity and magnetization experiments. A.O. and Y.G. performed quantum chemical calculations. All authors contributed in writing the manuscript. P.H. and Y.G. supervised the project. All authors have given approval to the final version of the manuscript.
Acknowledgments
The authors acknowledge Horst Borrmann and Steffen Hückmann for collecting laboratory PXRD data, Gudrun Auffermann, Anja Völzke, and Ulrike Schmidt for performing chemical analysis, Marcus Schmidt and Susanne Scharsach for conducting DTA/TG measurements, and Ralf Koban for carrying out electrical resistivity measurements. European Synchrotron Radiation Facility (ESRF, Grenoble, France) is acknowledged for providing beamtime at the ID22 beamline managed by Andrew N. Fitch.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, W.X.; Ishikawa, R.; Breedlove, B.; Yamashita, M. Single-chain magnets: Beyond the Glauber model. RSC Adv. 2013, 3, 3772–3798. [Google Scholar] [CrossRef]
- Ma, X.S.; Dakic, B.; Naylor, W.; Zeilinger, A.; Walther, P. Quantum simulation of the wavefunction to probe frustrated Heisenberg spin systems. Nat. Phys. 2011, 7, 399–405. [Google Scholar] [CrossRef]
- Bauer, B.; Cincio, L.; Keller, B.P.; Dolfi, M.; Vidal, G.; Trebst, S.; Ludwig, A.W.W. Chiral spin liquid and emergent anyons in a Kagome lattice Mott insulator. Nat. Commun. 2014, 5, 5137. [Google Scholar] [CrossRef] [PubMed]
- Modic, K.A.; Smidt, T.E.; Kimchi, I.; Breznay, N.P.; Biffin, A.; Choi, S.; Johnson, R.D.; Coldea, R.; Watkins-Curry, P.; McCandless, G.T.; et al. Realization of a three-dimensional spin-anisotropic harmonic honeycomb iridate. Nat. Commun. 2014, 5, 4203. [Google Scholar] [CrossRef] [PubMed]
- Pereiro, M.; Yudin, D.; Chico, J.; Etz, C.; Eriksson, O.; Bergman, A. Topological excitations in a Kagome magnet. Nat. Commun. 2014, 5, 4815. [Google Scholar] [CrossRef] [PubMed]
- Van Well, N.; Foyevtsova, K.; Gottlieb-Schönmeyer, S.; Ritter, F.; Manna, R.S.; Wolf, B.; Meven, M.; Pfleiderer, C.; Lang, M.; Assmus, W.; et al. Low-temperature structural investigations of the frustrated quantum antiferromagnets Cs2Cu(Cl4-xBrx). Phys. Rev. B 2015, 91, 035124. [Google Scholar] [CrossRef]
- Bisogni, V.; Kourtis, S.; Monney, C.; Zhou, K.J.; Kraus, R.; Sekar, C.; Strocov, V.; Büchner, B.; van den Brink, J.; Braicovich, L.; et al. Femtosecond Dynamics of Momentum-Dependent Magnetic Excitations from Resonant Inelastic X-Ray Scattering in CaCu2O3. Phys. Rev. Lett. 2014, 112, 147401. [Google Scholar] [CrossRef] [PubMed]
- Pak, C.; Kamali, S.; Pham, J.; Lee, K.; Greenfield, J.T.; Kovnir, K. Chemical Excision of Tetrahedral FeSe2 Chains from the Superconductor FeSe: Synthesis, Crystal Structure, and Magnetism of Fe3Se4(en)2. J. Am. Chem. Soc. 2013, 135, 19111–19114. [Google Scholar] [CrossRef] [PubMed]
- Kniep, R.; Höhn, P. 2.06—Low-Valency Nitridometalates. In Comprehensive Inorganic Chemistry II, 2nd ed.; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 137–160. [Google Scholar]
- Bailey, M.S.; Obrovac, M.N.; Baillet, E.; Reynolds, T.K.; Zax, D.B.; DiSalvo, F.J. Ca6(Cr2N6)H, the first quaternary nitride–Hydride. Inorg. Chem. 2003, 42, 5572–5578. [Google Scholar] [CrossRef] [PubMed]
- Höhn, P.; Ballé, J.T.; Fix, M.; Prots, Y.; Jesche, A. Single Crystal Growth and Anisotropic Magnetic Properties of Li2Sr[Li1-xFexN]2. Inorganics 2016, 4, 4040042. [Google Scholar] [CrossRef]
- STOE & Cie GmbH. WinXPow; STOE & Cie GmbH: Darmstadt, Germany, 2003. [Google Scholar]
- Altomare, A.; Camalli, M.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R. EXPO2009: Structure solution by powder data in direct and reciprocal space. J. Appl. Crystallogr. 2009, 42, 1197–1202. [Google Scholar] [CrossRef]
- Petřiček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Kristallogr. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Honda, K. Die thermomagnetischen Eigenschaften der Elemente. Ann. Phys. 1910, 337, 1027–1063. [Google Scholar] [CrossRef]
- Koepernik, K.; Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 1999, 59, 1743–1757. [Google Scholar] [CrossRef]
- Jepsen, O.; Burkhardt, A.; Andersen, O.K. The Stuttgart Tight-Binding LMTO-ASA Program; Version 4.7; Max-Planck-Institut für Festkörperforschung: Stuttgart, Germany, 1998. [Google Scholar]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Von Barth, U.; Hedin, L. Local exchange-correlation potential for spin polarized case: 1. J. Phys. C Solid State 1972, 5, 1629–1642. [Google Scholar] [CrossRef]
- Andersen, O.K. Linear Methods in Band Theory. Phys. Rev. B 1975, 12, 3060–3083. [Google Scholar] [CrossRef] [Green Version]
- Dronskowski, R.; Blöchl, P.E. Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Kohout, M. DGRID 4.6; Springer: Radebeul, Germany, 2011. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory, 2003 ed.; Clarendon Press: Oxford, UK, 1994; Volume 22, pp. 1–456. [Google Scholar]
- Ludwig, M.; Niewa, R.; Kniep, R. Dimers [Al2N6]12− and chains 1∞[AlN4/23−] in the crystal structures of Ca6[Al2N6] and Ba3[Al2N4]. Z. Naturforsch. B 1999, 54, 461–465. [Google Scholar] [CrossRef]
- Blase, W.; Cordier, G.; Ludwig, M.; Kniep, R. Sr3[Al2N4]: Ein Nitridoaluminat mit gewellten Tetraederketten 1∞[AlN4/23−]. Z. Naturforsch. B 1994, 49, 501–505. [Google Scholar] [CrossRef]
- Clarke, S.J.; DiSalvo, F.J. Synthesis and structure of one-, two-, and three-dimensional alkaline earth metal gallium nitrides: Sr3Ga2N4, Ca3Ga2Ne4, and Sr3Ga3N5. Inorg. Chem. 1997, 36, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Yamane, H.; DiSalvo, F.J. Ba3Ga2N4. Acta Crystallogr. C 1996, 52, 760–761. [Google Scholar] [CrossRef]
- Park, D.G.; Gál, Z.A.; DiSalvo, F.J. Sr3GeMgN4: New quaternary nitride containing Mg. J. Alloy. Compd. 2003, 360, 85–89. [Google Scholar] [CrossRef]
- Park, D.G.; DiSalvo, F.J. A Structural Comparison between a New Quaternary Nitride, Ba3GeMgN4, and Its Isostructural Sr analogue. Bull. Korean Chem. Soc. 2011, 32, 353–355. [Google Scholar] [CrossRef]
- Gudat, A.; Kniep, R.; Rabenau, A.; Bronger, W.; Ruschewitz, U. Li3FeN2, a ternary nitride with 1∞[FeN3−4/2] chains: Crystal structure and magnetic properties. J. Less Common Met. 1990, 161, 31–36. [Google Scholar] [CrossRef]
- Bérar, J.F.; Lelann, P. E.s.d.s and estimated probable-error obtained in Rietveld refinements with local correlations. J. Appl. Crystallogr. 1991, 24, 1–5. [Google Scholar] [CrossRef]
- Tennstedt, A.; Röhr, C.; Kniep, R. Sr3[MnN3] and Ba3[MnN3], the First Nitridomanganates(III): Trigonal-Planar Anions [MnIIIN3]6−. Z. Naturforsch. B 1993, 48, 794–796. [Google Scholar] [CrossRef]
- Yamada, T.; Fujii, Y. The Crystal Structure of α-Mn Reexamined on Single Crystal Specimens. J. Phys. Soc. Jpn. 1970, 28, 1503–1507. [Google Scholar] [CrossRef]
- Shoemaker, C.B.; Shoemaker, D.P.; Hopkins, T.E.; Yindepit, S. Refinement of the structure of b-manganese and of a related phase in the Mn-Ni-Si system. Acta Crystallogr. B 1978, 34, 3573–3576. [Google Scholar] [CrossRef]
- Aoki, M.; Yamane, H.; Shimada, M.; Kajiwara, T. Single crystal growth of Mn2N using an In-Na flux. Mater. Res. Bull. 2004, 39, 827–832. [Google Scholar] [CrossRef]
- Juza, R.; Deneke, K.; Puff, H. Ferrimagnetismus der Mischkristalle von Mn4N mit Chrom, Eisen und Nickel—41. Mitteilung über Metallnitride und -amide. Z. Elektrochem. 1959, 63, 551–557. [Google Scholar]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. B 1991, 47, 192–197. [Google Scholar] [CrossRef]
- Wintenberger, M.; Guyader, J.; Maunaye, M. Étude cristallographique et magnétique de MnGeN2 par diffraction neutronique. Solid State Commun. 1972, 11, 1485–1488. [Google Scholar] [CrossRef]
- Grins, J.; Käll, P.O.; Svensson, G. Synthesis and structural characterisation of MnWN2 prepared by ammonolysis of MnWO4. J. Mater. Chem. 1995, 5, 571–575. [Google Scholar] [CrossRef]
- Niewa, R.; Wagner, F.R.; Schnelle, W.; Hochrein, O.; Kniep, R. Li24[MnN3]3N2 and Li5[(Li1-xMnx)N]3, Two Intermediates in the Decomposition Path of Li7[MnN4] to Li2[(Li1-xMnx)N]: An Experimental and Theoretical Study. Inorg. Chem. 2001, 40, 5215–5222. [Google Scholar] [CrossRef] [PubMed]
- Coey, J.M.D.; Viret, M.; von Molnar, S. Mixed-valence manganites. Adv. Phys. 1999, 48, 167–293. [Google Scholar] [CrossRef]
- Nuss, J.; Dasari, P.L.V.K.; Jansen, M. K5Mn3O6 and Rb8Mn5O10, New Charge Ordered Quasi One-Dimensional Oxomanganates (II, III). Z. Anorg. Allg. Chem. 2015, 641, 316–321. [Google Scholar] [CrossRef]
- Michel, C.; Baranovskii, S.D.; Klar, P.J.; Thomas, P.; Goldlucke, B. Strong non-Arrhenius temperature dependence of the resistivity in the regime of traditional band transport. Appl. Phys. Lett. 2006, 89, 112116. [Google Scholar] [CrossRef]
- Ming, X.; Wang, X.L.; Du, F.; Han, B.; Wang, C.Z.; Chen, G. Unusual intermediate spin Fe3+ ion in antiferromagnetic Li3FeN2. J. Appl. Phys. 2012, 111, 063704. [Google Scholar] [CrossRef]
- Bronger, W.; Baranov, A.; Wagner, F.R.; Kniep, R. Atom Volumina and Charge Distributions in Nitridometalates. Z. Anorg. Allg. Chem. 2007, 633, 2553–2557. [Google Scholar] [CrossRef]
- Jiang, H.C.; Kruger, F.; Moore, J.E.; Sheng, D.N.; Zaanen, J.; Weng, Z.Y. Phase diagram of the frustrated spatially-anisotropic S = 1 antiferromagnet on a square lattice. Phys. Rev. B 2009, 79, 174409. [Google Scholar] [CrossRef]
- Ovchinnikov, A.; Bobnar, M.; Prots, Y.; Borrmann, H.; Sichelschmidt, J.; Grin, Y.; Höhn, P. Ca12[Mn19N23] and Ca133[Mn216N260]: Structural complexity by 2D intergrowth. Angew. Chem. 2018. [Google Scholar] [CrossRef]
- Niewa, R.; Zherebtsov, D.A.; Schnelle, W.; Wagner, F.R. Metal-metal bonding in ScTaN2. A new compound in the system ScN-TaN. Inorg. Chem. 2004, 43, 6188–6194. [Google Scholar] [CrossRef] [PubMed]
- Bolvin, H.; Wagner, F.R. Case of a Strong Antiferromagnetic Exchange Coupling Induced by Spin Polarization of a Mn-Mn Partial Single Bond. Inorg. Chem. 2012, 51, 7112–7118. [Google Scholar] [CrossRef] [PubMed]
- Höhn, P.; Niewa, R. Nitrides of Non-Main Group Elements. In Handbook of Solid State Chemistry, Part 1. Materials and Structure of Solids; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; pp. 251–359. [Google Scholar]
Figure 1.
Synchrotron PXRD pattern of Ba4[Mn3N6] (λ = 0.35434 Å) with experimental points shown in black, calculated pattern after Rietveld refinement in red, and difference curve in blue. Tick marks denote the calculated positions of Bragg reflections.
Figure 1.
Synchrotron PXRD pattern of Ba4[Mn3N6] (λ = 0.35434 Å) with experimental points shown in black, calculated pattern after Rietveld refinement in red, and difference curve in blue. Tick marks denote the calculated positions of Bragg reflections.

Figure 2.
Crystal structure of Ba4[Mn3N6] viewed along (a) [001] and (b) [100], for comparison, chains of edge-sharing tetrahedra in (c) Sr3[Ga2N4] [26] and (d) Li3[FeN2] [30] are drawn on the same scale.

Figure 3.
Coordination environment of (a) barium, (b) nitrogen, and (c) manganese atoms in Ba4[Mn3N6]. Distances are given in Å.
Figure 3.
Coordination environment of (a) barium, (b) nitrogen, and (c) manganese atoms in Ba4[Mn3N6]. Distances are given in Å.

Figure 4.
Temperature dependence of electrical resistivity for polycrystalline Ba4[Mn3N6]. Inset: ln(ρ/ρ295) vs. T−1 plot (circles) with a linear fit (red line).
Figure 4.
Temperature dependence of electrical resistivity for polycrystalline Ba4[Mn3N6]. Inset: ln(ρ/ρ295) vs. T−1 plot (circles) with a linear fit (red line).

Figure 5.
Temperature dependence of magnetic susceptibility for Ba4[Mn3N6] up to 400 K measured in different fields. Inset: dχT/dT(T) plot emphasizing the AFM transition.
Figure 5.
Temperature dependence of magnetic susceptibility for Ba4[Mn3N6] up to 400 K measured in different fields. Inset: dχT/dT(T) plot emphasizing the AFM transition.

Figure 6.
LSDA electronic density of states for Ba4[Mn3N6] in the AFM1 structure. Positive and negative DOS values correspond to major and minor spin channels, respectively.
Figure 6.
LSDA electronic density of states for Ba4[Mn3N6] in the AFM1 structure. Positive and negative DOS values correspond to major and minor spin channels, respectively.

Figure 7.
COHP curves for the Mn–Mn nearest neighbor interactions in Ba4[Mn3N6]: (a) Mn2+–Mn4+; (b) Mn4+–Mn4+. For symmetry reasons, the COHP curves for the Mn4+–Mn4+ spin up and spin down contacts overlap.
Figure 7.
COHP curves for the Mn–Mn nearest neighbor interactions in Ba4[Mn3N6]: (a) Mn2+–Mn4+; (b) Mn4+–Mn4+. For symmetry reasons, the COHP curves for the Mn4+–Mn4+ spin up and spin down contacts overlap.

Figure 8.
Schematic representation of the possible valence d-electron distribution along the chains in Ba4[Mn3N6]. Unpaired electrons on Mn2+ and Mn4+ are shown in blue and pink, respectively. Electron pairs representing Mn–Mn bonds are shown in cyan.
Figure 8.
Schematic representation of the possible valence d-electron distribution along the chains in Ba4[Mn3N6]. Unpaired electrons on Mn2+ and Mn4+ are shown in blue and pink, respectively. Electron pairs representing Mn–Mn bonds are shown in cyan.

Figure 9.
Mn–N and Mn–Mn interactions in nitridomanganates and manganes nitrides from 0D to 3D systems: (a) Li7[MnN4] [40]; (b) Ba3[MnN3] [32]; (c) Ba4[Mn3N6]; (d) Ca12[Mn19N23] [47]; (e) Mn4N [36].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystallographic Data and Experimental Details for Ba4[Mn3N6] at 298 K.
| Composition | Ba4Mn3N6 |
|---|---|
| Molecular weight/g mol−1 | 798.16 |
| Space group | Pbcn (#60) |
| Lattice parameters 1 | |
| a/Å | 9.9930(1) |
| b/Å | 6.17126(8) |
| c/Å | 14.4692(2) |
| V/Å3 | 892.31(2) |
| Z | 4 |
| ρcalcd/g cm−3 | 5.94 |
| T/K | 298 |
| Device | beamline ID22, ESRF |
| Radiation, λ/Å | 0.35434 |
| 2Θ max/o | 38 |
| 2Θ step/o | 0.002 |
| RI/Rp | 0.044/0.058 |
| Residual electron density peaks/e Å−3 | +1.10, −0.94 |
1 The standard deviations include the Bérar–Lelann’s correction [31].
Table 2.
Atomic Positions and Isotropic (Equivalent) Displacement Parameters (Å2) for Ba4[Mn3N6] 1.
| Atom | Site | x | y | z | Uiso*/Ueq |
|---|---|---|---|---|---|
| Ba1 | 8d | 0.82617(7) | 0.68848(10) | 0.19158(4) | 0.00687(19) |
| Ba2 | 8d | 0.65129(6) | 0.93009(10) | 0.94504(4) | 0.0077(2) |
| Mn1 | 4c | 0 | 0.1719(4) | 1/4 | 0.0066(6)* |
| Mn2 | 8d | 0.00844(13) | 0.0660(3) | 0.08193(10) | 0.0055(4)* |
| N1 | 8d | 0.9009(7) | 0.8512(11) | 0.0291(5) | 0.0067(11)* 2 |
| N2 | 8d | 0.1212(7) | 0.9716(10) | 0.1690(5) | 0.0067* 2 |
| N3 | 8d | 0.9288(7) | 0.3008(12) | 0.1347(5) | 0.0067* 2 |
1 The standard deviations include the Bérar–Lelann’s correction [31]; 2 Uiso(N1) = Uiso(N2) = Uiso(N3) constrained.
Table 3.
Anisotropic Displacement Parameters (Å2) for Ba4[Mn3N6] 1.
| Atom | U11 | U22 | U33 | U12 | U13 | U23 |
|---|---|---|---|---|---|---|
| Ba1 | 0.0088(3) | 0.0061(3) | 0.0058(3) | 0.0003(4) | 0.0016(3) | −0.0008(3) |
| Ba2 | 0.0066(3) | 0.0079(3) | 0.0085(4) | 0.0004(3) | −0.0015(4) | −0.0011(4) |
1 The standard deviations include the Bérar-Lelann’s correction [31].
Table 4.
Results of the LSDA calculations for magnetic structures AFM1 and AFM2 (FPLO).
| Structure | AFM1 | AFM2 |
|---|---|---|
| energy E with respect to AFM1 (meV f.u.−1) | 0 | 0.7 |
| electronic bandgap Eg (eV) | 0.17 | 0.18 |
| magnetic moment on Mn2+ (μB) | 2.96 | 2.96 |
| magnetic moment on Mn4+ (μB) | 1.10 | 1.09 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ovchinnikov, A.; Bobnar, M.; Prots, Y.; Schnelle, W.; Höhn, P.; Grin, Y. Ba4[Mn3N6], a Quasi-One-Dimensional Mixed-Valent Nitridomanganate (II, IV). Crystals 2018, 8, 235. https://doi.org/10.3390/cryst8060235
AMA Style
Ovchinnikov A, Bobnar M, Prots Y, Schnelle W, Höhn P, Grin Y. Ba4[Mn3N6], a Quasi-One-Dimensional Mixed-Valent Nitridomanganate (II, IV). Crystals. 2018; 8(6):235. https://doi.org/10.3390/cryst8060235
Chicago/Turabian StyleOvchinnikov, Alexander, Matej Bobnar, Yurii Prots, Walter Schnelle, Peter Höhn, and Yuri Grin. 2018. "Ba4[Mn3N6], a Quasi-One-Dimensional Mixed-Valent Nitridomanganate (II, IV)" Crystals 8, no. 6: 235. https://doi.org/10.3390/cryst8060235
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.