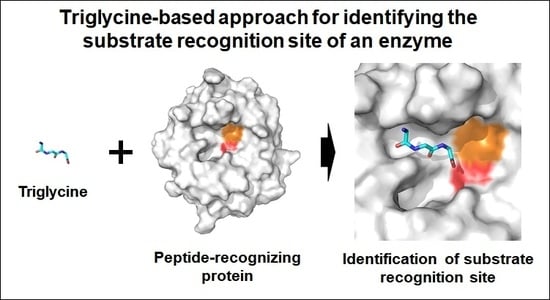
Triglycine-Based Approach for Identifying the Substrate Recognition Site of an Enzyme


1
Division of Biotechnology, Korea University, Seoul 02481, Korea
2
Institute of Life Science and Natural Resources, Korea University, Seoul 02481, Korea
Crystals 2019, 9(9), 444; https://doi.org/10.3390/cryst9090444
Submission received: 10 August 2019
/
Revised: 23 August 2019
/
Accepted: 24 August 2019
/
Published: 26 August 2019
(This article belongs to the Special Issue Crystallographic Studies of Enzymes)
Abstract
:Various peptides or non-structural amino acids are recognized by their specific target proteins, and perform a biological role in various pathways in vivo. Understanding the interactions between target protein and peptides (or non-structural amino acids) provides key information on the molecular interactions, which can be potentially translated to the development of novel drugs. However, it is experimentally challenging to determine the crystal structure of protein–peptide complexes. To obtain structural information on the substrate recognition of the peptide-recognizing enzyme, X-ray crystallographic studies were performed using triglycine (Gly-Gly-Gly) as the main-chain of the peptide. The crystal structure of Parengyodontium album Proteinase K in complex with triglcyine was determined at a 1.4 Å resolution. Two different bound conformations of triglycine were observed at the substrate recognition site. The triglycine backbone forms stable interactions with β5-α4 and α5-β6 loops of the main-chain. One of the triglycine-binding conformations was identical to the binding mode of a peptide-based inhibitor from a previously reported crystal structure of Proteinase K. Triglycine has potential application in X-ray crystallography in order to identify the substrate recognition sites in the peptide binding enzymes.
1. Introduction
Protein-protein interactions (PPIs) regulate most cellular processes, such as enzyme activities, intracellular localization, and/or physical interactions [1,2]. PPIs can be broadly classified into two types, protein-protein interactions (between domains of two proteins) and protein-peptide interactions (between a protein and linear sequence of other protein) [3]. In PPIs, proteins recognize the short peptide sequences in either a sequence-dependent or -independent manner [4]. Peptides bind to the cavities, grooves, or pockets in either the active site or substrate recognition sites on the proteins. Alternately, the peptides interact with the β-strands on the protein surface [4]. An investigation of the PPIs is necessary in order to understand the mechanistic details of the biological processes, and to develop novel drugs [5,6,7]. Among the various methods for PPI-based drug development, a structure-based approach has been widely employed, as in fragment-based drug discovery (FBDD) [8], so as to identify potential lead compounds for drug development. Although the initial success is usually less than that obtained by high-throughput screening (HTS) techniques, FBDD is considered to be more efficient in the optimization phase [9,10].
Peptide-based fragments can be used to understand the structural and mechanistic features of PPIs, and to obtain information on FBDD. I hypothesized that a crystallographic study using triglycine (Gly-Gly-Gly or glycyl-glycyl-glycine) molecules could provide information on the main-chain interactions of PPIs. Because triglycine, unlike other amino acid fragments, had a high degree of freedom in the torsion angle of the main chain, it was expected that the triglycine could be applied for the identification of the peptide binding site in peptide-recognizing proteins. To demonstrate this hypothesis, proteinase K (EC 3.4.21.64) from fungus Parengyodontium album (formerly Tritirachium album), named PaProK, was used as a model in this experiment. This enzyme is a broad-spectrum serine protease, and predominantly cleaves the internal peptide bonds adjacent to the carboxyl group of aliphatic and aromatic hydrophobic amino acids [11]. The active site consists of a catalytic triad Asp39-His69-Ser224, and the substrate recognition site is formed by Gly100-Tyr104 and Ser132-Gly136, which form a triple antiparallel β-strand with the substrate [12].
To investigate the structural features of PPI, here, I attempted X-ray crystallographic study using triglycine, and determined the crystal structure of the PaProK-triglycine complex at a 1.4 Å resolution. The triglycine was bound in two conformations to the substrate recognition site between β5–α4 and α5–β6 loops in PaProK. The conformations of the bound triglycine were compared with previously reported crystal structures of PaProK complexed with a peptide-based inhibitor. This structural investigation of the triglycine-PaProK interaction could provide useful information for understanding PPI.
2. Materials and Methods
2.1. Sample Preparation
Proteinase K from Parengyodontium album was purchased from Geogiachem (PR3050). The protein was dissolved and resuspended in 50 mM HEPES-NaOH (pH 7.0), and crystallized at 22 °C using the hanging-drop vapor diffusion method. The crystallization drops were set by mixing 1 µl of a PaProK (30 mg/ml) solution with 1 µl of a reservoir solution (0.1 M Tris-HCl at pH 8.0 and 2.0 M ammonium sulfate), and incubated with 0.5 ml of a reservoir solution. Suitable crystals (0.15 × 0.10 × 0.10 mm) for X-ray diffraction were obtained within two weeks. For the PaProK-triglycine complex crystals, 2 μl of a 0.3 M triglycine solution was added to the crystallization drop and was incubated for 30 min.
2.2. X-Ray Diffraction and Data Processing
The crystals were transferred to a reservoir buffer containing 25% (v/v) glycerol as cryoprotectant. The diffraction data were collected at 100 K on beamline 11C at the Pohang Light Source II (PLS-II, Pohang, South Korea) using a Pilatus 6 M detector [13]. The diffraction data sets were processed and reduced using the HKL2000 program [14]. Data collection statistics are presented in Table 1
2.3. Structure Determination
The PaProK structure was solved by the molecular replacement method using MOLREP [15] from the CCP4 program suite [16], with the coordinates from the Proteinase K structure (PDB: 1IC6) [17], as the search model. Manual model building and refinement were performed using COOT [18] and phenix.refinement in PHENIX [19], respectively. The refinement statistics are presented in Table 1. The figures were generated with PyMOL [20]. The final structures were validated by MolProbity [21]. The coordinates and structural factors have been deposited in the Protein Data Bank with an accession code (6KKF).
3. Results
3.1. Overall Structure
The crystals of the PaProK-triglycine complex belonged to the monoclinic space group P21, containing one molecule in the asymmetric unit. The electron density map was clear for the interpretation from Ala106 to Gln383. Bragg peaks were observed even at the around 1.1 Å resolution, but did not satisfy reliable data completeness, thus the diffraction data was processed to a 1.4 Å resolution. The final structure of PaProK-triglycine was refined to a 1.4 Å resolution, with Rwork and Rfree values of 18.22 and 21.81%, respectively.
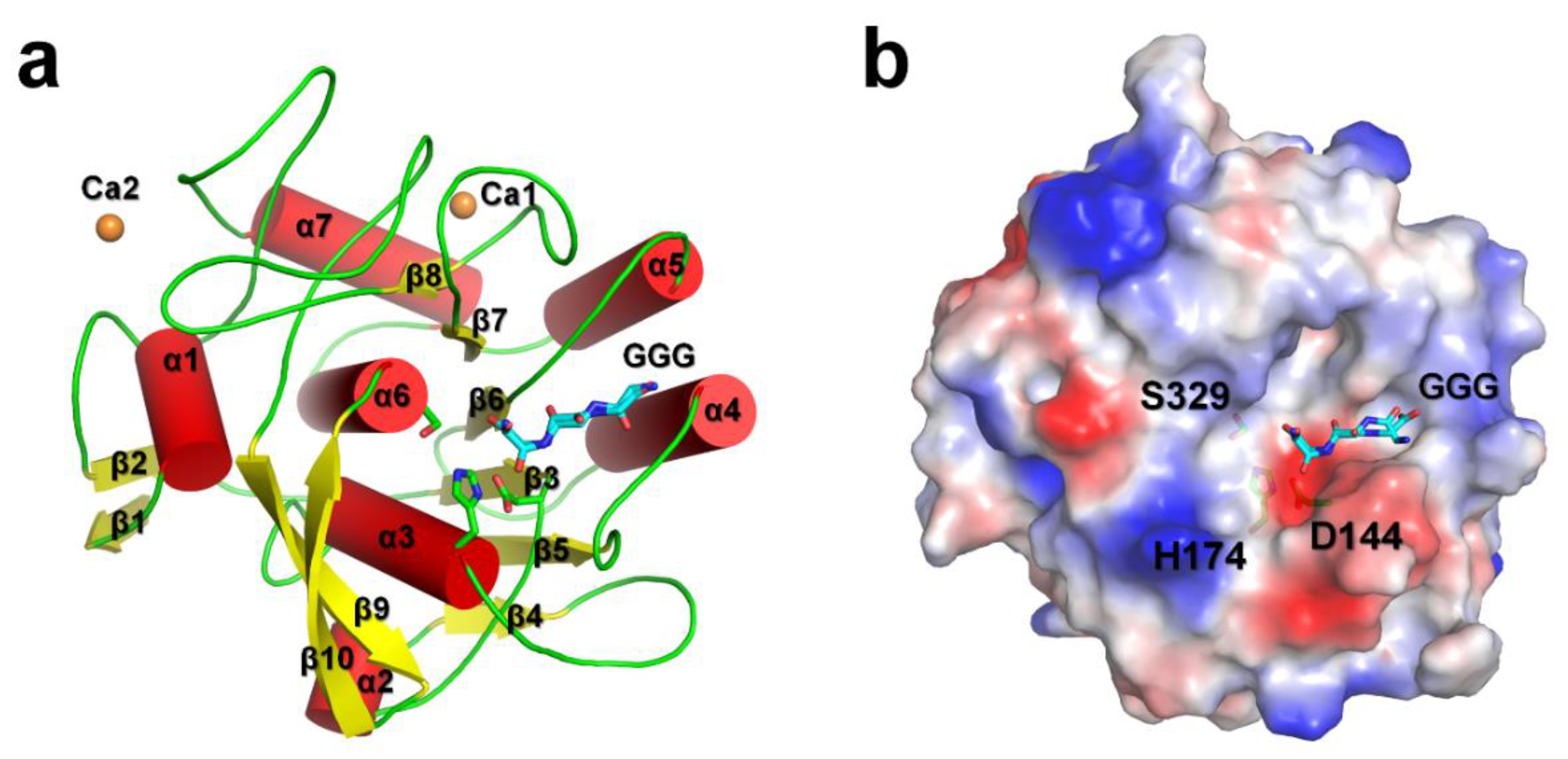
PaProK displayed a typical αβ-hydrolase fold with seven α-helices and ten β-strands. The six central β-strands (β3–β8) are surrounded by five peripheral α-helices (α3–α7; Figure 1a). This structure is similar to the previously reported crystal structures of PaProK (PDB code: 1IC6) [17], with a root-mean-square deviation (RMSD) of 0.2892 Å. The Ramachandran plot revealed that the geometry of Asp144 on the β3–α2 loop is distorted because of its torsion angles, phi (Φ) and psi (Ψ), at −172° and −142°, respectively. The Asp144 residue is part of the catalytic triad with His222 and Ser329. This distorted geometry of the catalytic triad is often observed in the serine proteases or esterase [22,23,24,25]. The Oδ1 and Oδ2 atoms of the Asp144 residue (charge-relay) are stabilized by hydrogen bonds with the Nδ atom of His174 (proton carrier), which are at a distance of 2.70 and 3.03 Å, respectively.
The electron density map clearly shows that PaProK contains two Ca2+ ions (named, Ca1 and Ca2; Figure 1a). These Ca2+ ions are involved in the thermal stability, but not in the proteolytic activity [12]. The B-factors for the entire protein, Ca1, and Ca2, at 11.89 Å2, 7.4 Å2, and 28.0 Å2, respectively, indicate that Ca1 binds the protein more strongly than Ca2. Ca1 was octahedrally coordinated by carbonyl groups of Pro280 (2.42 Å) and Val282 (2.39 Å), two carboxylate oxygens Asp305 (2.65 Å and 2.40 Å for Oδ1 and Oδ2, respectively), and four water molecules (2.33–2.51 Å). Ca2 was coordinated by carbonyl groups of Thr121 (2.49 Å), Asp365 (2.44 Å and 2.36 Å for Oδ1 and Oδ2, respectively), and one water molecule (2.46 Å). The triglycine is located on the negatively charged substrate binding pocket around the catalytic triad (Figure 1b).
3.2. Triglycine Binding Site on PaProK
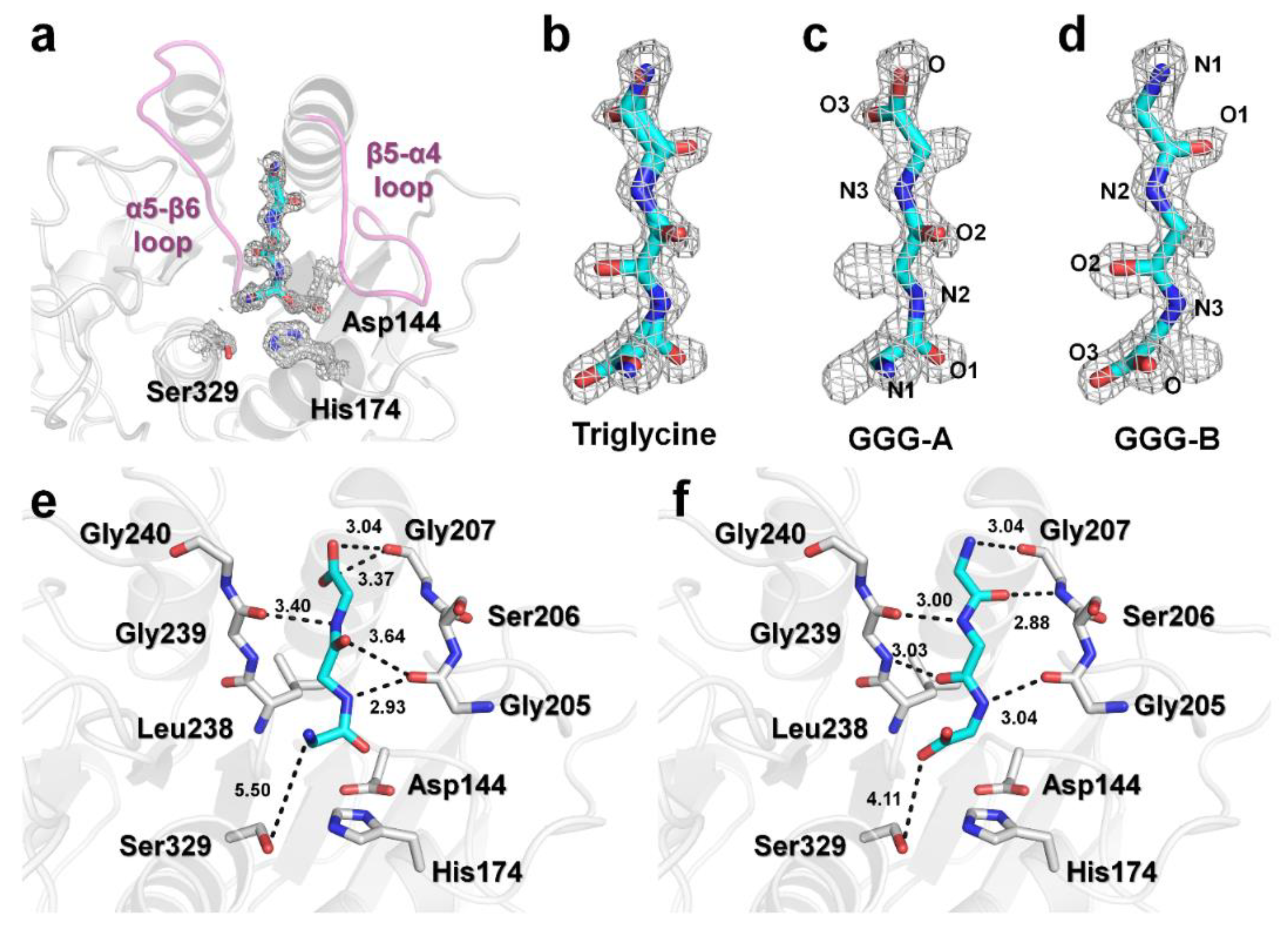
The electron density for triglycine was found at the substrate binding pocket between the β5–α4 and α5–β6 loops around the active site (Figure 2a and Supplementary Figure S1) [17]. The electron density for the triglycine molecule was not observed at any other positions, indicating that triglycine can bind only to the peptide binding site on PaProK. Interestingly, the electron density map of triglycine reveals two binding conformations, GGG-A and GGG-B (Figure 2b); while the amino-terminus of triglycine is directed toward PaProK active site in GGG-A (Figure 2c), the carboxy-terminus is directed toward the active site in GGG-B (Figure 2d). Refinement shows that GGG-A and GGG-B had a 0.56:0.44 ratios of occupancy on PaProK. The N2, N3, O2, O3, and carboxyl oxygen atoms of GGG-A interact with the oxygen atoms of Gly205 (2.93 Å), Gly239 (3.40 Å), Gly205 (3.64 Å), Gly207 (3.37 Å), and Gly207 (3.04 Å), respectively (Figure 2e). The N1 atom of triglycine is 5.5 Å away from the hydroxyl group of Ser329. The N1, O1, N2, O2, and N3 atoms of GGG-B interact with the oxygen atom of Gly207 (3.04 Å), nitrogen atom of Gly207 (2.88 Å), oxygen atom of Gly239 (3.00 Å), nitrogen atom of Gly239 (3.03 Å), and the oxygen atom of Gly207 (3.04 Å), respectively (Figure 2f). The O3 atom of triglycine is 4.2 Å away from the hydroxyl group of Ser329. As a result, the triglycine backbone of both conformations can be stably maintained by hydrogen bonds on the substrate binding site. In particular, GGG-B forms a triple antiparallel β-strand with the β5-α4 and α5-β6 loops. While the proteins involved in PPI generally recognize specific amino acid sequences of the substrate, triglycine, because of the lack of side chains, is considered to bind the substrate binding site with a lower affinity compared with the general substrates of PaProK.
3.3. Structural Comparison of PaProK-Triglycine Complex with Other ProK-Inhibitor Peptide
The X-ray crystal structure confirmed that triglycine binds at the PaProK substrate recognition site in two different conformations. To confirm whether the main-chain interactions between PaProK and triglycine provide information for FBDD, the PaProK-triglycine was compared with a previously reported crystal structure of PaProK complexed with a peptide-based inhibitor (Figure 3 and Supplementary Figure S2). In PDB, five proteinase K structures in complex with peptide-based inhibitors (PDB code 1P7V, 1P7W, 1PEK, 1PFG, and 3PRK [26,27,28]) were found. These structures show structural similarity to PaProK-triglycine with an RMSD of 0.192-0.437 Å. In PaProK-1P7V (Pro-Ala-Pro-Phe-Ala-Ala-Ala) and PaProK-1P7W (Pro-Ala-Pro-Phe-Ala-Ser-Ala), the heptapeptide inhibitor is located in the cleft around the active site, the substrate binding pocket (Supplementary Figure S2). The N-terminal Pro-Ala-Pro residues of these peptide inhibitors lie in the same substrate-recognition cleft as PaProK-triglycine and C-terminal residues are proximal to the β5–α4 loop (Supplementary Figure S2). The peptide-inhibitors in ProK-1PEK (N-Ac-Pro-Ala-Pro-Phe-DAla-Ala-NH2; hexapeptide inhibitor) and ProK-1PFG (N-Ac-Pro-Ala-Pro-Phe-DAla-Ala-Ala-Ala-NH2; octapeptide inhibitor), include N-terminal modification and D-form amino acids. These structures show the cleavage forms of the inhibitor. The N-terminal Pro-Ala-Pro residues of 1PEK and 1PFG are placed in the substrate recognition site, and the cleaved C-terminal fragments are located in the vicinity of the His174 residue (Supplementary Figure S2). Collectively, the N-terminal Pro-Ala-Pro residues of the inhibitors in all PaProK complex structures (1P7V, 1P7W, 1PEK, and 1PFG) were located in the same substrate binding position between β5–α4 and α5–β6 loops, similar to that observed with triglycine. However, the four Pro-Ala-Pro residues in the peptide inhibitor not only showed a different backbone geometry, but also had no similarity with the backbone interaction of triglycine. The different geometry of the backbone of these peptides is considered to be determined by the C-terminal amino acid sequence of Pro-Ala-Pro or the presence of L/D form of amino acid (Figure 3a).
In PaProK-3PPK (methoxysuccinyl-Ala-Ala-Pro-Ala-chloromethyl ketone), the peptide-inhibitor was covalently modified and showed in transition state in a crystal structure (Figure 3b). In this crystal structure, the N atom of Ala281 (number in PDB of ProK3PPK), O atom of Ala281, N3, and nitrogen atom of Ala282, and O atom of Ala282 of peptide-inhibitor interact with the O atom of Gly207 (2.72 Å), N atom of Gly207 (3.47 Å), the O atom of Gly239 (3.14 Å) and the N atom of Gly239 (3.07 Å), respectively. Notably, in ProK-3PPK, the Ala-Ala-Pro residues located at the substrate binding site between β5–α4 and α5–β6 loops exhibited almost identical backbone geometry to the GGG-B form of triglycine (Figure 3c). Meanwhile, the GGG-A of triglycine was not similar to the backbone of the peptide-based inhibitors.
4. Discussion
Understanding the mechanistic and structural characteristics of the substrate recognition in PPI could provide very useful insights for FBDD. To understand the mechanism underlying substrate recognition by the target protein, X-ray crystallography-based structural investigation is generally performed using peptide fragments of a substrate or similar sequence. Here, a triglycine molecule corresponding to the peptide backbone was employed to identify the substrate binding site on PaProK. Triglycine with its two configurations was correctly placed on the substrate binding site in PaProK. This approach can provide structural information on the recognition of the main chain of a peptide-recognizing protein. Furthermore, it could provide potential information for the initial FBDD. Comparison with the PaProK-peptide-based inhibitor structures revealed that one of the configurations of triglycine was found to have almost the same back-bone binding configuration as the peptide inhibitor in PaProK-3PPK. This indicates that the substrate recognition evidence obtained through triglycine can be translated to design novel peptide inhibitors. Further, the existence of two different conformations of triglycine in the substrate binding site of PaProK was confirmed. These results suggest that peptide-based inhibitors can be designed with two different conformations. However, this can provide information on nonspecific binding. Therefore, further studies on peptide interactions are needed, which could lead to improved products. To conclude, triglycine provided information on the substrate recognition mechanism in protein-protein or protein-peptide interactions that can be employed in FBDD.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/9/9/444/s1: Figure S1: 2Fo-Fc electron density map (blue, counted 1.5 σ) and Fo-Fc electron density map (green, counted 3 σ; red, counted -3 σ) of proteinase K complexd with triglycine (cyan). Figure S2: Comparison of substrate binding site of PaProK complexed with (a) triglycine or peptide based inhibitors from (b) PaProK-1P7V, (c) PaProK-1P7W, (d) PaProK-1PEK, (e) PaProK-1PFG, and (f) PaProK-3PPK. The transparent box in orange indicates the substrate binding site.
Funding
This research was funded by the National Research Foundation of Korea (NRF-2017R1D1A1B03033087 and 2017M3A9F6029736). This work was supported by Korea UMSIT and PAL (Korea), and the Korea University Grant.
Acknowledgments
The author thanks the staff of macromolecule crystallography beamlines at Pohang Accelerator Laboratory for assistance in data collection.
Conflicts of Interest
The author declares no conflict of interest.
References
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-Based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angew Chem. Int. Ed. Eng. 2015, 54, 8896–8927. [Google Scholar] [CrossRef] [PubMed]
- Milroy, L.G.; Grossmann, T.N.; Hennig, S.; Brunsveld, L.; Ottmann, C. Modulators of protein-protein interactions. Chem. Rev. 2014, 114, 4695–4748. [Google Scholar] [CrossRef] [PubMed]
- Nevola, L.; Giralt, E. Modulating protein-protein interactions: The potential of peptides. Chem. Commun. (Camb) 2015, 51, 3302–3315. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, R.L.; Wilson, I.A. Protein-Peptide interactions. Curr. Opin. Struct. Biol. 1995, 5, 103–113. [Google Scholar] [CrossRef]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Tounge, B.A.; Parker, M.H. Designing a diverse high-quality library for crystallography-based FBDD screening. Methods Enzymol. 2011, 493, 3–20. [Google Scholar]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef]
- Carr, R.A.; Congreve, M.; Murray, C.W.; Rees, D.C. Fragment-Based lead discovery: Leads by design. Drug Discov. Today 2005, 10, 987–992. [Google Scholar] [CrossRef]
- Ebeling, W.; Hennrich, N.; Klockow, M.; Metz, H.; Orth, H.D.; Lang, H. Proteinase K from tritirachium album limber. Eur. J. Biochem. 1974, 47, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Hinrichs, W.; Wolf, W.M.; Saenger, W. Crystal structure of calcium-free proteinase K at 1.5-A resolution. J. Biol. Chem. 1994, 269, 23108–23111. [Google Scholar] [PubMed]
- Park, S.Y.; Ha, S.C.; Kim, Y.G. The protein crystallography beamlines at the pohang light Source II. Biodesign 2017, 5, 30–34. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [PubMed]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Betzel, C.; Gourinath, S.; Kumar, P.; Kaur, P.; Perbandt, M.; Eschenburg, S.; Singh, T.P. Structure of a serine protease proteinase K from tritirachium album limber at 0.98 A resolution. Biochemistry 2001, 40, 3080–3088. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-Building tools for molecular graphics. Acta Cryst. D Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.8; Schrödinger: New York, NY, USA, 2010. Available online: http://pymol.sourceforge.net/overview/index.htm (accessed on 26 August 2019).
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Nam, K.H.; Kim, M.Y.; Kim, S.J.; Priyadarshi, A.; Kwon, S.T.; Koo, B.S.; Yoon, S.H.; Hwang, K. Structural and functional analysis of a novel hormone-sensitive lipase from a metagenome library. Proteins 2009, 74, 1036–1040. [Google Scholar] [CrossRef]
- Nam, K.H.; Kim, M.Y.; Kim, S.J.; Priyadarshi, A.; Lee, W.H.; Hwang, K.Y. Structural and functional analysis of a novel EstE5 belonging to the subfamily of hormone-sensitive lipase. Biochem. Biophys. Res. Commun 2009, 379, 553–556. [Google Scholar] [CrossRef]
- Nam, K.H.; Kim, S.J.; Priyadarshi, A.; Kim, H.S.; Hwang, K.Y. The crystal structure of an HSL-homolog EstE5 complex with PMSF reveals a unique configuration that inhibits the nucleophile Ser144 in catalytic triads. Biochem. Biophys. Res. Commun. 2009, 389, 247–250. [Google Scholar] [CrossRef]
- Nam, K.H.; Park, S.H.; Lee, W.H.; Hwang, K.Y. Biochemical and structural analysis of hormone-sensitive lipase homolog EstE7: Insight into the stabilized dimerization of HSL-Homolog proteins. Bull. Korean Chem. Soc. 2010, 31, 2627–2632. [Google Scholar] [CrossRef]
- Saxena, A.K.; Singh, T.P.; Peters, K.; Fittkau, S.; Betzel, C. Strategy to design peptide inhibitors: structure of a complex of proteinase K with a designed octapeptide inhibitor N-Ac-Pro-Ala-Pro-Phe-DAla-Ala-Ala-Ala-NH2 at 2.5 A resolution. Protein Sci. 1996, 5, 2453–2458. [Google Scholar] [CrossRef]
- Betzel, C.; Singh, T.P.; Visanji, M.; Peters, K.; Fittkau, S.; Saenger, W.; Wilson, K.S. Structure of the complex of proteinase K with a substrate analogue hexapeptide inhibitor at 2.2-A resolution. J. Biol. Chem. 1993, 268, 15854–15858. [Google Scholar]
- Wolf, W.M.; Bajorath, J.; Muller, A.; Raghunathan, S.; Singh, T.P.; Hinrichs, W.; Saenger, W. Inhibition of proteinase K by methoxysuccinyl-Ala-Ala-Pro-Ala-chloromethyl ketone. An x-ray study at 2.2-A resolution. J. Biol. Chem. 1991, 266, 17695–17699. [Google Scholar]
Figure 1.
Crystal structure of PaProK complexed with triglycine (GGG). (a) Cartoon and (b) electrostatic potential surface representation of Proteinase K. Triglycine (GGG) and Ca ions are indicated as sticks (cyan) and orange spheres, respectively.
Figure 1.
Crystal structure of PaProK complexed with triglycine (GGG). (a) Cartoon and (b) electrostatic potential surface representation of Proteinase K. Triglycine (GGG) and Ca ions are indicated as sticks (cyan) and orange spheres, respectively.

Figure 2.
Binding conformations of triglycine on PaProK. (a–d) 2Fo-Fc electron density of a triglycine on PaProK (grey colored mesh, 1.0 σ). (a) Triglycine is located between β5–α4 and α5–β6 loops (pink). (b) Electron density maps reveal two conformations of triglycine. (c) GGG-A and (d) GGG-B forms of triglycine molecules fitted into the electron density map. Interactions between PaProK with (e) GGG-A and (f) GGG-B.
Figure 2.
Binding conformations of triglycine on PaProK. (a–d) 2Fo-Fc electron density of a triglycine on PaProK (grey colored mesh, 1.0 σ). (a) Triglycine is located between β5–α4 and α5–β6 loops (pink). (b) Electron density maps reveal two conformations of triglycine. (c) GGG-A and (d) GGG-B forms of triglycine molecules fitted into the electron density map. Interactions between PaProK with (e) GGG-A and (f) GGG-B.

Figure 3.
Structural comparison of the PaProK-triglycine complex with PaProK complexed with other peptide-based inhibitors. (a) Superimposition of PaProK-triglycine with partial sequence (Pro-Ala-Pro) from PaProK complexed with peptide-based inhibitors (PDB codes 1P7V, 1P7W, 1PEK, and 1PFG). (b) Inhibitor binding configuration of PaProK-3PPK. (c) Superimposition of the backbone of triglycine and Pro-Ala-Ala from the peptide-based inhibitor in PaProK-3PPK.
Figure 3.
Structural comparison of the PaProK-triglycine complex with PaProK complexed with other peptide-based inhibitors. (a) Superimposition of PaProK-triglycine with partial sequence (Pro-Ala-Pro) from PaProK complexed with peptide-based inhibitors (PDB codes 1P7V, 1P7W, 1PEK, and 1PFG). (b) Inhibitor binding configuration of PaProK-3PPK. (c) Superimposition of the backbone of triglycine and Pro-Ala-Ala from the peptide-based inhibitor in PaProK-3PPK.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Data collection and refinement statistics.
| Data Collection | PaProK-Triglcyine |
|---|---|
| Resolution | 50.0–1.40 (1.42–1.40) |
| Space group | P21 |
| Unique reflections | 38738 (1831) |
| Unit cell parameter | |
| a, b, and c (Å) | 38.51, 67.68, and 43.59 |
| α, β, and γ (o) | 90.00, 111.39, and 90.00 |
| Completeness (%) | 94.8 (89.4) |
| Multiplicity | 3.8 (3.7) |
| I/σ(I) | 25.65 (5.17) |
| Rmerge | 0.166 (0.529) |
| Rpim | 0.096 (0.313) |
| Refinement | |
| Resolution | 40.59–1.40 |
| Rfactor | 18.22 |
| Rfree | 21.81 |
| Average B factor (Å2) | |
| Protein | 11.95 |
| Triglycine | 19.00 |
| Calcium | 18.60 |
| Water | 22.03 |
| Real space correlation coefficient | |
| Triglycine form A | 0.83 |
| Triglycine form B | 0.83 |
| R.m.s. deviations | |
| Bonds | 0.008 |
| Angles | 0.993 |
| Ramachandran | |
| Preferred | 96.53 |
| Allowed | 3.09 |
| Outliers | 0.39 |
Values in the parentheses refer to the highest resolution shell.
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nam, K.H. Triglycine-Based Approach for Identifying the Substrate Recognition Site of an Enzyme. Crystals 2019, 9, 444. https://doi.org/10.3390/cryst9090444
AMA Style
Nam KH. Triglycine-Based Approach for Identifying the Substrate Recognition Site of an Enzyme. Crystals. 2019; 9(9):444. https://doi.org/10.3390/cryst9090444
Chicago/Turabian StyleNam, Ki Hyun. 2019. "Triglycine-Based Approach for Identifying the Substrate Recognition Site of an Enzyme" Crystals 9, no. 9: 444. https://doi.org/10.3390/cryst9090444
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.