Improving Kinetics of “Click-Crosslinking” for Self-Healing Nanocomposites by Graphene-Supported Cu-Nanoparticles
by
 , , and
, , and
Neda Kargarfard
1,2 ,
,
Norman Diedrich
1,
Harald Rupp
1,
Diana Döhler
1,* and
Wolfgang H. Binder
1,* 1
Faculty of Natural Science II, Martin Luther University Halle-Wittenberg, Von-Danckelmann-Platz 4, D-06120 Halle (Saale), Germany
2
Leibniz-Institut für Polymerforschung Dresden e. V., Abteilung Reaktive Verarbeitung, Hohe Str. 6, D-01069 Dresden, Germany
*
Authors to whom correspondence should be addressed.
Polymers 2018, 10(1), 17; https://doi.org/10.3390/polym10010017
Submission received: 5 December 2017
/
Revised: 20 December 2017
/
Accepted: 21 December 2017
/
Published: 24 December 2017
(This article belongs to the Special Issue Graphene-Polymer Composites)
Abstract
:Investigation of the curing kinetics of crosslinking reactions and the development of optimized catalyst systems is of importance for the preparation of self-healing nanocomposites, able to significantly extend their service lifetimes. Here we study different modified low molecular weight multivalent azides for a capsule-based self-healing approach, where self-healing is mediated by graphene-supported copper-nanoparticles, able to trigger “click”-based crosslinking of trivalent azides and alkynes. When monitoring the reaction kinetics of the curing reaction via reactive dynamic scanning calorimetry (DSC), it was found that the “click-crosslinking” reactivity decreased with increasing chain length of the according azide. Additionally, we could show a remarkable “click” reactivity already at 0 °C, highlighting the potential of click-based self-healing approaches. Furthermore, we varied the reaction temperature during the preparation of our tailor-made graphene-based copper(I) catalyst to further optimize its catalytic activity. With the most active catalyst prepared at 700 °C and the optimized set-up of reactants on hand, we prepared capsule-based self-healing epoxy nanocomposites.
1. Introduction
Self-healing approaches do have a significant potential in polymeric materials, especially those based on embedded capsule systems [1]. The molecular design of such self-healing materials requires fast and efficient crosslinking processes, which often are afforded by catalytic reactions using homogeneous and heterogeneous chemistry [2]. In the past, a plethora of such processes has been reported, based on e.g., ring-opening metathesis polymerization (ROMP)- (….) [1,3,4,5,6,7,8,9,10], copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC)- (….) [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27], isocyanate- [28,29,30,31,32,33,34,35,36,37], thiol- [38,39,40,41,42,43,44,45,46] and hydrosilylation-chemistry [47,48], many of them using metal-catalysis.
In the field of ROMP-based self-healing [7,9], the curing behavior of renewable norbornenyl-functionalized isosorbide monomers in the copolymerization with dicyclopentadiene (DCPD) was investigated exhibiting a higher reactivity, consequently facilitating low temperature ROMP. Additionally, higher crosslinking densities were observed, resulting in improved thermal and mechanical properties highlighting the potential of renewable ROMP-monomers towards self-healing applications [9].
In particular, the search for graphene-supported catalysts [12,19,21,22,23] is ongoing, effecting Ru-based crosslinking within nanocomposites. Thus, graphene oxide (GO)-supported Grubb’s catalysts (GO-HG1 and GO-HG2) have been prepared and investigated in the ROMP of 5-ethylidene-2-norbornene at 40 to 60 °C. While showing catalytic activity the amount of catalyst was reduced from 5.0 to 0.5 wt % [7].
We recently reported on a “click”-based crosslinking chemistry useful as a principle for optimized self-healing materials [12]. In this particular system, multivalent azides and alkynes are crosslinked by the use of a Cu(I)-catalyst, acting as the known essential catalytic system for “click”-chemistry [12,49,50,51,52,53]. Of particular importance is the ability to tune the temperature at which crosslinking takes place, thus enabling self-healing at room temperature or even below [11,17]. The central principal of action is the use of encapsulated low molecular weight azides [19,22] or azide-functionalized polymers [11,14,15,17,20,27], and low molecular weight alkynes [15,19,20,22] or alkyne-functionalized polymers [11,14], embedded within capsules sized from 100 nm up to microns [13,15,19,54]. Whereas the uncatalyzed process is conventionally taking place at temperatures close to ~160 °C [55,56], the use of Cu(I) as catalyst can significantly lower the crosslinking temperature, together with an increase in crosslinking efficiency [49,50,51]. The kinetics of “click-crosslinking“ reactions was studied by us [12] and others [24,25] and the huge potential of the CuAAC towards self-healing materials is highlighted by the observation of autocatalytic effects in the melt state. Thus, the formed 1,2,3-triazole rings can act as internal ligands consequently significantly increasing the reaction rate [11], well known before for the addition of external triazole-containing ligands [57]. By additionally designing chelation assisting azides, the self-healing temperature could be tuned, and efficient network formation was even observed below room temperature (T = 10 °C) [17].
When probing a large variety of homogeneous and heterogeneous catalysts, the use of finely dispersed Cu2O on graphene-oxide (TRGO-Cu2O) displayed high catalytic activity in various “click-crosslinking” reactions in the melt, easy and efficient recyclability in solution experiments as well as high stability against oxygen [21]. Consequently, the catalyst was used to generate graphene-based nanocomposites via the CuAAC without addition of a base or any reducing agents. As the cheap and easy up-scalable catalyst acted as reinforcing filler, the mechanical, thermal and conductive properties of the final resin were improved [22], opening the possibility to generate self-healing capsule-based epoxy nanocomposites showing quantitative healing at room temperature within 36 h [19].
For a capsule-based self-healing approach, the encapsulation of the azides required a careful tuning of their hydrophobicity. It was unclear though, how and whether small changes in the azide-monomer would change its reactivity within the same click-system. Thus, a modification of the azide-monomer to a more hydrophobic surrounding deemed interesting, most of all to facilitate its encapsulation via emulsion-processes. Therefore, in this study we investigate the influence of small substitutions within the trivalent azides on the reaction kinetics investigated via DSC as well as in view of different homogeneous and heterogeneous copper(I) catalysts.
2. Materials and Methods
2.1. Materials
Trimethylol propane (purity > 97%), trimethylol propane triglycidyl ether (technical grade, 1H-NMR spectrum in Supplementary Materials Figure S1), propargyl bromide (80 wt % solution in toluene), sodium sulphate (Na2SO4, purity > 99%), sodium azide (NaN3, purity 99.5%), 4-dimethylaminopyridine (DMAP, purity 99%), graphite (synthetic grade), potassium permanganate (KMnO4, analytical grade), copper(II) acetate hydrate (purity 98%), acetic anhydride (purity > 99%) and decanoyl chloride (purity > 98%) were purchased from Sigma Aldrich (Taufkirchen, Germany) and were used as received. Sodium hydroxide (NaOH, purity > 99%), ammonium chloride (NH4Cl, purity > 99%), concentrated sulphuric acid (H2SO4, 95–98%), hydrochloric acid (HCl, 37%) and N,N-dimethylformamide (DMF, purity > 99%) were ordered from Grüssing (Filsum, Germany) and DMF was freshly distilled from CaH2 under nitrogen atmosphere before use. Butyryl chloride (purity > 98%) and calcium hydride (CaH2, purity > 92%) were obtained from Alfa Aesar (Karlsruhe, Germany). Dichloromethane (DCM, purity > 98%) was purchased from Overlack (Mönchengladbach, Germany), chloroform (CHCl3, purity > 98%) was received from VWR (Darmstadt, Germany) and methanol (MeOH, purity > 99.9%) was obtained from Brenntag (Mühlheim an der Ruhr, Germany) and all solvents were distilled prior use. Tetra-n-butylammonium bromide (TBAB, purity > 99%) was purchased from TCI (Eschborn, Germany) and sodium chloride (NaCl, purity > 99%), o-phosphoric acid (H3PO4, 85%), hydroxide peroxide (H2O2, 30 wt %) were received from Carl Roth (Karlsruhe, Germany) and were used as received. The synthesis of trimethylolpropane tripropargyl ether (TMPTE, 1) was done according to literature [22,58] and was further optimized to yield exclusively 1 (see Supplementary Materials, Scheme S1). The synthesis of (azidated trimethylolpropane triglycidyl ether, N3TMPTE, 2) was done according to literature [22,59] with slight modifications (see Supplementary Materials Scheme S2).
2.2. Methods
Nuclear magnetic resonance (NMR) spectroscopy: All NMR spectra were recorded on a Varian Gemini 400 spectrometer from Agilent Technologies (Waldbronn, Germany) at 400 MHz at 27 °C. Deuterated chloroform (CDCl3, purity > 99.8%, stabilized with Ag) was used as solvent and was purchased from Chemotrade (Düsseldorf, Germany). The chemical shifts were recorded in ppm and all the coupling constants in Hz. MestRec v.4.9.9.6 (Mestrelab Research, A Coruña, Spain, 2016) was used for data interpretation.
Attenuated total reflection Fourier transformed infrared (ATR-FTIR) spectroscopy: All ATR-FTIR spectra were recorded on a Bruker Tensor VERTEX 70 from Bruker Optics GmbH (Leipzig, Germany) equipped with a heatable Golden Gate Diamond ATR plate from Specac (Orpington, Kent, UK). Opus 6.5 (Bruker Optik GmbH, Leipzig, Germany, 2008) and OriginPro 8G (Version 8.0951, OriginLab Corporation, Northampton, MA, USA, 2008) was used for data interpretation.
Thin layer chromatography (TLC): TLC was performed with TLC aluminum sheets (silica gel 60 F254) obtained from Merck (Darmstadt, Germany). Spots on the TLC plates were visualized by UV light (254 or 366 nm) or by oxidizing agents like “blue stain” consisting of Ce(SO4)2∙4H2O (1.0 g, analytical grade, Sigma Aldrich (Taufkirchen, Germany)), (NH4)6Mo7O24∙4H2O (2.5 g, analytical grade, Sigma Aldrich (Taufkirchen, Germany)), dissolved in concentrated H2SO4 (6.0 mL, 95–97%, Grüssing (Filsum, Germany)) and distilled water (90.0 mL).
Electrospray ionization time of flight mass spectrometry (ESI-TOF MS): ESI-TOF MS was performed with a micro TOF focus from Bruker Daltonics GmbH (Bremen, Germany) with an electrospray ionization source (ESI source). Samples were dissolved in CHCl3 (HPLC grade, VWR (Darmstadt, Germany)) or MeOH (HPLC grade, VWR (Darmstadt, Germany)) and sodium iodide (purity > 99.9%, Sigma Aldrich (Taufkirchen, Germany)), 20 mg∙mL−1 acetone (HPLC grade, Sigma Aldrich (Taufkirchen, Germany)) was added. The analyte was injected with a 180 µL∙h−1 flow rate at 180 °C.
Differential scanning calorimetry (DSC): DSC was performed on a differential scanning calorimeter 204F1/ASC Phönix from Netzsch (Selb, Germany). Crucibles and lids made of aluminum were used. Measurements were performed in a temperature range from −20 to 250 °C using heating rates of 5, 10, 15 and 20 K∙min−1. As purge gas, a flow of dry nitrogen (20 mL∙min−1) was used. For evaluation of data, the Proteus Thermal Analysis Software (Version 5.2.1, NETZSCH-Geraetebau GmbH, Selb, Germany, 2011) and OriginPro 8G (Version 8.0951, OriginLab Corporation, Northampton, MA, USA, 2008) was used.
Rheology: In situ rheology was performed on an oscillatory plate rheometer MCR 101/SN 80753612 from Anton Paar (Graz, Austria). All measurements were performed within the linear viscoelastic regime (LVE) using a PP08 measuring system. For evaluation of data, the RheoPlus/32 software (V 3.40, Anton Paar Germany GmbH, Ostfildern, Germany, 2008) and OriginPro 8G (Version 8.0951, OriginLab Corporation, Northampton, MA, USA, 2008) were used.
Glass tube furnace: The thermal reduction of GO-Cu(II) was performed in a glass tube furnace (Mod. RSR-B120/750/11) from Nabertherm GmbH (Lilienthal, Germany).
Freeze drying: Freeze drying was performed on a LyoQuest freeze dryer from Telstar (Utrecht, The Netherlands) operating at −80 °C and 0.18 mbar.
Ultrasonicator: For the dispersion of the GO-species via ultrasonication a sonication tip Vibra Cell VCX500 from Zinsser Analytic (Frankfurt, Germany) was used.
Flame atomic absorption spectroscopy (FAAS): FAAS was performed on a novAA 350 #113A0641 Tech: Flamme spectrometer from Analytik Jena AG (Jena, Germany) using Aspect LS 1.4.1.0 (Analytik Jena AG, Jena, Germany) as software. Therefore, external calibration and calibration via doping were performed. To determine the copper-content within TRGO-Cu2O the samples were burned to ash at 800 °C under atmospheric conditions and were dispersed in nitric acid (2 M). This solution was diluted in a 1:1 ratio with a potassium chloride solution (0.2%) and a mixture containing 25 mL of the dispersed nitric sample solution and 25 mL of the potassium chloride solution.
Transmission electron microscopy (TEM): TEM investigations were performed using a EM 900 transmission electron microscope from Carl Zeiss Microscopy GmbH (Oberkochen, Germany) and the images were taken with a SSCCD SM-1k-120 camera from TRS (Moorenweis, Germany). For sample preparation, TRGO-Cu2O was dispersed in water and sprayed on a carbon-layered copper grid. After one minute, the excess solution was removed with filter paper and the samples were dried at room temperature.
X-ray diffraction (XRD): XRD-measurements were performed on a D8 X-ray diffractometer from Bruker AXS GmbH (Karlsruhe, Germany). For analysis of the raw data, Diffrac. Suite EVA 3.1 (Bruker AXS GmbH, Karlsruhe, Germany) with an integrated database for the determination of the phases was used as software. For sample preparation, TRGO-Cu2O was rubbed in the presence of isopropanol and was put on a glass slide. For evaluation of data, OriginPro 8G (Version 8.0951, OriginLab Corporation, Northampton, MA, USA, 2008) was used.
2.3. General Synthesis Procedure for the Preparation of Trivalent Azides
The synthesis was carried out under a dry atmosphere of nitrogen. A two-necked round-bottom flask equipped with magnetic stir bar, rubber septum and gas tap was heated under vacuum and flushed with nitrogen several times. 4-Dimethylaminopyridine (0.2 eq) was added to 2 (1.0 eq) dissolved in dry DMF and the solution was stirred for ten minutes at room temperature. Afterwards, the desired anhydride or acid chloride (6.0–8.0 eq) was added dropwise to the reaction mixture and the solution was stirred at room temperature. After finishing the reaction, the crude product was either purified by extraction or column chromatography and the obtained product was dried in high vacuo. As determined via NMR-spectroscopy, final products contain up to 25% impurities such as free epichlorohydrine and bivalent residues as trimethylolpropane triglycidyl ether was used in technical grade (see 1H-NMR spectrum in Supplementary Materials Figure S1).
3. Results
3.1. Synthesis of Trivalent Alkyne and Trivalent Azides
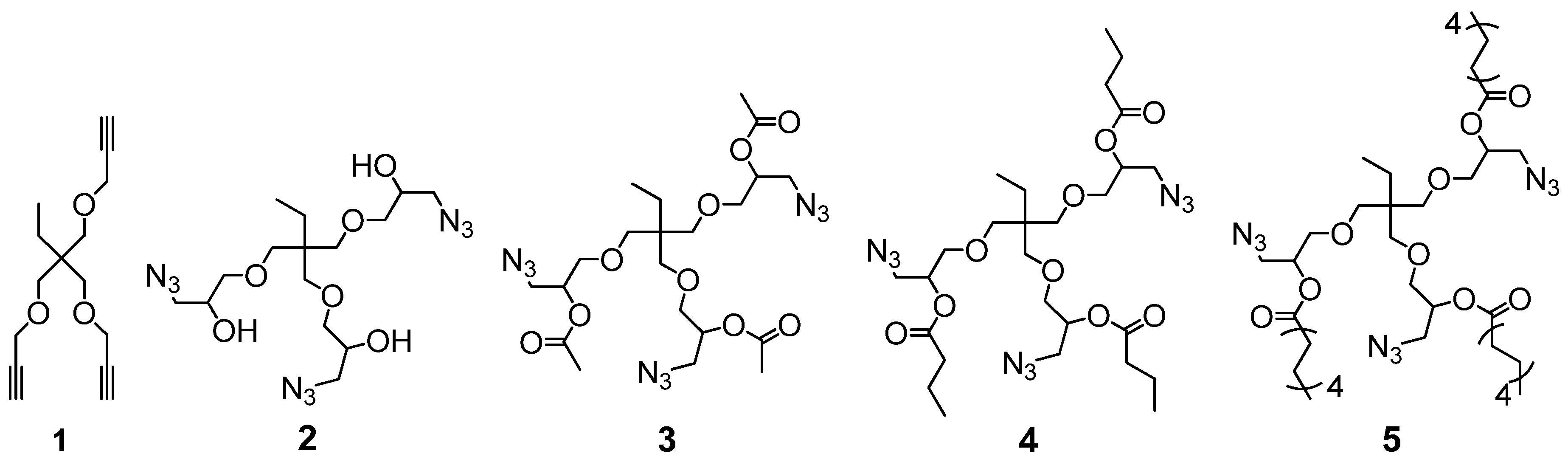
In Figure 1, an overview of the synthesized trivalent alkyne 1 and the trivalent azides is given. Trivalent alkyne 1 and trivalent azide 2 have been synthesized according to literature [22,58,59].
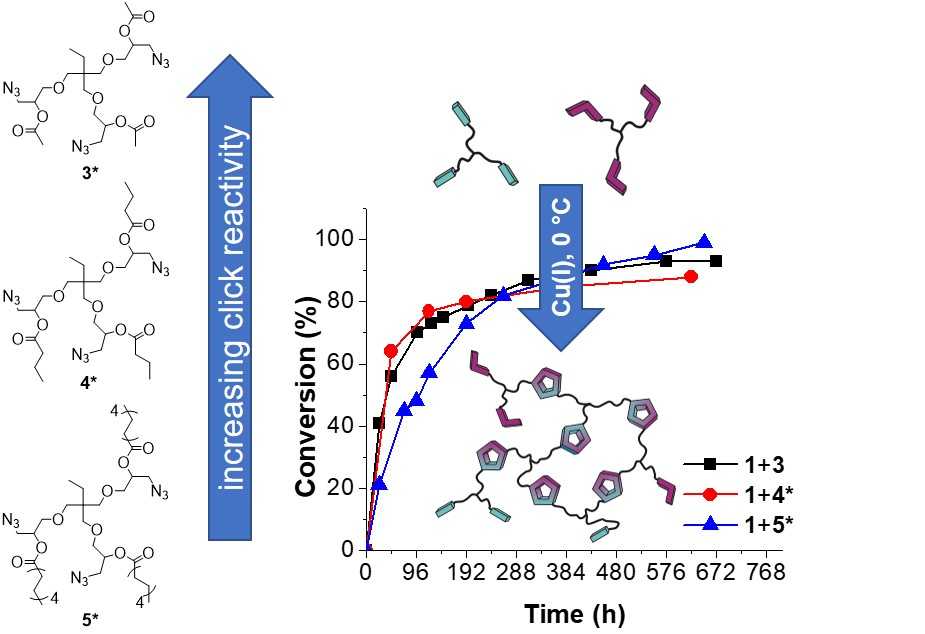
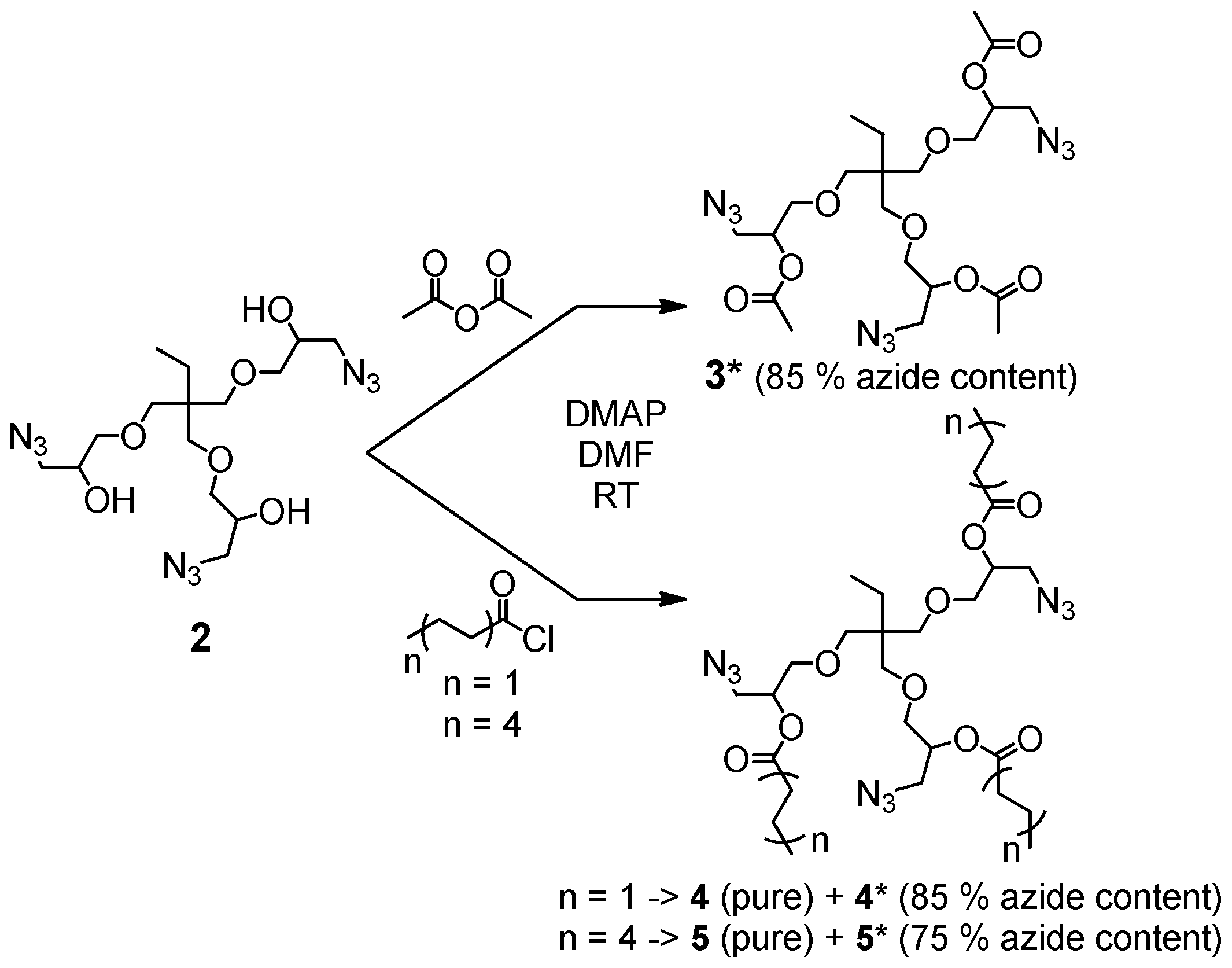
The synthesis of the trivalent azides 3*, 4, 4*, 5 and 5* is presented in Scheme 1, and details are given in the general synthesis procedure in Section 2.3 and in the Supplementary Materials. In brief, the trivalent azide 2 was either converted with acetic anhydride or the desired acid chloride at room temperature in dry DMF and in the presence of DMAP as a nucleophilic basic catalyst.
The trivalent azide 3* was obtained as a light-yellow, viscous liquid (85% azide content) containing the bivalent azide and free epichlorohydrine present in the starting material (trimethylolpropane triglycidyl ether (technical grade)). This mixture was further studied without purification to investigate its suitability for easy preparable and up-scalable room temperature-based self-healing nanocomposites relying on “click-crosslinking“ reactions. For the synthesis of the azides 4 and 5, a pure trivalent compound (4 and 5) and a mixture of bi- and trivalent product (4* and 5*, 85 and 75% azide content, respectively) were obtained and further separated via column chromatography. All prepared multivalent azide- and alkyne-functionalized compounds were characterized via NMR- and IR-spectroscopy as well as ESI-TOF mass spectrometry proving their purity and functional group content (for more details see Supplementary Materials Figures S2–S5: 1H- and 13C-NMR spectra of trivalent azides 4 and 5).
3.2. DSC Investigation of “Click-Crosslinking” Trivalent Alkyne and Trivalent Azides
Thermal analysis by differential scanning calorimetry (DSC) provides useful information about the relationship between the extent of a (crosslinking) reaction and the required time of curing at a certain temperature. Furthermore, information about kinetic parameters can be retained. Thus, DSC analysis is helpful to obtain a wide range of data of the investigated “click-crosslinking” reactions of trivalent alkyne 1 and trivalent azides 3*, 4, 4*, 5 and 5* such as the enthalpy of the reaction (ΔH), the onset temperature (Tonset), the temperature at the maximum of the DSC curve (Tp) and the apparent activation energy of the reaction (Ea, app). It should be mentioned, that the experimentally determined activation energies are indicated as apparent activation energies, since it is well known that the physical conditions during crosslinking are at least partially restricted by the mass and heat transport in the solid phase, consequently influencing the internal energy as well as the vibrational states of the investigated reactants [60,61]. Furthermore, it should be emphasized that our prepared TRGO-Cu2O catalyst is composed of several graphene sheets showing a lack in dispersibility with the reactants due to missing functional groups of increasing polarity. Thus, a relatively high apparent activation energy is expected—a phenomenon also observed in graphene oxide nanocomposite epoxy coatings [62].
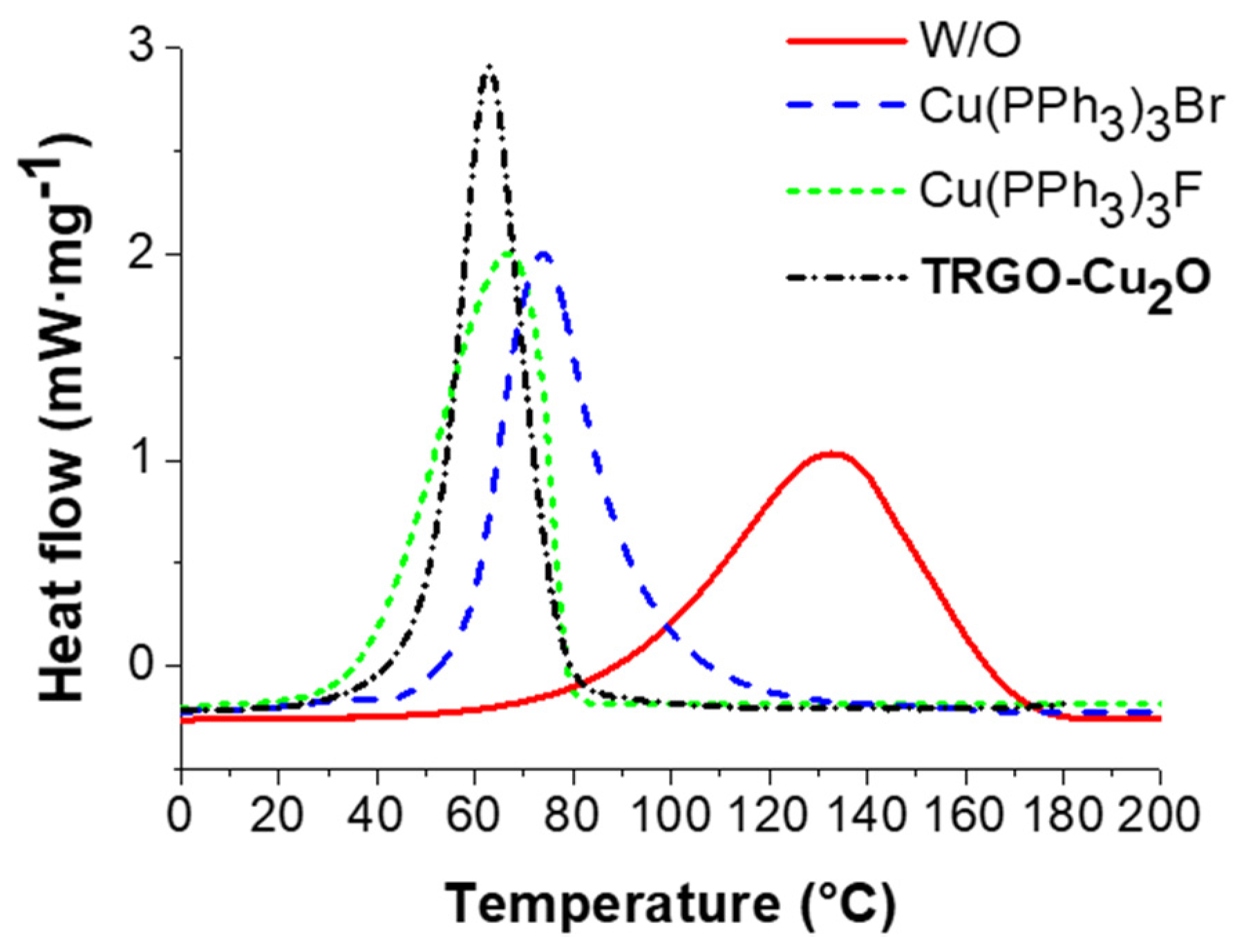
Via DSC investigations, the “click-crosslinking” reaction conversion can be estimated with respect to a determined reference value, a maximum ΔH value (262 kJ∙mol−1) for a reference click reaction between phenylacetylene and benzyl azide when quantitatively forming one triazole unit and therefore being representative of one successful “click” reaction. This reference value is in line with reported literature values for “click” reactions ranging between 210 to 270 kJ∙mol−1 [63]. DSC measurements for the “click-crosslinking” reaction of trivalent alkyne 1 with trivalent azide 3* investigated at a heating rate of 5 K∙min−1 in the presence of different homo- and heterogenous copper(I) catalysts (1 mol % per functional group) as well as without catalyst (W/O) are plotted in Figure 2 and the obtained results are summarized in Table 1.
For the Huisgen cycloaddition of trivalent alkyne 1 and trivalent azide 3* (Table 1, Entry 1) 78% conversion was achieved corresponding to an enthalpy of 205 kJ∙mol−1. Crosslinking took place at high temperatures and Tonset and Tp were observed at 91 and 133 °C, respectively. In the case of the homogenous catalysts (Cu(PPh3)3F and Cu(PPh3)3Br, Table 1, Entry 2 and 3), the observed enthalpies were 191 and 185 kJ∙mol−1, consequently showing a conversion of 73 and 71%, respectively. By using TRGO-Cu2O as a catalyst for “click-crosslinking” 1 and 3* (Table 1, Entry 4), an enthalpy of 177 kJ∙mol−1 was observed corresponding to 67% conversion. Moreover, the lowest apparent activation energy (55 kJ∙mol−1) and the lowest maximum peak temperature Tp (63 °C) were achieved in the presence of TRGO-Cu2O and the lowest Tonset (39 °C) by using Cu(PPh3)3F. According to these results, TRGO-Cu2O and Cu(PPh3)3F were the best catalysts for the “click-crosslinking“ reaction of trivalent alkyne 1 and trivalent azide 3*.
To investigate the activity of the pure trivalent azides 4 and 5 in comparison to the partially functionalized trivalent azides 4* and 5* (85 and 75% azide content) together with trivalent alkyne 1 in the CuAAC crosslinking reaction, DSC measurements were run by using TRGO-Cu2O as a catalyst (1 mol % per functional group). The DSC thermograms at 5 K∙min−1 with TRGO-Cu2O as a catalyst are plotted in Figure 3a,b and the obtained results are summarized in Table 1.
For “click-crosslinking” trivalent alkyne 1 with trivalent azide 4 (Table 1, Entry 5), the observed enthalpy was 233 kJ∙mol−1, corresponding to 89% conversion. In comparison, the observed enthalpy for “click-crosslinking” trivalent alkyne 1 with trivalent azide 4* (Table 1, Entry 9) showed a slightly lower enthalpy value of 211 kJ∙mol−1 and a conversion of 81%, related to the presence of the bivalent byproduct consequently lowering the conversion. In contrast, the reaction temperatures (Tp and Tonset) were decreased for “click-crosslinking” trivalent alkyne 1 with trivalent azide 5*, mainly attributed to a lower viscosity and therefore, to a faster diffusion. Thus, an enthalpy of 248 kJ∙mol−1 was observed for “click-crosslinking” trivalent alkyne 1 with trivalent azide 5 (Table 1, Entry 10) at relatively high reaction temperatures corresponding to 95% conversion. In comparison, a lower enthalpy of 127 kJ∙mol−1 (48% conversion) was measured for the “click-crosslinking” reaction of trivalent alkyne 1 with trivalent azide 5* (Table 1, Entry 14) while the reaction temperatures were reduced (Tonset = 32 °C, Tp = 56 °C). Consequently, further investigations towards easily up-scalable room temperature-based self-healing nanocomposites were continued by using trivalent azides 4* and 5*.
DSC thermograms of “click-crosslinking” trivalent alkyne 1 with trivalent azides 4* or 5* without catalyst (Huisgen cycloaddition) as well as with different catalysts (1 mol % per functional group) are illustrated in Figure 3c,d, respectively. For the uncatalyzed crosslinking reaction of trivalent alkyne 1 with trivalent azide 4* (Table 1, Entry 6), a high reaction enthalpy of 227 kJ∙mol−1 and high reaction temperatures (Tonset = 94 °C, Tp = 125 °C) were observed, corresponding to a conversion of 87%. For the homogenous catalyst Cu(PPh3)3F the lowest enthalpy was observed, while the click reaction happened at relatively low temperatures (Table 1, Entry 8). The determined apparent activation energy for the Huisgen cycloaddition was 75 kJ∙mol−1, while an enhanced apparent activation energy was detected for the click reaction using TRGO-Cu2O (97 kJ∙mol−1). In contrast, a lower apparent activation energy was determined for Cu(PPh3)3F and Cu(PPh3)3Br, showing values of 61 and 70 kJ∙mol−1 (Table 1, Entry 7 and 8), respectively. When reacting trivalent alkyne 1 with trivalent azide 5*, lower conversions were observed for the (uncatalyzed) Huisgen cycloaddition reaction as well as for all “click-crosslinking” reactions. Thus, a conversion of 66% was achieved for the uncatalyzed reaction (Table 1, Entry 11), corresponding to an enthalpy of 174 kJ∙mol−1, while “click-crosslinking” happened at high temperatures (Tonset and Tp of 102 and 141 °C). In comparison to the other catalysts, TRGO-Cu2O (Table 1, Entry 14) resulted in the lowest enthalpy of 127 kJ∙mol−1, which was achieved at Tonset and Tp of 32 and 56 °C, respectively. In the case of the homogeneous catalysts (Cu(PPh3)3F and Cu(PPh3)3Br) a similar conversion of 53 to 57% was observed, while lower reaction temperatures were achieved in the presence of Cu(PPh3)3F (Table 1, Entry 13, Tonset = 38 °C, Tp = 62 °C). According to the obtained results, Cu(PPh3)3F turned out to be the best catalyst for the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azides 4* or 5*.
3.3. DSC Investigation of “Click-Crosslinking” Trivalent Alkyne 1 and Trivalent Azides 3*, 4* and 5* at 0 °C
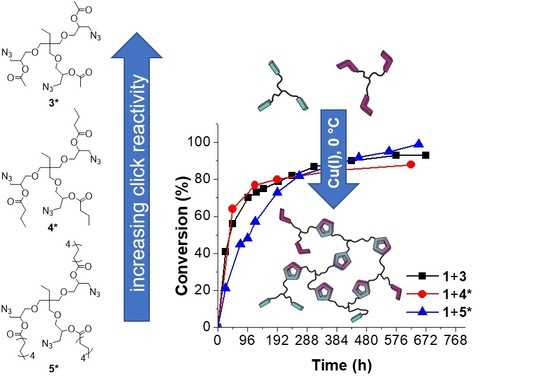
We were interested in quantifying “click-crosslinking” reactions during preparation and mixing of the components, thus understanding whether “click” reactions at 0 °C play an essential role. Therefore, DSC measurements were applied to investigate the kinetic behavior of the “click-crosslinking” reaction between trivalent alkyne 1 and trivalent azides 3*, 4* and 5* with different chain lengths to find a suitable and fast catalytic system for the CuAAC. As the usage of Cu(PPh3)3F resulted in high enthalpies at low crosslinking temperatures in the previously performed experiments, Cu(PPh3)3F was chosen as catalyst to check the activity of the multivalent azides and alkynes within “click-crosslinking” at 0 °C. Therefore, 1:1 mixtures of trivalent alkyne 1 and different trivalent azides (3*, 4* or 5*) together with Cu(PPh3)3F (1 mol % per functional group) were prepared. Immediately after preparation of the mixtures, DSC measurements were run at a heating rate of 5 K∙min−1 to observe the enthalpy of the “click-crosslinking” reaction at time zero (ΔH0). Afterwards, the mixtures were stored at 0 °C and further DSC investigations were conducted in defined time intervals and the conversion was determined (for more details see Supplementary Materials Figure S4 and Table S1).
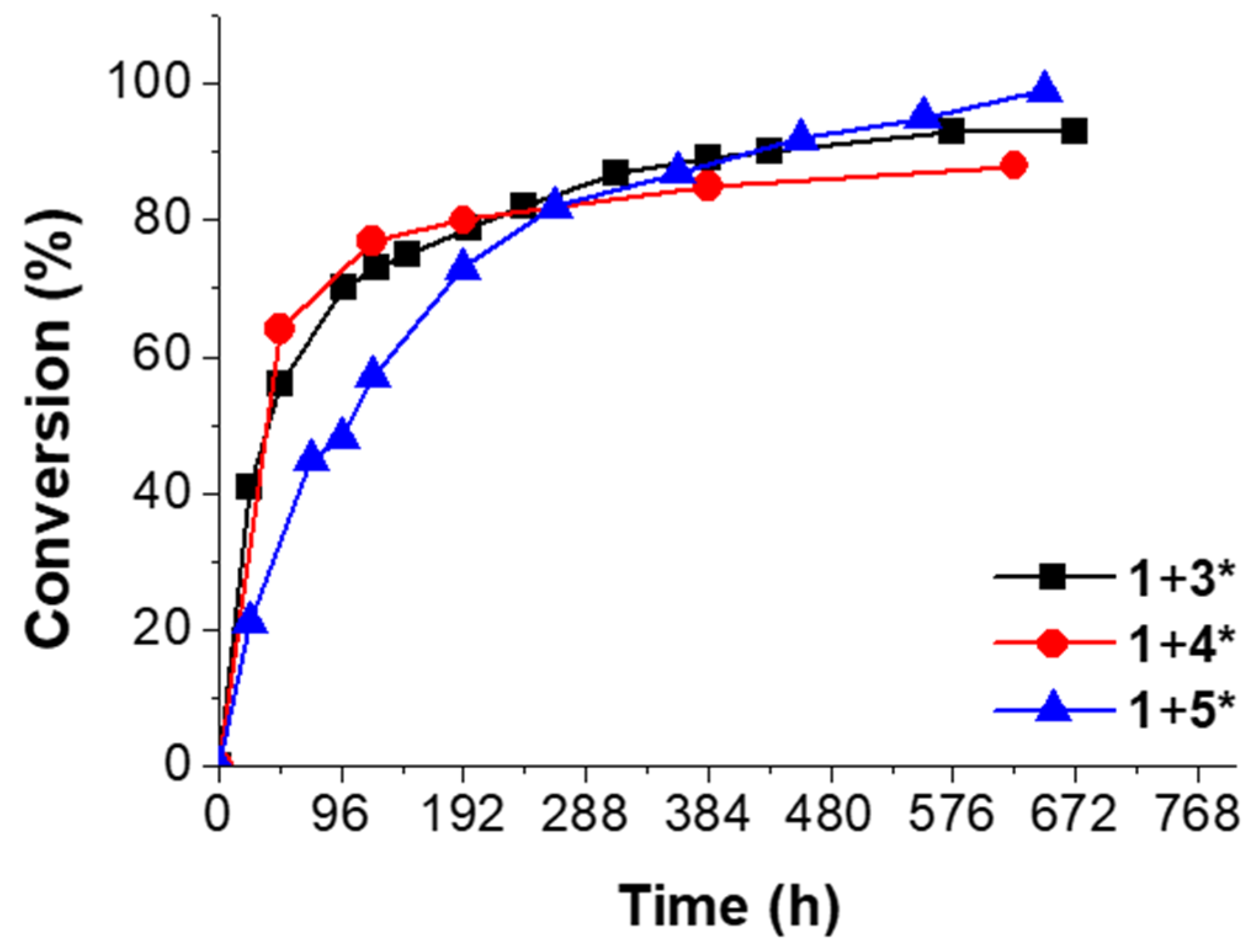
Immediately after mixing trivalent alkyne 1 and trivalent azide 3*, the measured enthalpy of the “click-crosslinking” reaction was 186 kJ∙mol−1, and Tonset and Tp were 39 and 67 °C, respectively (Figure 4, black squares; Supplementary Materials, Table S1, Entry 1). After 48 h storage at 0 °C, the “click-crosslinking” enthalpy decreased around one half of its initial value and reached 82 kJ∙mol−1, corresponding to 56% conversion (Supplementary Materials, Table S1, Entry 3). After 312 h, the “click-crosslinking” enthalpy decreased further to 24 kJ∙mol−1 (13 days, 87% conversion, Supplementary Materials, Table S1, Entry 9). Afterwards, the conversion did not show any significant increase, and the “click-crosslinking” reaction between alkyne 1 and azide 3* reached its maximum conversion of 93% at 0 °C after 576 h (24 days, Supplementary Materials, Table S1, Entry 12).
The mixture of trivalent alkyne 1 and trivalent azide 4* showed a high enthalpy of 222 kJ∙mol−1 immediately after sample preparation, and Tonset and Tp were 37 and 54 °C, respectively (Figure 4, red circles; Supplementary Materials, Table S1, Entry 14). After 48 h, the “click-crosslinking” enthalpy decreased to 80 kJ∙mol−1 and a conversion of 64% was observed (Supplementary Materials, Table S1, Entry 15). After 384 h, the conversion of the “click-crosslinking” reaction at 0 °C increased further to 85%, corresponding to a reaction enthalpy of 33 kJ∙mol−1 (Supplementary Materials, Table S1, Entry 18). In comparison, the conversion after 624 h did not significantly change and reached a constant value of 88% (27 kJ∙mol−1, Supplementary Materials, Table S1, Entry 19). Thus, it was concluded that the maximum “click-crosslinking” conversion obtainable by converting trivalent alkyne 1 and trivalent azide 4* at 0 °C is below 90%.
Immediately after preparation of the reaction mixture of trivalent alkyne 1 and trivalent azide 5*, a “click-crosslinking” enthalpy of 172 kJ∙mol−1 was observed, and Tonset and Tp were 63 and 86 °C, respectively (Figure 4, blue curve; Supplementary Materials, Table S1, Entry 20). After 96 h, the reaction enthalpy of the “click-crosslinking” reaction decreased to 89 kJ∙mol−1, related to a conversion of 48% (Supplementary Materials, Table S1, Entry 23). After 552 h, the enthalpy decreased further to 8 kJ∙mol−1 (95% conversion), and Tonset was not detectable anymore due to the very low reaction enthalpy. After 648 h, a complete “click-crosslinking” conversion was achieved, and no reaction peak was observed.
Moreover, in all crosslinking experiments investigated at 0 °C, the peak temperature Tp and the onset temperature Tonset increased in comparison to their initial values. This increase of the crosslinking temperatures is mainly attributed to the early network formation taking place at 0 °C, and is therefore related to a slowed down monomer diffusion.
To sum up the DSC investigations at 0 °C, the “click-crosslinking” reactions of trivalent alkyne 1 with trivalent azides 3* and 4* at 0 °C were faster within the first 100 h than the corresponding “click-crosslinking” reaction in the presence of trivalent azide 5*. This phenomenon was mainly attributed to the increasing chain length of the trivalent azide. Thus, under the same conditions, molecules with shorter side chains show faster “click-crosslinking” in comparison to the molecules with the longer chain length, mainly attributed to lower viscosity. Nevertheless, slightly higher conversions of the “click-crosslinking” reaction were observed on long timescales for the trivalent azides with increased chain length.
3.4. Rheology Investigation of “Click-Crosslinking” Trivalent Alkyne 1 and Trivalent Azides 4* and 5*
The viscoelastic and the kinetic behavior of the “click-crosslinking” reaction between trivalent alkyne 1 and trivalent azides 4* and 5* (1:1 ratio of azide and alkyne) and the resulting self-healing capability were investigated via in situ rheology. Therefore, the isothermal “click-crosslinking” reaction was directly performed on a rheometer plate at 20 °C using Cu(PPh3)3F (1 mol % per functional group) as a catalyst. The observed crossover times for the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azides 4* and 5* with Cu(PPh3)3F were 190 and 1445 minutes, respectively (see Supplementary Materials, Figure S6). By comparing these times with the crossover time of the “click-crosslinking” reaction of trivalent azide 3* with the trivalent alkyne 1 which was 35 minutes [22], it was concluded that with increasing chain length of the azide the crossover time increased. This observation was in line with the DSC investigations proving a decreased “click-crosslinking“ reactivity with increasing chain length.
3.5. Synthesis and Characerization of TRGO-Cu2O Prepared at Different Temperatures
TRGO-Cu2O was prepared via thermal reduction of copper(II)-modified graphene oxide in a glass tube furnace according to a previously published procedure [21,64,65], while the reduction temperature was varied between 300 to 800 °C, finally obtaining six different batches of the desired heterogeneous copper(I) catalyst (for more details see Scheme S6 in the Supplementary Materials) to optimize the synthesis procedure in terms of the catalytic activity since DSC. Investigations for “click-crosslinking“ trivalent alkyne 1 with trivalent azide 3*, 4* or 5* revealed slightly higher reaction temperatures (Tonset and Tp) in comparison to the homogeneously catalyzed “click-crosslinking“ reactions in the presence of Cu(PPh3)3F.
The prepared TRGO-based copper(I) catalysts were investigated via XRD-measurements (see Supplementary Materials Figure S7a) and for all prepared TRGO-Cu2O catalysts the characteristic reflex of GO at 2θ = 11° has disappeared due to successful reduction. Furthermore, for all prepared catalysts, reflexes at 2θ = 38° and 2θ = 43° were observed related to the formed copper species (pure copper as well as copper(I)). For the TRGO-Cu2O samples prepared at 700 and 800 °C, two additional reflexes at 2θ = 51° and 2θ = 26° were detected as characteristic reflexes for pure copper and graphite, respectively. Thus, it could be concluded, that some of the oxidic groups have been eliminated during thermal reduction partially resulting in graphite-like structures. The broad signal at 2θ = 25° observed for all prepared TRGO-based copper(I) catalysts was caused by an interference with the sample holder.
The prepared TRGO-Cu2O samples were further analyzed via TEM investigations (see Supplementary Materials Figure S7b–g) in which the formed nanosized copper(I) particles were visualized. The size of the particles was investigated via Image J. The particle size increased with increasing preparation temperature of TRGO-Cu2O, and average particle-diameters between 25 to 150 nm were determined. Furthermore, it was observed, that TRGO-based catalysts prepared at 700 and 800 °C displayed a more disperse distribution of nanosized copper(I) particles, which may be attributed to the formation of pure copper interacting with graphite-like structures detected in XRD investigations.
3.6. Crosslinking Reactions of Alkynes and Azides in the Presence of TRGO-Cu2O Prepared at Different Temperatures
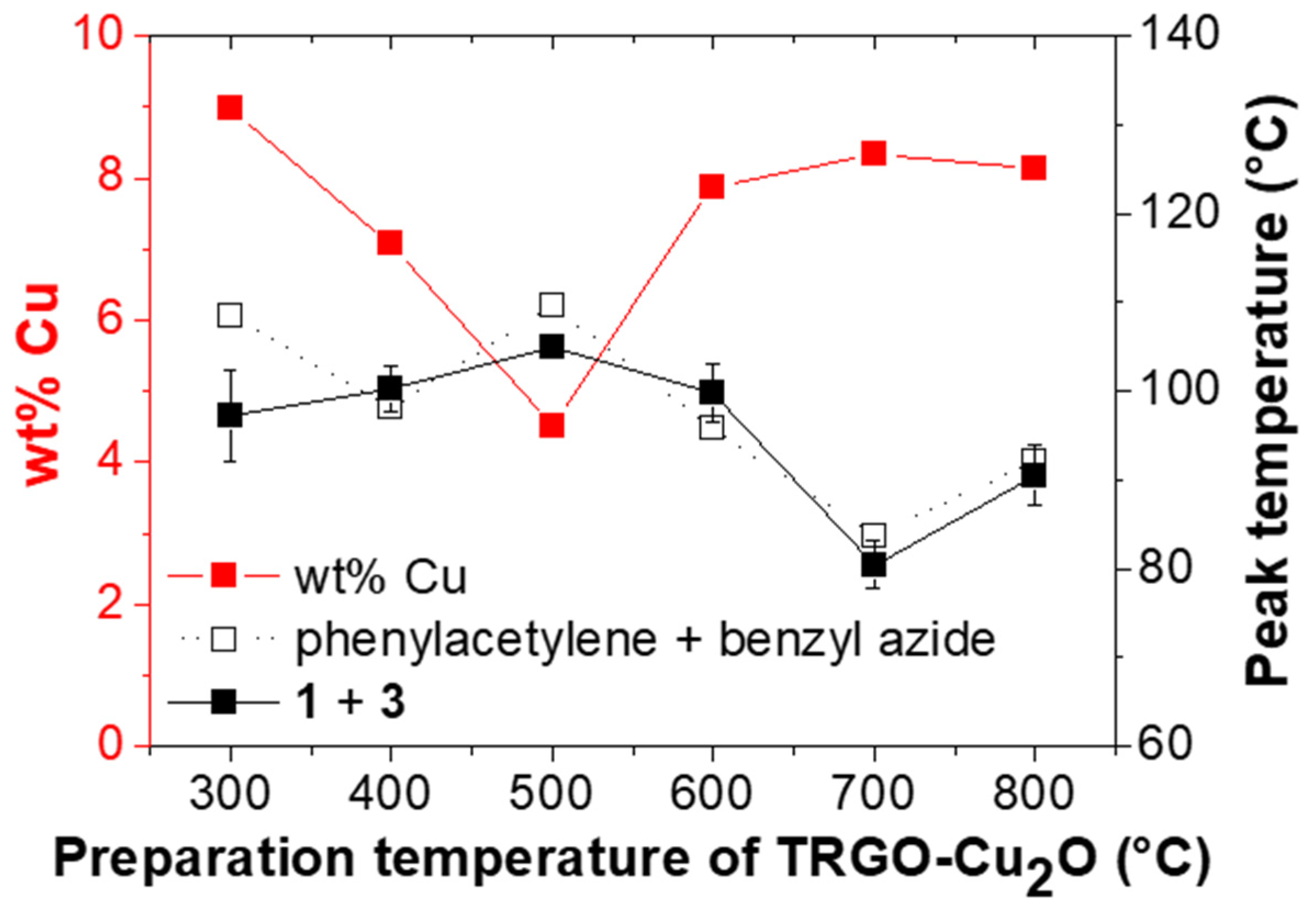
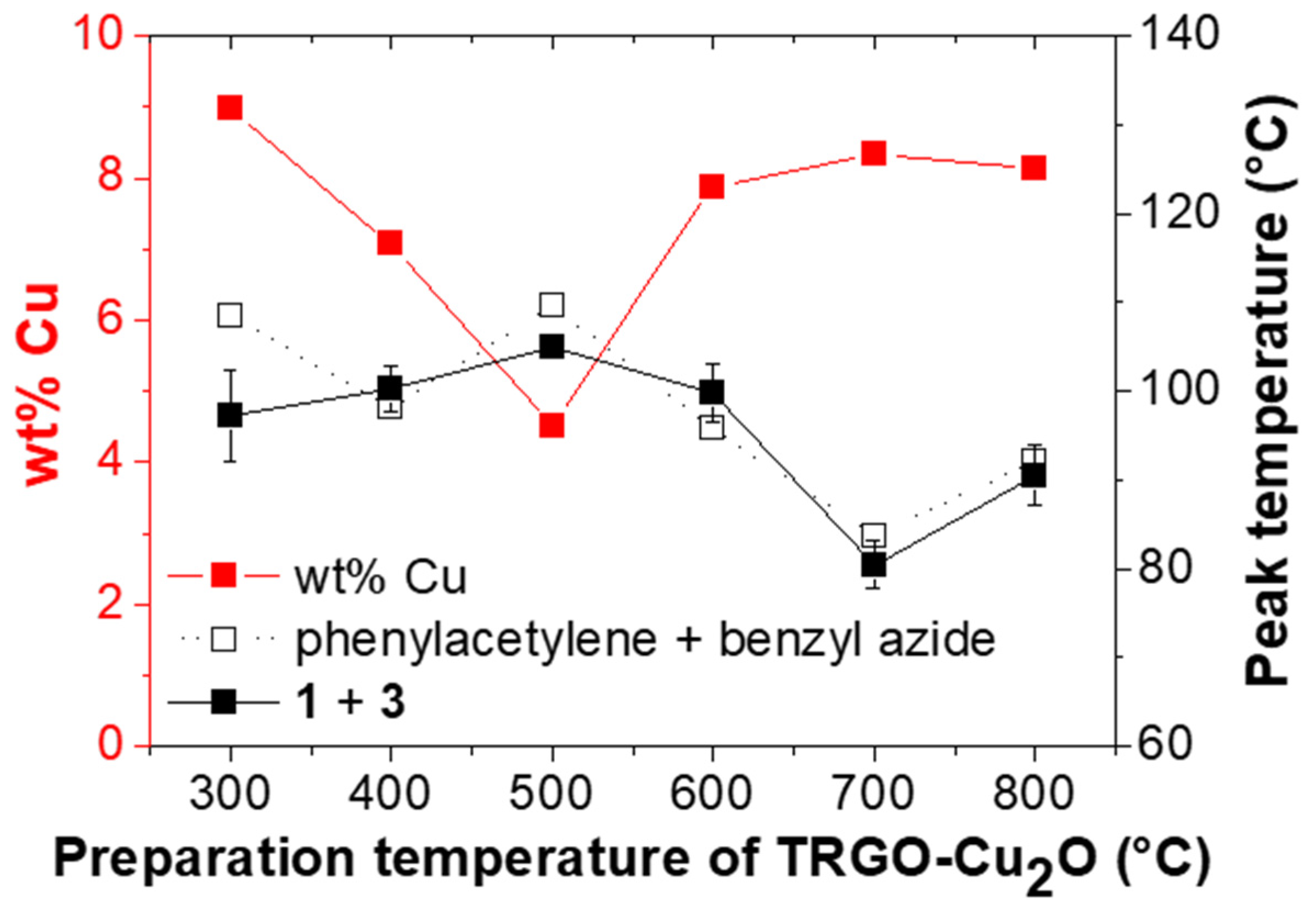
The catalytic activity of the TRGO-Cu2O catalysts (prepared at different temperatures) towards “click-crosslinking” was investigated via DSC investigations. Therefore, in the first step, a model reaction between phenylacetylene and benzyl azide (1:1 ratio of azide and alkyne) was investigated at a heating rate of 5 K∙min−1, and the reaction temperatures (Tonset and Tp), the reaction enthalpy (ΔH) and the conversion were recorded (for more information see Supplementary Materials Figure S8 and Table S2). While comparing the different catalysts prepared at 300 to 800 °C, it was observed that Tonset and Tp decreased with increasing temperature applied during the reduction of copper(II)-modified graphene oxide towards TRGO-Cu2O, in line with the expectation and the increasing size of the formed copper particles. Thereby, one exception was observed, and the catalyst prepared at 500 °C showed the highest peak temperature. Thus, FAAS measurements were performed to determine the loading of TRGO with immobilized copper nanoparticles. While most of the prepared catalysts displayed around 8 wt % of copper, a strong decrease was noted for the TRGO-Cu2O synthesized at 500 °C, directly linked to the observed reduced catalytic activity during the click reaction of phenylacetylene and benzyl azide.
In the next step, the catalytic activity was tested in a more complex system suitable for the preparation of room-temperature based self-healing epoxy nanocomposites. “Click-crosslinking“ of trivalent alkyne 1 and trivalent azide 3* (1:1 ratio of azide and alkyne, assuming 66% azide content of 3*) in the presence of different TRGO-Cu2O catalysts prepared from 300 to 800 °C (5 wt %) at a heating rate of 5 K∙min−1 (see Supplementary Materials Figure S10) was investigated, and the obtained reaction temperatures (Tonset and Tp), the reaction enthalpies (∆H) and the conversions are summarized in Table 2. For comparison, the non-catalyzed reaction between trivalent alkyne 1 and trivalent azide 3* was repeated, showing similar reaction temperatures (Tonset = 96 vs. 91 °C, Tp = 130 vs. 133 °C; Table 2, Entry 1) as described before, but a reduced conversion due to the higher amount of bivalent residues (66 vs. 75% azide content, see Supplementary Materials Figure S7). All DSC measurements were performed in three independent experiments to ensure their reproducibility and to approximate the expected error. Thereby, differences especially in the peak shape were observed to be related to sample preparation and limited blending of the reactants as well as a limited diffusion to the catalyst surface being typical for a reaction directly catalyzed by a solid support [60,61].
For “click-crosslinking” trivalent alkyne 1 and trivalent azide 3*, a similar trend was observed as in the model reaction between phenylacetylene and benzyl azide. Thus, the reaction temperatures (Tonset and Tp) decreased with increasing reduction temperature applied during the preparation of the different TRGO-Cu2O catalysts. Thereby, the lowest Tonset and Tp of 64 and 80 °C, respectively, were observed for “click-crosslinking” trivalent alkyne 1 and trivalent azide 3* in the presence of TRGO-Cu2O prepared at 700 °C while the catalyst prepared at 500 °C showed the worst result.
The obtained peak temperatures for the click model reaction of phenylacetylene and benzyl azide as well as for the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3* were correlated to the preparation temperature of TRGO-Cu2O and the corresponding amount of copper within these catalysts (see Figure 5).
The already mentioned trends within the catalytic activity of TRGO-Cu2O were well highlighted for both click reactions. The catalyst prepared at 500 °C showed a low amount of copper and consequently a high peak temperature during the click reactions, while the best results in terms of copper loading as well as peak temperature were observed for TRGO-Cu2O prepared at 700 °C. The decrease of the copper loading for the catalyst prepared at 500 °C may be related to the decrease of oxygen-functional groups with increasing reduction temperature known to act as reactive sites for the nucleation and growth of metal nanoparticles [66,67]. Following this argumentation, a further decrease in the copper loading together with a decreased catalytic activity would be expected, which was not observed in this particular study. We therefore assume that either the formation of pure copper indicated by XRD investigations is boosting the catalytic activity or that the diffusion of metal atoms, especially favored at higher temperatures, leads to the formation of non-stable metal clusters influencing the catalytic activity [68].
Thus, for the preparation of self-healing nanocomposites according to a previously published procedure [19], the optimized TRGO-Cu2O catalyst prepared at 700 °C was used. Thereby, the trivalent alkyne 1 together with this particular TRGO-Cu2O were directly distributed within the epoxy matrix together with µm-sized capsules filled with trivalent azide 3*. Further self-healing investigations of our optimized healing system as well as the determination of self-healing efficiencies are ongoing in our laboratories and will be part of a future publication.
4. Conclusions
Different low molecular weight, multivalent azides with small structural changes were synthesized and their crosslinking kinetics was investigated in a CuAAC-based curing reaction. Therefore, different homogeneous and heterogeneous copper(I) catalysts were screened and the kinetic parameters such as the reaction temperatures, the enthalpy of the reaction as well as the apparent activation energies were recorded via DSC investigations. We observed, that the “click-crosslinking” reactivity decreased with increasing chain length of the azide. Furthermore, a significant click reactivity of all investigated azides could be proven already at 0 °C.
The reaction conditions for the preparation of our home-made TRGO-Cu2O catalyst were optimized: When increasing the reaction temperature to 700 °C, the resulting copper(I) catalyst displayed the highest catalytic activity as shown in model click reactions as well as in “click-crosslinking“ reactions between trivalent alkyne 1 and trivalent azide 3*.
The tuned catalyst was subsequently dispersed in an epoxy matrix together with the trivalent alkyne 1 and the encapsulated trivalent azide 3* (µm-sized capsules). Further self-healing investigations of the so prepared capsule-based self-healing graphene-supported epoxy nanocomposites are ongoing and will be part of a future publication.
Supplementary Materials
The following are available online at www.mdpi.com/2073-4360/10/1/17/s1. Scheme S1: Synthesis of trimethylolpropane tripropargyl ether (TMPTPE, 1); Scheme S2: Synthesis of azidated trimethylolpropane tripropargyl ether (N3TMPTPE, 2); Scheme S3: Synthesis of ((2-((2-acetoxy-3-azidopropoxy)methyl)-2-ethylpropane-1,3-diyl) bis (oxy))bis(3-azidopropane-1,2-diyl) diacetate 3; Scheme S4: Synthesis of ((2-((3-azido-2-(butyryloxy)propoxy)methyl)-2-ethylpropane-1,3-diyl) bis(oxy))bis(3-azidopropane-1,2-diyl) dibutyrate 4; Scheme S5: Synthesis of ((2-((3-azido-2-(decanoyloxy)propoxy)methyl)-2-ethylpropane-1,3-diyl) bis (oxy))bis(3-azidopropane-1,2-diyl) bis(decanoate) 5; Scheme S6: Synthesis of modified, thermally reduced graphene oxide TRGO-Cu2O; Figure S1: 1H-NMR spectrum of trimethylolpropane triglycidyl ether; Figure S2: 1H-NMR spectrum of 4; Figure S3: 13C-NMR spectrum of 4; Figure S4: 1H-NMR spectrum of 5; Figure S5: 13C-NMR spectrum of 5; Figure S6: Rheological behavior of trialkyne 1 and (a) triazide 4* or (b) triazide 5* applying Cu(PPh3)3F as a catalyst at 20 °C; Figure S7: (a) XRD measurements of GO-Cu(II) and TRGO-Cu2O prepared at different temperatures (300 °C–800 °C). Reflexes of Cu, Cu2O, GO and graphite are shown for comparison. TEM images of TRGO-Cu2O prepared at (b) 300 °C, (c) 400 °C, (d) 500 °C, (e) 600 °C, (f) 700 °C and (g) 800 °C; Figure S8: DSC measurements of the click reaction of phenylacetylene and benzyl azide with TRGO-Cu2O as a catalyst at a heating rate of 5 K∙min−1; Figure S9: DSC measurements of the crosslinking reaction of trivalent alkyne 1 and trivalent azide 3* without catalyst (W/O) at a heating rate of 5 K⋅min−1; Figure S10: DSC measurements of the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3* with TRGO-Cu2O as a catalyst prepared at (a) 300 °C, (b) 400 °C, (c) 500 °C, (d) 600 °C, (e) 700 °C and (f) 800 °C at a heating rate of 5 K⋅min−1; Table S1: Conversion of the “click-crosslinking“ reaction of trivalent alkyne 1 and trivalent azide 3*, 4* or 5* at 0 °C with Cu(PPh3)3F as a catalyst at a heating rate of 5 K∙min−1: Reaction temperatures (Tonset and Tp), reaction enthalpies (ΔH) and conversions; Table S2: Thermal properties of the click reaction of phenyl acetylene and benzyl azide with TRGO-Cu2O as a catalyst (prepared at different temperatures) at a heating rate of 5 K∙min−1: Reaction temperatures (Tonset and Tp), reaction enthalpies (ΔH) and conversions.
Acknowledgments
The authors thank Anette Meister for providing TEM images, Thomas Heymann for providing FAAS measurements and Toni Buttlar for providing XRD measurements. The authors are grateful for Grant DFG BI 1337/8-1 and BI 1137/8-2 of the German Science Foundation within the framework of the SPP 1568 (“Design and Generic Principles of Self-Healing Materials”). Diana Döhler additionally acknowledges the support from the DFG Young Research GRANT SHE-STARS within the SPP 1568. Furthermore, this project has received funding from the European Union’s Seventh Framework Program for research, technological development and demonstration under grant agreement no. 313978 which is thankfully acknowledged.
Author Contributions
Neda Kargarfard and Norman Diedrich prepared all described compounds and performed all described characterizations and measurements if not otherwise mentioned. Harald Rupp, Diana Döhler and Wolfgang H. Binder designed the paper and wrote parts of the paper. Diana Döhler and Wolfgang H. Binder designed the work and provided funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- White, S.R.; Sottos, N.R.; Geubelle, P.H.; Moore, J.S.; Kessler, M.R.; Sriram, S.R.; Brown, E.N.; Viswanathan, S. Autonomic healing of polymer composites. Nature 2001, 409, 794–817. [Google Scholar] [CrossRef] [PubMed]
- Michael, P.; Döhler, D.; Binder, W.H. Improving autonomous self-healing via combined chemical/physical principles. Polymer 2015, 69, 216–227. [Google Scholar] [CrossRef]
- Huang, G.C.; Lee, J.K.; Kessler, M.R. Evaluation of norbornene-based adhesives to amine-cured epoxy for self-healing applications. Macromol. Mater. Eng. 2011, 296, 965–972. [Google Scholar] [CrossRef]
- Aïssa, B.; Haddad, E.; Jamroz, W.; Hassani, S.; Farahani, R.D.; Merle, P.G.; Therriault, D. Micromechanical characterization of single-walled carbon nanotube reinforced ethylidene norbornene nanocomposites for self-healing applications. Smart Mater. Struct. 2012, 21, 105028. [Google Scholar] [CrossRef]
- Mauldin, T.C.; Leonard, J.; Earl, K.; Lee, J.K.; Kessler, M.R. Modified rheokinetic technique to enhance the understanding of microcapsule-based self-healing polymers. ACS Appl. Mater. Interfaces 2012, 4, 1831–1837. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, M.; Guadagno, L. Healing efficiency of epoxy-based materials for structural applications. Polym. Compos. 2013, 34, 1525–1532. [Google Scholar] [CrossRef]
- Mariconda, A.; Longo, P.; Agovino, A.; Guadagno, L.; Sorrentino, A.; Raimondo, M. Synthesis of ruthenium catalysts functionalized graphene oxide for self-healing applications. Polymer 2015, 69, 330–342. [Google Scholar] [CrossRef]
- Monfared Zanjani, J.S.; Okan, B.S.; Letofsky-Papst, I.; Menceloglu, Y.; Yildiz, M. Repeated self-healing of nano and micro scale cracks in epoxy based composites by tri-axial electrospun fibers including different healing agents. RSC Adv. 2015, 5, 73133–73145. [Google Scholar] [CrossRef]
- Wang, B.; Mireles, K.; Rock, M.; Li, Y.; Thakur, V.K.; Gao, D.; Kessler, M.R. Synthesis and preparation of bio-based ROMP thermosets from functionalized renewable isosorbide derivative. Macromol. Chem. Phys. 2016, 217, 871–879. [Google Scholar] [CrossRef]
- Longo, P.; Mariconda, A.; Calabrese, E.; Raimondo, M.; Naddeo, C.; Vertuccio, L.; Russo, S.; Iannuzzo, G.; Guadagno, L. Development of a new stable ruthenium initiator suitably designed for self-repairing applications in high reactive environments. J. Ind. Eng. Chem. 2017, 54, 234–251. [Google Scholar] [CrossRef]
- Döhler, D.; Michael, P.; Binder, W.H. Autocatalysis in the room temperature copper(I)-catalyzed alkyne-azide “click” cycloaddition of multivalent poly(acrylate)s and poly(isobutylene)s. Macromolecules 2012, 45, 3335–3345. [Google Scholar] [CrossRef]
- Döhler, D.; Michael, P.; Binder, W.H. CuAAC-based click chemistry in self-healing polymers. Acc. Chem. Res. 2017, 50, 2610–2620. [Google Scholar] [CrossRef] [PubMed]
- Döhler, D.; Rana, S.; Rupp, H.; Bergmann, H.; Behzadi, S.; Crespy, D.; Binder, W.H. Qualitative sensing of mechanical damage by a fluorogenic “click” reaction. Chem. Commun. 2016, 52, 11076–11079. [Google Scholar] [CrossRef] [PubMed]
- Döhler, D.; Zare, P.; Binder, W.H. Hyperbranched polyisobutylenes for self-healing polymers. Polym. Chem. 2014, 5, 992–1000. [Google Scholar] [CrossRef]
- Gragert, M.; Schunack, M.; Binder, W.H. Azide/Alkyne-“Click”-Reactions of encapsulated reagents: Toward self-healing materials. Macromol. Rapid Commun. 2011, 32, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Michael, P.; Binder, W.H. A mechanochemically triggered “click” catalyst. Angew. Chem. Int. Ed. 2015, 54, 13918–13922. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Döhler, D.; Ströhl, D.; Binder, W.H. Chelation-assisted CuAAC of star-shaped polymers enables fast self-healing at low temperatures. Polym. Chem. 2016, 7, 2342–2351. [Google Scholar] [CrossRef]
- Raimondo, M.; De Nicola, F.; Volponi, R.; Binder, W.H.; Michael, P.; Russo, S.; Guadagno, L. Self-repairing CFRPs targeted towards structural aerospace applications. Int. J. Struct. Integr. 2016, 7, 656–670. [Google Scholar] [CrossRef]
- Rana, S.; Döhler, D.; Nia, A.S.; Nasir, M.; Beiner, M.; Binder, W.H. “Click”-triggered self-healing graphene nanocomposites. Macromol. Rapid Commun. 2016, 37, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Schunack, M.; Gragert, M.; Döhler, D.; Michael, P.; Binder, W.H. Low-temperature Cu(I)-catalyzed “click” reactions for self-healing polymers. Macromol. Chem. Phys. 2012, 213, 205–214. [Google Scholar] [CrossRef]
- Shaygan Nia, A.; Rana, S.; Döhler, D.; Noirfalise, X.; Belfiore, A.; Binder, W.H. Click chemistry promoted by graphene supported copper nanomaterials. Chem. Commun. 2014, 50, 15374–15377. [Google Scholar] [CrossRef] [PubMed]
- Shaygan Nia, A.; Rana, S.; Döhler, D.; Osim, W.; Binder, W.H. Nanocomposites via a direct graphene-promoted “click” reaction. Polymer 2015, 79, 21–28. [Google Scholar] [CrossRef]
- Shaygan Nia, A.; Rana, S.; Döhler, D.; Jirsa, F.; Meister, A.; Guadagno, L.; Koslowski, E.; Bron, M.; Binder, W.H. Carbon-supported copper nanomaterials: Recyclable catalysts for huisgen [3 + 2] cycloaddition reactions. Chem. Eur. J. 2015, 21, 10763–10770. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Mauldin, T.C.; Kessler, M.R. Kinetics of bulk azide/alkyne “click” polymerization. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 4093–4102. [Google Scholar] [CrossRef]
- Sheng, X.; Rock, D.M.; Mauldin, T.C.; Kessler, M.R. Evaluation of different catalyst systems for bulk polymerization through “click” chemistry. Polymer 2011, 52, 4435–4441. [Google Scholar] [CrossRef]
- Vasiliu, S.; Kampe, B.; Theil, F.; Dietzek, B.; Döhler, D.; Michael, P.; Binder, W.H.; Popp, J. Insights into the mechanism of polymer coating self-healing using raman spectroscopy. Appl. Spectrosc. 2014, 68, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Döhler, D.; Peterlik, H.; Binder, W.H. A dual crosslinked self-healing system: Supramolecular and covalent network formation of four-arm star polymers. Polymer 2015, 69, 264–273. [Google Scholar] [CrossRef]
- Cao, S.; Li, S.; Li, M.; Xu, L.; Ding, H.; Xia, J.; Zhang, M.; Huang, K. A thermal self-healing polyurethane thermoset based on phenolic urethane. Polym. J. 2017, 49, 775–781. [Google Scholar] [CrossRef]
- Haghayegh, M.; Mirabedini, S.M.; Yeganeh, H. Preparation of microcapsules containing multi-functional reactive isocyanate-terminated-polyurethane-prepolymer as healing agent, part II: Corrosion performance and mechanical properties of a self-healing coating. RSC Adv. 2016, 6, 50874–50886. [Google Scholar] [CrossRef]
- Haghayegh, M.; Mirabedini, S.M.; Yeganeh, H. Microcapsules containing multi-functional reactive isocyanate-terminated polyurethane prepolymer as a healing agent. Part 1: Synthesis and optimization of reaction conditions. J. Mater. Sci. 2016, 51, 3056–3068. [Google Scholar] [CrossRef]
- Hillewaere, X.K.D.; Teixeira, R.F.A.; Nguyen, L.-T.T.; Ramos, J.A.; Rahier, H.; Du Prez, F.E. Autonomous self-healing of epoxy thermosets with thiol-isocyanate chemistry. Adv. Funct. Mater. 2014, 24, 5575–5583. [Google Scholar] [CrossRef]
- Keller, M.W.; Hampton, K.; McLaury, B. Self-healing of erosion damage in a polymer coating. Wear 2013, 307, 218–225. [Google Scholar] [CrossRef]
- McIlroy, D.A.; Blaiszik, B.J.; Caruso, M.M.; White, S.R.; Moore, J.S.; Sottos, N.R. Microencapsulation of a reactive liquid-phase amine for self-healing epoxy composites. Macromolecules 2010, 43, 1855–1859. [Google Scholar] [CrossRef]
- Park, J.I.; Choe, A.; Kim, M.P.; Ko, H.; Lee, T.H.; Noh, S.M.; Kim, J.C.; Cheong, I.W. Water-adaptive and repeatable self-healing polymers bearing bulky urea bonds. Polym. Chem. 2018, 9, 11–19. [Google Scholar] [CrossRef]
- Schüssele, A.C.; Nübling, F.; Thomann, Y.; Carstensen, O.; Bauer, G.; Speck, T.; Mülhaupt, R. Self-healing rubbers based on NBR blends with hyperbranched polyethylenimines. Macromol. Mater. Eng. 2012, 297, 411–419. [Google Scholar] [CrossRef]
- Ying, H.; Zhang, Y.; Cheng, J. Dynamic urea bond for the design of reversible and self-healing polymers. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zechel, S.; Geitner, R.; Abend, M.; Siegmann, M.; Enke, M.; Kuhl, N.; Klein, M.; Vitz, J.; Gräfe, S.; Dietzek, B.; et al. Intrinsic self-healing polymers with a high E-modulus based on dynamic reversible urea bonds. NPG Asia Mater. 2017, 9, 420. [Google Scholar] [CrossRef]
- Akiyama, T.; Ushio, A.; Itoh, Y.; Kawaguchi, Y.; Matsumoto, K.; Jikei, M. Synthesis and healing properties of poly(arylether sulfone)-poly(alkylthioether) multiblock copolymers containing disulfide bonds. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3545–3553. [Google Scholar] [CrossRef]
- Billiet, S.; Van Camp, W.; Hillewaere, X.K.D.; Rahier, H.; Du Prez, F.E. Development of optimized autonomous self-healing systems for epoxy materials based on maleimide chemistry. Polymer 2012, 53, 2320–2326. [Google Scholar] [CrossRef]
- Chakma, P.; Rodrigues Possarle, L.H.; Digby, Z.A.; Zhang, B.; Sparks, J.L.; Konkolewicz, D. Dual stimuli responsive self-healing and malleable materials based on dynamic thiol-Michael chemistry. Polym. Chem. 2017, 8, 6534–6543. [Google Scholar] [CrossRef]
- Kuhl, N.; Geitner, R.; Vitz, J.; Bode, S.; Schmitt, M.; Popp, J.; Schubert, U.S.; Hager, M.D. Increased stability in self-healing polymer networks based on reversible Michael addition reactions. J. Appl. Polym. Sci. 2017, 134, 44805. [Google Scholar] [CrossRef]
- Pepels, M.; Filot, I.; Klumperman, B.; Goossens, H. Self-healing systems based on disulfide-thiol exchange reactions. Polym. Chem. 2013, 4, 4955–4965. [Google Scholar] [CrossRef]
- Yoon, J.A.; Kamada, J.; Koynov, K.; Mohin, J.; Nicolaÿ, R.; Zhang, Y.; Balazs, A.C.; Kowalewski, T.; Matyjaszewski, K. Self-healing polymer films based on thiol–disulfide exchange reactions and self-healing kinetics measured using atomic force microscopy. Macromolecules 2011, 45, 142–149. [Google Scholar] [CrossRef]
- Yue, H.-B.; Fernández-Blázquez, J.P.; Beneito, D.F.; Vilatela, J.J. Real time monitoring of click chemistry self-healing in polymer composites. J. Mater. Chem. A 2014, 2, 3881–3887. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.H.; Vuluga, D.; Lecamp, L.; Burel, F. Photoinitiated thiol-epoxy addition for the preparation of photoinduced self-healing fatty coatings. RSC Adv. 2016, 6, 32098–32105. [Google Scholar] [CrossRef]
- Zhu, D.Y.; Cao, G.S.; Qiu, W.L.; Rong, M.Z.; Zhang, M.Q. Self-healing polyvinyl chloride (PVC) based on microencapsulated nucleophilic thiol-click chemistry. Polymer 2015, 69, 1–9. [Google Scholar] [CrossRef]
- Chung, U.S.; Min, J.H.; Lee, P.-C.; Koh, W.-G. Polyurethane matrix incorporating PDMS-based self-healing microcapsules with enhanced mechanical and thermal stability. Colloids Surf. A 2017, 518, 173–180. [Google Scholar] [CrossRef]
- Wei, K.; Gao, Z.; Liu, H.; Wu, X.; Wang, F.; Xu, H. Mechanical activation of platinum—acetylide complex for olefin hydrosilylation. ACS Macro Lett. 2017, 6, 1146–1150. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Peptides, the Wave of the Future; American Peptide Society: San Diego, CA, USA, 2001. [Google Scholar]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Binder, W.H.; Sachsenhofer, R. ‘Click’ Chemistry in polymer and materials science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. “Click”-chemistry in polymer and material science: An update. Macromol. Rapid Commun. 2008, 29, 952–981. [Google Scholar] [CrossRef]
- Zhao, Y.; Döhler, D.; Lv, L.-P.; Binder, W.H.; Landfester, K.; Crespy, D. Facile phase-separation approach to encapsulate functionalized polymers in core–shell nanoparticles. Macromol. Chem. Phys. 2014, 215, 198–204. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem. Int. Ed. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Huisgen, R. Kinetics and reaction mechanisms: Selected examples from the experience of forty years. Pure Appl. Chem 1989, 61, 613–628. [Google Scholar] [CrossRef]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef] [PubMed]
- Gorman, I.E.; Willer, R.L.; Kemp, L.K.; Storey, R.F. Development of a triazole-cure resin system for composites: Evaluation of alkyne curatives. Polymer 2012, 53, 2548–2558. [Google Scholar] [CrossRef]
- Kantheti, S.; Sarath, P.S.; Narayan, R.; Raju, K.V.S.N. Synthesis and characterization of triazole rich polyether polyols using click chemistry for highly branched polyurethanes. React. Funct. Polym. 2013, 73, 1597–1605. [Google Scholar] [CrossRef]
- Bond, G.C.; Keane, M.A.; Kral, H.; Lercher, J.A. Compensation phenomena in heterogeneous catalysis: General principles and a possible explanation. Cat. Rev. 2000, 42, 323–383. [Google Scholar] [CrossRef]
- Mulokozi, A.M. Kinetic parameters in heterogeneous kinetics. Thermochim. Acta 1992, 197, 363–372. [Google Scholar] [CrossRef]
- Nonahal, M.; Rastin, H.; Saeb, M.R.; Sari, M.G.; Moghadam, M.H.; Zarrintaj, P.; Ramezanzadeh, B. Epoxy/PAMAM dendrimer-modified graphene oxide nanocomposite coatings: Nonisothermal cure kinetics study. Prog. Org. Coat. 2018, 114, 233–243. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Kyotani, T.; Suzuki, K.-Y.; Yamashita, H.; Tomita, A. Formation of carbon-metal composites from metal ion exchanged graphite oxide. Tanso 1993, 1993, 255–265. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Kamat, P.V. Graphene-based nanoarchitectures. anchoring semiconductor and metal nanoparticles on a two-dimensional carbon support. J. Phys. Chem. Lett. 2010, 1, 520–527. [Google Scholar] [CrossRef]
- Goncalves, G.; Marques, P.A.A.P.; Granadeiro, C.M.; Nogueira, H.I.S.; Singh, M.K.; Grácio, J. Surface modification of graphene nanosheets with gold nanoparticles: The role of oxygen moieties at graphene surface on gold nucleation and growth. Chem. Mater. 2009, 21, 4796–4802. [Google Scholar] [CrossRef]
- Gan, Y.; Sun, L.; Banhart, F. One- and two-dimensional diffusion of metal atoms in graphene. Small 2008, 4, 587–591. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structures of trivalent alkyne 1 and trivalent azides 2, 3, 4 and 5.
Scheme 1.
Synthesis of trivalent azides 3*, 4, 4*, 5 and 5*.
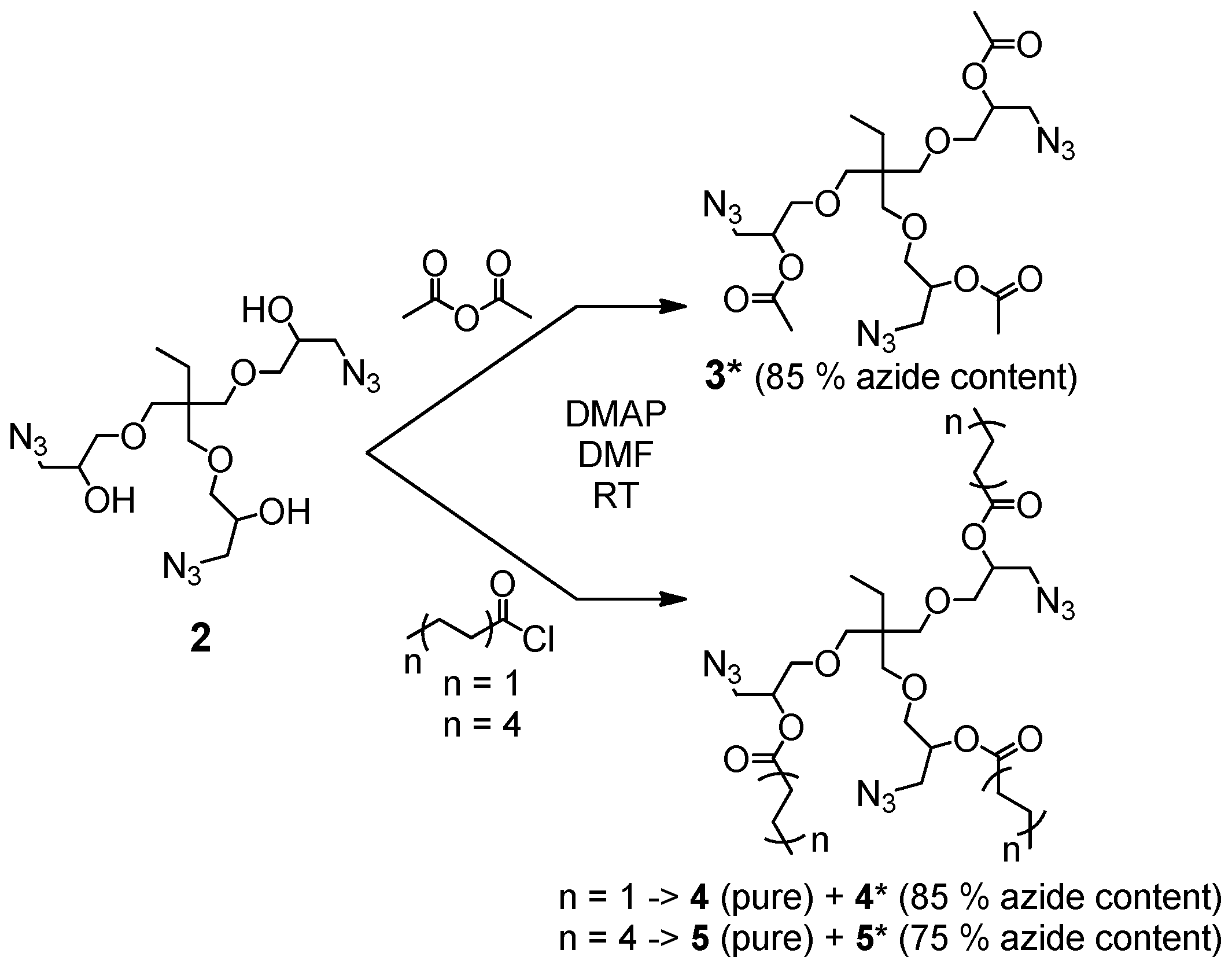
Figure 2.
Dynamic scanning calorimetry (DSC) measurements of the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3* with Cu(PPh3)3Br, Cu(PPh3)3F, Cu2O on graphene-oxide (TRGO-Cu2O, prepared at 600 °C) as well as without catalyst (W/O) at a heating rate of 5 K∙min−1.
Figure 2.
Dynamic scanning calorimetry (DSC) measurements of the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3* with Cu(PPh3)3Br, Cu(PPh3)3F, Cu2O on graphene-oxide (TRGO-Cu2O, prepared at 600 °C) as well as without catalyst (W/O) at a heating rate of 5 K∙min−1.

Figure 3.
DSC measurements of the “click-crosslinking” reaction of trivalent alkyne 1 and (a) trivalent azides 4 (red curve) or 4* (black curve) with TRGO-Cu2O, (b) trivalent azides 5 (red curve) or 5* (black curve) with TRGO-Cu2O (prepared at 600 °C), (c) trivalent azide 4* with different catalysts as well as without catalyst (W/O), (d) trivalent azide 5* with different catalysts as well as without catalyst (W/O) (all at a heating rate of 5 K∙min−1).
Figure 3.
DSC measurements of the “click-crosslinking” reaction of trivalent alkyne 1 and (a) trivalent azides 4 (red curve) or 4* (black curve) with TRGO-Cu2O, (b) trivalent azides 5 (red curve) or 5* (black curve) with TRGO-Cu2O (prepared at 600 °C), (c) trivalent azide 4* with different catalysts as well as without catalyst (W/O), (d) trivalent azide 5* with different catalysts as well as without catalyst (W/O) (all at a heating rate of 5 K∙min−1).
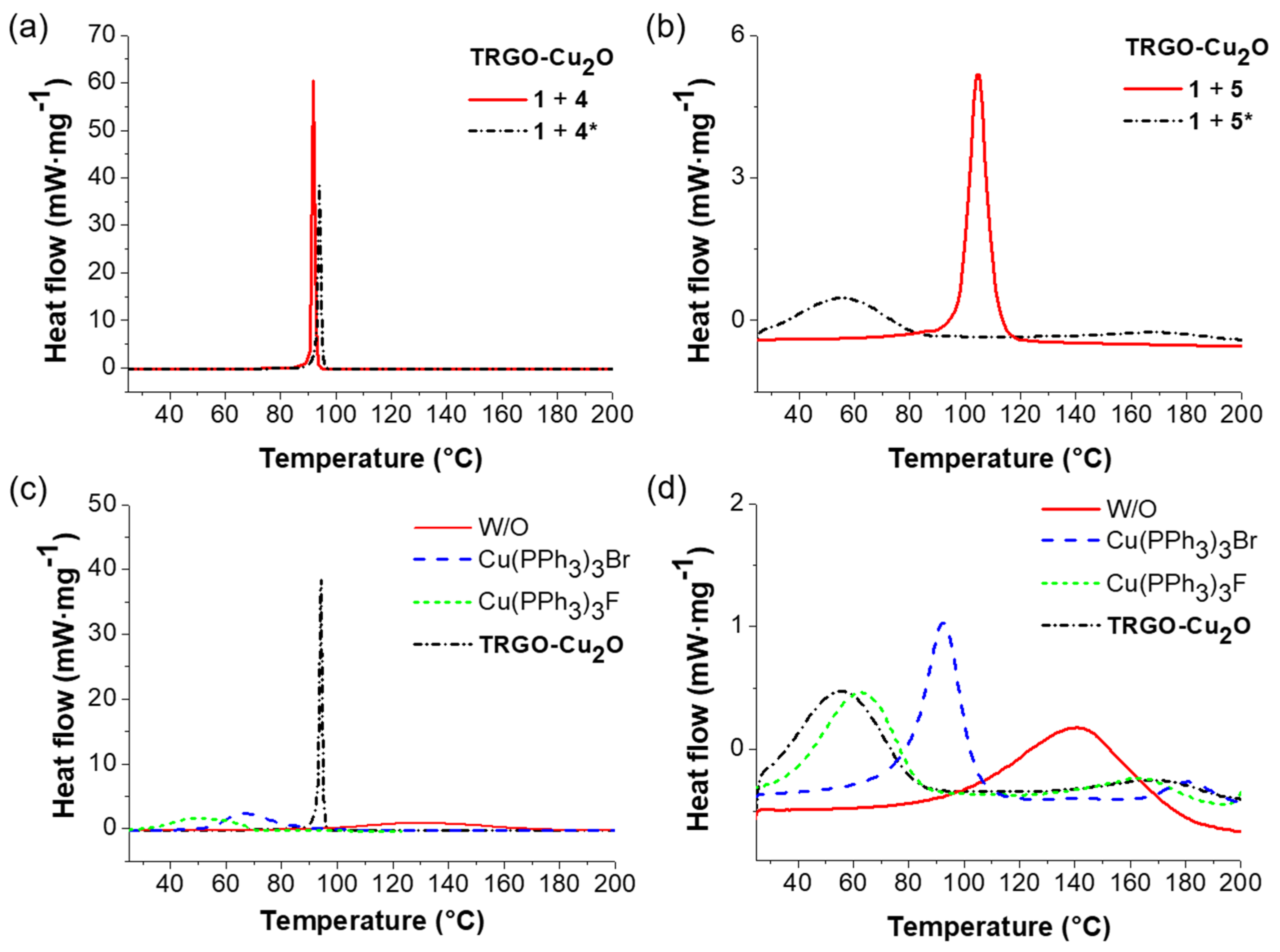
Figure 4.
Conversion vs. time of the “click-crosslinking“ reaction of trivalent alkyne 1 and trivalent azides 3* (black squares), 4* (red circles) or 5* (blue triangles) at 0 °C with Cu(PPh3)3F as catalysts at a heating rate of 5 K∙min−1.
Figure 4.
Conversion vs. time of the “click-crosslinking“ reaction of trivalent alkyne 1 and trivalent azides 3* (black squares), 4* (red circles) or 5* (blue triangles) at 0 °C with Cu(PPh3)3F as catalysts at a heating rate of 5 K∙min−1.
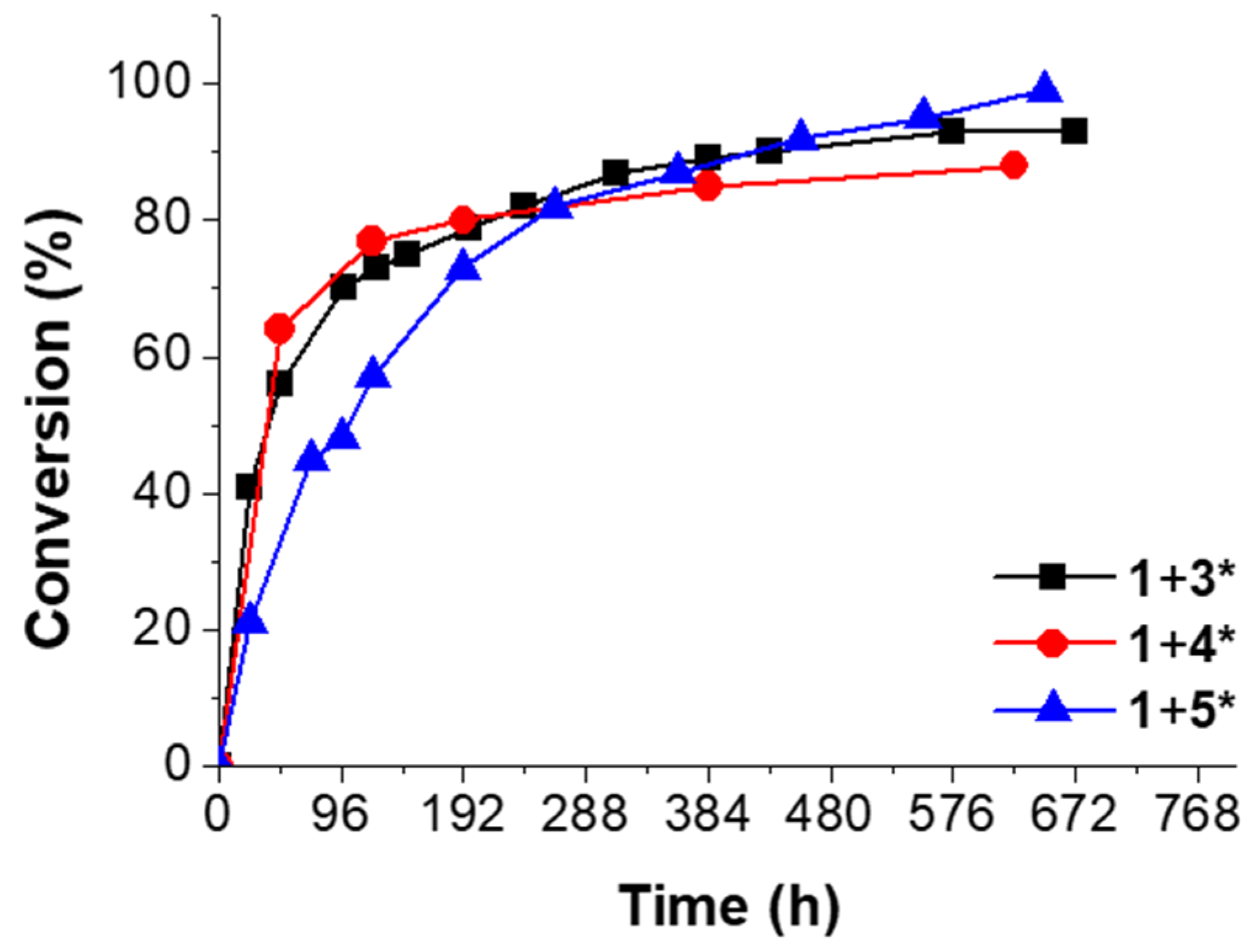
Figure 5.
Amount (wt %) of copper within TRGO-Cu2O and peak temperature (Tp) of the click reaction vs. the preparation temperature of TRGO-Cu2O for the click reaction of phenylacetylene and benzyl azide and for the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3*. Please note that the lines between the measuring points are drawn to guide the eye.
Figure 5.
Amount (wt %) of copper within TRGO-Cu2O and peak temperature (Tp) of the click reaction vs. the preparation temperature of TRGO-Cu2O for the click reaction of phenylacetylene and benzyl azide and for the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3*. Please note that the lines between the measuring points are drawn to guide the eye.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Thermal properties, reaction temperatures (Tonset and Tp), reaction enthalpies (ΔH), apparent activation energies (Ea, app) and conversions of the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3*, 4, 4*, 5 or 5* with different catalysts (1 mol %) as well as without catalyst (W/O) at a heating rate of 5 K∙min−1. TRGO-Cu2O was prepared at 600 °C.
Table 1.
Thermal properties, reaction temperatures (Tonset and Tp), reaction enthalpies (ΔH), apparent activation energies (Ea, app) and conversions of the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3*, 4, 4*, 5 or 5* with different catalysts (1 mol %) as well as without catalyst (W/O) at a heating rate of 5 K∙min−1. TRGO-Cu2O was prepared at 600 °C.
| Entry | Azide | Catalyst | Mass (%) | Tonset 1 (°C) | Tp 1 (°C) | ΔH 2 (kJ∙mol−1) | Ea, app (kJ∙mol−1) | Conversion 3 (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 3* | W/O | - | 91 | 133 | 205 | 83 | 78 |
| 2 | Cu(PPh3)3Br | 3.4 | 59 | 74 | 185 | 87 | 71 | |
| 3 | Cu(PPh3)3F | 3.2 | 39 | 66 | 191 | 83 | 73 | |
| 4 | TRGO-Cu2O | 7.4 | 51 | 63 | 177 | 55 | 67 | |
| 5 | 4 | TRGO-Cu2O | 6.7 | 91 | 92 | 233 | 104 | 89 |
| 6 | 4* | W/O | - | 94 | 125 | 227 | 76 | 87 |
| 7 | Cu(PPh3)3Br | 2.9 | 54 | 66 | 223 | 70 | 85 | |
| 8 | Cu(PPh3)3F | 3.2 | 32 | 50 | 192 | 61 | 73 | |
| 9 | TRGO-Cu2O | 6.9 | 93 | 94 | 211 | 97 | 81 | |
| 10 | 5 | TRGO-Cu2O | 5.0 | 98 | 105 | 248 | 133 | 95 |
| 11 | 5* | W/O | - | 102 | 141 | 174 | 100 | 66 |
| 12 | Cu(PPh3)3Br | 2.2 | 80 | 93 | 149 | 55 | 57 | |
| 13 | Cu(PPh3)3F | 2.4 | 38 | 62 | 140 | 103 | 53 | |
| 14 | TRGO-Cu2O | 5.2 | 32 | 56 | 127 | 85 | 48 |
1 According to our previous publication [22] and our experience, the error is typically ≈ ±5 K. 2 According to our previous publication [22], the error is typically ≈ ±6 kJ·mol−1. 3 Calculated with respect to the enthalpy for 100% click conversion which is ΔH = 262 kJ·mol−1 for the reference reaction of phenylacetylene and benzyl azide with 1 mol % of Cu(PPh3)3Br as catalyst.
Table 2.
Thermal properties of the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3* with TRGO-Cu2O as a catalyst (prepared at different temperatures) at a heating rate of 5 K∙min−1: Reaction temperatures (Tonset and Tp), reaction enthalpies (ΔH) and conversions.
Table 2.
Thermal properties of the “click-crosslinking” reaction of trivalent alkyne 1 and trivalent azide 3* with TRGO-Cu2O as a catalyst (prepared at different temperatures) at a heating rate of 5 K∙min−1: Reaction temperatures (Tonset and Tp), reaction enthalpies (ΔH) and conversions.
| Entry | Catalyst | Tonset 1 (°C) | Tp 1 (°C) | ∆H 2 (kJ⋅mol−1) | Conversion 3 (%) |
|---|---|---|---|---|---|
| 1 | W/O | 96 | 130 | 75 | 29 |
| 2 | TRGO-Cu2O (300 °C) | 82 | 97 | 108 | 41 |
| 3 | TRGO-Cu2O (400 °C) | 80 | 100 | 101 | 38 |
| 4 | TRGO-Cu2O (500 °C) | 90 | 105 | 107 | 41 |
| 5 | TRGO-Cu2O (600 °C) | 91 | 100 | 115 | 44 |
| 6 | TRGO-Cu2O (700 °C) | 64 | 80 | 107 | 41 |
| 7 | TRGO-Cu2O (800 °C) | 72 | 91 | 113 | 43 |
1 Measurements were performed thrice, and the error is ≈ ±5 K. 2 Measurements were performed thrice and the error is ≈ ±6 kJ⋅mol−1. 3 Calculated with respect to the enthalpy for 100% click conversion, which is ΔH = 262 kJ⋅mol−1 for the reference reaction of phenylacetylene and benzyl azide with 1 mol % of Cu(PPh3)3Br as catalyst.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kargarfard, N.; Diedrich, N.; Rupp, H.; Döhler, D.; Binder, W.H. Improving Kinetics of “Click-Crosslinking” for Self-Healing Nanocomposites by Graphene-Supported Cu-Nanoparticles. Polymers 2018, 10, 17. https://doi.org/10.3390/polym10010017
AMA Style
Kargarfard N, Diedrich N, Rupp H, Döhler D, Binder WH. Improving Kinetics of “Click-Crosslinking” for Self-Healing Nanocomposites by Graphene-Supported Cu-Nanoparticles. Polymers. 2018; 10(1):17. https://doi.org/10.3390/polym10010017
Chicago/Turabian StyleKargarfard, Neda, Norman Diedrich, Harald Rupp, Diana Döhler, and Wolfgang H. Binder. 2018. "Improving Kinetics of “Click-Crosslinking” for Self-Healing Nanocomposites by Graphene-Supported Cu-Nanoparticles" Polymers 10, no. 1: 17. https://doi.org/10.3390/polym10010017
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.