Thermo-Responsive Starch-g-(PAM-co-PNIPAM): Controlled Synthesis and Effect of Molecular Components on Solution Rheology


Engineering and Technology Institute Groningen, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands
*
Author to whom correspondence should be addressed.
Polymers 2018, 10(1), 92; https://doi.org/10.3390/polym10010092
Submission received: 7 December 2017
/
Revised: 12 January 2018
/
Accepted: 16 January 2018
/
Published: 19 January 2018
(This article belongs to the Collection Polysaccharides)
Abstract
:A series of highly branched random copolymers of acrylamide (AM) and N-isopropylacrylamide (NIPAM) have been prepared from a waxy potato starch-based macroinitiator by aqueous Cu0-mediated living radical polymerization (Cu0-mediated LRP). The NIPAM intake in the copolymer was varied between 0% and 50 mol % to evaluate the influence of chain composition on the aqueous rheological properties as well as their low critical solution temperature (LCST). The viscosity of the copolymer was found to increase with the NIPAM intake and an LCST can be observed when the NIPAM content is high enough (e.g., 50 mol %). In addition, thermo-thickening behavior was observed at a low shear rate (γ ≤ 10 s−1) and higher NIPAM content was found to shift the onset of thermo-thickening behavior to a lower temperature. However, the absolute increase in viscosity values is reduced with the NIPAM intake. Besides this, an interesting significant thermo-thickening behavior was also observed on highly branched starch-g-polyacrylamide at high temperatures (>80 °C), which has not been previously reported. Rheology tests also revealed a good salt-resistant property in copolymers with low NIPAM content (e.g., <25 mol %). Considering the viscosity profile in saline as compared to that in pure water, this NIPAM intake seems to represent an optimum balance of viscosity and salt-resistance performance.
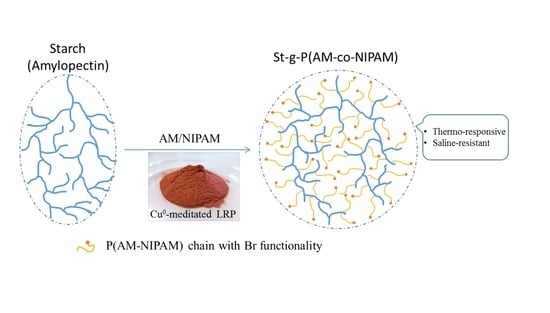
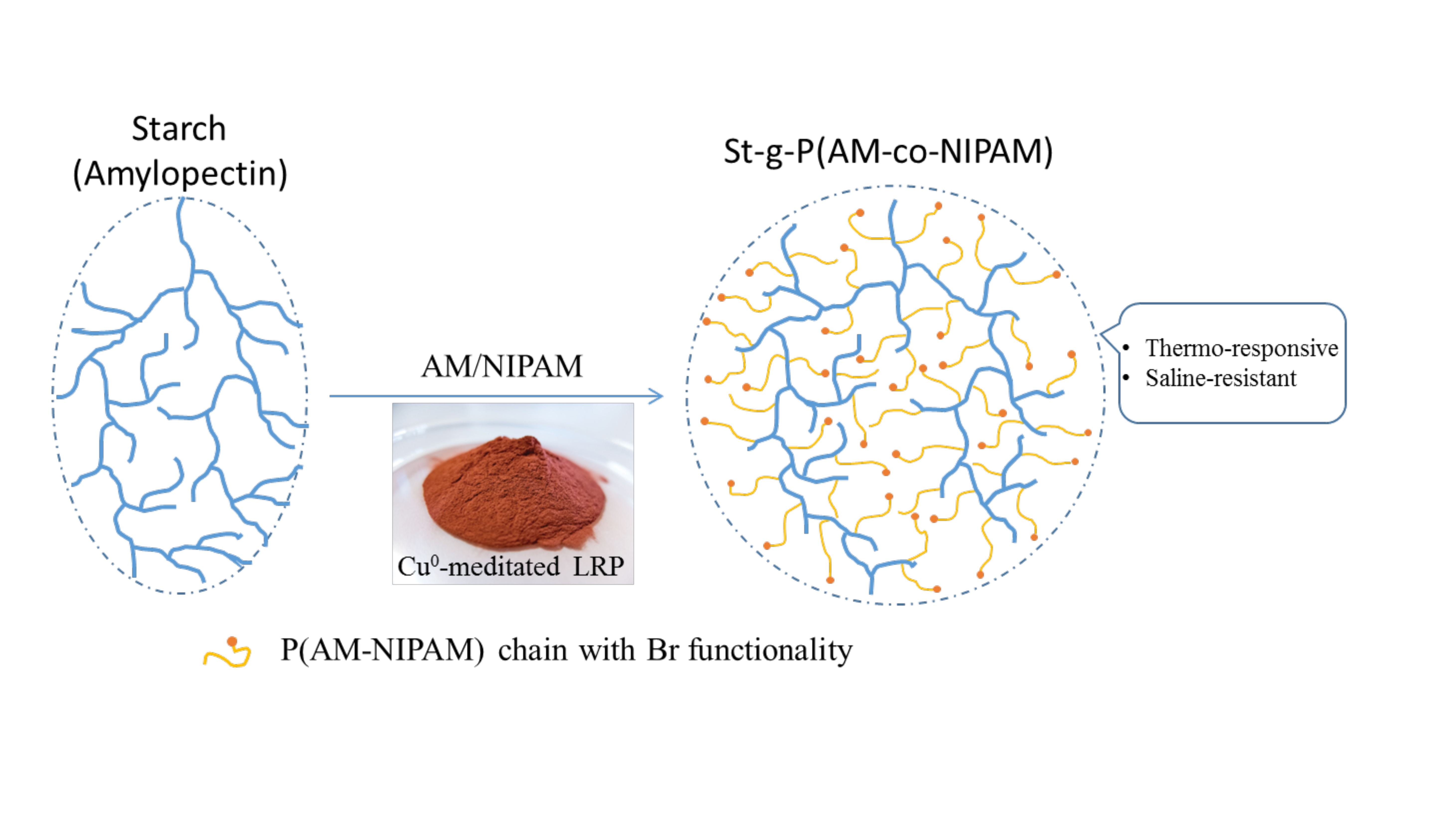
1. Introduction
Stimuli-responsive polymers have been of great interest in the past decades due to their responsive abilities to a variety of factors, like pH [1], temperature [2], ultrasound [3], light [4], and electric/magnetic fields [5,6]. Among these polymers, thermo-responsive (or thermo-sensitive) ones are still the most widely studied subgroup since the earliest report on poly(N-isopropylacrylamide) (PNIPAM) [7]. According to the response of solubility to the change of temperature, generally, two different types of thermo-responsive polymer can be distinguished. One type is represented by polymers, like polyzwitterions and poly(ethylene oxide), which have an upper critical solution temperature (UCST). This means that their solubility in a given solvent increases with temperature above a given critical value, i.e., the UCST [8]. The other type comprises polymers like PNIPAM that display the opposite responsive behavior, i.e., the solubility decreases with temperature below a given critical value (LCST). Since the discovery of thermo-responsive polymers, application fields have been expanded to a variety of subjects: drug delivery [3,9], bioengineering [2,10], sensors [11,12], functional coatings [13,14], and enhanced oil recovery (EOR) [15,16].
In the above-mentioned applications, most of the polymers are supposed to perform their thermo-responsive functions in the range of 20 °C to 50 °C [2,3,4,14,17,18]. As the phase transition temperature possessed by linear PNIPAM is in the range of 31 °C to 33 °C (these values being irrespective of the polymer concentration, though slightly affected by the average molecular weight) [16,19], which falls within the described application windows’ (e.g., drug carrier and functional coating) temperature range [2,3,14], the synthesis of PNIPAM (co)polymers has been an attractive subject since the first report about its LCST phenomenon in 1968 [20]. From then on, many investigations have focused on manipulating the LCST window of PNIAPM. To achieve this, several factors should be considered including the molecular weight [19], properties of end groups [19,21], and chemical structure (molecular composition and architecture) [16,22]. Thanks to the development of controlled polymerization techniques (Atom-transfer radical-polymerization (ATRP), Reversible addition–fragmentation chain-transfer (RAFT), etc.), these kinds of studies can be carried out in a convenient and more accurate manner.
According to previous findings on PNIPAM homopolymers, the LCST can only be tuned by the structure of the end groups when the molecular weight is low enough (degree of polymerization (DP) below 200) [19,23] or the tacticity is well controlled [24]. Thus, most of the efforts have been spent on the synthesis of PNIPAM (block, random, graft) copolymers and polymers with differing architecture [22,25]. For these polymers, not only do the composition and architecture affect the LCST, but their solution rheological behavior can also be manipulated due to the inter-/intra-molecular thermo-reversible aggregation of the hydrophobic NIPAM moieties above the LCST. Although varieties of PNIPAM copolymers with different architecture have been synthesized, relatively few reports focus on their thermo-thickening properties. To the best of our knowledge, only block and graft PNIPAM copolymers have been studied so far [15,16,26,27].
In the case of grafted comb-like polymers, compared with side chains with a block structure, it has been reported that random PNIPAM copolymer side chains could endow the product with better thermo-thickening behavior (above 50 °C) and solubility in water for applications like EOR [16,28]. From this point of view, the synthesis of branched PNIPAM copolymers will be helpful to fully understanding the influence of the structure on polymers’ properties. In this case, waxy potato starch, which contains more than 95% amylopectin with a highly branched structure that is composed of 105–106 anhydroglucose units (AGU), will be an interesting candidate as the core due to its inherent high molecular weight and highly branched structure (see also Figure S1 illustration and Graphical abstract) [29,30,31,32].
Based on this, in the present work, random copolymers of acrylamide and N-isopropylacrylamide were grafted from a waxy potato starch backbone at the molecular level by Cu0-mediated LRP in aqueous solution. The obtained highly branched copolymer of starch-g-poly(acrylamide-co-N-isopropylacrylamide) (St-g-(PAM-co-PNIPAM) was characterized by 1H-NMR and FTIR. The influence of chain composition on the highly branched polymers’ rheological properties (at both room temperature and high temperature), LCST, and their response to salinity was studied and compared with the comb-like copolymer reported [16].
2. Materials and Methods
2.1. Materials
Waxy potato starch (>95% amylopectin, molecular weight in the range 107–109 Da and roughly 5% of α (1–6) branching points) was kindly donated by Avebe (Veendam, The Netherlands) and dried under vacuum at 60 °C for 48 h before use. Lithium chloride was purchased from Sigma-Aldrich and dried under vacuum at 80 °C for 24 h before use. Anhydrous N,N-dimethylacetamide (DMAc) was purchased from Sigma-Aldrich in Sure/Seal™ (Steinheim, Germany). 2-bromopropionyl bromide (BpB), formaldehyde solution (37%), and formic acid (>95%) were purchased from Sigma-Aldrich and used as received. Tris(2-aminoethyl)amine (Tren) was purchased from Tokyo Chemical Industry Co., Ltd. (TCI, Tokyo, Japan) and used as received. Tris[2-(dimethylamino)ethyl]amine (Me6Tren) was synthesized following the procedures reported [33]. N-Isopropylacrylamide (NIPAM, stabilized with 4-Methoxyphenol (MEHQ)) was purchased from TCI and recrystallized from acetone to remove the inhibitor. Acrylamide (AM) was purchased from Sigma-Aldrich and used as received. Copper powder (<75 μm) was purchased from Sigma-Aldrich and stored under an N2 atmosphere.
2.2. Characterization
NMR spectra were recorded on a Varian Mercury Plus 400 MHz spectrometer (Varian, Inc., Palo Alto, CA, USA) using deuterated solvents purchased from Sigma-Aldrich. Fourier Transform Infrared (FTIR) spectra were recorded with attenuated total reflection (ATR) accessories on an IRTracer-100 SHIMADZU Fourier Transform Infrared Spectrophotometer (Shimadzu Corp., Kyoto, Japan) and data were processed with LabSolutions IR software (Version 2.11, Shimadzu, Kyoto, Japan, 2014). Aqueous gel permeation chromatography (GPC) was conducted on an Agilent 1200 system (Agilent, Santa Clara, CA, USA) equipped with a differential refractive index (DRI) detector and column set (PSS SUPREMA 100 Å, 1000 Å, 3000 Å) from Polymer Standard Service GmbH (PSS, Mainz, Germany). The mobile phase used was 0.05 M NaNO3. Column oven and detector temperatures were regulated to 40 °C, with a flow rate of 1 mL/min. Polyacrylamide standards from PSS were used for calibration. Samples were filtered through a membrane with 0.22 μm pore size before injection. Experimental molar mass and polydispersity index (PDI) values of synthesized polymers were determined by conventional calibration using PSS WinGPC UniChrom GPC/SEC software (Version 8.20, Polymer Standards Service GmbH, Mainz, Germany, 1992–2014).
Rheological properties were measured using a HAAKE Mars III (Thermo Scientific, Waltham, MA, USA) rheometer equipped with a cone-and-plate geometry (diameter 60 mm, angle 2°). Solution viscosity was measured as a function of shear rate (0.1 to 1750 s−1, T = 20 °C), salt concentration (5000~100,000 ppm of NaCl, T = 20 °C, shear rate 10 s−1) and temperature (10 °C to 90 °C, shear rate 1 s−1, 3 s−1, 10 s−1 and 30 s−1), respectively.
The intrinsic viscosity was determined according to the Martin equation [34]:
where is the reduced viscosity, is the specific viscosity, c is the polymer concentration, [η] is the intrinsic viscosity, and . is a constant dependent on the polymer–solvent system.
The relaxation time () was determined according to the “Carreau–Yasuda” model [35,36,37]:
where η is the viscosity, is the zero shear rate viscosity, is the viscosity at the infinite shear rate, is the critical shear rate for the onset of shear thinning, n − 1 is the power law index, and represents the transition region between and the power law region.
The cloud point of the different polymers was determined by UV–vis analysis. A JASCO V-730 UV–vis spectrophotometer (JASCO, Easton, MD, USA) equipped with a temperature-controlled six-position sample holder was used. The transmittance of the polymer solutions (1.2 wt %) was recorded at 350 nm at temperature ranges from 20 °C to 95 °C against a reference sample containing demineralized water. Temperature was manually controlled with the software, and each measurement was taken after the temperature was stabilized within ±0.5 °C for 30 s.
2.3. Synthesis of Starch-Based Macroinitiator (StBr)
Waxy potato starch (2.59 g, 16 mmol) and lithium chloride (1.02 g, 24 mmol) were added to a 250 mL three-necked flask (dried overnight at 100 °C before use) connected with a mechanical stirrer. The system was vacuumed under heat and backfilled with N2 three times to remove residual water. Anhydrous DMAc (100 mL) was transferred to the flask and the mixture was stirred at 130 °C for about 1 h under an N2 atmosphere. A transparent solution formed when the mixture cooled down to room temperature naturally. The solution was cooled down with an ice bath and then 0.42 mL (4 mmol) BpB was added dropwise within 30 min under the protection of N2. The mixture was then warmed up naturally to room temperature and stirred for 3 h. The final products were precipitated out with tenfold acetone and then filtered, washed, and dried under vacuum at 45 °C for 24 h. The resulting white powder was then purified by Soxhlet extraction with ethanol as the solvent for 24 h (final yield: 87%). The obtained degree of substitution (DS) represents a convenient compromise as it allows a proper characterization (not possible for lower values where spectroscopic data are difficult to identify), while, at the same time, does not compromise the solubility in water (for too high DS values).
2.4. Synthesis of St-g-(PAM-co-PNIPAM) by Aqueous Cu0-Mediated LRP
Typical Polymerization Protocol: H2O (100 mL), StBr (48.6 mg, 0.04 mmol), a mixture of AM and NIPAM (240 mmol in total), and Me6TREN (23 μL, 0.08 mmol) were charged to a 250 mL three-neck round-bottom flask with a magnetic stirrer bar and rubber septum. The solution was deoxygenated by three freeze–pump–thaw cycles. Cu powder (5.2 mg, 0.08 mmol) was then added with rapid stirring under the protection of nitrogen. The mixture was allowed to react for 15 min at room temperature. The resulting solution was freeze-dried and followed by Soxhlet extraction with ethanol as the solvent for 48 h. The product was then vacuum-dried at 65 °C for 48 h. For the purposes of brevity and clarity, taking the grafted product with no NIPAM content as an example, the sample was named St-g-PNIPAM-P0; 0 here stands for the fact that the mole percentage of NIPAM in the feeding AM/NIPAM monomer mixture is 0%.
2.5. Cleaving of Graft Polymer Chains from the Starch Backbone
The starch-based copolymer (0.25 g) was dissolved in 25 mL Milli-Q water in a round-bottom flask, and 0.25 mL concentrated hydrochloric acid was then added. The mixture was stirred and refluxed at 100 °C for 3 h. The resulting free polyacrylamide (PAM) was precipitated out with methanol, then filtered and washed with methanol three times. The product was dried under vacuum at 60 °C for 24 h.
3. Results and Discussion
The successful synthesis of water-soluble waxy potato starch-based macroinitiator (StBr) was proved by FTIR and NMR (1H-NMR, 13C-NMR, and gHSQC) characterization. Details can be seen in the supporting information (Figures S2 and S3).
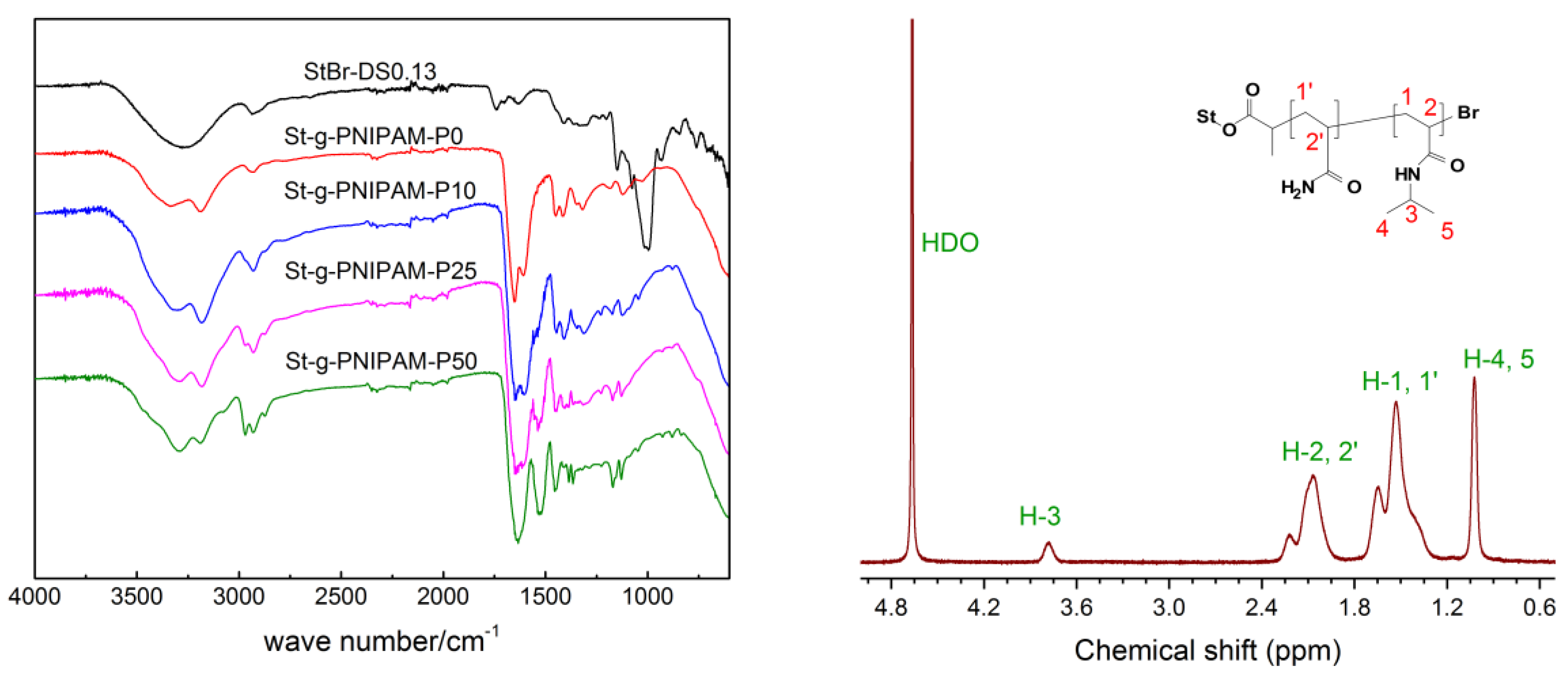
A series of St-g-(PAM-co-PNIPAM) with different NIPAM molar intakes were then synthesized by Cu0-mediated LRP with StBr as the initiator and Cu powder/Me6Tren as the catalyst system. According to our previous work, the target DP for all the samples was set to 6000 to achieve satisfactory viscosity values, for example, for EOR applications. The polymer was characterized by FTIR (Figure 1, Left) and 1H-NMR (Figure 1, Right). The absorption peak around 3188 cm−1 in the FTIR spectrum was attributed to the stretch of the N–H bond in the amide group. For the St-g-PNIAPM-P0, typical amide group peaks at 1652 cm−1 (amide I) and 1610 cm−1 (amide II) could also be seen in the spectrum. With increasing NIPAM intake in the copolymer, the amide I and II peaks gradually shifted to 1628 cm−1 and 1530 cm−1, respectively [38]. The 1H-NMR spectrum of the copolymer is shown in Figure 1 (Right), in which the peak around 1.0 ppm was attributed to the methyl protons in the NIPAM unit. The peak at 3.8 ppm originates from the tertiary carbon protons in the NIPAM amide group. The signals in the range of 1.9–2.3 ppm and 1.2–1.8 ppm were assigned to the tertiary and secondary carbon protons in the copolymer backbone, respectively.
As shown in Table 1, the NIPAM content in the monomer mixture was varied from 0% to 50% (mol %) while the overall target DP was set to 6000 for all the polymers. 1H-NMR was used to determine the mole percentage of the NIPAM unit in the product according to the following equation:
where A is the sum of the integrals of peaks H-2,2′ and H-1,1′ when the integral of peak H-3 was set to 1 (Figure 1, Right). Clearly, the composition of the resulting copolymer is quite similar to that of the feed, as shown in Table 1. This clearly suggests almost equal reactivity ratios for AM and NIPAM [39].
Due to the high polymerization rate and the corresponding high solution viscosity, it was not possible to take samples to monitor the polymerization kinetics. Nevertheless, at the end, the PAM homopolymer was thus cleaved from the St-g-PNIPAM-P0 backbone and characterized by GPC to determine whether it is a controlled polymerization. As shown in Table 1 and Figure S4, compared with the grafted starch copolymer with PDI of 2.15, a narrower molecular weight distribution (PDI = 1.36) that indicated a well-controlled polymerization was observed after the hydrolyzation of the starch backbone. It is very difficult to estimate whether all initiation sites actually contributed to the grafting of the chains (especially for the copolymers, where no GPC data after cleavage can be collected) even if it is clear that those sites that reacted resulted in polymeric chains with relatively narrow PDI values. This might stem from a different reactivity of the individual starch macromolecules and actually explain the shoulder in the GPC traces (Figure S4).
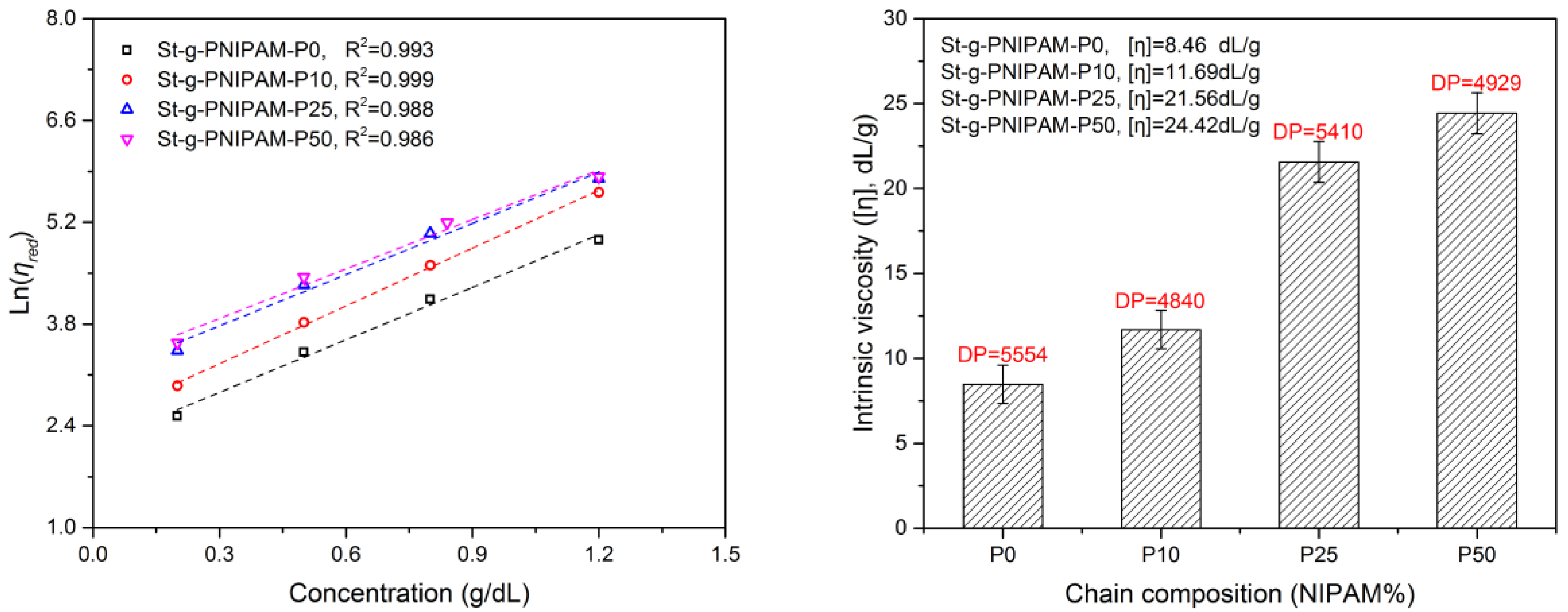
To investigate the influence of chain composition on copolymer solution properties, a series of rheology tests were carried out. For a polymer with a given molecular structure, the intrinsic viscosity is an indication of polymer’s hydrodynamic volume [40,41]. The intrinsic viscosity can be obtained by extrapolating the plot of against polymer concentration to c = 0 (see Experimental section and Figure 2).
As can be observed in Figure 2, the intrinsic viscosity increased with the NIPAM content in the grafted copolymer. Considering the chain length of PAM-co-PNIPAM, the intrinsic viscosity of St-g-PNIPAM-P25 and St-g-PNIPAM-P50 is significantly higher than that of St-g-PNIPAM-P0 and St-g-PNIPAM-P10, respectively. This means that the incorporation of the NIPAM unit expands the hydrodynamic volume of St-g-PAM in water, which can be explained by the breaking of the strong intra-hydrogen bond between AM units. This could also explain why St-g-PNIPAM-P0 has poor solubility in cold water compared with the rest of the prepared copolymers.
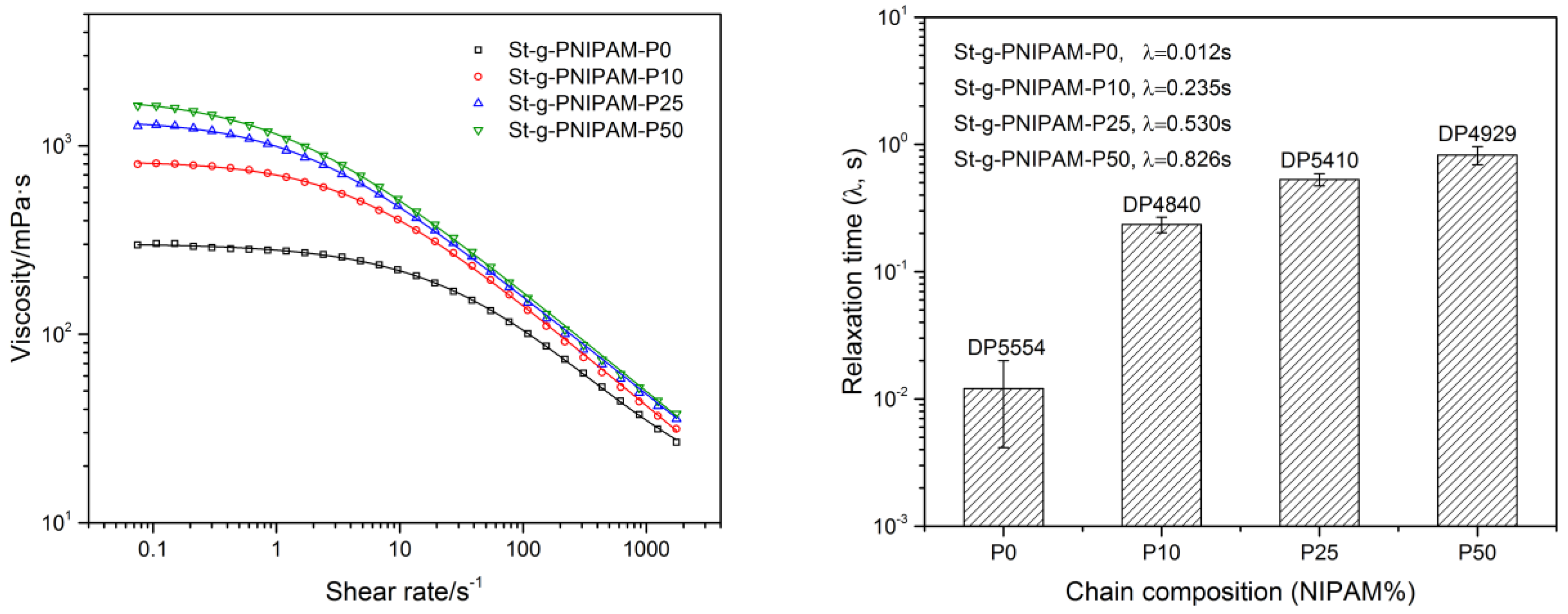
The influence of chain composition on solution viscosity as a function of shear rate was also evaluated at the same polymer concentration (1.2 wt %), the result of which is shown in Figure 3 (Left). It is clear that the viscosity of copolymers with a larger NIPAM content is higher than that of those with a lower NIPAM ratio in composition (at the same overall DP), especially in the low shear rate region. This was attributed to the expansion of molecular hydrodynamic volume, as indicated in the evaluation of the intrinsic viscosity. Furthermore, these data were also fitted with the “Carreau–Yasuda” model (see Experimental section) to study the influence of chain composition on the copolymer’s relaxation time () (Figure 3, Right). As indicated in Figure 3 (Right), a higher NIPAM unit content leads to higher and, thus, a lower critical shear thinning rate () should be observed in the flow curve. This is in line with Figure 3 (Left), which shows a shift in the onset of shear thinning behavior towards lower shear rate regions as the mole ratio of NIPAM deceases.
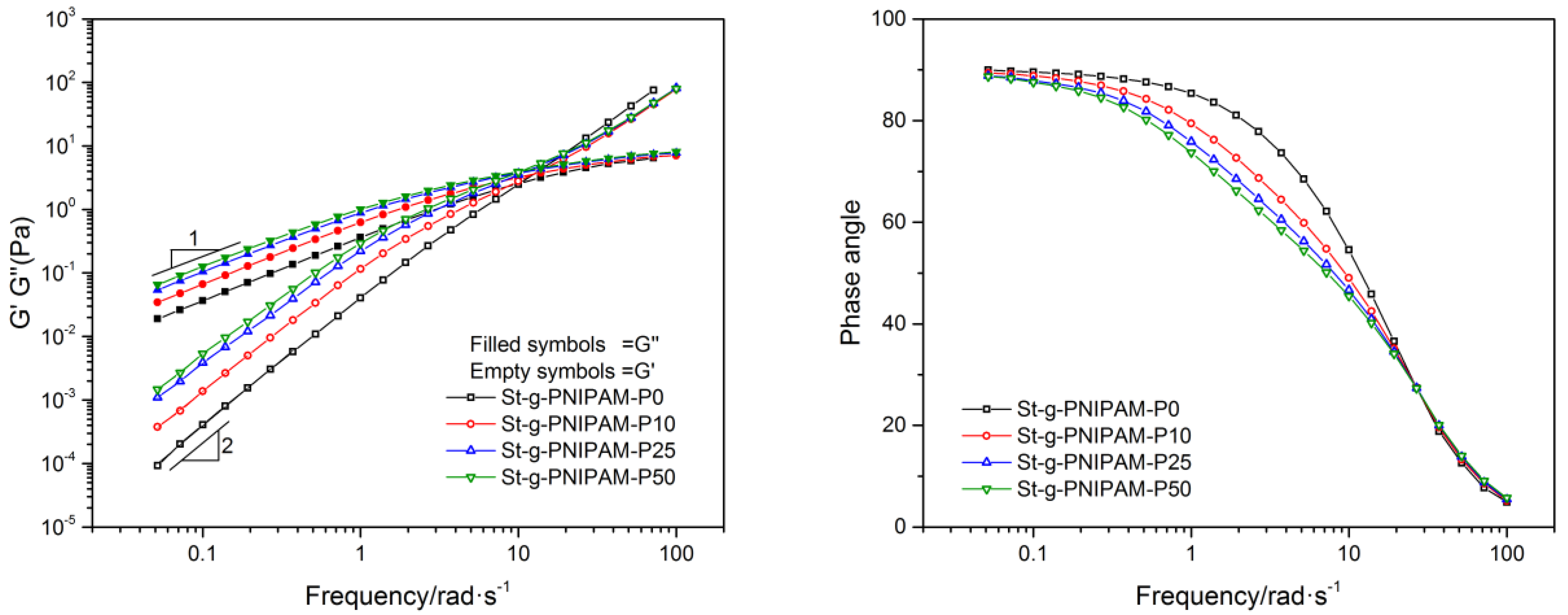
Besides viscosity, the viscoelastic property is another important factor that effects the applications of polymers. For example, it has been reported that higher elasticity is beneficial for improving the sweep efficiency in EOR [42,43]. The influence of composition on the copolymers’ viscoelastic properties is shown in Figure 4. As can be observed, both the storage modulus (G′) and the loss modulus (G′′) increased as the NIPAM ratio in the copolymer increased. In the terminal zone (low frequencies region), for all the samples, entangled polymer solution flow behavior with G′′ directly proportional to the frequency (ω) (slope = 1) and G′ proportional to ω2 (slope = 2) was indicated [44]. A comparison of phase angles (ω < 20 rad/s) at equal polymer concentrations demonstrates that the copolymer with a higher NIPAM ratio displays a more pronounced elastic response, especially when compared with that of copolymers with a similar grafted chain length (e.g., St-g-PNIPAM-P0 and St-g-PNIPAM-P10).
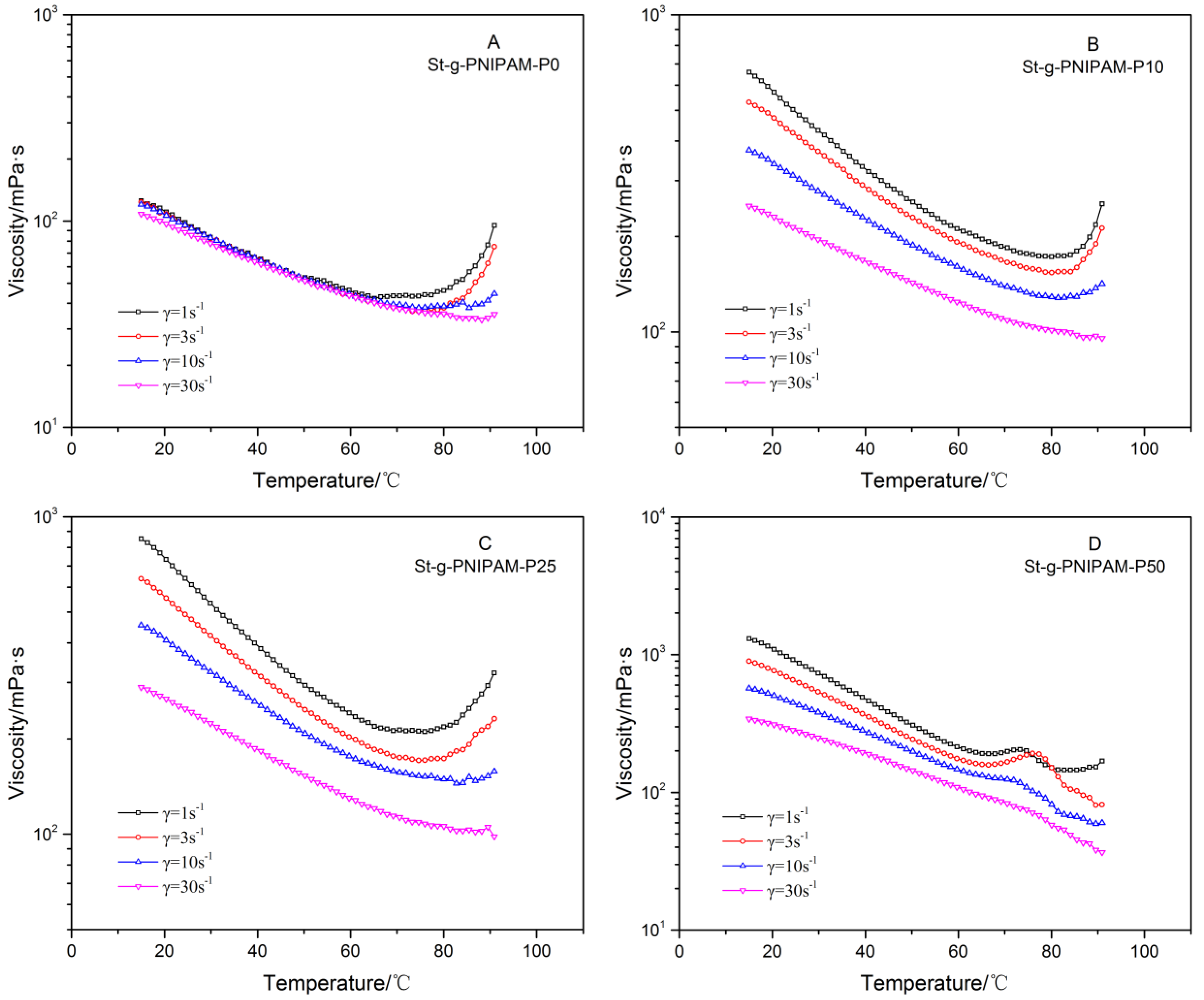
For NIPAM (co)polymers, the influence of structure and composition on their thermo-responsive behavior is also of great interest for applications. The viscosity (solutions with the same polymer concentrations) as a function of temperature was measured at different shear rates, and the results are displayed in Figure 5.
Linear PAM is not a thermo-responsive polymer in solution and neither is PAM with the comb-like structure according to previous reports [16,35]. However, an interesting thermo-thickening behavior was observed for the starch-based highly branched PAM when the temperature is above 80 °C (Figure 5A), especially at a low shear rate. During measurements, the cone and plate were covered with a cap to avoid water evaporation. Water evaporation as a cause for the increase in viscosity can be ruled out also based on the fact that this was not observed for linear PAM (Mn = 35,200) (see Figure S5). Considering the lower viscosity at room temperature compared with copolymers containing NIPAM (vide supra), this thermo-responsive behavior was attributed to the breakdown of strong intra-molecular hydrogen bonds at high temperatures, which is beneficial for the expansion of hydrodynamic volume. At a high shear rate, the inter-molecular interaction (physical entanglement, hydrogen bonding, and hydrophobic association in the case of the NIPAM copolymer) is broken down so that the thermo-thickening effect is suppressed. Thermo-responsive polymers are of interest for many applications. For example, in EOR, this shear-sensitive thermo-thickening behavior is favorable because it endows the solution with a relatively low viscosity (in turn, beneficial for energy saving) in a wellbore due to the high flow rate (thus, high shear rate). In a reservoir, however, higher viscosity will be displayed because of the low shear rate resulting from the porous structure of the oil formation—in turn, generating elastic instabilities and turbulent flow. Figure 5 also reveals that when NIPAM is copolymerized with acrylamide, the intra-molecular hydrogen bonding is inherently weakened and, thus, the incremental ratio of viscosity from the onset of thermal-thickening decreases at higher NIPAM ratios. Further, comparing the viscosity profiles at the shear rate of 1 s−1, we notice that the onset of thermal-thickening shifted to a lower temperature gradually as the ratio of NIPAM increased in the copolymer.
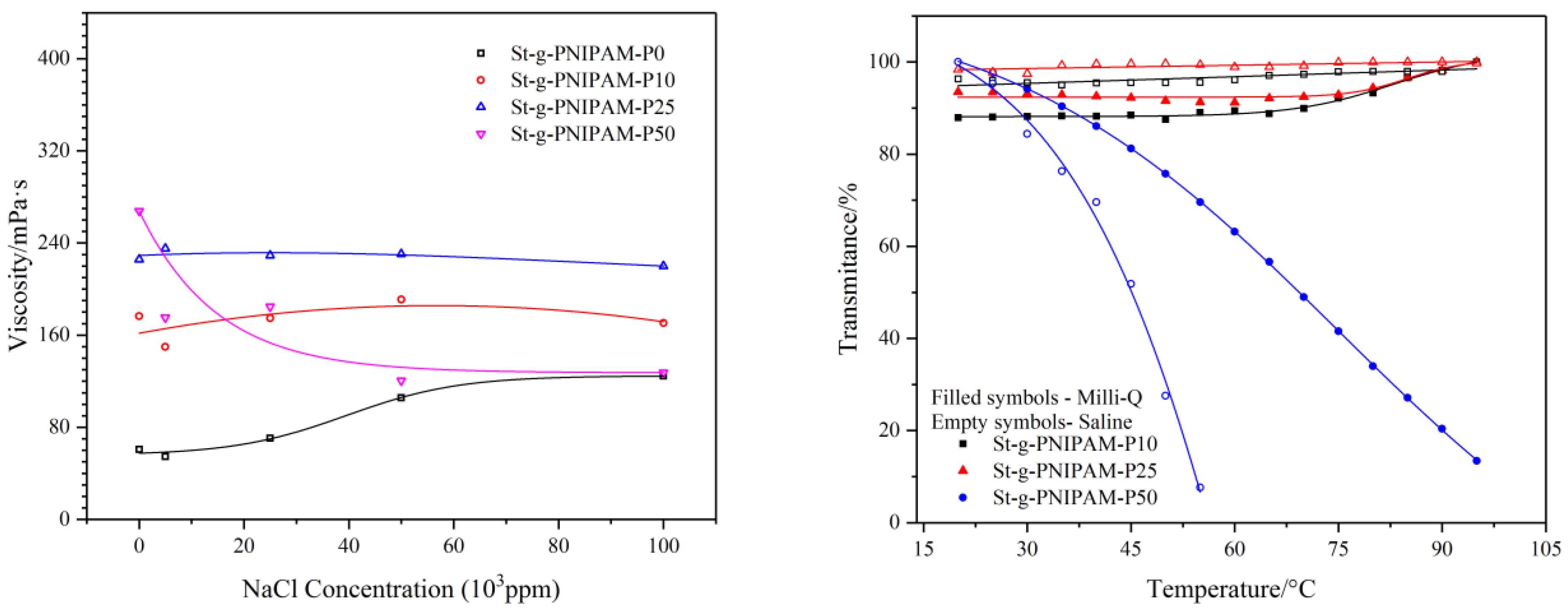
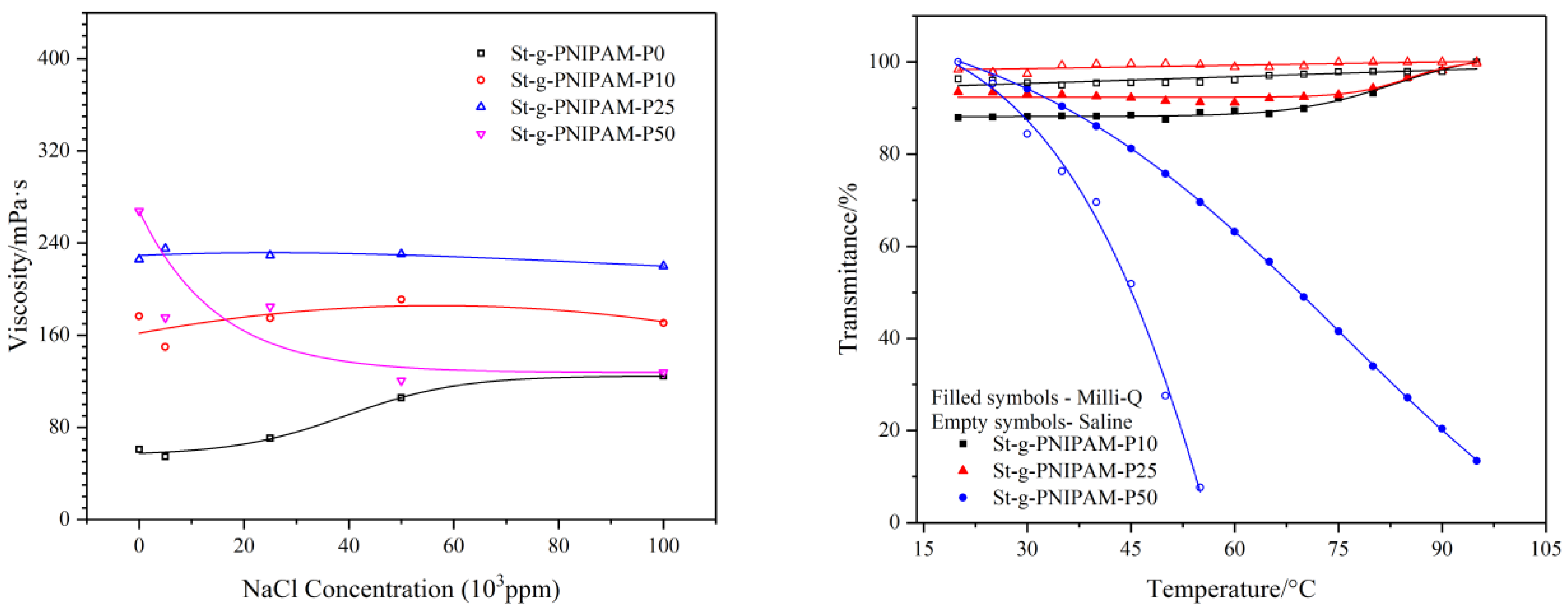
Not only temperature effects the performance of NIPAM (co)polymers; salinity also has an impact on their solution properties. The viscosity as a function of salt (NaCl) concentration was measured at a shear rate of 10 s−1. The result is shown in Figure 6 (Left). Besides this, the influence of salt (0 ppm and 100,000 ppm) on the copolymer’s cloud point was also studied and the result is shown in Figure 6 (Right).
As can be seen in Figure 6, an increasing trend in the viscosity of St-g-PNIPAM-P0 was implied as the salt concentration increased. This could be explained by the “structure” change of water (e.g., the decrease of “free water”) due to the addition of salt [45]. Contrasting with the behavior of St-g-PNIPAM-P0, copolymers containing NIPAM in a low mole ratio (10 mol % and 25 mol % in this case) displayed a relatively stable viscosity profile versus salt concentration. This is very different from partially hydrolyzed polyacrylamide (HPAM), the viscosity of which normally displays an abrupt drop due to the collapse of molecular hydrodynamic volume resulting from the salt screening of the repulsive electrostatic forces between charged carboxyl groups [46]. For copolymers containing a high mole ratio of NIPAM (50% for example), a decreasing trend can be observed with the increase of salt concentration. This was blamed on the “salt out” effect of NaCl on NIPAM units in the copolymer, which caused shrinking of the polymer hydrodynamic volume and thus reduced the viscosity [47].
In the case of the cloud point, as shown in Figure 6 (Right), copolymers with a low NIPAM ratio (10 mol % and 25 mol %) displayed no phase transition in the temperature range of 20 °C to 95 °C. The addition of salt has no significant influence on their cloud point. St-g-PNIPAM-P50, however, has a cloud point at around 68 °C (50 mol % of the transmittance) due to high NIPAM content. The addition of NaCl (100,000 ppm) shifted the cloud point to 45 °C.
4. Conclusions
Different highly branched random copolymers of acrylamide (AM) and N-isopropylacrylamide (NIPAM) were prepared with a water-soluble waxy potato starch-based macroinitiator by aqueous Cu0-mediated living radical polymerization (Cu0-mediated LRP) at room temperature. The mole ratio of NIPAM was varied in the range of 0% to 50% to investigate the influence of chain composition on the polymers’ aqueous rheological properties as well as their LCST. The viscosity of the grafted copolymer was found to increase as the NIPAM ratio increased, due to the breakdown of the intra-molecular hydrogen bond. Compared with commercially available linear partially hydrolyzed polyacrylamide, the viscosity of the highly branched AM/NIPAM copolymer also displayed good stability in saline water. However, too much NIPAM content is not preferable for the copolymers’ salt-resistance performance, which can also be proved by the phase transition behavior (LCST) of the different copolymers.
As expected, the viscosity versus temperature profile revealed the low shear rate ( 10 s−1) thermo-thickening phenomenon of AM/NIPAM copolymers. Unlike the reported comb-like random copolymer [16], however, a high NIPAM ratio (50 mol %) doesn’t endow the highly branched copolymer with as significant a thermo-thickening property as that of a low NIPAM content copolymer. Surprisingly, a pronounced thermo-thickening behavior was also observed on highly branched starch-g-polyacrylamide at high temperatures (>80 °C), which has not been reported before. This was attributed to the disruption of the strong intramolecular hydrogen bonds originating from the highly branched structure at high temperatures.
Considering the cheapness of the starch, the ease of synthesis, and the copolymers’ saline resistance and thermo-thickening behavior in water, the highly branched random AM/NIPAM copolymers could be good candidates for applications like EOR.
Supplementary Materials
The following are available online at www.mdpi.com/2073-4360/10/1/92/s1, Figure S1: Illustration for the structure of amylopectin and grafted copolymer, Scheme S1: Synthesis of waxy potato starch-based ATRP macroinitiator and St-g-PAM, Figure S2: FTIR spectra of Starch-Br with different DS, Figure S3: 1H-NMR (a), 13C-NMR (b) and gHSQC (c) spectra of St-Br (DS = 0.13) in D2O, Figure S4: Molecular weight distribution of St-Br, St-g-PNIPAM-P0 before and after hydrolysis (St-g-PNIPAM-P0-H), Figure S5: Viscosity versus temperature of PAM (Mn = 35200) with 1.5 wt % copolymer concentration (γ = 1 s−1).
Acknowledgments
This work is performed under the financial support from the China Scholarship Council (CSC) with Grant Number 201406380107. We thank Avebe (Veendam, The Netherlands) for the donation of the waxy potato starch.
Author Contributions
Yifei Fan and Francesco Picchioni conceived and designed the experiments; Yifei Fan and Nadia Boulif performed the experiments; Yifei Fan analyzed the data; Yifei Fan and Francesco Picchioni wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kocak, G.; Tuncer, C.; Bütün, V. Ph-responsive polymers. Polym. Chem. 2017, 8, 144–176. [Google Scholar] [CrossRef]
- Wang, B.; Xu, Q.; Ye, Z.; Liu, H.; Lin, Q.; Nan, K.; Li, Y.; Wang, Y.; Qi, L.; Chen, H. Copolymer brushes with temperature-triggered, reversibly switchable bactericidal and antifouling properties for biomaterial surfaces. ACS Appl. Mater. Interfaces 2016, 8, 27207–27217. [Google Scholar] [CrossRef] [PubMed]
- Zardad, A.-Z.; Choonara, Y.; du Toit, L.; Kumar, P.; Mabrouk, M.; Kondiah, P.; Pillay, V. A review of thermo- and ultrasound-responsive polymeric systems for delivery of chemotherapeutic agents. Polymers 2016, 8, 359. [Google Scholar] [CrossRef]
- Zhang, X.; Pint, C.L.; Lee, M.H.; Schubert, B.E.; Jamshidi, A.; Takei, K.; Ko, H.; Gillies, A.; Bardhan, R.; Urban, J.J.; et al. Optically- and thermally-responsive programmable materials based on carbon nanotube-hydrogel polymer composites. Nano Lett. 2011, 11, 3239–3244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Choi, H. Stimuli-responsive polymers and colloids under electric and magnetic fields. Polymers 2014, 6, 2803–2818. [Google Scholar] [CrossRef]
- Thevenot, J.; Oliveira, H.; Sandre, O.; Lecommandoux, S. Magnetic responsive polymer composite materials. Chem. Soc. Rev. 2013, 42, 7099–7116. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Brooks, W.L.; Sumerlin, B.S. New directions in thermoresponsive polymers. Chem. Soc. Rev. 2013, 42, 7214–7243. [Google Scholar] [CrossRef] [PubMed]
- Seuring, J.; Agarwal, S. Polymers with upper critical solution temperature in aqueous solution: Unexpected properties from known building blocks. ACS Macro Lett. 2013, 2, 597–600. [Google Scholar] [CrossRef]
- Badi, N. Non-linear peg-based thermoresponsive polymer systems. Prog. Polym. Sci. 2017, 66, 54–79. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Nie, X.; Wen, T.; Ji, Y.; Wu, X.; Zhao, Y.; Chen, C. Near infrared laser-induced targeted cancer therapy using thermoresponsive polymer encapsulated gold nanorods. J. Am. Chem. Soc. 2014, 136, 7317–7326. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, S. Responsive polymers for detection and sensing applications: Current status and future developments. Macromolecules 2010, 43, 8315–8330. [Google Scholar] [CrossRef]
- Zhang, Q.; Vancoillie, G.; Mees, M.A.; Hoogenboom, R. Thermoresponsive polymeric temperature sensors with broad sensing regimes. Polym. Chem. 2015, 6, 2396–2400. [Google Scholar] [CrossRef]
- Rotzetter, A.C.; Schumacher, C.M.; Bubenhofer, S.B.; Grass, R.N.; Gerber, L.C.; Zeltner, M.; Stark, W.J. Thermoresponsive polymer induced sweating surfaces as an efficient way to passively cool buildings. Adv. Mater. 2012, 24, 5352–5356. [Google Scholar] [CrossRef] [PubMed]
- Nagase, K.; Onuma, T.; Yamato, M.; Takeda, N.; Okano, T. Enhanced wettability changes by synergistic effect of micro/nanoimprinted substrates and grafted thermoresponsive polymer brushes. Macromol. Rapid Commun. 2015, 36, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- Van Mastrigt, F.; Stoffelsma, T.; Wever, D.A.Z.; Picchioni, F. Thermoresponsive comb polymers as thickeners for high temperature aqueous fluids. Mater. Today Commun. 2017, 10, 34–40. [Google Scholar] [CrossRef]
- Wever, D.A.Z.; Riemsma, E.; Picchioni, F.; Broekhuis, A.A. Comb-like thermoresponsive polymeric materials: Synthesis and effect of macromolecular structure on solution properties. Polymer 2013, 54, 5456–5466. [Google Scholar] [CrossRef]
- Zhao, Y.; Bai, T.; Shao, Q.; Jiang, S.; Shen, A.Q. Thermoresponsive self-assembled nipam-zwitterion copolymers. Polym. Chem. 2015, 6, 1066–1077. [Google Scholar] [CrossRef]
- Tran, N.T.; Truong, N.P.; Gu, W.; Jia, Z.; Cooper, M.A.; Monteiro, M.J. Timed-release polymer nanoparticles. Biomacromolecules 2013, 14, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Furyk, S.; Zhang, Y.; Ortiz-Acosta, D.; Cremer, P.S.; Bergbreiter, D.E. Effects of end group polarity and molecular weight on the lower critical solution temperature of poly(n-isopropylacrylamide). J. Polym. Sci. Part A Polym. Chem. 2006, 44, 1492–1501. [Google Scholar] [CrossRef]
- Heskins, M.; Guillet, J.E. Solution properties of poly(n-isopropylacrylamide). J. Macromol. Sci. Part A Chem. 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Xia, Y.; Yin, X.; Burke, N.A.D.; Stöver, H.D.H. Thermal response of narrow-disperse poly(n-isopropylacrylamide) prepared by atom transfer radical polymerization. Macromolecules 2005, 38, 5937–5943. [Google Scholar] [CrossRef]
- Xue, N.; Qiu, X.-P.; Chen, Y.; Satoh, T.; Kakuchi, T.; Winnik, F.M. Effect of chain architecture on the phase transition of star and cyclic poly(n-isopropylacrylamide) in water. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 2059–2068. [Google Scholar] [CrossRef]
- Xia, Y.; Burke, N.A.D.; Stöver, H.D.H. End group effect on the thermal response of narrow-disperse poly(N-isopropylacrylamide) prepared by atom transfer radical polymerization. Macromolecules 2006, 39, 2275–2283. [Google Scholar] [CrossRef]
- Ito, M.; Ishizone, T. Living anionic polymerization ofn-methoxymethyl-n-isopropylacrylamide: Synthesis of well-defined poly(N-isopropylacrylamide) having various stereoregularity. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 4832–4845. [Google Scholar] [CrossRef]
- Lanzalaco, S.; Armelin, E. Poly(n-isopropylacrylamide) and copolymers: A review on recent progresses in biomedical applications. Gels 2017, 3, 36. [Google Scholar] [CrossRef]
- Cheaburu, C.N.; Ciocoiu, O.-N.; Staikos, G.; Vasile, C. Thermoresponsive sodium alginate-g-poly(N-isopropylacrylamide) copolymers iii. Solution properties. J. Appl. Polym. Sci. 2013, 127, 3340–3348. [Google Scholar] [CrossRef]
- Díaz-Silvestre, S.E.; St Thomas, C.; Rivera-Vallejo, C.; Cadenas-Pliego, G.; Pérez-Alvarez, M.; de León-Gómez, R.D.; Jiménez-Regalado, E.J. Concentration effect of n-isopropylacrylamide on viscoelastic properties of hydrosoluble thermo-thickening copolymers. Polym. Bull. 2017, 74, 4009–4021. [Google Scholar] [CrossRef]
- Li, X.E.; Xu, Z.; Yin, H.; Feng, Y.; Quan, H. Comparative studies on enhanced oil recovery: Thermoviscosifying polymer versus polyacrylamide. Energy Fuels 2017, 31, 2479–2487. [Google Scholar] [CrossRef]
- Jane, J.-L. Starch: Structure and properties. In Chemical and Functional Properties of Food Saccharides; Tomasik, P., Ed.; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Yoo, S.-H.; Jane, J.-L. Molecular weights and gyration radii of amylopectins determined by high-performance size-exclusion chromatography equipped with multi-angle laser-light scattering and refractive index detectors. Carbohydr. Polym. 2002, 49, 307–314. [Google Scholar] [CrossRef]
- Zobel, H.F. Molecules to granules: A comprehensive starch review. Starch Stärke 1988, 40, 44–50. [Google Scholar] [CrossRef]
- Shrestha, A.K.; Halley, P.J. Starch modification to develop novel starch-biopolymer blends: State of art and perspectives. In Starch Polymers: From Genetic Engineering to Green Applications, 1st ed.; Newnes: Oxford, OK, 2014; pp. 105–143. [Google Scholar]
- Icke, R.N.; Wisegarver, B.B.; Alles, G.A. B-phenylethyldimethylamine. Org. Synth. 1945, 25, 89–92. [Google Scholar]
- Masuelli, M.A.; Sansone, M.G. Hydrodynamic properties of gelatin-studies from intrinsic viscosity measurements. In Products and Applications of Biopolymers; InTech: London, UK, 2012. [Google Scholar]
- Wever, D.A.Z.; Polgar, L.M.; Stuart, M.C.A.; Picchioni, F.; Broekhuis, A.A. Polymer molecular architecture as a tool for controlling the rheological properties of aqueous polyacrylamide solutions for enhanced oil recovery. Ind. Eng. Chem. Res. 2013, 52, 16993–17005. [Google Scholar] [CrossRef]
- Yasuda, K.; Armstrong, R.; Cohen, R. Shear flow properties of concentrated solutions of linear and star branched polystyrenes. Rheol. Acta 1981, 20, 163–178. [Google Scholar] [CrossRef]
- Carreau, P.J. Rheological equations from molecular network theories. Trans. Soc. Rheol. 1972, 16, 99–127. [Google Scholar] [CrossRef]
- Beattie, D.A.; Addai-Mensah, J.; Beaussart, A.; Franks, G.V.; Yeap, K.Y. In situ particle film atr ftir spectroscopy of poly (N-isopropyl acrylamide) (pnipam) adsorption onto talc. Phys. Chem. Chem. Phys. 2014, 16, 25143–25151. [Google Scholar] [CrossRef] [PubMed]
- Chiklis, C.K.; Grasshoff, J.M. Swelling of thin films. I. Acrylamide–n-isopropylacrylamide copolymers in water. J. Polym. Sci. Part A-2 Polym. Phys. 1970, 8, 1617–1626. [Google Scholar] [CrossRef]
- Arvidson, S.A.; Rinehart, B.T.; Gadala-Maria, F. Concentration regimes of solutions of levan polysaccharide from bacillus sp. Carbohydr. Polym. 2006, 65, 144–149. [Google Scholar] [CrossRef]
- Lee, J.; Tripathi, A. Intrinsic viscosity of polymers and biopolymers measured by microchip. Anal. Chem. 2005, 77, 7137–7147. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Wang, D.; Zhong, H. Study on flow behavoirs of viscoelastic polymer solution in micropore with dead end. In SPE Annual Technical Conference and Exhibition; Society of Petroleum Engineers: Richardson, TX, USA, 2006. [Google Scholar]
- Zhu, D.; Zhang, J.; Han, Y.; Wang, H.; Feng, Y. Laboratory study on the potential eor use of hpam/ves hybrid in high-temperature and high-salinity oil reservoirs. J. Chem. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Picout, D.R.; Ross-Murphy, S.B. Thermoreversible and Irreversible Physical Gels from Biopolymers; Marcel Dekker, Inc.: New York, NY, USA, 2002. [Google Scholar]
- Saito, S. Salt effect on polymer solutions. J. Polym. Sci. Part A Polym. Chem. 1969, 7, 1789–1802. [Google Scholar] [CrossRef]
- Gou, S.; He, Y.; Ma, Y.; Luo, S.; Zhang, Q.; Jing, D.; Guo, Q. A water-soluble antimicrobial acrylamide copolymer containing sulfitobetaine for enhanced oil recovery. RSC Adv. 2015, 5, 51549–51558. [Google Scholar] [CrossRef]
- Du, H.; Wickramasinghe, R.; Qian, X. Effects of salt on the lower critical solution temperature of poly(N-isopropylacrylamide). J. Phys. Chem. B 2010, 114, 16594–16604. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
FTIR spectra of StBr and Starch-g-poly(acrylamide-co-N-isopropylacrylamide) (St-g-(PAM-co-PNIPAM)) with different N-isopropylacrylamide (NIPAM) mole ratio (Left) and 1H-NMR spectra of St-g-(PAM-co-PNIPAM) in D2O (Right).
Figure 1.
FTIR spectra of StBr and Starch-g-poly(acrylamide-co-N-isopropylacrylamide) (St-g-(PAM-co-PNIPAM)) with different N-isopropylacrylamide (NIPAM) mole ratio (Left) and 1H-NMR spectra of St-g-(PAM-co-PNIPAM) in D2O (Right).

Figure 2.
Reduced viscosity as a function of copolymer concentration (Martin’s equation) (Left) and corresponding intrinsic viscosity (Right).
Figure 2.
Reduced viscosity as a function of copolymer concentration (Martin’s equation) (Left) and corresponding intrinsic viscosity (Right).

Figure 3.
Viscosity as a function of shear rate (Left, 1.2 wt % copolymer solution) and corresponding relaxation time from the “Carreau–Yasuda” model (Right).
Figure 3.
Viscosity as a function of shear rate (Left, 1.2 wt % copolymer solution) and corresponding relaxation time from the “Carreau–Yasuda” model (Right).

Figure 4.
The storage (G′), loss storage (G′′) (Left) and phase angle (Right) as a function of frequency at 1.2 wt % copolymer concentration.
Figure 4.
The storage (G′), loss storage (G′′) (Left) and phase angle (Right) as a function of frequency at 1.2 wt % copolymer concentration.

Figure 5.
Viscosity versus temperature of (A) St-g-PNIPAM-P0; (B) St-g-PNIPAM-P10; (C) St-g-PNIPAM-P25; and (D) St-g-PNIPAM-P50 with 1.2 wt % copolymer concentration.
Figure 5.
Viscosity versus temperature of (A) St-g-PNIPAM-P0; (B) St-g-PNIPAM-P10; (C) St-g-PNIPAM-P25; and (D) St-g-PNIPAM-P50 with 1.2 wt % copolymer concentration.

Figure 6.
Influence of composition and salt concentration on polymers’ viscosity (γ = 10 s−1) (Left) and cloud point (Right) with 1 wt % copolymer solution.
Figure 6.
Influence of composition and salt concentration on polymers’ viscosity (γ = 10 s−1) (Left) and cloud point (Right) with 1 wt % copolymer solution.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Experimental data for St-g-(PAM-co-PNIPAM) synthesized by Cu0-mediated living radical polymerization (LRP).
Table 1.
Experimental data for St-g-(PAM-co-PNIPAM) synthesized by Cu0-mediated living radical polymerization (LRP).
| Sample | Monomer Ratio a (AM:NIPAM) | Time/min | Conversion/% b | Ratio c (NIPAM) | DP d | PDI e | |
|---|---|---|---|---|---|---|---|
| AM | NIPAM | ||||||
| St-g-PNIPAM-P0 | 100:0 | 12 | 91.56 | - | 0 | 5554 | 1.36 |
| St-g-PNIPAM-P10 | 90:10 | 12 | 80.66 | 80.66 | 10 | 4840 | 1.64 |
| St-g-PNIPAM-P25 | 75:25 | 15 | 92.57 | 82.95 | 23 | 5410 | - f |
| St-g-PNIPAM-P50 | 50:50 | 12 | 87.08 | 77.22 | 47 | 4929 | - f |
a Overall ratio for polymerization [M]/[I]/[Cu0]/[L]= 6000:1:2:2; b Monomer conversion determined by mass and NMR; c NIPAM unit ratio in the copolymer, determined according to 1H-NMR; d Degree of polymerization (DP) determined by mass and NMR; e Polydispersity index (PDI) values for (co)polymer cleaved from the starch backbone; f Not available due to potential high intermolecular association, leading to retention of the polymer inside the gel permeation chromatography (GPC) column.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fan, Y.; Boulif, N.; Picchioni, F. Thermo-Responsive Starch-g-(PAM-co-PNIPAM): Controlled Synthesis and Effect of Molecular Components on Solution Rheology. Polymers 2018, 10, 92. https://doi.org/10.3390/polym10010092
AMA Style
Fan Y, Boulif N, Picchioni F. Thermo-Responsive Starch-g-(PAM-co-PNIPAM): Controlled Synthesis and Effect of Molecular Components on Solution Rheology. Polymers. 2018; 10(1):92. https://doi.org/10.3390/polym10010092
Chicago/Turabian StyleFan, Yifei, Nadia Boulif, and Francesco Picchioni. 2018. "Thermo-Responsive Starch-g-(PAM-co-PNIPAM): Controlled Synthesis and Effect of Molecular Components on Solution Rheology" Polymers 10, no. 1: 92. https://doi.org/10.3390/polym10010092
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.