Coordination Polymers Based on Phthalic Acid and Aminopyrazine Ligands: On the Importance of N–H···π Interactions

 , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments
2.3. Preparations
2.3.1. General Procedure
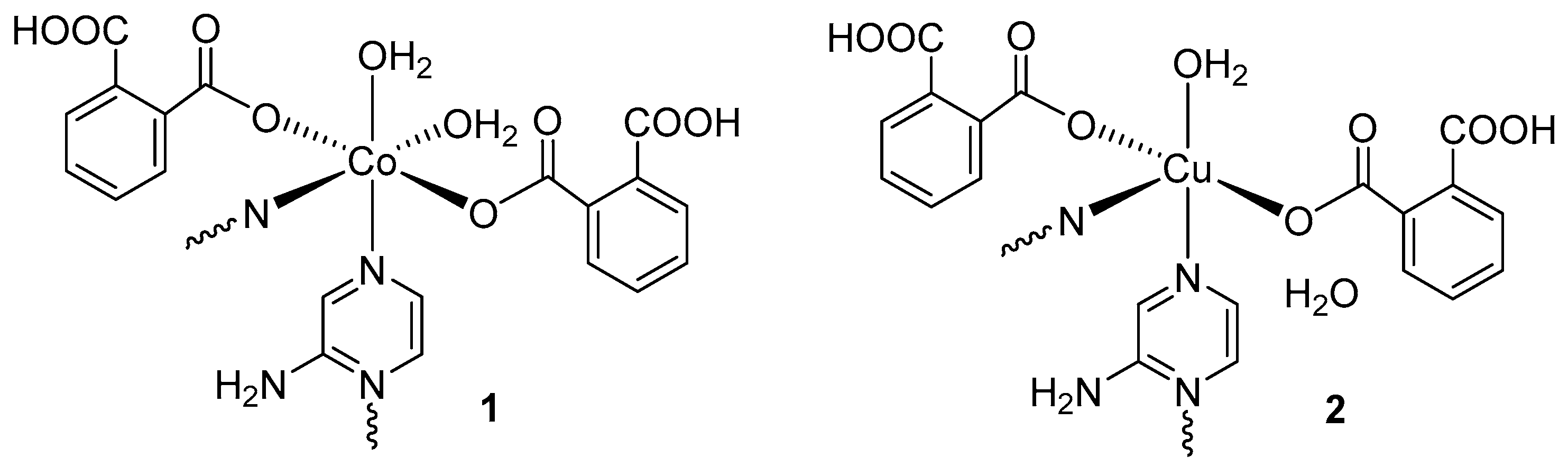
2.3.2. {Co(HL1)2(μ-L2)(H2O)2}n (1)
2.3.3. {[Cu(HL1)2(μ-L2)H2O]·H2O}n (2)
2.4. X-ray Details
2.5. Theoretical Methods
3. Results and Discussion
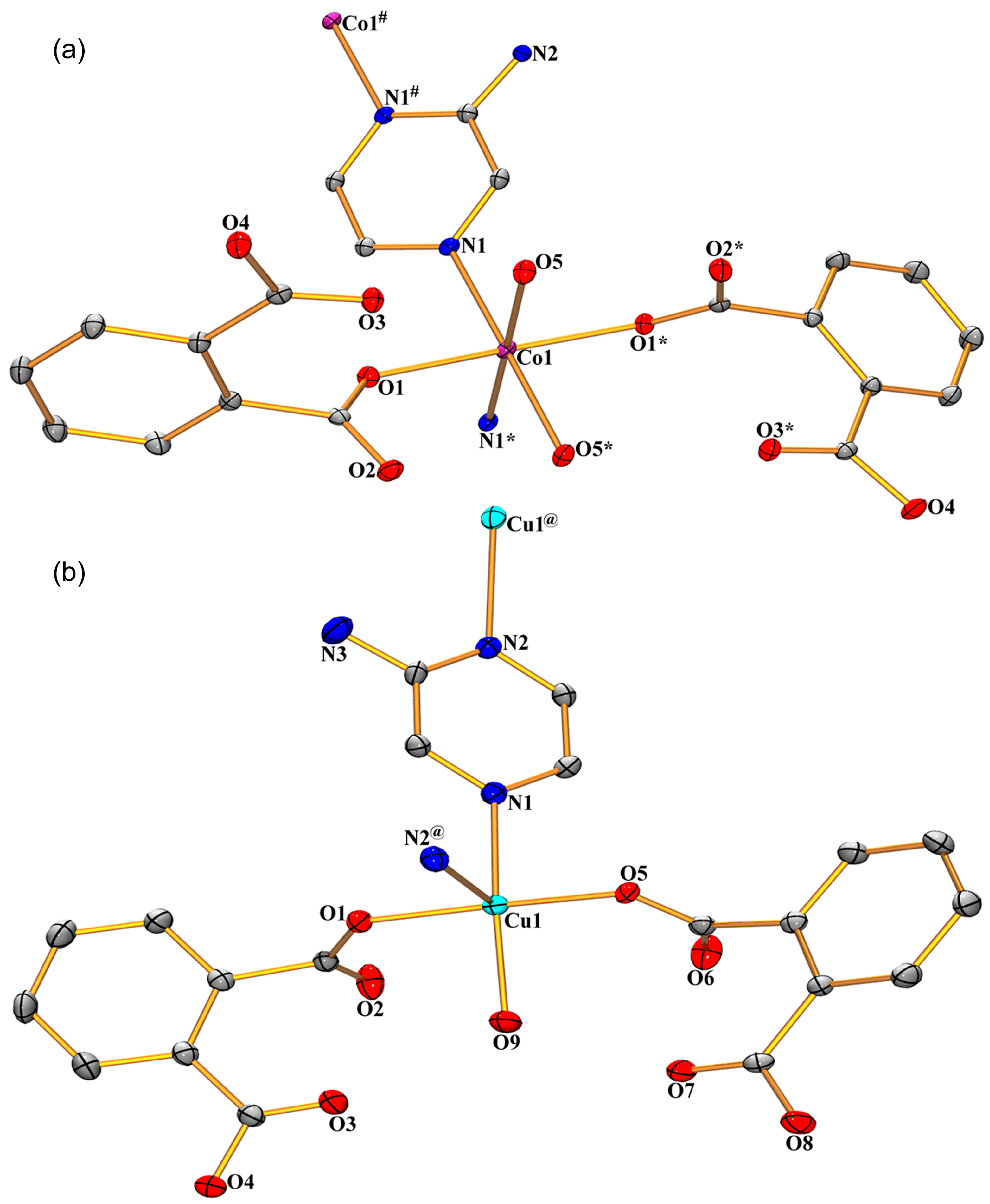
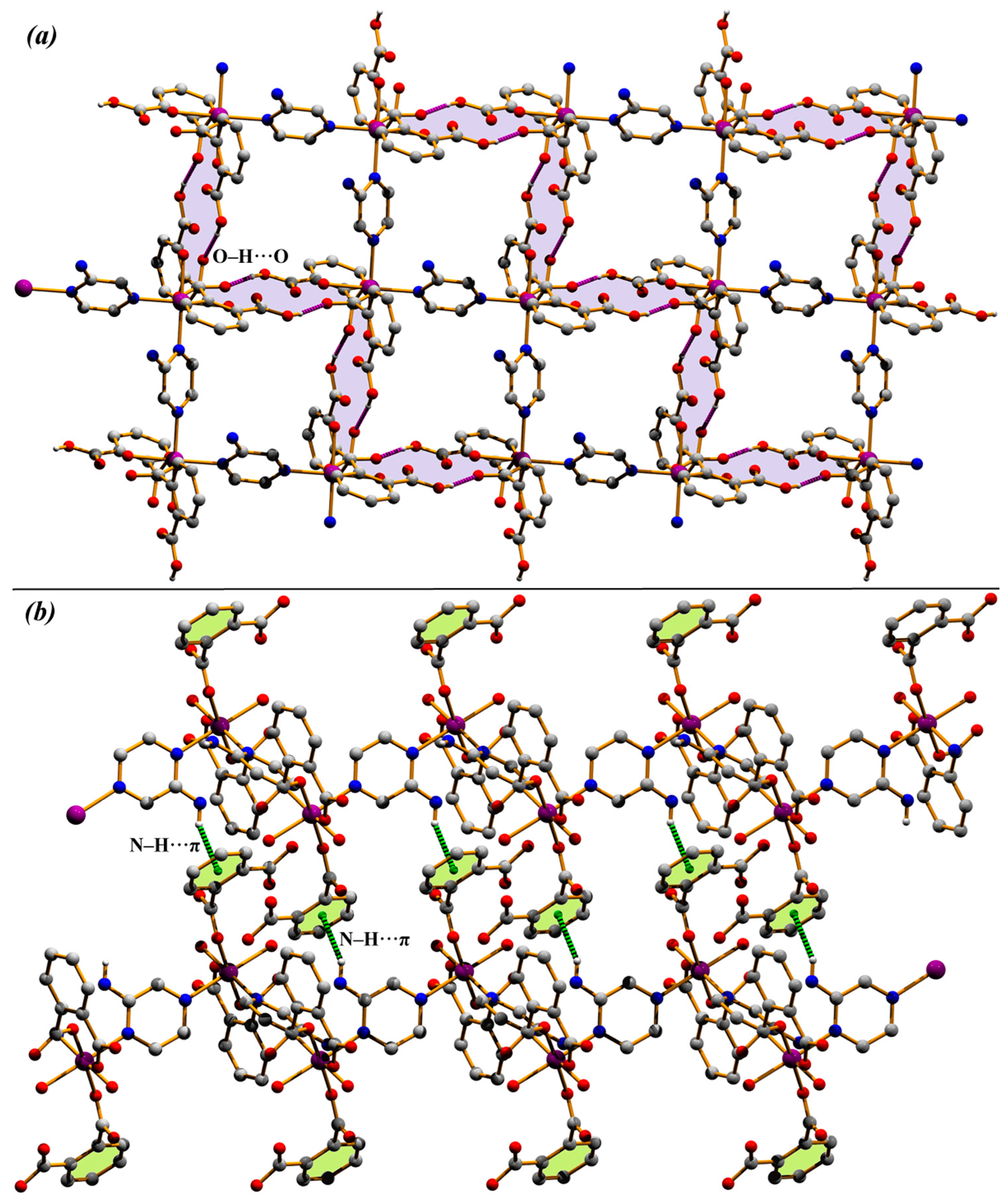
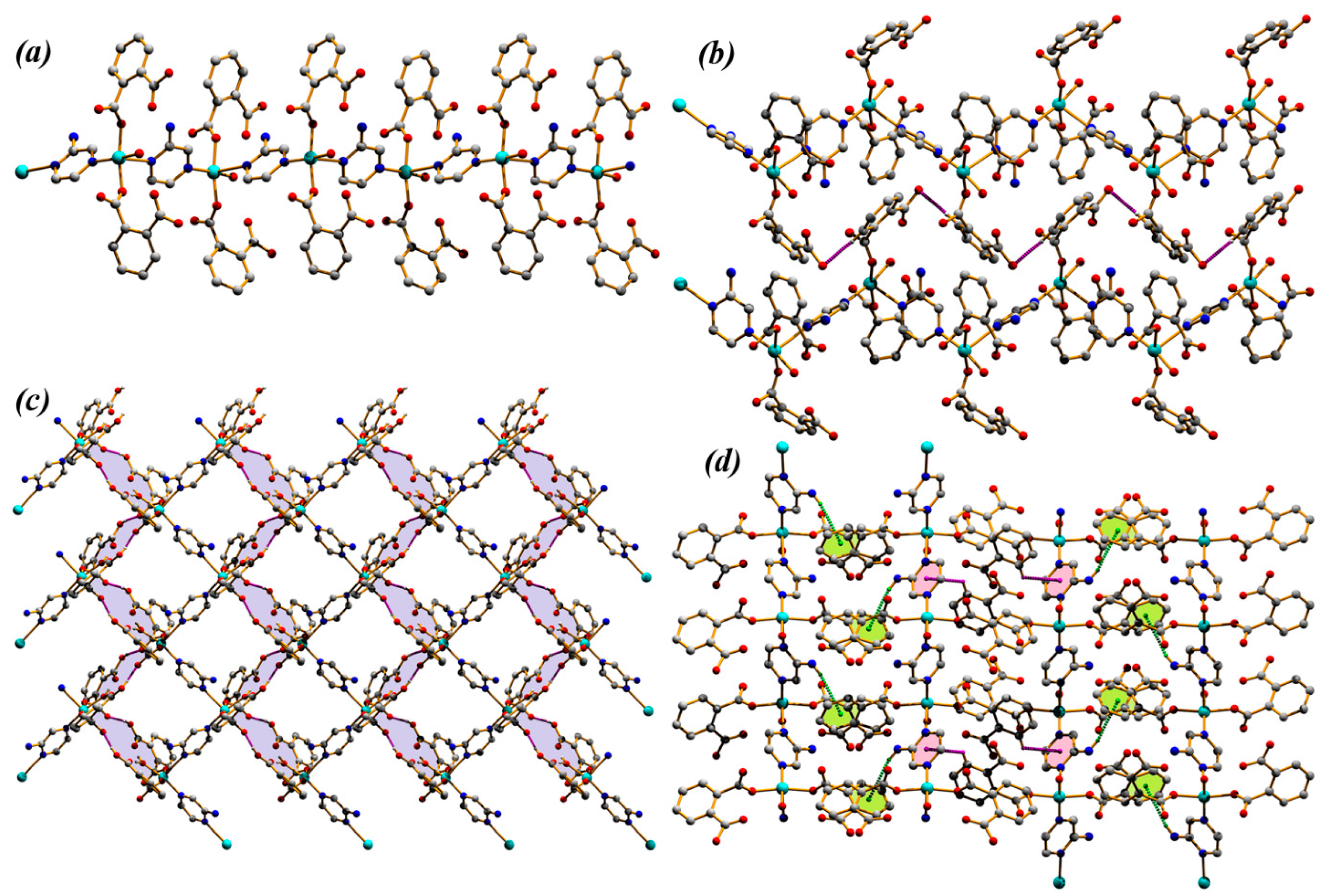
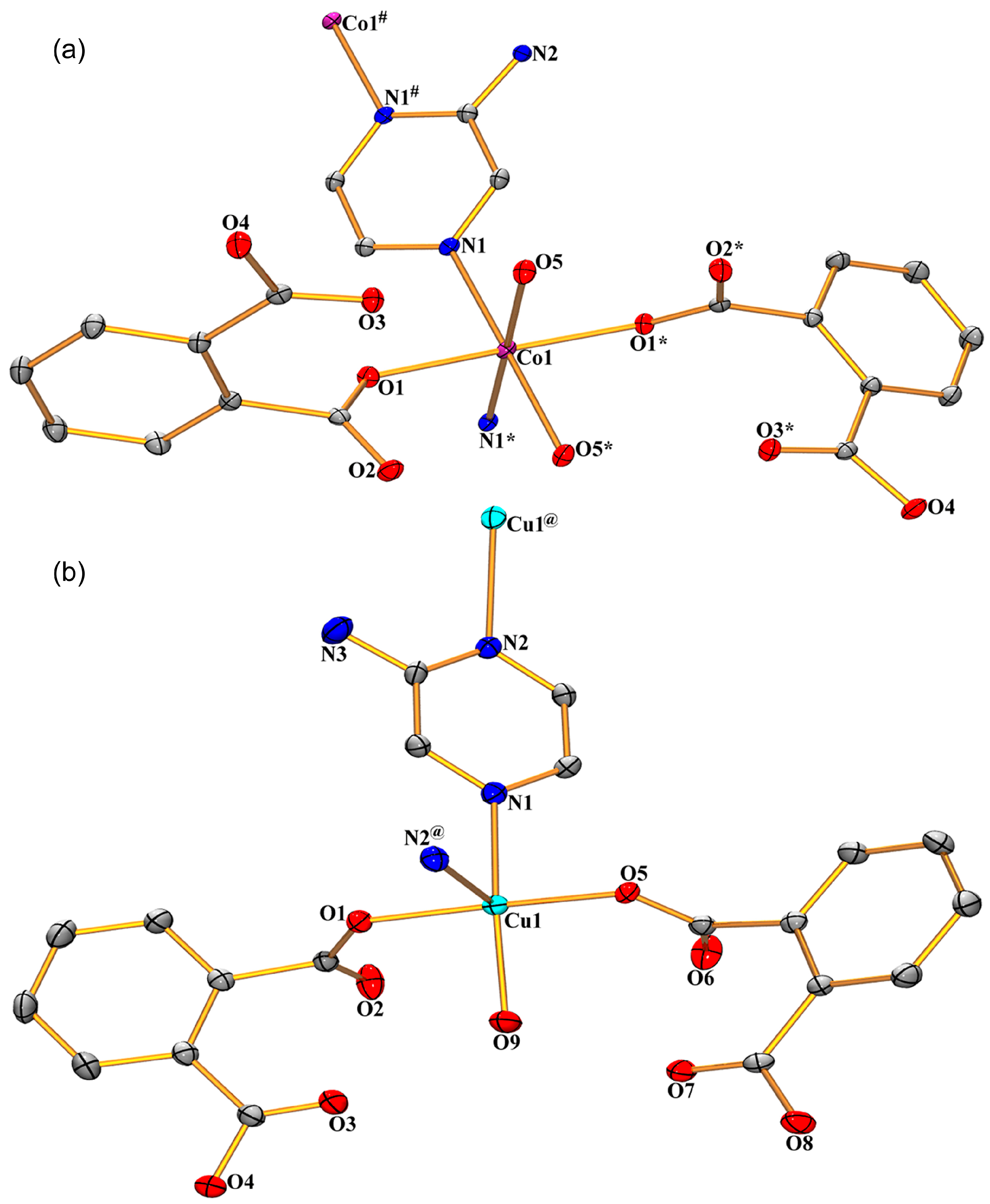
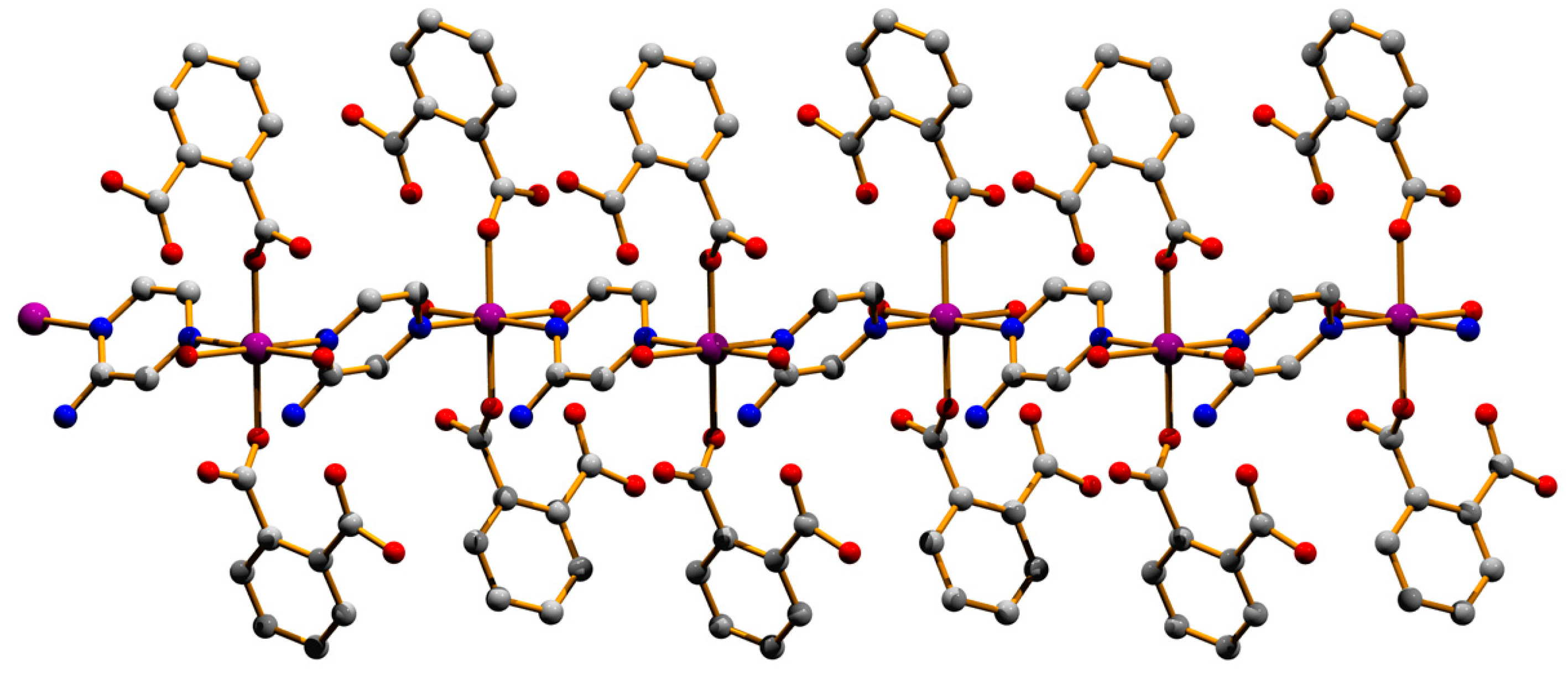
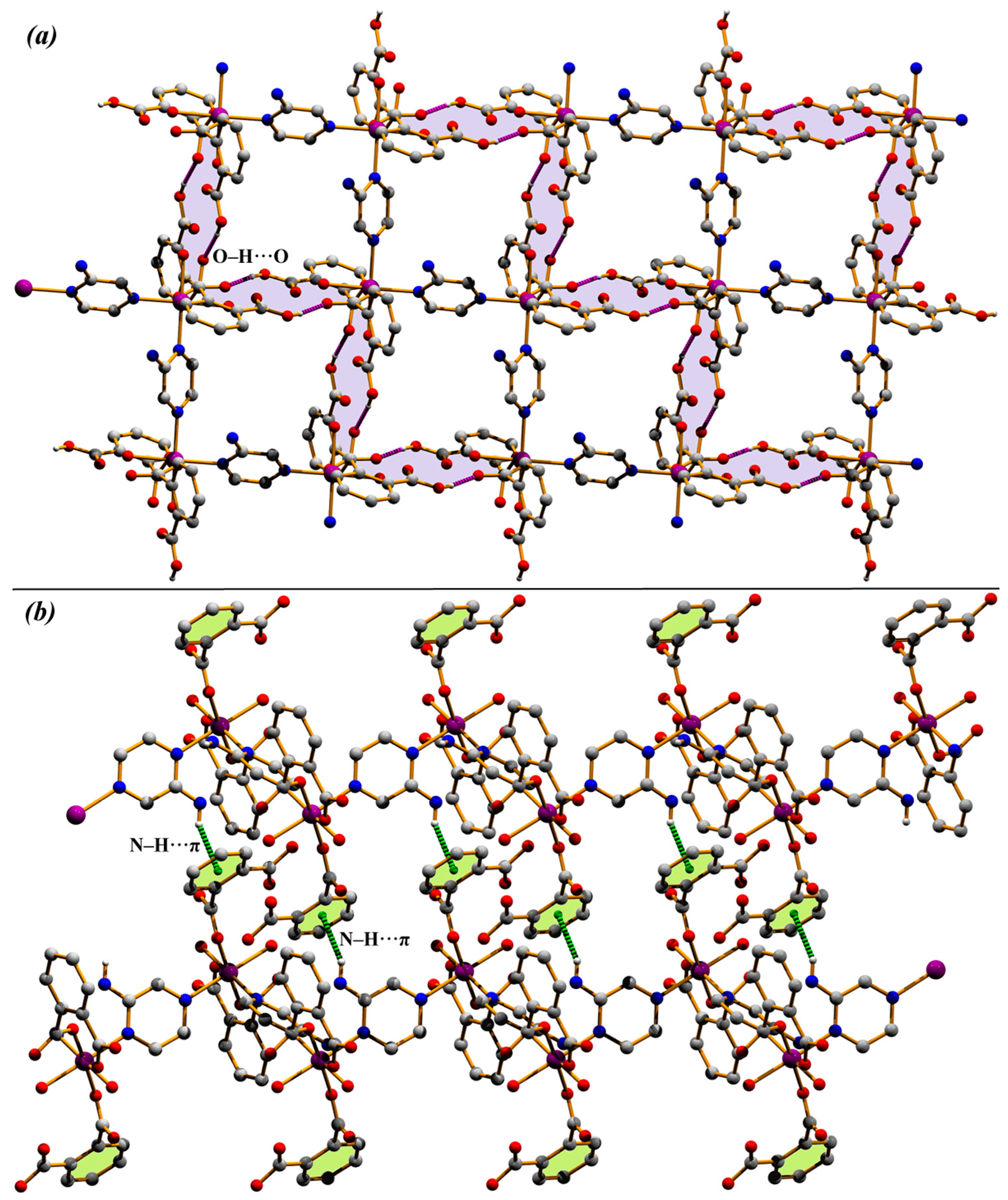
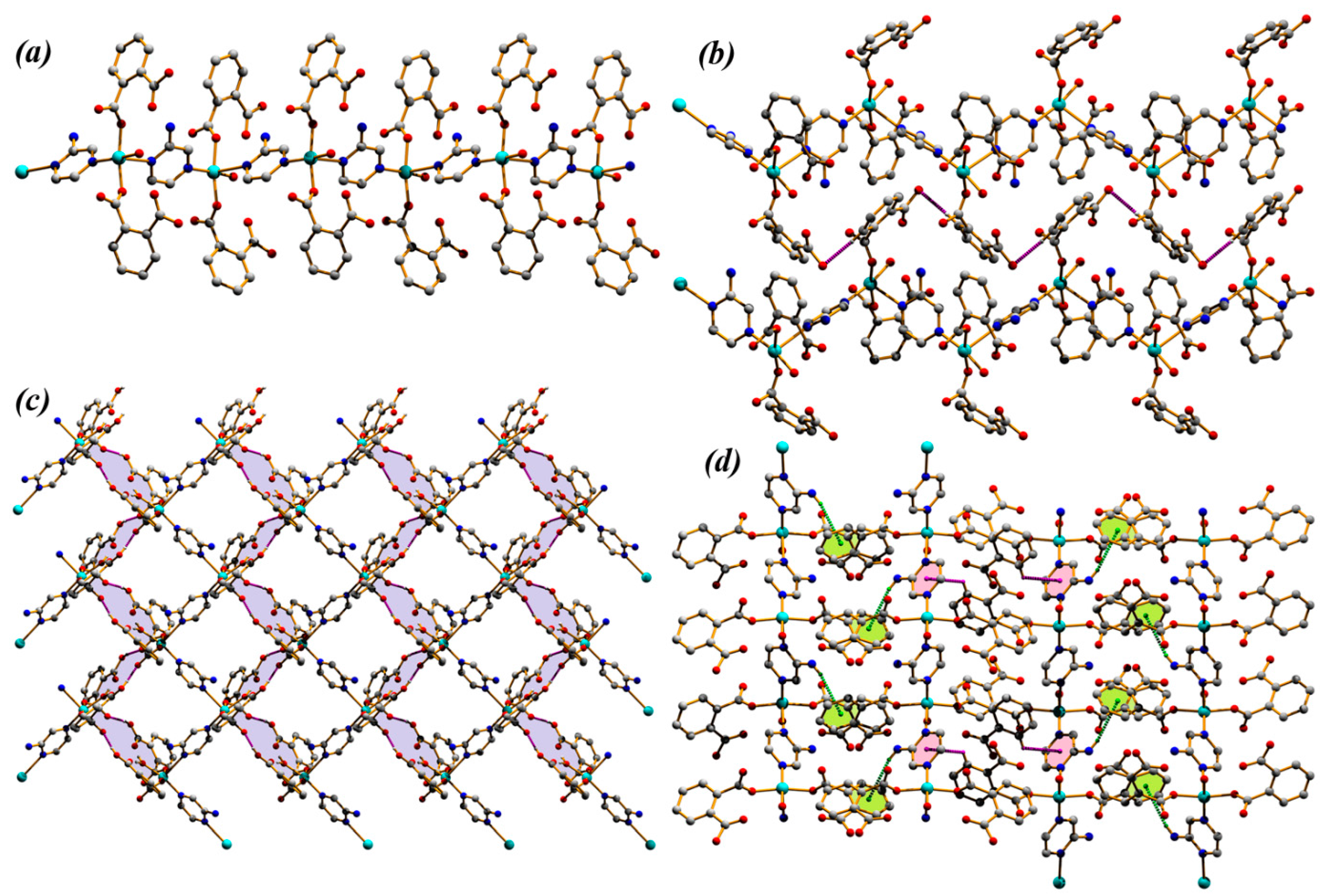
3.1. Structural Description of Compounds 1 and 2
3.2. Thermal Analysis
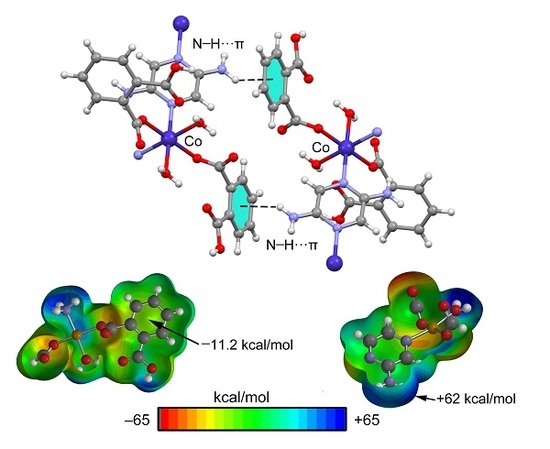
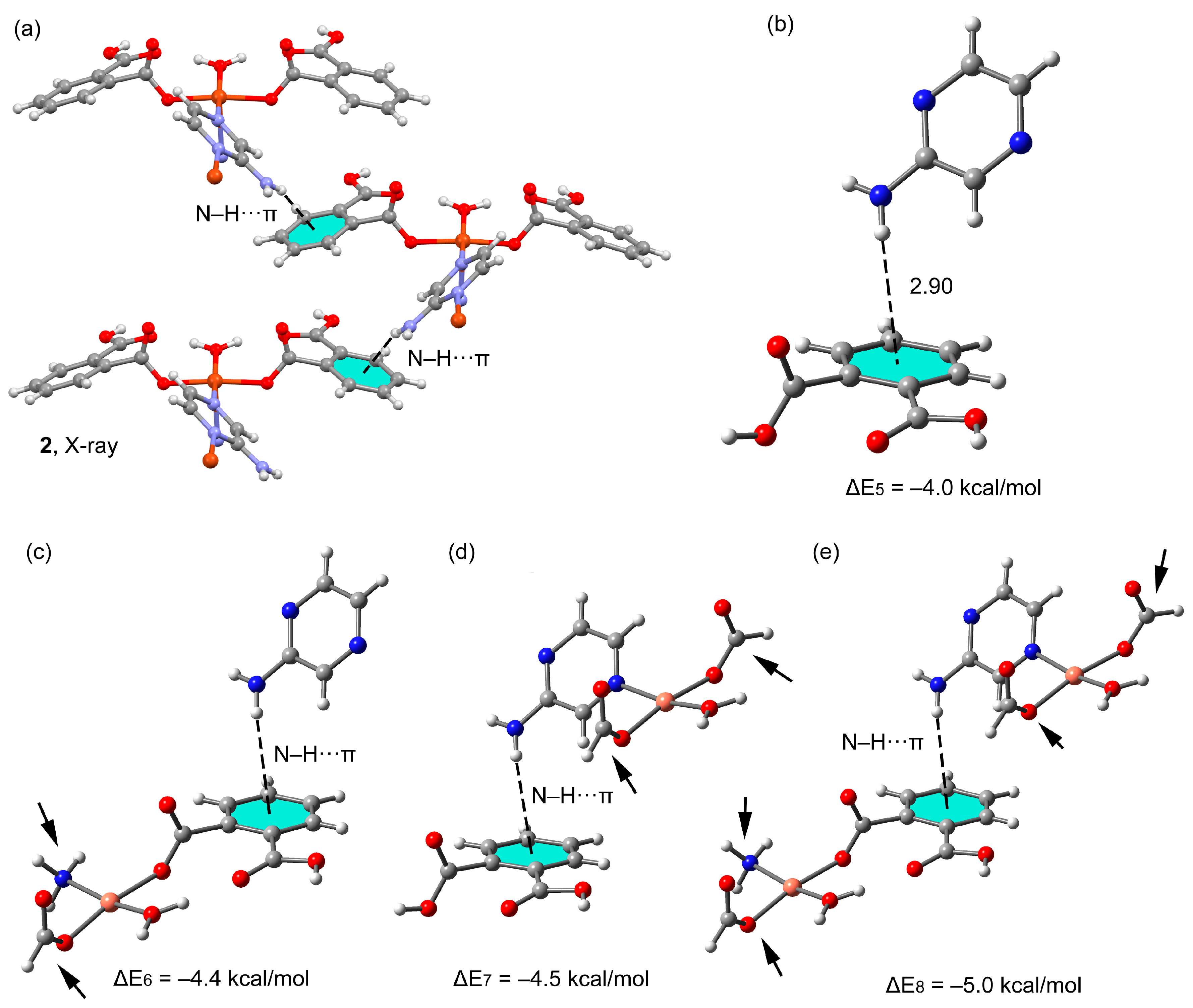
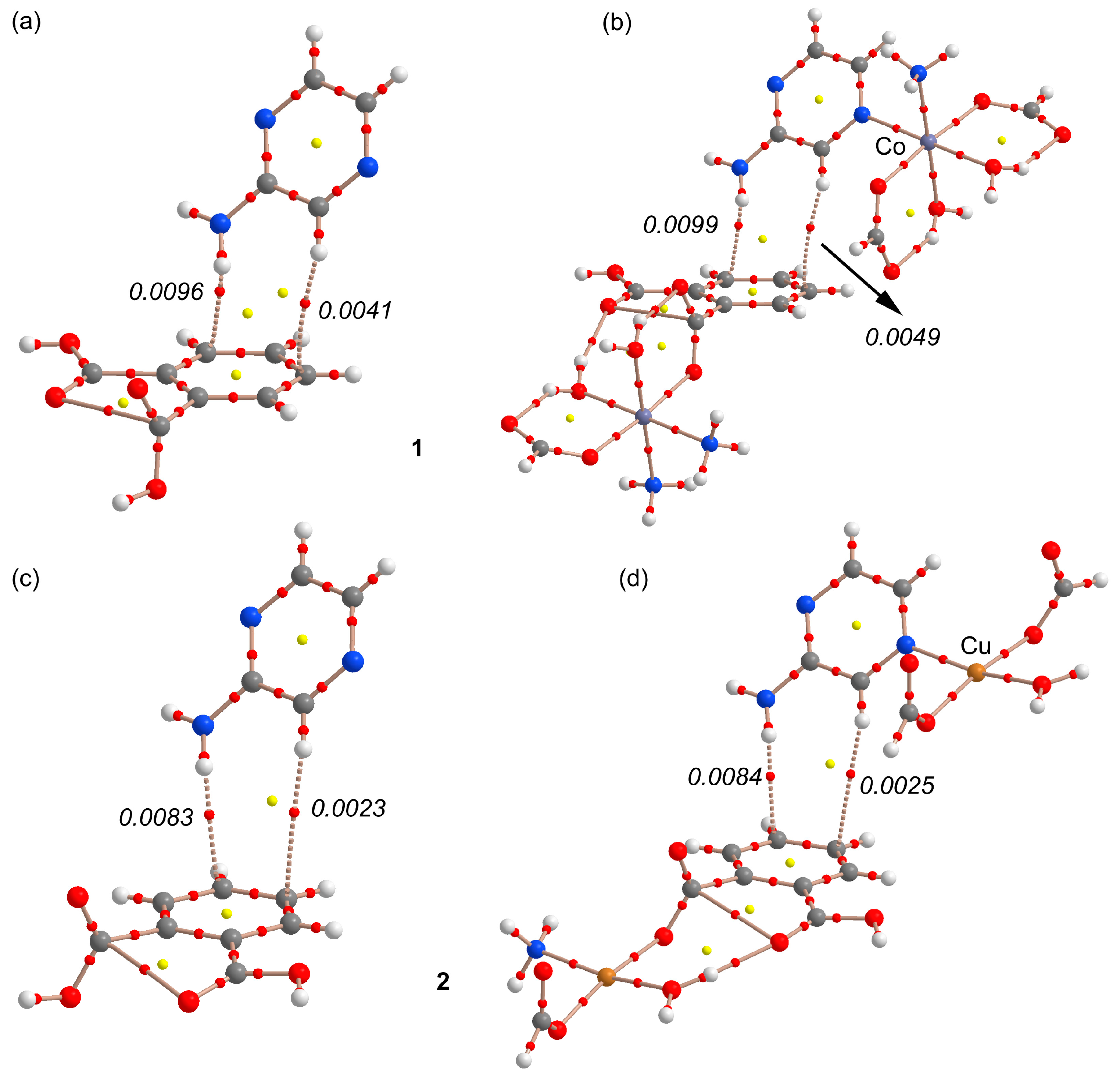
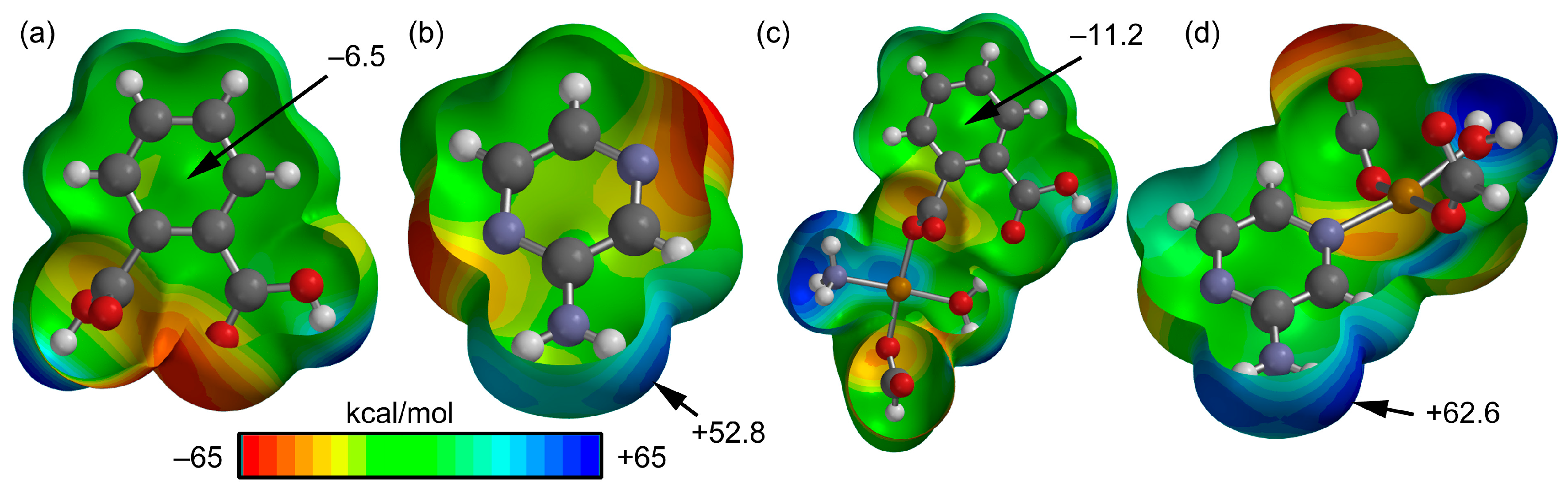
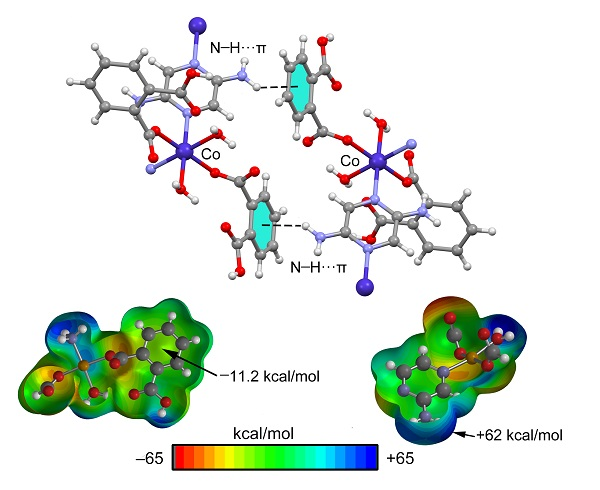
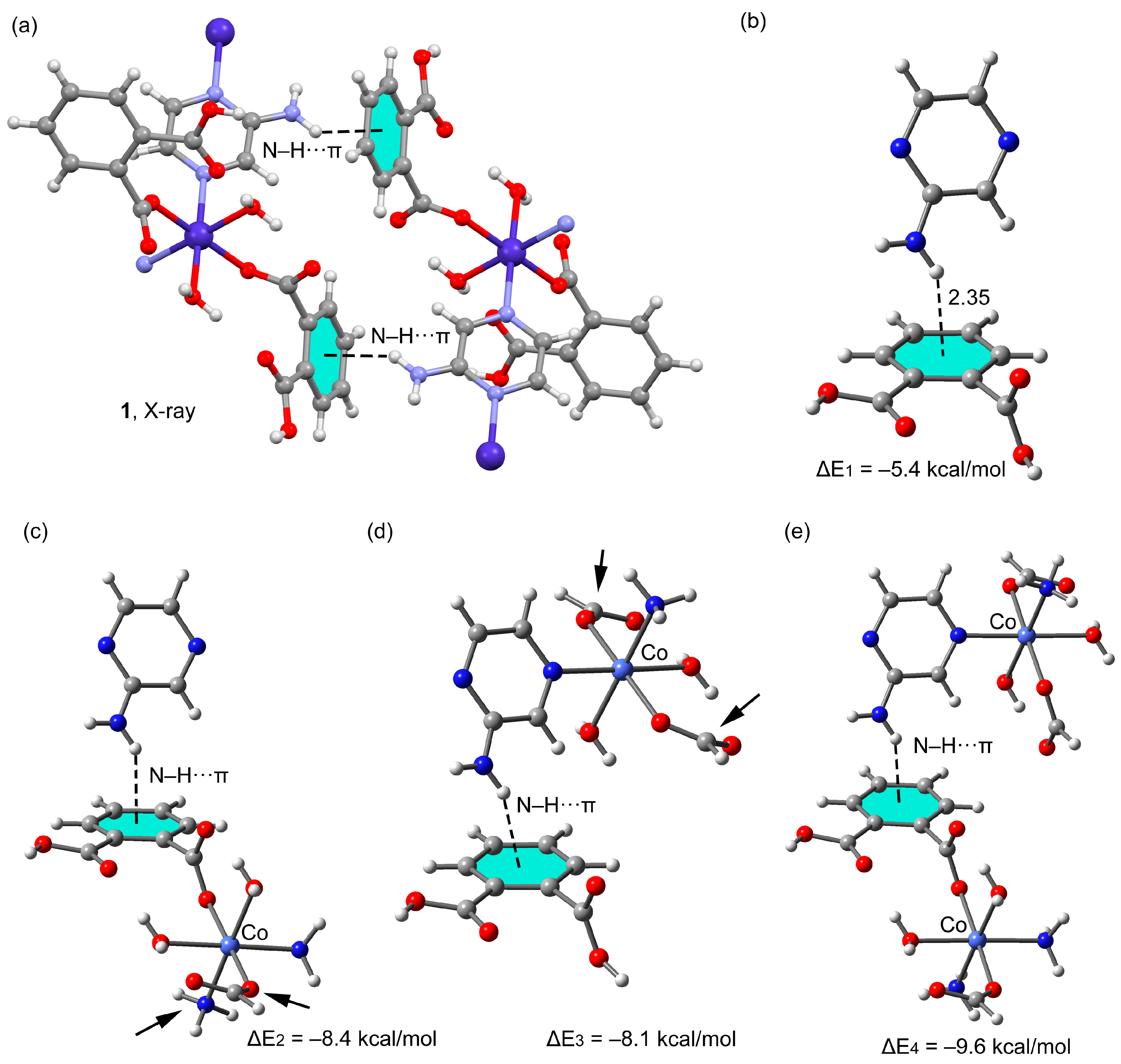
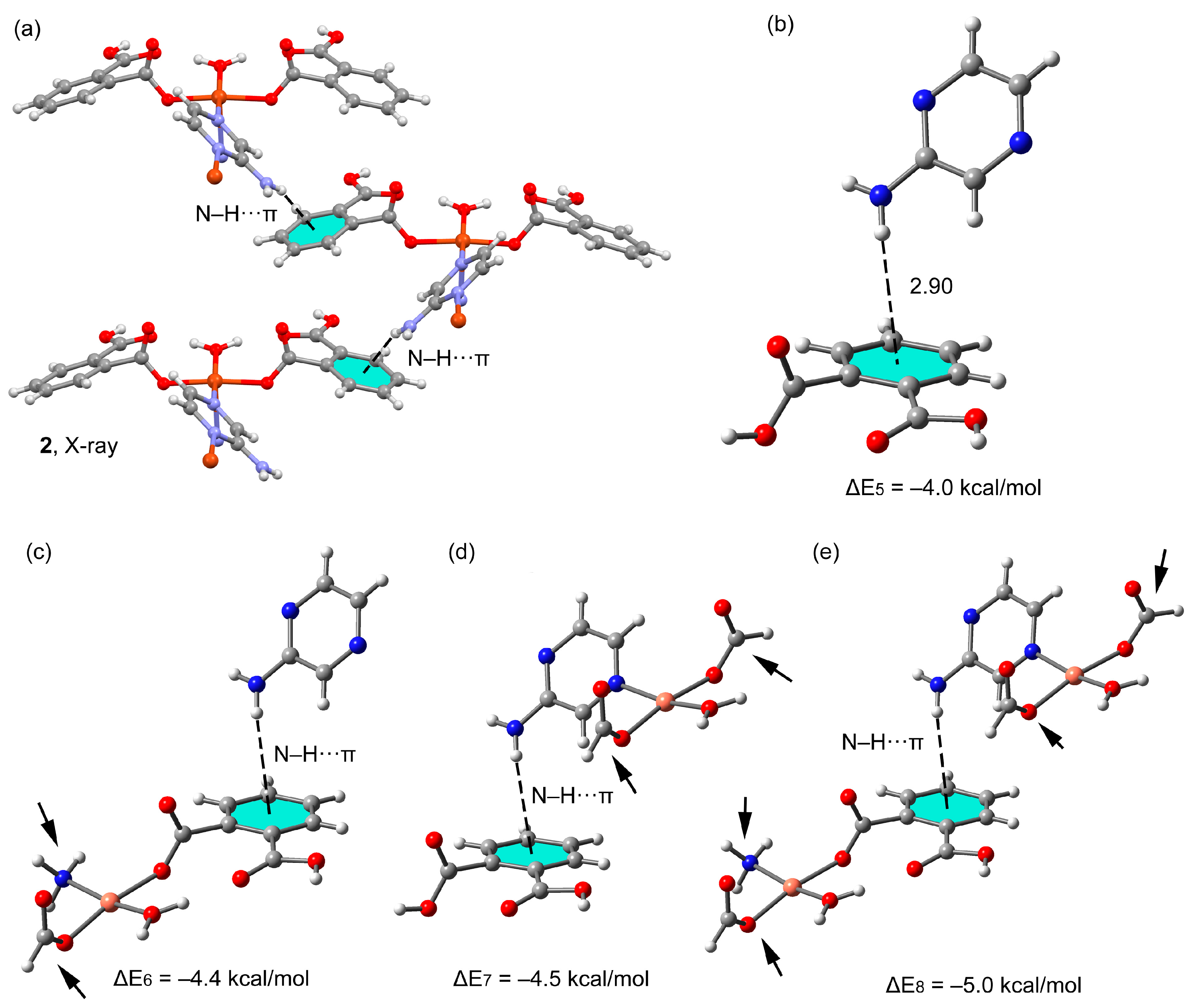
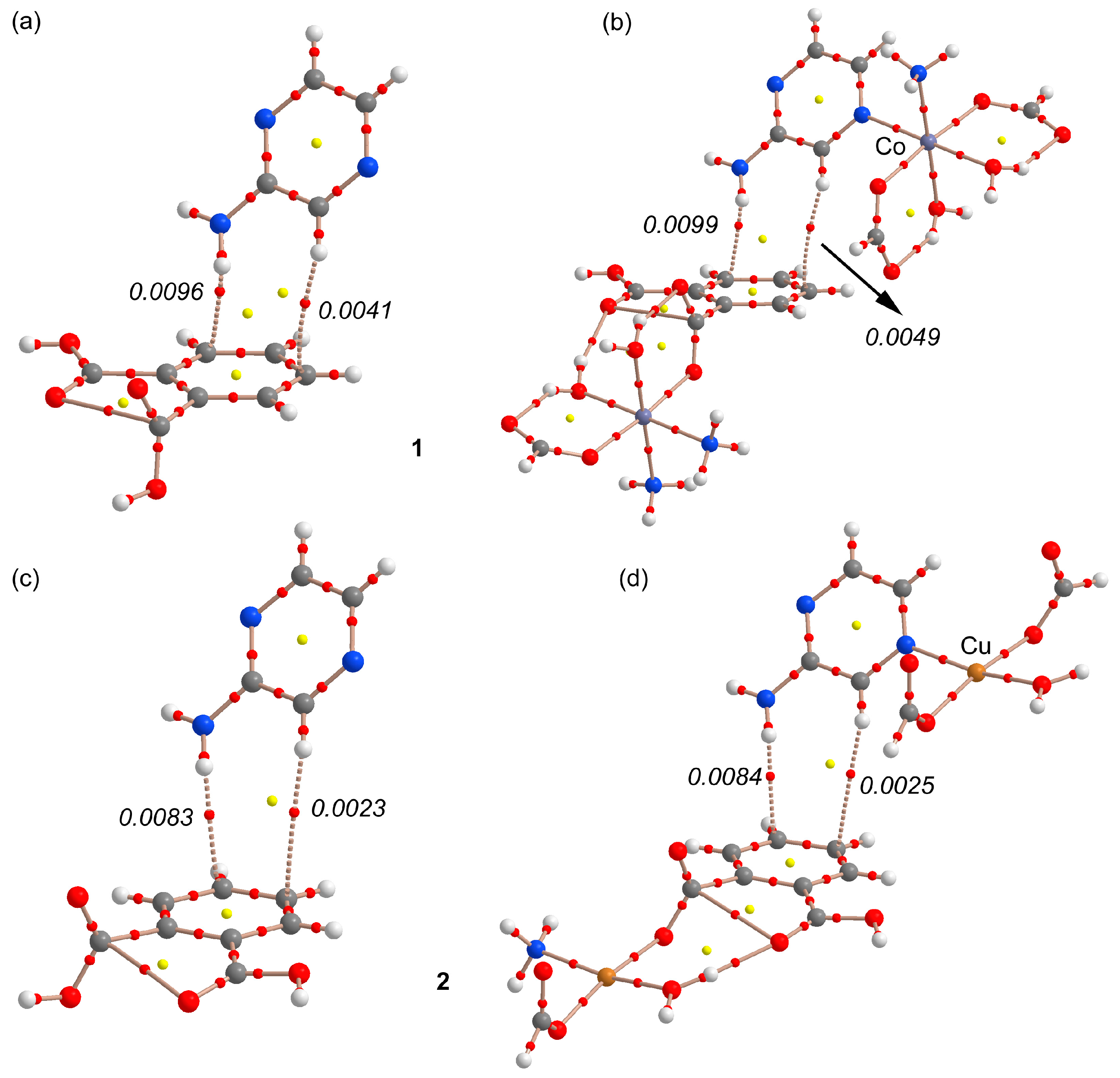
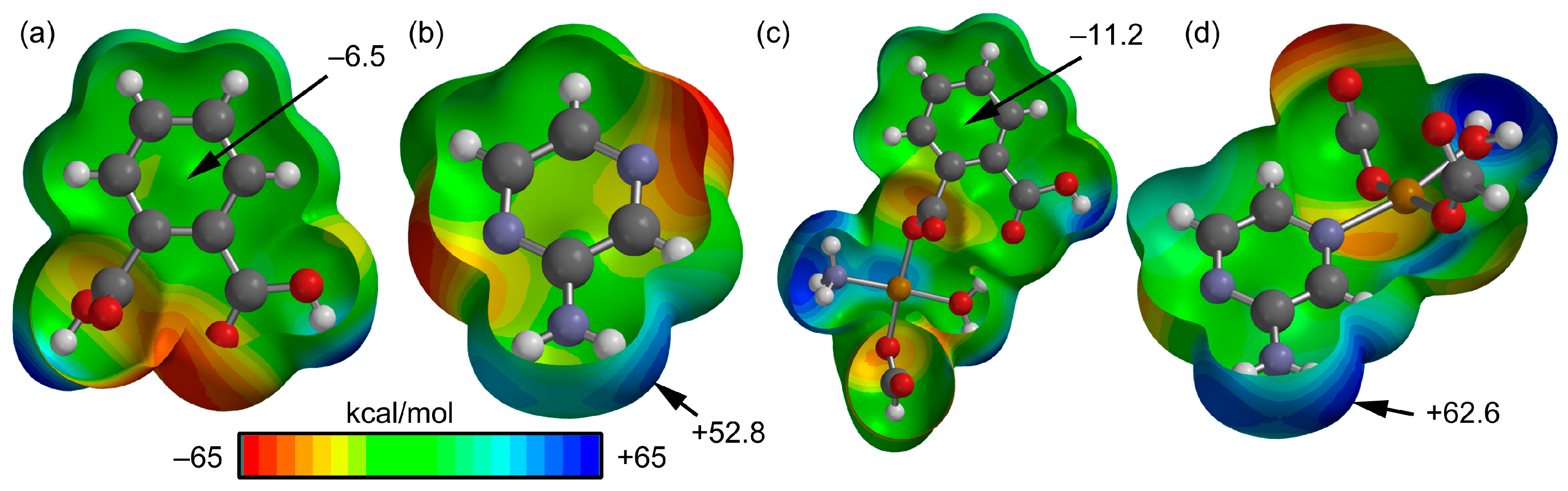
3.3. Theoretical Study
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tiekink, E.R.T.; Vittal, J.J. Frontiers in Crystal Engineering; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Yaghi, O.M.; Li, H.; Davis, C.; Richardson, D.; Groy, T.L. Synthetic strategies, structure patterns, and emerging properties in the chemistry of modular porous solids. Acc. Chem. Res. 1998, 31, 474–484. [Google Scholar] [CrossRef]
- Batten, S.R.; Neville, S.M.; Turner, D.R. Coordination Polymers: Design, Analysis and Application; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- Carlucci, L.; Ciani, G.; Proserpio, D.M.; Mitina, T.G.; Blatov, V.A. Entangled two-dimensional coordination networks: A general survey. Chem. Rev. 2014, 114, 7557–7580. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.-C.; Chen, L. Design and Construction of Coordination Polymers; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Amo-Ochoa, P.; Alexandre, S.S.; Hribesh, S.; Galindo, M.A.; Castillo, O.; Gómez-García, C.J.; Pike, A.J.; Soler, J.M.; Houlton, A.; Zamora, F. Coordination Chemistry of 6-Thioguanine Derivatives with Cobalt: Toward Formation of Electrical Conductive One-Dimensional Coordination Polymers. Inorg. Chem. 2013, 52, 5290–5299. [Google Scholar] [CrossRef] [PubMed]
- Amo-Ochoa, P.; Castillo, O.; Alexandre, S.S.; Welte, L.; de Pablo, P.J.; Rodríguez-Tapiador, M.I.; Gómez-Herrero, J.; Zamora, F. Synthesis of Designed Conductive One-Dimensional Coordination Polymers of Ni(II) with 6-Mercaptopurine and 6-Thioguanine. Inorg. Chem. 2009, 48, 7931–7936. [Google Scholar] [CrossRef] [PubMed]
- Amo-Ochoa, P.; Zamora, F. Coordination polymers with nucleobases: From structural aspects to potential applications. Coord. Chem. Rev. 2014, 276, 34–58. [Google Scholar] [CrossRef]
- Seth, P.; Bauzá, A.; Frontera, A.; Massera, C.; Gamez, P.; Ghosh, A. Analysis of the contribution of the π-acidity of the s-tetrazine ring in the crystal packing of coordination polymers. CrystEngComm 2013, 15, 3031–3039. [Google Scholar] [CrossRef]
- Bloom, J.W.G.; Wheeler, S.E. Taking the Aromaticity out of Aromatic Interactions. Angew. Chem. Int. Ed. 2011, 50, 7847–7849. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.K.; Manna, P.; Singh, N.J.; Mitra, M.; Jana, A.D.; Das, A.D.; Choudhury, S.R.; Kar, T.; Mukhopadhyay, S.; Kim, K.S. Molecular architecture using novel types of noncovalent π-interactions involving aromatic neutrals, aromatic cations and π-anions. CrystEngComm 2013, 15, 1285–1288. [Google Scholar] [CrossRef]
- Manna, P.; Seth, S.K.; Mitra, M.; Das, A.; Singh, N.J.; Choudhury, S.R.; Kar, T.; Mukhopadhyay, S. A successive layer-by-layer assembly of supramolecular frameworks driven by a novel type of face-to-face π+–π+ interactions. CrystEngComm 2013, 15, 7879–7886. [Google Scholar] [CrossRef]
- Ma, J.C.; Dougherty, D.A. The cation-π interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Gamez, P.; Mascal, M.; Mooibroek, T.J.; Reedijk, J. Putting Anion–π Interactions Into Perspective. Angew. Chem. Int. Ed. 2011, 50, 9564–9583. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Seth, S.K.; Mitra, M.; Choudhury, S.R.; Bauzá, A.; Frontera, A.; Mukhopadhyay, S. Experimental and Computational Study of Counterintuitive ClO4−···ClO4− Interactions and the Interplay between π+−π and Anion···π+ Interactions. Cryst. Growth Des. 2014, 14, 5812–5821. [Google Scholar] [CrossRef]
- Manna, P.; Seth, S.K.; Bauzá, A.; Mitra, M.; Choudhury, S.R.; Frontera, A.; Mukhopadhyay, S. pH Dependent Formation of Unprecedented Water−Bromide Cluster in the Bromide Salts of PTP Assisted by Anion−π Interactions: Synthesis, Structure, and DFT Study. Cryst. Growth Des. 2014, 14, 747–755. [Google Scholar] [CrossRef]
- Mitra, M.; Manna, P.; Bauzá, A.; Ballester, P.; Seth, S.K.; Choudhury, S.R.; Frontera, A.; Mukhopadhyay, S. 3-Picoline Mediated Self-Assembly of M(II)−Malonate Complexes (M = Ni/Co/Mn/Mg/Zn/Cu) Assisted by Various Weak Forces Involving Lone Pair−π, π−π, and Anion···π−Hole Interactions. J. Phys. Chem. B 2014, 118, 14713–14726. [Google Scholar] [CrossRef] [PubMed]
- Notash, B.; Safari, N.; Khavasi, H.R. Anion-controlled structural motif in one-dimensional coordination networks via cooperative weak noncovalent interactions. CrystEngComm 2012, 14, 6788–6796. [Google Scholar] [CrossRef]
- Seth, S.K.; Saha, I.; Estarellas, C.; Frontera, A.; Kar, T.; Mukhopadhyay, S. Supramolecular Self-Assembly of M-IDA Complexes Involving Lone-Pair···π Interactions: Crystal Structures, Hirshfeld Surface Analysis, and DFT Calculations [H2IDA = iminodiacetic acid, M = Cu(II), Ni(II)]. Cryst. Growth Des. 2011, 11, 3250–3265. [Google Scholar] [CrossRef]
- Manna, P.; Seth, S.K.; Das, A.; Hemming, J.; Prendergast, R.; Helliwell, M.; Choudhury, S.R.; Frontera, A.; Mukhopadhyay, S. Anion Induced Formation of Supramolecular Associations Involving Lone pair−π and Anion−π Interactions in Co(II) Malonate Complexes: Experimental Observations, Hirshfeld Surface Analyses and DFT Studies. Inorg. Chem. 2012, 51, 3557–3571. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Blanco, F.; Deya, P.M.; Elguero, J.; Estarellas, C.; Frontera, A.; Quinonero, D. Cooperativity in multiple unusual weak bonds. Theor. Chem. Acc. 2010, 126, 1–14. [Google Scholar] [CrossRef]
- Steed, J.W.; Turner, D.R.; Wallace, K.J. Core Concepts in Supramolecular Chemistry and Nanochemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2007; pp. 194–228. [Google Scholar]
- Bruker SAINT, version 6.36a; Bruker-AXS Inc.: Madison, WI, USA, 2002.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suit for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Hacer, M.; Horn, H.; Kömel, C. Electronic structure calculations on workstation computers: The program system Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Boys, S.B.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, version 13.05.06; TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Benbellat, N.; Gavrilenko, K.S.; Gal, Y.L.; Cador, O.; Golhen, S.; Gouasmia, A.; Fabre, J.; Ouahab, L. Co(II)−Co(II) Paddlewheel Complex with a Redox-Active Ligand Derived from TTF. Inorg. Chem. 2006, 45, 10440–10442. [Google Scholar] [CrossRef] [PubMed]
- Devereux, M.; Shea, D.O.; Kellett, A.; McCann, M.; Walsh, M.; Egan, D.; Deegan, C.; Kedziora, K.; Rosair, G.; Bunz, H.M. Synthesis, X-ray crystal structures and biomimetic and anticancer activities of novel copper(II)benzoate complexes incorporating 2-(4′-thiazolyl)benzimidazole(thiabendazole), 2-(2-pyridyl)benzimidazole and 1,10-phenanthroline as chelating nitrogen donor ligands. J. Inorg. Biochem. 2007, 101, 881–892. [Google Scholar] [PubMed]
- Khan, S.; Masum, A.A.; Islam, M.M.; Drew, M.G.B.; Bauzá, A.; Frontera, A.; Chattopadhyay, S. Observation of π-hole interactions in the solid state structures of three new copper(II) complexes with a tetradentate N4 donor Schiff base: Exploration of their cytotoxicity against MDA-MB 468 cells. Polyhedron 2017, 123, 334–343. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Seth, S.K.; Bauzá, A.; Frontera, A. Screening polymorphism in a Ni(II) metal–organic framework: Experimental observations, Hirshfeld surface analyses and DFT studies. CrystEngComm 2018, 20, 746–754. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | 1 | 2 |
|---|---|---|
| Empirical formula | C20H18CoN3O10 | C20H19CuN3O10 |
| Formula Weight | 519.30 | 524.92 |
| Temperature (K) | 100(2) | 293(2) |
| Wavelength (Å) | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Orthorhombic |
| space group | C2/c | Pbca |
| a, b, c (Å) | 17.3919(8), 11.7007(5), 10.3379(5) | 11.1679(4), 10.7968(4), 35.0988(11) |
| α, β, γ (°) | 90, 93.5973(14), 90 | 90, 90, 90 |
| Volume (Å3) | 2099.59(17) | 4232.1(3) |
| Z/Density (calcd.) (Mg/m3) | 4/1.643 | 8/1.648 |
| Absorption coefficient (mm−1) | 0.883 | 1.097 |
| F(000) | 1064 | 2152 |
| Crystal size (mm3) | 0.15 × 0.11 × 0.06 | 0.16 × 0.10 × 0.07 |
| Limiting indices | −16 ≤ h ≤ 25 | −13 ≤ h ≤ 13 |
| −16 ≤ k ≤ 16 | −12 ≤ k ≤ 12 | |
| −14 ≤ l ≤ 15 | −41 ≤ l ≤ 41 | |
| Reflections collected/unique | 12747/3496 [R(int) = 0.0293] | 56686/3700 [R(int) = 0.1005] |
| Completeness to θ (%) | 99.7 | 99.4 |
| Absorption correction | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. and min. transmission | 0.917 and 0.864 | 0.93 and 0.87 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/parameters | 3496/176 | 3700/324 |
| Goodness-of-fit on F2 | 1.060 | 1.110 |
| Final R indices [I > 2σ(I)] | R1 = 0.0351, wR2 = 0.0756 | R1 = 0.0505, wR2 = 0.1247 |
| R indices (all data) | R1 = 0.0463, wR2 = 0.0800 | R1 = 0.0614, wR2 = 0.1292 |
| Largest diff. peak and hole (e·Å−3) | 0.478 and −0.312 | 0.820 and −0.627 |
| D–H∙∙∙A | d(D–H) | d(H∙∙∙A) | d(D∙∙∙A) | D–H∙∙∙A | Symmetry |
|---|---|---|---|---|---|
| Compound 1 | |||||
| O(5)–H(5O1)∙∙∙O(3) | 0.84 | 1.97 | 2.779(2) | 159 | 1 − x, y, 1/2 − z |
| O(5)–H(5O2)∙∙∙O(2) | 0.84 | 1.94 | 2.676(2) | 144 | --- |
| O(4)–H(4)∙∙∙O(2) | 0.84 | 1.74 | 2.579(2) | 174 | x, 1 − y, −1/2 + z |
| N(2)–H(2B)∙∙∙Cg(2) | 2.34 | 3.087(3) | 142 | −1/2 + x, 1/2 − y, −1/2 + z | |
| N(2’)–H(2’B)∙∙∙Cg(2) | 2.40 | 3.169(2) | 146 | −1/2 + x, 1/2 − y, −1/2 + z | |
| Compound 2 | |||||
| N(3)–H(3A)∙∙∙O(1) | 0.86 | 2.14 | 2.863(7) | 142 | 3/2 − x, −1/2 + y, z |
| O(4)–H(4)∙∙∙O(2) | 0.82 | 1.80 | 2.616(4) | 170 | 1/2 − x, 1/2 + y, z |
| O(8)–H(8)∙∙∙O(6) | 0.82 | 1.81 | 2.625(4) | 177 | 1/2 − x, 1/2 + y, z |
| O(9)–H(9A)∙∙∙O(3) | 0.94 | 1.84 | 2.751(4) | 164 | --- |
| O(9)–H(9B)∙∙∙O(7) | 0.95 | 1.77 | 2.716(4) | 170 | --- |
| O(10)–H(10A)∙∙∙O(7) | 0.88 | 2.59 | 3.310(6) | 139 | 1/2 + x, 3/2 − y, 1 − z |
| O(10)–H(10B)∙∙∙O(6) | 0.88 | 2.15 | 2.846(5) | 136 | 1 − x, 1 − y, 1 − z |
| O(10)–H(10B)∙∙∙O(2) | 0.89 | 1.92 | 2.797(5) | 166 | 1 − x, 1 − y, 1 − z |
| C(3)–H(3)∙∙∙O(4) | 0.93 | 2.53 | 3.388(5) | 154 | 1 − x, −1/2 + y, 3/2 − z |
| N(3)–H(3B)∙∙∙Cg(2) | 2.89 | 3.720(7) | 164 | 1/2 − x, y, 3/2 − z | |
| C(14)–H(14)∙∙∙Cg(1) | 2.97 | 3.558(4) | 122 | 1/2 + x, 3/2 − y, 1 − z | |
| N(3’)–H(3’B)∙∙∙Cg(3) | 2.28 | 2.979(2) | 139 | 1 − x, 1 − y, 1 − z | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, A.; Seth, S.K.; Bauzá, A.; Mukhopadhyay, S.; Frontera, A. Coordination Polymers Based on Phthalic Acid and Aminopyrazine Ligands: On the Importance of N–H···π Interactions. Polymers 2018, 10, 182. https://doi.org/10.3390/polym10020182
Hossain A, Seth SK, Bauzá A, Mukhopadhyay S, Frontera A. Coordination Polymers Based on Phthalic Acid and Aminopyrazine Ligands: On the Importance of N–H···π Interactions. Polymers. 2018; 10(2):182. https://doi.org/10.3390/polym10020182
Chicago/Turabian StyleHossain, Anowar, Saikat Kumar Seth, Antonio Bauzá, Subrata Mukhopadhyay, and Antonio Frontera. 2018. "Coordination Polymers Based on Phthalic Acid and Aminopyrazine Ligands: On the Importance of N–H···π Interactions" Polymers 10, no. 2: 182. https://doi.org/10.3390/polym10020182