Advantageous Microwave-Assisted Suzuki Polycondensation for the Synthesis of Aniline-Fluorene Alternate Copolymers as Molecular Model with Solvent Sensing Properties


 , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
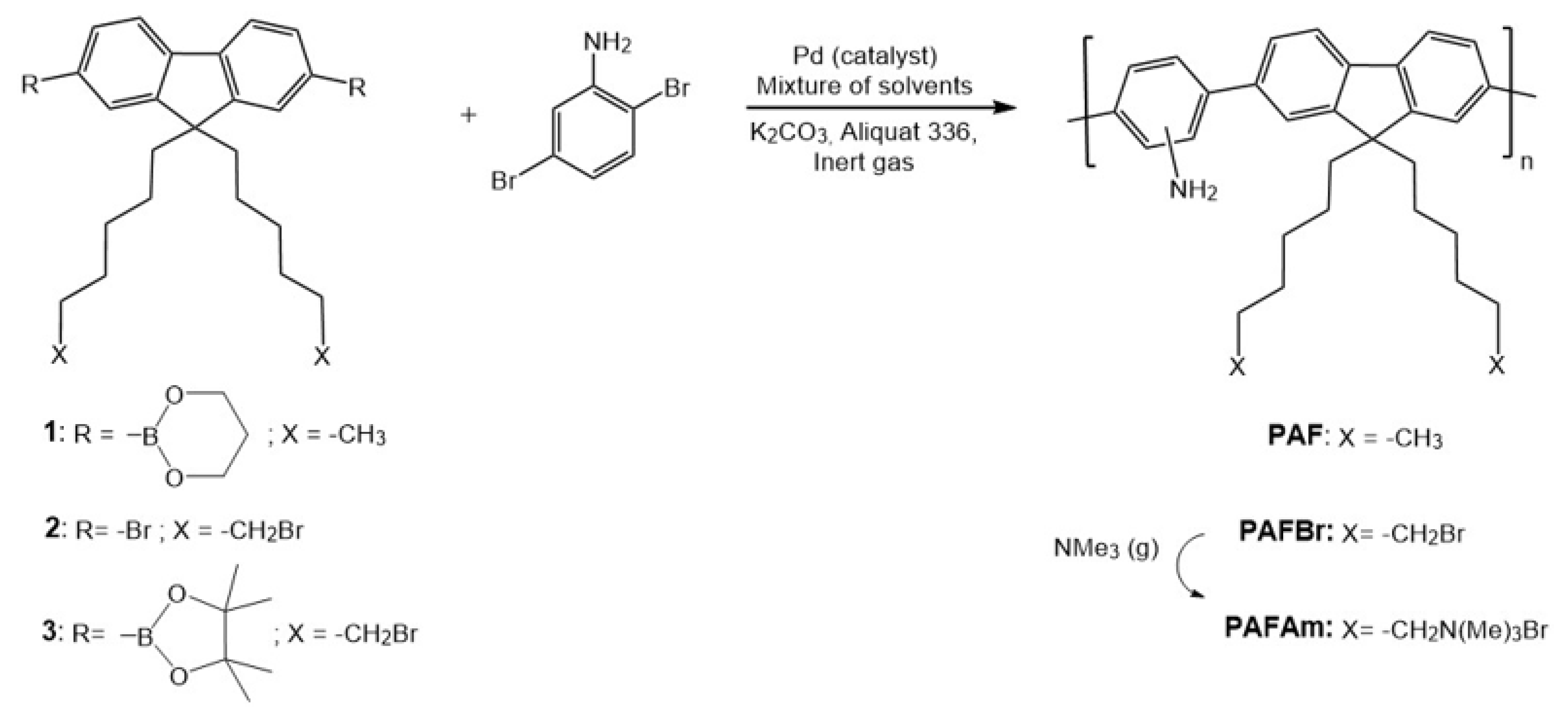
2.3. Synthesis and Characterization
3. Results and Discussion
3.1. Optimization of the Synthesis of PAF and Characterization of the Different Batches
3.1.1. General Considerations
3.1.2. Characterization of PAF Batches
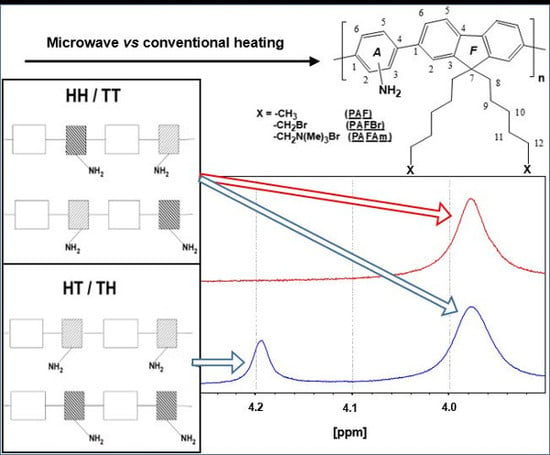
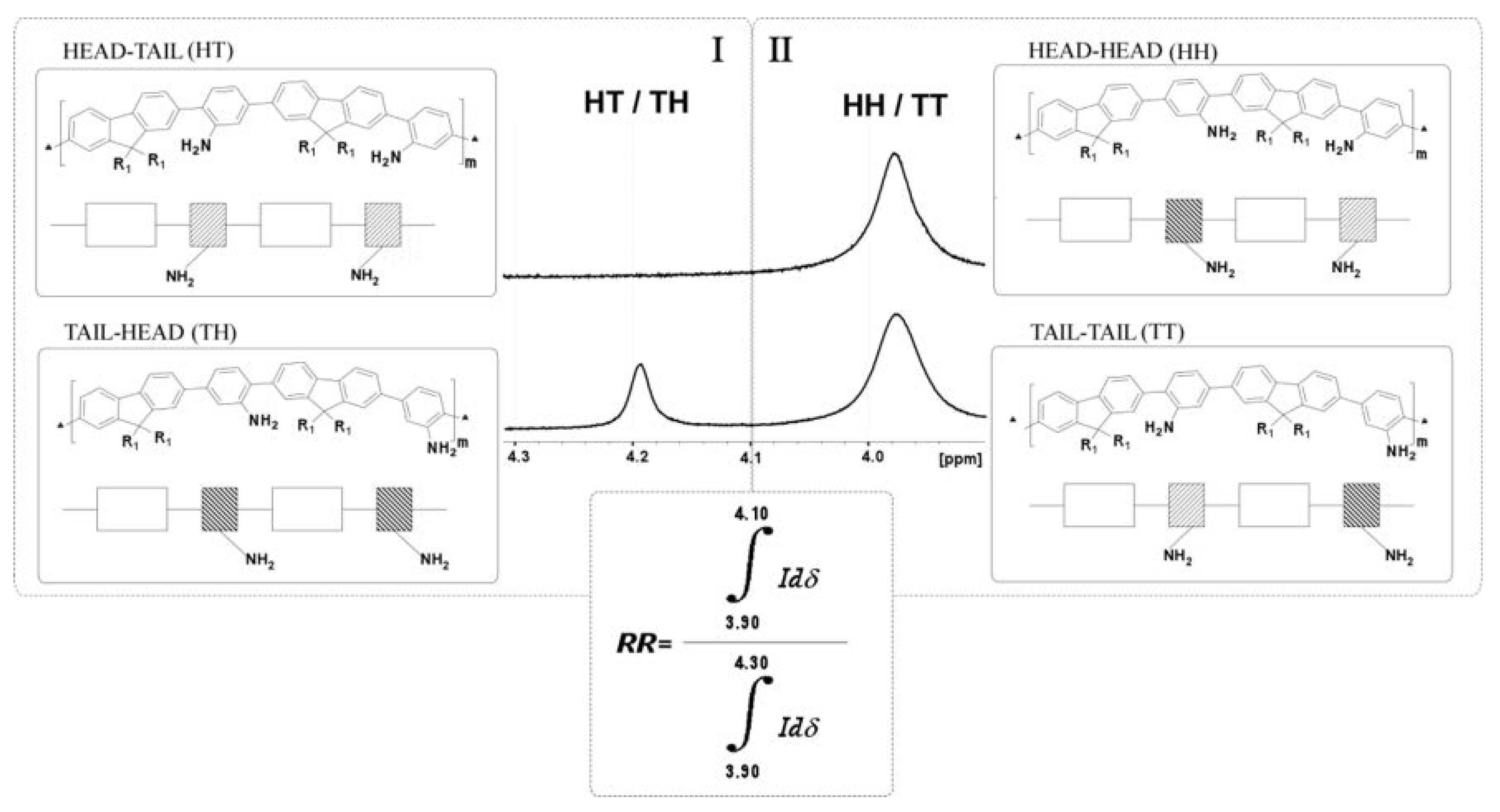
3.1.3. Study of the Regioregularity of Synthesized PAF by 1H-NMR
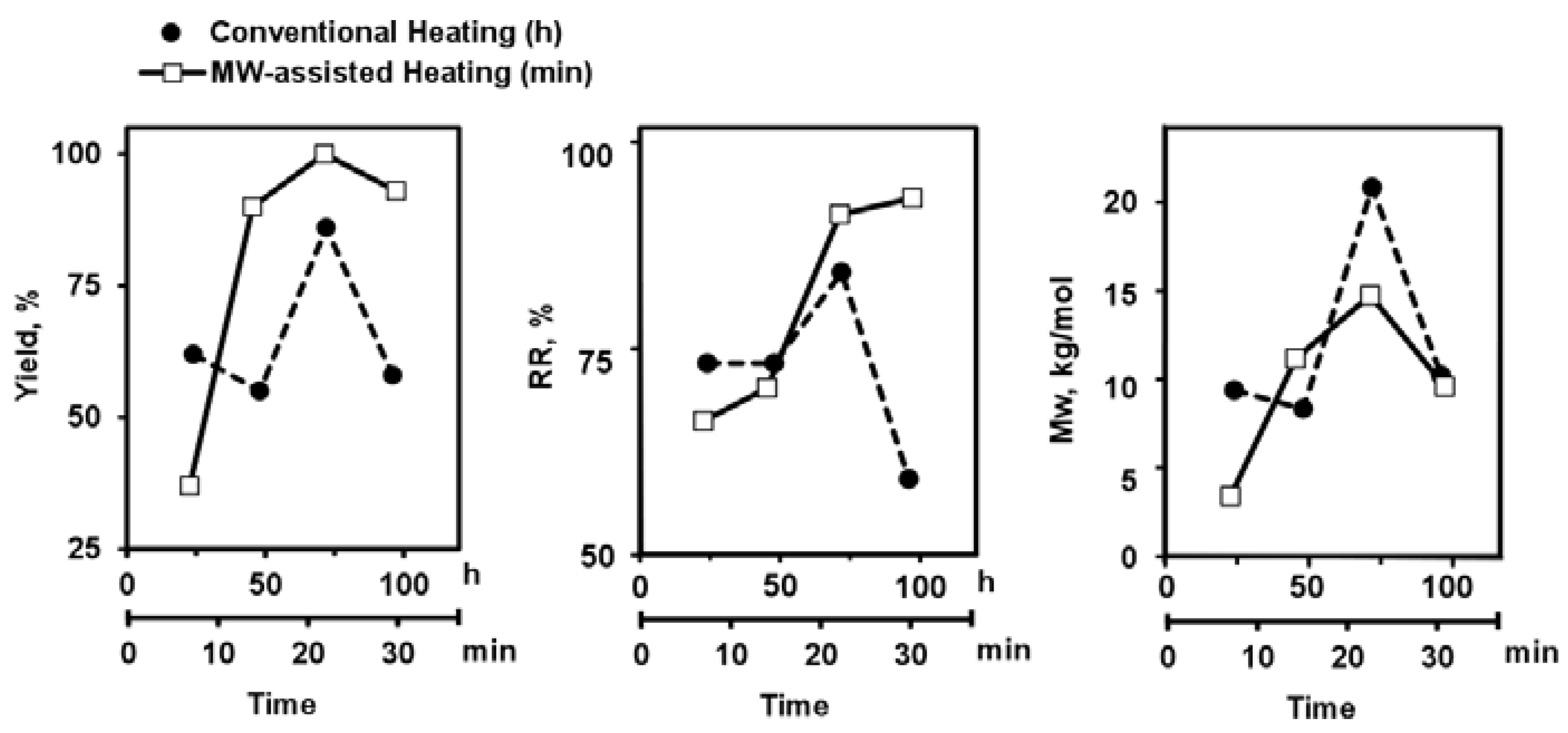
3.1.4. Synthesis Optimization of PAF
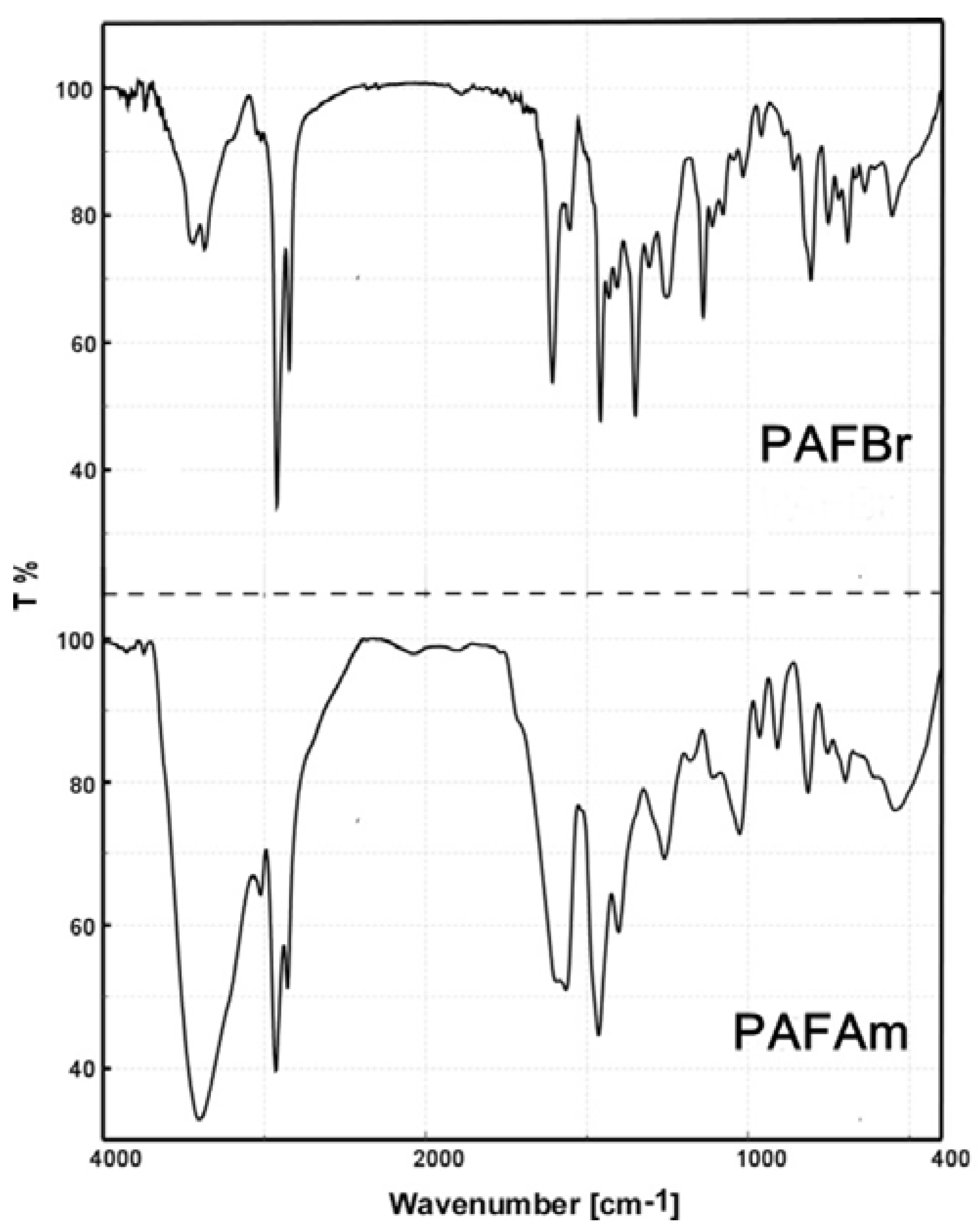
3.2. Synthesis of a New Cationic Polyelectrolyte (PAFAm)
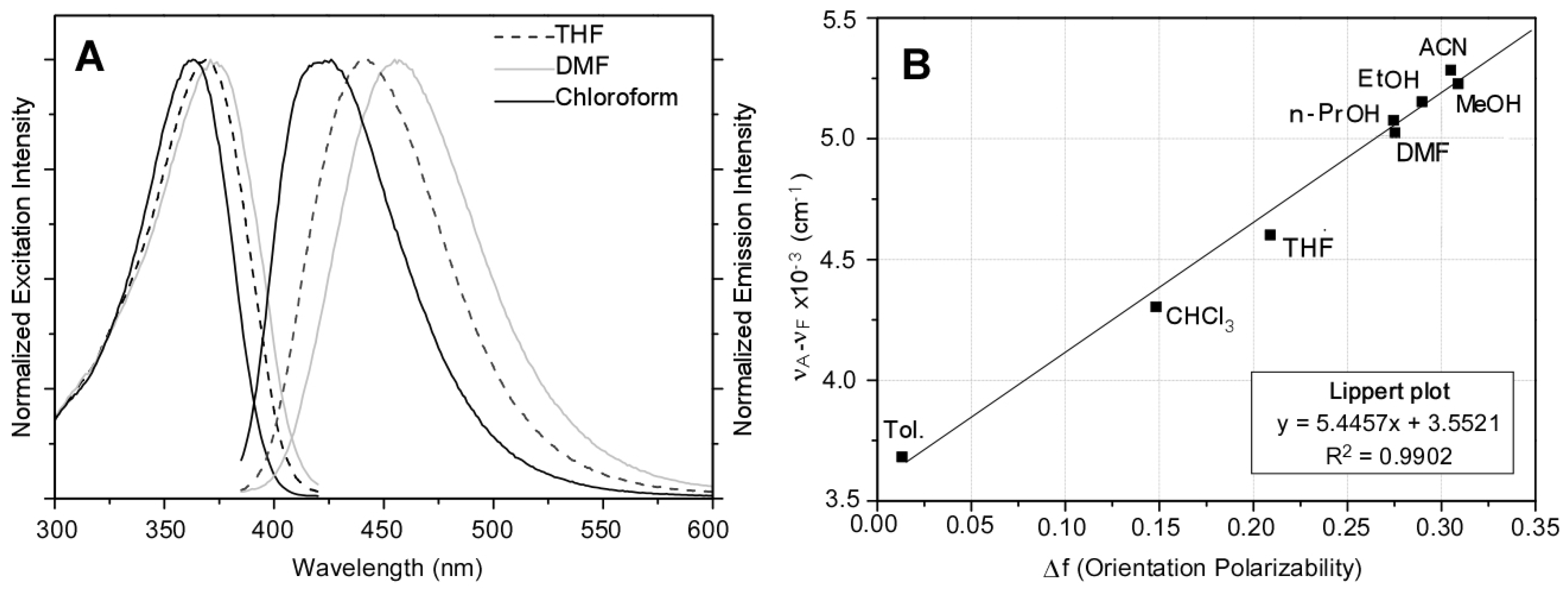
3.3. Fluorescence Solvatochromism of PAF
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bazan, G.C. Novel organic materials through control of multichromophore interactions. J. Org. Chem. 2007, 72, 8615–8635. [Google Scholar] [CrossRef] [PubMed]
- Guenes, S.; Neugebauer, H.; Sariciftci, N.S. Conjugated polymer-based organic solar cells. Chem. Rev. 2007, 107, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.W., III; Joly, G.D.; Swager, T.M. Chemical sensors based on amplifying fluorescent conjugated polymers. Chem. Rev. 2007, 107, 1339–1386. [Google Scholar] [CrossRef] [PubMed]
- Hoven, C.V.; Garcia, A.; Bazan, G.C.; Thuc-Quyen, N. Recent applications of conjugated polyelectrolytes in optoelectronic devices. Adv. Mater. 2008, 20, 3793–3810. [Google Scholar] [CrossRef]
- Scherf, U.; Gutacker, A.; Koenen, N. All-conjugated block copolymers. Acc. Chem. Res. 2008, 41, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.J.; Yang, S.H.; Hsu, C.S. Synthesis of conjugated polymers for organic solar cell applications. Chem. Rev. 2009, 109, 5868–5923. [Google Scholar] [CrossRef] [PubMed]
- Grimsdale, A.C.; Chan, K.L.; Martin, R.E.; Jokisz, P.G.; Holmes, A.B. Synthesis of light-emitting conjugated polymers for applications in electroluminescent devices. Chem. Rev. 2009, 109, 897–1091. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Taranekar, P.; Reynolds, J.R.; Schanze, K.S. Conjugated polyelectrolytes: Synthesis, photophysics, and applications. Angew. Chem. Int. Ed. 2009, 48, 4300–4316. [Google Scholar] [CrossRef] [PubMed]
- Tapia, M.J.; Montserin, M.; Valente, A.J.; Burrows, H.D.; Mallavia, R. Binding of polynucleotides to conjugated polyelectrolytes and its applications in sensing. Adv. Colloid Interface Sci. 2010, 158, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.B.; Sariciftci, N.S.; Grote, J.G. Bio-organic optoelectronic devices using DNA. Org. Electron. 2010, 223, 189–212. [Google Scholar]
- Duarte, A.; Pu, K.-Y.; Liu, B.; Bazan, G.C. Recent advances in conjugated polyelectrolytes for emerging optoelectronic applications. Chem. Mater. 2011, 23, 501–515. [Google Scholar] [CrossRef]
- Bian, L.Y.; Zhu, E.W.; Tang, J.; Tang, W.H.; Zhang, F.J. Recent progress in the design of narrow bandgap conjugated polymers for high-efficiency organic solar cells. Prog. Polym. Sci. 2012, 37, 1292–1331. [Google Scholar] [CrossRef]
- Zhu, C.; Liu, L.; Yang, Q.; Lv, F.; Wang, S. Water-soluble conjugated polymers for imaging, diagnosis, and therapy. Chem. Rev. 2012, 112, 4687–4735. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhu, C.; Yuan, H.; Liu, L.; Lv, F.; Wang, S. Conjugated polymer nanoparticles: Preparation, properties, functionalization and biological applications. Chem. Soc. Rev. 2013, 42, 6620–6633. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Baumgarten, M.; Mullen, K. Designing pi-conjugated polymers for organic electronics. Prog. Polym. Sci. 2013, 38, 1832–1908. [Google Scholar] [CrossRef]
- Rochat, S.; Swager, T.M. Conjugated amplifying polymers for optical sensing applications. ACS Appl. Mater. Interfaces 2013, 5, 4488–4502. [Google Scholar] [CrossRef] [PubMed]
- Blayney, A.J.; Perepichka, I.F.; Wudl, F.; Perepichka, D.F. Advances and challenges in the synthesis of poly(p-phenylene vinylene)-based polymers. Isr. J. Chem. 2014, 54, 674–688. [Google Scholar] [CrossRef]
- Li, K.; Liu, B. Polymer-encapsulated organic nanoparticles for fluorescence and photoacoustic imaging. Chem. Soc. Rev. 2014, 43, 6570–6597. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Grimsdale, A.C.; Jones, D.J.; Watkins, S.E.; Holmes, A.B. Polyfluorenes without monoalkylfluorene defects. J. Am. Chem. Soc. 2007, 129, 11910–11911. [Google Scholar] [CrossRef] [PubMed]
- Schluter, A.D. The tenth anniversary of suzuki polycondensation (spc). J. Polym. Sci. A 2001, 39, 1533–1556. [Google Scholar] [CrossRef]
- Sakamoto, J.; Rehahn, M.; Wegner, G.; Schlueter, A.D. Suzuki polycondensation: Polyarylenes a la carte. Macromol. Rapid Commun. 2009, 30, 653–687. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Islam, M.M.; Islam, S.M. Suzuki-miyaura reaction by heterogeneously supported pd in water: Recent studies. RSC Adv. 2015, 5, 42193–42221. [Google Scholar] [CrossRef]
- Scherf, U.; List, E.J.W. Semiconducting polyfluorenes—Towards reliable structure-property relationships. Adv. Mater. 2002, 14, 477–487. [Google Scholar] [CrossRef]
- Becker, K.; Scherf, U.; Neher, D. Polyfluorenes; Springer: Berlin, Germmany, 2008. [Google Scholar]
- Xie, L.H.; Yin, C.R.; Lai, W.Y.; Fan, Q.L.; Huang, W. Polyfluorene-based semiconductors combined with various periodic table elements for organic electronics. Prog. Polym. Sci. 2012, 37, 1192–1264. [Google Scholar] [CrossRef]
- Kowalski, S.; Allard, S.; Zilberberg, K.; Riedl, T.; Scherf, U. Direct arylation polycondensation as simplified alternative for the synthesis of conjugated (co)polymers. Prog. Polym. Sci. 2013, 38, 1805–1814. [Google Scholar] [CrossRef]
- Mercier, L.G.; Leclerc, M. Direct (hetero)arylation: A new tool for polymer chemists. Acc. Chem. Res. 2013, 46, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, A.E.; Thompson, B.C. Optimization of direct arylation polymerization (DARP) through the identification and control of defects in polymer structure. J. Polym. Sci. A 2015, 53, 135–147. [Google Scholar] [CrossRef]
- Morin, P.O.; Bura, T.; Sun, B.; Gorelsky, S.I.; Li, Y.N.; Leclerc, M. Conjugated polymers a la carte from time-controlled direct (hetero)arylation polymerization. ACS Macro Lett. 2015, 4, 21–24. [Google Scholar] [CrossRef]
- Zhang, W.; Lu, P.; Wang, Z.; Ma, Y. Microwave-assisted suzuki coupling reaction for rapid synthesis of conjugated polymerpoly(9,9-dihexylfluorene)s as an example. J. Polym. Sci. A 2013, 51, 1950–1955. [Google Scholar] [CrossRef]
- Gao, X.; Lu, P.; Ma, Y. Ultrasound-assisted suzuki coupling reaction for rapid synthesis of polydihexylfluorene. Polymer 2014, 55, 3083–3086. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Z.; Zhang, Y.; Lu, P.; Liu, L.; Ma, Y. Microwave-assisted FeCl3-mediated rapid synthesis of poly(9,9-dihexylfluorene) with high molecular weight. Polymer 2014, 55, 5346–5349. [Google Scholar] [CrossRef]
- Mallavia, R.; Montilla, F.; Pastor, I.; Velasquez, P.; Arredondo, B.; Alvarez, A.L.; Mateo, C.R. Characterization and side chain manipulation in violet-blue poly- (9,9-dialkylfluoren-2,7-diyl)-alt-co-(benzen-1,4-diyl) backbones. Macromolecules 2005, 38, 3185–3192. [Google Scholar] [CrossRef]
- Molina, R.; Gomez-Ruiz, S.; Montilla, F.; Salinas-Castillo, A.; Fernandez-Arroyo, S.; del Mar Ramos, M.; Micol, V.; Mallavia, R. Progress in the synthesis of poly(2,7-fluorene-alt-1,4-phenylene), pfp, via suzuki coupling. Macromolecules 2009, 42, 5471–5477. [Google Scholar] [CrossRef]
- Knaapila, M.; Lyons, B.P.; Hase, T.P.A.; Pearson, C.; Petty, M.C.; Bouchenoire, L.; Thompson, P.; Serimaa, R.; Torkkeli, M.; Monkman, A.P. Influence of molecular weight on the surface morphology of aligned, branched side-chain polyfluorene. Adv. Funct. Mater. 2005, 15, 1517–1522. [Google Scholar] [CrossRef]
- Abbel, R.; Schenning, A.; Meijer, E.W. Molecular weight optimum in the mesoscopic order of chiral fluorene (co)polymer films. Macromolecules 2008, 41, 7497–7504. [Google Scholar] [CrossRef]
- Murage, J.; Eddy, J.W.; Zimbalist, J.R.; McIntyre, T.B.; Wagner, Z.R.; Goodson, F.E. Effect of reaction parameters on the molecular weights of polymers formed in a suzuki polycondensation. Macromolecules 2008, 41, 7330–7338. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Jiang, H.; Schanze, K.S. Polymer chain length dependence of amplified fluorescence quenching in conjugated polyelectrolytes. Macromolecules 2008, 41, 3422–3428. [Google Scholar] [CrossRef]
- Knaapila, M.; Monkman, A.P. Methods for controlling structure and photophysical properties in polyfluorene solutions and gels. Adv. Mater. 2013, 25, 1090–1108. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed]
- Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis—A review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- De la Hoz, A.; Diaz-Ortiz, A.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Dallinger, D.; Kappe, C.O. Microwave-assisted synthesis in water as solvent. Chem. Rev. 2007, 107, 2563–2591. [Google Scholar] [CrossRef] [PubMed]
- Wiesbrock, F.; Hoogenboom, R.; Schubert, U.S. Microwave-assisted polymer synthesis: State-of-the-art and future perspectives. Macromol. Rapid Commun. 2004, 25, 1739–1764. [Google Scholar] [CrossRef]
- Kappe, C.O.; Dallinger, D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Discov. 2006, 5, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.A.; Strauss, C.R. Toward rapid,“green”, predictable microwave-assisted synthesis. Acc. Chem. Res. 2005, 38, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Galbrecht, F.; Buennagel, T.W.; Scherf, U.; Farrell, T. Microwave-assisted preparation of semiconducting polymers. Macromol. Rapid Commun. 2007, 28, 387–394. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Schubert, U.S. Microwave-assisted polymer synthesis: Recent developments in a rapidly expanding field of research. Macromol. Rapid Commun. 2007, 28, 368–386. [Google Scholar] [CrossRef]
- Ebner, C.; Bodner, T.; Stelzer, F.; Wiesbrock, F. One decade of microwave-assisted polymerizations: Quo vadis? Macromol. Rapid Commun. 2011, 32, 254–288. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, I.; Mizoguchi, N.; Sato, M. Self-doped polyphenylenes containing electron-accepting viologen side group. Macromolecules 2009, 42, 4416–4425. [Google Scholar] [CrossRef]
- Chen, T.A.; Wu, X.M.; Rieke, R.D. Regiocontrolled synthesis of poly(3-alkylthiophenes) mediated by rieke zinc—Their characterization and solid-state properties. J. Am. Chem. Soc. 1995, 117, 233–244. [Google Scholar] [CrossRef]
- Yamaguchi, I.; Makishi, S. Synthesis and chemical properties of electrochromic-conjugated polyphenylenes with pendant viologen-tcnq salts. J. Appl. Polym. Sci. 2013, 129, 397–403. [Google Scholar] [CrossRef]
- Liu, B.; Bazan, G.C. Synthesis of cationic conjugated polymers for use in label-free DNA microarrays. Nat. Protoc. 2006, 1, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Mallavia, R.; Martinez-Perez, D.; Chmelka, B.F.; Bazan, G.C. Blue fluorescent films based on poly-2,7-fluorene-phenylene derivatives. Bol. Soc. Española Cerám. Vidrio 2004, 43, 327–330. [Google Scholar] [CrossRef]
- Klaerner, G.; Miller, R.D. Polyfluorene derivatives: Effective conjugation lengths from well-defined oligomers. Macromolecules 1998, 31, 2007–2009. [Google Scholar] [CrossRef]
- Kappe, C.O. Microwave dielectric heating in synthetic organic chemistry. Chem. Soc. Rev. 2008, 37, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Qing Han, L.; Yong, D.; Gang, Z.; Zhen, Z.; Song, M. Suzuki-miyaura cross-coupling reaction catalyzed by supported palladium under microwave irradiation. Curr. Org. Synth. 2017, 14, 462–476. [Google Scholar]
- Herrero, M.A.; Kremsner, J.M.; Kappe, C.O. Nonthermal microwave effects revisited: On the importance of internal temperature monitoring and agitation in microwave chemistry. J. Org. Chem. 2008, 73, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Kluwer Academic/Plenum: New York, NY, USA, 1999; p xxiii 698. [Google Scholar]
- Pina, J.; de Melo, J.S.; Breusov, D.; Scherf, U. Donor-acceptor-donor thienyl/bithienyl-benzothiadiazole/quinoxaline model oligomers: Experimental and theoretical studies. Phys. Chem. 2013, 15, 15204–15213. [Google Scholar] [CrossRef] [PubMed]
- Ooyama, Y.; Ito, G.; Kushimoto, K.; Komaguchi, K.; Imae, I.; Harima, Y. Synthesis and fluorescence and electrochemical properties of d-pi-a structural isomers of benzofuro[2,3-c]oxazolo[4,5-a]carbazole-type and benzofuro[2,3-c]oxazolo[5,4-a]carbazole-type fluorescent dyes. Org. Biomol. Chem. 2010, 8, 2756–2770. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. Handbook of Chemistry and Physics; CRCPress: Boca Raton, FL, USA, 1999. [Google Scholar]
- Melhuish, W.H. Quantum efficiencies of fluorescence of organic substances-effect of solvent and concentration of fluorescent solute. J. Phy. Chem. 1961, 65, 229–235. [Google Scholar] [CrossRef]

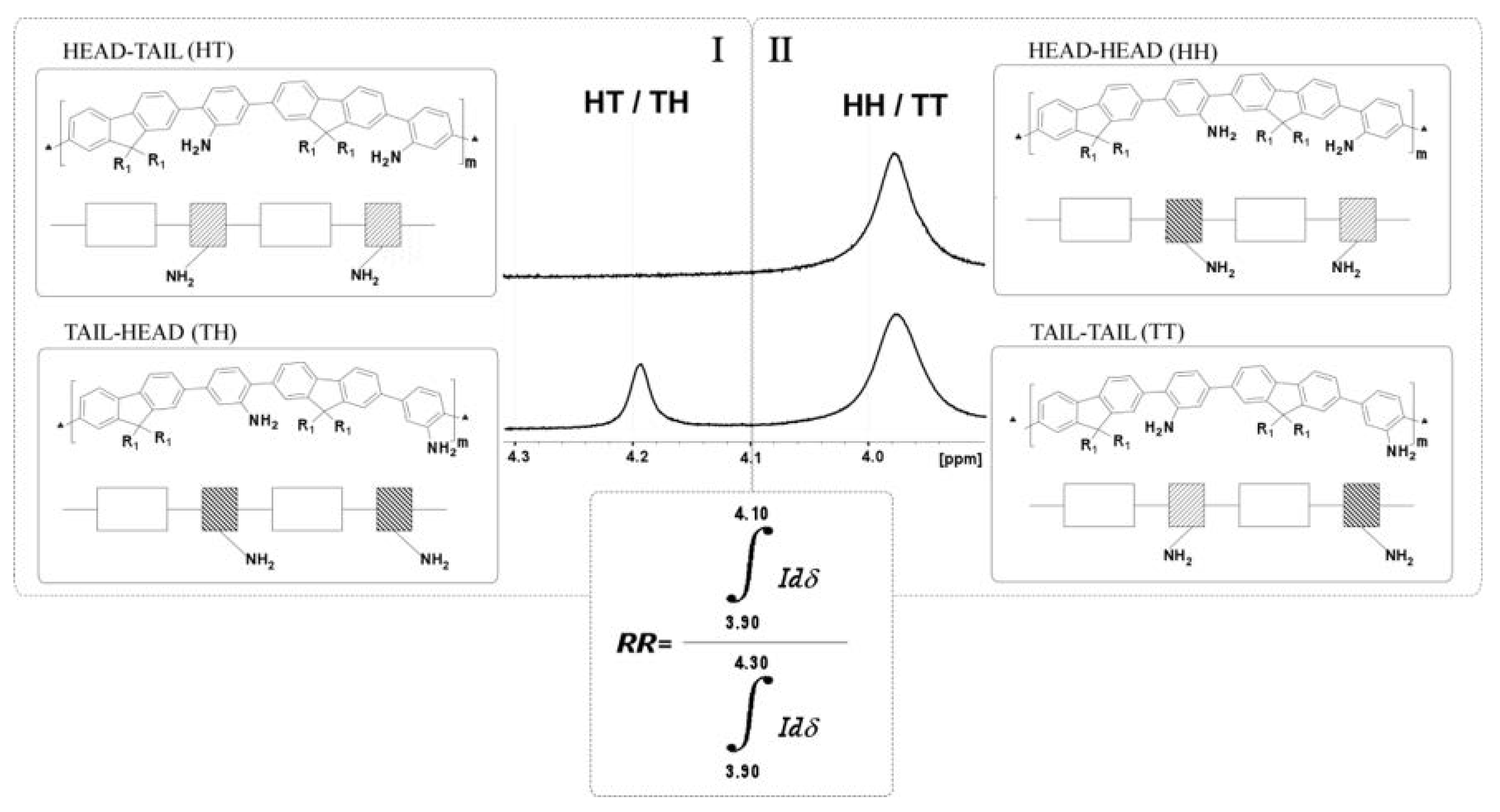





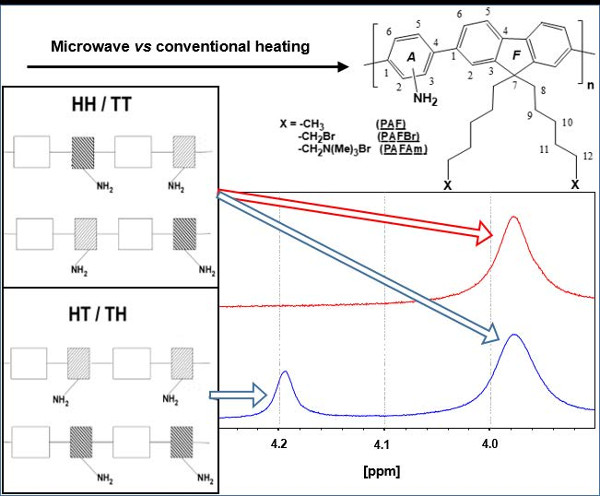{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Heating conditions | Yield (%) | Mw c (kg/mol) | PDI d | n d | % RR by NMR e | |||
|---|---|---|---|---|---|---|---|---|---|
| 1 | [Pd(PPh3)4] | Oil bath;conventional flask | 80 °C | 24 h | 62 | 9.90 | 1.9 | 22 | 75 |
| 2 | 72 h | 86 | 20.77 | 1.6 | 47 | 87 | |||
| 3 | Oil bath;cressure vessel | 135 °C | 22 min | 72 | 6.39 | 2.1 | 14 | 83 | |
| 4 | 24 h | 34 | 3.54 | 1.5 | 8 | 78 | |||
| 5 | µW;Dynamicmodeb | 135 °C | 14 min | 57 | 6.91 | 2.0 | 16 | 70 | |
| 6 | 22 min | 73 | 11.80 | 1.9 | 27 | 74 | |||
| 7 | µW;SPS mode b | 135 °C | 14 min | 90 | 11.60 | 2.0 | 26 | 73 | |
| 8 | 22 min | 99 | 15.02 | 2.1 | 34 | 94 | |||
| 9 | [Pd(Amphos)2Cl2] | 14 min | 71 | 5.43 | 1.9 | 12 | 86 | ||
| 10 | cis-[PdCl2(dppf)] | 14 min | 47 | 6.26 | 1.8 | 14 | 64 | ||
| # | Heating Mode | [Pd(PPh3)4] (%) | Yield (%) | Mw c (kg/mol) | PDI d | nd | % RR by NMR e |
|---|---|---|---|---|---|---|---|
| 11 | 1 | 90 | 10.70 | 1.9 | 24 | 93 | |
| 2 | Oil bath a | 3 | 86 | 20.77 | 1.6 | 47 | 87 |
| 12 | 6 | 86 | 36.49 | 2.7 | 83 | 86 | |
| 13 | 1 | 85 | 5.73 | 1.7 | 13 | 84 | |
| 7 | µW SPS mode b | 3 | 90 | 11.60 | 2.0 | 26 | 72 |
| 14 | 6 | 100 | 6.45 | 1.6 | 15 | 69 |
| Solvent | ∆f a | λAmax(nm) | λEmax(nm) | νA − νE(cm−1) | ФPL b |
|---|---|---|---|---|---|
| Toluene | 0.0132 | 363 | 419 | 3682 | 0.50 ± 0.05 |
| Chloroform | 0.1482 | 360 | 426 | 4303 | 0.49 ± 0.06 |
| THF | 0.2089 | 366 | 443 | 4601 | 0.55 ± 0.05 |
| n-Propanol | 0.2746 | 361 | 442 | 5076 | 0.18 ± 0.02 |
| DMF | 0.2754 | 371 | 456 | 5024 | 0.54 ± 0.03 |
| Ethanol | 0.2898 | 362 | 445 | 5152 | 0.16 ± 0.02 |
| Acetonitrile | 0.3050 | 357 | 440 | 5284 | 0.11 ± 0.02 |
| Methanol | 0.3089 | 363 | 448 | 5227 | 0.10 ± 0.02 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Guilló, R.; Falco, A.; Martínez-Tomé, M.J.; Mateo, C.R.; Herrero, M.A.; Vázquez, E.; Mallavia, R. Advantageous Microwave-Assisted Suzuki Polycondensation for the Synthesis of Aniline-Fluorene Alternate Copolymers as Molecular Model with Solvent Sensing Properties. Polymers 2018, 10, 215. https://doi.org/10.3390/polym10020215
Vázquez-Guilló R, Falco A, Martínez-Tomé MJ, Mateo CR, Herrero MA, Vázquez E, Mallavia R. Advantageous Microwave-Assisted Suzuki Polycondensation for the Synthesis of Aniline-Fluorene Alternate Copolymers as Molecular Model with Solvent Sensing Properties. Polymers. 2018; 10(2):215. https://doi.org/10.3390/polym10020215
Chicago/Turabian StyleVázquez-Guilló, Rebeca, Alberto Falco, M. José Martínez-Tomé, C. Reyes Mateo, María Antonia Herrero, Ester Vázquez, and Ricardo Mallavia. 2018. "Advantageous Microwave-Assisted Suzuki Polycondensation for the Synthesis of Aniline-Fluorene Alternate Copolymers as Molecular Model with Solvent Sensing Properties" Polymers 10, no. 2: 215. https://doi.org/10.3390/polym10020215