Biosynthetic Pathway and Genes of Chitin/Chitosan-Like Bioflocculant in the Genus Citrobacter

Abstract
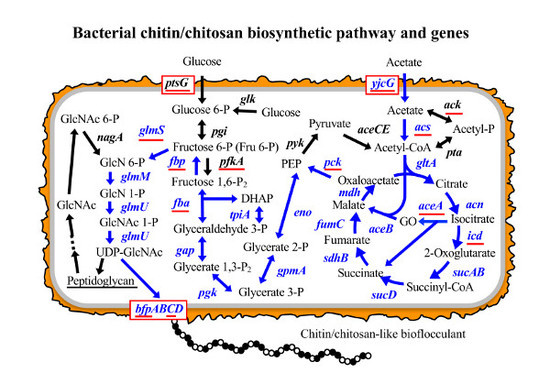
:
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Media, and Culture Conditions
2.2. Measurement of Flocculation Activity and Determination of Flocculation Titer
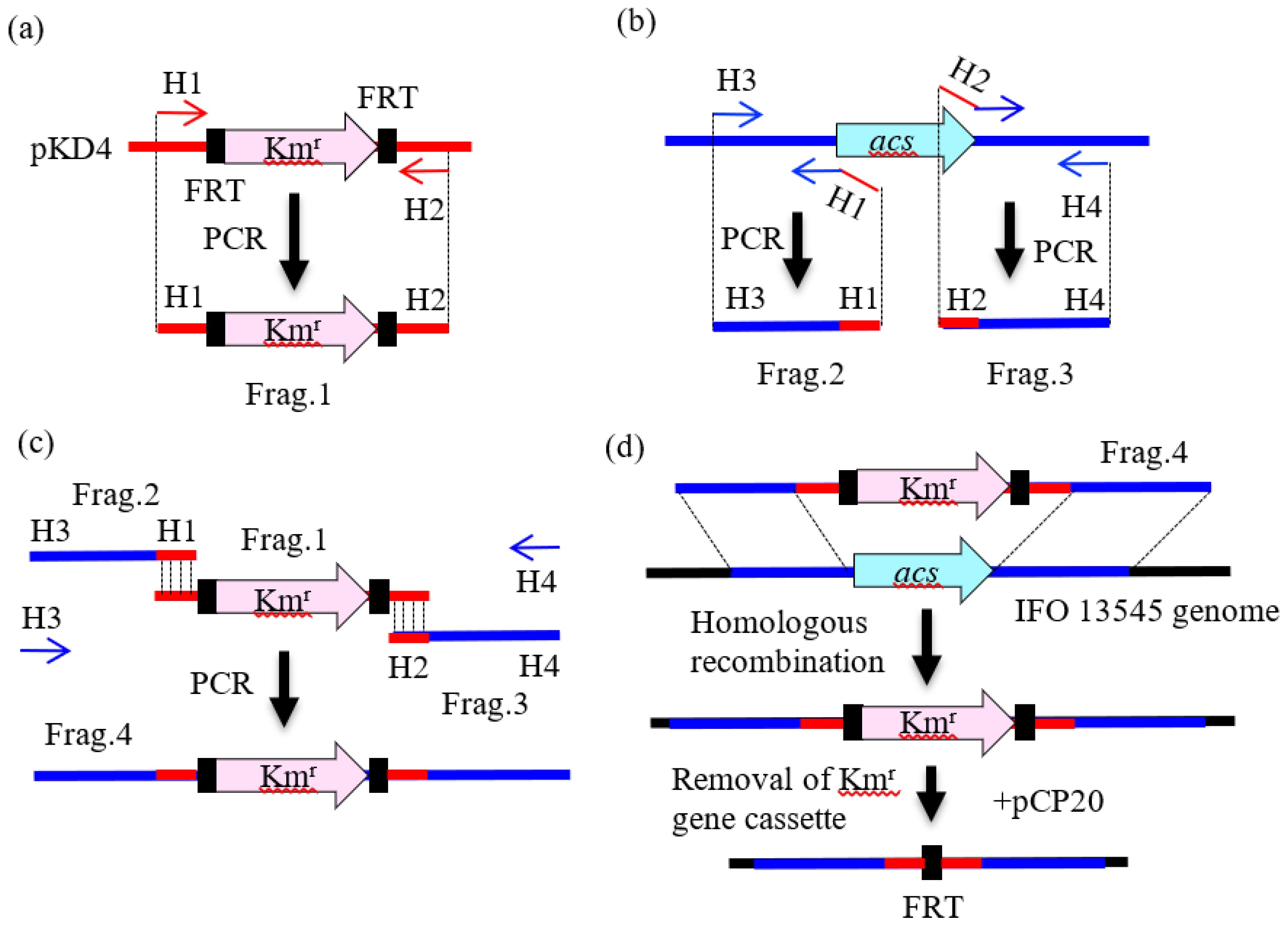
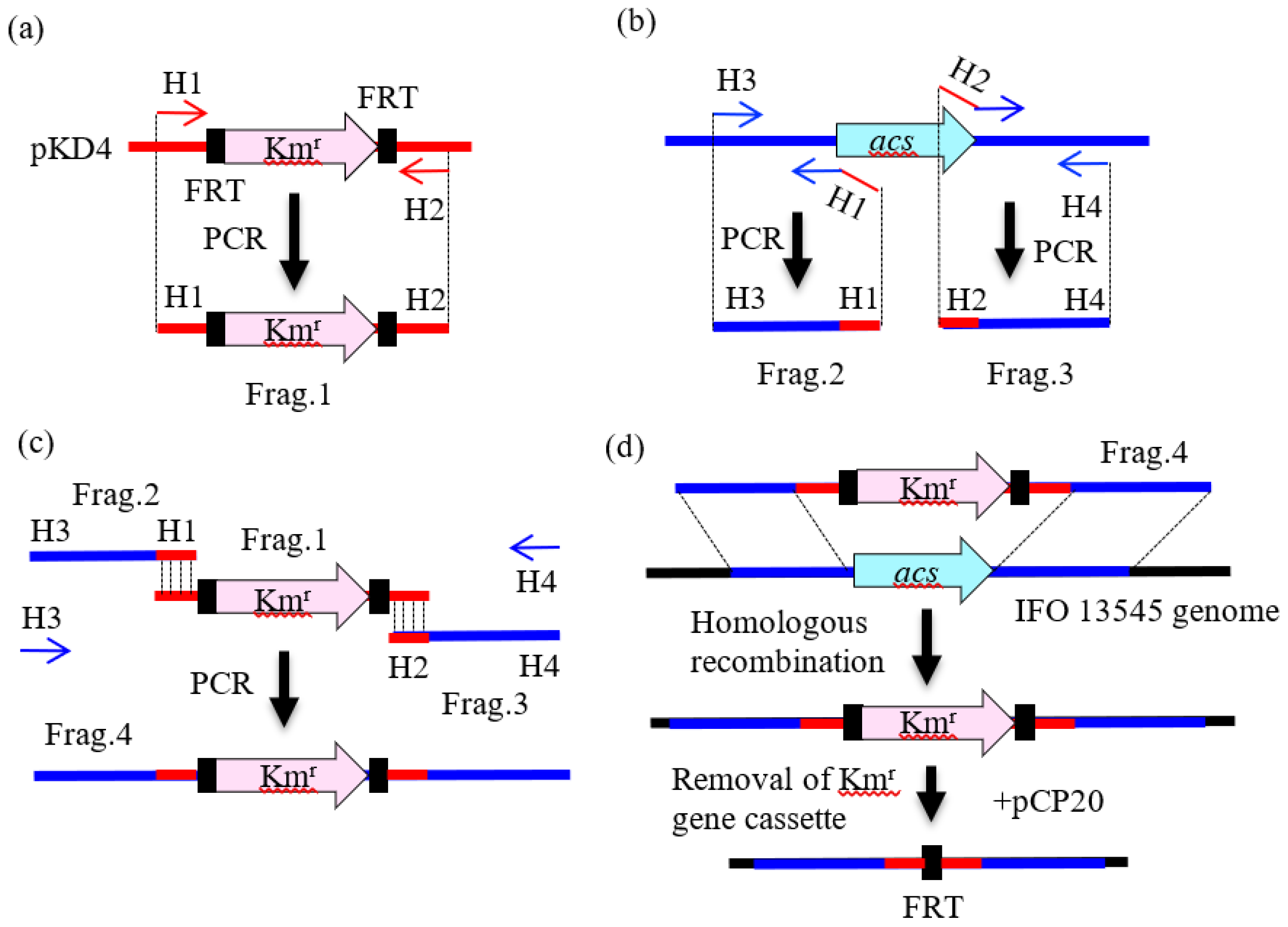
2.3. Preparation of DNA Fragments by PCR for Gene Disruption
2.4. Gene Disruption by Homologous Recombination
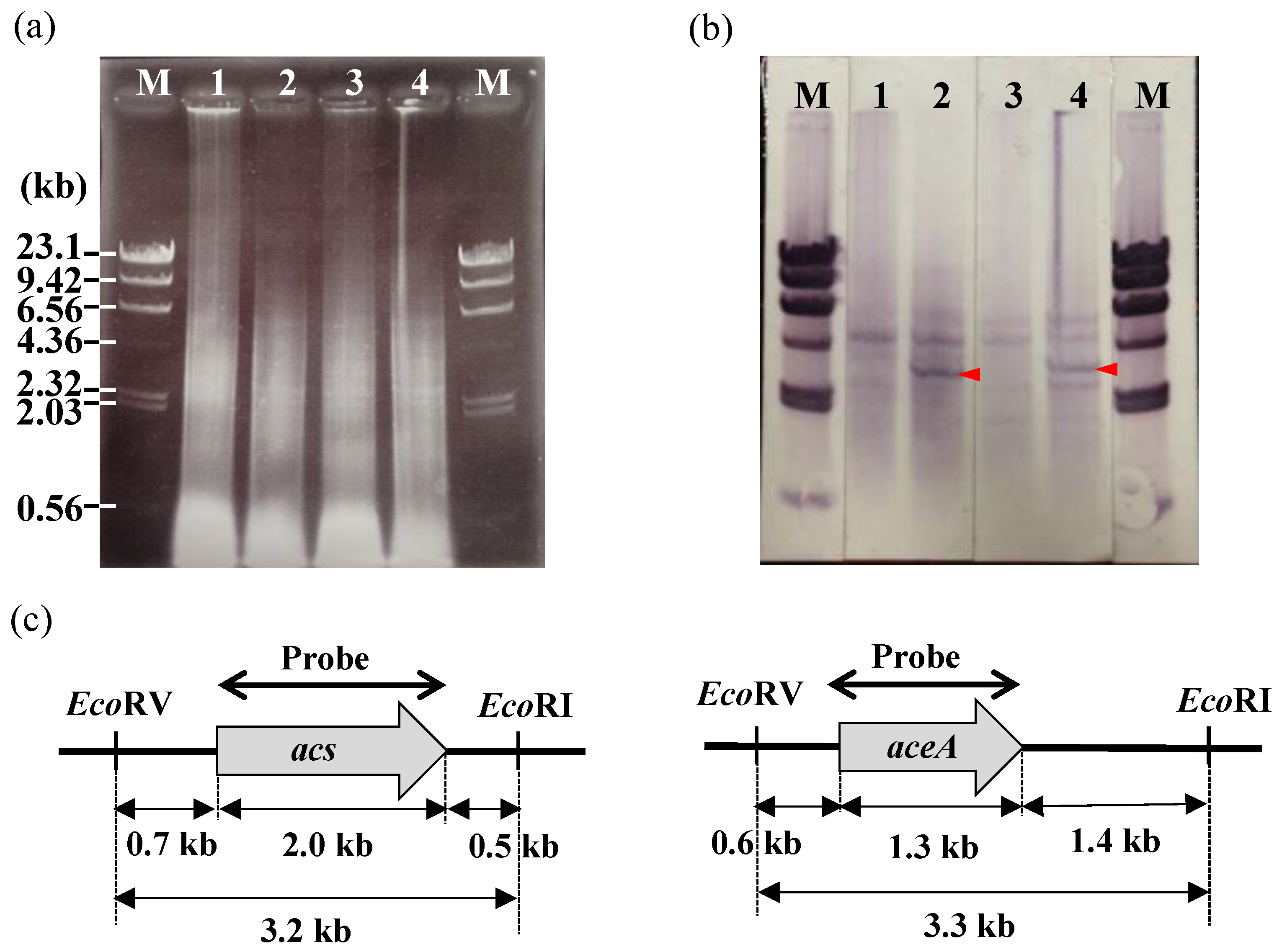
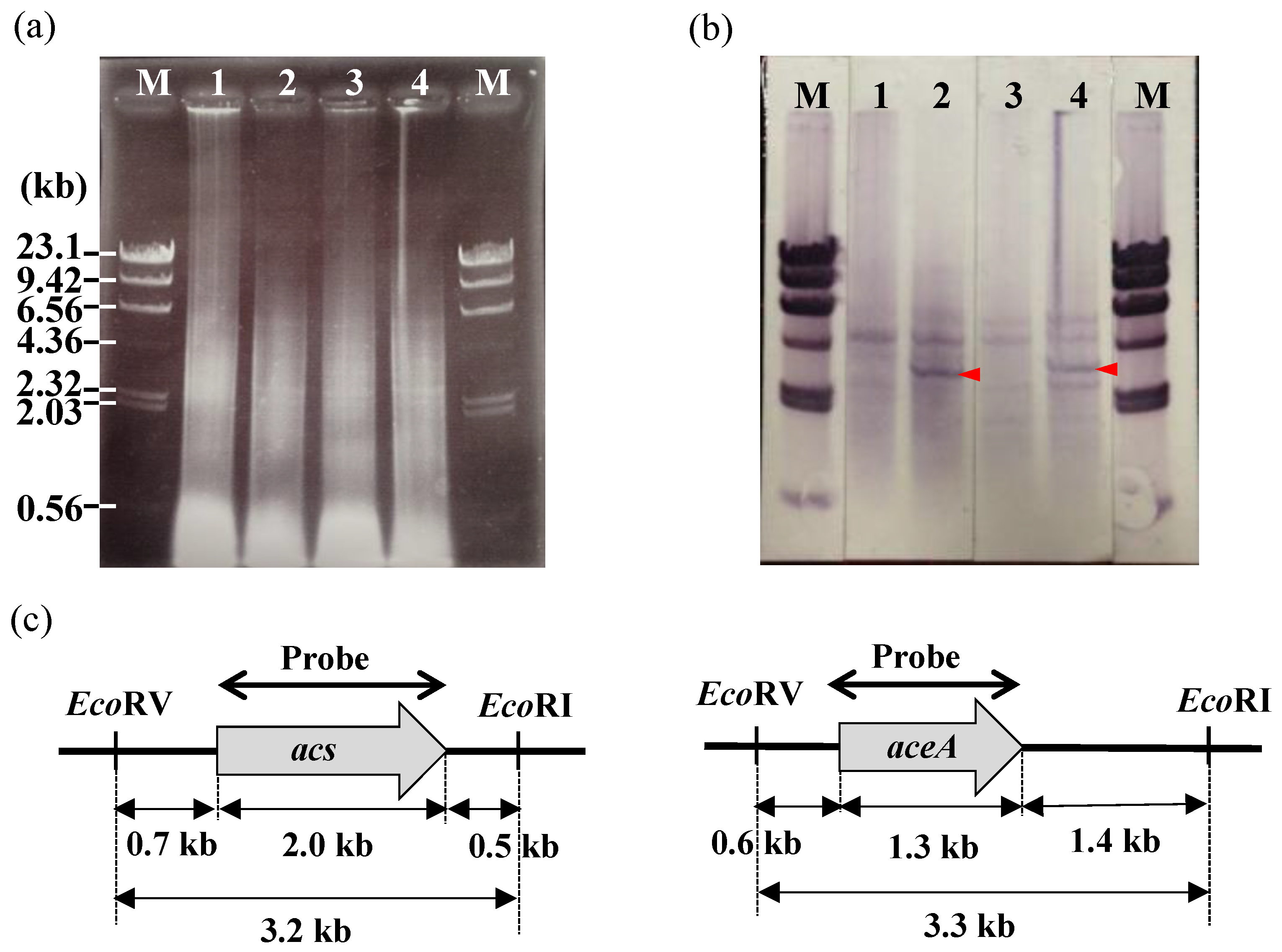
2.5. Southern Hybridization
2.6. Batch Cultivation in a Mini-Jar Fermentor
2.7. Quantitative Reverse-Transcription-PCR (qRT-PCR)
2.8. Nucleotide Sequences Registered in the Databases
3. Results
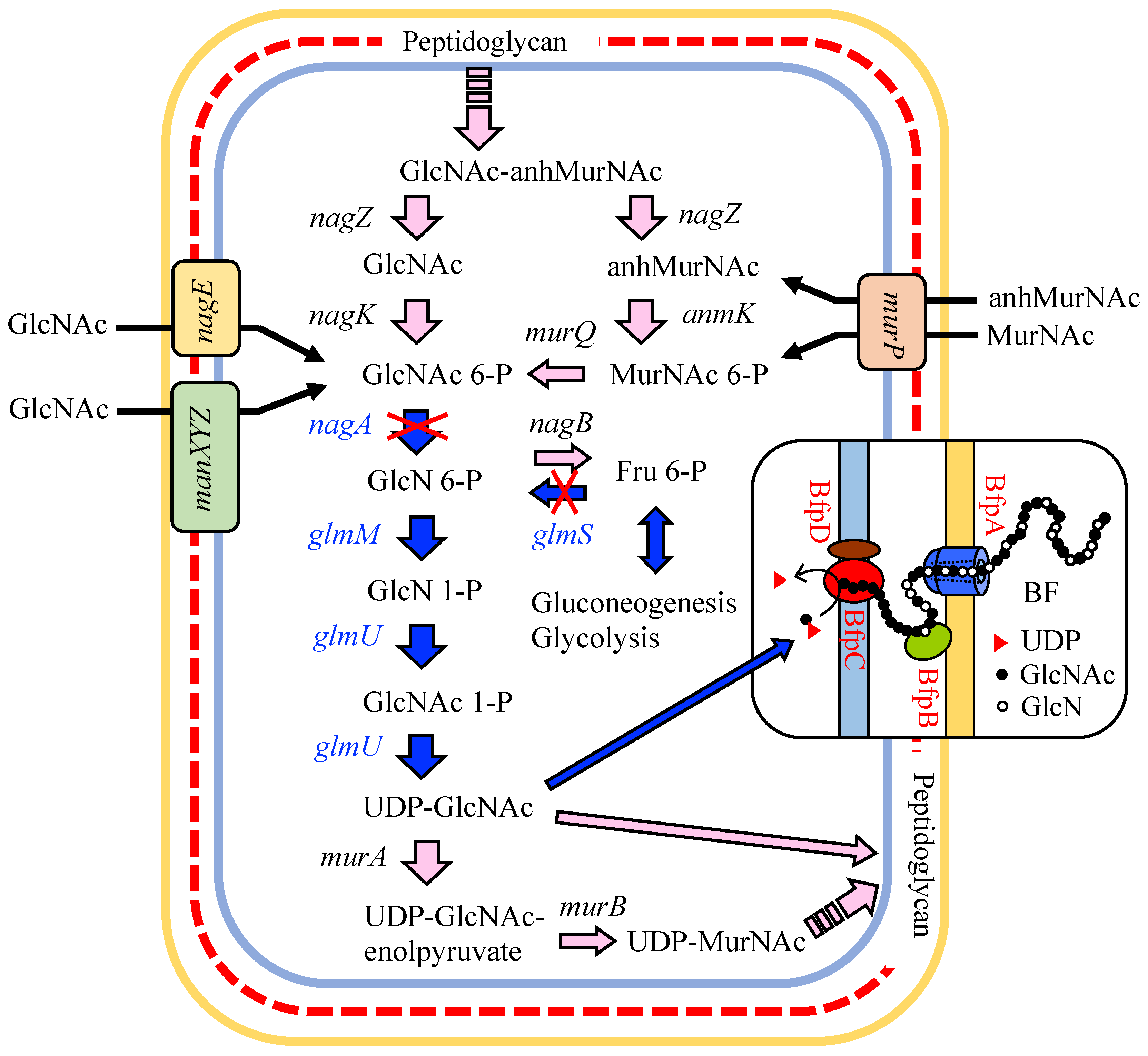
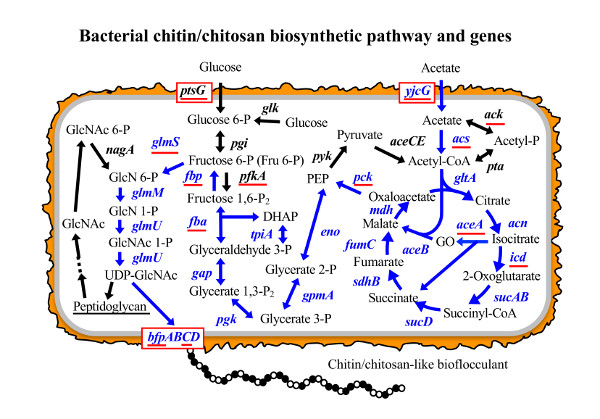
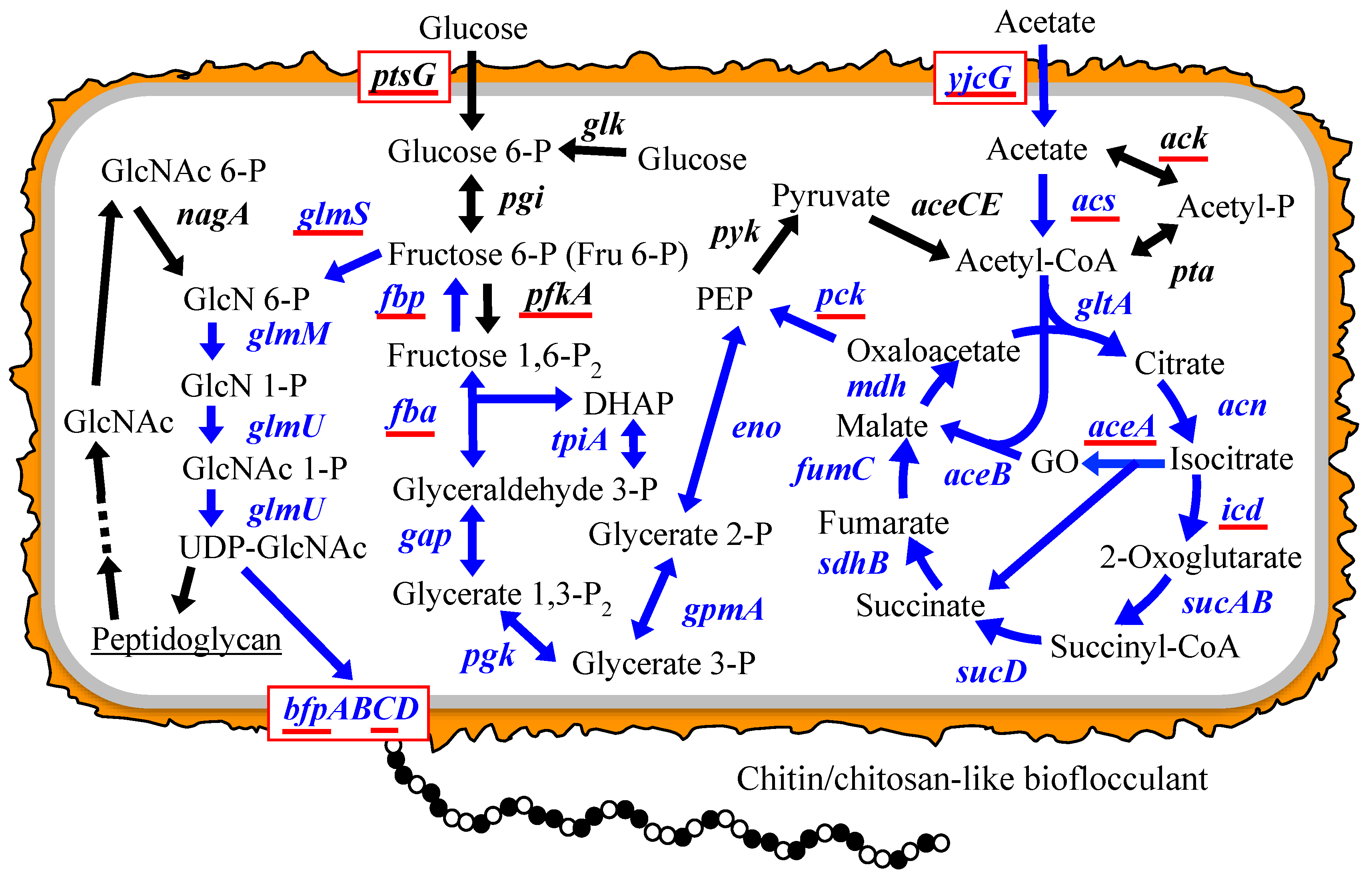
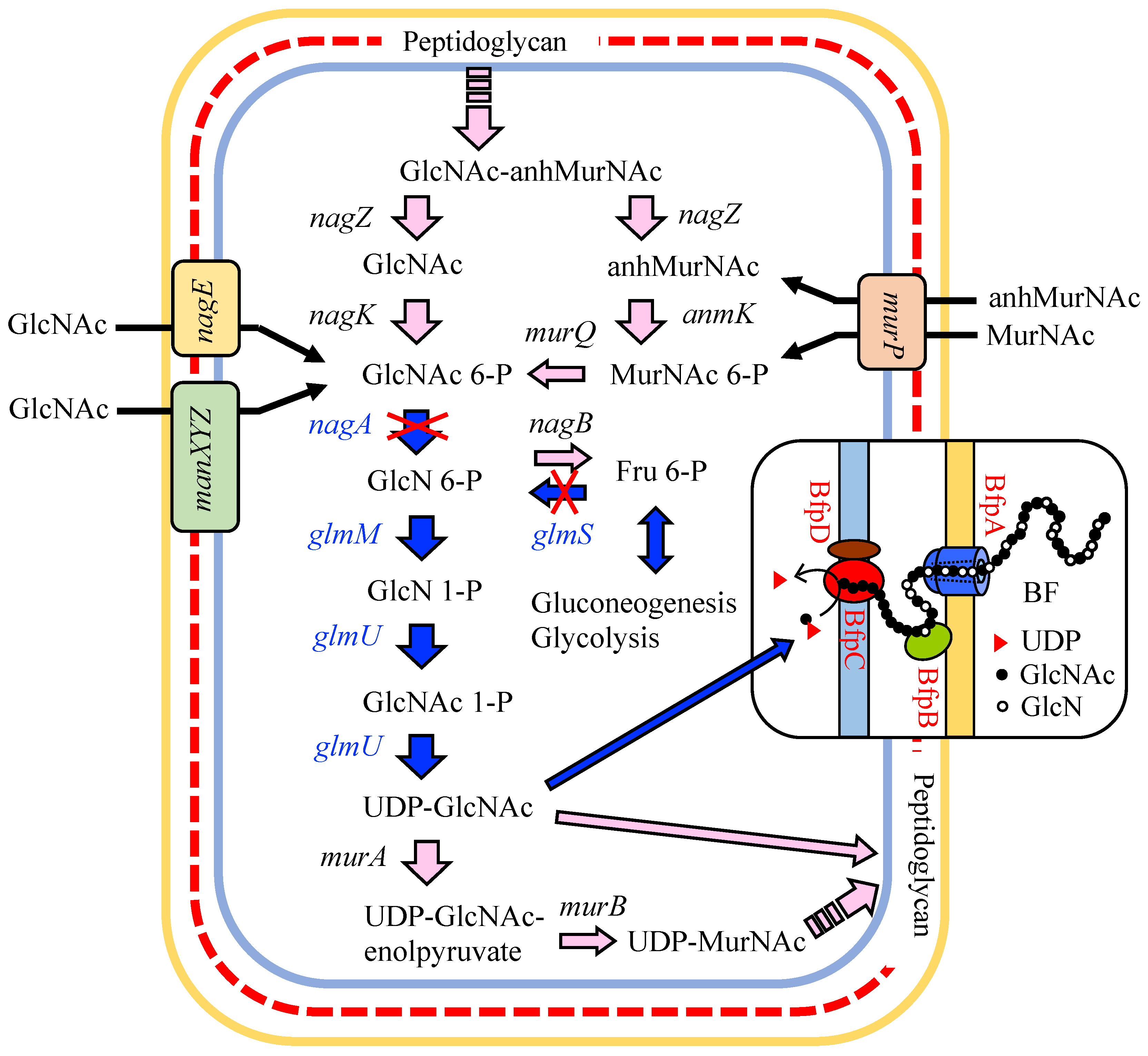
3.1. Putative BF Synthetic Pathway of C. freundii IFO 13545 and Its Related Genes
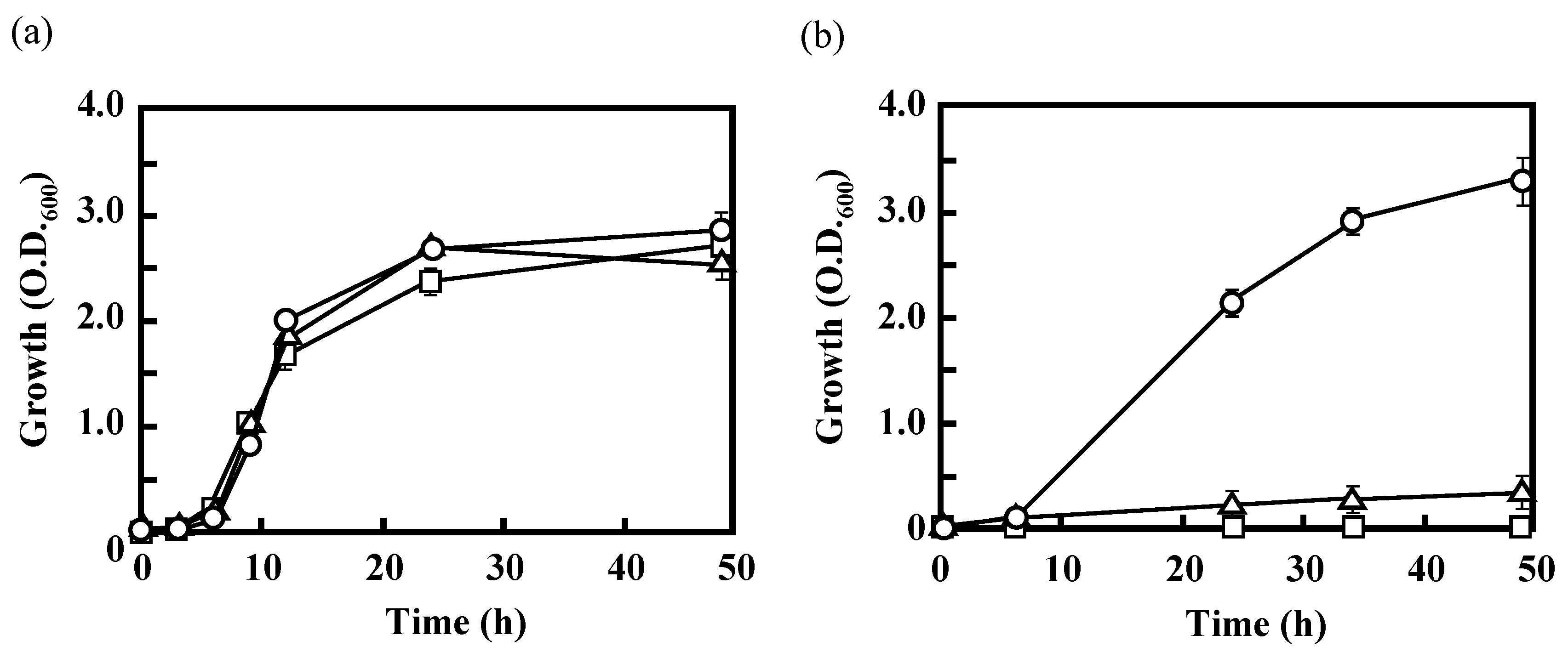
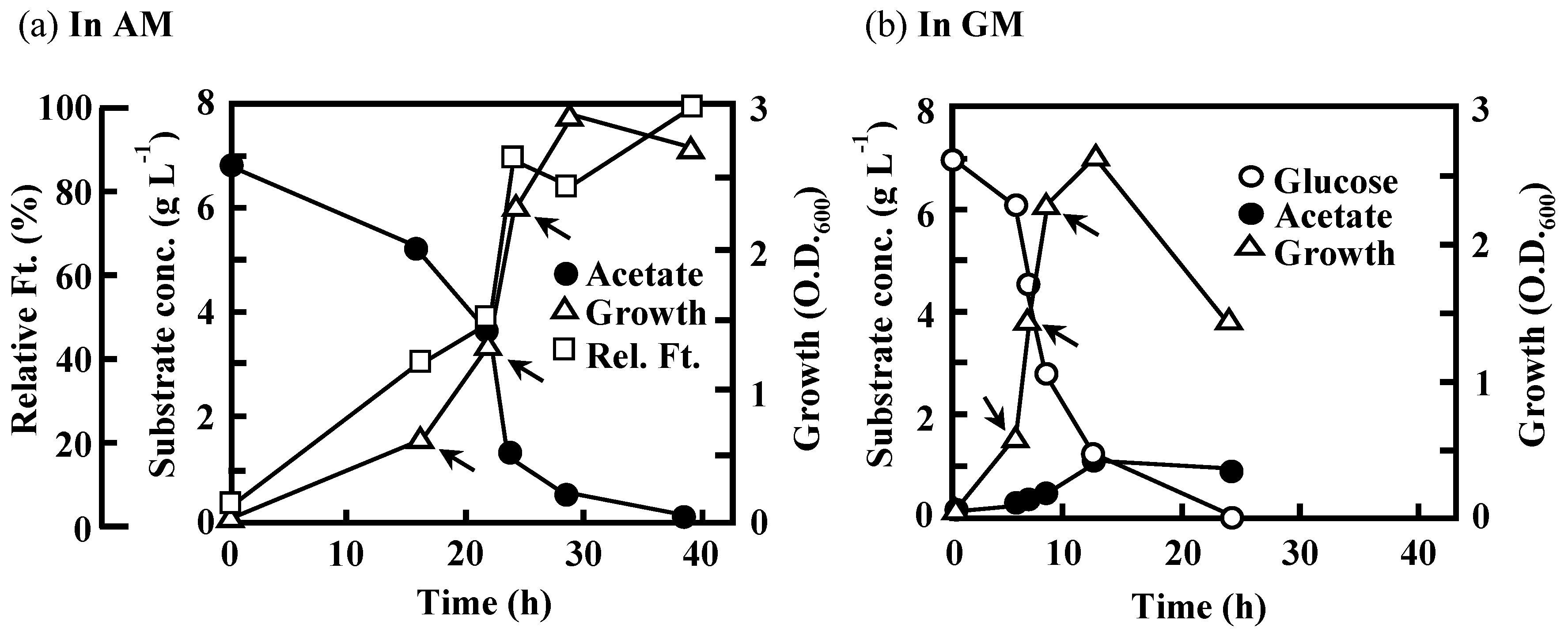
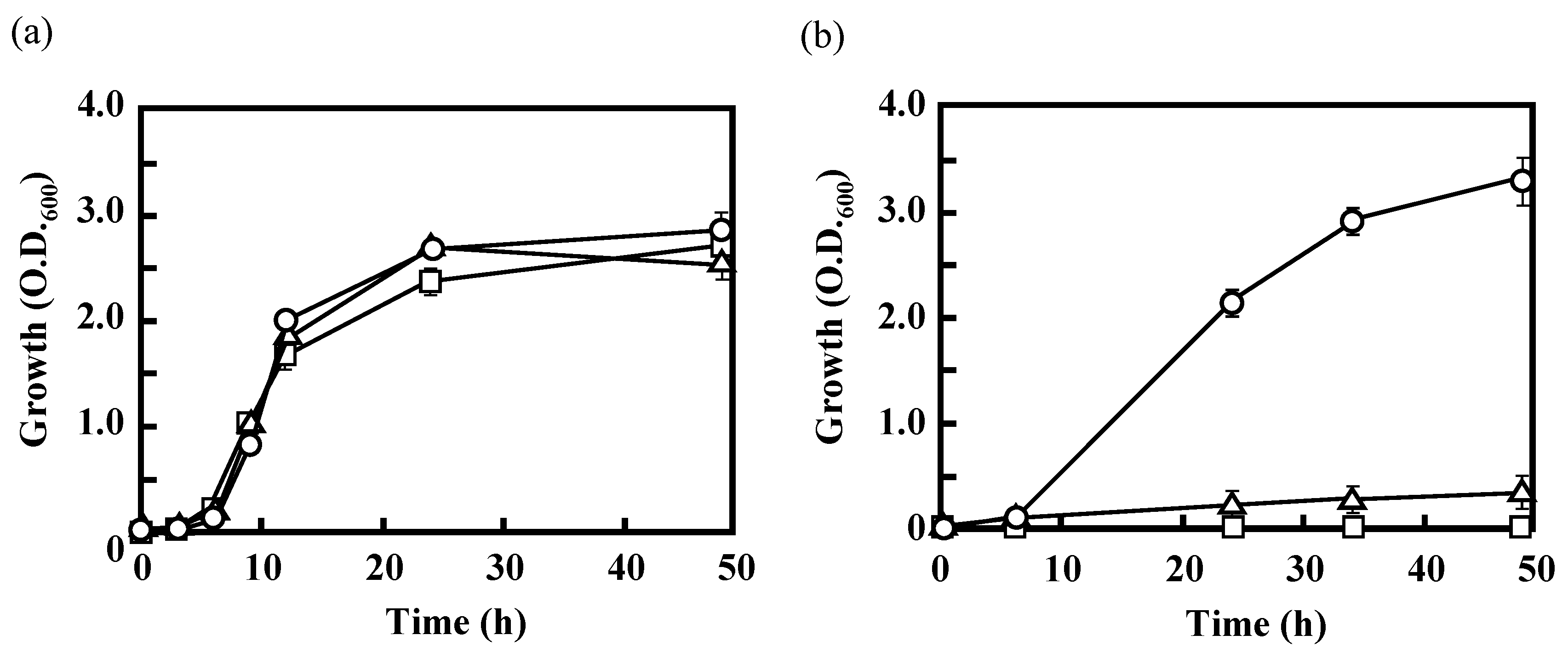
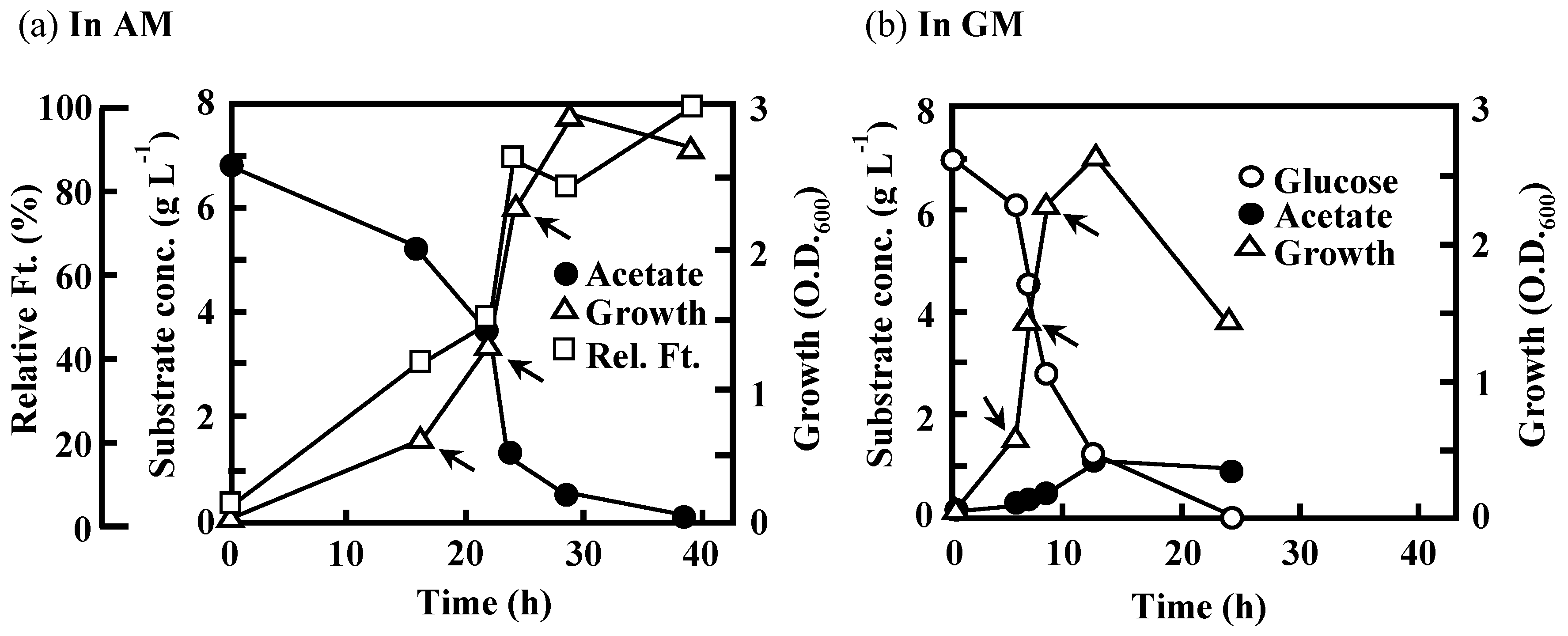
3.2. Effects of the Disruption of Acs or AceA on the Growth of IFO 13545 on Acetate
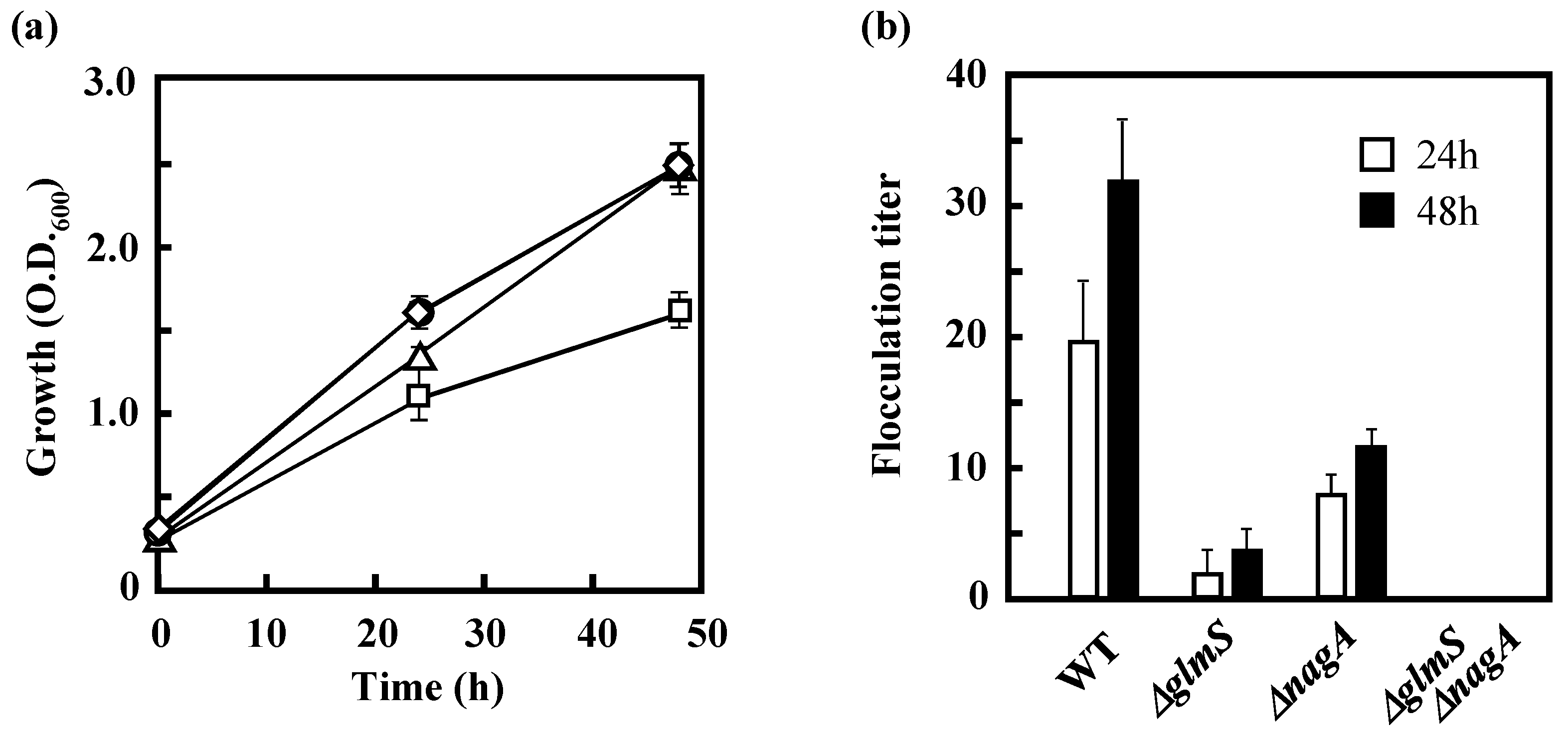
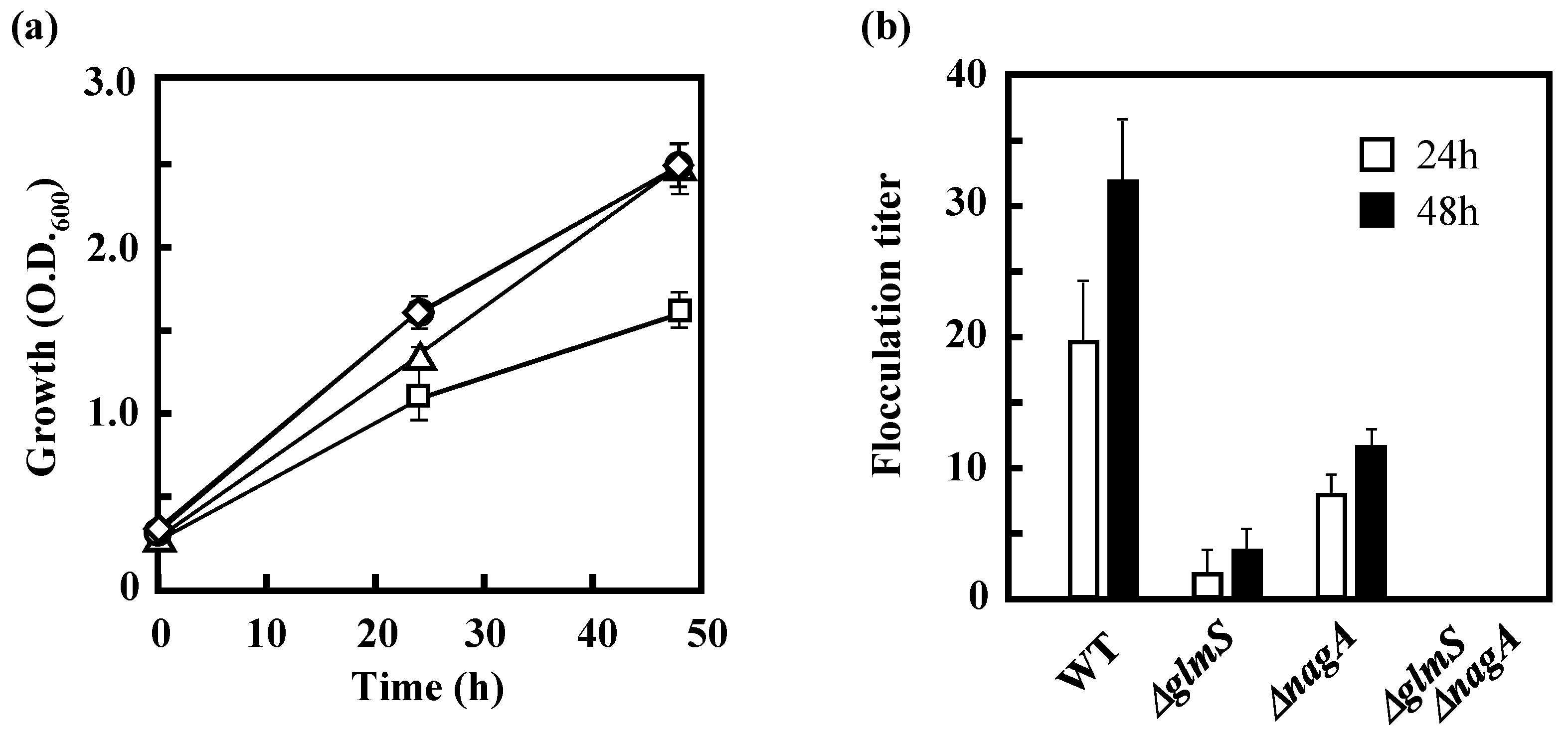
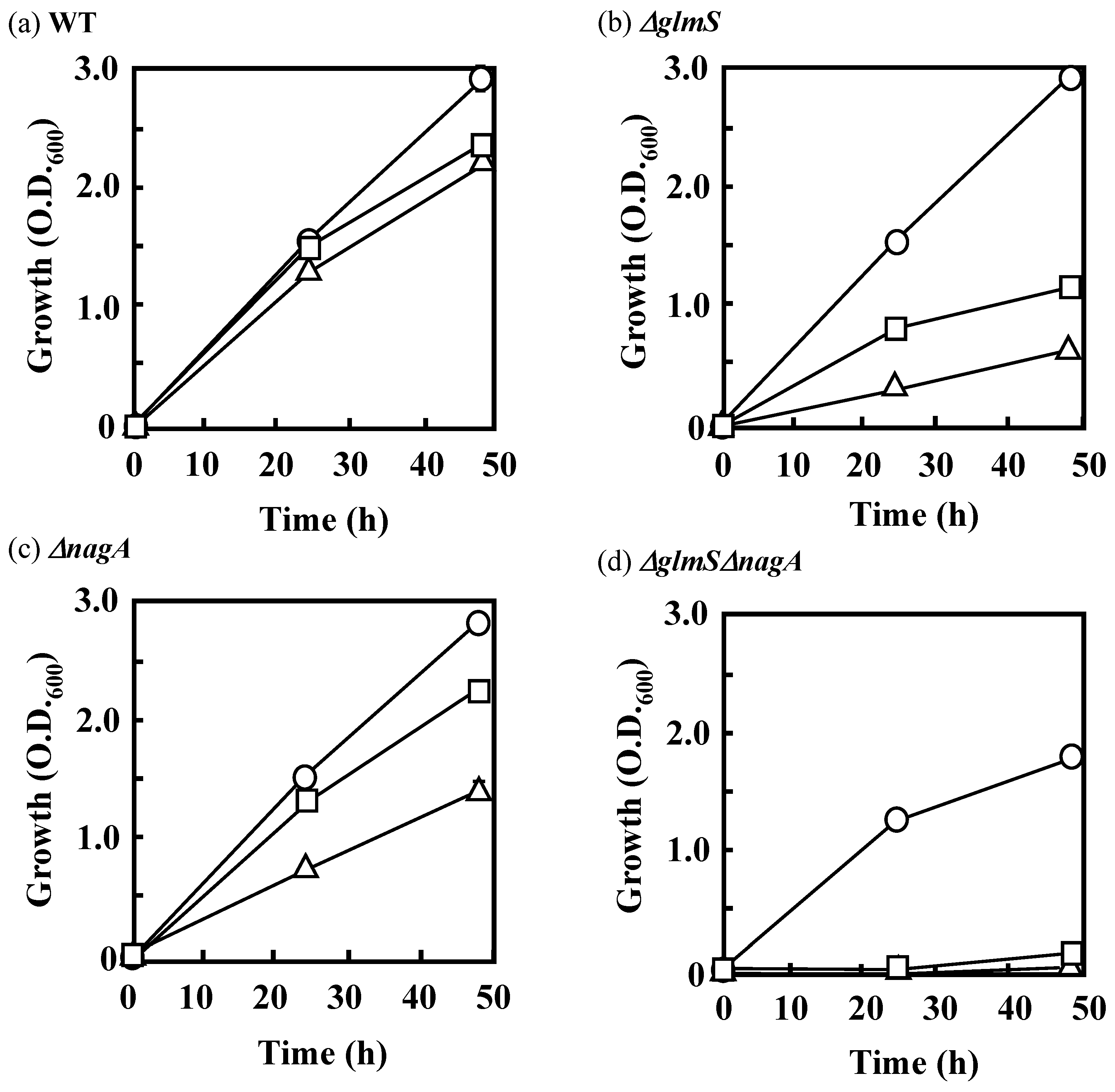
3.3 Effects of the Disruption of GlmS and/or NagA on the Growth of IFO 13545 on Acetate and on Its Flocculation Activity
3.4. Identification of Genes that Are Involved in the Polymerization and Secretion of the Chitin/Chitosan-Like BF
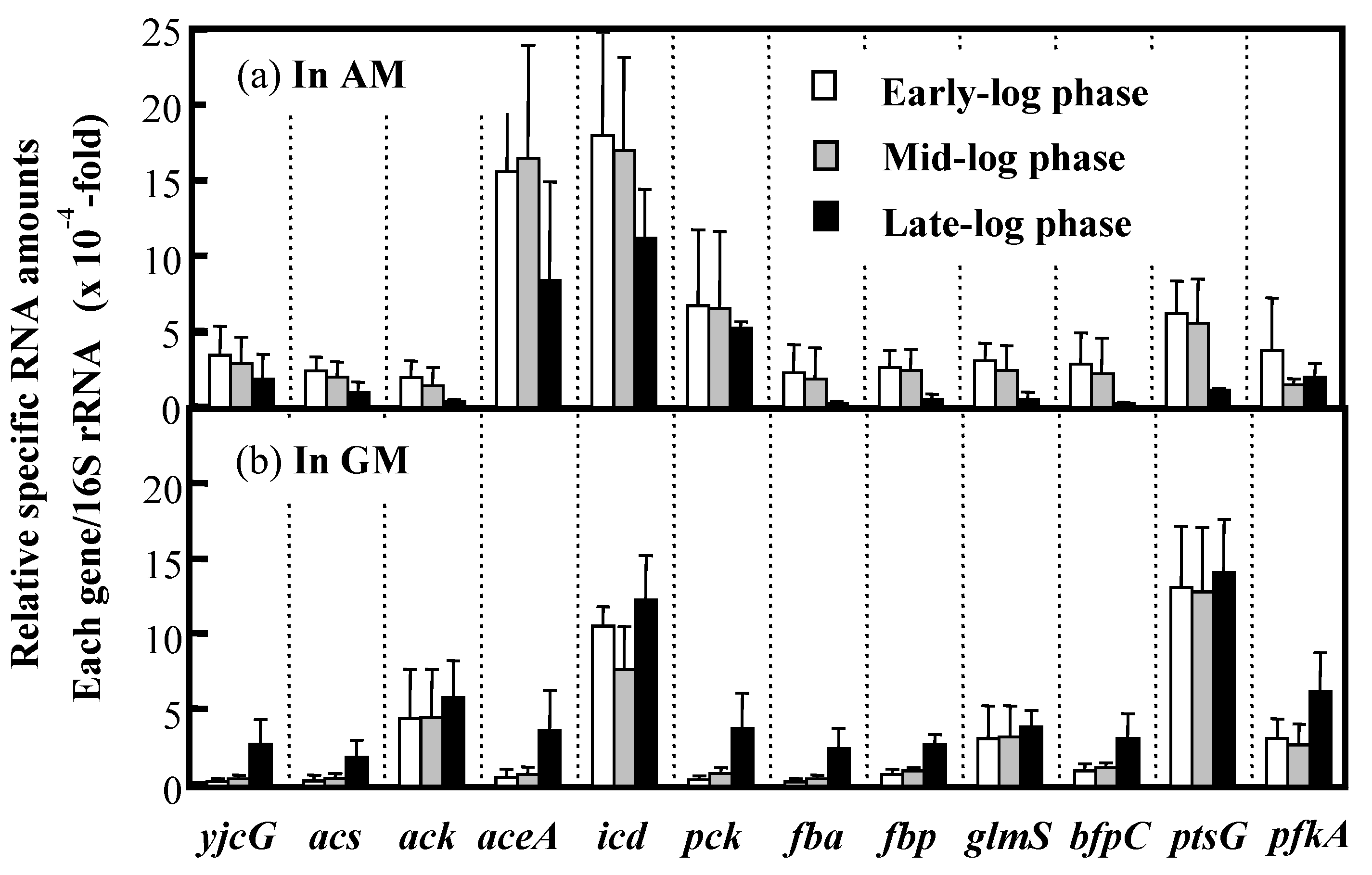
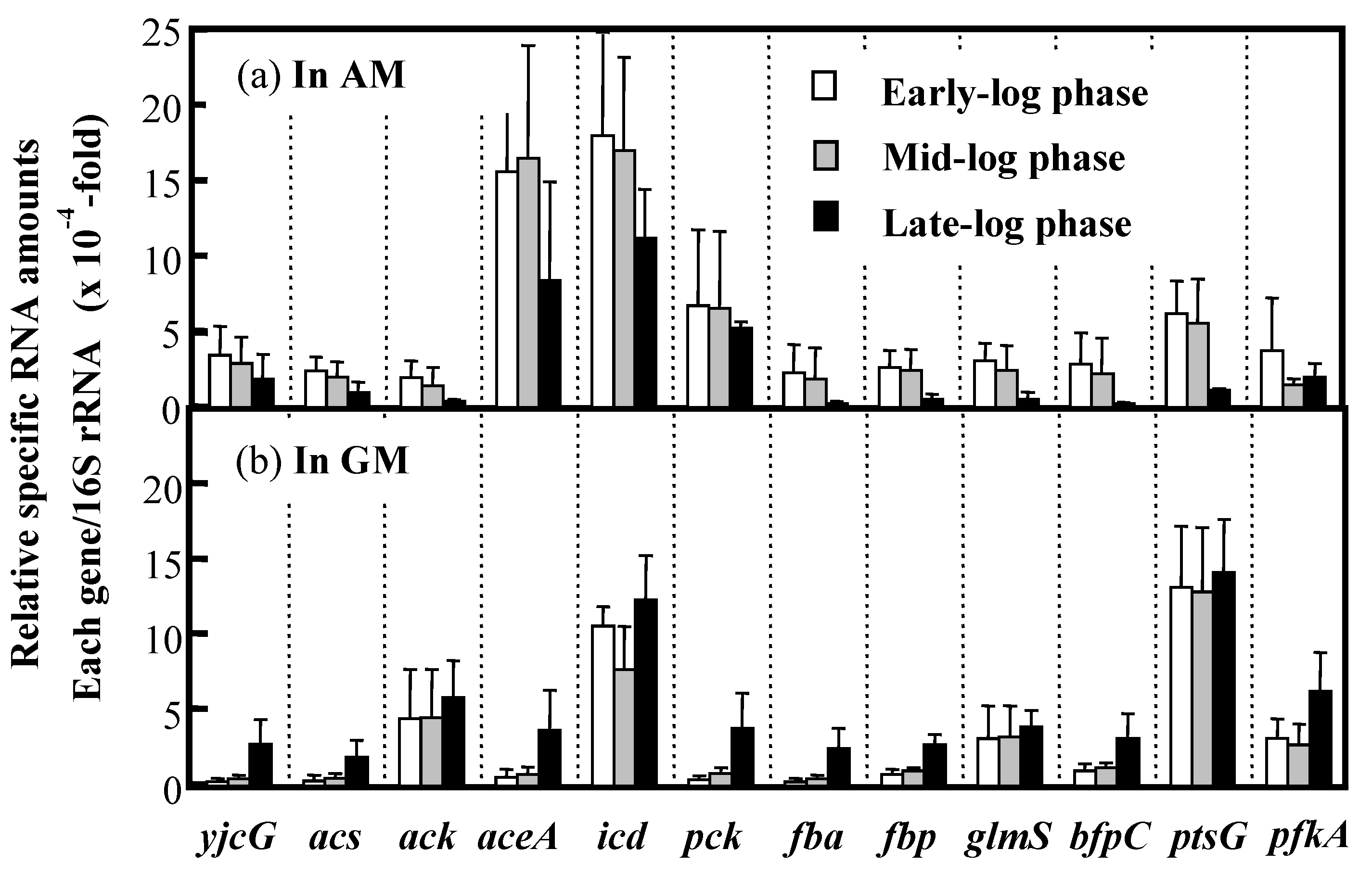
3.5. Expression of Key Genes Involved in Acetate Assimilation and BF Production in IFO 13545
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, Q.; Dunn, E.; Grandmaison, E.W.; Goosen, M.F.A. Applications and properties of chitosan. J. Bioact. Compat. Polym. 1992, 7, 370–397. [Google Scholar] [CrossRef]
- Struszczyk, M.H. Chitin and chitosan, Part I properties and production. Polimery 2002, 47, 316–325. [Google Scholar]
- Hamed, I.; Özogul, F.; Regenstein, J.M. Industrial application of crustacean by-products (chitin, chitosan, and chitooligosaccharides): A review. Trends Food Sci. Technol. 2016, 48, 40–50. [Google Scholar] [CrossRef]
- Struszczyk, M.H. Chitin and chitosan, Part III some aspects of biodegradation and bioactivity. Polimery 2002, 47, 619–629. [Google Scholar]
- Prashanth, K.V.H.; Tharanathan, R.N. Chitin/chitosan: Modifications and their unlimited application potential-an overview. Trends Food Sci. Technol. 2007, 18, 117–131. [Google Scholar] [CrossRef]
- Struszczyk, M.H. Chitin and chitosan, Part II applications of chitosan. Polimery 2002, 47, 396–403. [Google Scholar]
- Knezevic-Jugovic, Z.; Petronijevic, Z.; Smelcerovic, A. Chitin, Chitosan, Oligosaccharides and Their Derivatives: Biological Activities and Applications; Kim, S.-K., Ed.; CRC Press: New York, NY, USA, 2010; Chapter 3; pp. 24–36. ISBN 978-1-4398-1604-2. [Google Scholar]
- Fujita, M.; Ike, M.; Tachibana, S.; Kitada, G.; Kim, S.M.; Inoue, Z. Characterization of a bioflocculant produced by Citrobacter sp. TKF04 from acetic and propionic acids. J. Biosci. Bioeng. 2000, 89, 40–46. [Google Scholar] [CrossRef]
- Son, M.K.; Shin, H.D.; Huh, T.L.; Jang, J.H.; Lee, Y.H. Novel cationic microbial polyglucosamine biopolymer from new Enterobacter sp. BL-2 and its bioflocculation efficacy. J. Microbiol. Biotechnol. 2005, 15, 626–632. [Google Scholar]
- Kim, L.S.; Hong, S.J.; Son, M.K.; Lee, Y.H. Polymeric and compositional properties of novel extracellular microbial polyglucosamine biopolymer from new strain of Citrobacter sp. BL-4. Biotechnol. Lett. 2006, 28, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Inoue, T.; Kato, D.; Negoro, S.; Ike, M.; Takeo, M. Distribution of chitin/chitosan-like bioflocculant-producing potential in the genus Citrobacter. Appl. Microbiol. Biotechnol. 2013, 97, 9569–9577. [Google Scholar] [CrossRef] [PubMed]
- Cozzone, A.J. Regulation of acetate metabolism by protein phosphorylation in enteric bacteria. Annu. Rev. Microbiol. 1998, 52, 127–164. [Google Scholar] [CrossRef] [PubMed]
- Barreteau, H.; Kovac, A.; Boniface, A.; Sova, M.; Gobec, S.; Blanot, D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 168–207. [Google Scholar] [CrossRef] [PubMed]
- Atlas, R.M. Handbook of Microbiological Media, 4th ed.; CRC Press: New York, NY, USA, 2010; pp. 807, 934. ISBN 978-1-4398-0406-3. [Google Scholar]
- Yamamoto, S.; Izumiya, H.; Morita, M.; Arakawa, E.; Watanabe, H. Application of lambda Red recombination system to Vibrio cholerae genetics: Simple methods for inactivation and modification of chromosomal genes. Gene 2009, 438, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.C. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 1998, 180, 2063–2071. [Google Scholar] [PubMed]
- DIG Application Manual for Filter Hybridization, in ROCHE Homepage. Available online: http://lifescience.roche.com/wcsstore/RASCatalogAssetStore/Articles/05353149001_08.08.pdf (accessed on 6 December 2017).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Beatty, C.M.; Browning, D.F.; Busby, S.J.; Simel, E.J.; Hovel-Miner, G.; Wolfe, A.J. Regulation of acetyl coenzyme A synthetase in Escherichia coli. J. Bacteriol. 2000, 182, 4173–4179. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Uehara, T. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol. Mol. Biol. Rev. 2008, 72, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Plumbridge, J. An alternative route for recycling of N-acetylglucosamine from peptidoglycan involves the N-acetylglucosamine phosphotransferase system in Escherichia coli. J. Bacteriol. 2009, 191, 5641–5647. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Rice, J.D.; Goller, C.; Pannuri, A.; Taylor, J.; Meisner, J.; Beveridge, T.J.; Preston, J.F., 3rd; Romeo, T. Roles of pgaABCD genes in synthesis, modification, and export of the Escherichia coli biofilm adhesin poly-β-1,6-N-acetyl-d-glucosamine. J. Bacteriol. 2008, 190, 3670–3680. [Google Scholar] [CrossRef] [PubMed]
- Reeves, P.R.; Cunneen, M.M. Biosynthesis of O-antigen chains and assembly. In Microbial Glycobiology; Moran, A.P., Holst, O., Brennan, P.J., von Itzstein, M., Eds.; Academic Press: Amsterdam, The Netherland, 2009; Chapter 18; pp. 319–335. ISBN 978-0-1237-4546-0. [Google Scholar]
- Reid, A.N.; Szymanski, C.M. Biosynthesis and assembly of capsular polysaccharides. In Microbial Glycobiology; Moran, A.P., Holst, O., Brennan, P.J., von Itzstein, M., Eds.; Academic Press: Amsterdam, The Netherland, 2009; Chapter 20; pp. 351–373. ISBN 978-0-1237-4546-0. [Google Scholar]
- Izano, E.A.; Sadovskaya, I.; Vinogradov, E.; Mulks, M.H.; Velliyagounder, K.; Ragunath, C.; Kher, W.B.; Ramasubbu, N.; Jabbouri, S.; Perry, M.B.; et al. Poly-N-acetylglucosamine mediates biofilm formation and antibiotic resistance in Actinobacillus pleuropneumoniae. Microb. Pathog. 2007, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Wang, X.; Hinnebusch, B.J.; Preston, J.F., 3rd; Romeo, T. Depolymerization of β-1,6-N-acetyl-d-glucosamine disrupts the integrity of diverse bacterial biofilms. J. Bacteriol. 2005, 187, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.A.; Davies, G.J.; Bulone, V.; Henrissat, B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 1997, 326, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Gerke, C.; Kraft, A.; Sussmuth, R.; Schweitzer, O.; Gotz, F. Characterization of the N-acetylglucosaminyltransferase activity involved in the biosynthesis of the Staphylococcus epidermidis polysaccharide intercellular adhesin. J. Biol. Chem. 1998, 273, 18586–18593. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, G.N.; Altman, E.; Sangurdekar, D.P.; Khodursky, A.B.; Eiteman, M.A. Overflow metabolism in Escherichia coli during steady-state growth: Transcriptional regulation and effect of the redox ratio. Appl. Environ. Microbiol. 2006, 72, 3653–3661. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.D.K.; Jones-Mortimer, M.C.; Kornbero, H.L. The enzyme interconversion of acetate and acetyl-coenzyme A in Escherichia coli. J. Gen. Microbiol. 1977, 102, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Luli, G.W.; Strohl, W.R. Comparison of growth, acetate production, and acetate inhibition of Escherichia coli strains in batch and fed-batch fermentations. Appl. Eniviron. Microbiol. 1990, 56, 1004–1011. [Google Scholar]
- Oh, M.K.; Rohlin, L.; Kao, K.C.; Liao, J.C. Global expression profiling of acetate-grown Escherichia coli. J. Biol. Chem. 2002, 277, 13175–13183. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Lee, S.E.; Park, B.S.; Son, M.K.; Jung, Y.M.; Yang, S.O.; Choi, H.K.; Hur, S.H.; Yum, J.H. Proteomic analysis on acetate metabolism in Citrobacter sp. BL-4. Int. J. Sci. 2012, 8, 66–78. [Google Scholar] [CrossRef]
- Son, M.K.; Hong, S.J.; Lee, Y.H. Acetate-mediated pH-stat fed-batch cultivation of transconjugant Enterobacter sp. BL-2S over-expressing glmS gene for excretive production of microbial polyglucosamine PGB-1. J. Ind. Microbiol. Biotechnol. 2007, 34, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.D.; Severson, D.K.; Grund, A.D.; Wassink, S.L.; Burlingame, R.P.; Berry, A.; Running, J.A.; Kunesh, C.A.; Song, L.; Jerrell, T.A.; et al. Metabolic engineering of Escherichia coli for industrial production of glucosamine and N-acetylglucosamine. Metab. Eng. 2005, 7, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.D.; Grund, A.D.; Wassink, S.L.; Peng, S.S.; Nielsen, K.L.; Huckins, B.D.; Burlingame, R.P. Directed evolution and characterization of Escherichia coli glucosamine synthase. Biochimie 2006, 88, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, G.; Andrianopoulos, K.; Hobbs, M.; Reeves, P.R. Organization of the Escherichia coli K-12 gene cluster responsible for production of the extracellular polysaccharide colanic acid. J. Bacteriol. 1996, 178, 4885–4893. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Preston, J.F., 3rd; Romeo, T. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J. Bacteriol. 2004, 186, 2724–2734. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Gene | Enzyme | Strain with Homologous Gene | Homologous Gene Accession No. | Identity at nt Sequence Level (%) | Identity at aa Sequence Level (%) |
|---|---|---|---|---|---|---|
| AB823554 | aceA | isocitrate lyase | E. coli MG1655 | U00096 | 86 | 96 |
| AB823555 | aceB | malate synthase | E. coli MG1655 | U00096 | 84 | 92 |
| AB823558 | acn | aconitate hydratase I | E. coli MG1655 | U00096 | 87 | 96 |
| AB823559 | acs | acetyl-CoA synthetase | E. coli MG1655 | U00096 | 85 | 94 |
| LC020545 | ack | acetate kinase | E. coli MG1655 | U00096 | 91 | 96 |
| AB823580 | bfpA | PGA export porin | C. werkmanii | KF057877 | 91 | 98 |
| AB823581 | bfpB | PGA N-deacetylase | C. freundii CFNIH1 | CP007557 | 90 | 97 |
| AB823582 | bfpC | PGA synthase | C. werkmanii strain BF-6 | KF057878 | 94 | 99 |
| AB823583 | bfpD | PGA biosynthesis protein | C. freundii CFNIH1 | CP007557 | 88 | 95 |
| LC020058 | cpsA | undecaprenyl-phosphate glucose phosphotransferase | C. freundii MTCC1658 | CP007557 | 86 | 100 |
| AB823561 | eno | enolase | E. coli MG1655 | U00096 | 93 | 97 |
| AB823562 | fba | fructose bisphosphate aldolase | E. coli MG1655 | U00096 | 84 | 97 |
| LC027370 | fbp | fructose 1,6-bisphosphatase | E. coli MG1655 | U00096 | 96 | 97 |
| AB823563 | fumC | fumarate hydratase | Salmonella enterica | CP007530 | 81 | 93 |
| AB823564 | g6pd | glucose 6-phosphate dehydrogenase | E. coli MG1655 | U00096 | 86 | 97 |
| AB823565 | gap | glyceraldehyde 3-phosphate dehydrogenase | E. albertii KF-1 | CP007025 | 79 | 91 |
| AB823566 | glk | glucokinase | E. coli MG1655 | U00096 | 80 | 93 |
| AB823568 | glmM | phosphoglucosamine mutase | E. coli MG1655 | U00096 | 85 | 96 |
| AB823569 | glmS | glucosamine-6-phosphate synthase | E. coli MG1655 | U00096 | 87 | 95 |
| AB823570 | glmU | uridyltransferase/glucosamine-1-phosphate acetyltransferase | E. coli MG1655 | U00096 | 83 | 91 |
| AB823571 | gltA | type II citrate synthase | E. coli MG1655 | U00096 | 86 | 96 |
| AB823572 | gpmA | phosphoglyceromutase | E. coli MG1655 | U00096 | 88 | 96 |
| AB823573 | icd | isocitrate dehydrogenase | E. coli MG1655 | U00096 | 88 | 97 |
| AB823574 | mdh | malate dehydrogenase | E. coli MG1655 | U00096 | 87 | 97 |
| LC363529 | nagA | N-acetylglucosamine 6-phosphate deacetylase | E. coli MG1655 | U00096 | 84 | 91 |
| AB823577 | pck | phosphoenolpyruvate carboxykinase | E. coli MG1655 | U00096 | 86 | 93 |
| AB823579 | pfkA | phosphofructokinase I | E. coli MG1655 | U00096 | 87 | 95 |
| AB823585 | pgk | phosphoglycerate kinase | E. coli MG1655 | CP007025 | 79 | 90 |
| LC020430 | pta | phosphotransacetylase | C. freundii MTCC1658 | EKS56947 | 98 | 100 |
| AB823586 | ptsG | PTS system glucose-specific transporter subunits IIBC | E. coli MG1655 | U00096 | 89 | 97 |
| AB823587 | pyk | pyruvate kinase | E. albertii KF1 | CP007025 | 85 | 95 |
| AB823588 | sdhB | succinate dehydrogenase iron-sulfur subunit | E. coli MG1655 | U00096 | 87 | 96 |
| AB823589 | sucA | 2-oxoglutarate dehydrogenase E1 component | E. coli MG1655 | U00096 | 89 | 94 |
| AB823590 | sucB | dihydrolipoamide succinyltransferase | E. coli MG1655 | U00096 | 87 | 94 |
| AB823591 | sucD | succinyl-CoA synthetase subunit alpha | E. coli MG1655 | U00096 | 88 | 95 |
| AB823592 | tpiA | triosephosphate isomerase | E. coli MG1655 | U00096 | 90 | 95 |
| LC018665 | yjcG | acetate permease | E. coli strain ST2747 | CP007394 | 86 | 96 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeo, M.; Kimura, K.; Mayilraj, S.; Inoue, T.; Tada, S.; Miyamoto, K.; Kashiwa, M.; Ikemoto, K.; Baranwal, P.; Kato, D.; et al. Biosynthetic Pathway and Genes of Chitin/Chitosan-Like Bioflocculant in the Genus Citrobacter. Polymers 2018, 10, 237. https://doi.org/10.3390/polym10030237
Takeo M, Kimura K, Mayilraj S, Inoue T, Tada S, Miyamoto K, Kashiwa M, Ikemoto K, Baranwal P, Kato D, et al. Biosynthetic Pathway and Genes of Chitin/Chitosan-Like Bioflocculant in the Genus Citrobacter. Polymers. 2018; 10(3):237. https://doi.org/10.3390/polym10030237
Chicago/Turabian StyleTakeo, Masahiro, Kazuyuki Kimura, Shanmugam Mayilraj, Takuya Inoue, Shohei Tada, Kouki Miyamoto, Masami Kashiwa, Keishi Ikemoto, Priyanka Baranwal, Daiichiro Kato, and et al. 2018. "Biosynthetic Pathway and Genes of Chitin/Chitosan-Like Bioflocculant in the Genus Citrobacter" Polymers 10, no. 3: 237. https://doi.org/10.3390/polym10030237